
Synthesis and catalytic application of a donor-free bismuthenium cation†
Nilanjana
Sen
a,
Pallavi
Sarkar
b,
Yadram
Meena
a,
Srinu
Tothadi
 c,
Swapan K.
Pati
*b and
Shabana
Khan
*a
c,
Swapan K.
Pati
*b and
Shabana
Khan
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research Pune, Dr Homi Bhabha Road, Pashan, Pune-411008, India. E-mail: shabana@iiserpune.ac.in
bTheoretical Sciences Unit, School of Advanced Materials (SAMat), Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore-560064, India. E-mail: swapan.jnc@gmail.com
cCSIR-Central Salt and Marine Chemicals Research (AcSIR), Ghaziabad-201002, UP, India
First published on 7th June 2024
Abstract
Herein, we report the synthesis and catalytic application of a new N,N′-dineopentyl-1,2-phenylenediamine-based bismuthenium cation (3). 3 has been synthesized via the treatment of chlorobismuthane LBiCl [L = 1,2-C6H4{N(CH2tBu)}2] (2) with AgSbF6, and was further used as a robust catalyst for the cyanosilylation of ketones under mild reaction conditions. Experimental studies and DFT calculations were performed to understand the mechanistic pathway.
The unique role of precious transition metals in homogeneous catalysis has been well-known for years. However, there is a pressing demand for sustainable alternatives for precious transition metals because of their toxicity, cost, and limited abundance. Recent years have witnessed a surge in surrogating transition metals by more earth-abundant and cost-effective main group elements.1,2 One of the desirable classes of the p-block catalysts is Lewis-acidic cationic compounds of the pnictogen family, known for their versatile electrophilic nature and ease of synthesis.3 Among the group 15 elements, bismuth has gained significant popularity in recent years due to its capability to access redox cycles.4 Given our interest in this area,5,6 we were interested in synthesizing Bi(III) cations and utilizing them in catalysis. Although there are a few low-coordinate bismuthenium ions available in the literature,7 no catalysis is explored with them. Recently, we have used stibenium cations as an efficient cyanosilylation catalyst, which is a well-established organic transformation of carbonyl moieties to the cyanohydrin product.6 Several main group compounds have been utilized as catalysts in the cyanosilylation of carbonyls,1a but there are only three reports on compounds with group 15 elements.6,8,9
Surprisingly, only one Bi-based compound (Bi-siloxane) has been utilized for the cyanosilylation reaction to date, which has minimal reactivity as a catalyst.9 Hence, the potential of bismuthenium cations remains unexplored as the catalysts for the cyanosilylation of carbonyls. In this work, we have mainly focused on synthesizing a donor-free mono-cationic bismuthenium compound with an N,N′-dineopentyl-1,2-phenylenediamine ligand (3) and employed it in the cyanosilylation of ketones for the first time. Compound 3 shows excellent catalytic efficiency with a high TON, superseding the analogous stibenium cation. Our results are discussed below.
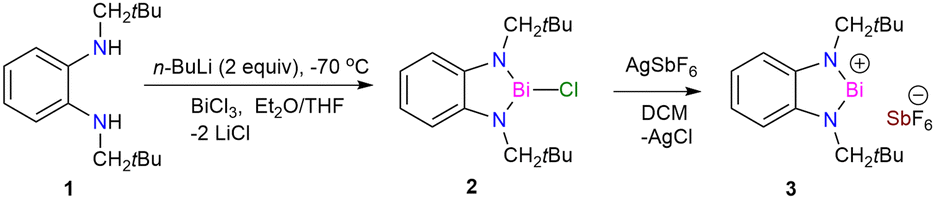
The lithiated salt of N,N′-dineopentyl-1,2-phenylenediamine (1) was treated with one equiv. of BiCl3 in an Et2O/THF mixture at −70 °C, which afforded the desired compound 2 (Scheme 1). The formation of compound 2 was confirmed by NMR spectroscopic studies and single crystal X-ray diffraction studies. 2 crystallizes in the monoclinic space group P1 (see ESI† Fig. S1a). To generate the target bismuthenium cation (3), we treated compound 2 with 1 equiv. of AgSbF6 in DCM, which afforded 3 as a reddish-purple colored solid in ∼66% yield. Routine analytical techniques and single crystal X-ray diffraction studies are used to establish the chemical composition of 3. The 19F NMR spectrum exhibits a peak at −169.42 ppm, corresponding to the SbF6 anionic moiety. 3 crystallizes in the monoclinic space group C2/c. The molecular structure of 3 (Fig. 1) unveils the presence of a Bi(III) cationic center along with a discrete SbF6 anionic counterpart. The Bi–N bond distances in 3 are found to be 2.117(2) and 2.163(2) Å, which are comparable to those reported for the bis[N,N′,N′-tris(trimethylsilyl)hydrazino]bismuthenium cation (2.150(2), 2.115(2) Å) by Schulz and coworkers,7d but are longer than that of [Bi(NON)Ar][AlCl4] (2.113(12), 2.111(12) Å) by Coles and coworkers.7c The N–Bi–N bond angle of 3 is observed to be 77.79(8)°, which is found to be much smaller than those reported for bismuthenium cations mentioned above (100.28(5)° and 111.9(1)°, respectively).7c,7d The molecular structure of 3 reveals that the bismuth center displays an intermolecular π-arene interaction in η3 fashion, leading to π-stacked dimers that are antiparallel to each other (see ESI,† Fig. S1b). The cationic Bi(III) center in 3 also displays two long contacts with the fluorine atoms of SbF6 anions. The Bi⋯F interactions in 3 display bond distances of 2.848 and 2.973 Å, which are similar to those reported for the carbodicarbene stabilized bismuthenium ion (2.904(2) Å) by Gilliard and coworkers.10 We have also recorded the UV-Vis absorption spectra for the compounds 2 and 3 (see ESI,† Fig. S7) and corroborated it with the TD-DFT calculation [B3LYP-D3/lanl2dz(Bi), 6-311+G(d,p) for lighter atoms]. The theoretical calculation reveals that the absorption peaks around ∼500 nm for both of them are mainly due to the π-electron charge transfer from the aromatic ring to the vacant p-orbital of the bismuth atom (see ESI,† Fig. S8, Table S4) (Fig. 2).
 | ||
| Scheme 1 Syntheses of complexes 2–3. | ||
 | ||
| Fig. 1 The molecular structure of 3 with anisotropic displacement parameters is depicted at the 50% probability level. Hydrogen atoms are not shown for clarity. Selected bond lengths (Å) for 3: Bi1–N1 2.117(2), Bi1–N2 2.163(2), Bi1–F6 6.066(2). Selected bond angles (°) for 3: N1–Bi1–N2 77.79(6), Bi–N1–C1 114.69, Bi1–N2–C2 112.91. | ||
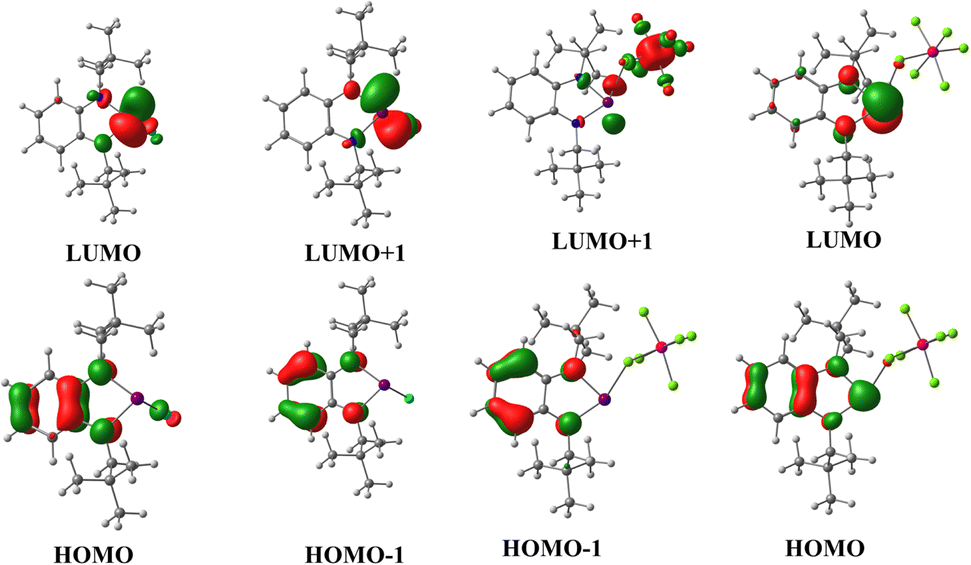 | ||
| Fig. 2 Frontier molecular orbitals of 2 and 3 [B3LYP-D3/lanl2dz(Bi), 6-311+G(d,p) for lighter atoms]. | ||
Furthermore to estimate the Lewis acidity of 2 and 3, we followed the Gutmann–Beckett method11 and recorded the 31P NMR spectra of 2 and 3 in the presence of OPEt3. For compound 2, the 31P NMR spectrum shows a peak at δ 52.25 ppm; while in the case of compound 3, it is observed at δ 59.65 ppm (Fig. 3). It is evident from the chemical shifts of the 31P NMR spectra that 3 is more Lewis acidic than 2. With this information in hand, we explored the catalytic application of 3 for the cyanosilylation of carbonyl compounds (Scheme 2), which has been a lucrative catalytic reaction for the main group catalysts.12 The optimization of the reaction conditions was performed using acetophenone as the model substrate in the presence of 3 as a catalyst. We carried out a brief screening of the catalyst loading, temperature, and time for cyanosilylation of acetophenone using 1.2 equivalent TMSCN to get the best conversion of the product. A 0.1 mol% of the catalyst loading in 15 min led to product formation in 99% yield (entry 3, Table S3, ESI†), which is the most suitable reaction condition for further substrate scope. With these optimization conditions, we began to explore the catalytic efficiency of 3 on various substrates of ketones (Chart 1).
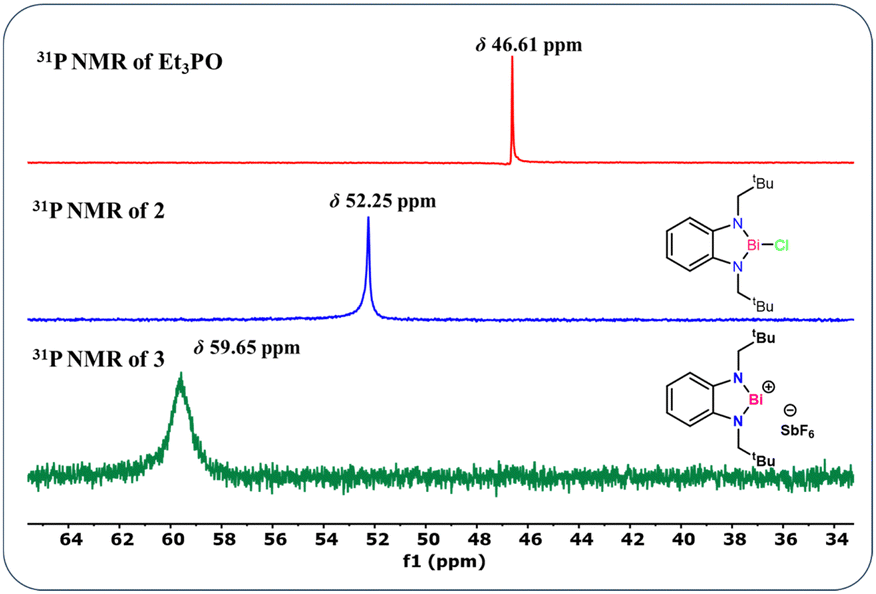 | ||
| Fig. 3 Stacked 31P NMR spectra of 2–3 in the presence of Et3PO in CDCl3 (Gutmann–Beckett method). | ||
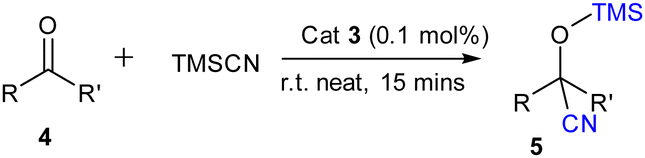 | ||
| Scheme 2 Cyanosilylation of ketones. | ||
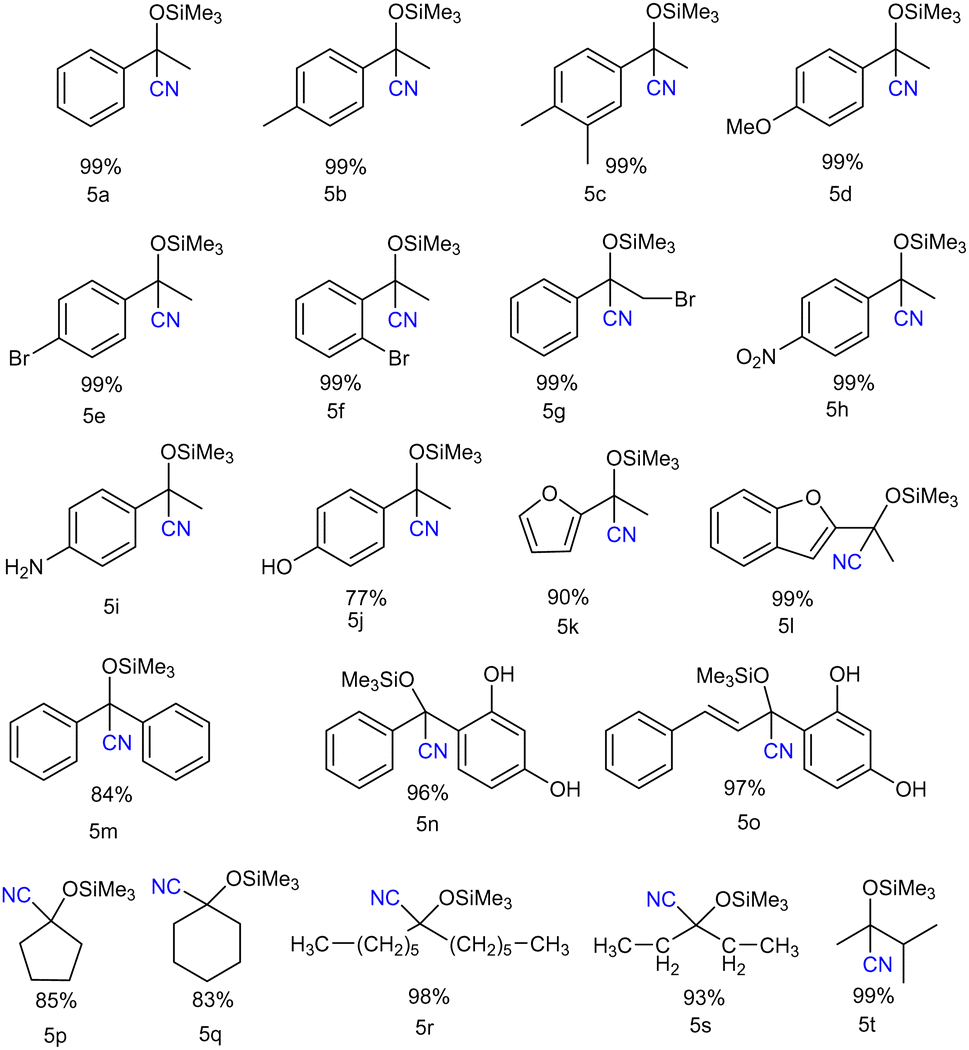 | ||
| Chart 1 Substrate scope for cyanosilylation of ketones using catalyst 3. Reaction conditions: ketones (0.25 mmol), TMSCN (0.38 mmol, 1.5 equiv.), neat reaction at room temperature. The catalyst loading is 0.1 mol%. Reaction time: 15 min. Yield (%) determined by 1H NMR spectroscopy using mesitylene as an internal standard. | ||
We started with the derivatives of acetophenone. Both electron-donating and electron-withdrawing group-substituted acetophenone derivatives underwent cyanosilylation to form the corresponding cyanohydrin product in moderate to good yields (Chart 1). Along with the aromatic ketones, the aliphatic ketones also showed good product conversion under the same reaction conditions. Chart 1 delineates the scope of ketones we have studied in this work. The commonly available aromatic ketones with electron-donating groups like 4-methylacetophenone (5b), and 3,4-dimethylacetophenone (5c), as well as with electron-withdrawing groups like bromoacetophenone (5e–5f) gave excellent product conversion. We employed 2-acetylfuran (5k) and 2-acetylbenzofuran (5l), and in both cases, we observed an excellent product conversion. Since the cyanosilylation of aliphatic ketones is more challenging, we focused on the aliphatic ketones after the substrate scope study of the aromatic ketones. It is important to note that there are only a few reports on the cyanosilylation of aliphatic ketones with the main group catalysts.13 Interestingly, all the aliphatic ketones e.g. dihexyl ketone (5r), diethyl ketone (5s), and 3-methylbutanone (5t) along with the cyclic ketones (cyclopentanone (5p) and cyclohexanone (5q)) afforded their corresponding cyanohydrin products in good to excellent yield. The cyanosilylation of benzophenone (5m) and benzoresorcinol (5n) also gave a good product yield percentage. The TON and TOF for this reaction (TON = 990; TOF = 66 min−1) were found to be much higher than those reported for the group-15 compounds, stibenium cation (TON = 99; TOF = 3.3 min−1)6 and bismuth-siloxane (TON = 82.5; TOF = 5.5 min−1),9 reflecting the better catalytic performance of 3.
We have also investigated the kinetic study of our cyanosilylation reaction for which we have calculated the initial rates kobs of the reaction from the product versus time plot (see ESI,† Fig. S51a and S52a) followed by the plot of log10(kobs) versus log10x (x = varying conc. of acetophenone and TMSCN) from which we determined the order of the reaction. For acetophenone, the order of the reaction was found to be 0.241 (see ESI,† Fig. S51b), which indicates the fractional order of the reaction.13 In the case of TMSCN, the order is found to be 0.564 (see ESI,† Fig. S52b), representing that TMSCN plays a significant role in the cyanosilylation of ketones in the presence of the catalyst.14 We have also performed the experiments to get the Arrhenius plot [ln(k) vs. 1/T (T = temperature)] and calculated the activation energy from the slope of the plot (Fig. S52c and d, ESI†). The activation energy is found to be 26.54 kcal mol−1, significantly lower than that of the theoretical results (31.3 kcal mol−1) but in line with the experimental conditions.12e This energy difference is presumably due to the approximation used in DFT calculation (see ESI† for the details).
We further performed the solution-state NMR studies to understand the mechanistic pathway of the cyanosilylation reaction catalyzed by 3. The 1H, 13C, and 29Si NMR spectra of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric reaction of the catalyst and TMSCN were recorded to study the changes. The 29Si NMR spectrum of the reaction mixture exhibits a new peak at δ 7.33 ppm, which is downfield shifted as compared to the pure TMSCN (δ −11.31 ppm) (Fig. S57, ESI†). The 1H NMR spectrum also shows a new peak at δ 0.05 ppm along with some residual TMSCN (δ 0.21 ppm), confirming the formation of an intermediate species. In the 13C NMR spectrum, we observed the appearance of a new peak at δ 2.00 ppm (–CH3), which differs from that of the free TMSCN (δ −2.26 ppm) (see Fig. S53–S55, ESI†). The IR spectrum of the reaction mixture showed a new peak at ν = 1603 cm−1 (Fig. S56, ESI†), which confirms the lowering in the stretching frequency of the CN bond of TMSCN upon coordination with the cationic Bi(III) center of 3.
:1 stoichiometric reaction of the catalyst and TMSCN were recorded to study the changes. The 29Si NMR spectrum of the reaction mixture exhibits a new peak at δ 7.33 ppm, which is downfield shifted as compared to the pure TMSCN (δ −11.31 ppm) (Fig. S57, ESI†). The 1H NMR spectrum also shows a new peak at δ 0.05 ppm along with some residual TMSCN (δ 0.21 ppm), confirming the formation of an intermediate species. In the 13C NMR spectrum, we observed the appearance of a new peak at δ 2.00 ppm (–CH3), which differs from that of the free TMSCN (δ −2.26 ppm) (see Fig. S53–S55, ESI†). The IR spectrum of the reaction mixture showed a new peak at ν = 1603 cm−1 (Fig. S56, ESI†), which confirms the lowering in the stretching frequency of the CN bond of TMSCN upon coordination with the cationic Bi(III) center of 3.
Theoretical studies were performed to gain insight into the mechanistic pathway and support our experimental data (M06-2X/lanl2dz(Bi), 6-311+G(d,p) for lighter atoms). A model catalyst 3m, where CH2tBu groups are replaced by the methyl groups, was used for the density functional theory (DFT) calculation (see ESI† for the details). It is proposed that the first step of the mechanistic cycle is the coordination of TMSCN to the bismuth center (Fig. 4). The lone pair of the nitrogen atom of TMSCN is donated to the vacant p-orbital of the cationic bismuth center, resulting in the formation of INT13m (Bi–N distance 2.611 Å, ESI† Fig. S64). Upon addition of the substrate acetophenone, a four-membered transition state TS3m is formed, where the oxygen atom of the substrate coordinates with the silicon center of –SiMe3 of TMSCN, and the carbon atom of the cyanide group attaches to the carbonyl carbon of acetophenone. The interaction between the Bi center and TMSCN is stronger in TS3m than INT13m, as revealed by the lowering of the Bi–N distance (Bi–N distance 2.420 Å). Finally, in the next step, rearrangement takes place to form INT23m, where the –SiMe3 moiety gets transferred to the oxygen atom of the substrate, and the –CN group forms a bond with the carbonyl carbon of the substrate. In INT23m, the anchoring of the catalyst becomes much weaker, as reflected by the Bi–N bond distance of 2.769 Å, which is much longer than those in TS3m and INT13m. The final product dissociates from INT23m with the regeneration of the catalyst (3m).
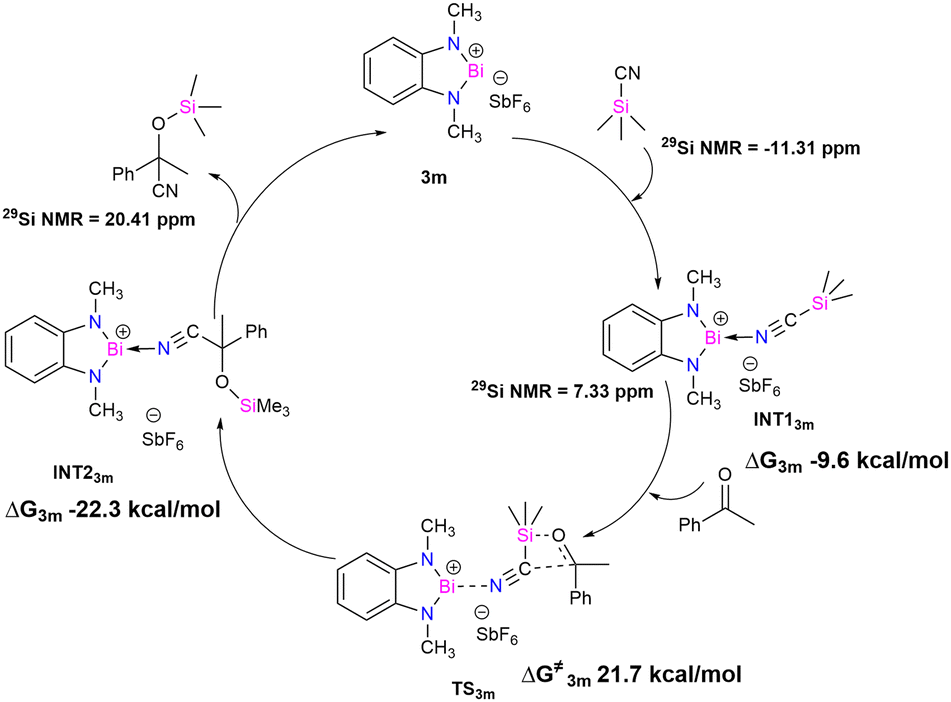 | ||
| Fig. 4 Proposed catalytic cycle for cyanosilylation of acetophenone using model catalyst 3m. | ||
In summary, we have synthesized a donor-free mono-cationic bismuth compound (3) and utilized it as a catalyst for the cyanosilylation of ketones. Compound 3 displays excellent catalytic activity toward the cyanosilylation of ketones with the TON and TOF of 990 and 66 min−1, which is much higher than the previously reported stibenium cation and bismuth-siloxane. These new findings pave the way for exploring p-block cations as sustainable catalysts in several important organic transformations.
SK thanks the SERB-CRG grant (CRG/2021/000395) for the financial support. SK also thanks DST-FIST for the single-crystal X-ray diffractometer. NS thanks DST-INSPIRE for the fellowship. NS thanks Moushakhi Ghosh for the cif of compound 2 and Jabed Hossain for helping with the crystal refinement. PS acknowledges the Council of Scientific and Industrial Research (CSIR), Govt. of India, for the Senior Research Fellowship (SRF). SKP also acknowledges J. C. Bose National Fellowship and DST, Govt. of India, for the financial support. ST thanks AESD, CIF and CSIR-CSMCRI for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) S. Pahar, G. Kundu and S. S. Sen, ACS Omega, 2020, 5, 25477–25484 CrossRef CAS PubMed
; (b) L. Fohlmeister and A. Stasch, Chem. – Eur. J., 2016, 22, 10235–10246 CrossRef CAS PubMed
-
(a) N. Sen and S. Khan, Chem. – Asian J., 2021, 16, 705–719 CrossRef CAS PubMed
-
(a) C. Lichtenberg, Chem. Commun., 2021, 57, 4483 RSC
-
(a) A. D. Obi, D. A. Dickie, W. Tiznado, G. Frenking, S. Pan and R. J. Gilliard, Inorg. Chem., 2022, 61, 19452–19462 CrossRef CAS PubMed
- R. Dasgupta, S. Das, S. Hiwase, S. K. Pati and S. Khan, Organometallics, 2019, 38(7), 1429–1435 CrossRef CAS
- N. Sen, P. Gothe, P. Sarkar, S. Das, S. Tothadi, S. K. Pati and S. Khan, Chem. Commun., 2022, 58, 10380–10383 RSC
-
(a) R. J. Schwamm, B. M. Day, M. P. Coles and C. M. Fitchet, Inorg. Chem., 2014, 53, 3778–3787 CrossRef CAS PubMed
- W.-B. Wu, X.-P. Zeng and J. Zhou, J. Org. Chem., 2020, 85, 14342–14350 CrossRef CAS PubMed
- Y. Li, J. Wang, Y. Wu, H. Zhu, P. P. Samuel and H. W. Roesky, Dalton Trans., 2013, 42, 13715–13722 RSC
- J. E. Walley, L. S. Warring, G. Wang, D. A. Dickie, S. Pan, G. Frenking and R. J. Gilliard, Angew. Chem., Int. Ed., 2021, 60, 6682–6690 CrossRef CAS PubMed
-
(a) M. A. Beckett, G. C. Strickland, J. R. Holland and K. S. Varma, Polymer, 1996, 37, 4629–4631 CrossRef CAS
-
(a) S. Yadav, R. Dixit, K. Vanka and S. S. Sen, Chem. – Eur. J., 2018, 24, 1269–1273 CrossRef CAS PubMed
-
(a) M. K. Sharma, S. Sinhababu, G. Mukherjee, G. Rajaraman and S. Nagendran, Dalton Trans., 2017, 46, 7672–7676 RSC
- J. Hossain, B. K. Shah and S. Khan, ACS Catal., 2023, 13, 13577–13587 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, UV-Vis data, X-ray data, and theoretical details. CCDC 2322619 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01805b |
| This journal is © The Royal Society of Chemistry 2024 |