
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ near-ambient pressure X-ray photoelectron spectroscopy reveals the effects of water, oxygen and light on the stability of PM6:Y6 photoactive layers†
Qilun
Zhang
 *ab,
Yongzhen
Chen
a,
Xianjie
Liu
a and
Mats
Fahlman
ab
*ab,
Yongzhen
Chen
a,
Xianjie
Liu
a and
Mats
Fahlman
ab
aLaboratory of Organic Electronics, Department of Science and Technology (ITN), Linköping University, SE-60174, Norrköping, Sweden. E-mail: qilun.zhang@liu.se
bWallenberg Wood Science Center, Department of Science and Technology (ITN), Linköping University, SE-60174, Norrköping, Sweden
First published on 1st February 2023
Abstract
The power conversion efficiency of organic solar cells (OSCs) has taken a further leap in the past three years owing to the emergence of Y6; however, their inferior stability hinders commercialization. Understanding the ambient degradation mechanism of photovoltaic materials is a key component to address this challenge. In this study, we first used in situ near-ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) to investigate the effects of water, oxygen and absorbed photons on the stability of PM6 and Y6. The studied materials PM6 and Y6 show instability to oxygen and water, respectively, possibly due to the weak interaction between PM6 backbone sulphur and oxygen, and Y6 end cyano groups show instability to water. In addition, the stability of Y6 in blended PM6:Y6 films is enhanced, which is confirmed by the performance of OSCs with blended or quasi-bilayer PM6:Y6 photoactive layers. Our findings reveal PM6 and Y6 degradation on ambient exposure and predict a possible way to prevent the degradation of Y6 in OSCs.
Introduction
Transitioning the energy requirement of human civilization away from fossil fuels and towards renewable solar energy is a world priority, and potentially offers solutions to the problems of energy crisis, carbon neutrality and environmental pollution. Organic solar cells (OSCs) based on organic semiconductors have attracted increasing attention owing to the advantages of low cost, lightweight, flexibility, ease of fabrication and synthesis-based tailoring depending on the purpose.1–4 Progress has been made in their research and development to pursue higher power conversion efficiency (PCE) by using strategies such as exploiting novel donor and non-fullerene acceptor materials,5–10 improving the morphology of photoactive layers (PALs),10–13 integrating ternary components into bulk heterojunction (BHJ) blends,14–17 optimizing the device structure with novel charge transport layers and different fabrication processes.18–21 As a result, the PCE of OSCs has been boosted from 10% to over 19% in recent years. Nevertheless, the relatively low stability of OSCs is perhaps the most prominent obstacle for commercialization.The origins of the instability or degradation in OSCs are related to a series of factors, which mostly come from the operation of cells. Organic photo-active semiconductors are easily affected by absorbed water and oxygen, resulting in a change in the electronic structure even without strong chemical interactions.22,23 Moreover, water- and oxygen-induced oxidation will affect the electrical and optical properties, e.g., decrease in conductivity and bleaching of films.24,25 The low work function electrodes (e.g., Ca and Al) also suffer from performance loss due to oxygen/water exposure. Insulating metal oxide layers can be formed by water- and oxygen-induced oxidation, which significantly hinders charge extraction in OSCs.26,27 Light and heat are also considered to be significant factors in causing performance degradation in OSCs. Specifically, the morphology of the PAL is easily changed by the accumulation of heat, whereas ultraviolet light is even much more harmful than heat, as it will induce irreversible decomposition of organic semiconductor materials.28–30 A deeper understanding of the degradation mechanism clearly is the key to improving OSC stability and needs to be further explored.
As 2,2′-((2Z,2′Z)-((12,13-bis(2-ethylhexyl)-3,9-diundecyl-12,13-dihydro-[1,2,5]thiadiazolo[3,4-e]thieno[2′′,3′′:4′,5′]thieno [2′,3′:4,5]pyrrolo[3,2-g]thieno[2′,3′:4,5]thieno[3,2-b]indole-2,10-diyl)bis(methanylylidene))bis(5,6-difluoro-3-oxo-2,3-dihydro-1H-indene-2,1-diylidene))dimalononitrile (Y6) is the “star” fused-ring electron acceptor (FREA) material, and to a large degree, contributes to the top PCE exceeding 19%. Y6 is therefore a material of particular interest for studying the stability of state-of-the-art OSCs. Indeed, A–D–A type FREAs have an intrinsic instability due to the strong electron-deficient characteristic of exocyclic vinyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) as conjugated bridges to link the D unit with the A unit.31–34 Different types of photochemical reactions have been demonstrated in ITIC-based FREAs by a combination of UV-vis and IR spectroscopies.25 In this work, to further assess the influence of ambient atmosphere on advanced OSC PALs’ stability, we monitor the O2 gas, water vapor and photon flux interaction with poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] (PM6) and Y6 films by in situ NAP-XPS, in which the in situ response of organic materials to the external environment can be detected. With the NAP-XPS method, we can precisely control the environment of the organic films including the type of gas, pressure and illumination while tracking the chemical modification (core levels) and the corresponding effect on the electronic structure (valence region). Our studies show, first, that water and oxygen interact weakly with the backbone sulphur and cyano groups of PM6 and Y6, respectively. Second, Y6 films are more sensitive to O2 gas and water vapor than PM6:Y6 blend films typically featured in BHJ OSCs. We tested our results and conclusions on OPV devices based on blended PM6:Y6 PALs and quasi-bilayer PM6/Y6 PALs, and the expected degradation effects are confirmed.
C) as conjugated bridges to link the D unit with the A unit.31–34 Different types of photochemical reactions have been demonstrated in ITIC-based FREAs by a combination of UV-vis and IR spectroscopies.25 In this work, to further assess the influence of ambient atmosphere on advanced OSC PALs’ stability, we monitor the O2 gas, water vapor and photon flux interaction with poly[(2,6-(4,8-bis(5-(2-ethylhexyl-3-fluoro)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] (PM6) and Y6 films by in situ NAP-XPS, in which the in situ response of organic materials to the external environment can be detected. With the NAP-XPS method, we can precisely control the environment of the organic films including the type of gas, pressure and illumination while tracking the chemical modification (core levels) and the corresponding effect on the electronic structure (valence region). Our studies show, first, that water and oxygen interact weakly with the backbone sulphur and cyano groups of PM6 and Y6, respectively. Second, Y6 films are more sensitive to O2 gas and water vapor than PM6:Y6 blend films typically featured in BHJ OSCs. We tested our results and conclusions on OPV devices based on blended PM6:Y6 PALs and quasi-bilayer PM6/Y6 PALs, and the expected degradation effects are confirmed.
Results and discussion
The chemical structures of the donor PM6, acceptor Y6 and their UPS spectra are shown in Fig. S1 (ESI†). The light-absorption spectra and energy levels of PM6 and Y6 are well matched for use as PALs in OSCs. The stability of PM6 and Y6 neat films was investigated by sequentially exposing to O2 gas and water vapor at a pressure of about 1–2 mbar in the darkness or under (non-UV) illumination. The exposure time was controlled between 20 and 60 min before each spectrum was recorded at the same pressure, and there was negligible change observed in the core levels between the exposures (Fig. S2, ESI†). The corresponding results of the core-level spectra are shown in Fig. 1: C 1s and S 2p for both PM6 and Y6, and N 1s and F 1s spectra in addition for Y6. The data for the fresh samples are in good agreement with the core-level spectra expected from the chemical structures of PM6 and Y6.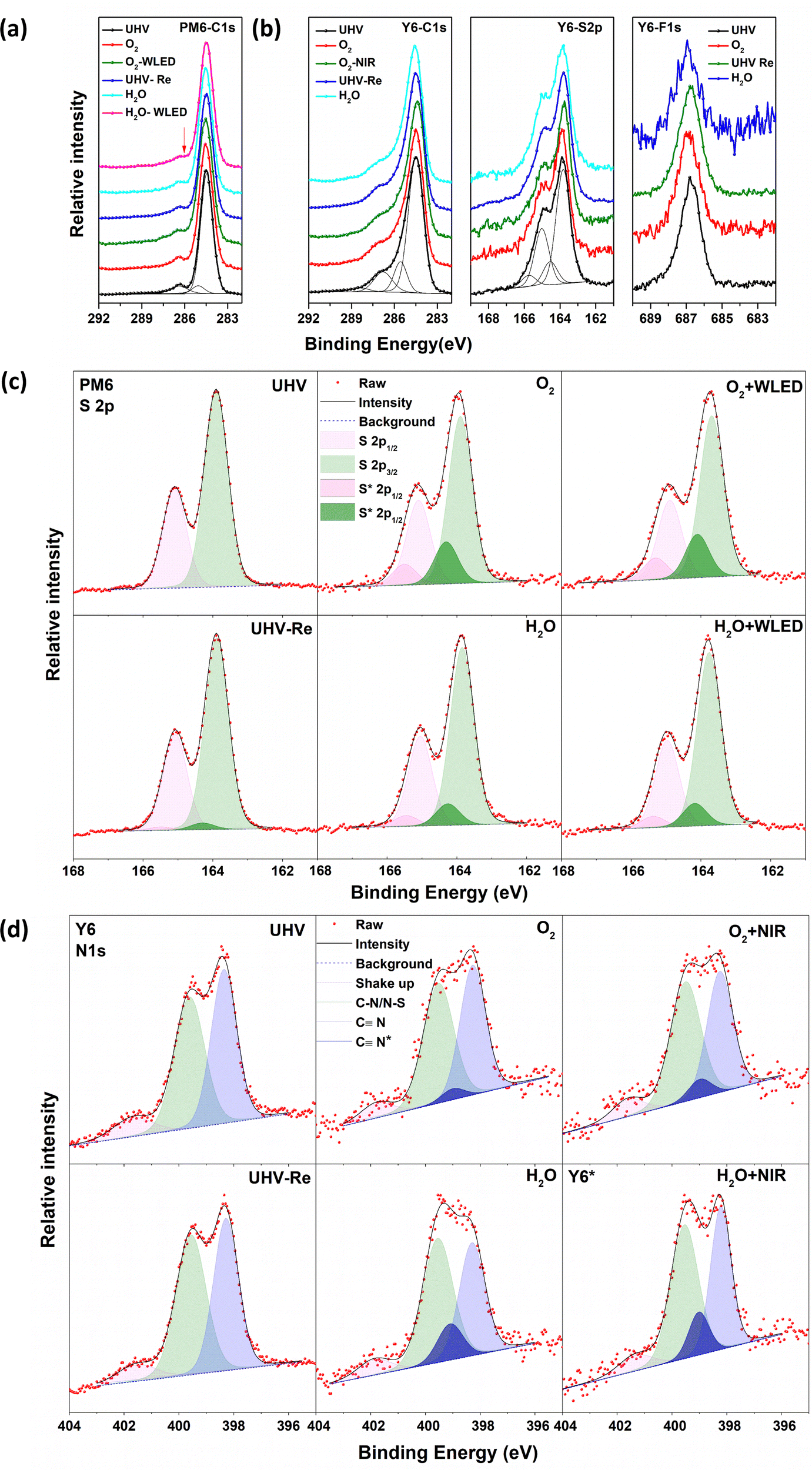 | ||
| Fig. 1 Evolution of core-level spectra: (a) C 1s of the PM6 film and (b) C 1s, S 2p, and F 1s of the Y6 film under different exposure conditions. Deconvolution of the core-level spectral evolution of (c) XPS S 2p of the PM6 film and (d) N 1s of the Y6 film upon different exposure conditions. | ||
The C 1s spectra with fitted peaks of neat PM6 films (left panel in Fig. 1a) measured in UHV show a symmetric hydrocarbon-like main component at 284.5 eV due to C–C bonding in the side chains and aromatic rings and few contributions from the C–S bond. We assigned the weak component at approximately 286.3 eV as a shake-up feature. The contributions from the CO and C–F bonds are not clearly resolved as they overlap at approximately 288.5 eV. When the PM6 films are exposed to O2, the main peak at 284.5 eV shows no shift but there is a slight flattening of the shoulder at about 286.3 eV. This spectral modification is more obvious after the additional 10 min illumination by a white light-emitting diode (WLED, wavelength 400–700 nm), indicating that the chemical environment of some of the C–S sites has been changed, causing a shift in their binding energy. The C 1s spectra cannot fully be recovered to the pristine PM6 state even after pumping out O2 reaching 10−10 mbar. Exposure to water vapor shows similar phenomena to O2, i.e., modification of some of the C–S sites.
The S 2p spectra show the S 2p3/2–2p1/2 doublet with the S 2p3/2 component at 163.9 eV, typical of sulphur in C–S bonds as expected from the PM6 chemical structure. For films exposed to O2 or water vapor, we add to the S 2p spectra additional doublet peaks at a higher binding energy with reasonable fitting, in agreement with the C 1s core-level evolution. We assign this additional doublet to the existence of S interacting with water or O2. The experimental S 2p spectra and the fitted peaks are shown in Fig. 1c, where the O2/water exposure-induced doublet peaks are marked with S*. The relative peak areas are summarized in Table 1 (fitting parameters are given in the ESI†). After 20 min of O2 exposure, an S* doublet with about 20.1% of the total intensity can be observed, reaching 21.2% after WLED illumination. After pumping O2 away, the S* doublet feature decreases to 3.4%, which implies that the O2 interaction is largely reversible and, hence, weak. PM6 films are less sensitive to water vapor exposure, as the fitting results show about 10.8% and 11.6% total intensity of S* films upon exposure to water vapor and WLED illumination, respectively. It should be noted that no feature at ∼168 eV is observed, indicating that sulphone groups (–SO3−) are not formed at these timescales of O2/water exposure under non-UV white-light illumination.
| Film | Element | UHV (%) | O2 (%) | O2-light (%) | UHV-Re (%) | H2O (%) | H2O-light (%) |
|---|---|---|---|---|---|---|---|
| PM6 | S 2p | 0 | 20.1 | 21.2 | 3.4 | 10.8 | 11.6 |
| Y6 | N 1s | 0 | 7.2 | 12.2 | 0 | 23.8 | 23.6 |
| PM6:Y6 | N 1s | 0 | 3.8 | 11.8 | 0 | 12.5 | 30.5 |
For Y6 films, a similar main component in PM6 can be found in the C 1s spectra from C–C bonding in the side chains and aromatic rings (Fig. 1b). However, the weak feature at ∼286.3 eV of the Y6 films is slightly different with a more extended shoulder, assigned to contribution from C–S bonds, shake-up and the various bonds between C and N. The tail feature at approximately 288.5 eV is from CO and C–F bonds. Unlike the PM6 films, the C 1s spectrum shows negligible change of the shoulder peak after exposure to O2, water and near-Infrared range (NIR, wavelength 800–950 nm) illumination. Two doublets are needed to fit the S 2p spectra of the Y6 films, with S 2p3/2 components at 163.8 and 164.5 eV for S–C and S–N bonds respectively. We see only slight changes in the S 2p spectrum upon exposure to water, O2 and illumination. Moreover, the F 1s spectra at 686.8 eV also show no change. All the observed results indicate that there is no detectable interaction of C, S, and F in Y6 to O2/water exposure and illumination.
As shown in Fig. 1d, the N 1s spectrum of pristine Y6 films at UHV can be deconvoluted with two main components at 398.4 and 399.6 eV, respectively, and a satellite peak at 401.7 eV. Based on the chemical structure of the Y6 molecule, the filled light blue peak is assigned to cyano groups (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N), and the other filled green peak is assigned to C–N and N–S bonds. The green peak has slightly broader FWHM but similar spectral weight as the blue one. The pink satellite feature at 401.7 eV is assigned to shake-up.35 Upon exposure to O2, water and illumination, the N 1s spectrum undergoes obvious changes in the spectral features. One additional component located between the two main features is needed to fit the N 1s spectra, which we use to track the influence of the various types of exposures on the Y6 film. The extra peak is labelled as CN* as appearance of this feature coincides with a decrease of the original N 1s cyano feature, indicating that water interacts preferentially with this group. The relative percentages of CN* are summarized in Table 1. The percentage of CN* represents the relative change of the N 1s spectrum, i.e., the percentage of N atoms that has been influenced by O2/water exposure or light illumination. When the Y6 film is exposed to O2 and additional illumination, the CN* peak percentage reaches 7.2% and 12.2% respectively. After pumping O2 down to UHV, this feature disappears, indicating a reversible and hence weak interaction between Y6 and O2. Water exposure induces a larger CN* percentage, 21.1%. The addition of illumination slightly increases CN* further to 22.7%. This result indicates that the Y6 film is more sensitive to water atmosphere than to O2. In addition, we suspect that modification of the cyano groups can affect the linked exocyclic vinyl groups, contributing to the breakage of the D–A bridge, as reported in other FREA materials.31–34
N), and the other filled green peak is assigned to C–N and N–S bonds. The green peak has slightly broader FWHM but similar spectral weight as the blue one. The pink satellite feature at 401.7 eV is assigned to shake-up.35 Upon exposure to O2, water and illumination, the N 1s spectrum undergoes obvious changes in the spectral features. One additional component located between the two main features is needed to fit the N 1s spectra, which we use to track the influence of the various types of exposures on the Y6 film. The extra peak is labelled as CN* as appearance of this feature coincides with a decrease of the original N 1s cyano feature, indicating that water interacts preferentially with this group. The relative percentages of CN* are summarized in Table 1. The percentage of CN* represents the relative change of the N 1s spectrum, i.e., the percentage of N atoms that has been influenced by O2/water exposure or light illumination. When the Y6 film is exposed to O2 and additional illumination, the CN* peak percentage reaches 7.2% and 12.2% respectively. After pumping O2 down to UHV, this feature disappears, indicating a reversible and hence weak interaction between Y6 and O2. Water exposure induces a larger CN* percentage, 21.1%. The addition of illumination slightly increases CN* further to 22.7%. This result indicates that the Y6 film is more sensitive to water atmosphere than to O2. In addition, we suspect that modification of the cyano groups can affect the linked exocyclic vinyl groups, contributing to the breakage of the D–A bridge, as reported in other FREA materials.31–34
XPS core-level spectra of PM6:Y6 blend films were studied as well. The samples are divided into two groups, with and without annealing, since annealing post treatments commonly used in OSCs will strongly affect the morphology of the films, which may further make a difference in the interaction between water/O2 species and OSC materials. The C 1s and S 2p spectra for annealed PM6:Y6 films are shown in Fig. S4a (ESI†). All spectra show mixed features from both PM6 and Y6. The changes in the C 1s and S 2p spectra under different exposures are weak and very similar to the neat Y6 film. We focus on the evolution of the N 1s spectrum, as shown in Fig. 2, since it best indicates the stability of Y6 in the blended film. We carefully analysed the N 1s spectra, following the same guidelines of the neat Y6 film. The two main peaks remain at similar binding energies to the neat Y6 film at 398.32 and 399.6 eV in UHV, respectively, indicating that most Y6 molecules are unaffected by exposure. There is an additional N 1s feature (CN*) appearing upon O2/water exposure in the fitted spectra. The relative percentage of this new feature is summarized in Table 1. The N 1s spectra of PM6:Y6 films show significantly smaller influence by O2 and water compared to the neat Y6 films with values of 3.8% and 12.5% respectively for the CN* feature. We propose that the surrounding PM6 polymer phase will partially block the interaction between water/O2 and Y6. However, the interaction is strongly affected by NIR illumination, whereupon the CN* percentage reaches 11.8% and 30.5% for O2 and water exposure, respectively. Similar phenomena are observed in the as-cast un-annealed PM6:Y6 film, as shown in Fig. S4b (ESI†).
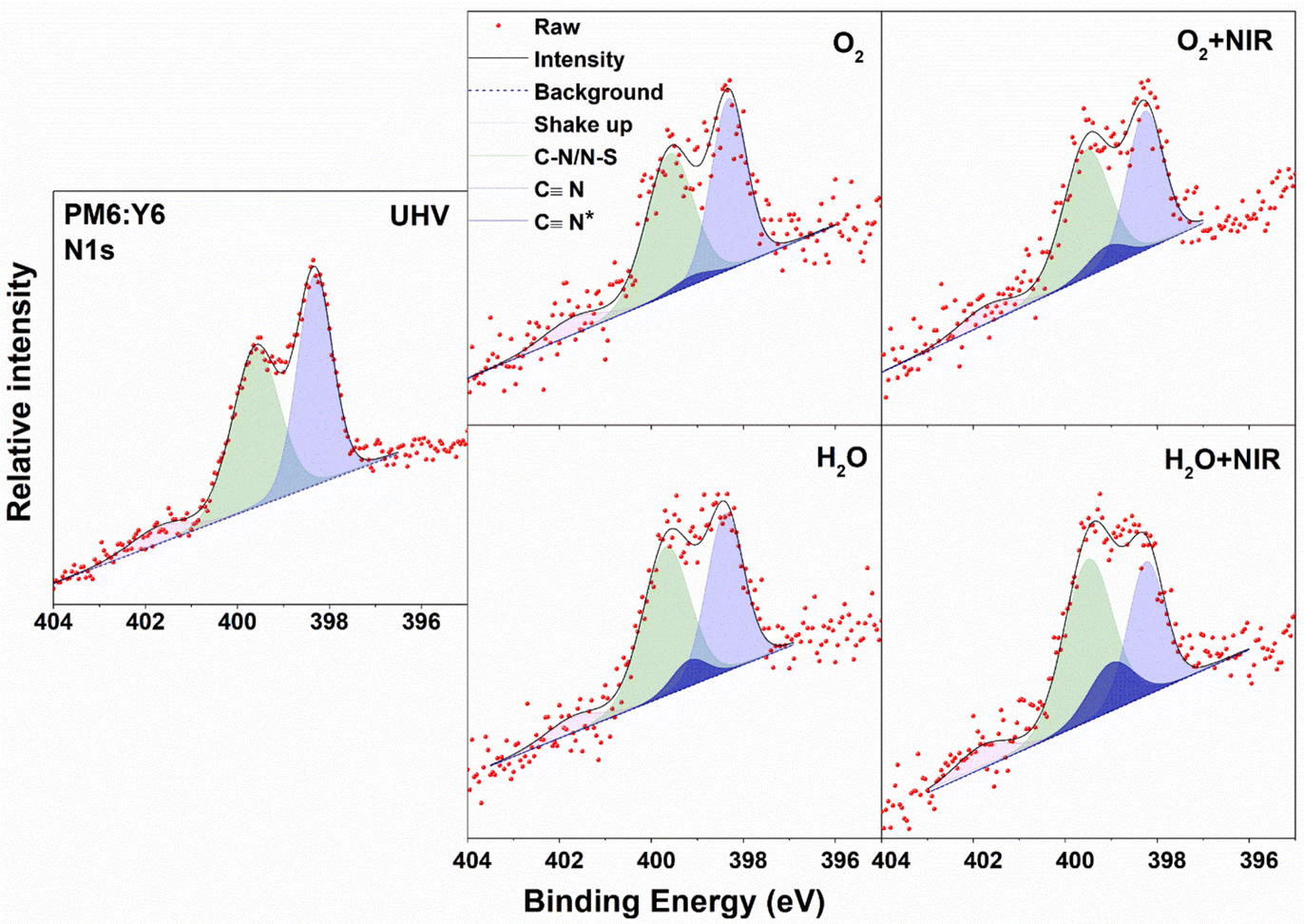 | ||
| Fig. 2 Deconvolution of the N 1s core-level spectra of the PM6:Y6 blend film upon exposure to O2, water and illumination. | ||
We now explore if the modifications in the chemical environment observed from the core-level spectra affect the frontier electronic structures of PM6, Y6 and blend films. The evolution of the valence band (VB) spectra upon different types of exposures is shown in Fig. 3. The valence band spectra of neat PM6 films show a strong and well-resolved feature at ∼3.5 eV, whereas the frontier feature derived from the HOMO is weak and lacks a sharp edge. When exposed to O2 and illumination, the frontier VB features all undergo modification, most clearly visible in the 3.5 eV feature that becomes broader with decreased intensity, indicating enhanced energy disorder in the PM6 electronic structure. For O2 exposure in the darkness, we still observe a slight broadening of the 3.5 eV feature, but the corresponding effect in the already initially weak and less well-defined VB edge is not clearly visible. Nevertheless, the broadening/smearing out of the frontier VB features is indicative of increased energetic disorder. Like in the core-level spectra, the second frontier peak is partially recovered when pumping down back to UHV, suggesting that the energetic disorder is induced by weak electrostatic interactions between O2 and sulphur groups rather than changes in morphology. Water exposure has a much weaker effect on the frontier valence electronic structure of PM6, essentially introducing no further modification, in agreement with the PM6 film core level results. In contrast, the valence band features located at 1.20, 2.18 and 3.38 eV for the Y6 films are clearly significantly affected by O2, becoming smeared out when exposed to O2 and even more so upon light illumination. However, by pumping down to UHV, the original VB structure can be almost completely recovered. This is in agreement with the Y6 N 1s core level results. Water exposure also induces significant smearing of the Y6 VB features, and the original spectrum is only partially recovered when pumping out water. This follows the evolution of the N 1s core level, where the original cyano feature at 398.4 eV remains suppressed even after pumping out water down to UHV (Fig. S5, ESI†). For the blended films, the valence band spectra show only slight energetic broadening of the second frontier feature upon O2 exposure, with stronger modification in the spectra when exposed to water and NIR illumination, consistent with our observations from the N 1s spectra of blended films.
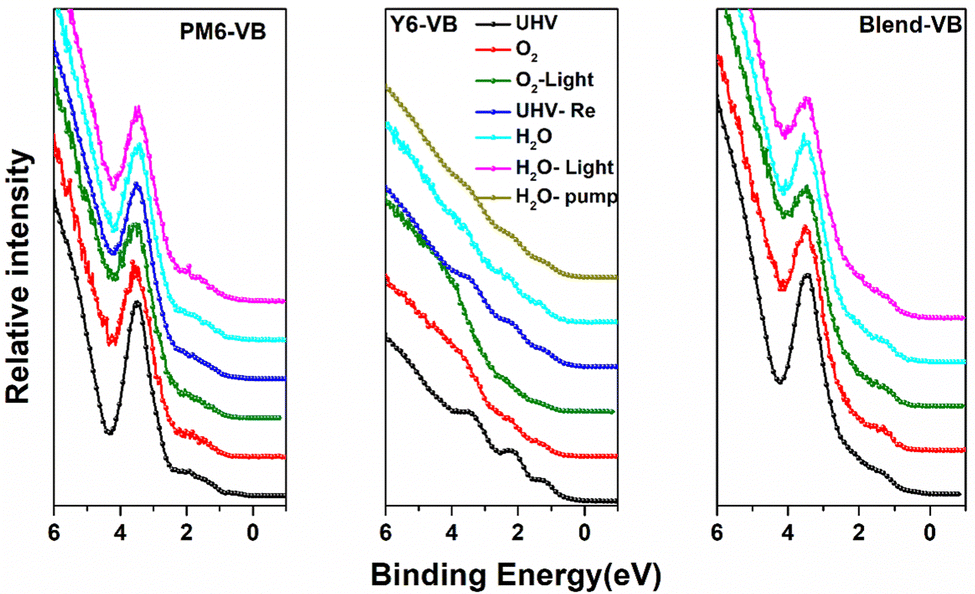 | ||
| Fig. 3 Evolution of the VB spectra of PM6,Y6 and blend films upon exposure to O2, water and illumination. | ||
Based on the combined core level and valence band region results, we propose that O2 exposure primarily causes (reversible) p-type doping36,37 that is enhanced by illumination and that the effect is more pronounced for the PM6 donor than the Y6 acceptor. O2 is unlikely to affect p-type doped Y6 in the absence of illumination due to its deep highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), but the presence of O2 in nanovoids can still locally modify the electronic structure and induce trap states. Water exposure effects, however, are more significant for the Y6 acceptor than for the PM6 donor, possibly due to Y6 being a FREA material with intrinsic fragile exocyclic vinyl groups. Note that blending Y6 with PM6 improves the stability, with blended PM6:Y6 PAL having better stability to both O2 and water than that of the “best” individual component films under the same conditions. Furthermore, the results suggest that short-term exposure does not lead to significant irreversible degradation.
To explore if the effects on the electronic structure observed by NAP-XPS modify device performance, OSCs were fabricated with different architectures of blended and quasi-bilayer PALs. As shown in Fig. 4a, the devices adopt a conventional configuration of indium tin oxide (ITO)/poly(3,4-ethylenedioxythiophene): poly(styrene sulfonate) (PEDOT:PSS)/PAL/N,N′-bis{3-[3(dimethylamino)propylamino]propyl}perylene-3,4,9,10-tetracarboxylic diimide (PDINN)/Ag, where PEDOT:PSS and PDINN served as the anode and cathode interlayers, respectively. To explore the device degradation caused by water and O2, OSCs without encapsulation were separately stored in the darkness in a glovebox or a fume hood, respectively.
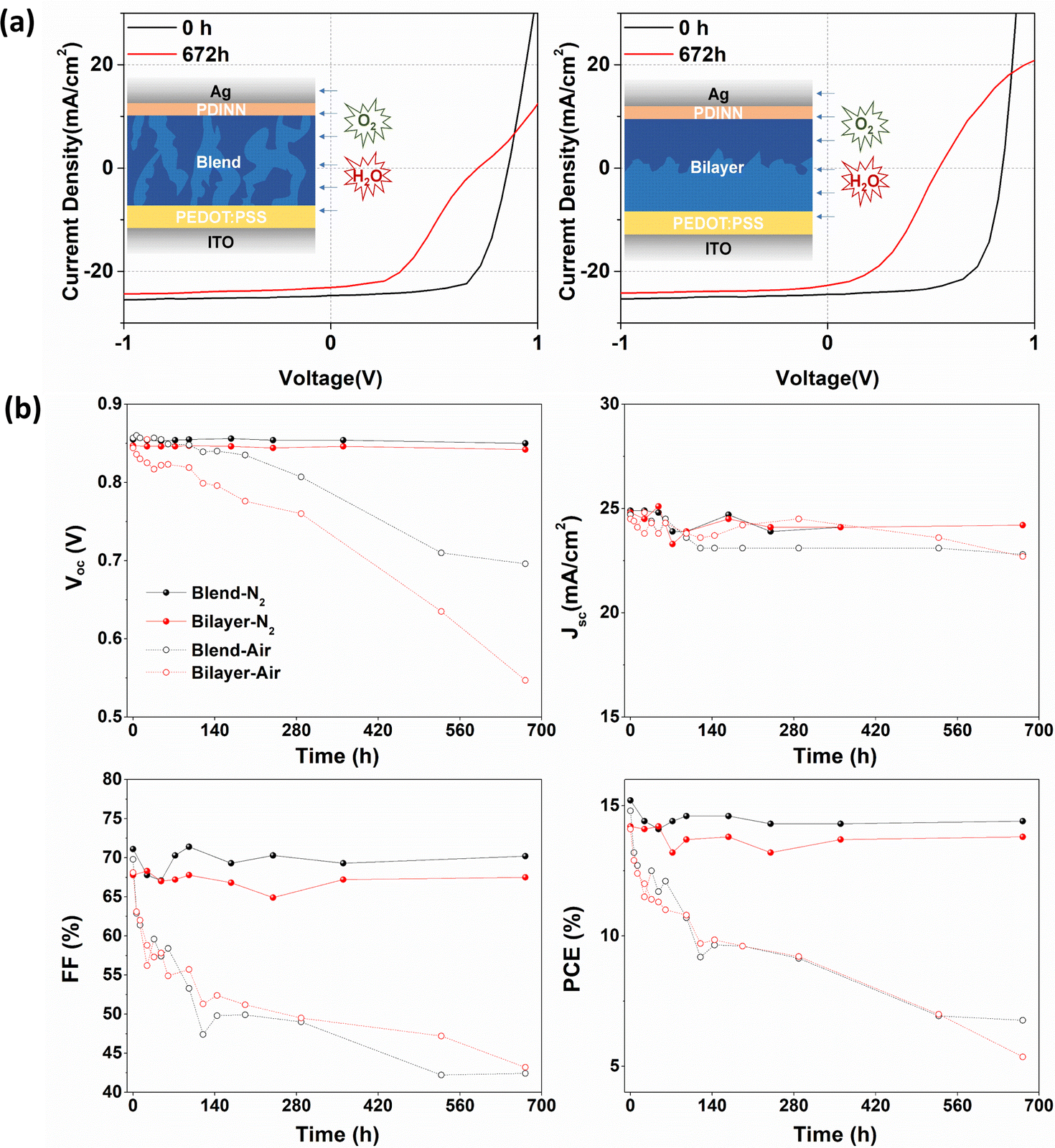 | ||
| Fig. 4 (a) Scheme of water and O2 diffusion into the OSC, and the corresponding J–V curves with blended and bilayer PALs stored in air. (b) Performance evolution of OSCs with blended (black curve) and bilayer (red curve) PALs stored in a glovebox (N2 – solid curve) and fume hood (air – dashed curve) for 672 hours. | ||
The devices stored in a fume hood show extremely strong degradation compared to the ones in the glovebox, as observed from the summary of the time-dependent photovoltaic parameters and current density–voltage (J–V) curves (Fig. 4a, b, and Fig. S6, ESI†). In Fig. 4b, the data on the performance of N2-stored and air-stored devices are drawn with solid and open circles respectively, and the devices with blended PALs and bilayer PALs are drawn with black and red curves, respectively. The glovebox-stored devices retained more than 95% of their photovoltaic performance with only slight degradation of open-circuit voltage (VOC), short-circuit current (JSC) and fill factor (FF) compared to the fresh state, whereas the fume hood stored devices show more than 50% reduction of PCE. We assume that most of the degradation is caused by the diffusion of water and O2 into the PALs from the atmosphere. The blended PAL-based devices show better VOC stability towards air exposure, in agreement with the core level and VB results. The VOC depends on the energy gap between the HOMO level of the donor and the LUMO level of the acceptor in the PAL. However, the inhomogeneous broadening of HOMO or LUMO levels (energetic disorder) reduces the VOC due to the accumulation of charge carries in the tail of the electronic density of states.38–40 The bilayer PAL system with a neat Y6 phase at the top layer suffers from more VOC loss caused by water/O2-induced energetic disorders as expected, given the VB and N 1s core-level spectra results. The Jsc and FF evolve in the same way for the blended and bilayer PAL-based devices.
Conclusions
In summary, we conducted in situ NAP-XPS to track the changes in the OSC materials PM6 and Y6 under O2/water exposure and/or illumination in real time. The results indicated that the modification of the spectral features originates from the interaction between PM6, Y6 molecules and exposed O2 or water species, with relatively strong interaction of O2 to PM6 and water to Y6. The main observation from the NAP-XPS results is that sulphur is the main interaction site for PM6 and cyano nitrogen for Y6. We propose that the evolution of the core-level and VB spectra indicates reversible “p-doping” of PM6 for both O2/water exposure, accelerated by illumination. In contrast, O2 exposure has a significantly weaker effect on Y6, but water exposure leads to partially irreversible modifications of chemical and VB structures for Y6. Additional results on PM6:Y6 blend films revealed an enhanced stability of Y6 in the darkness, which is consistent with the device data, where blend OSCs better retain the original VOC. However, our results indicated that the stability is still not sufficient to obtain high PCE devices of PM6:Y6 PALs that have been exposed to air even under dark conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the Knut and Alice Wallenberg Foundation (KAW) through the Wallenberg Wood Science Center. The work in addition was in part supported by the Swedish Energy Agency (grant. no. 45411-1), by the Swedish Research Council (project grants 2016-05498, 2016-05990, 2020-04538, and 2018-06048), by the STINT grant (CH2017-7163) and by the Swedish Government Strategic Research Area in Materials Science on Functional Materials at Linköping University (Faculty Grant SFO Mat LiU no. 2009 00971).Notes and references
- J. Hou, O. Inganäs, R. H. Friend and F. Gao, Nat. Mater., 2018, 17, 119–128 CrossRef CAS PubMed.
- M. Kaltenbrunner, M. S. White, E. D. Głowacki, T. Sekitani, T. Someya, N. S. Sariciftci and S. Bauer, Nat. Commun., 2012, 3, 1–7 Search PubMed.
- G. Li, R. Zhu and Y. Yang, Nat. Photonics, 2012, 6, 153–161 CrossRef CAS.
- C. Sun, F. Pan, H. Bin, J. Zhang, L. Xue, B. Qiu, Z. Wei, Z.-G. Zhang and Y. Li, Nat. Commun., 2018, 9, 1–10 CrossRef PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Yuan, Y. Zhang, L. Zhou, G. Zhang, H.-L. Yip, T.-K. Lau, X. Lu, C. Zhu, H. Peng and P. A. Johnson, Joule, 2019, 3, 1140–1151 CrossRef CAS.
- P. Bi, S. Zhang, Z. Chen, Y. Xu, Y. Cui, T. Zhang, J. Ren, J. Qin, L. Hong and X. Hao, Joule, 2021, 5, 2408–2419 CrossRef CAS.
- Y. Cui, H. Yao, J. Zhang, K. Xian, T. Zhang, L. Hong, Y. Wang, Y. Xu, K. Ma and C. An, Adv. Mater., 2020, 32, 1908205 CrossRef CAS PubMed.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao and K. Sun, Sci. Bull., 2020, 65, 272–275 CrossRef CAS PubMed.
- H. Yao, Y. Cui, D. Qian, C. S. Ponseca Jr, A. Honarfar, Y. Xu, J. Xin, Z. Chen, L. Hong and B. Gao, J. Am. Chem. Soc., 2019, 141, 7743–7750 CrossRef CAS PubMed.
- Z. Wang, K. Gao, Y. Kan, M. Zhang, C. Qiu, L. Zhu, Z. Zhao, X. Peng, W. Feng and Z. Qian, Nat. Commun., 2021, 12, 1–14 CrossRef CAS PubMed.
- L. Ma, H. Yao, J. Wang, Y. Xu, M. Gao, Y. Zu, Y. Cui, S. Zhang, L. Ye and J. Hou, Angew. Chem., Int. Ed., 2021, 60, 15988–15994 CrossRef CAS PubMed.
- Q. Wang, Y. Qin, M. Li, L. Ye and Y. Geng, Adv. Energy Mater., 2020, 10, 2002572 CrossRef CAS.
- H. Chen, R. Zhang, X. Chen, G. Zeng, L. Kobera, S. Abbrent, B. Zhang, W. Chen, G. Xu and J. Oh, Nat. Energy, 2021, 6, 1045–1053 CrossRef CAS.
- L. Zuo, S. B. Jo, Y. Li, Y. Meng, R. J. Stoddard, Y. Liu, F. Lin, X. Shi, F. Liu and H. W. Hillhouse, Nat. Nanotechnol., 2022, 17, 53–60 CrossRef CAS PubMed.
- C. Wang, X. Liu, Y. Xiao, J. Bergqvist, X. Lu, F. Gao and M. Fahlman, Sol. RRL, 2020, 4, 2000261 CrossRef CAS.
- Q. L. Zhang, H. T. Zhang, Z. Wu, C. F. Wang, R. Zhang, C. Y. Yang, F. Gao, S. Fabiano, H. Y. Woo, M. Ek, X. J. Liu and M. Fahlman, Sol. RRL, 2022, 6.9, 2200381 CrossRef.
- J. Yao, B. Qiu, Z.-G. Zhang, L. Xue, R. Wang, C. Zhang, S. Chen, Q. Zhou, C. Sun and C. Yang, Nat. Commun., 2020, 11, 1–10 CrossRef PubMed.
- Y. Lin, B. Adilbekova, Y. Firdaus, E. Yengel, H. Faber, M. Sajjad, X. Zheng, E. Yarali, A. Seitkhan and O. M. Bakr, Adv. Mater., 2019, 31, 1902965 CrossRef CAS PubMed.
- Z. Wu, C. Sun, S. Dong, X.-F. Jiang, S. Wu, H. Wu, H.-L. Yip, F. Huang and Y. Cao, J. Am. Chem. Soc., 2016, 138, 2004–2013 CrossRef CAS PubMed.
- L. Zhan, S. Li, X. Xia, Y. Li, X. Lu, L. Zuo, M. Shi and H. Chen, Adv. Mater., 2021, 33, 2007231 CrossRef CAS PubMed.
- C. Wang, S. F. Ni, S. Braun, M. Fahlman and X. J. Liu, J. Mater. Chem. C, 2019, 7, 879–886 RSC.
- Q. Bao, X. Liu, S. Braun and M. Fahlman, Adv. Energy Mater., 2014, 4, 1301272 CrossRef.
- H. K. H. Lee, A. M. Telford, J. A. Röhr, M. F. Wyatt, B. Rice, J. Wu, A. de Castro Maciel, S. M. Tuladhar, E. Speller and J. McGettrick, Energy Environ. Sci., 2018, 11, 417–428 RSC.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. Tang, C. Sun, Z. Luo and K. Chang, J. Mater. Chem. A, 2019, 7, 25088–25101 RSC.
- M. Wang, F. Xie, J. Du, Q. Tang, S. Zheng, Q. Miao, J. Chen, N. Zhao and J.-B. Xu, Sol. Energy Mater. Sol. Cells, 2011, 95, 3303–3310 CrossRef CAS.
- K. Kawano, R. Pacios, D. Poplavskyy, J. Nelson, D. D. Bradley and J. R. Durrant, Sol. Energy Mater. Sol. Cells, 2006, 90, 3520–3530 CrossRef CAS.
- I. Sachs-Quintana, T. Heumüller, W. R. Mateker, D. E. Orozco, R. Cheacharoen, S. Sweetnam, C. J. Brabec and M. D. McGehee, Adv. Funct. Mater., 2014, 24, 3978–3985 CrossRef CAS.
- A. Classen, T. Heumueller, I. Wabra, J. Gerner, Y. He, L. Einsiedler, N. Li, G. J. Matt, A. Osvet and X. Du, Adv. Energy Mater., 2019, 9, 1902124 CrossRef CAS.
- O. R. Yamilova, I. V. Martynov, A. S. Brandvold, I. V. Klimovich, A. H. Balzer, A. V. Akkuratov, I. E. Kusnetsov, N. Stingelin and P. A. Troshin, Adv. Energy Mater., 2020, 10, 1903163 CrossRef CAS.
- X. Zhu, L. Hu, W. Wang, X. Jiang, L. Hu and Y. Zhou, ACS Appl. Energy Mater., 2019, 2, 7602–7608 CrossRef CAS.
- H. Liu, W. Wang and Y. Zhou, J. Mater. Chem. A, 2021, 9, 1080–1088 RSC.
- L. Hu, S. Xiong, W. Wang, L. Sun, F. Qin and Y. Zhou, J. Phys. Chem. C, 2020, 124, 2307–2312 CrossRef CAS.
- S. Xiong, L. Hu, L. Hu, L. Sun, F. Qin, X. Liu, M. Fahlman and Y. Zhou, Adv. Mater., 2019, 31, 1806616 CrossRef PubMed.
- M. J. Capitán, J. Álvarez and C. Navio, Phys. Chem. Chem. Phys., 2018, 20, 10450–10459 RSC.
- A. Weu, J. A. Kress, F. Paulus, D. Becker-Koch, V. Lami, A. A. Bakulin and Y. Vaynzof, ACS Appl. Energy Mater., 2019, 2, 1943–1950 CrossRef CAS.
- J. M. Zhuo, L. H. Zhao, R. Q. Png, L. Y. Wong, P. J. Chia, J. C. Tang, S. Sivaramakrishnan, M. Zhou, E. C. W. Ou and S. J. Chua, Adv. Mater., 2009, 21, 4747–4752 CAS.
- L. Perdigón-Toro, L. Q. Phuong, F. Eller, G. Freychet, E. Saglamkaya, J. I. Khan, Q. Wei, S. Zeiske, D. Kroh and S. Wedler, Adv. Energy Mater., 2022, 12, 2103422 CrossRef.
- S. D. Collins, C. M. Proctor, N. A. Ran and T. Q. Nguyen, Adv. Energy Mater., 2016, 6, 1501721 CrossRef.
- J. C. Blakesley and D. Neher, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 075210 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc04378e |
| This journal is © The Royal Society of Chemistry 2023 |