
Tuning properties of biocatalysis using protein cage architectures
Yang
Wang
 and
Trevor
Douglas
*
and
Trevor
Douglas
*
Department of Chemistry, Indiana University, 800 E Kirkwood Ave, Bloomington, IN 47405, USA. E-mail: trevdoug@indiana.edu
First published on 31st March 2023
Abstract
Compartmentalization of cellular activities is an extremely important mechanism within cells, across all domains of life, for high efficiency of cell function. Bacterial microcompartments are exemplary protein-based cage structures that act as subcellular compartments encapsulating biocatalysts. They are able to achieve segregation of metabolic reactions from the bulk environment, which can alter the properties (including efficiency and selectivity) of biochemical processes and enhance overall cell function. By mimicking these naturally occurring compartments using protein cage platforms, synthetic catalytic materials have been made to achieve well-defined biochemical catalysis with desired and enhanced activities. This Perspective reviews the study in the past decade or so on artificial nanoreactors developed based on protein cage architectures, and summarizes the effects of protein cages on the properties of encapsulated enzymatic catalysis, including reaction efficiency and substrate selectivity. Given the significance of metabolic pathways in living systems and its inspiration in biocatalysis, our perspectives are also presented on cascade reactions, which are illustrated from three aspects: the technical challenges of controlling molecular diffusion to achieve the desired properties of multistep biocatalysis, the solutions to these challenges presented by nature, and how biomimetic approaches have been adopted in the design of biocatalytic materials using protein cage architectures.
Yang Wang received his BS in Pharmaceutical Sciences from Tianjin University in 2017, where he worked with Prof. Kenneth Woycechowsky from 2015 to 2018. In early 2017, he was also an exchange student in Prof. Marcia Haigis’ Lab at Harvard Medical School. Subsequently, he joined Indiana University in 2018 and is now a PhD candidate in Chemistry working with Prof. Trevor Douglas on biomimetic materials. |
Trevor Douglas graduated from the University of California, San Diego with a BS in Chemistry. He earned a PhD in Inorganic Chemistry from Cornell University in 1991, where he worked with Klaus Theopold. He was a postdoctoral fellow with Stephen Mann (Bath University), where he worked in the area of biomineralization. Before moving to Indiana University, he was a member of the faculty in the Department of Chemistry and Biochemistry at Montana State University where he was the Regents Professor. In 2014, he became the Earl Blough Professor of Chemistry at Indiana University where his lab continues to explore the area of biomimetic materials chemistry. |
1. Introduction
Compartmentalization is a central feature of biology. Complex organisms are highly compartmentalized across multiple length scales: organs, tissues, cells, and subcellular structures.1,2 This spatial organization enhances the overall efficiency of life by hierarchically dividing complex activities into multiple more specific and straightforward tasks, which can be completed by simpler but specialized structures with isolated environments to diminish the potential interference between individual tasks.3 The basic level of hierarchy of compartmentalization are subcellular compartments, which are usually defined by a particular physical boundary and delimit spaces where certain fundamental biochemical processes can take place. Membrane-bound organelles (such as mitochondria and chloroplasts) were once believed to be the only subcellular compartments.3 However, the discovery of bacterial microcompartments has demonstrated that proteins which form cage-like assemblies can also act as subcellular compartments for specialized biochemical processes.4 For instance, carboxysomes, found in many cyanobacteria and chemoautotrophs, are exemplary subcellular compartments whose structures are only made up of protein shells.4 They work as nanoreactors, capable of carbon fixation, by encapsulating the enzymes essential for the anabolic pathway, significantly enhancing overall cell function. In fact, protein cage architectures are ubiquitous in all domains of life,1 and intensive effort has been applied to understand their roles in biocatalysis.4,5 The knowledge obtained from these investigations have also guided us to employ the naturally occurring characteristics of protein cages to modulate various biocatalytic processes.Protein cages show some similarities with lipid bilayer-based compartments, given that both compartments are at the molecular level in the hierarchy of the biological compartmentalization. They are able to entrap biomacromolecules (often in very large numbers) inside their segregated environments.5,6 The entrapment limits the movement of molecules within the small volume of the compartment cavity from free diffusion in bulk solution, which creates an elevated local concentration of the molecules, known as confinement effects.7 The molecules also experience crowding effects after encapsulation due to the exclusion from the volume occupied by co-encapsuled molecules, which enforces inter- and intramolecular interactions.8 Furthermore, the local microenvironment inside the compartments might show different biophysical and biochemical properties from the bulk environment, such as pH,9,10 potentially altering the properties of encapsulated cargos. Simultaneously, there are distinct contrasts between protein cage architectures and membrane compartments. While membrane compartments are often heterogeneous and fluidic in morphology,1 the protein cages usually self-assemble from a limited, defined number of different proteins, resulting in modular structures with high symmetry, high homogeneity in shape and size, and potential to disassemble under altered biophysical or biochemical environments.11 Pores, formed at the symmetry axes of the assembled protein cage architectures, control the permeability of the compartments,11 which is different from membrane compartments that are mostly associated with membrane transport proteins.12 Understanding these properties helps us investigate how the protein cage architectures are utilized in nature to tune biocatalysis, and design biomimetic nanoreactors with different functionalities using the diverse architectures of protein cages.
So far, many fundamental studies have been done on naturally occurring protein cages with catalytic functions, while many synthetic nanoreactors have been designed by enzyme encapsulation inside protein cages using various approaches. This Perspective focuses on drawing the connections between the phenomena observed from the nanoreactors and the underlying mechanisms, as well as the challenges in biomimetics, by discussing the effects of protein cages on biocatalysis from three major aspects (Fig. 1): the catalysts (enzymes; Section 3), the catalyzed molecules (substrates and products; Section 4), and their interactions (cascade reactions; Section 5). We hope the views can provide insights and inspiration into more effective design and development of functional biocatalytic materials in the future.
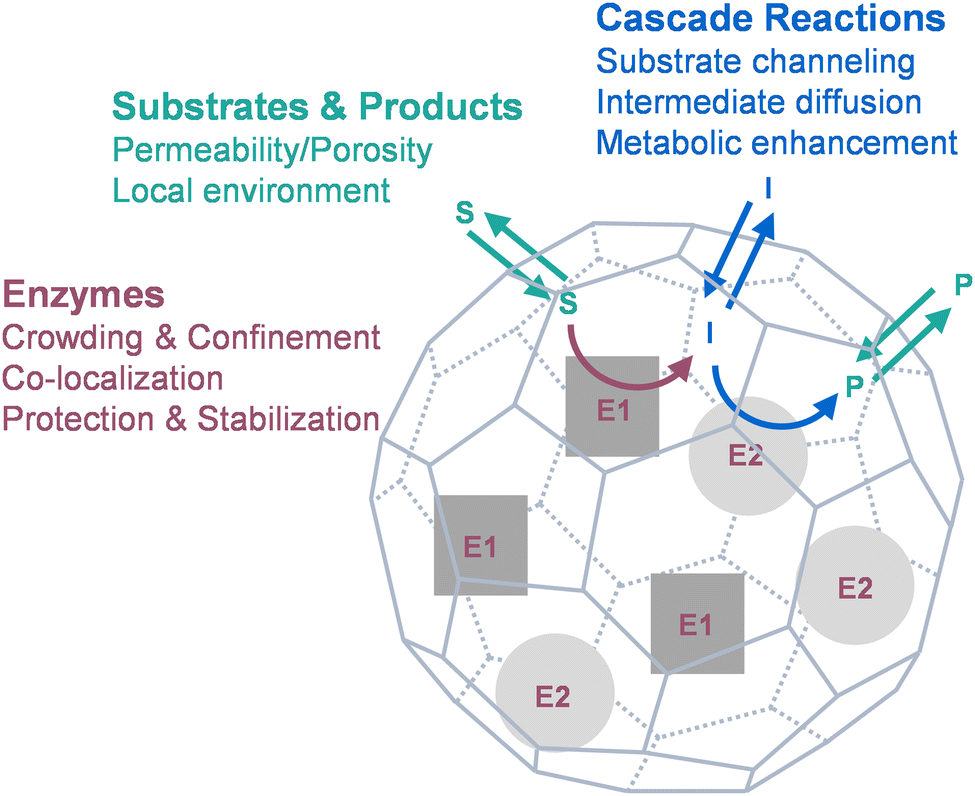 | ||
| Fig. 1 The properties of biocatalysis can be tuned by encapsulation of enzymes inside protein cages, which alters enzyme behaviors, substrate access and product release, and efficiency of cascade reactions. E, enzyme; S, substrate; I, intermediate; P, product. | ||
2. Ensemble measurements: average behavior of the entire population
The approaches and tools, that are essential for property determination, have unique advantages and limitations that influence the scope of the studies and therefore require thorough discussion. Most studies have used straightforward ensemble measurements to investigate the properties of protein cage-associated biocatalysis. These data can reveal important overall function of the whole population, but might miss the details of individual particle behavior due to heterogeneity that likely exists at multiple levels.The function of most proteins arises from their three-dimensional structures, and protein misfolding can cause loss of the activity. It is not surprising that different levels of misfolding can take place in some protein molecules within a population, resulting in activity heterogeneity, which has been confirmed in some cases by single-molecule experiments.13,14 This poses a technical challenge in studying engineered protein cages that are functionalized by protein encapsulation, since it is difficult to know how active each individual species is encapsulated inside a single cage. Many studies have shown that proteins encapsulated inside protein cages can be completely inactive,15 or gradually lose some or all the activity after sample preparation while the protein cage structure is unaltered.16,17 Encapsulation of enzymes at different maturation stages can result in nanoreactors with various activities: the more correctly folded enzymes, the more active the nanoreactors.18,19 Some work has also probed the denaturation of encapsulated enzymes inside intact protein cages.16,17 These results suggest the existence of partially or completely non-functional proteins inside the engineered protein cages. The absolute homogeneity of any preparation is not certain, which means that within an ensemble there might be a number of imperfect particles, in addition to particles with a range of cargo loading. Therefore, we need to bear in mind that most studies in the literature only employed methods applicable to ensemble measurements of properties to characterize the protein cages, and the generated data thus only reflect the overall average behavior of the samples. Simultaneously, single-particle techniques and research (such as native mass spectrometry,20,21 atomic force microscopy,22,23 and single particle fluorescence22,24) have been under intensive development to give insight into the distribution of the protein cages at different levels including their formation and functionality.
3. Direct effects: changes in endogenous properties of enzymes
3.1. Encapsulated enzymes exhibit altered apparent kinetic behavior
Changes in catalytic activity are sometimes observed after encapsulation of enzymes, which is usually attributed to the crowded and confined microenvironment inside the protein cages. Most examples suggest catalytic activity of encapsulated enzymes slow down compared to the enzymes that are free in solution, likely due to limited structural dynamics required for catalysis, as a result of crowding. There are reports where an enhanced activity was observed after encapsulation, but usually very little conclusive mechanistic understanding was made.Cornelissen and coworkers investigated the effects of crowding and confinement, where different copies of a lipase were encapsulated inside VLPs derived from Cowpea chlorotic mottle virus (CCMV).25 The enzyme exhibited a higher turnover number after encapsulation in general, but a trend of decrease in turnover was observed with an increase in enzyme loading density. With the same enzyme loading, the activity of the enzyme slowed down when green fluorescent protein was co-encapsulated as a crowding agent. Work by our group has studied the separate effects of crowding and confinement.26 Using an in vitro assembly approach, an alcohol dehydrogenase was encapsulated inside VLPs derived from P22 bacteriophage with a gradient of loading density (Fig. 2). When the enzyme was the sole cargo encapsulated inside the VLPs, the kcat of the enzyme was almost the same regardless of the loading density, suggesting that self-crowding of the enzyme inside the protein cage does not lead to alteration in catalytic behavior (Fig. 2(b)). Simultaneously, a catalytically inert protein as a non-self crowding agent, was co-encapsulated inside the nanoreactors with different enzyme loading densities, where a nearly constant overall loading density (of the two proteins) was maintained inside the capsids. In comparison to nanoreactors only loaded with the enzyme, the apparent turnover rate of the enzyme decreased as the amount of the inert protein increased, which indicates the crowding effect originated from other molecules can cause the alteration of the catalytic behavior (Fig. 2(c)). Furthermore, the kcat of the encapsulated enzyme was found to be significantly lower than that of free enzyme at the same low total concentration of the enzyme, but similar to the activity of free enzyme at high concentration (Fig. 2(d)). This observation demonstrates the confinement effect resulting from encapsulation leads to a high local concentration, which may change the catalytic behavior of enzymes compared to them being free in diluted solution. Although this work was only performed with one particular model enzyme and may not be able to completely and independently separate the effects of crowding and confinement, it is clear that self-crowding, crowding from foreign molecules, and confinement can independently, and together, impact the catalytic behavior of enzymes. Possibly, self-packing is highly evolved to form quaternary structures that can maintain or even facilitate enzyme function, while foreign crowding agents can potentially disrupt enzyme structure and alter the catalytic behavior.
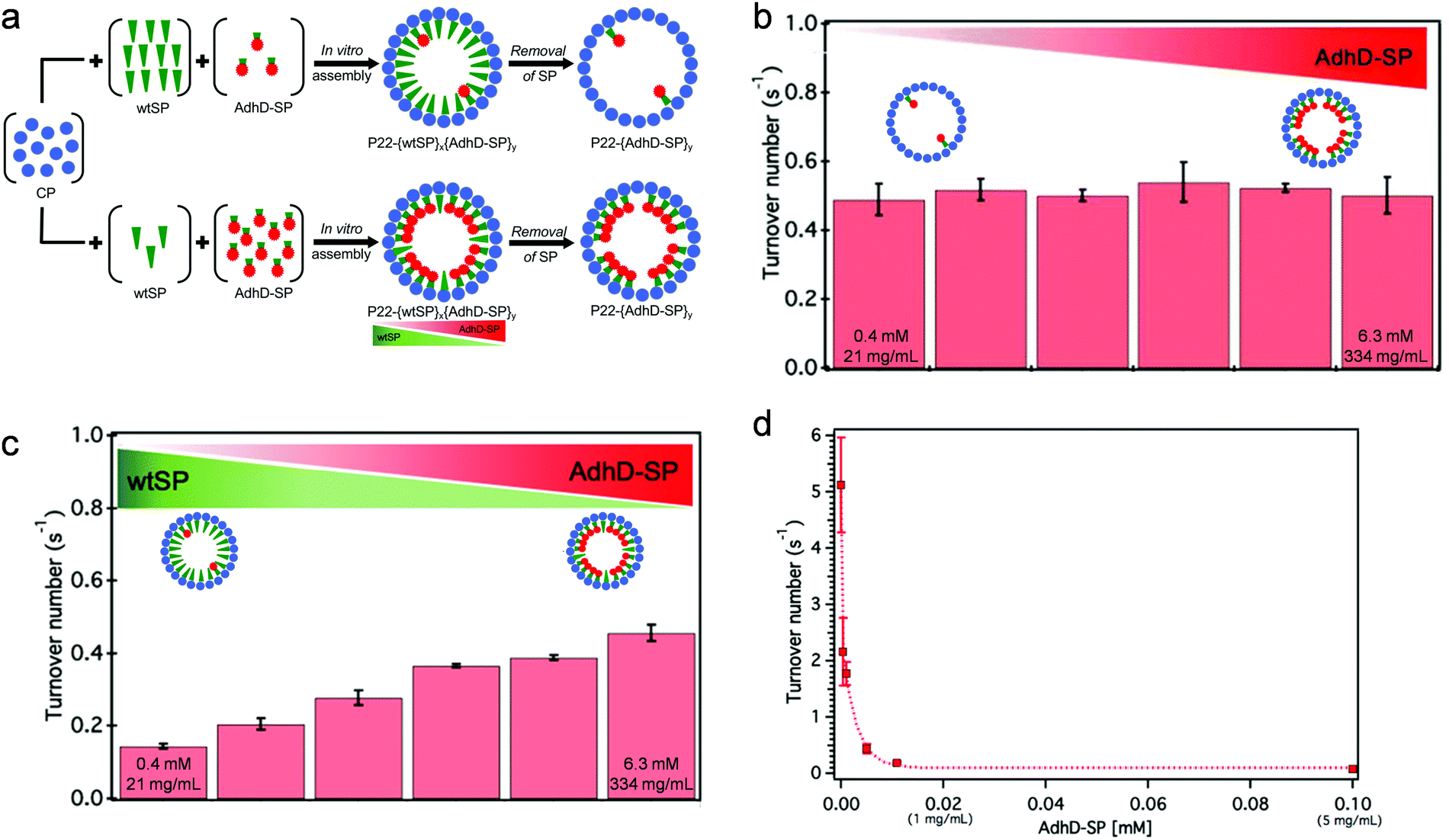 | ||
| Fig. 2 Crowding and confinement effects on the catalytic activity of alcohol dehydrogenase D (AdhD) after encapsulation inside P22 VLPs. (a) In vitro assembly allows controlled loading of AdhD inside P22 VLPs by co-encapsulation of wild-type scaffold protein (wtSP), which can be removed after particle formation. (b) Self crowding does not have an effect on AdhD activity. (c) wtSP as an inert, non-self crowding agent changes the catalytic activity of AdhD. The extreme local concentrations of AdhD inside the VLPs are labeled in (b) and (c). (d) Activity of free (unencapsulated) AdhD at different concentrations. Reproduced from ref. 26 with permission from the Royal Society of Chemistry. | ||
3.2. Co-localization of proteins enhances intermolecular communication
Co-localization of multiple copies of proteins is an outcome induced by crowding and confinement inside protein cage cavities. The co-localization increases the proximity between macromolecules, and therefore reinforces their physical interactions. Encapsulation leads to an increased local concentration of molecules and therefore a higher probability of intermolecular communication, which has been demonstrated by some works showing encapsulation of protein FRET pairs inside protein cages enhances the FRET efficiency significantly.27,28 This effect has been used to boost the activity of enzymes that are formed by weak oligomerization of several subunits. For example, an active Hyd-1 hydrogenase requires the formation of quaternary structure comprising a heterodimer of two subunits that undergo a weak, dynamic equilibrium in dilute solution (Fig. 3(a)). Once both subunits were encapsulated inside P22 VLPs (Fig. 3(b)), the weak inter-subunit association was reinforced, resulting in an improved catalytic efficiency of the enzyme (Fig. 3(c)).19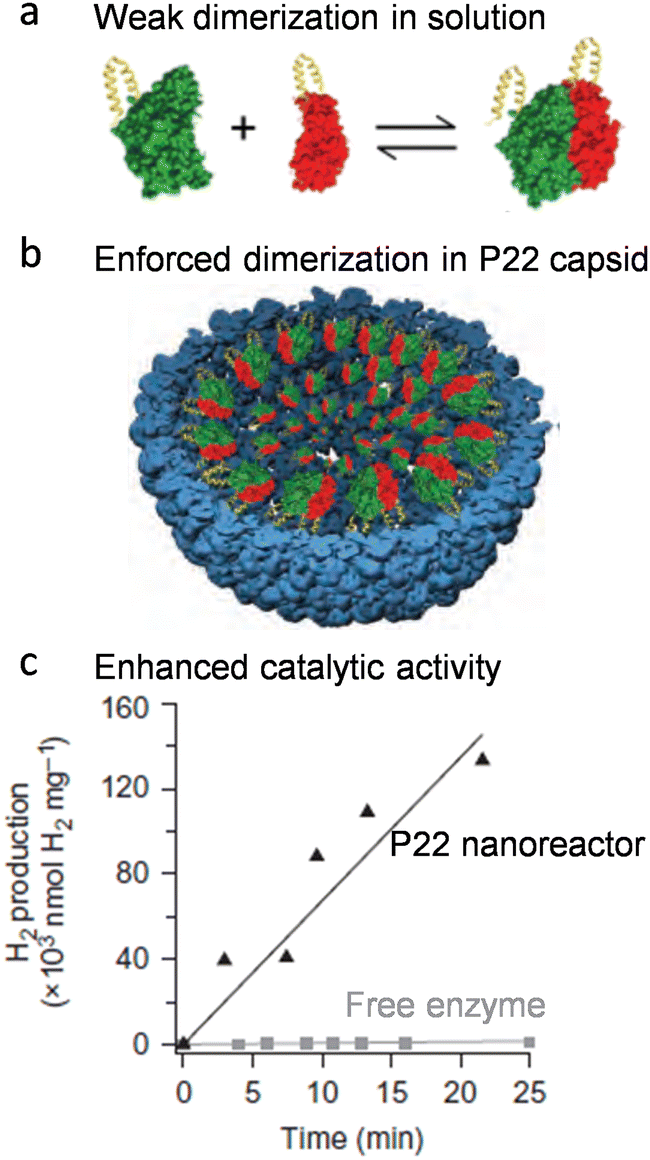 | ||
| Fig. 3 Activity enhancement of the heterodimeric Hyd-1 hydrogenase using P22 VLPs. (a) The two subunits exhibit only weak dimerization when free in solution. (b) Co-localization of the two subunits inside P22 VLPs enhances the dimerization. (c) The P22 nanoreactor showed enhanced catalytic activity compared to the free enzyme subunits. Reprinted with permission from ref. 2. Copyright 2022 American Chemical Society. | ||
3.3. Protein cages can act as enzyme stabilizers
Enzymes often show higher stability against thermal and chemical stimuli after encapsulation in protein cages. Finn et al. used Qβ VLPs as a model of protein cages to systematically investigate the influence of protein cages on the stability of encapsulated enzymes and the underlying mechanisms.17 The VLPs were found to show a universal protective effect on enzymes in response to challenges including heat, organic solvents, and chaotropic agents. By monitoring the intrinsic fluorescence of the enzymes during the denaturation, they found the VLPs effectively inhibited the enzyme unfolding due to the interior crowding, although other mechanisms for the protection could not be ruled out such as acceleration of refolding and shifting folding equilibrium. Using a dye-based thermal shift assay, our group showed the melting temperatures were higher when enzymes are encapsulated inside P22 VLPs.16 Since this assay directly probes the hydrophobic regions exposed during thermal denaturation, the results echo and support the mechanism of unfolding inhibition imposed by protein cages.Protein cages have also been used as molecular chaperones to stabilize enzymes that are prone to aggregation.29 For example, encapsulation of an α-galactosidase inside P22 VLPs during recombinant expression resulted in active nanoreactors, which prevents the enzyme from forming inactive, insoluble inclusion body when it is expressed alone.30 Similarly, T4 lysozyme is also an unstable enzyme and easily aggregates in the presence of negatively charged molecules. However, the enzyme activity was sustained after encapsulation inside CCMV VLPs, likely due to the stabilization effect resulting from charge complementation between the enzyme and the negatively charged VLP lumen.31
4. Selectivity alteration: flux of substrates and products
4.1. Capsid pores influence molecular diffusion
The permeability of protein cages regulates the molecular diffusion between the bulk environment and the capsid cavity, and can potentially control the selectivity of the reactions catalyzed by the encapsulated enzymes. Carboxysome is an exemplary and inspirational, naturally occurring protein cage that exhibits selectivity in permeability: the capsid pores allow passive diffusion of bicarbonate into the protein cage and achieve retention of carbon dioxide produced in situ by encapsulated carbonic anhydrase (CA), while excluding molecular oxygen (Fig. 4(a)).32–34 This mechanism only presents CO2 to the encapsulated ribulose-1,5-bisphosphate carboxylase–oxygenase (RuBisCO), prompting this promiscuous enzyme to only display carboxylase activity, i.e., the reaction selectivity of RuBisCO is regulated by the controlled flux of substrates across the capsid of carboxysome. (The inspiration for cascade reactions from this system is further discussed in Section 5). Utilizing the ability to block oxygen entry, artificially nanoreactors have been designed with encapsulation of oxygen-sensitive hydrogenases in carboxysome for enhanced enzyme stability and activity.35 These systems demonstrate that capsid permeability plays an important role in the overall reactivity of the protein cage nanoreactors.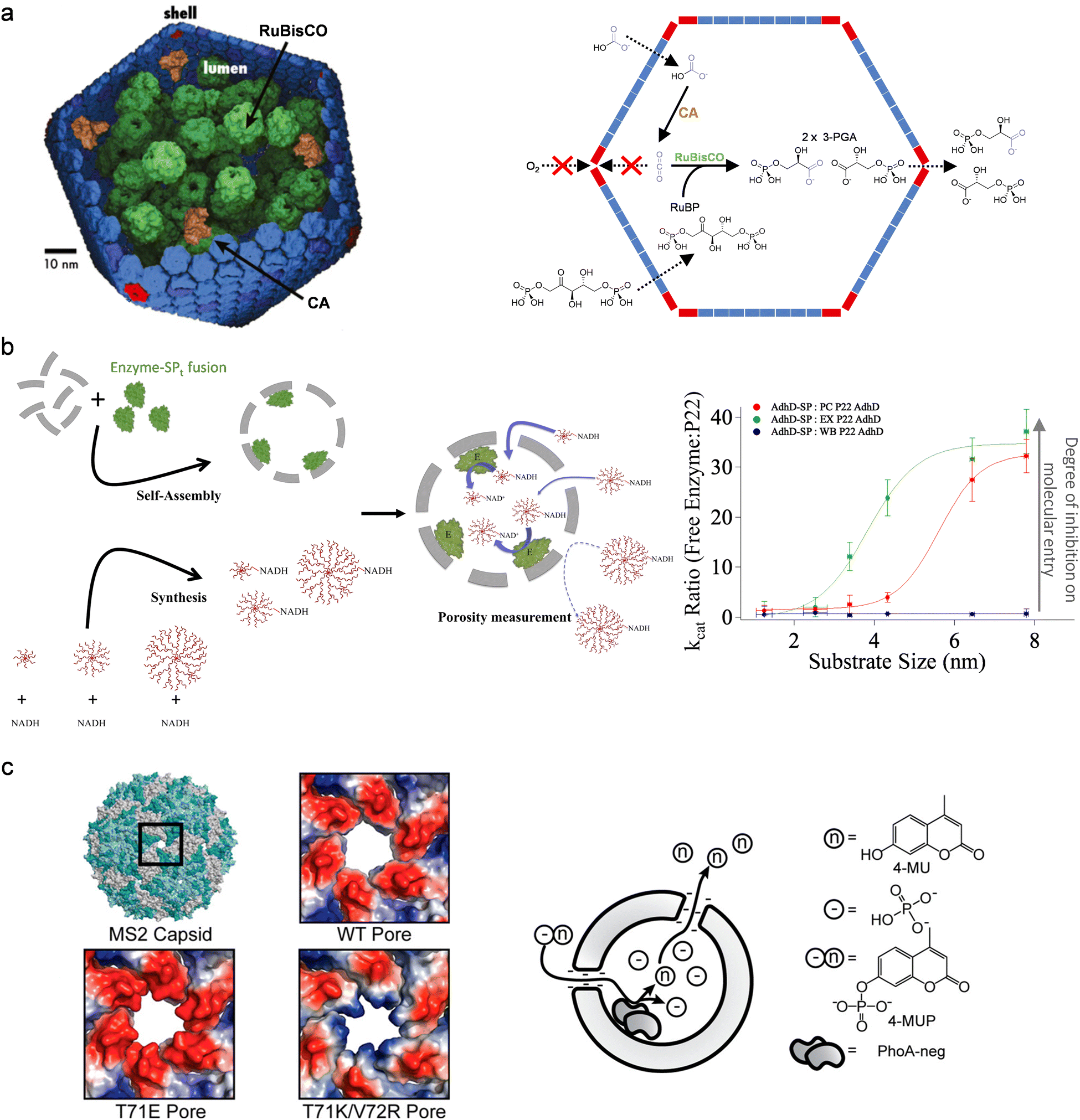 | ||
| Fig. 4 Alteration of biocatalysis using the porosity of protein cages. (a) Left: The carboxysome co-encapsulates carbonic anhydrase (CA; a diffusion-limited enzyme) and ribulose-1,5-bisphosphate carboxylase–oxygenase (RuBisCO). Reproduced from ref. 34. Right: The capsid retains CO2 produced in situ by CA to maintain an environment with high local concentration of the metabolic intermediate, while O2 is excluded from access to RuBisCO. This mechanism prompts the promiscuous RuBisCO to use CO2 as substrate instead of O2, enhancing the metabolic pathway of carbon fixation. Reproduced from ref. 63 under the terms of the Creative Commons CC BY license. (b) The porosity of P22 VLPs with different morphologies (PC, procapsid; EX, expanded; WB, wiffle ball) was probed using an enzymatic reaction and synthetic substrates with different sizes. The apparent activity of the encapsulated enzyme, when compared to the free enzyme, is indictive of the substrate entry into the VLPs and therefore the porosity of capsid. Reproduced with permission from ref. 36. Copyright 2021 The Authors. Published by Springer Nature under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/). (c) The electrostatics of pores on MS2 VLPs can be changed by mutations of pore-lining residues, which was shown to alter the product accumulation inside the capsid and therefore the activity of the encapsulated enzyme. Reprinted with permission from ref. 37. Copyright 2015 American Chemical Society. | ||
The diversity of capsid pores has been reviewed comprehensively in a recent work, but their potential as gating systems to alter the selectivity of biocatalysis remains to be fully uncovered.11 We have probed the porosity of P22 VLPs in the context of a biocatalytic reaction within the cage (Fig. 4(b)).36 In this system, an alcohol dehydrogenase was encapsulated inside P22 VLPs to form nanoreactors, while the substrate of the enzyme was modified in size and charges by synthetic fusion to different dendrimers. Feeding the nanoreactors with the modified substrates showed that the capsid pores impose a certain size threshold in allowing molecular diffusion: they do not pose a barrier in the diffusion of the substrates much smaller than the threshold to access the encapsulated enzymes, while entry of bulky molecules bigger than the pores is significantly inhibited. Interestingly, electrostatics start to exert influence on the diffusion of the substrates when they have sizes similar to the pore size: the residues close to the pores create an electric field which facilitates the transport of negatively charged molecules into the nanoreactors. This work explicitly suggests that both sterics and electrostatics are important mechanisms behind the substrate selectivity of protein cage nanoreactors, and also provides an approach to study the role of capsid pores in protein cage nanoreactors.
Modification of capsid pores can change the selectivity of the nanoreactors. In the just mentioned P22 VLP system, the electrostatic effect can be tuned by genetic engineering of residues close the pore.36 The work by Tullman-Ercek et al. on MS2 VLP-based nanoreactors also demonstrates that coulombic barriers, built by the pores for molecular diffusion, can be enhanced by mutations to pore-lining residues (Fig. 4(c)).37 This work also shows the reaction of the nanoreactors is regulated not only by the influx of substrates but also the efflux of the products, as the encapsulated enzymes can be inhibited by product accumulated inside the capsid. More studies have been done recently to engineer capsid pores, using either genetic38 or chemical39 methods, to investigate the effects on molecular diffusion. The resultant structures and knowledge present great potential for developing protein cage nanoreactors for selective biocatalysis.
4.2. Local environment of capsid cavity can change substrate selectivity
The nano-environment of the protein cage cavity can be significantly different from the bulk solution, resulting from the side chains of the amino acid residues displayed on the capsid interior surface. For example, the pH value of the capsid cavity was probed experimentally for CCMV40 and DNA binding protein from starved cells (Dps),41 and found about 0.5 and 1.2 unit different from the bulk environment, respectively. Protein cages can also be genetically engineered to alter the cavity environment, such as an engineered lumazine synthase protein cage possessing highly charged luminal surface (Fig. 5).42 Encapsulation of an enzyme inside this engineered protein cage resulted in a nanoreactor with selectivity over substrates with different charges, due to the electrostatic attraction and repulsion between the substrates and the charged interior of the protein cage. The highly charged local environment can also be created as an emergent property by fabricating higher-order assembly of protein cage nanoreactors that have highly homogenous structure and surface charges, which has been used to tune substrate selectivity of biocatalytic processes.43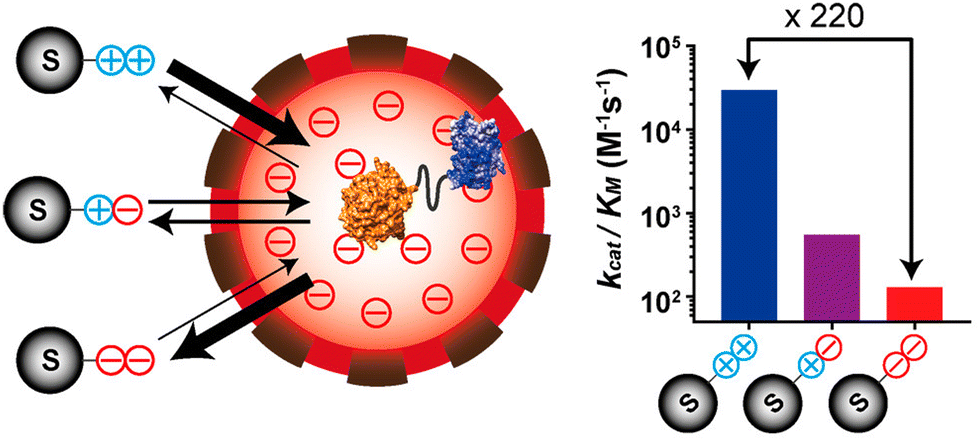 | ||
| Fig. 5 The access of charged substrates to an enzyme was altered using the highly negatively charged luminal surface of an engineered lumazine synthase protein cage. Reprinted with permission from ref. 42. Copyright 2017 American Chemical Society. | ||
5. Cascade reactions: why and how to induce substrate channeling
Inspired by bacterial microcompartments, many artificial metabolons have been designed by encapsulation of functionally coupled enzymes (or catalytic domains) that catalyze metabolic pathways. These naturally occurring complexes and synthetic mimics have offered us opportunities to investigate and utilize the enzyme-substrate/product relations that govern the efficient multistep biochemical processes.5.1. Proximity alone cannot ensure substrate channeling due to fast diffusion of small molecules
Substrate channeling is a well-known mechanism that rationalizes the high efficiency of the metabolism in organisms. It describes the direct passage of metabolic intermediates from an enzyme to the subsequent one in a metabolic pathway, without being released into the bulk solution.44 In nature, this phenomenon takes place in some multifunctional enzymes with multiple catalytic domains, as well as some multienzyme complexes recognized as metabolons.45,46 It was initially believed that simple close packing of functional coupled enzymes would automatically lead to a kinetic advantage via substrate channeling to raise the efficiency of metabolic pathways. However, a growing body of evidence suggests it is highly unlikely to achieve channeling by only enforcing close proximity between enzymes.Theoretical analyses have compared the catalytic efficiency of enzymes and the diffusion rate of small molecules, given that intermediates in pathways experience competition between diffusion to the active site of the downstream enzymes and diffusion into the bulk solution. Most enzymes operate at rates (average kcat/KM ∼ 105 M−1 s−1) that are 3–4 orders of magnitude slower than diffusion rates for collision between enzymes and small molecule metabolites in dilute solution (∼108 M−1 s−1).47 Even inside the crowded intracellular environment, the diffusion rate only reduced to a quarter to a third compared to dilute solutions.46 Therefore, diffusion of small molecules occurs at a much shorter timescale than the enzyme turnover, i.e., metabolic reactions are mostly reaction-limited rather than diffusion-limited. From another perspective, substrate channeling enhances multistep efficiency by significantly raising the local concentration of intermediates that are directly accessible to the second enzyme, compared to the total intermediate concentration in solution. Under most conditions (where diffusion is about 100–1000 times higher than turnover, as mentioned above), there is little to no difference in the effective local concentration of a small molecule metabolite within 5 μm of the active site where it is produced, and only beyond this distance does the effective concentration of intermediate drop.44 However, in most practical in vitro synthetic biocatalytic systems, the inter-enzyme distance is already less than 5 μm in an isotropic solution even when no inter-enzyme proximity is enforced.† Therefore, enforcing inter-enzyme proximity cannot increase the local concentration of intermediate to any appreciable extent to speed up the second metabolic reaction. (That being said, micron-scale distances, where diffusion becomes a limiting factor for cascade reactions, have been investigated48 in the demonstrated enhancement of multistep efficiency in vivo,49 and also employed for making micro-sized reactors.50‡)
Experimental results on the artificial metabolons made of protein cages, consistent with the theoretical calculations mentioned above, suggest that proximity between functionally coupled enzymes or enzymatic domains does not necessarily induce kinetic advantages for multistep transformations. Our lab has found that the efficiency of a sugar metabolic pathway remained the same, whether the paired enzymes of the pathway were encapsulated separately in different P22 VLPs or were co-encapsulated together in the same P22 VLP (Fig. 6(a)).51 We also made a P22 VLP nanoreactor capable of the two-step glutathione biosynthesis by encapsulation of a bifunctional enzyme possessing two catalytic domains (Fig. 6(b)).16 No channeling was observed for either the free enzyme or the enzyme encapsulated nanoreactor, suggesting that high proximity, induced by either connecting the active sites within a single molecule (intramolecular proximity) or packing the enzyme to near crystalline densities (high intermolecular proximity), cannot surpass the challenge of fast diffusion of the intermediate. Hilvert and coworkers synthesized a carboxysome mimic by encapsulation of CA and RuBisCO inside an engineered protein cage based on lumazine synthase, but this artificial nanoreactor did not enhance the carbon fixation pathway as seen in the natural carboxysomes (Fig. 4(a)), also demonstrating that enzyme proximity is not the core determinant for the increased efficiency of carboxysomes (see Section 4.1 and 5.2).52 In addition to more studies on cascade reactions,53 fast diffusion of small molecules is also observed in experiments on protein cage nanoreactors catalyzing other reactions.54
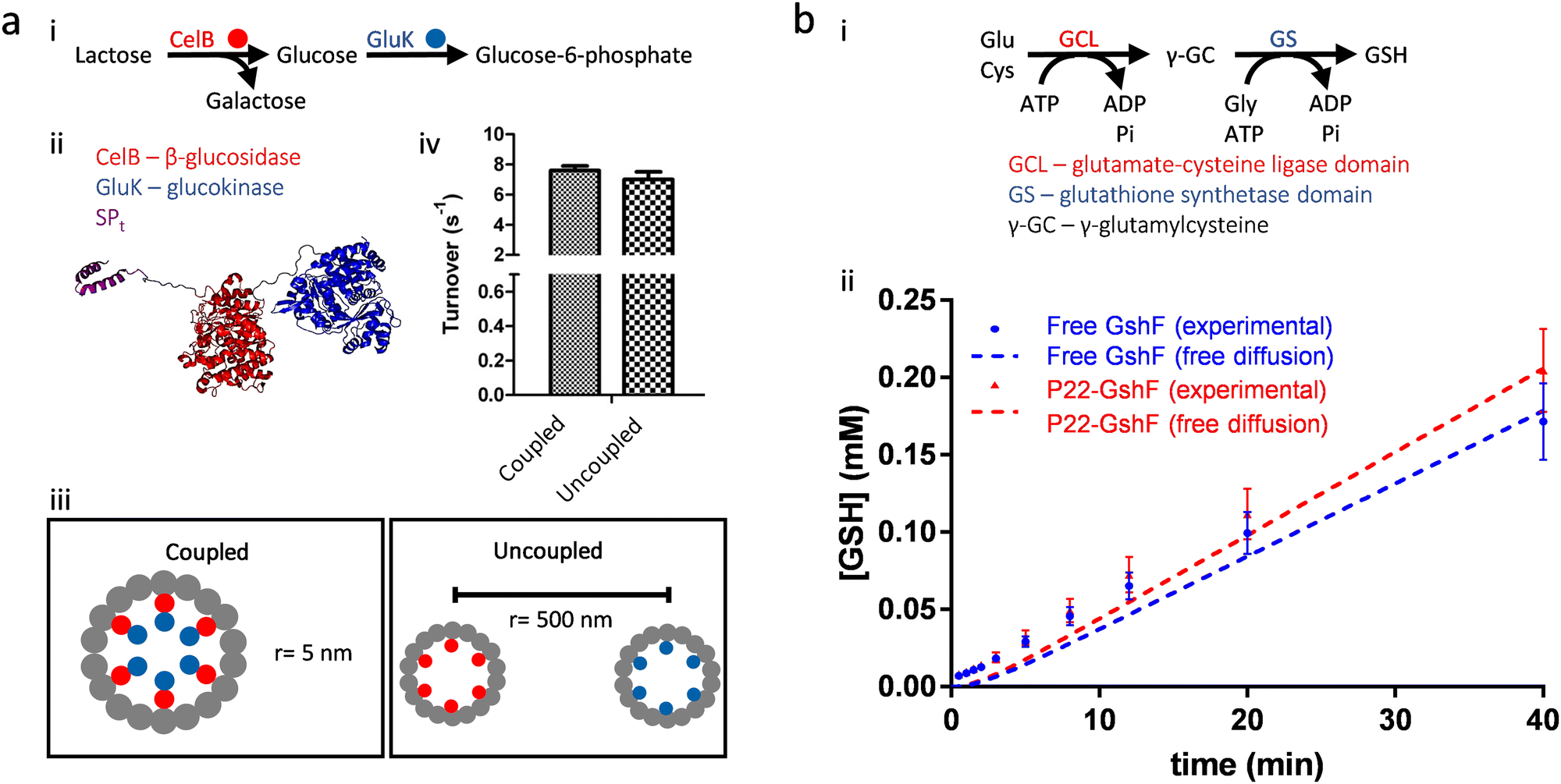 | ||
| Fig. 6 Proximity induced by encapsulation does not enhance the efficiency of multistep reactions. (a) A functionally coupled enzyme pair capable of a sugar metabolic pathway (i) was co-encapsulated inside P22 VLPs by genetic fusion (ii). However, this nanoreactor capable of two-step reaction showed similar overall efficiency, compared to the mixture of the two nanoreactors capable of each individual step (iii and iv). (b) The bifunctional glutathione full synthetase (GshF), possessing two catalytic domains (GCL and GS), catalyzes the complete glutathione (GSH) biosynthetic pathway (i). Neither free GshF in solution nor GshF encapsulated inside P22 VLPs (P22-GshF) showed increased pathway efficiency from the free diffusion scenario. Adapted with permission from ref. 2 and 16. Copyright 2022 and 2020 American Chemical Society. | ||
The effect of proximity on cascade reactions has also been computationally modeled.55 Simulations on Brownian dynamics of reaction intermediate at 298 K in aqueous solution suggest that the probability of the second reaction is dependent on not only the inter-enzyme distance, but also their relative location: when the active sites are in a face-to-face orientation, the probability of channeling is higher. However, even if the orientation is optimal, any channeling effect diminishes significantly once the active sites are more than 1 nm apart, which is difficult to achieve even in the densely packed solid state (crystalline). In summary, close proximity alone between functionally coupled enzymes should not be used as the primary design principle in synthetic approaches to the construction of acellular biocatalytic materials to achieve efficiency enhancement of enzymatic cascade reactions.§ However, inter-enzyme proximity (which results in co-localization) has been used as an auxiliary means to induce substrate channeling together with other primary mechanisms that can control the diffusion of cascade intermediates, both naturally and synthetically (see Section 5.2 and 5.3).
5.2. Naturally occurring metabolons formed by protein cages induce substrate channeling via various mechanisms for enhanced overall metabolism inside organisms
Controlling the diffusion of reaction intermediates is the key to substrate channeling. Examples of substrate channeling in nature can be categorized into regulation of diffusion direction (such as molecular tunnels and electrostatic guidance) and limitation of diffusible space (such as swing arm and spatial confinement).44 The carboxysome is exemplary in which the reaction intermediate produced in situ cannot pass through the capsid pores to escape the protein cage, resulting in channeling between the encapsulated enzymes (see Section 4.1).32 Similarly, the Pdu (propanediol utilization) microcompartments also have pores with selective permeability, which restrain the diffusion of the intermediate out of the protein cage where the associated enzymes are encapsulated.4 In these cases, the protein cages do not directly participate in catalysis, but offer a segregated environment that allows co-localization of enzymes and limits intermediate diffusion to enhance catalysis. In a contrasting example of pyruvate dehydrogenase complex (PDHc, Fig. 7(a)), the catalytic E2 subunits form a cage-like core to which the other catalytic subunits, E1 and E3, are bound through strong non-covalent interactions.56 This structure is different from the bacterial microcompartmemts where the biocatalysts are all encapsulated inside the non-catalytic protein cages, but it also achieves co-localization of multiple functionally coupled biocatalysts. As part of the E1 activity, the intermediate of this multistep reaction is covalently tethered to E2 through the lipoamide swing arm so that the intermediate (acetyl group) cannot diffuse into bulk solution and can instead be channeled from the active site of E1 to that of E2 for catalysis (Fig. 7(b)).57 Similar structures to PDHc are also found in 2-oxoglutarate dehydrogenase complex (OGDHc) and branched-chain 2-oxo acid dehydrogenase complex (BCDHc), which all demonstrate substrate channeling of intermediates by covalent attachment between co-localized active sites.58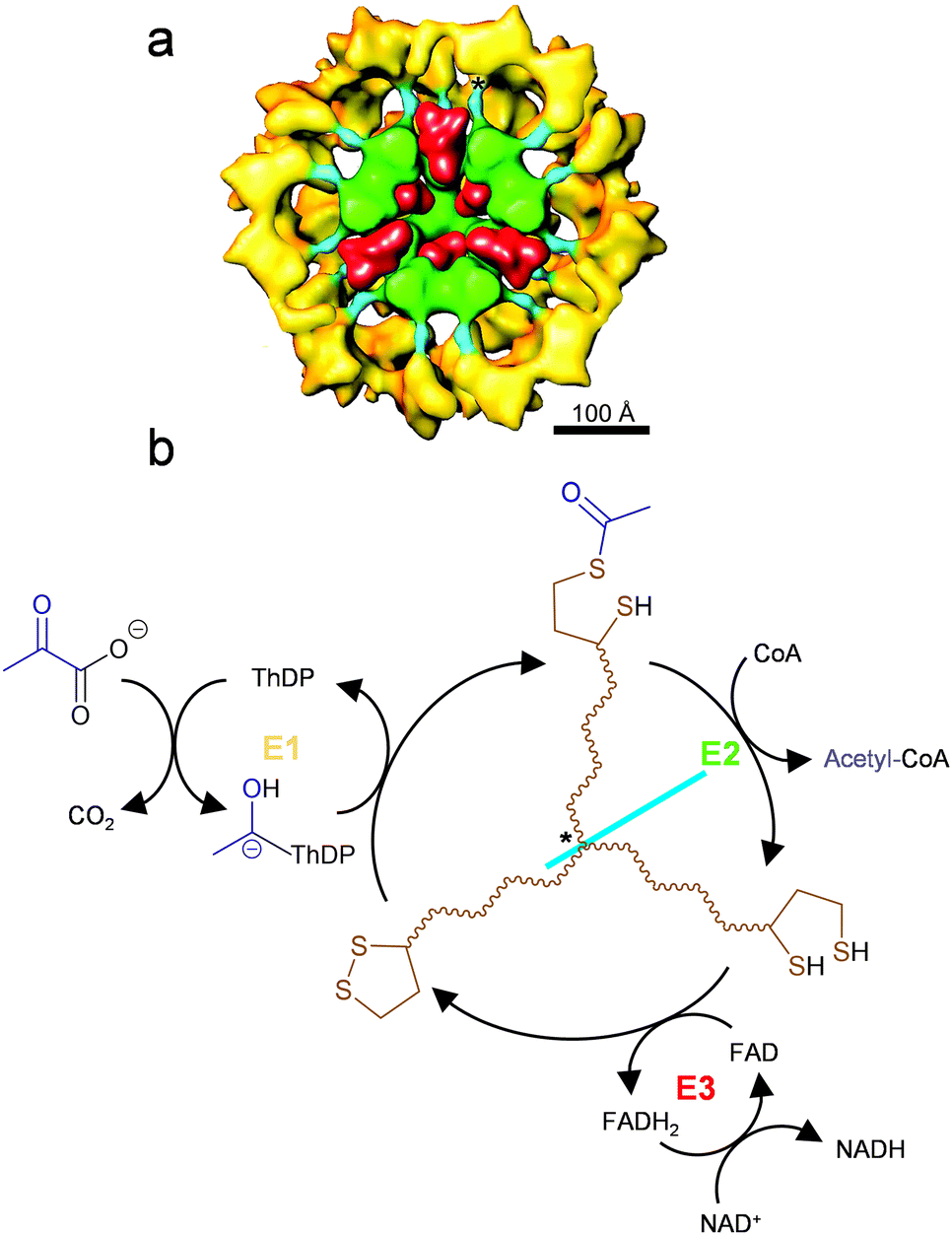 | ||
| Fig. 7 Substrate channeling in pyruvate dehydrogenase complex (PDHc). (a) Cut-away structure model of PDHc viewed on its 3-fold axis. The catalytic domain of the E2 subunits forms a cage-like core (green), which an intramolecular linker (cyan) connects to the domain where the E1 subunits bind (yellow). The E3 homodimer (red) is located at the pentagonal opening of the E2 core (green). The anchor for the lipoyl domains to pivot is indicated by asterisk. Reproduced with permission from ref. 56. Copyright (2001) National Academy of Sciences. (b) The scheme of the cascade reaction catalyzed by PDHc. The lipoamide swing arm (brown) on E2 subunit facilitates the transfer of acetyl group (blue) from thiamine diphosphate (ThDP) to coenzyme A (CoA) by substrate channelling. The lipoic acid cofactor is regenerated by E3. | ||
Substrate channeling induced by the protein cage-based metabolons can enhance the overall function of organisms. Toxic and labile intermediates can be prevented from being leaked into the bulk environment of the organisms, highlighted by the Pdu microcompartment which sequesters and processes a toxic aldehyde intermediate.4 Also, substrate channeling can guide enzymes and metabolites for the downstream reactions of interest when several different pathways are possible. For example, the carboxysome enforces RuBisCO, a promiscuous enzyme that can use both CO2 and O2 as substrates, to only exhibit carboxylase activity (CO2 as substrate) rather than oxygenase activity (O2 as substrate), ensuring the efficiency of carbon fixation which is vital to some organisms (see Section 4.1).32 Moreover, substrate channeling by a swing arm mechanism can enhance active-site coupling and thus the multistep efficiency by changing substrate specificity and raising the effective local concentration of the intermediates. As demonstrated by PDHc and OGDHc, lipoic acid cofactor is attached to the enzyme complexes by swing arms, so that the cofactor (as multistep intermediate) can be directly shuttled between between co-localized active sites and regenerated in situ, without diffusing into the bulk solution.44
5.3. Taking full advantage of costly cofactors is one plausible rationale for developing in vitro systems that can induce substrate channeling
Inspired by the naturally occurring structures, biocatalytic complexes have been designed to induce substrate channeling using biomimetic approaches. A significant amount of research on metabolic engineering has concentrated on developing modified organisms for metabolite production;59 inducing substrate channeling in vivo might provide us with metabolic benefits mentioned in Section 5.2.60Some beneficial effects mentioned in Section 5.2 provides the rationale and methods for the development of in vitro systems capable of substrate channeling as biocatalytic materials. Enzyme promiscuity and metabolite toxicity are often not concerns in in vitro systems, since they have a much cleaner composition compared to in vivo systems and are not necessarily involved with living systems. However, the efficient utilization of labile intermediates is a valid rationale to induce channeling in synthetic biocatalytic materials. It might also be of great value in more complex system to use substrate channeling as switches between different pathways. There are works done based on these rationales,61 but the utility of substrate channeling in these areas needs to be further explored.
Most current research on designing in vitro biocatalytic systems intends to utilize the kinetic advantage of substrate channeling to make downstream reactions operate at high velocities at the early stage of cascade reactions (Fig. 8(a)), when the total turnover count of the first step is still low. This advantage shortens the lag phase prior to achieving the steady state phase (reflected as transient time τ), and has been thought to enhance the efficiency of cascade reactions.44,45,62 However, even without channeling, τ can be tuned and even reduced to zero by changing the rates of each step, which is usually not difficult in vitro by changing enzyme amount. In other words, there is little compelling reason to induce channeling in vitro for classical single-direction cascade reaction (Fig. 8(a)). Nevertheless, this mechanism, where effective concentration of the intermediate to enzymes can be elevated by substrate channeling, is still valuable and inspirational for designing biocatalytic materials. One valid rationale to use this mechanism in vitro is to recycle enzyme cofactors in situ by channeling them as reaction intermediates between functionally coupled enzymes, where a cofactor becomes the intermediate of each individual step of an “infinite-step” reaction (circular cascade reaction, Fig. 8(b)). This idea can allow cofactor-dependent cascade reactions to proceed at high overall speeds by only investing very small quantities of the precious cofactors in the system.
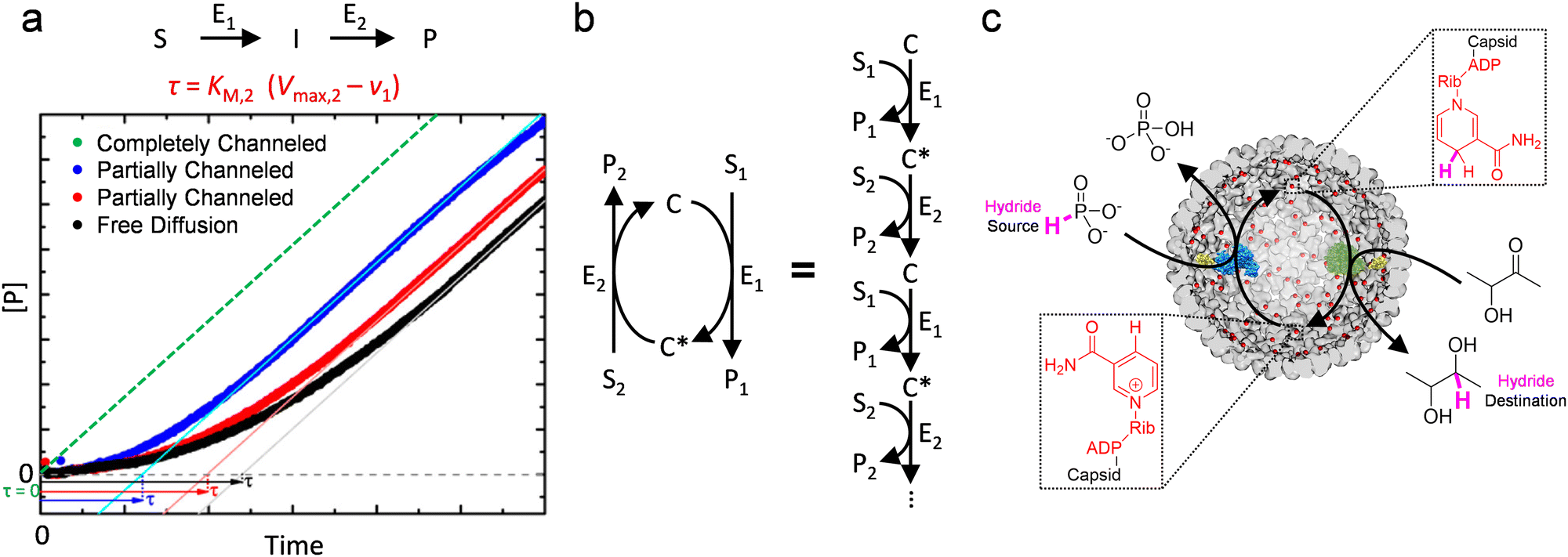 | ||
| Fig. 8 Effective utilization of substrate channeling for in vitro biocatalysis. (a) Substrate channeling reduces the lag phage, reflected by transient time τ, of a classical two-step reaction. The transient time is dependent on rate of the first step (v1), and the Michaelis constant (KM,2) and the maximum rate (Vmax,2) of the second step. Adapted and modified with permission from ref. 62. Copyright 2017 American Chemical Society. (b) A cofactor-dependent circular cascade reaction can be depicted as an “infinite-step” reaction where the different forms of the cofactor (C and C*) are regarded as intermediates alternately. E, enzyme; S, substrate; I, intermediate; P, product. (c) Covalent tethering of NAD on the luminal surface of P22 VLP with a swing arm allowed channeling of the cofactor between an enzyme couple encapsulated inside the VLP, which enhanced the efficiency of the two-step hydride transfer reaction. Reproduced from ref. 63 under the terms of the Creative Commons CC BY license. | ||
We have developed nanoreactors based on P22 VLPs to enhance nicotinamide adenine dinucleotide (NAD)-mediated cascade reactions (Fig. 8(c)).63 In this system, two functionally coupled enzymes were co-encapsulated inside the VLP to induce proximity between the enzymes, as in the carboxysomes. However, different from the carboxysomes, the capsid pores on P22 do not impose any barrier to the diffusion of small molecules including NAD.36 Instead, the NAD cofactor was covalent tethered to the interior lumen of the capsid by mimicking the swing arm structure of PDHc (see Section 5.2). Thus, the immobilized NAD could shuttle between the co-localized enzyme pair without escape into the bulk solution, and be continuously recycled in situ. This design made the cascade reaction operate with enhanced efficiency even with a very small amount of the cofactor. As a consequence, the expensive cofactor was taken advantage of more effectively compared to the in vitro systems without substrate channeling. This work highlights several effects of protein cages on biocatalysis as discussed above. The confinement and crowding inside the protein cage induced the co-localization of the molecules that participated in the cascade reaction. The roles of capsid pores were also considered: substrates and products could be easily accessed and released given their free diffusion through the capsids, while controlling the diffusion of NAD was realized using a biomimetic swing-arm tethering approach. Furthermore, given the modularity of P22 VLPs, these nanoreactors were disassembled and reassembled with enzymes capable of different NAD-dependent reactions, demonstrating another advantage of the protein cage architectures as templates for the construction of artificial nanoreactors.
6. Conclusions
Protein cage architectures have been used both in nature and artificially to tune the properties of biocatalysis, including changing the properties of enzymes, regulating the diffusion of substrates and products, and modulating the communication between enzymes and small molecule metabolites. Most of the effects originate from the segregated, limited space defined by the cage structure, which can lead to molecular confinement and crowding, a local environment with different biophysical and biochemical properties from the solution, and a potential barrier that may influence the inside–outside molecular exchange. Investigation on the naturally occurring protein cages and their mimics have broadened our perspectives about the underlying mechanisms behind biocatalysis, with regard to efficiency, selectivity, catalyst stability, and molecular interactions. This knowledge offers us fundamental basis and inspiration for design and development of functional biocatalytic materials, as well as applications in synthetic biology such as metabolic engineering.The structures of protein cages offer a great range of platforms for developing biocompatible catalytic materials with a variety of functions, which conform to the growing need for renewable and environmentally friendly green chemistry materials. Given the emergent use of protein cage architectures for tunable biocatalysis, future biocatalytic materials might contribute to advanced functionalities including enhanced efficiency (including rate and yield) and lifetime of biocatalysts, as well as selectivity of chemical processes. This will likely require more fundamental understanding about the structure–function relationships of the protein cages, and state-of-the-art techniques for refinement (for example, in silico design, directed evolution, high throughput screening) and detailed characterization (for example, at single-particle level) of the biocatalytic protein cages. In addition, recent progress in higher-order assemblies of protein cages shows their great potential in the field of heterogenous biocatalysis and biocatalyst preservation.2,64 With respect to applications, enzymatic cascade reactions are of special interest, since they realize multistep chemistries in one pot with high activity, stereoselectivity, and little intermediate residue, compared to traditional chemical synthesis.65,66 The employment of protein cage architectures can potentially endow cascade reactions with further advantageous features, such as efficiency enhancement, substrate selectivity, enzyme stabilization and longevity, controlled diffusion of metabolic intermediates, cofactor regeneration, heterogenous catalysis, and catalyst recovery.2,67 Simultaneously, the utility of protein cages is also growing in whole-cell catalysis and metabolic engineering.35,68–70 Therefore, translational outcomes of protein cage-associated biocatalysis can be expected in industry including pharmaceutical/fine chemical manufacturing, biofuel/agricultural production, and environmental remediation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by grants from the National Science Foundation (CMMI-1922883), and the Human Frontier Science Program (HFSPO RGP0012/2018). YW was partially supported by Robert & Marjorie Mann Fellowship from Department of Chemistry, Indiana University. The authors would also like to thank Pawel Kraj (Indiana University) and Nathasha D. Hewagama (Indiana University) for their suggestions.Notes and references
- Y. Diekmann and J. B. Pereira-Leal, Biochem. J., 2013, 449, 319–331 CrossRef CAS PubMed.
- Y. Wang and T. Douglas, Acc. Chem. Res., 2022, 55, 1349–1359 CrossRef CAS PubMed.
- A. H. Chen and P. A. Silver, Trends Cell Biol., 2012, 22, 662–670 CrossRef CAS PubMed.
- S. Frank, A. D. Lawrence, M. B. Prentice and M. J. Warren, J. Biotechnol., 2013, 163, 273–279 CrossRef CAS PubMed.
- H. Kirst and C. A. Kerfeld, BMC Biol., 2019, 17, 79 CrossRef PubMed.
- P. A. Monnard and P. Walde, Life, 2015, 5, 1239–1263 CrossRef CAS PubMed.
- S. Oehler and B. Müller-Hill, J. Mol. Biol., 2010, 395, 242–253 CrossRef CAS PubMed.
- R. J. Ellis, Trends Biochem. Sci., 2001, 26, 597–604 CrossRef CAS PubMed.
- N. Demaurex, News Physiol. Sci., 2002, 17, 1–5 CAS.
- R. J. Nap, A. L. Božič, I. Szleifer and R. Podgornik, Biophys. J., 2014, 107, 1970–1979 CrossRef CAS PubMed.
- N. Tasneem, T. N. Szyszka, E. N. Jenner and Y. H. Lau, ACS Nano, 2022, 16, 8540–8556 CrossRef CAS PubMed.
- W. Sadée, V. Drübbisch and G. L. Amidon, Pharm. Res., 1995, 12, 1823–1837 CrossRef PubMed.
- D. M. Rissin, H. H. Gorris and D. R. Walt, J. Am. Chem. Soc., 2008, 130, 5349–5353 CrossRef CAS PubMed.
- X. Li, Y. Jiang, S. Chong and D. R. Walt, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8346–8351 CrossRef CAS PubMed.
- J. Sharma, M. Uchida, H. M. Miettinen and T. Douglas, Nanoscale, 2017, 9, 10420–10430 RSC.
- Y. Wang, M. Uchida, H. K. Waghwani and T. Douglas, ACS Synth. Biol., 2020, 9, 3298–3310 CrossRef CAS PubMed.
- S. Das, L. Zhao, K. Elofson and M. G. Finn, Biochemistry, 2020, 59, 2870–2881 CrossRef CAS PubMed.
- D. P. Patterson, C. Hjorth, A. Hernandez Irias, N. Hewagama and J. Bird, ACS Synth. Biol., 2022, 11, 2956–2968 CrossRef PubMed.
- P. C. Jordan, D. P. Patterson, K. N. Saboda, E. J. Edwards, H. M. Miettinen, G. Basu, M. C. Thielges and T. Douglas, Nat. Chem., 2016, 8, 179–185 CrossRef CAS PubMed.
- S. Tamara, M. A. den Boer and A. J. R. Heck, Chem. Rev., 2022, 122, 7269–7326 CrossRef CAS PubMed.
- T. P. Wörner, T. M. Shamorkina, J. Snijder and A. J. R. Heck, Anal. Chem., 2021, 93, 620–640 CrossRef PubMed.
- K. Strobl, E. Selivanovitch, P. Ibáñez-Freire, F. Moreno-Madrid, I. A. T. Schaap, R. Delgado-Buscalioni, T. Douglas and P. J. de Pablo, Small, 2022, 18, e2200059 CrossRef PubMed.
- P. J. de Pablo, Semin. Cell Dev. Biol., 2018, 73, 199–208 CrossRef CAS PubMed.
- M. Comellas-Aragonès, H. Engelkamp, V. I. Claessen, N. A. Sommerdijk, A. E. Rowan, P. C. Christianen, J. C. Maan, B. J. Verduin, J. J. Cornelissen and R. J. Nolte, Nat. Nanotechnol., 2007, 2, 635–639 CrossRef PubMed.
- I. J. Minten, V. I. Claessen, K. Blank, A. E. Rowan, R. J. Nolte and J. J. Cornelissen, Chem. Sci., 2011, 2, 358–362 RSC.
- J. Sharma and T. Douglas, Nanoscale, 2020, 12, 336–346 RSC.
- A. O'Neil, P. E. Prevelige, G. Basu and T. Douglas, Biomacromolecules, 2012, 13, 3902–3907 CrossRef PubMed.
- N. H. Dashti, R. S. Abidin and F. Sainsbury, ACS Nano, 2018, 12, 4615–4623 CrossRef CAS PubMed.
- S. Chakraborti, A. Korpi, M. Kumar, P. Stępień, M. A. Kostiainen and J. G. Heddle, Nano Lett., 2019, 19, 3918–3924 CrossRef CAS PubMed.
- D. P. Patterson, B. LaFrance and T. Douglas, Chem. Commun., 2013, 49, 10412–10414 RSC.
- L. Schoonen, S. Maassen, R. J. M. Nolte and J. C. M. van Hest, Biomacromolecules, 2017, 18, 3492–3497 CrossRef CAS PubMed.
- J. S. Borden and D. F. Savage, Curr. Opin. Microbiol., 2021, 61, 58–66 CrossRef CAS PubMed.
- J. H. Hennacy and M. C. Jonikas, Annu. Rev. Plant Biol., 2020, 71, 461–485 CrossRef CAS PubMed.
- W. Bonacci, P. K. Teng, B. Afonso, H. Niederholtmeyer, P. Grob, P. A. Silver and D. F. Savage, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 478–483 CrossRef CAS PubMed.
- T. Li, Q. Jiang, J. Huang, C. M. Aitchison, F. Huang, M. Yang, G. F. Dykes, H. L. He, Q. Wang, R. S. Sprick, A. I. Cooper and L. N. Liu, Nat. Commun., 2020, 11, 5448 CrossRef CAS PubMed.
- E. Selivanovitch, B. LaFrance and T. Douglas, Nat. Commun., 2021, 12, 2903 CrossRef CAS PubMed.
- J. E. Glasgow, M. A. Asensio, C. M. Jakobson, M. B. Francis and D. Tullman-Ercek, ACS Synth. Biol., 2015, 4, 1011–1019 CrossRef CAS PubMed.
- L. S. R. Adamson, N. Tasneem, M. P. Andreas, W. Close, E. N. Jenner, T. N. Szyszka, R. Young, L. C. Cheah, A. Norman, H. I. MacDermott-Opeskin, M. L. O'Mara, F. Sainsbury, T. W. Giessen and Y. H. Lau, Sci. Adv., 2022, 8, eabl7346 CrossRef CAS PubMed.
- R. Gao, H. Tan, S. Li, S. Ma, Y. Tang, K. Zhang, Z. Zhang, Q. Fan, J. Yang, X. E. Zhang and F. Li, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2104964119 CrossRef CAS PubMed.
- S. J. Maassen, P. van der Schoot and J. J. L. M. Cornelissen, Small, 2018, 14, e1802081 CrossRef PubMed.
- H. K. Waghwani and T. Douglas, J. Mater. Chem. B, 2021, 9, 3168–3179 RSC.
- Y. Azuma, D. L. V. Bader and D. Hilvert, J. Am. Chem. Soc., 2018, 140, 860–863 CrossRef CAS PubMed.
- E. Selivanovitch, M. Uchida, B. Lee and T. Douglas, ACS Nano, 2021, 15, 15687–15699 CrossRef CAS PubMed.
- I. Wheeldon, S. D. Minteer, S. Banta, S. C. Barton, P. Atanassov and M. Sigman, Nat. Chem., 2016, 8, 299–309 CrossRef CAS PubMed.
- L. J. Sweetlove and A. R. Fernie, Nat. Commun., 2018, 9, 2136 CrossRef PubMed.
- L. J. Sweetlove and A. R. Fernie, Annu. Rev. Plant Biol., 2013, 64, 723–746 CrossRef CAS PubMed.
- A. Bar-Even, E. Noor, Y. Savir, W. Liebermeister, D. Davidi, D. S. Tawfik and R. Milo, Biochemistry, 2011, 50, 4402–4410 CrossRef CAS PubMed.
- O. Idan and H. Hess, ACS Nano, 2013, 7, 8658–8665 CrossRef CAS PubMed.
- M. Castellana, M. Z. Wilson, Y. Xu, P. Joshi, I. M. Cristea, J. D. Rabinowitz, Z. Gitai and N. S. Wingreen, Nat. Biotechnol., 2014, 32, 1011–1018 CrossRef CAS PubMed.
- Y. Xiong, S. Tsitkov, H. Hess, O. Gang and Y. Zhang, ACS Nano, 2022, 16, 10383–10391 CrossRef CAS PubMed.
- D. P. Patterson, B. Schwarz, R. S. Waters, T. Gedeon and T. Douglas, ACS Chem. Biol., 2014, 9, 359–365 CrossRef CAS PubMed.
- R. Frey, S. Mantri, M. Rocca and D. Hilvert, J. Am. Chem. Soc., 2016, 138, 10072–10075 CrossRef CAS PubMed.
- M. Brasch, R. M. Putri, M. V. de Ruiter, D. Luque, M. S. Koay, J. R. Castón and J. J. Cornelissen, J. Am. Chem. Soc., 2017, 139, 1512–1519 CrossRef CAS PubMed.
- D. P. Patterson, P. E. Prevelige and T. Douglas, ACS Nano, 2012, 6, 5000–5009 CrossRef CAS PubMed.
- P. Bauler, G. Huber, T. Leyh and J. A. McCammon, J. Phys. Chem. Lett., 2010, 1, 1332–1335 CrossRef CAS PubMed.
- Z. H. Zhou, D. B. McCarthy, C. M. O'Connor, L. J. Reed and J. K. Stoops, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14802–14807 CrossRef CAS PubMed.
- E. M. Ciszak, L. G. Korotchkina, P. M. Dominiak, S. Sidhu and M. S. Patel, J. Biol. Chem., 2003, 278, 21240–21246 CrossRef CAS PubMed.
- I. Skalidis, C. Tüting and P. L. Kastritis, Cell Commun. Signaling, 2020, 18, 136 CrossRef CAS PubMed.
- B. M. Woolston, S. Edgar and G. Stephanopoulos, Annu. Rev. Chem. Biomol. Eng., 2013, 4, 259–288 CrossRef CAS PubMed.
- Y. H. Zhang, Biotechnol. Adv., 2011, 29, 715–725 CrossRef CAS PubMed.
- G. Ke, M. Liu, S. Jiang, X. Qi, Y. R. Yang, S. Wootten, F. Zhang, Z. Zhu, Y. Liu, C. J. Yang and H. Yan, Angew. Chem., Int. Ed., 2016, 128, 7609–7612 CrossRef.
- Y. Liu, D. P. Hickey, J. Y. Guo, E. Earl, S. Abdellaoui, R. D. Milton, M. S. Sigman, S. D. Minteer and S. Calabrese Barton, ACS Catal., 2017, 7, 2486–2493 CrossRef CAS.
- Y. Wang, E. Selivanovitch and T. Douglas, Adv. Sci., 2023, e2206906 CrossRef PubMed.
- W. M. Aumiller, M. Uchida and T. Douglas, Chem. Soc. Rev., 2018, 47, 3433–3469 RSC.
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
- L. F. Bugada, M. R. Smith and F. Wen, ACS Catal., 2018, 8, 7898–7906 CrossRef CAS.
- D. J. Glover and D. S. Clark, ACS Cent. Sci., 2016, 2, 438–444 CrossRef CAS PubMed.
- W. Kang, X. Ma, D. Kakarla, H. Zhang, Y. Fang, B. Chen, K. Zhu, D. Zheng, Z. Wu, B. Li and C. Xue, Angew. Chem., Int. Ed., 2022, 61, e202214001 CAS.
- L. C. Cheah, T. Stark, L. S. R. Adamson, R. S. Abidin, Y. H. Lau, F. Sainsbury and C. E. Vickers, ACS Synth. Biol., 2021, 10, 3251–3263 CrossRef CAS PubMed.
- T. W. Giessen and P. A. Silver, ChemBioChem, 2016, 17, 1931–1935 CrossRef CAS PubMed.
- H. P. Erickson, Biol. Proced. Online, 2009, 11, 32–51 CrossRef CAS PubMed.
- Y. Zhang, S. Tsitkov and H. Hess, Nat. Commun., 2016, 7, 13982 CrossRef CAS PubMed.
- J. Fu, M. Liu, Y. Liu, N. W. Woodbury and H. Yan, J. Am. Chem. Soc., 2012, 134, 5516–5519 CrossRef CAS PubMed.
Footnotes |
| † Based on theoretical calculations, in a homogeneous solution, a spacing of less than 5 μm between enzymes corresponds to an enzyme concentration of over 13 pM.71 Only under conditions where the total enzyme concentration is lower than 13 pM (i.e. inter-enzyme distance over 5 μm in homogeneous solution), can enforcing inter-enzyme proximity possibly decrease the inter-enzyme distance to less than 5 μm, which in turn leads to an increase in local concentration of intermediates. However, an enzyme concentration of less than 13 pM is much lower than that used in almost all in vitro biocatalytic systems, where the inter-enzyme distance is already less than 5 μm so that local concentration of intermediates cannot be possibly raised in theory. |
| ‡ Some cases of multistep enhancement at the microscale might need further investigation. For valuable in vitro biocatalytic materials that can catalyze cascade reactions with enhanced efficiency, the local concentration of the intermediates should be elevated significantly. In the control experiments of some studies, however, the local concentration of the intermediates was decreased (by increasing the inter-enzyme distance to over 5 μm). This led to a scenario where the local concentration of intermediate in the developed materials was apparently higher compared to the control experiments, but actually unchanged compared to most in vitro biocatalytic systems (where inter-enzyme distance is already less than 5 μm; see the last footnote). The comparison can be very beneficial for understanding the relationship between enzyme cascades and their intermediates, as well as the enhanced metabolic efficiency in vivo, but it might not be a useful approach for making acellular biocatalytic materials with enhanced multistep reactions. |
| § Inter-enzyme proximity might induce some additional effects that can alter the diffusion of intermediates and consequently substrate channeling, such as pH alteration for optimal enzymatic efficiency72 and favorable molecular interactions within the hydration shell of the proximal enzymes.73 A recent study showed the efficiency of a cascade reaction was enhanced by inter-enzyme proximity, which was induced by co-localization of the cascade enzymes on the exterior surface of a protein cage.68 The molecular mechanism behind the increased efficiency, however, needs to be further investigated in the future. |
| This journal is © The Royal Society of Chemistry 2023 |