
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of oxidation-induced ordering on the electrical and mechanical properties of a polythiophene co-processed with bistriflimidic acid†
Sandra
Hultmark
a,
Mariavittoria
Craighero
a,
Sepideh
Zokaei
a,
Donghyun
Kim
 b,
Emmy
Järsvall
a,
Furqan
Farooqi
a,
Sara
Marina
c,
Renee
Kroon
bd,
Jaime
Martin
c,
Igor
Zozoulenko
bd and
Christian
Müller
*ae
b,
Emmy
Järsvall
a,
Furqan
Farooqi
a,
Sara
Marina
c,
Renee
Kroon
bd,
Jaime
Martin
c,
Igor
Zozoulenko
bd and
Christian
Müller
*ae
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296, Göteborg, Sweden. E-mail: christian.muller@chalmers.se
bLaboratory of Organic Electronics, Linköping University, 60174, Norrköping, Sweden
cPOLYMAT, University of the Basque Country UPV/EHU, 20018, San Sebastián, Spain
dWallenberg Wood Science Center, Linköping University, 60174, Norrköping, Sweden
eWallenberg Wood Science Center, Chalmers University of Technology, 41296, Göteborg, Sweden
First published on 23rd November 2022
Abstract
The interplay between the nanostructure of a doped polythiophene with oligoether side chains and its electrical as well as mechanical properties is investigated. The degree of order of the polymer is found to strongly vary when co-processed with bistriflimidic acid (H-TFSI). The neat polythiophene as well as strongly oxidized material are largely disordered while intermediate concentrations of H-TFSI give rise to a high degree of π-stacking. The structural disorder of strongly oxidized material correlates with a decrease in the kinetic fragility with H-TFSI concentration, suggesting that positive interactions between TFSI anions and the polymer reduce the ability to crystallize. The electrical conductivity as well as the Young's modulus first increase upon the addition of 4–10 mol% of H-TFSI, while the loss of π-stacking observed for strongly oxidized material more significantly affects the latter. As a result, material comprising 25 mol% H-TFSI displays an electrical conductivity of 58 S cm−1 but features a relatively low Young's modulus of only 80 MPa. Decoupling of the electrical and mechanical properties of doped conjugated polymers may allow the design of soft conductors that are in high demand for wearable electronics and bioelectronics.
10th Anniversary StatementThe Journal of Materials Chemistry C is a prominent platform that disseminates some of the most important new insights within the field of, e.g., organic electronics. We appreciate the journal because it is forward-looking and intends to serve the research community, emphasizing its support for young researchers and emerging topics. We wish the journal all the best for the coming 10 years, which will no doubt bring about tremendous scientific advances and help address some of the most important challenges of our time. Hopefully, during the coming years an increasing number of articles will not only continue to report exciting fundamental insights and technological advances but also describe truly sustainable materials chemistry. |
Introduction
Molecular doping of conjugated polymers is used to optimize the performance of a wide range of electronic devices from thin-film transistors to organic solar cells and it is essential for improving their thermoelectric properties.1–3 Conjugated polymers must display both excellent electrical and mechanical properties to enable the fabrication of mechanically robust and flexible devices.4,5 However, the impact of doping on the mechanical properties has not yet received much attention. The elastic modulus E of conjugated polymers tends to decrease upon doping if the neat polymer is stiff, i.e. E ≫ 100 MPa, likely due to a plasticization type effect.6–9 Instead, doping of softer polymers with E < 100 MPa has been found to increase the modulus due to an increase in crystalline order.10,11Conjugated polymers with oligoether side chains currently receive considerable interest for energy harvesting12–14 and storage15,16 as well as bioelectronics.17,18 In case of both p- and n-type polymers it has been argued that the selection of suitable oligoether side chains can improve the compatibility with molecular dopants because of favorable dopant/side-chain interactions.13,19–21 Recently, a conductivity of up to 550 S cm−1 has been achieved for a polythiophene with mixed oligoether and alkoxy side chains.14 We have investigated how the mechanical properties of the polythiophene p(g42T-T), which carries tetraethylene glycol side chains (see Fig. 1a for chemical structure), changes upon doping with 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ).11 Doping with F4TCNQ raised the conductivity of p(g42T-T) to 52 S cm−1 and at the same time both strongly shifted the glass transition temperature Tg from −46 to 3 °C and induced significant crystallization of the initially poorly ordered polymer.11 As a result, the Young's modulus strongly increased from E = 8 to 232 MPa. Doping with 2,5-difluoro-7,7,8,8-tetracyanoquinodimethane (F2TCNQ) resulted in an even higher tensile elastic modulus of up to 377 MPa. Intriguingly, the elastic modulus of F4TCNQ-doped p(g42T-T) increased in parallel with the electrical conductivity, a trend that has also been observed for doped conjugated polymer-based fibers22 including poly(3,4-ethylenedioxythiophene):poly(styrene sulfonate) (PEDOT:PSS) fibers.23 Hence, it is challenging to design highly doped materials that nevertheless feature a relatively low elastic modulus, complicating the design of soft electrical conductors, which are in high demand for wearable electronics.
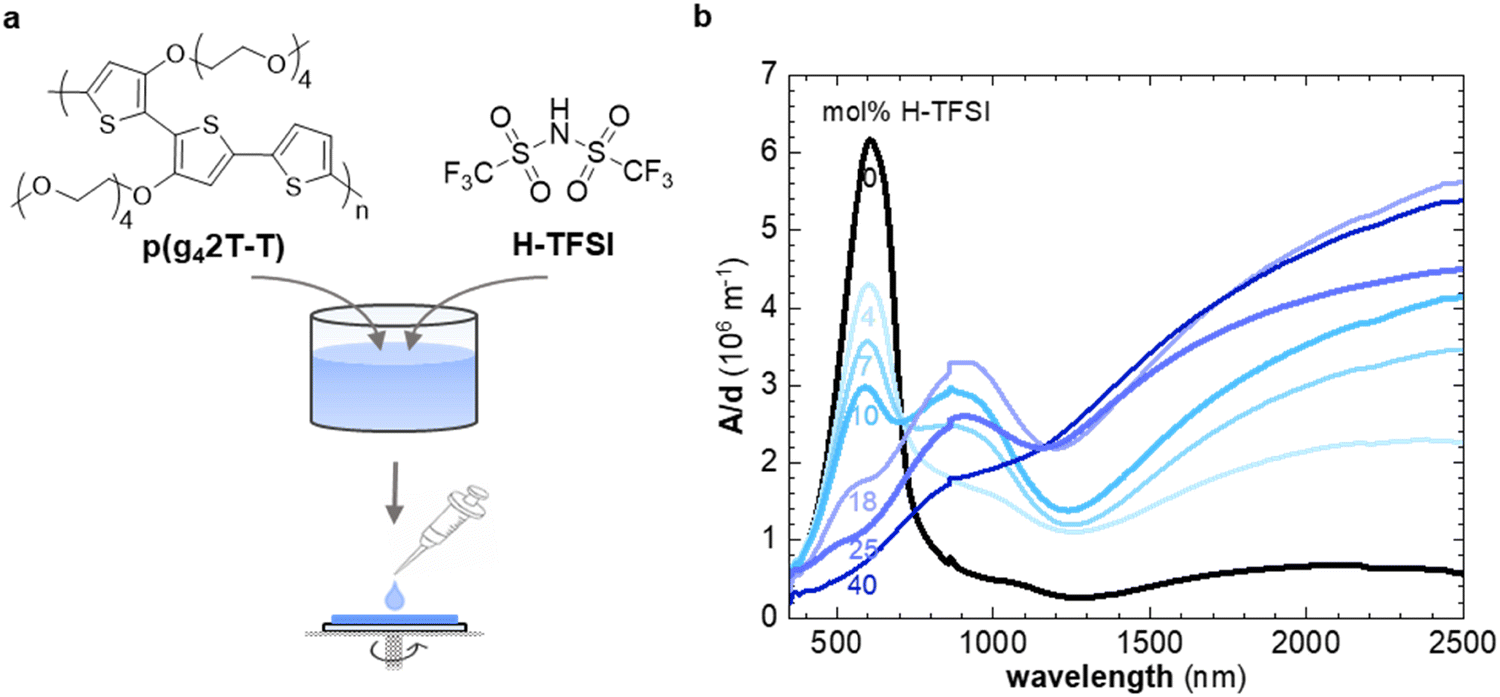 | ||
| Fig. 1 (a) Molecular structures of p(g42T-T) and H-TFSI and schematic of co-processing of polymer and dopant. (b) UV-Vis spectra of thin films of p(g42T-T) co-processed with H-TFSI and aged for 2 days (mol% of H-TFSI calculated with respect to the number of thiophene rings). | ||
The polymer p(g42T-T) can be doped through the addition of acids such as bistriflimidic acid (H-TFSI) and sulfonic acids, resulting in an electrical conductivity of up to 120 S cm−1 and a high degree of stability at elevated temperatures.24 Acid doping of p(g42T-T) may at least in part lead to protonation of the backbone, similar to other polythiophenes,25–28 but is also thought to involve an acid mediated oxidation of the polymer through O2, in agreement with the observation that a high electrical conductivity is only obtained when the material is exposed to air.24,29 Further, ageing of p(g42T-T):H-TFSI at ambient conditions for several days results in a significant increase in the electrical conductivity.24 Intriguingly, doping of poly(3-hexylthiophene) (P3HT) with ethylbenzene sulfonic acid (EBSA) slightly decreases the Tg of the polymer from 30 to 15 °C, i.e. the dopant has a plasticization type effect,8 which suggests that acid dopants may be a good choice for the design of highly doped polymers that are to remain relatively soft.
Here, we study how doping of p(g42T-T) with H-TFSI affects the mechanical properties. We find that the elastic modulus initially increases with the doping level due to doping induced crystallization, while the Tg is much less strongly affected as compared to F4TCNQ-doped p(g42T-T). For higher dopant levels above 20 mol% H-TFSI, the degree of order decreases again, leading to a highly doped material with an electrical conductivity of 58 S cm−1 but a modulus of only about 87 MPa. Moreover, we determine the impact of doping on the kinetic fragility of p(g42T-T), which gradually decreases with H-TFSI content, consistent with a reduced tendency of the polymer to crystallize.
Results and discussion
In a first set of experiments, we used UV-vis spectroscopy to investigate the extent of oxidation of thin films of p(g42T-T) co-processed with H-TFSI after storage in air for 2 days (Fig. 1b). We focused on aged films because the electrical conductivity of p(g42T-T) doped with H-TFSI has been found to increase upon air exposure.24 We ensured that co-processing with H-TFSI had not irreversibly altered the polymer by reducing aged films under alkaline conditions, which recovered the UV-vis absorption spectrum of neat p(g42T-T) as well as its solubility in chloroform (ESI† Fig. S1). Neat p(g42T-T) shows an absorption peak at around 600 nm as well as shallow absorption features at higher wavelengths due to adventitious oxygen doping of the polymer. The absorption peak at 600 nm successively decreases with increasing concentration of H-TFSI (Fig. 1b). Instead, two polaronic absorption bands emerge around 900 nm and in the infrared part of the spectra. Comparison of the recorded UV-vis spectra with spectra predicted for the same polymer by a density functional theory (DFT) study30 indicates that co-processing with 40 mol% H-TFSI results in a strongly oxidized polymer that likely features both polarons and bipolarons/polaron pairs. We note that the polaronic absorption at 900 nm reaches a maximum at a H-TFSI concentration of 18 mol% but then decreases in intensity for higher H-TFSI concentrations. The polaronic absorption in the infrared instead increases continuously. An isosbestic point at 700 nm is observed for up to 10 mol% H-TFSI, which indicates the presence of two major species, i.e., neat backbone segments that are increasingly replaced by oxidized segments with one positive charge (polarons). A new isosbestic point emerges at 1170 nm for higher concentrations of H-TFSI, which we explain with the emergence of bipolarons/polaron pairs upon further oxidation.Grazing-incidence wide-angle X-ray scattering (GIWAXS) was used to determine the degree of ordering of p(g42T-T) co-processed with H-TFSI (see ESI† Fig. S2 for GIWAXS patterns). The neat polymer shows a distinct out-of-plane diffraction peak at q100 ≈ 0.35 Å−1, characteristic of lamellar stacking and an edge-on texture, while no q010 diffraction can be discerned (Fig. 2a). Upon the addition of 4 mol% H-TFSI the q100 diffraction instead appears in-plane, indicating a change in texture to a predominately face-on orientation (note that the same polymer adopts an edge-on texture when doped with F4TCNQ).11 The q100 diffraction also shifts to a lower q value due to intercalation of TFSI counterions between the side chains of the polymer. Moreover, a clear out-of-plane diffraction peak q010 ≈ 1.7 Å−1 emerges due to π-stacking of the polymer backbone, indicating that co-processing with small amounts of 4 and 7 mol% H-TFSI leads to a significant increase in order. An increase in the degree of π-stacking upon doping has also been observed for regio-random poly(3-hexylthiophene).31,32 A further increase in H-TFSI concentration leads to a q100 diffraction that is present in both in-plane and out-of-plane diffractograms, indicating a mixed face-on and edge-on texture. Further, we observe a significant decrease in the intensity of the q010 diffraction and a shift to lower q values. As a result, films that contain a large amount of, e.g., 25 and 40 mol% H-TFSI give rise to GIWAXS diffractograms that are more comparable to neat p(g42T-T) with an out-of-plane q100 diffraction, which albeit remains at a lower q value of about 0.25 Å−1, as well as a weak q010 diffraction (Fig. 2a).
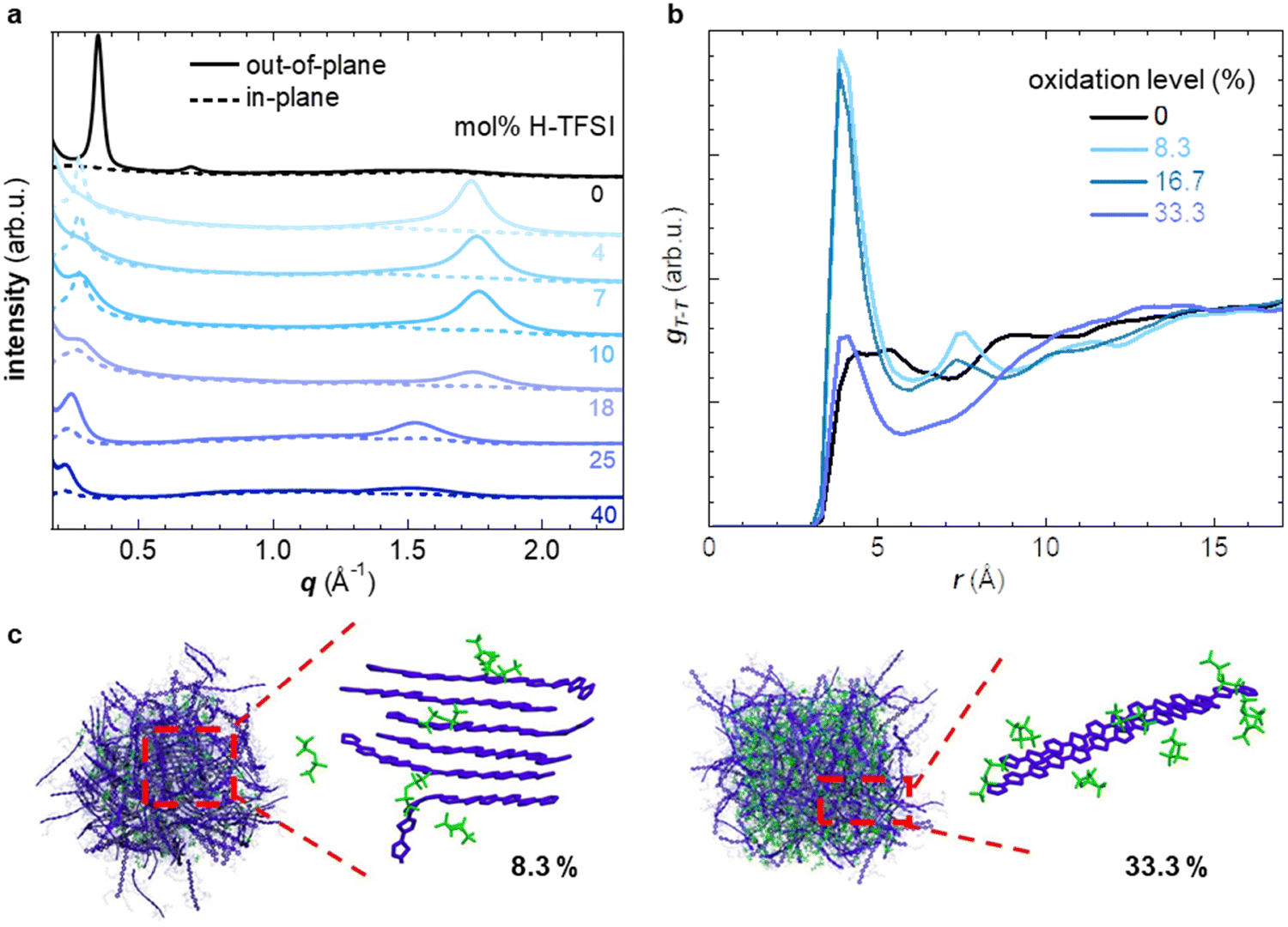 | ||
| Fig. 2 (a) In-plane (dashed lines) and out-of-plane GIWAXS diffractograms (solid lines) of p(g42T-T) co-processed with different amounts of H-TFSI. (b) Radial distribution functions of the distance between the center of mass of thiophene rings that are part of different chains (gT–T) from molecular dynamics (MD) simulations. (c) Snapshots of p(g42T-T) with oxidation levels of 8.3 and 33.3% showing representative crystallites/aggregates (blue) and TFSI anions (green). | ||
Molecular dynamics (MD) simulations allowed to study how the nanostructure changes upon oxidation in more detail. We created a simulation box comprising 200 oligomers, consisting of four g42T-T repeat units each (12 thiophene rings), and compared a system of neat oligomers with three systems where each oligomer carried a charge of +1e, +2e or +4e (oxidation level of 8.3%, 16.7% or 33.3%) with the charge balanced by TFSI anions (see experimental section for details). The radial distribution function gT–C (r) of the distance between the thiophene rings of the oligomer backbone and TFSI anions features a peak that shifts from r ≈ 6.8 Å to 5.2 Å as the oxidation level increases from 8.3% to 33.3% (ESI† Fig. S3b), indicating that the anions move closer to the backbone. The radial distribution function of the distance between the center of mass of thiophene rings gT–T (r) is featureless in case of neat oligomers and does not exhibit any indication of π-stacking (Fig. 2b). As the oxidation level increases to 8.3% and further to 16.7% a pronounced peak in gT–T (r) appears at r ≈ 4 Å, corresponding to the π-stacking distance, accompanied by smaller and broader peaks at near multiples of the π-stacking distance. A high degree of π-stacking is also evident from snapshots of the MD simulated nanostructure, which feature ordered domains composed of several chains (cf. presence of crystallites in MD snapshots in Fig. 2c and ESI† Fig. S3). Instead, for an oxidation level of 33.3% gT–T (r) only features a minor π-stacking peak at r ≈ 4 Å (Fig. 2b) and snapshots of the nanostructure do not feature extended ordered domains (Fig. 2c and ESI† Fig. S3), which suggests a low degree of π-stacking, consistent with the GIWAXS analysis of p(g42T-T):H-TFSI films (Fig. 2a). Moreover, we find that the average end-to-end distance of oligomer chains is larger for intermediate oxidation levels of 8.3% and 16.7% than for neat oligomers or an oxidation level of 33.3% (ESI† Fig. S3c), which agrees with the deduced changes in the degree of π-stacking (see Fig. 2a). Hence, both MD simulations and GIWAXS indicate that the degree of π-stacking increases for weakly oxidized p(g42T-T), while the polymer reverts to a less ordered material similar to neat p(g42T-T) in case of high oxidation levels.
We employed thermal analysis to explore the ability of oxidized p(g42T-T) to crystallize. The neat polymer features a very low glass transition temperature Tg = −59 °C.11 We used differential scanning calorimetry (DSC) at a scan rate of 10 °C min−1 to explore the change in Tg of p(g42T-T) co-processed with different amounts of H-TFSI, both of as-cast material and material aged for 14 days at ambient conditions. We find that the Tg increases to not more than −32 °C for aged material comprising 40 mol% H-TFSI (Fig. 3). Since processing is carried out at ambient conditions, i.e. at T ≫ Tg, the observed changes in Tg cannot explain why strongly oxidized p(g42T-T) is unable to develop a high degree of order.
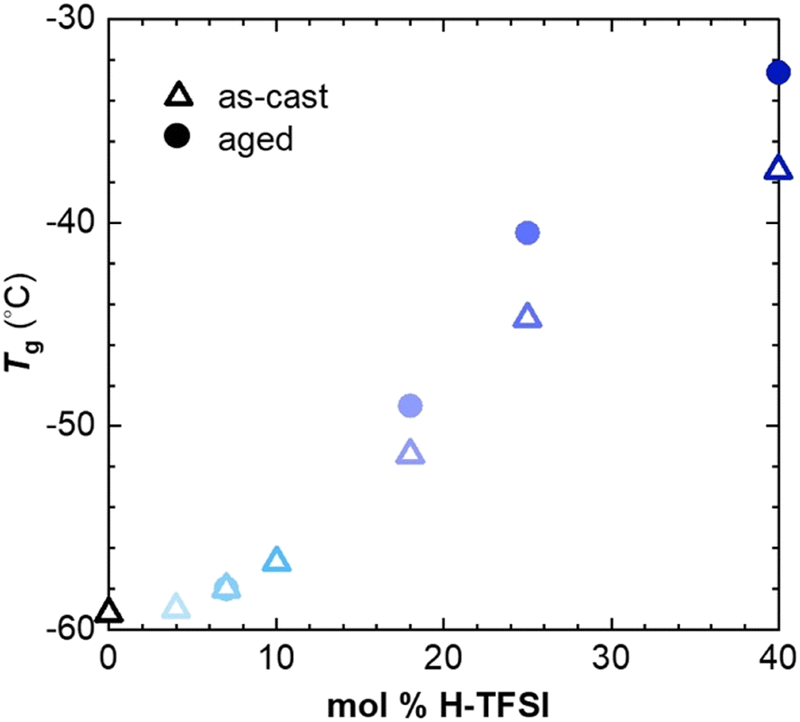 | ||
| Fig. 3 Glass transition temperature Tg of p(g42T-T) co-processed with H-TFSI corresponding to the midpoint in DSC thermograms recorded at a heating rate of 10 °C min−1 for as-cast samples (open triangles) and samples aged for 14 days at ambient conditions (solid circles). | ||
We have previously observed that doping of p(g42T-T) with F4TCNQ results in a more significant increase in Tg to 3 °C for an oxidation level of 16.8% (20 mol% F4TCNQ).11 We rule out that the size of the anion has an impact on the Tg – as is the case for poly(ethylene oxide) blended with Li-TFSI or NaI33 – because H-TFSI and F4TCNQ have a similar molar mass of 280 and 276 g mol−1, respectively. Another possibility is that the presence of water, which may form during acid mediated oxidation by O2, acts as a plasticizer. An FTIR spectrum of p(g42T-T) co-processed with 18 mol% H-TFSI does not feature a peak from hydroxyl groups around 3300 cm−1 (ESI† Fig. S4), which suggests that only a minimal amount of water is present in the here studied samples. The reaction scheme proposed by Mammone and MacDiarmid29 for acid mediated oxidation by O2 would result in a maximum of about 0.6 wt% water in case of p(g42T-T) co-processed with 18 mol% H-TFSI, assuming that one H2O molecule is formed per two polarons, consuming two H-TFSI molecules in the process. In case of polyamides, for example, the absorption of 0.5 wt% water decreases the Tg by about 15 °C compared to dry polyamide.34 Hence, we argue it is unlikely that the presence of water can alone explain the low Tg of p(g42T-T) co-processed with H-TFSI. We propose that changes in the conformation of the polymer, possibly due to protonation in case of doping with H-TFSI, as well as the location of the anion relative to the polymer backbone results in the observed differences in Tg. Differences in anion location are suggested by MD simulations, which show that TFSI anions are positioned further away from the polymer backbone than F4TCNQ anions, as evidenced by the calculated difference in the onset in gT–C (r) at r ≈ 4–5 Å and 3.5 Å, respectively (see ESI† Fig. S3b and ref. 11).
Fast scanning calorimetry (FSC) was used to study the relaxation kinetics of the polymer in more detail. We used FSC to measure the impact of the cooling rate q on the fictive temperature 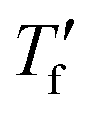 , which is the temperature where the material freezes into a glass. The material was first heated to 50 °C, then cooled at −0.1 to −1000 K s−1 and finally heated again at 2000 K s−1 (Fig. 4a). To determine
, which is the temperature where the material freezes into a glass. The material was first heated to 50 °C, then cooled at −0.1 to −1000 K s−1 and finally heated again at 2000 K s−1 (Fig. 4a). To determine 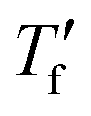 we used the area matching method established by Moynihan et al. for fast cooling rates,35 and an extrapolation method for slow cooling rates,36,37 for which we observe that
we used the area matching method established by Moynihan et al. for fast cooling rates,35 and an extrapolation method for slow cooling rates,36,37 for which we observe that 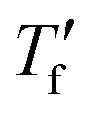 lies below the onset of the glass transition (Fig. 4b; see experimental section for details). FSC heating thermograms display a pronounced enthalpy overshoot that increases in size as the absolute value of q decreases (Fig. 4b; see ESI† Fig. S5 and S6 for raw data). This is because in case of slow cooling the material has more time to relax and hence requires more energy during re-heating to regain its mobility and reach the liquid state.
lies below the onset of the glass transition (Fig. 4b; see experimental section for details). FSC heating thermograms display a pronounced enthalpy overshoot that increases in size as the absolute value of q decreases (Fig. 4b; see ESI† Fig. S5 and S6 for raw data). This is because in case of slow cooling the material has more time to relax and hence requires more energy during re-heating to regain its mobility and reach the liquid state.
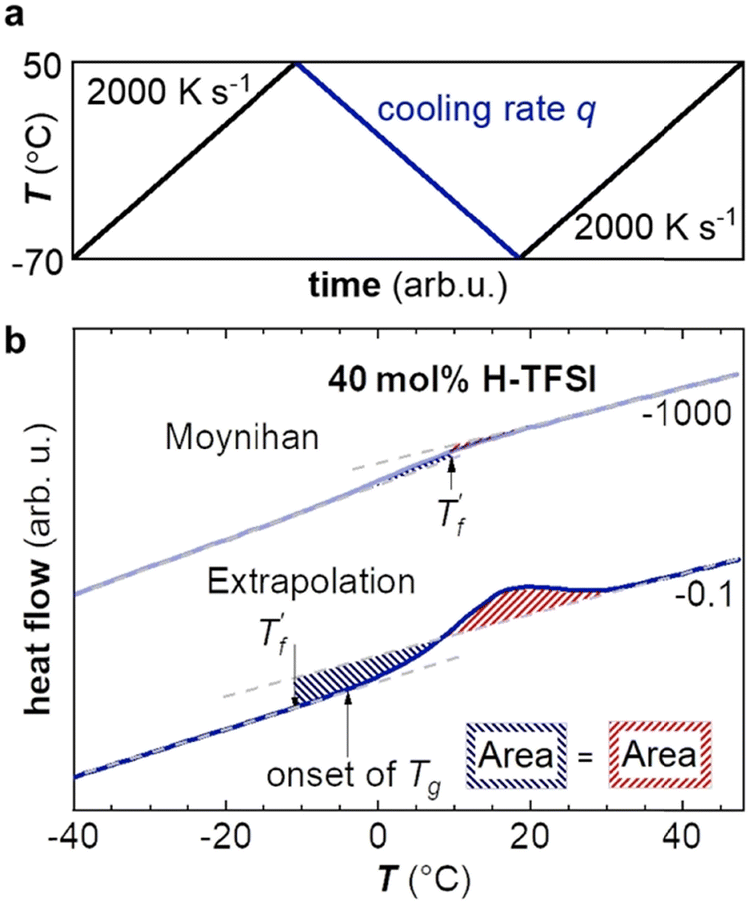 | ||
| Fig. 4 (a) Fast scanning calorimetry (FSC) protocol: the sample is first heated to 50 °C, then cooled at rate q ranging from −0.1 to −1000 K s−1, and finally heated again at 2000 K s−1. (b) FSC heating thermograms after fast (q = −1000 K s−1; light blue) and slow (q = −0.1 K s−1; dark blue) cooling of p(g42T-T) co-processed with 40 mol% H-TFSI and aged for 2 days. | ||
We calculated the kinetic fragility, m, a measure for the rate of change in relaxation time of a material with temperature around its Tg,38,39 by plotting −log|q| against 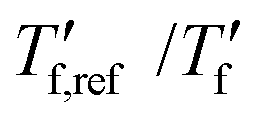 where
where 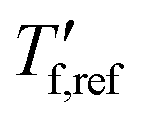 is obtained from DSC heating thermograms at 0.17 K s−1 (Fig. 5a; see ESI† Fig. S7 and S8 for fragility plots and Table S1 for
is obtained from DSC heating thermograms at 0.17 K s−1 (Fig. 5a; see ESI† Fig. S7 and S8 for fragility plots and Table S1 for 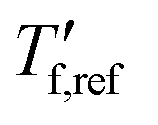 values). The fragility was then extracted from the slope around
values). The fragility was then extracted from the slope around 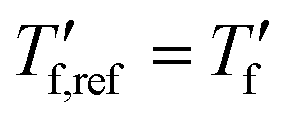
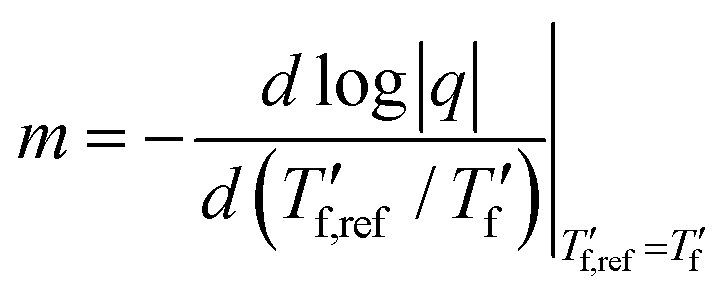 | (1) |
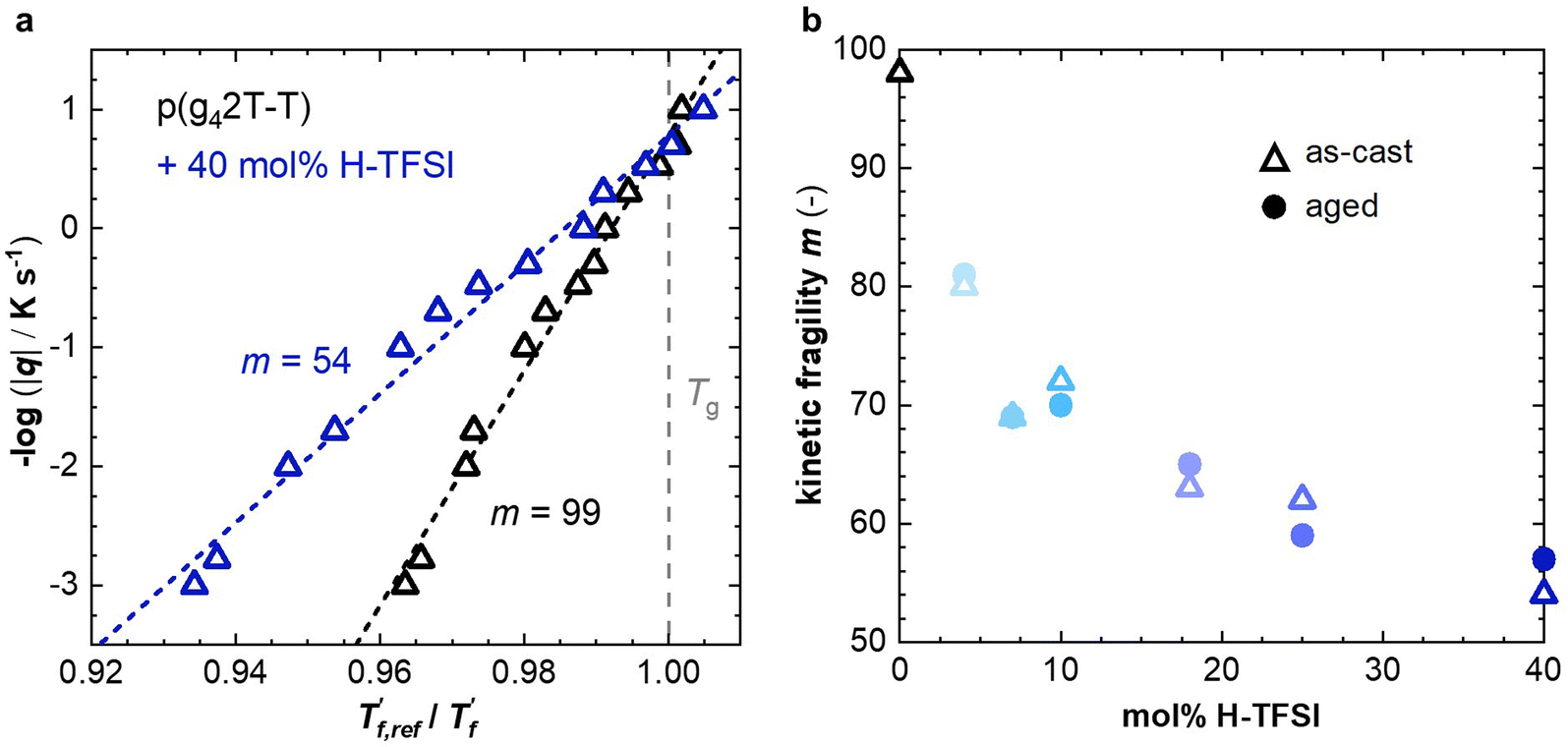 | ||
Fig. 5 (a) Fragility plot with −log|q| vs.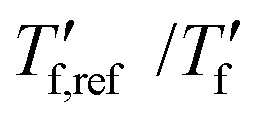 of neat p(g42T-T) (black open triangles) and as-cast p(g42T-T) co-processed with 40 mol% H-TFSI (open blue triangles); of neat p(g42T-T) (black open triangles) and as-cast p(g42T-T) co-processed with 40 mol% H-TFSI (open blue triangles); 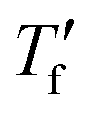 was determined with FSC for q = −0.1 to −1000 K s−1 and was determined with FSC for q = −0.1 to −1000 K s−1 and 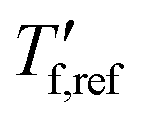 was measured with DSC for q = 0.17 K s−1 (10 °C min−1). (b) Kinetic fragility as a function of mol% H-TFSI for as-cast samples (open triangles) and samples aged for 2 days (solid circles). was measured with DSC for q = 0.17 K s−1 (10 °C min−1). (b) Kinetic fragility as a function of mol% H-TFSI for as-cast samples (open triangles) and samples aged for 2 days (solid circles). | ||
For neat p(g42T-T) we obtain a fragility of m = 99 while co-processing with 40 mol% H-TFSI results in a much lower fragility of m = 54. The kinetic fragility decreases with the H-TFSI concentration reaching the lowest values for the most strongly oxidized samples, which implies that oxidized p(g42T-T) is a stronger glass former than neat p(g42T-T) (Fig. 5b). We explain the decrease in fragility with the presence of positive interactions between TFSI anions and the oligoether side chains and/or the positively charged backbone, which reduce the mobility of oxidized polymer chains. A reduced ability to undergo conformational changes can be expected to decrease the ability to crystallize, which is consistent with the lower degree of crystalline order inferred from GIWAXS diffractograms (see Fig. 2a). We would like to point out that the GIWAXS samples were analyzed at room temperature, i.e., above the Tg. Nevertheless, highly oxidized p(g42T-T) remains disordered even though samples were kept above the Tg for a long time.
In a further set of experiments, we investigated the impact of the H-TFSI concentration on the electrical and mechanical properties of p(g42T-T). The conductivity was determined for thin films with a thickness of 40–140 nm after 2 days of ageing at ambient conditions. The conductivity increases with the dopant concentration up to 25 mol% H-TFSI, where it reaches a conductivity of (58 ± 3) S cm−1, followed by a slight decrease to (20 ± 1) S cm−1 (Fig. 6a). The conductivity decreases for 40 mol% H-TFSI despite a further increase in the oxidation level, as evidenced by UV-vis spectroscopy (cf.Fig. 1) and corroborated by a further reduction in the Seebeck coefficient (ESI† Fig. S9). The here measured conductivities are similar to those reported by Hofmann et al.,24 indicating that ageing for 2 days was sufficient for the thin films to reach a steady state.
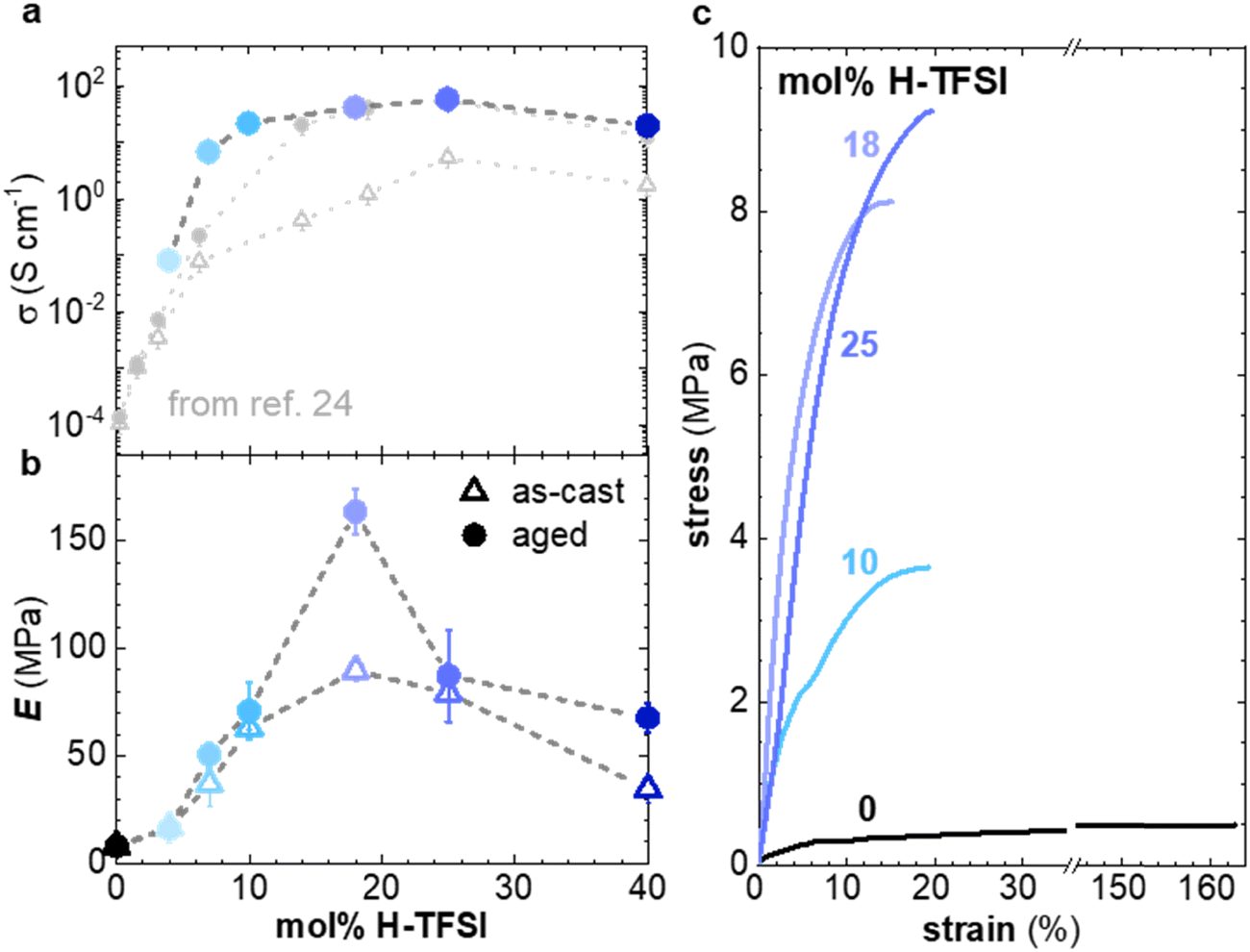 | ||
| Fig. 6 (a) Electrical conductivity σ (top) of films of p(g42T-T) co-processed with H-TFSI and aged for 2 days (solid blue circles), as well as as-cast films (open grey triangles) and films aged for 3 days from ref. 24 (full grey circles). (b) Young's modulus E of as-cast samples (open triangles) and samples aged for 14 days (solid circles). (c) Stress–strain curves recorded at room temperature during tensile deformation of samples aged for 14 days. | ||
To facilitate mechanical measurements, thick samples are needed. We prepared 20–70 μm free-standing films by drop casting and increased the ageing time to a minimum of 14 days. Tensile deformation of both as-cast and aged samples was carried out (Fig. 6b, c and ESI† Fig. S10). The neat polymer displays a low Young's modulus of only 8 ± 2 MPa.11 For as-cast samples the elastic modulus increases up to E = (90 ± 5) MPa at 18 mol% H-TFSI, and then decreases at higher concentrations of H-TFSI (Fig. 6b). Aged samples display a similar trend with the Young's modulus reaching a maximum of (164 ± 11) MPa at 18 mol% H-TFSI, followed by a drop in modulus at higher concentrations. The Young's modulus of aged samples is higher than for as-cast ones, especially at H-TFSI concentrations higher than 9 mol%, indicating a gradual modification of the nanostructure due to slow kinetics of the doping process. We measured an electrical conductivity of (40 ± 5) S cm−1 for a several-months old thick sample doped with 18 mol% H-TFSI, that we had previously used for mechanical testing, which confirms that bulk samples can retain a high conductivity over long periods of time.
Oxidation modifies the nanostructure of p(g42T-T), i.e. leads to π-stacking of the polymer (see Fig. 2 and 3) and the addition of anions in between the oligoether side chains. Ordered domains can be expected to benefit both charge transport and increase the Young's modulus of the material. It is interesting that samples co-processed with 10 mol% and 25 mol% of H-TFSI show similar elastic moduli but different electrical properties, i.e., a conductivity of σ = (22 ± 1) S cm−1 and (58 ± 3) S cm−1, respectively (Fig. 6a and b). It appears that the mechanical and electrical properties, which usually correlate with each other because they are both affected by interchain interactions, can be decoupled to a certain extent.
Conclusions
Ordering of the polymer p(g42T-T) co-processed with H-TFSI strongly depends on the oxidation level. A combination of GIWAXS and MD simulations has shown that the neat polymer as well as strongly oxidized material display a low degree of order, while an intermediate concentration of 4–10 mol% H-TFSI results in considerable π-stacking. Both the electrical and mechanical properties are found to depend on the H-TFSI concentration and are influenced by the degree of π-stacking. The electrical conductivity strongly increased with H-TFSI concentration, reaching a maximum of 58 S cm−1 for 25 mol% H-TFSI, followed by a slight decrease for larger H-TFSI concentrations. Likewise, the Young's modulus reached a peak value of 164 MPa, albeit for 18 mol% H-TFSI, and decreased again for higher H-TFSI concentrations. As a result, the material containing 25 mol% H-TFSI showed a lower Young's modulus of only 80 MPa. Evidently, co-processing with an acid can be used to modulate both the electrical and mechanical properties of soft polythiophenes and may aid the design of soft conductors that are of interest for wearable electronics and bioelectronics.Experimental section
Materials
p(g42T-T) was synthesized according to a previously reported procedure (number-average molecular weight Mn = 24 kg mol−1; polydispersity index PDI = 3.3).19 Chloroform (purity > 99.8%), anhydrous acetonitrile (purity 99.8%), H-TFSI and potassium hydroxide (purity > 85%) were purchased from Sigma Aldrich, dimethyl sulfoxide (purity > 99%) was purchased from VWR Chemicals. All chemicals were used as received. P(g42T-T) was dissolved at 6 g L−1 in a mixture of chloroform and anhydrous acetonitrile (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v:v) while H-TFSI was dissolved at 10 g L−1 in anhydrous acetonitrile. Appropriate amounts of these two solutions were mixed to prepare solutions containing different amounts of H-TFSI and p(g42T-T). All solutions were placed on a hotplate at 35 °C with a magnetic stirrer to ensure dissolution. The molar percentage of H-TFSI is calculated with respect to the number of thiophene rings.
:2, v:v) while H-TFSI was dissolved at 10 g L−1 in anhydrous acetonitrile. Appropriate amounts of these two solutions were mixed to prepare solutions containing different amounts of H-TFSI and p(g42T-T). All solutions were placed on a hotplate at 35 °C with a magnetic stirrer to ensure dissolution. The molar percentage of H-TFSI is calculated with respect to the number of thiophene rings.
UV-Vis spectroscopy
Films with a thickness of 40 to 140 nm were wire-bar coated onto glass substrates with a K Control Coater from RK Print Coat Instruments Ltd and aged for 2 days at ambient conditions. Measurements were performed with a PerkinElmer Lambda 1050 spectrometer.Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR)
Thick samples were drop cast onto cleaned glass slides, removed from the substrate with a sharp blade, and finally aged for about 14 days at ambient conditions. Measurements were performed with a PerkinElmer Frontier FTIR spectrometer.Atomic force microscopy (AFM)
The thickness of thin films was measured using a Digital Instrument Dimension 3000 Large Sample AFM with a type G Scanner. A standard silicon tip was used in tapping mode for the analysis.Grazing incidence wide angle X-ray scattering (GIWAXS)
Films were prepared by drop-casting onto cleaned silicon wafers. GIWAXS diffractograms were recorded at the beamline NCD – SWEET of the Alba synchrotron light source facility using an X-ray wavelength of 1 Å and a sample-detector distance of 201.17 cm.Differential scanning calorimetry (DSC)
Material was solidified by drop casting onto cleaned glass slides, dried in a vacuum oven for 1 h at 30 °C or aged for 14 days at ambient conditions, removed from the glass slides, and placed into 40 μL Al crucibles. DSC measurements were conducted under nitrogen at a rate of 10 °C min−1 with a Mettler Toledo DSC2 equipped with a Gas controller GC 200. The glass transition temperature Tg and reference fictive temperature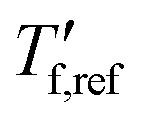 were extracted from second DSC heating thermograms.
were extracted from second DSC heating thermograms.
Fast scanning calorimetry (FSC)
Samples were prepared by drop casting directly onto FSC chip sensors followed by ageing for 2 days at ambient conditions, were applicable. Measurements were performed with a Mettler Toledo Flash DSC 1. Samples were first heated to 50 °C, then cooled to −70 °C with cooling rates ranging from −0.1 to −1000 K s−1, followed by heating with 2000 K s−1. To calculate the limiting fictive temperature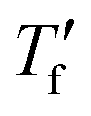 , we used Moynihan's area matching method if
, we used Moynihan's area matching method if 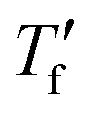 was above the onset of devitrification, which is equal to Richardson's method in the Mettler Toledo software:
was above the onset of devitrification, which is equal to Richardson's method in the Mettler Toledo software:where Cpl is the heat capacity of the liquid, Cpg is the heat capacity of the glass and Cp is the apparent heat capacity of the sample. If
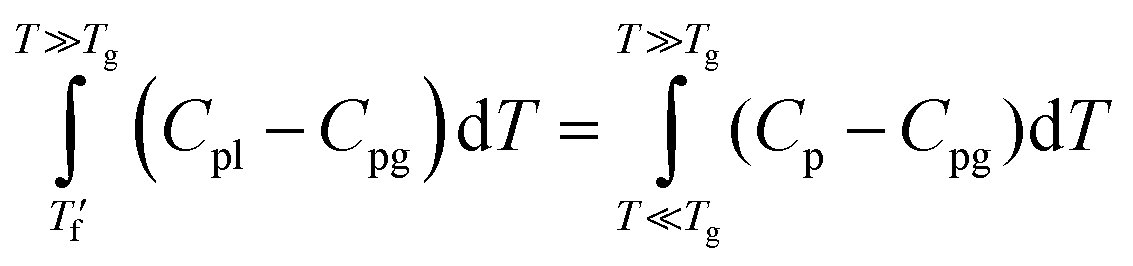
 was below the onset of devitrification, a simplified extrapolation method was used:
was below the onset of devitrification, a simplified extrapolation method was used: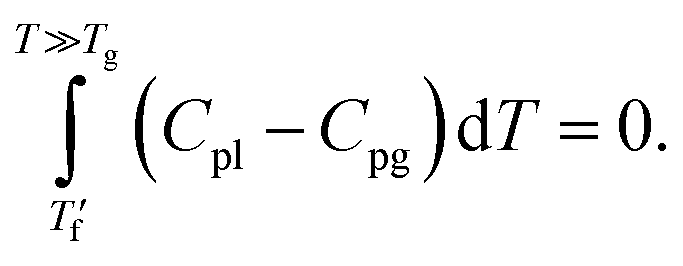
Molecular dynamics simulations
Classical molecular dynamics (MD) simulations were carried out using the LAMMPS package.40 General AMBER force field (GAFF)41 as implemented in moltemplate code42 was employed for bonded and non-bonded interactions. The cutoff was set to 1.1 nm for both Lennard–Jones and Coulombic interactions, and the k-mean scheme of particle-particle particle-mesh43–45 was used for long range Coulombic interactions. Initially, a computational box of 30 × 30 × 30 nm3 was prepared that included 200 oligomer chains consisting of four g42T-T units, i.e. a total of 12 thiophene rings per chain. The oligomer chains had a total charge of 0, +1e, +2e or +4e corresponding to oxidation levels of 0, 8.3, 16.7 or 33.3%, respectively. In addition, TFSI anions (with a charge of −1e) were randomly placed in the box to achieve charge neutrality. Additional information can be found in the ESI.†Electrical characterization
Films with a thickness of 40–140 nm were wire-bar coated onto glass substrates, pre-cleaned with acetone and isopropanol, or poly(ethylene terephthalate) foil for measurements of the electrical resistivity or Seebeck coefficient, respectively, followed by ageing for 2 days at ambient conditions. The electrical resistivity was measured with a four-point probe set-up from Jandel Engineering. The Seebeck coefficient was measured with a SB1000 instrument (samples cut into pieces of around 1 mm × 5 mm and mounted on the sample stage with silver paste) equipped with a K2000 temperature controller using a thermal load of 1–2 K and a constantan wire as the internal reference. The electrical resistivity of a thick sample doped with 18mol% H-TFSI, fixated with silver paint from Agar Scientific Ltd on a glass slide, was measured in 2-point configuration using a U1253B multimeter from Keysight Technologies.Dynamic mechanical analysis (DMA)
Free-standing films with a thickness of 20–70 μm were drop cast onto cleaned glass slides, dried in vacuum oven for 1.5 hours at 30 °C, removed from the substrate with a sharp blade, and finally aged for about 14 days at ambient conditions, where applicable. Thicknesses were measured with a caliper. Tensile deformation was performed with a Q800 dynamic mechanical analyzer from TA Instruments using a gauge length of 3.7–4.2 mm with a force rate of 0.005 N min−1 in a controlled force mode.Author contributions
S. H. and F. F. carried out thermal analysis, M. C. and S. Z. carried out electrical and mechanical measurements, D. K. and I. Z. carried out molecular dynamics simulations, E. J. carried out UV-vis spectroscopy, S. M. and J. M. carried out GIWAXS measurements, R. K. synthesized the polymer p(g42T-T), C. M. supervised the study and wrote the manuscript together with S. H., M. C. and S. Z.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from the Swedish Research Council through grant agreements no. 2016-05990 and 2018-03824, the European Union's Horizon 2020 research and innovation programme through the Marie Skłodowska-Curie grant agreement no. 955837 (HORATES) and the Knut and Alice Wallenberg Foundation through a Wallenberg Academy Fellowship Prolongation grant. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and HPC2N. We thank Anders Mårtensson for carrying out the AFM measurements.References
- B. Lüssem, C. M. Keum, D. Kasemann, B. Naab, Z. N. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef.
- M. Massetti, F. Jiao, A. J. Ferguson, D. Zhao, K. Wijeratne, A. Wurger, J. L. Blackburn, X. Crispin and S. Fabiano, Chem. Rev., 2021, 121, 12465–12547 CrossRef CAS PubMed.
- A. D. Scaccabarozzi, A. Basu, F. Anies, J. Liu, O. Zapata-Arteaga, R. Warren, Y. Firdaus, M. I. Nugraha, Y. B. Lin, M. Campoy-Quiles, N. Koch, C. Müller, L. Tsetseris, M. Heeney and T. D. Anthopoulos, Chem. Rev., 2022, 122, 4420–4492 CrossRef CAS PubMed.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- R. X. Xie, R. H. Colby and E. D. Gomez, Adv. Electron. Mater., 2018, 4, 1700356 CrossRef.
- Y. Cao, P. Smith and A. J. Heeger, Polymer, 1991, 32, 1210–1218 CrossRef CAS.
- J. Hynynen, E. Jarsvall, R. Kroon, Y. D. Zhang, S. Barlow, S. R. Marder, M. Kemerink, A. Lund and C. Müller, ACS Macro Lett., 2019, 8, 70–76 CrossRef CAS PubMed.
- R. Kroon, A. I. Hofmann, L. Y. Yu, A. Lund and C. Müller, Chem. Mater., 2019, 31, 2770–2777 CrossRef CAS PubMed.
- J. Mun, J. Kang, Y. Zheng, S. C. Luo, Y. L. Wu, H. X. Gong, J. C. Lai, H. C. Wu, G. Xue, J. B. H. Tok and Z. N. Bao, Adv. Electron. Mater., 2020, 6, 2000251 CrossRef CAS.
- J. Moulton and P. Smith, Synth. Met., 1991, 40, 13–22 CrossRef CAS.
- S. Zokaei, D. Kim, E. Järsvall, A. M. Fenton, A. R. Weisen, S. Hultmark, P. H. Nguyen, A. M. Matheson, A. Lund, R. Kroon, M. L. Chabinyc, E. D. Gomez, I. Zozoulenko and C. Müller, Mater. Horiz., 2022, 9, 433–443 RSC.
- D. Kiefer, A. Giovannitti, H. D. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. A. Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS.
- J. Liu, L. Qiu, R. Alessandri, X. K. Qiu, G. Portale, J. J. Dong, W. Talsma, G. Ye, A. A. Sengrian, P. C. T. Souza, M. A. Loi, R. C. Chiechi, S. J. Marrink, J. C. Hummelen and L. J. A. Koster, Adv. Mater., 2018, 30, 1704630 CrossRef.
- H. Li, Z. Xu, J. Song, H. Y. Chai, L. L. Wu and L. D. Chen, Adv. Funct. Mater., 2022, 32, 2110047 CrossRef CAS.
- A. V. Volkov, H. D. Sun, R. Kroon, T. P. Ruoko, C. Y. Che, J. Edberg, C. Müller, S. Fabiano and X. Crispin, ACS Appl. Energy Mater., 2019, 2, 5350–5355 CrossRef CAS.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- C. B. Nielsen, A. Giovannitti, D. T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. McCulloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS.
- R. Kroon, D. Kiefer, D. Stegerer, L. Y. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef.
- B. X. Dong, C. Nowak, J. W. Onorato, T. Z. Ma, J. Niklas, O. G. Poluektov, G. Grocke, M. F. DiTusa, F. A. Escobedo, C. K. Luscombe, P. F. Nealey and S. N. Patel, Chem. Mater., 2021, 33, 741–753 CrossRef CAS.
- D. Scheunemann, E. Järsvall, J. Liu, D. Beretta, S. Fabiano, M. Caironi, M. Kemerink and C. Müller, Chem. Phys. Rev., 2022, 3, 021309 CrossRef CAS.
- A. Lund, N. M. van der Velden, N. K. Persson, M. M. Hamedi and C. Müller, Mater. Sci. Eng., R, 2018, 126, 1–29 CrossRef.
- R. Sarabia-Riquelme, R. Andrews, J. E. Anthony and M. C. Weisenberger, J. Mater. Chem. C, 2020, 8, 11618–11630 RSC.
- A. I. Hofmann, R. Kroon, L. Y. Yu and C. Müller, J. Mater. Chem. C, 2018, 6, 6905–6910 RSC.
- S. A. Chen and M. Y. Hua, Macromolecules, 1993, 26, 7108–7110 CrossRef CAS.
- S. N. Patel, A. M. Glaudell, D. Kiefer and M. L. Chabinyc, ACS Macro Lett., 2016, 5, 268–272 CrossRef CAS PubMed.
- E. M. Thomas, E. C. Davidson, R. Katsumata, R. A. Segalman and M. L. Chabinyc, ACS Macro Lett., 2018, 7, 1492–1497 CrossRef CAS.
- M. Arvind, C. E. Tait, M. Guerrini, J. Krumland, A. M. Valencia, C. Cocchi, A. E. Mansour, N. Koch, S. Barlow, S. R. Marder, J. Behrends and D. Neher, J. Phys. Chem. B, 2020, 124, 7694–7708 CrossRef CAS PubMed.
- R. J. Mammone and A. G. Macdiarmid, J. Chem. Soc., Faraday Trans., 1985, 81, 105–112 RSC.
- I. Sahalianov, J. Hynynen, S. Barlow, S. R. Marder, C. Müller and I. Zozoulenko, J. Phys. Chem. B, 2020, 124, 11280–11293 CrossRef CAS PubMed.
- P. Y. Yee, D. T. Scholes, B. J. Schwartz and S. H. Tolbert, J. Phys. Chem. Lett., 2019, 10, 4929–4934 CrossRef CAS.
- E. Lim, A. M. Glaudell, R. Miller and M. L. Chabinyc, Adv. Electron. Mater., 2019, 5, 1800915 CrossRef.
- N. A. Stolwijk, C. Heddier, M. Reschke, M. Wiencierz, J. Bokeloh and G. Wilde, Macromolecules, 2013, 46, 8580–8588 CrossRef CAS.
- A. Sambale, M. Kurkowski and M. Stommel, Thermochim. Acta, 2019, 672, 150–156 CrossRef CAS.
- C. T. Moynihan, A. J. Easteal, M. A. D. Bolt and J. Tucker, J. Am. Ceram. Soc., 1976, 59, 12–16 CrossRef CAS.
- S. Y. Gao, Y. P. Koh and S. L. Simon, Macromolecules, 2013, 46, 562–570 CrossRef CAS.
- R. Tao, E. Gurung, M. M. Cetin, M. F. Mayer, E. L. Quitevis and S. L. Simon, Thermochim. Acta, 2017, 654, 121–129 CrossRef CAS.
- R. Böhmer, K. L. Ngai, C. A. Angell and D. J. Plazek, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef.
- C. Rodriguez-Tinoco, M. Gonzalez-Silveira, M. A. Ramos and J. Rodriguez-Viejo, Riv. Nuovo Cimento, 2022, 45, 325–406 CrossRef CAS.
- D. C. Prieve and W. B. Russel, J. Colloid Interface Sci., 1988, 125, 1–13 CrossRef CAS.
- W. Wang and R. Wolf, DA J. Comput. Chem., 2004, 25, 92 CrossRef PubMed.
- A. Jewett, Moltemplate http://www.moltemplate.org.
- R. W. Hockney and J. W. Eastwood, Computer simulation using particles, crc Press, 2021 Search PubMed.
- A. Y. Toukmaji and J. A. Board Jr., Comput. Phys. Commun., 1996, 95, 73–92 CrossRef CAS.
- E. Pollock and J. Glosli, Comput. Phys. Commun., 1996, 95, 93–110 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2tc03927c |
| This journal is © The Royal Society of Chemistry 2023 |