
ROS-responsive thermosensitive polypeptide hydrogels for localized drug delivery and improved tumor chemoimmunotherapy†
Fujiang
Li
ab,
Junfeng
Ding
ab,
Zhenyu
Li
 ab,
Yan
Rong
*a,
Chaoliang
He
*ab and
Xuesi
Chen
ab
ab,
Yan
Rong
*a,
Chaoliang
He
*ab and
Xuesi
Chen
ab
aCAS Key Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China. E-mail: yrong@ciac.ac.cn; clhe@ciac.ac.cn
bSchool of Applied Chemistry and Engineering, University of Science and Technology of China, Hefei 230026, China
First published on 24th April 2024
Abstract
In this study, we developed a ROS-responsive thermosensitive poly(ethylene glycol)-polypeptide hydrogel loaded with a chemotherapeutic drug, doxorubicin (Dox), an antiviral imidazoquinoline, resiquimod (R848), and antibody targeting programmed cell death protein 1 (aPD-1) for local chemoimmunotherapy. The hydrogel demonstrated controllable degradation and sustained drug release behavior according to the concentration of ROS in vitro. Following intratumoral injection into mice bearing B16F10 melanoma, the Dox/R848/aPD-1 co-loaded hydrogel effectively inhibited tumor growth, prolonged animal survival time and promoted anti-tumor immune responses with low systemic toxicity. In the postoperative model, the Dox/R848/aPD-1 co-loaded hydrogel exhibited enhanced tumor recurrence prevention and long-term immune memory effects. Thus, the hydrogel-based local chemoimmunotherapy system demonstrates potential for effective anti-tumor treatment and suppression of tumor recurrence.
1. Introduction
Cancer ranks among the top causes of death worldwide and its impact continues to grow.1 Chemotherapy, radiotherapy, surgery, and immunotherapy are commonly employed in the treatment of cancer.2,3 However, relying on a single treatment often proves challenging to achieve desired outcomes.4 For example, after surgical resection, tumor recurrence presents a significant concern.5 The application of chemotherapy and radiotherapy often carries potential risks of serious systemic side effects.6,7 Immune checkpoint blockade (ICB) therapy is associated with suboptimal objective response rates (ORRs) and severe immune-related adverse events (irAEs).8 Recently, the combination of chemotherapy and immunotherapy has been considered to be a promising treatment strategy.9,10 Doxorubicin (Dox) is a conventional chemotherapy drug against tumors that can trigger immunogenic cell death (ICD) of tumor cells and elicit the production of tumor-associated antigens.11,12 Resiquimod (R848), a small molecule agonist of TLR 7/8, plays a significant role in activating antigen-presenting cells and stimulating T-lymphocyte proliferation.13,14 The combination of Dox and the immune adjuvant R848 effectively promotes dendritic cells (DCs) antigenic presentation and stimulates T cells activation and proliferation.13,15 They are then treated in combination with immune checkpoint blockers such as programmed cell death protein 1 antibody (aPD-1) to block PD-1/PD-L1 pathways, thereby further enhancing the T cell response and achieving better anti-tumor efficacy.14,16,17Due to the specificity of tumor growth, the anti-tumor drugs cannot accurately reach the site of the tumor through intravenous injections, and drug accumulation can lead to serious systemic side effects.9,18 Therefore, many laboratories are actively conducting research on drug delivery systems to enhance the potency of anticancer medications, improve the efficiency of targeted drug delivery and mitigate the systemic toxicity of drugs.19–23 A hydrogel is a three-dimensional network of polymers that possesses the capacity to retain substantial quantities of water.24,25 These naturally derived or synthetic polymers with biocompatibility can be made into aqueous solutions and easily mixed with drugs in vitro.26,27 After being injected into the body, they can transform into hydrogels which effectively encapsulate the drugs and directly deliver to the tumor site.28 Thus, the hydrogel can be used as a good local delivery system for single or multiple anti-tumor drugs.29,30 Meanwhile, the strategy requires only one injection and can significantly prolong the local residence of the drug with minimally invasive administration.26,31,32
In order to improve drug targeting, stimulation-responsive delivery systems have become an important direction in current anti-tumor research, and they can be stimulated externally (light, magnetic field, or ultrasound) or internally (pH, temperature, enzyme, or redox potential) to achieve rapid degradation and sustained drug release.33–36 Reactive oxygen species (ROS) are widely found in various solid tumors and are one of the unique markers of cancer cells compared to normal cells.37L-Methionine is an important antioxidant existing in organisms.38 It can be oxidized by ROS to methionine sulfide, making it an ideal material for synthesizing polymers with ROS responsiveness.39,40
In this study, we present a ROS-responsive thermosensitive hydrogel based on a methoxy poly(ethylene glycol)-block-poly(L-methionine) copolymer (mPEG-b-PMet) as a drug depot for the co-delivery of Dox, R848, and aPD-1 for in situ tumor chemoimmunotherapy. We synthesized three mPEG-b-PMet copolymers with different degrees of polymerization. The test tube inversion method had confirmed that all three polymers have thermally responsive sol–gel phase transition, and mPEG-b-PMet22 with a suitable polymerization degree was selected for subsequent experiments. In vitro degradation and drug release were conducted to evaluate the ROS responsiveness of synthetic polypeptide hydrogels. To confirm the immunogenic cell death (ICD) effect of Dox-loaded hydrogels, in vitro experiments were conducted to measure calreticulin (CRT) expression and adenosine triphosphate (ATP) release. Meanwhile, the promotion of R848-loaded hydrogels on DC activation and macrophage M1 polarization was studied. In order to confirm the efficacy of anti-tumor activity and suppressing postoperative recurrence of tumors in vivo, we intratumorally administered Dox/R848/aPD-1 co-delivery hydrogels in a B16F10 melanoma treatment model and a postoperative model, respectively. The assessment of the tumor-specific immune response was conducted utilizing enzyme-linked immunosorbent assay (ELISA) and flow cytometry, which encompassed an examination of the infiltration of immune cells within tumors and the subsequent secretion of inflammatory cytokines (Scheme 1).
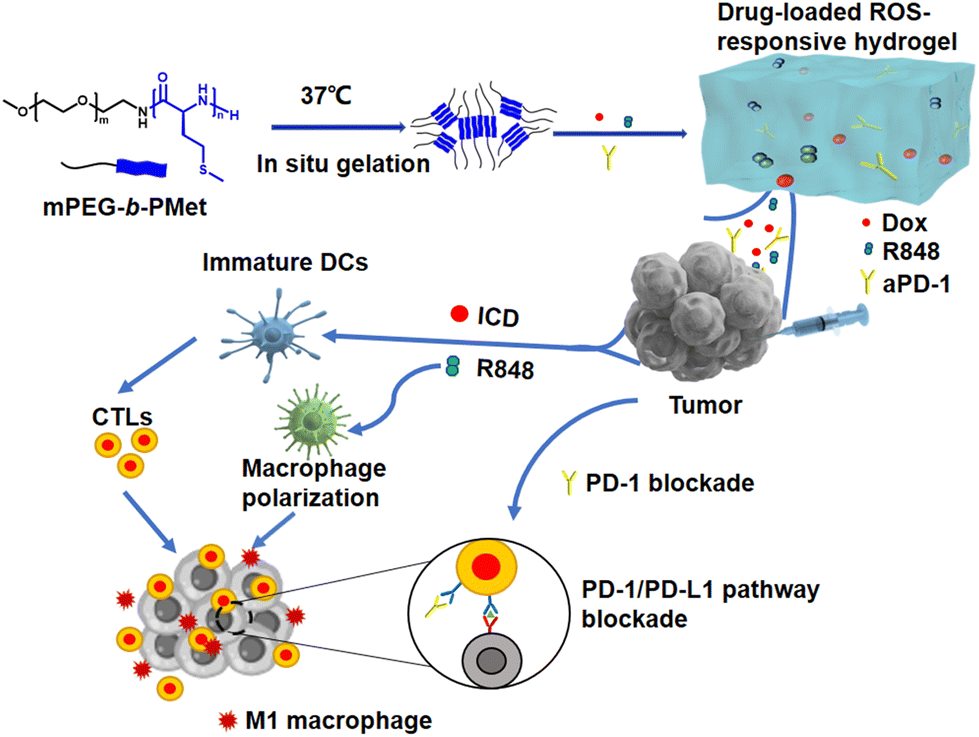 | ||
| Scheme 1 Schematic figure of a drug-loaded hydrogel in situ delivery system for chemoimmunotherapy on tumors. | ||
2. Results and discussion
2.1. Copolymer preparation and hydrogel formation
As described in our previous report,33 the mPEG-b-PMet diblock copolymer was synthesized through the ring-opening polymerization (ROP) of L-methionine N-carboxyanhydride (Met NCA) using mPEG-NH2 as a macromolecular initiator (Scheme S1, ESI†). The confirmation of the synthesis of mPEG-b-PMet was obtained through the 1H NMR spectra and all peaks of the spectrum had a reasonable attribution by analyzing the chemical structure of the copolymer (Fig. 1A). The polymerization degrees of the obtained three copolymers were 11, 22 and 32, respectively, by comparing the integral area of the methylene proton in Met with the integral area of the methylene proton in mPEG (Fig. S1, ESI†). The monomodal distribution of gel permeation chromatography (GPC) also confirmed the successful synthesis (Fig. S2 and Table S1, ESI†).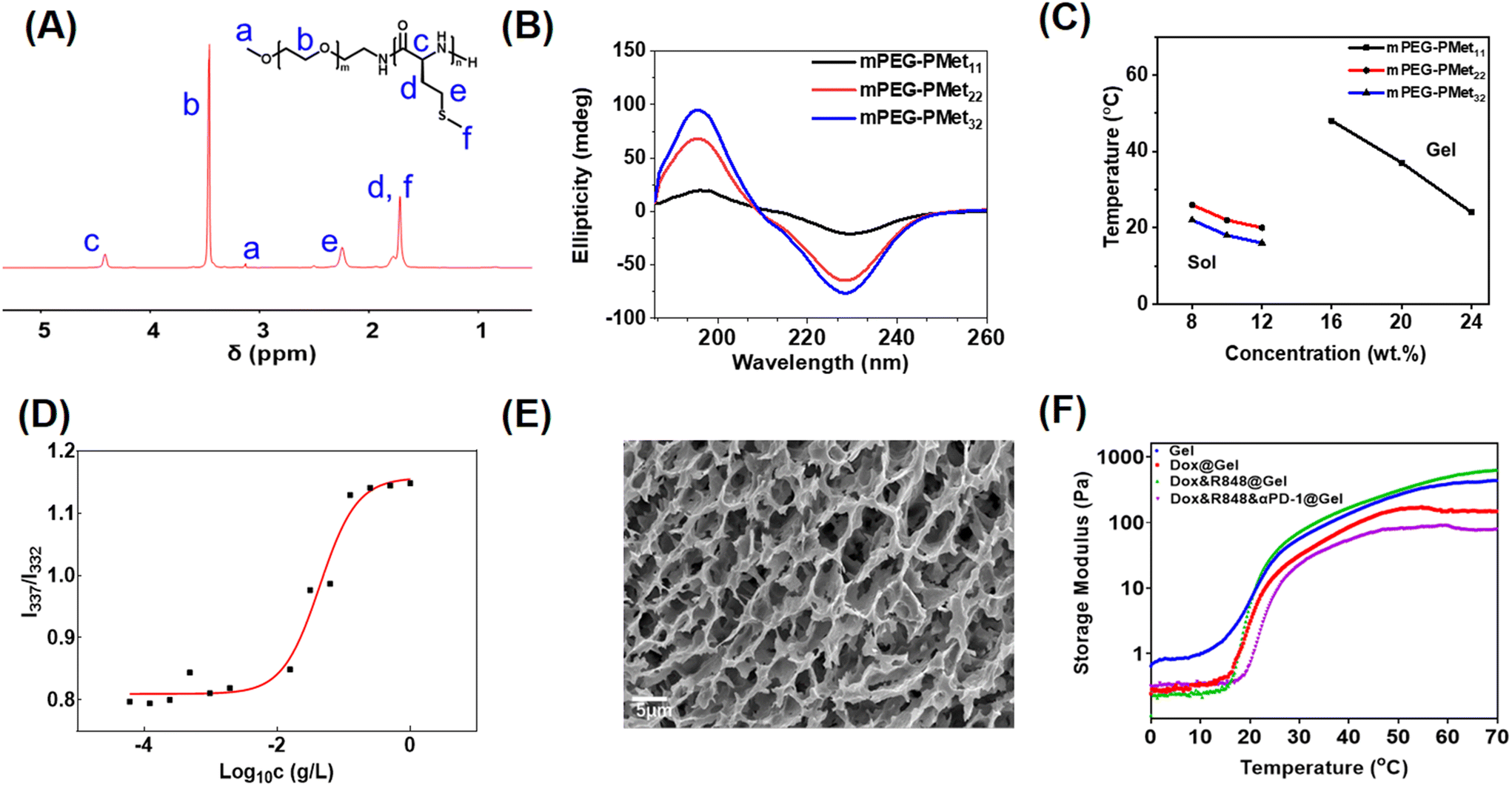 | ||
| Fig. 1 (A) The 1H NMR spectra of the copolymer. (B) Circular dichroism spectra of three different copolymers. (C) Temperature-dependent sol–gel phase diagram of copolymers with three different polymerization degrees of copolymers. (D) The intensity ratios (I337/I332) of mPEG-b-PMet22. (E) SEM image of lyophilized hydrogels of 8.0 wt% mPEG-b-PMet22. (F) The rheological characteristics of the hydrogels formed by 8.0 wt% mPEG-b-PMet22 with or without drugs. | ||
Circular dichroism (CD) spectra and Fourier transform infrared (FTIR) spectra were used to analyze the secondary structures of block copolymers in the aqueous state and solid solution, respectively. The positive band at 195 nm and the negative band at 220–230 nm indicated that the secondary structure of this block polymer mainly adopted an α-helix conformation, and the characteristics become more pronounced with the growth of PMet chain segments (Fig. 1B). The findings were also indicated in the FTIR spectra (Fig. S3, ESI†). Meanwhile, we had studied the change of polypeptide fragments with temperature through CD spectra. The results showed that the CD band intensities of different copolymers at 220–230 nm all displayed a slight decrease as the temperature increased, indicating that the secondary structure of the polypeptide fragment was affected by temperature (Fig. S4, ESI†).
The sol–gel phase transition of mPEG-b-PMet in PBS solution was studied by the tube inversion method and the phase diagram is shown in Fig. 1C. The mPEG-b-PMet solutions with different polymerization degrees can complete the sol–gel phase transition at specific temperatures and concentrations, respectively. Among them, mPEG-b-PMet22 is the more ideal material for subsequent experiments because it exhibited a suitable transition temperature (20–26 °C) and a lower gelation concentration (Fig. S6, ESI†). Subsequent experiments were carried out on mPEG-b-PMet22.
In the block polymer, there are hydrophilic mPEG and relatively hydrophobic PMet segments, which tend to form rod-like micelles by self-assembly to maintain thermodynamic balance.41 In the excitation spectra of pyrene, the absorption peak shows a significant blue shift from 337 nm to 332 nm with the gradual dilution of mPEG-b-PMet22 solution, which results from the increase in hydrophilicity. The intensity ratio at 337 nm to 332 nm serves as the basis for this analysis (I337/I332); we calculated the critical micelle concentration (CMC) of mPEG-b-PMet22 as 10.0 mg L−1 (Fig. 1D). In addition, SEM and TEM were used to observe the microscopic morphology of micelles that were generated by mPEG-b-PMet22 block copolymers, and a substantial number of rod-shaped or spherical micelles were detected (Fig. S5, ESI†). The microscopic morphology of the 8 wt% mPEG-b-PMet22 hydrogel was observed by SEM and we can observe the porous structure of the lyophilized hydrogel, which may contribute to the encapsulation and delivery of drugs (Fig. 1E).
To examine the mechanical properties of the hydrogel, the storage modulus (G′) of the hydrogel with or without drugs incorporation was assessed using rheometry. When the temperature rose to about 20 °C, the G′ of the blank hydrogel increased dramatically, which is consistent with the results obtained through tube inversion method previously, confirming the temperature sensitivity of the hydrogel (Fig. 1F). In addition, the encapsulation of Dox, R848 and aPD-1 had no significant effect on the G′ of the hydrogel, indicating that the hydrogel may be a good drug delivery system.
2.2. ROS-triggered release and degradation behaviors in vitro and degradation in vivo
Hydrogen peroxide (H2O2) is an endogenous ROS and a common metabolic byproduct of multicellular organisms.42 The in vitro degradation behaviors of the mPEG-b-PMet22 hydrogel (8 wt%) at different H2O2 concentrations were determined. The mass loss of the hydrogel was mainly caused by gel erosion, which was significantly enhanced by the oxidation of amino acid residues caused by ROS.33 As shown in Fig. 2A, the mass loss was about 50% after incubation of the hydrogel in phosphate buffer saline (PBS) without H2O2 for 22 days, and then the hydrogel mass was almost no longer lost. When incubated in PBS with 1 mM H2O2, the hydrogel was completely degraded in 16 days. Meanwhile, when the hydrogel was treated in PBS with 10 mM H2O2, the degradation time was dramatically reduced to 5 days. In addition, the oxidized copolymers were obtained by lyophilizing the degradation medium with different H2O2 concentrations (Scheme S2, ESI†). The structural changes of post-oxidation copolymers were characterized by FTIR and CD spectra (Fig. S7, ESI†). The results showed that with the increase of H2O2 concentration, the typical absorption intensity of sulfonyl groups assigned at ≈530 cm−1 in the FTIR spectrum increased significantly, which indicated that the methionine residues in block polymers have been oxidized by H2O2. The CD spectrum showed that the α helix content increased significantly with the increase of oxidation degree, consistent with previous studies.33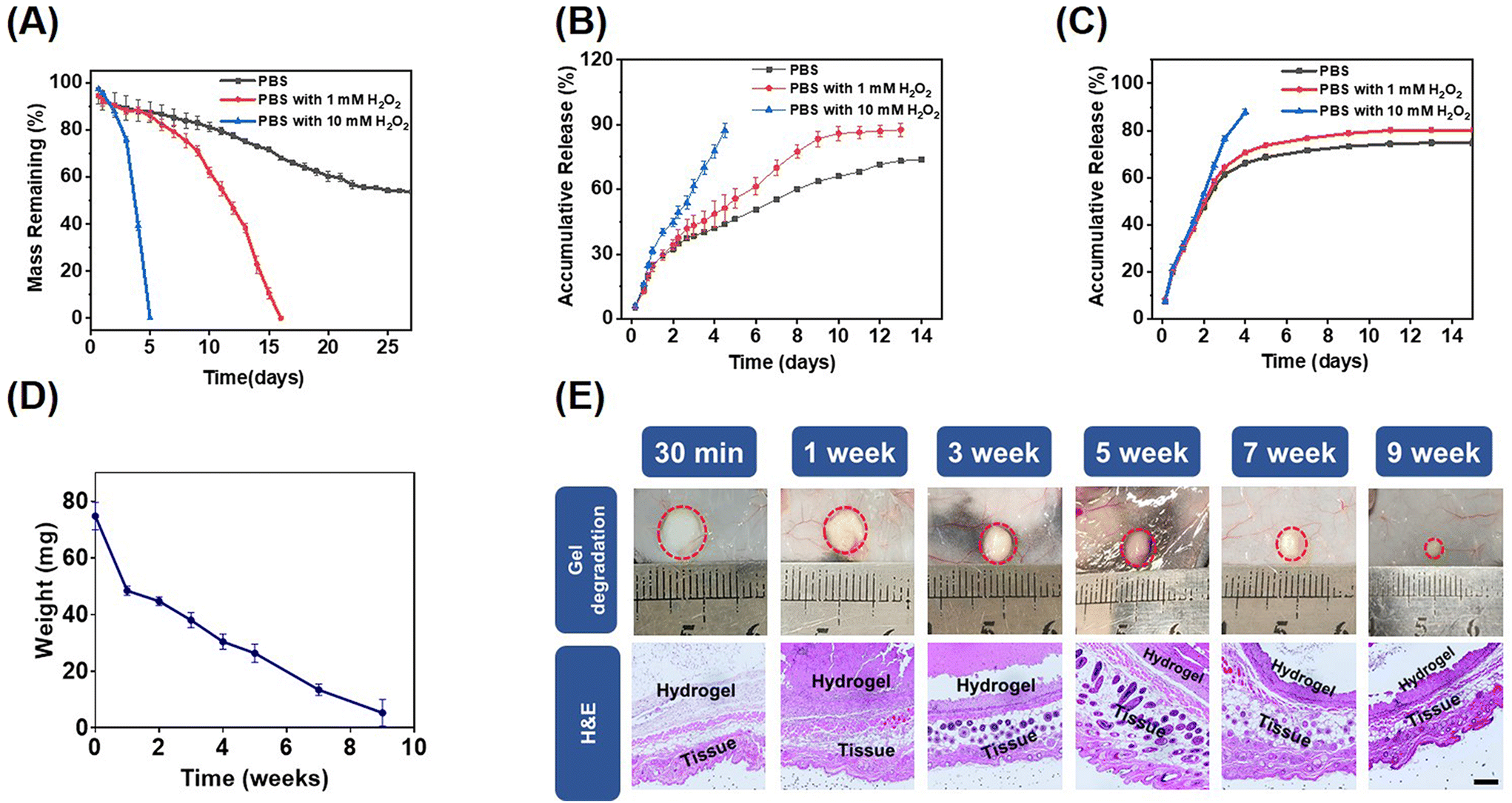 | ||
| Fig. 2 (A) The degradation characteristic of the hydrogel formed by 8.0 wt% mPEG-b-PMet22 evaluated at varying concentrations of hydrogen peroxide in vitro (n = 3). The release behaviors of Dox (B) and R848 (C) from the hydrogel formed by 8.0 wt% mPEG-b-PMet22in vitro (n = 3). (D) The degradation characteristic of the hydrogel formed by 8.0 wt% mPEG-b-PMet22in vivo (n = 3). (E) The biocompatibility of the hydrogel formed by 8.0 wt% mPEG-b-PMet22in vivo (scale bar: 100 μm). | ||
To evaluate the ROS response release behavior of the drug-encapsulated mPEG-b-PMet22 hydrogel, in vitro releases of the hydrogel loaded with Dox or R848 at different H2O2 concentrations were studied, as shown in Fig. 2B and C, respectively. Dox and R848 showed similar release behavior, with over 80% of the drugs released at 10 mM H2O2 concentration within 5 days. The accumulative release also remained higher at a low concentration of H2O2 than that without H2O2. This phenomenon was more evident in the Dox release model, which may be attributed to the fact that Dox is present in the form of hydrochloride and its good hydrophilicity allows it to mix evenly with the polymer solution while spreading more easily during release. Meanwhile, we used IgG as a model antibody to simulate the in vitro release of aPD-1. Within PBS containing 1 or 10 mM H2O2, the accumulative release amount reached over 85% within 13 days or 5 days, which was higher than that without H2O2 (less than 75%) (Fig. S8, ESI†).
The biocompatibility and degradation of the hydrogel in vivo were studied by injecting the copolymer solutions into the subcutaneous layer of C57BL/6 mice (Fig. 2D). The hydrogel underwent gradual degradation in mice and completely degraded within a period of 10 weeks, which may be attributed to the hydrogel erosion and enzymatic cleavage of polypeptide fragments.43,44 In addition, there were no significant chronic inflammation, hemorrhage, hyperemia or necrosis in the tissues surrounding the hydrogel observed by H&E staining, which indicated that the hydrogel was biocompatible (Fig. 2E).
2.3. Cytotoxicity and immune modulation activity in vitro
To evaluate the cell compatibility of hydrogel precursor block copolymers, L929 cells were subjected to culture with varying concentrations of mPEG-b-PMet22in vitro and then measured with cell counting kit-8 (CCK8) tests. As shown in Fig. 3A, the cells remained more than 80% alive even in polymer solutions with a concentration of 1 mg mL−1, indicating that the synthesized polymer had good cellular compatibility.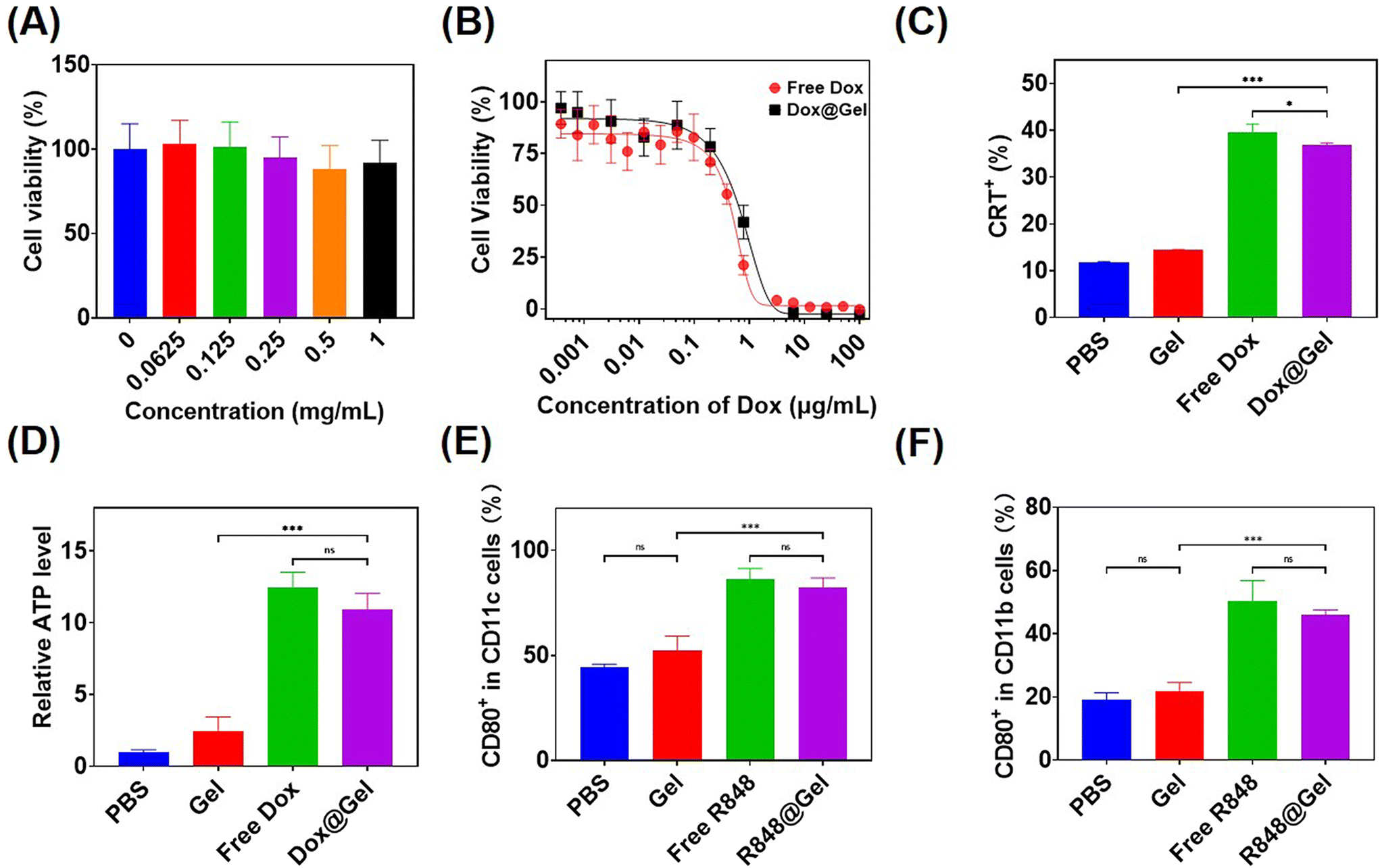 | ||
| Fig. 3 (A) The in vitro cytotoxicity of the copolymer (n = 5). (B) The in vitro cytotoxicity on B16F10 cells of free Dox and Dox@Gel (n = 5). (C) Quantitative analysis of CRT expression by flow cytometry on B16F10 cells following different treatments (n = 3). (D) The quantitative analysis of ATP release from B16F10 cells following different treatments (n = 5). (E) Quantification data of activated DCs following different treatments (n = 3). (F) Quantification data of M1 macrophages following different treatments (n = 4). Data are presented as the mean ± standard deviation (SD) and the statistical significance was assessed by one-way analysis of variance (ANOVA) with a Tukey post-test. The statistical significance was denoted as *p < 0.05, **p < 0.01 and ***p < 0.001. ns: no significance. | ||
Free Dox and Dox-loaded hydrogels, referred to as Dox@Gel, were incubated with B16F10 tumor cells for 24 h, respectively, to detect the tumor-killing efficiency. It can be seen that both of them had similar killing efficiency, and the IC50 of the Dox@Gel group (0.74 μg mL−1) was slightly higher than that of the free Dox group (0.53 μg mL−1), which may be attributed to the slow release of Dox by the hydrogel (Fig. 3B).
Anthracycline drugs, such as Dox, have the ability to induce immunogenic death of tumor cells (ICD).12 ICD has been shown to cause damage-associated molecular patterns (DAMPs) release, including the release of ATP, exposure of calreticulin (CRT) on cell membranes and the release of high mobility group box 1 (HMGB1).45,46 Incubation of B16F10 cells with free Dox and Dox@Gel was conducted, respectively, followed by the analysis of CRT expression and ATP release in the cells after ICD through flow cytometry and ATP test kit. The results showed that B16F10 cells treated with free Dox and Dox@Gel showed over 35% CRT expression, instead of less than 15% CRT expression in PBS and the blank gel. Meanwhile, the ATP release level in the PBS group was only 8% of that in the Dox group and 10% of that in the Dox@Gel group (Fig. 3C and D). These results confirmed that Dox@Gel was effective in inducing B16F10 immunogenic death.
It has been proved that the immune adjuvant R848 can promote the maturation of dendritic cells and the M1 polarization of macrophages.14,47 Therefore, in this study, free R848 and R848-loaded hydrogel (donated as R848@Gel) were incubated with bone marrow-derived dendritic cells (BMDCs) and bone marrow-derived macrophages (BMDMs) for 24 h, respectively. DC maturity markers and BMDM M1 polarization markers were analyzed by flow cytometry. The results showed that the expression of CD80, the marker of DC maturation, was close to 90% in the last two groups, but only about 40% in the PBS group. Meanwhile, the expression of CD80, the marker of M1 polarization in BMDMs, had no significant difference between the free R848 and R848@Gel groups, and both of which were over 40%, while that of the PBS group was less than 20% (Fig. 3E and F). These results showed that both free R848 and R848@Gel can significantly contribute to DC maturation and macrophage M1 polarization.
2.4. Hydrogel-mediated local combination therapy in vivo
In order to evaluate the anti-tumor effect of local therapy with the hydrogel, we built a monolateral tumor treatment model in mice. In short, B16F10 tumor cells were subcutaneously injected into the right side of C57BL/6 mice, and the mice were randomly divided into 5 groups (n = 5) when the tumor volume grew up to about 80 mm3. Then, different drug-containing systems were intratumorally injected, including PBS, a solution of free Dox, R848 and aPD-1 (denoted as free Dox/R848/aPD-1), a Dox-loaded hydrogel (Dox@Gel), a Dox/R848 co-loaded hydrogel (Dox/R848@Gel) and a Dox/R848/aPD-1 co-loaded hydrogel (Dox/R848/aPD-1@Gel). The Dox/R848/aPD-1@Gel group demonstrated a significantly greater anti-tumor effect compared to the other groups and the average tumor volume was less than 300 mm3. The results showed that the sustained release of Dox, R848 and aPD-1 encapsulated in the hydrogel at the tumor site could significantly enhance the anti-tumor effect (Fig. 4B and C). Meanwhile, it was observed that none of the local treatments had a significant impact on the body weight of the mice. The toxic and side effects were also evaluated by H&E staining of the heart, liver, spleen, lungs and kidneys (Fig. S9, ESI†). No significant histopathology changes were observed after receiving the treatments. Therefore, the results substantiated that the local administration of hydrogels, which contain either a single drug or multiple anti-tumor drugs, can lead to low systemic toxicity.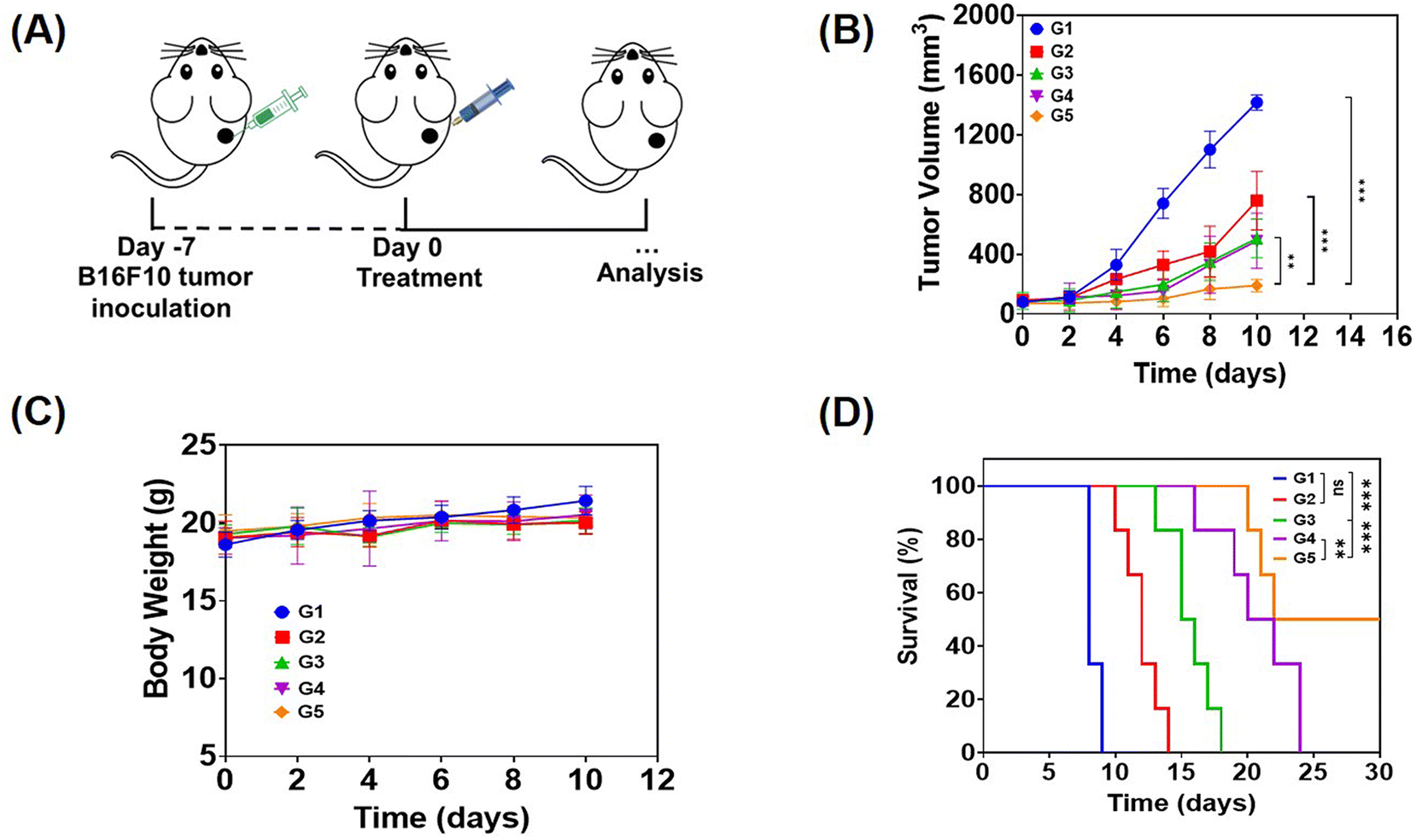 | ||
| Fig. 4 (A) Schematic of the in vivo anti-tumor effect. (B) The weight of the mouse body throughout the treatment period (n = 5). (C) Average tumor volume curves during the experiment time (n = 5). (D) The survival curves of mice analyzed in various groups (n = 6). G1: PBS, G2: free Dox/R848/aPD-1, G3: Dox@Gel, G4: Dox/R848@Gel and G5: Dox/R848/aPD-1@Gel. Data are presented as the mean ± standard deviation (SD) and the statistical significance was assessed by one-way analysis of variance (ANOVA), respectively, using the least significant difference (LSD) post-test (B) or log-rank test (D) (*p < 0.05, **p < 0.01, and ***p < 0.001). ns: no significance. | ||
The efficacy of drug-loaded hydrogels in inhibiting tumor growth was confirmed through an independent experiment involving the survival of mice subjected to various treatments. All mice in the PBS group survived for no more than 10 days, while in the other four groups, it can be seen that the survival time of mice treated with the drug-loaded hydrogel was longer than that of mice treated with the free drugs, especially the Dox/R848/aPD-1@Gel group displayed a survival time exceeding 30 days according to Fig. 4D. The results demonstrated that the local co-administration of a chemotherapy drug and an immunotherapy agent in the hydrogel significantly enhanced the anti-tumor effect.
In the immune system, DCs, as antigen-presenting cells, activate acquired immunity by presenting antigens to T lymphocytes, which is one of the most effective ways to stimulate the immune system to resist cancer.48 M1 macrophages can secrete cytokines such as IL-6 and TNF-α with anti-tumor effects.49 T cells are pivotal to the anti-tumor immune response and come in different subtypes, each with distinct roles in cellular immunity. The most significant subtypes are CD8+ T cells, known as cytotoxic T-lymphocyte cells (CTL cells), which directly eliminate tumor cells and CD4+ T cells, which are critical components in regulating the immune response by producing cytokines.50,51 In order to analyze the various immune responses in the treated mice, the spleen and tumors of the treated mice were collected and the immune cells were analyzed by flow cytometry.
The changes of DCs, macrophages, CD8+ T cells and CD4+ T cells were detected in tumors of different treatment groups. In terms of DC activation, the higher proportions of mature DCs (CD11c+CD86+) were detected in tumors treated with Dox/R848 @Gel and Dox/R848/aPD-1@Gel, indicating that the localized chemoimmunotherapy enhanced the immune response in tumors (Fig. 5A). Meanwhile, the ratio of M1 macrophages (CD11b+CD80+) in tumors was the highest in the Dox/R848/aPD-1@Gel group, which potentially enhanced the killing efficacy of macrophages toward tumor cells (Fig. 5C). In the Dox/R848/aPD-1@Gel group, CD4+ T cells (CD3+CD4+) and CD8+ T cells (CD3+CD8+) were significantly higher than those in other groups (Fig. 5C and D), which showed that the infiltration of T cells in tumors was significantly enhanced after the treatment with Dox/R848/aPD-1@Gel. In addition, it can be seen from Fig. 5E and F that the expressions of the pro-inflammatory factor IFN-γ were notably elevated in both CD4+ T cells and CD8+ T cells within the spleen of mice in the Dox/R848/aPD-1@Gel group compared to those in the PBS group, which may promote apoptosis of tumor cells.
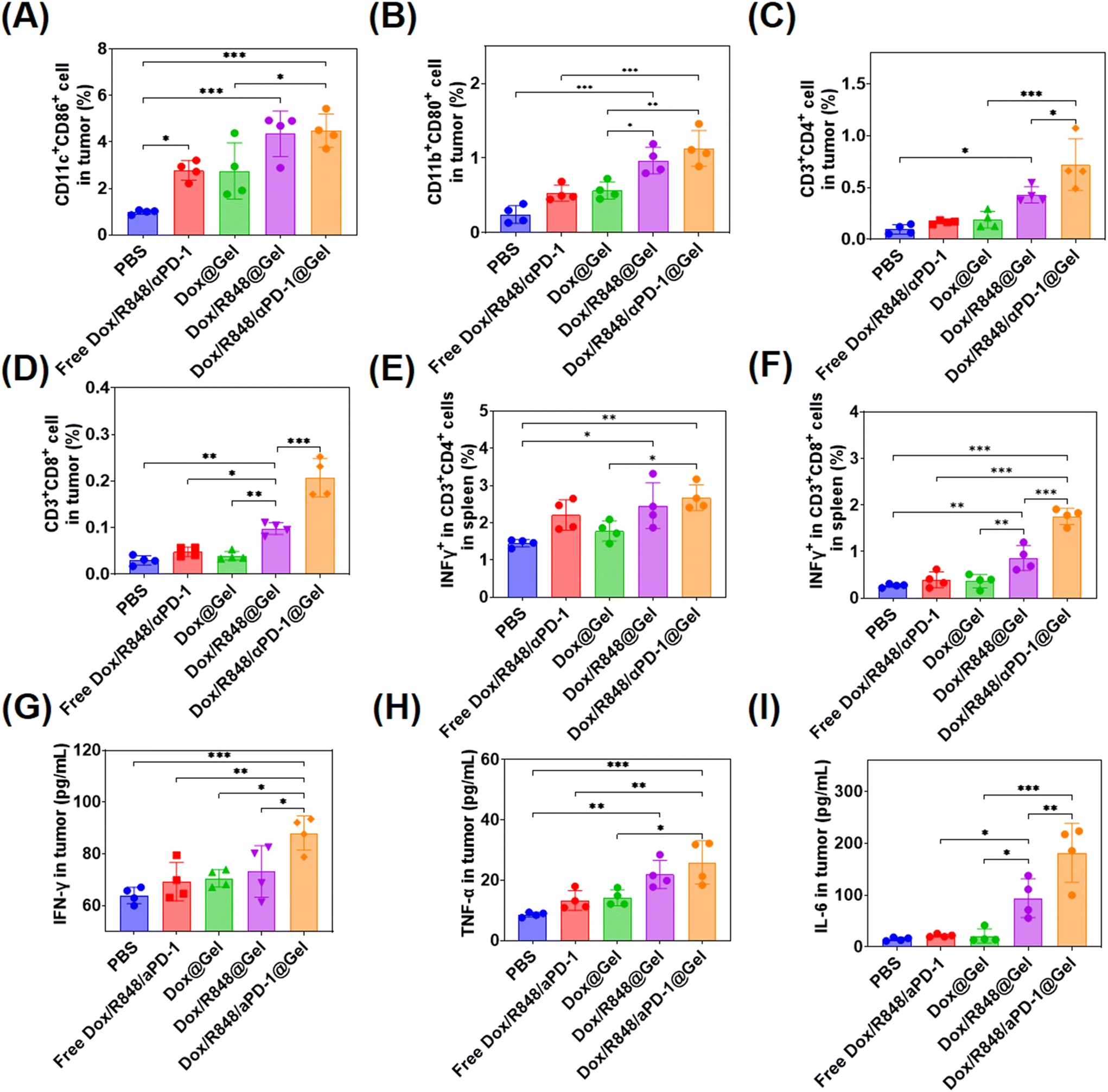 | ||
| Fig. 5 Quantitative analysis of mature dendritic cells (CD11c+CD86+) by flow cytometry (A), M1 macrophages (CD11b+CD80+) (B), CD3+CD4+ T cells (C), and CD3+CD4+ T cells (D) in tumors (n = 4). Quantitative analysis of IFN-γ+ in CD3+CD4+ cells (E) and IFN-γ+ in CD3+CD8+ cells (F) in the spleen (n = 4). Concentration of interferon-γ (IFN-γ) (G), tumor necrosis factor-α (TNF-α) (H), and interleukin-6 (IL-6) (I) in tumors (n = 4). Data were presented as the mean ± standard deviation and the statistical significance was assessed by one-way analysis of variance (ANOVA) with a Tukey post-test. The statistical significance was denoted as *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
Furthermore, the levels of several key cytokines such as IFN-γ, TNF-α, and IL-6, which serve as representative markers of the immune response, were analyzed in tumors with ELISA kits. IFN-γ plays a crucial role in anti-cancer immunity by increasing the activity of Th1 cells, CTLs, NK cells, DCs and macrophages and promoting antigen presentation.52 TFN-α is a cytokine secreted by macrophages that directly kills tumor cells without significant toxicity to normal cells.53 IL-6 has been shown to promote the proliferation and activation of T cells.54 The results showed that the expression levels of these three cytokines in the Dox/R848/aPD-1@Gel group were significantly higher than those in other groups after treatment (Fig. 5G–I). According to the above results, the local chemoimmunotherapy with Dox/R848/aPD-1@Gel induced the most robust immune response, which probably contributed to the most effective anti-tumor action.
In addition, we stained CD8+ T cells and CD11c+ cells of tumors in different groups and observed that CD8+ T cells and CD11c+ cells (Fig. S10, ESI†) were highest in the Dox/R848/aPD-1@Gel group than in other groups, indicating a stronger anti-tumor effect.
2.5. Prevention of postsurgical tumor recurrence
To simulate the clinical treatment of melanoma, we developed a mouse model using B16F10 melanoma cells. B16F10 cells were subcutaneously injected into the right side of C57BL/6 mice, and the visibile tumors were surgically removed completely and the skin was sutured when the tumor grew to 200–300 mm3. Then the mice were randomly divided into 5 groups for administration (n = 10), and were treated with PBS, free Dox/R848/aPD-1, Dox@Gel, Dox/R848@Gel and Dox/R848/aPD-1@Gel in turn (Fig. 6A). The results showed that even if the visible tumors were all removed, infiltration of residual tumor cells may also lead to tumor recurrence, and the survival of mice was significantly prolonged by treatment with Dox/R848/aPD-1@Gel (Fig. 6B). As shown in Fig. 6C and D, the recurrence rate of tumors in mice treated with PBS was up to 90%, and after treatment with free drugs, the recurrence rate decreased to 70%. However, it is noteworthy that the tumor recurrence was significantly inhibited in the last three groups, especially in the Dox/R848/aPD-1@Gel group, where only 10% of mice relapsed (Fig. 6G), while the Dox@Gel group and the Dox/R848@Gel group had a recurrence rate of 40% and 20%, respectively (Fig. 6E and F). The results confirmed that the localized administration of hydrogels, incorporating either a single drug or multiple anti-tumor drugs, effectively mitigates the recurrence of tumors.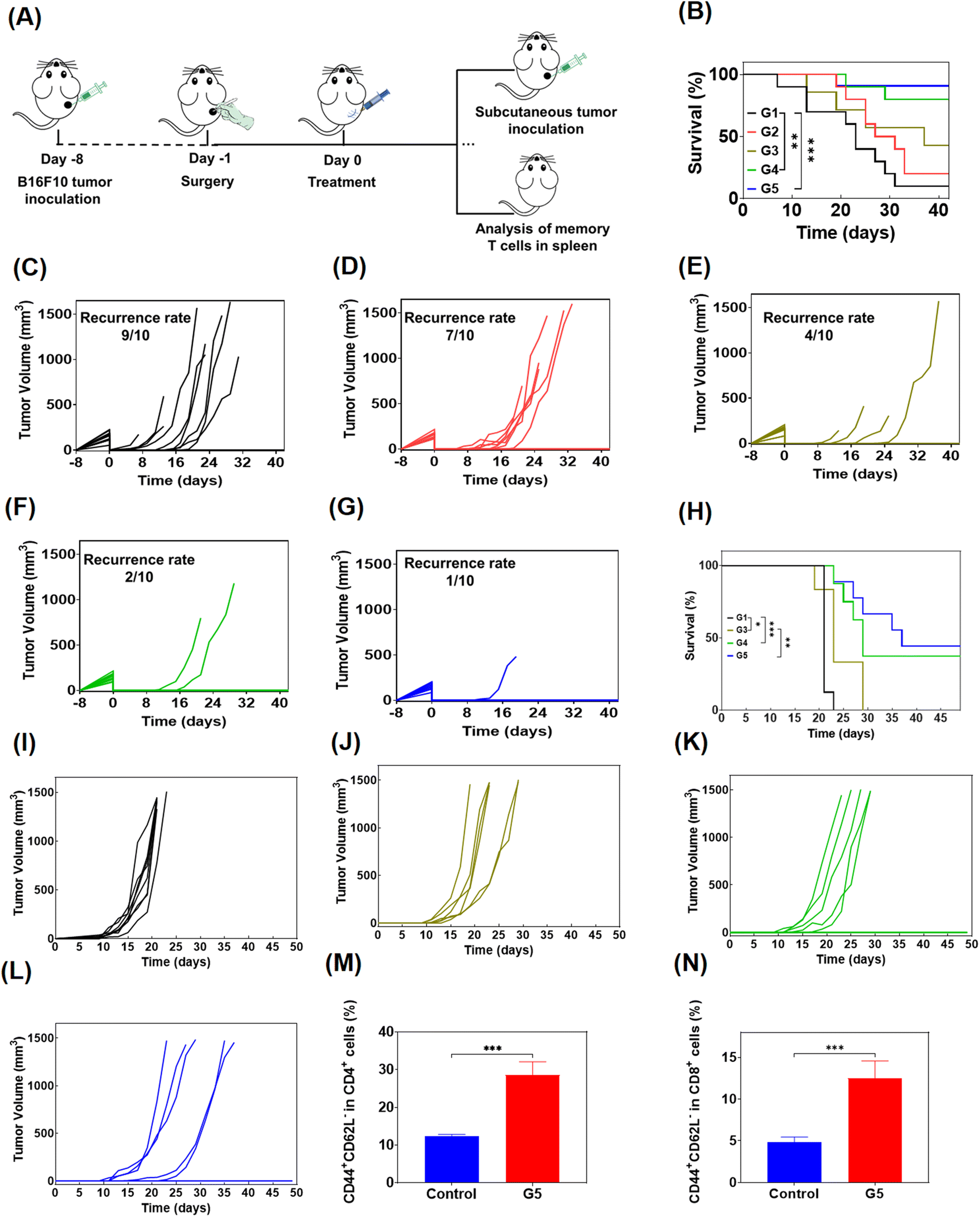 | ||
| Fig. 6 (A) Schematic of post-surgical treatment. (B) The survival curves of mice following different treatments (n = 10). Individual tumor volume curves of the mice in different treatments donated as G1 (C), G2 (D), G3 (E), G4 (F), and G5 (G) (n = 10). (H) The survival curves of mice subjected to tumor rechallenge with various treatment regimens (n = 8, 6, 8 or 9). The growth trajectories of individual recurrent tumors in mice in the control group (I), G3 (J), G4 (K), and G5 (L) (n = 8, 6, 8 or 9). (M) The quantitative analysis of CD44+CD62L− effector memory T cells in CD4+ cells by flow cytometry (n = 4). (N) The quantitative analysis of CD44+CD62L− effector memory T cells in CD8+ cells by flow cytometry (n = 4). G1: PBS, G2: free Dox/R848/aPD-1, G3: Dox@Gel, G4: Dox/R848@Gel and G5: Dox/R848/aPD-1@Gel. Data were presented as the mean ± standard deviation. Statistical significance was assessed using a log-rank test (B and H) or two tailed Student's t-test (M and N). The statistical significance was denoted as *p < 0.05, **p < 0.01 and ***p < 0.001. | ||
In order to further study the long-term immune memory effect following hydrogel-based chemoimmunotherapy, the mice cured in drug-loaded hydrogel groups were rechallenged by inoculating secondary B16F10 tumors (Fig. 6A) and the control group was healthy mice of similar age. As can be seen from the figure (Fig. 6H–L), the growth of tumors in the hydrogel group was significantly inhibited compared to the control group. Moreover, the chance of recurrence in cured mice was significantly suppressed even when tumor cells were re-injected, which was approximately 50% in the Dox/R848/aPD-1@Gel group. The results confirmed that the local chemoimmunotherapy based on the hydrogel produced long-term anti-tumor immune memory and prevented the recurrence of tumors.
In addition, we analyzed the effector memory T cells of mice treated with Dox/R848/αPD-1@Gel through flow cytometry to further study the immune memory effect of the treated mice. TEM cells offer immediate protection against the rechallenged pathogens.55 It was found that the proportions of CD4+ TEM and CD8+ TEM cells in the Dox/R848/aPD-1@Gel group were markedly superior to those observed in the control group, indicating that Dox/R848/aPD-1@Gel therapy can effectively induce a T cell memory response (Fig. 6M and N).
3. Conclusion
In this study, a combined treatment strategy for local anti-tumor chemoimmunotherapy was established by encapsulating Dox, R848 and aPD-1 in a ROS-responsive thermosensitive mPEG-b-PMet hydrogel. The mPEG-b-PMet hydrogel demonstrated an ROS-accelerated degradation behavior in vitro, which was concentration dependent. The sustained release of Dox, R848 or aPD-1 in hydrogels lasted for more than 13 days in vitro, and the presence of ROS can accelerate this process. The Dox released from hydrogels showed killing efficiency against B16F10 cells and was confirmed to cause the ICD of tumor cells by in vitro characterizing CRT expression and ATP release. Meanwhile, R848 released from hydrogels induced activation of DCs and M1 polarization of macrophages in vitro. In the B16F10 melanoma treatment model, the Dox/R848/aPD-1@Gel formulation demonstrated the highest level of tumor suppression and significantly increased the survival time of mice compared to other formulations. Moreover, the Dox/R848/aPD-1@Gel treatment significantly promoted anti-tumor immune responses, including the activation of DCs, infiltration of CD8+ and CD4+ T cells, M1 polarization of macrophages, and secretion of cytokine IFN-γ, IL-6 and TNF-α in tumors. In the mice postoperative model, Dox/R848/aPD-1@Gel significantly inhibited tumor recurrence and the growth of reinjected tumor cells, while being accompanied by a significant increase in effector memory T cells compared to healthy mice. In summary, the above results showed that this local chemoimmunotherapy hydrogel can be a promising platform for effective anti-tumor treatment and suppression of postsurgical tumor recurrence.4. Experimental section
4.1. Materials
Amino-terminated monomethyl poly (ethylene glycol) (mPEG-NH2, MW = 2000 Da) was purchased from Ponsure Biotech, Inc. (Shanghai, China). L-Methionine was obtained from Aladdin. Other chemical reagents were purchased from Anhui Senrise Technologies Co., Ltd (Anhui, China). Dox was procured from Sigma-Aldrich, while R848 was obtained from Zhengzhou JACS Chem Product Co., Ltd (Zhengzhou, China). InvivoMab anti-mouse PD-1 was sourced from BioXcell (West Lebanon, USA). The anti-CRT antibody was procured from Abcam (Cambridge, UK). The ATP assay kit was obtained from Meilunbio (Dalian, China). BMDM- and BMDC-induced cytokines, including M-CSF, GM-CSF, and IL-4, were procured from PeproTech (Rocky Hill, USA). Cytokine Elisa kits were purchased from Tianjin Anaerobic Biotechnology Co., Ltd (Tianjin, China). Cell media were purchased from Wuhan Servicebio Technology Co., Ltd. Various antibodies for flow cytometry were obtained from Biolegend (San Diego, USA).4.2. Characterization
The 1H NMR spectra of Met-NCA in CDCl3 and mPEG-b-PMet in CF3COOD were recorded using a 500 MHz Bruker instrument. The CD spectrum of 0.2 mg mL−1 mPEG-b-PMet aqueous solution was recorded using a Chirascan CD spectrometer (Applied Photophysics, Surry, UK). The CMC of mPEG-b-PMet was determined using a pyrene fluorescence probe, as measured using a Fluoromax-4 spectrophotometer (Horiba Ltd, Kyoto, Japan). The storage modulus was obtained using a MCR 301 rheometer (Anton Paar, Austria). The microscopic porous structure of the 8 wt% freeze-dried hydrogel and the microscopic morphology of 1 mg mL−1 of polymer aqueous solution dried at 37 °C were tested by SEM (Gemini 2, Carl Zeiss, Germany).4.3. Synthesis of mPEG-b-PMet
The preparation of the mPEG-b-PMet block copolymer was achieved through ring-opening polymerization of Met-NCA initiated by mPEG-NH2. Briefly, after weighing mPEG-NH2 (1 g, 0.5 mmol), toluene (60 mL) was added to remove water through azeotropic distillation for a period of 4 h at approximately 130 °C. After the toluene was completely evaporated and the system returned to room temperature, 40 mL of anhydrous DMF was added to dissolve mPEG-NH2 and Met-NCA (1.75 g, 10 mmol) was then added. It was reacted at room temperature for 3 days. The product was dissolved in a small amount of DMF and dialyzed against water (MWCO: 500 Da) for 3 days, and then collected by lyophilization.4.4. Phase diagram
The gelation of mPEG-b-PMet solutions of varying concentrations was assessed utilizing the tube inversion technique. In brief, the process involved configuring mPEG-b-PMet solutions with gradient concentrations in 2 mL vials and placing them in an ice bath for 48 h with continuous stirring. After mixing, the vials were placed in a water bath and the temperature was gradually increased (at a rate of 2 °C every 10 min). To identify the sol–gel phase transition temperature, the sample was considered to have reached the gel state when it showed no fluidity after tilting for 30 s.4.5. ROS-triggered degradation in vitro
The degradation media were PBS solutions containing 1 mM and 10 mM H2O2, respectively. The 8.0 wt% polymer solutions were placed into glass vials (300 μL for each vial) in an ice bath and stirred for 48 h, followed by incubation at 37 °C for 5 h to stabilize them into hydrogels. Then PBS solutions with 0, 1 or 10 mM H2O2 were added into the glass vials (1.6 mL for each vial). Subsequently, the samples were placed in a shaker set (50 rpm) at a temperature of 37 °C. The degradation media were periodically removed, and the remaining hydrogel mass was recorded. Subsequently, an equal volume of fresh degradation media was reintroduced. Three parallel samples were prepared for each concentration.4.6. ROS-triggered drug release
300 μg of Dox and R848 were dissolved in 300 μL of 8.0 wt% polymer solutions, respectively, in order to obtain polymer solutions with 1.0 mg mL−1 drugs and the vials were placed in an ice bath and stirred for 48 h. In addition, Cy3-IgG was mixed with 8.0 wt% polymer solutions and the final concentration of Cy3-IgG was 0.1 mg mL−1. The mixed solutions were incubated at 37 °C for 5 h to make them form hydrogels containing drugs. Then, PBS solutions with concentrations of 0, 1, and 10 mM H2O2 were added to glass vials with 1.6 mL for each vial. 1.2 ml of medium was collected and the same volume of fresh medium was added at specified time intervals. The quantification of Dox and R848 were tested using a UV-2401PC (Shimadzu Co. Japan) at 480 nm and 320 nm, respectively. The quantification of Cy3-IgG was tested using a microplate reader (Tecan Spark).4.7. Cytotoxicity of the hydrogel
10% FBS supplemented DMEM (200 μL) containing L929 cells (5 × 103) was added into each well of the 96-well plates and incubated at 37 °C for 24 h. Then, the fresh medium mixed with different concentrations of the copolymer was configured to replace the previous medium. After 24 h incubation, the cell activity was detected using the CCK-8 assay.Meanwhile, 1 × 104 B16F10 cells were added into each well of the 48-well plates and incubated in 10% FBS supplemented DMEM for 24 h. Various concentrations of free Dox and Dox-loaded hydrogels were added. After 24 h incubation, the cell activity was measured using the CCK-8 assay.
4.8. Induction of ICD in tumor cells
The CRT expression of B16F10 cells was analyzed by flow cytometry. The complete medium containing 3 × 105 B16F10 cells was added into each well of the 6-well plates and incubated overnight. Then, PBS, the hydrogel, free Dox (5 μg mL−1) and the Dox-loaded hydrogel (5 μg mL−1 Dox) were added into the medium with a subsequent 4 h incubation, respectively. Anti-CRT/AF488 was used for incubating each sample for 1 h and the cell membrane was stained for detection.ATP release caused by ICD in tumor cells were tested using ATP test kits. The complete medium containing 3 × 104 B16F10 cells were added to each well of the 24-well plates and incubated overnight. Then, PBS, the hydrogel, free Dox (5 μg mL−1) and the Dox-loaded hydrogel (5 μg mL−1 Dox) were added into the cell medium with a subsequent 24 h incubation, respectively. Finally, the collected supernatant was analyzed using an ATP test kit.
4.9. DC maturation and macrophage polarization in vitro
BMDCs and BMDMs were extracted according to previous reports, respectively.56,57 BMDCs were added into the 12-well plates with PBS, the hydrogel, free R848 (5 μg mL−1) or the R848-loaded hydrogel (5 μg mL−1 R848) and incubated for 24 h, respectively. The cells were collected and stained with antibodies (CD11c-FITC and CD80-APC) in order to detect mature dendritic cells by flow cytometry.BMDMs were treated with PBS, the hydrogel, free R848 or the R848-loaded hydrogel and incubated for 24 h, respectively. The cells were collected and stained with antibodies (CD11b-FITC and CD80-APC) and the M1 macrophages were detected by flow cytometry.
4.10. Degradation of hydrogels in vivo
C57BL/6 female mice (6–8 weeks) were used to study the degradation of hydrogels in vivo. 100 μL solution containing 8.0 wt% of copolymer solution was subcutaneously injected into the backs of each mouse. The mice were subsequently euthanized and the hydrogels were isolated and photographed at various time points. These time points included 30 minutes and 1, 2, 3, 4, 5, 7, and 9 weeks. Meanwhile, the skin tissues around the hydrogel were fixed with 4% poly-formaldehyde solution, subsequently sectioned, and H&E staining was performed for histopathological observation.4.11. Local treatment effect in vivo
In order to evaluate the anti-tumor effect of the drug-loaded hydrogel, a B16F10 melanoma-bearing mice model was constructed. B16F10 cell suspensions (50 μL, 1 × 106 cells per mouse) were injected into the right dorsal region of the female C57BL/6 mice (6–8 weeks old, Beijing Vital River Laboratory Animal Technology Co., Ltd). Approximately 7 days later, when the tumor reached a volume of about 80 mm3, the mice were randomly divided into 5 groups (n = 5) and intratumorally injected with PBS, free Dox/R848/aPD-1, Dox@Gel, Dox/R848@Gel and Dox/R848/aPD-1@Gel, respectively, for 50 μL per mouse (3 mg kg−1 Dox, 250 μg kg−1 R848, and 2 mg kg−1 aPD-1). The tumor volumes and body weights of the mice were recorded every two days, and the tumor volume was calculated using the formula V = 1/2 × length × width2. On the 10th day post-treatment, when the tumors of the mice in PBS Group were over 1500 mm3, the mice were sacrificed to harvest the tumors. Concurrently, survival rate experiment was conducted independently according to the procedures in anti-tumor treatment in vivo (n = 6). The mice were euthanized when the tumor grew over 1500 mm3. Furthermore, H&E staining was employed to analyze the histopathology of the vital organs.114.12. Immunological analysis in vivo
After 10 days of treatment, tumors, lymph nodes and spleen of the mice in each group were collected. Single-cell suspensions were prepared after grinding, filtration, etc. and stained with targeted antibodies for the flow cytometry.The tumor tissue, weighing 100 mg in each mouse, was finely pulverized and then subjected to centrifugation to obtain the supernatant. The supernatant was subsequently evaluated using enzyme-linked immunosorbent assay (ELISA) kits to quantify the concentrations of various cytokines, including interleukin-6 (IL-6), interferon gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α).
4.13. Post-surgical treatment effect in vivo
In order to assess the effectiveness of tumor recurrence inhibition following tumor resection, A B16F10 melanoma-bearing mouse model was constructed utilizing a similar methodology. After incubating melanoma for 13 days, C57BL/6 mice with tumors, having an average volume of approximately 200–300 mm3, were selected for the establishment of a tumor-resection model. The mice were randomly divided into 5 groups (n = 10), including PBS, free Dox/R848/aPD-1, Dox@Gel, Dox/R848@Gel and Dox/R848/aPD-1@Gel. The visible tumors were completely removed and each mouse was subcutaneously injected with 50 μL of the specified formulation at the site of surgery. The tumor volume was recorded every two days and if it exceeded 1500 mm3, the mice were euthanized.To study the long-term immune memory effect after treatment, the cured mice in Dox@Gel, Dox/R848@Gel, and Dox/R848/aPD-1@Gel therapeutic groups were re-injected with B16F10 cell suspensions (2 × 105 cells per mouse), and the healthy mice treated in the same way were used as controls. The tumor volume was recorded every two days and if it exceeded 1500 mm3, the mice were euthanized. Furthermore, the spleens in the Dox/R848/aPD-1@Gel group after 45 days treatment were collected and then ground, filtered, and lysed red blood cells to obtain cell suspensions, which were stained with CD3-FITC, CD4-APCcy7, CD8-APC, CD62L-PEcy7 and CD44-PE antibodies for analyzing the effector memory T cells by flow cytometry.
4.14. Animal procedure
The animal experiments were conducted in compliance with the guidelines provided by the Animal Experimental Center of Jilin University for the care and use of laboratory animals. All animal treatments were approved by the Animal Ethics Committee of the Changchun Institute of Applied Chemistry, Chinese Academy of Sciences.4.15. Statistical analysis
The data were presented as the mean ± standard deviation (SD). Statistical analysis was conducted using one-way analysis of variance (ANOVA) with the post hoc test of least significant difference (LSD) or Tukey, or two-tailed Student's t-test. The survival time of animals was plotted with a Kaplan-Meier curve, and significant differences were analyzed using the log-rank test. Statistical significance was denoted as *p < 0.05, **p < 0.01, and ***p < 0.001.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the National Key Research and Development Project of China (grant no. 2022YFC2403202), the National Natural Science Foundation of China (projects 51973218, 51833010, 52173147, and 52203202), and the Scientific and Technological Development Projects of Jilin Province (20210204136YY).References
- J. Ding, T. Wang, Z. Chen, Z. Lin, X. Chen and C. He, J. Polym. Sci., 2022, 60, 1595–1609 CrossRef CAS
.
- R. Baskar, K. A. Lee, R. Yeo and K. W. Yeoh, Int. J. Med. Sci., 2012, 9, 193–199 CrossRef PubMed
- M. Esposito, S. Ganesan and Y. B. Kang, Nat. Cancer, 2021, 2, 258–270 CrossRef PubMed
- L. Pang, Z. Zhao and X. Song, Chaos, Solitons Fractals, 2016, 87, 293–301 CrossRef
- D. Wang, J. Liu, J. Duan, H. Yi, J. Liu, H. Song, Z. Zhang, J. Shi and K. Zhang, Nat. Commun., 2023, 14, 4511 CrossRef CAS PubMed
- T. Wang, D. Wang, H. Yu, B. Feng, F. Zhou, H. Zhang, L. Zhou, S. Jiao and Y. Li, Nat. Commun., 2018, 9, 1532 CrossRef PubMed
- H. Zhao, Q. Song, C. Zheng, B. Zhao, L. Wu, Q. Feng, Z. Zhang and L. Wang, Adv. Funct. Mater., 2020, 30, 2005747 CrossRef CAS
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed
- C. L. He, Z. H. Tang, H. Y. Tian and X. S. Chen, Adv. Drug Delivery Rev., 2016, 98, 64–76 CrossRef CAS PubMed
- A. C. Palmer and P. K. Sorger, Cell, 2017, 171, 1678–1691 CrossRef CAS PubMed
- L. Bezu, L. C. Gomes-de-Silva, H. Dewitte, K. Breckpot, J. Fucikova, R. Spisek, L. Galluzzi, O. Kepp and G. Kroemer, Front. Immunol., 2015, 6, 187 Search PubMed
- L. Galluzzi, A. Buqué, O. Kepp, L. Zitvogel and G. Kroemer, Nat. Rev. Immunol., 2017, 17, 97–111 CrossRef CAS PubMed
- H. Phuengkham, C. Song and Y. T. Lim, Adv. Mater., 2019, 31, 1903242 CrossRef CAS PubMed
- J. Sun, Z. Liu, H. Yao, H. Zhang, M. Zheng, N. Shen, J. Cheng, Z. Tang and X. Chen, Adv. Mater., 2023, 35, 2207733 CrossRef CAS PubMed
- X. Dong, A. Yang, Y. Bai, D. Kong and F. Lv, Biomaterials, 2020, 230, 119659 CrossRef CAS PubMed
- Y. Shi, D. Li, C. He and X. Chen, Macromol. Biosci., 2021, 21, 2100049 CrossRef CAS PubMed
- K.-S. Wang, Y.-F. Jin, Q.-S. Tong, Y.-C. Huang, Z.-L. Gao, S.-J. Zheng, J.-Y. Zhang, J. Wang and J.-Z. Du, Chin. J. Polym. Sci., 2023, 41, 32–39 CrossRef CAS
- W. Mu, Q. Chu, Y. Liu and N. Zhang, Nano-Micro Lett., 2020, 12, 142 CrossRef CAS PubMed
- C. Sun, J. Lu, J. Wang, P. Hao, C. Li, L. Qi, L. Yang, B. He, Z. Zhong and N. Hao, J. Nanobiotechnol., 2021, 19, 14 CrossRef CAS PubMed
- S. Kim, W. Lee, H. Park and K. Kim, Gel 2023, 9, 319 Search PubMed.
- T. Yu, Y. Li, X. Gu and Q. Li, Front. Pharmacol., 2020, 11, 532457 CrossRef CAS PubMed
- Y. Shi, D. Li, C. He and X. Chen, Macromol. Biosci., 2021, 21, 2100049 CrossRef CAS PubMed
- H. Ma, C. He and X. Chen, Macromol. Biosci., 2021, 21, 2100039 CrossRef CAS PubMed
- A. Domiński, T. Konieczny, M. Godzierz, M. Musioł, H. Janeczek, A. Foryś, M. Domińska, G. Pastuch-Gawołek, T. Piotrowski and P. Kurcok, Pharmaceutics, 2022, 14, 2490 CrossRef PubMed
- W. Chen, K. Shi, J. Liu, P. Yang, R. Han, M. Pan, L. Yuan, C. Fang, Y. Yu and Z. Qian, Bioact. Mater., 2023, 23, 1–15 CAS
- M. Norouzi, B. Nazari and D. W. Miller, Drug Discovery Today, 2016, 21, 1835–1849 CrossRef CAS PubMed
- M. K. Nguyen and D. S. Lee, Macromol. Biosci., 2010, 10, 563–579 CrossRef CAS PubMed
- P. Z. Elias, G. W. Liu, H. Wei, M. C. Jensen, P. J. Horner and S. H. Pun, J. Controlled Release, 2015, 208, 76–84 CrossRef CAS PubMed
- X. Wu, C. He, Y. Wu and X. Chen, Biomaterials, 2016, 75, 148–162 CrossRef CAS PubMed
- T. Wang, J. Ding, S. Liang, Z. Lin, J. Yang, Z. Zhang, Z. Zou, G. Li, X. Chen and C. He, Adv. Funct. Mater., 2023, 2312360 Search PubMed
- J. Wu, Y. Qu, K. Shi, B. Chu, Y. Jia, X. Xiao, Q. He and Z. Qian, Chin. Chem. Lett., 2018, 29, 1819–1823 CrossRef CAS
- J. Shi, L. Yu and J. Ding, Acta Biomater., 2021, 128, 42–59 CrossRef CAS PubMed
- Q. Xu, C. He, K. Ren, C. Xiao and X. Chen, Adv. Healthcare Mater., 2016, 5, 1979–1990 CrossRef CAS PubMed
- Y. Shen, X. H. Fu, W. X. Fu and Z. B. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC
- Z. Lin, J. Ding, X. Chen and C. He, Chem. – Asian J., 2023, 18, e202300021 CrossRef CAS PubMed
- Y. Zhou, Z. Chen, D. Zhao, D. Li, C. He and X. Chen, Adv. Mater., 2021, 33, 2102044 CrossRef CAS PubMed
- B. Halliwell, M. V. Clement and L. H. Long, FEBS Lett., 2000, 486, 10–13 CrossRef CAS PubMed
- J. Moskovitz, S. Bar-Noy, W. M. Williams, B. S. Berlett and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12920–12925 CrossRef CAS PubMed
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef CAS PubMed
- S. Yu, C. Wang, J. Yu, J. Wang, Y. Lu, Y. Zhang, X. Zhang, Q. Hu, W. Sun, C. He, X. Chen and Z. Gu, Adv. Mater., 2018, 30, 1801527 CrossRef PubMed
- M. J. Webber and E. T. Pashuck, Adv. Drug Delivery Rev., 2021, 172, 275–295 CrossRef CAS PubMed
- M. Ren, D. Dong, Q. Xu, J. Yin, S. Wang and F. Kong, Talanta, 2021, 234, 122684 CrossRef CAS PubMed
- Q. Lv, S. J. Yu, F. L. Quan, C. L. He and X. S. Chen, Adv. Ther., 2020, 3, 1900165 CrossRef
- J. C. Ioannou, A. M. Donald and R. H. Tromp, Food Hydrocolloids, 2015, 46, 216–225 CrossRef CAS
- J. Fucikova, O. Kepp, L. Kasikova, G. Petroni, T. Yamazaki, P. Liu, L. Zhao, R. Spisek, G. Kroemer and L. Galluzzi, Cell Death Dis., 2020, 11, 1013 CrossRef CAS PubMed
- S.-J. Park, W. Ye, R. Xiao, C. Silvin, M. Padget, J. W. Hodge, C. Van Waes and N. C. Schmitt, Oral Oncol., 2019, 95, 127–135 CrossRef CAS PubMed
- A. Mohapatra, S. K. Rajendrakumar, K. Cherukula, M.-S. Park, S. Padmanaban, A. Vasukuty, A. Mohanty, J. Y. Lee, W. K. Bae and I.-K. Park, Biomater. Sci., 2023, 11, 1853–1866 RSC
- F. Veglia and D. I. Gabrilovich, Curr. Opin. Immunol., 2017, 45, 43–51 CrossRef CAS PubMed
- L. Tang, Y. Yin, Y. Cao, C. Fu, H. Liu, J. Feng, W. Wang and X.-J. Liang, Adv. Mater., 2023, 35, 2303835 CrossRef CAS PubMed
- H. Raskov, A. Orhan, J. P. Christensen and I. Gögenur, Br. J. Cancer, 2021, 124, 359–367 CrossRef CAS PubMed
- R. V. Luckheeram, R. Zhou, A. D. Verma and B. Xia, Clin. Dev. Immunol., 2012, 925135 Search PubMed
- K. Schroder, P. J. Hertzog, T. Ravasi and D. A. Hume, J. Leukocyte Biol., 2004, 75, 163–189 CrossRef CAS PubMed
- D.-I. Jang, A. H. Lee, H.-Y. Shin, H.-R. Song, J.-H. Park, T.-B. Kang, S.-R. Lee and S.-H. Yang, Int. J. Mol. Sci., 2021, 22, 2719 CrossRef CAS PubMed
- P. Uciechowski and W. C. M. Dempke, Oncology, 2020, 98, 131–137 CrossRef CAS PubMed
- R. Ma, T. Ji, H. Zhang, W. Dong, X. Chen, P. Xu, D. Chen, X. Liang, X. Yin, Y. Liu, J. Ma, K. Tang, Y. Zhang, Y. Peng, J. Lu, Y. Zhang, X. Qin, X. Cao, Y. Wan and B. Huang, Nat. Cell Biol., 2018, 20, 21–27 CrossRef CAS PubMed
- K. Inaba, M. Inaba, N. Romani, H. Aya, M. Deguchi, S. Ikehara, S. Muramatsu and R. M. Steinman, J. Exp. Med., 1992, 176, 1693–1702 CrossRef CAS PubMed
- S. Manzanero, Methods Mol. Biol., 2012, 844, 177–181 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4bm00241e |
| This journal is © The Royal Society of Chemistry 2024 |