
Cross-linkable star-hyperbranched unimolecular micelles for the enhancement of the anticancer activity of clotrimazole†
Mateusz
Gosecki
 a,
Piotr
Ziemczonek
a,
Monika
Gosecka
*a,
Malgorzata
Urbaniak
a,
Ewelina
Wielgus
a,
Monika
Marcinkowska
b,
Anna
Janaszewska
*b and
Barbara
Klajnert-Maculewicz
b
a,
Piotr
Ziemczonek
a,
Monika
Gosecka
*a,
Malgorzata
Urbaniak
a,
Ewelina
Wielgus
a,
Monika
Marcinkowska
b,
Anna
Janaszewska
*b and
Barbara
Klajnert-Maculewicz
b
aCentre of Molecular and Macromolecular Studies, Polish Academy of Sciences, Sienkiewicza 112, 90-363 Lodz, Poland. E-mail: mdybko@cbmm.lodz.pl
bDepartment of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, 141/143 Pomorska Street, 90-236 Lodz, Poland
First published on 21st February 2023
Abstract
Clotrimazole, a hydrophobic drug routinely used in the treatment of vaginal candidiasis, also shows antitumor activity. However, its use in chemotherapy has been unsuccessful to date due to its low solubility in aqueous media. In this work, new unimolecular micelles based on polyether star-hyperbranched carriers of clotrimazole are presented that can enhance solubility, and consequently the bioavailability, of clotrimazole in water. The amphiphilic constructs consisting of a hydrophobic poly(n-alkyl epoxide) core and hydrophilic corona of hyperbranched polyglycidol were synthesized in a three-step anionic ring-opening polymerization of epoxy monomers. The synthesis of such copolymers, however, was only possible by incorporating a linker to facilitate the elongation of the hydrophobic core with glycidol. Unimolecular micelles-clotrimazole formulations displayed significantly increased activity against human cervical cancer HeLa cells compared to the free drug, along with a weak effect on the viability of the normal dermal microvascular endothelium cells HMEC1. This selective activity of clotrimazole on cancer cells with little effect on normal cells was a result of the fact that clotrimazole targets the Warburg effect in cancer cells. Flow cytometric analysis revealed that the encapsulated clotrimazole significantly blocks the progression of the HeLa cycle in the G0/G1 phase and induces apoptosis. In addition, the ability of the synthesized amphiphilic constructs to form a dynamic hydrogel was demonstrated. Such a gel facilitates the delivery of drug-loaded single-molecule micelles to the affected area, where they can form a continuous, self-healing layer.
10th Anniversary StatementThis journal is an inspiring source of knowledge revealing the relationship between the chemical strategies for material construction and their functions. This approach requires, however, the interdisciplinary cooperation of specialists from different disciplines, which can support a great step forward in solving the biomedical problems facing mankind. |
Introduction
Amphiphilic copolymers able to form micelles are of great interest as carriers of hydrophobic drugs.1–3 Micellization is, however, an equilibrium process, i.e., beside self-assembled macromolecules in the form of micelles, free macromolecules are present in the aqueous solution, even above the critical micelle concentration, which can be problematic for in vivo applications.4,5 In addition, the stability of conventional micelles depends on many parameters, such as the temperature, pH, and ionic strength, which can trigger the disassembly of micelles.6,7 The dilution of the copolymer solution below its critical micelle concentration is also unfavourable. To overcome the instability of micellar-based self-assemblies, intrinsically stable single-molecular amphiphilic core–shell constructs called unimolecular micelles are generally synthesized, which can preserve their structure regardless of the concentration.8Among the unimolecular micelles, various architectures of copolymers, such as dendrimer-like, hyperbranched, star-shaped, and brush-like, can be distinguished.9 The synthesis of unimolecular micelles, however, is often based on numerous reactions that occur via step-by-step modifications,10 or step copolymerization,11 or involving both modification reactions and polymerizations.12,13 The suitable structure of unimolecular micelles can, however, endow them with intelligent behaviour, i.e. a pH decrease or increase in the temperature can lead to structural changes of the material, which can enhance drug release from the interior of the macromolecule.11–13
In recent years, there has been growing interest in unimolecular micelles capable of both encapsulating water-insoluble drugs and forming hydrogels.14,15 This combination of features offers great potential for unimolecular micelles for applications in topical therapies requiring the controlled, prolonged, but infrequent delivery of a drug that must be released at the target site.14,15 Hydrogels, because of their swollen state along with elastic properties, mimic the properties of the extracellular matrix of most tissues, with minimal irritation of the surrounding tissues when applied in vivo.16–18 Furthermore, the highly porous structure of hydrogels facilitates the encapsulation of significant amounts of drugs, making them of great interest as drug-delivery platforms. However, the significant discrepancy between the hydrophilic environment of the hydrogel and the hydrophobic nature of the drugs prevents the incorporation of such drug molecules into the hydrogel frame. Despite the beneficial impact of a prolonged interaction of the drug-loaded hydrogel with mucosa, the low water solubility of numerous active substances reduces the efficiency of such therapies due to the low bioavailability of the hydrophobic drugs enclosed in the frame of the hydrogel. Hence, an important challenge is the development of hydrogels for the delivery of commonly used hydrophobic drugs in intravaginal antimicrobial therapies.
Clotrimazole, a highly hydrophobic drug in antifungal therapies, whose action is based on the disruption of the calcium pump, Ca2+ transport in the cell, and the inhibition of ergosterol synthesis, which leads to damage to the fungal cell membrane,19 is routinely applied in the treatment of skin infections caused by dermatophytes, yeasts, moulds, and other species of fungi,20 as well as in the therapy of vulvovaginitis. Due to the very low solubility of clotrimazole in water (0.49 μg mL−1),21 it exhibits both a low bioavailability and therapeutic efficacy and thus is categorized as a class II drug in the Biopharmaceutical Classification System (BCS). In topical therapies, as little as 0.5% of clotrimazole is absorbed, which reduces its effectiveness.22
Recently, several reports have appeared showing clotrimazole's anticancer properties.23–26 These studies confirmed the strong effect of the drug on mammalian cancer cells’ viability through affecting the glycolytic enzymes related to the cytoskeleton and inhibiting cell glycolysis and ATP production23,24 and showed that clotrimazole might be a calmodulin (CaM) antagonist.25 Marinho-Carvalho et al., for instance, demonstrated that the drug inhibited cell glycolysis by the direct inhibition of phosphofructokinase (PFK) and EC 2.7.1.11 – the key regulatory enzyme of the glycolytic pathway and a CaM – binding protein.26 Compared to normal cells, cancer cells show an increased rate of glycolysis27 and their mitochondria metabolize (apart from pyruvate) glutamine. which enables the faster biosynthesis of lipids and amino acids, necessary for the construction of membranes and proteins.28 This glycolytic preference of cancer cells is called the Warburg effect.29 Clotrimazole acts directly on glycolytic enzymes, and thus targets the Warburg effect,30 and was tested to target cancer cells with little effect on normal cells. Unfortunately, it failed in chemotherapy due to its low solubility in hydrophilic media.31 Generally, anticancer therapies require new chemotherapeutic strategies. For example, cervical cancer, one of the most aggressive forms of cancer in women and the fourth most common cancer with increasing incidence worldwide, is resistant to conventional chemotherapy.32 Effective therapy, especially at the early stages of the disease, can be decisive in the efficiency of treatment.
Due to the beneficial Warburg effect observed for clotrimazole, it could be crucial to enhance the drug solubility in aqueous media to increase its potential in anticancer therapies. For this goal, unimolecular micelles are considered suitable candidates as the main building component for the construction of hydrogel carriers of hydrophobic drugs. Among the unimolecular micelles, stars, comb-like, hyperbranched, and dendritic, etc., constructs can be distinguished,33,34 Their structures ensure the creation of voids between the individual branches of the macromolecule, which can play the role of an efficient reservoir for hydrophobic drugs. Hyperbranched polyglycidol (HbPGL) is a premier polymer amongst known hyperbranched constructs applied in biomedical applications as nanocarriers for the delivery of therapeutic agents and theranostics, i.e. diagnostics combined with treatment.35 The high biomedical potential of HbPGL results from its excellent biocompatibility36–42 and low toxicity.43 In addition, thanks to its better thermal and oxidative stability, HbPGL is considered an alternative to PEG.44 The excessive hydrophilic nature of HbPGL, however, prevents its direct use for the delivery of hydrophobic drugs. Therefore, various strategies for HbPGL hydrophobization by the incorporation of hydrophobic moieties have been proposed. Recently, we synthesized hydrogels based on single-molecule micelles composed of hyperbranched polyglycidol, the inner part of which, directly under the 1,2-diol-rich corona of the macromolecules, was hydrophobized with phenyl groups.15 This was achieved by modifying the monohydroxyl groups of the linear constitutive units, while the 1,2-diol molecules remained intact and were used to form a hydrogel with polyacrylamide with 2-acrylamide-phenylboronic acid motifs. The water solubility of these constructs was, however, strictly dependent on the degree of hydrophobization, which reduced their biomedical potential. Moreover, with the increase in the hydrophobization degree of monohydroxyl groups, both the flow and self-healing ability of the hydrogels were reduced. Due to this fact, in the current study we report a new type of polyether unimolecular micelle constructed from a hyperbranched polyglycidol shell built on a star-shaped hydrophobic core.
In this work, we demonstrate the anticancer properties of our clotrimazole-loaded star-hyperbranched polyether copolymers. Since the mechanism of action of clotrimazole in cancer cells is well known, the main aim of this study was not only to confirm that the encapsulated clotrimazole was more toxic to cervical cancer HeLa cells than to non-cancerous HMEC1 cells compared to the free drug, but also to verify whether the improvement of its bioavailability would enable the selection of a system for the selective, and, more importantly, long-term delivery of clotrimazole to cervical cancer cells without impairing the mechanism of action of the drug. In addition, in this study, we focused on the rheological properties of hydrogels constructed of star-hyperbranched amphiphiles, such as the injectability, flow behaviour, and self-healing properties, which are important for intravaginal anticancer application.
To the best of our knowledge, this is the first report on the usage of polymer constructs to increase clotrimazole bioavailability towards the enhancement of anticancer treatment. Because of the high demand for the design of various carriers of hydrophobic drugs differing in the spectrum of biological activity, it was crucial to find the relationship between the structural characteristics of amphiphilic star-hyperbranched constructs, the encapsulation efficiency of a hydrophobic drug, its biological activity, and the rheological properties of the formed hydrogels. This study contributes to a better understanding of the construction strategy of such a material and its biological function.
Results and discussion
Synthesis and characteristics of star-hyperbranched amphiphilic core–shell based on polyethers
For the encapsulation of clotrimazole, we designed star-hyperbranched core–shell amphiphilic block copolymers consisting of hydrophobic poly(n-alkyl epoxides), i.e. 1,2-epoxybutane and 1,2-epoxyhexane, and hydrophilic hyperbranched polyglycidol (Scheme 1). HbPGL of low and moderate molecular weight, as targeted in this work, is commonly synthesized in bulk.45 The big difference between glycidol and poly(1,2-epoxybutane) and poly(1,2-epoxyhexane), however, did not allow the direct polymerization of glycidol onto the core macromolecules. To overcome this restriction, we decided to add a linker that could be polymerized at the poly(1,2-alkyl epoxide) chain-end and that was compatible with glycidol, thereby allowing its polymerization.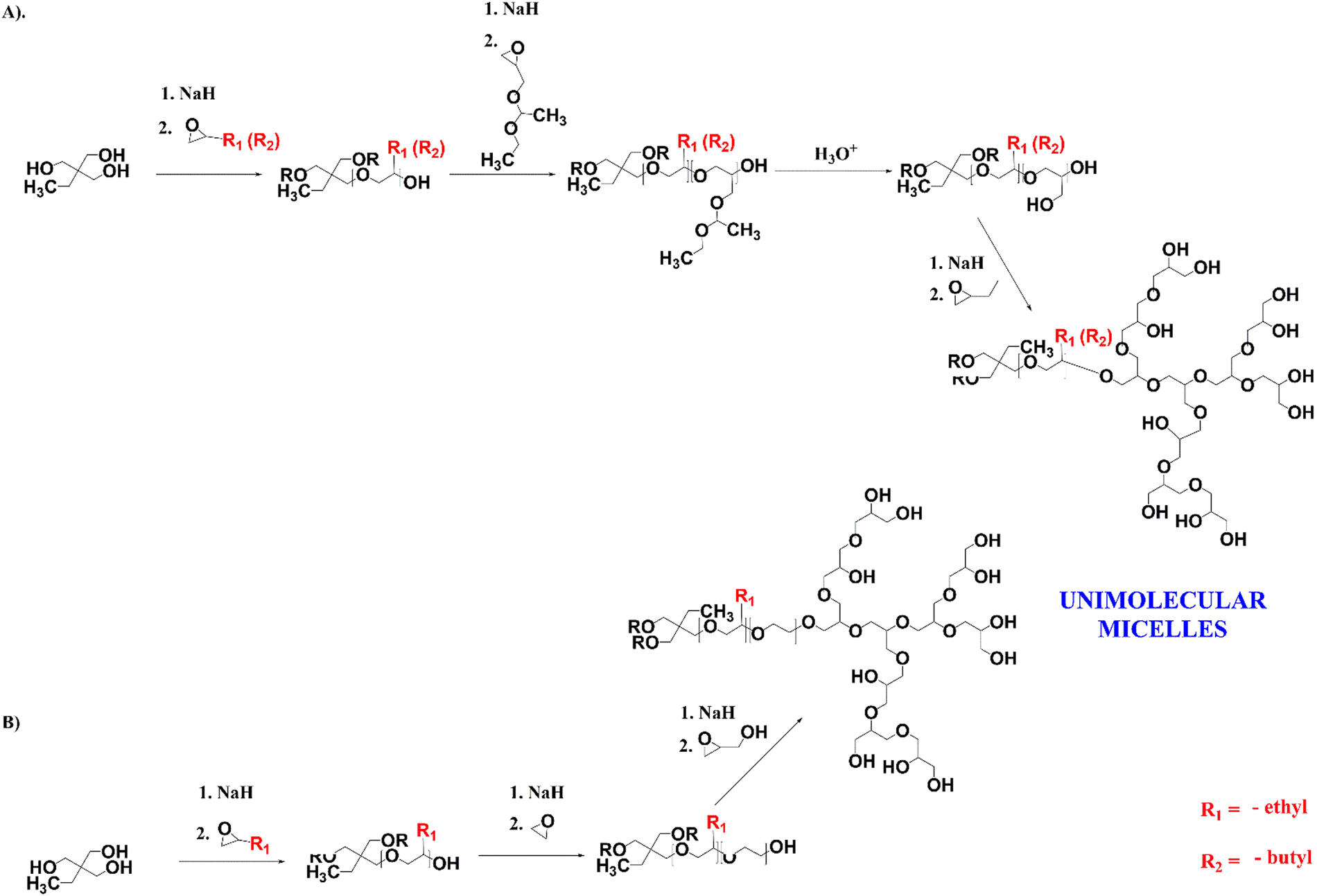 | ||
| Scheme 1 Synthetic route for the star-hyperbranched amphiphilic polyether-based copolymers. | ||
For a linker, we chose linear polyglycidol, which was obtained by the polymerization of hydrophobic ethoxyethyl glycidyl ether (EEGE) followed by deprotection of the monohydroxyl groups (Scheme 1(A)), which increased the hydrophilicity of the macroinitiator. In addition, after the deprotection and subsequent polymerization of glycidol, the linker was in fact indistinguishable from the HbPGL core. Because the monohydroxyl group present in each repeating group of the deprotected EEGE segment could be a potential initiating centre, glycidol polymerization on such a macroinitiator could lead to the formation of a dense HbPGL shell directly on the core, which could affect the encapsulation efficiency of hydrophobic drugs. Therefore, for poly(1,2-epoxybutane) with DPn = 15, apart from the poly(EEGE) linker, we synthesized two copolymers with linkers based on the ethylene oxide oligomers differing in length (DPn = 3 × 8 and 3 × 30) to separate the hydrophobic shell from the HbPGL core (Scheme 1(B)).
For the study, we synthesized poly(1,2-epoxybutane) tri-arm homopolymers containing 10 and 15 constitutional units per arm, and poly(1,2-epoxyhexane) tri-arm homopolymers composed of 10 constitutional units per arm. The length of the arms was confirmed based on the 1H NMR spectra recorded in chloroform (ESI,† Fig. S1, S7 and S17), by comparing the integration of the methyl protons of an initiator (δ in the range from 0.79–0.86 ppm) and the integration of the protons stemming from the methyl groups of 1,2-epoxybutane or 1,2-epoxyhexane, respectively (δ in the range from 0.88–0.99 ppm). In addition, the gel permeation chromatography (GPC; CH2Cl2) data for all the synthesized hydrophobic cores revealed their low dispersity index (Table 1).
| Copolymer | Symbol | Core | Linker typea | Shell | Unimolecular micelles | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of constitutional units per arm (1H NMR) | M w/Mn GPC (CH2Cl2) | Number of constitutional units (1H NMR data) | M w/Mn GPC (CH2Cl2) | M n (13C INVGATE) | M n (NMR) | M w/Mn GPC (DMF) | D h (DLS), nm | PdI | ||
| a Linker based on linear oligomers of ethoxyethyl glycidyl ether (EEGE) or ethylene oxide (EO). | ||||||||||
| Poly(1,2-epoxybutane)-based copolymers | A86 | 10 | 1.109 | EEGE (4) | 1.361 | 8260 | 9000 | 1.674 | 8.3 | 0.288 |
| A92 | 15 | 1.115 | EEGE (14) | 1.669 | 7500 | 8600 | 1.551 | 9.4 | 0.379 | |
| A165 | 15 | 1.086 | EO (8) | 1.202 | 8900 | 10300 | 1.237 | 10.4 | 0.183 | |
| A167 | 15 | 1.086 | EO (30) | 1.112 | 9400 | 11800 | 1.246 | 19.8 | 0.190 | |
| Poly(1,2-epoxyhexane)-based copolymer | A71 | 10 | 1.102 | EEGE (10) | 1.196 | 9300 | 10300 | 1.886 | 15.1 | 0.232 |
The degree of EEGE polymerization was calculated based on a comparison of the integration of the methyl protons of the initiator, and the methine protons of the EEGE polymers at 4.67–4.78 ppm (ESI,† Fig. S2, S8 and S18), and then monohydroxyl groups were deprotected, which was confirmed based on 1H NMR spectroscopy (ESI,† Fig. S3, S9 and S19). The degree of EO copolymerization was estimated based on the integrations of the protons coming from the initiator (methyl group), poly(n-alkyl epoxide) segment, and ethylene oxide chains (ESI,† Fig. S13). The structure of the core–shell polymers formed by glycidol polymerization on the synthesized three-armed core polymers was determined by 1H (ESI,† Fig. S4, S10, S14 and S20), 1H DOSY (ESI,† Fig. S6, S12, S16 and S22), and 13C NMR methods (ESI,† Fig. S5, S11, S15 and S21). The degree of glycidol polymerization was estimated from the 13C INVGATED spectra45 The molecular weights of all the synthesized copolymers’ cores were similar and ranged between 7500 and 9300. Interestingly, the copolymers with PEO showed a narrower distribution of molar weight, approximately 1.24, in comparison with those with the linear polyglycidol linker i.e. over 1.5. The difference may be due to not having a fast enough intermolecular chain transfer reaction between the potentially active chain ends of the macromolecules.
A dynamic light scattering study was performed for aqueous solutions of the synthesized copolymers and revealed nanometre-sized particles with a quite low dispersity of hydrodynamic diameters (Table 1), which could be ascribed to the molecular weight distribution of the copolymer. As shown in the TEM microscopic image (ESI,† Fig. S23), unimolecular micelles based on star-hyperbranched copolymers were roughly spherical in the aqueous solution. The average diameter of the unimolecular micelles was close to the result from the DLS measurement.
Drug loading and in vitro release
Clotrimazole is a highly hydrophobic drug, which in its structure contains non-conjugated three phenyl rings and an imidazole moiety. It is routinely applied in the treatment of candidiasis triggered by Candida albicans and other Candida species. Unfortunately, due to its poor solubility in water, its bioavailability is limited. Therefore, to unlock its anticancer properties, it is crucial to increase the solubility of clotrimazole. To estimate the potential of the synthesized amphiphilic unimolecular micelles for clotrimazole solubilization, encapsulation experiments at different drug-to-polymer ratios were carried out (Table 2). The process of clotrimazole solubilization was performed according to the ultrasound-assisted solvent evaporation method. Then the drug–polymer mixture was suspended in deionized water and filtrated to remove the insoluble fraction of clotrimazole that remained not encapsulated. As a reference, hyperbranched polyglycidol homopolymer was used, which did not display the ability for clotrimazole encapsulation. The incorporation of a hydrophobic poly(n-alkyl epoxy)-based core with DPn ranging from 10 to 15 into the hyperbranched polyglycidol structure provided a maximum encapsulation capacity of clotrimazole of approximately 5 drug molecules/macromolecule. This corresponds to a drug-loading capacity of 156.70 to 336 mg g−1 dependent on the molecular weight of the polymer carrier. Based on the encapsulation experiments performed, there was no evident effect of the length of the side chains in the poly(n-alkyl-epoxide) used or the type of linker introduced on the encapsulation capacity.| Copolymer | Drug-loaded copolymer | Drug-loading capacity, mg g−1 | Encapsulation efficiency, % |
|---|---|---|---|
| A86 | En170 | 15.0 | 83 |
| En171 | 36.0 | 80 | |
| En172 | 50.7 | 80 | |
| En137 | 229.0 | 83 | |
| A71 | En143 | 18.2 | 100 |
| En144 | 36.3 | 73 | |
| En145 | 58.0 | 90 | |
| En94 | 336 | 100 | |
| A92 | En30 | 111 | 88.6 |
| En77 | 179 | 100 | |
| En72 | 22.0 | 82 | |
| En82 | 92.8 | 29 | |
| A165 | En156 | 156.7 | 100 |
| En163 | 120 | 35 | |
| En157 | 25.6 | 4 |
The in vitro release profiles of clotrimazole-loaded copolymers were determined in order to investigate the potential use of the synthesized star-hyperbranched amphiphilic constructs as drug carriers at the conditions corresponding to the vagina environment, i.e. pH 5.6 and 37 °C. Drug-release experiments were performed on copolymer formulations containing the same number of encapsulated drug molecules.
Contrary to traditional micelles,22 for all the amphiphilic star-hyperbranched copolymers investigated here, an initial burst release of the drug was not observed (Fig. 1). The complete drug-release time differed amongst the investigated systems and ranged from 32 h for En 156 to 48 h for En 137. The most sustained and the slowest drug release was observed for the unimolecular micelles constructed of poly(1,2-epoxybutane) core of DPn = 10 per arm prolonged with glycidol via a linear glycidyl linker. Sustained drug release provides a constant drug dose and is of great importance in the case of quickly metabolized drugs.46
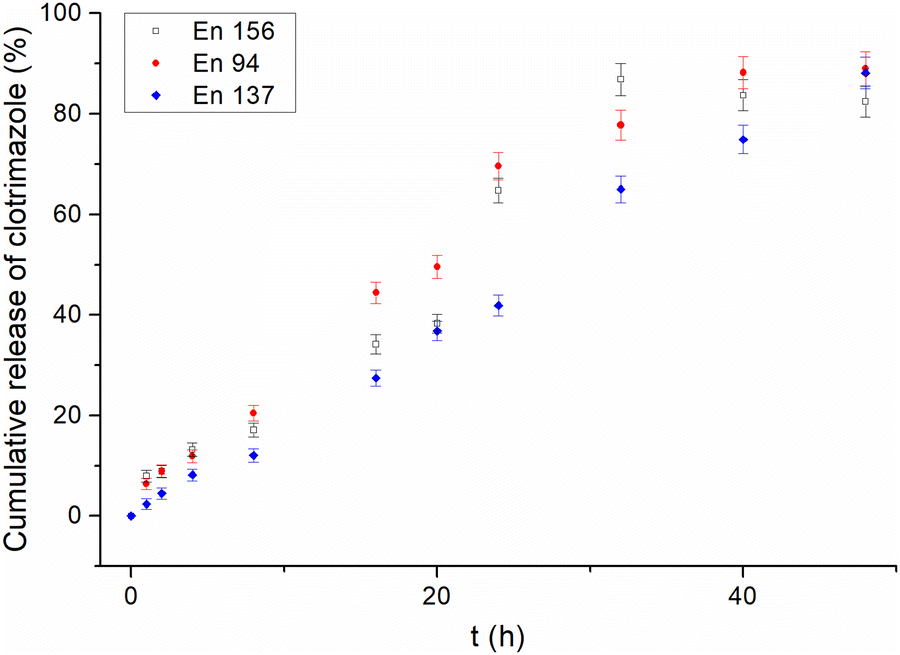 | ||
| Fig. 1 Release profiles of clotrimazole from different star-hyperbranched polyether-based unimolecular micelles. | ||
Effects of the unimolecular micelles and their clotrimazole-loaded constructs on cell viability
The synthesized star-hyperbranched unimolecular micelles were investigated in view of their cytotoxicity against two cell lines: cancerous HeLa and non-cancer HMEC1 cells. First, it was necessary to check whether the unimolecular micelles themselves were non-toxic carriers and at the same time whether they could effectively transport clotrimazole to the cells and release it there. Therefore, using the MTT assay, we compared the toxicity of free amphiphilic constructs with the corresponding structures containing various amounts of clotrimazole and with the drug itself. Measurements were carried out after 24 and 48 h incubation. The cytotoxicity profiles demonstrated that the unloaded copolymers were non-toxic over the concentration range tested for both cell lines, and only the A92 copolymer was slightly toxic to the HeLa cancer line (Fig. 2).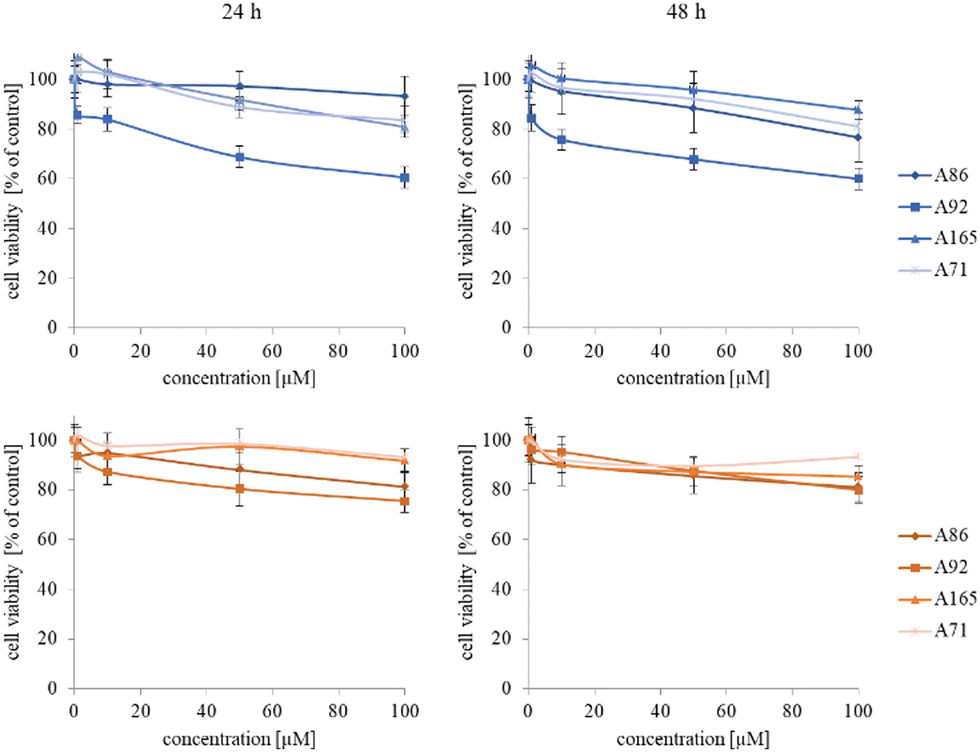 | ||
| Fig. 2 Influence of the neat polyether-based star-hyperbranched unimolecular micelles on HeLa (upper panel) and HMEC1 (lower panel) cells viability after 24 and 48 h incubation. Results are expressed as the mean ± SD (n = 16). | ||
In the next step, again using the MTT assay, the cytotoxicity of the unimolecular micelles with encapsulated clotrimazole was determined (Fig. 3–5). Since the systems contained varying amounts of encapsulated drug, the cell viability results were normalized by the clotrimazole content to allow a direct comparison with the free drug. On the basis of the A86 copolymer, constructs with encapsulated clotrimazole with the signatures En170, En171, and En172 were created, based on A92–En30; based on A165–En163, En157, En156; and based on A71–En143, En144, En145, respectively.
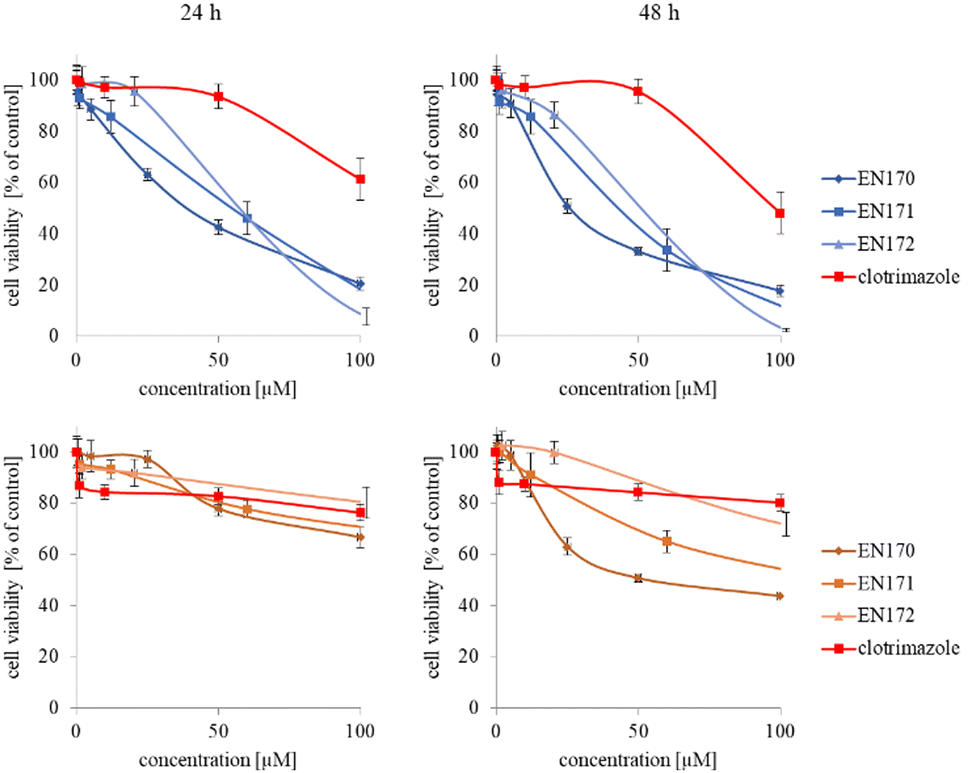 | ||
| Fig. 3 Influence of clotrimazole-loaded A86 unimolecular micelles (En170, En171, En172) on HeLa (upper panel) and HMEC1 (lower panel) cells viability after 24 and 48 h incubation. Results are expressed as the mean ± SD (n = 16). | ||
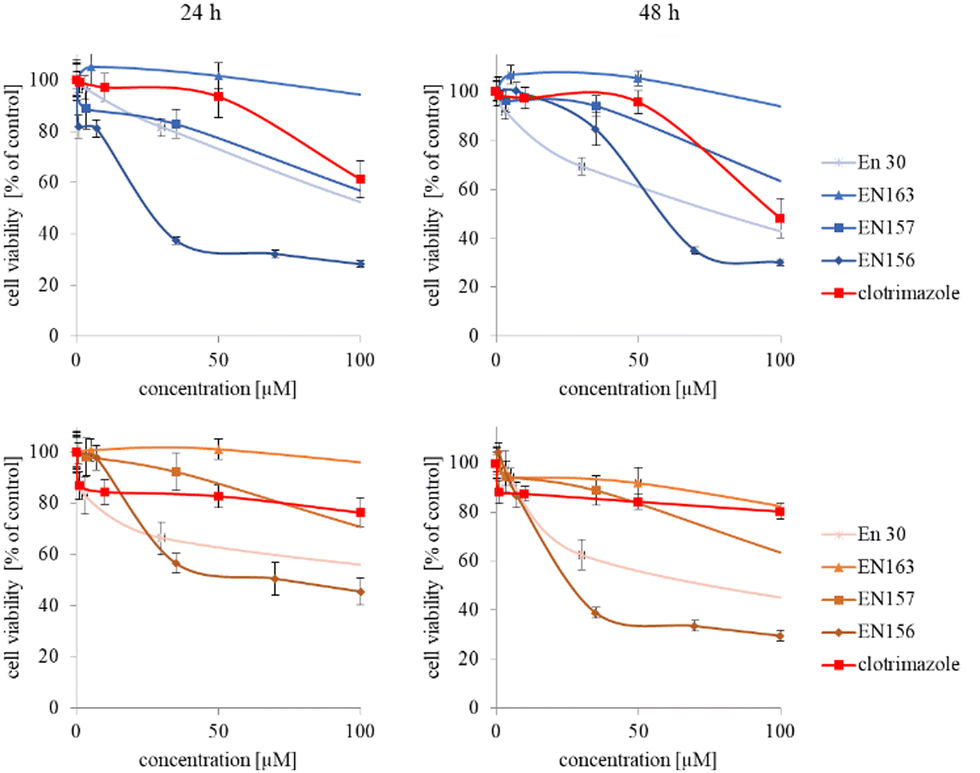 | ||
| Fig. 4 Influence of clotrimazole-loaded A92 (En30) and A165 (En163, En157, En156) unimolecular micelles on HeLa (upper panel) and HMEC1 (lower panel) cells viability after 24 and 48 h incubation. Results are expressed as the mean ± SD (n = 16). | ||
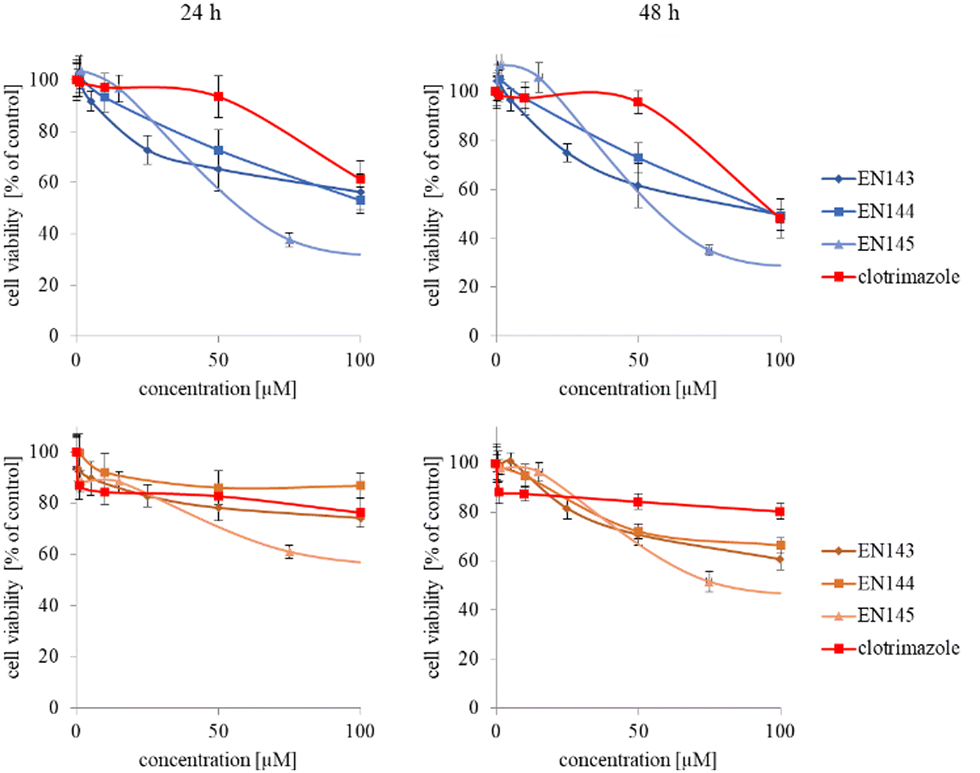 | ||
| Fig. 5 Influence of clotrimazole-loaded A71 unimolecular micelles (En143, En144, En145) on HeLa (upper panel) and HMEC1 (lower panel) cells viability after 24 and 48 hour incubation. Results are expressed as mean ± SD (n = 16). | ||
As expected (considering the previously mentioned Warburg effect29), free clotrimazole was more potent against cancer cells and had a weak effect on the viability of normal cells. However, in the tested concentration range, its anticancer effect on HeLa cells was not spectacular. After normalizing the results for the clotrimazole content, we noticed that the cytotoxicity of the drug-loaded unimolecular micelles was not correlated with the amount of drug trapped in the analyzed structures. The systems based on the A86 copolymer released clotrimazole more efficiently over a longer period (Fig. 1), and thus from the point of view of cancer cell biology, and due to its selectivity, was more toxic to cancer cells than to normal cells. In addition, despite using the same concentration of free clotrimazole and clotrimazole trapped in the structure, the one transported to the cell in the A86 structure was more toxic to cervical cancer cells than the free drug (Fig. 3). Interestingly, the structure from which the drug was released faster (e.g. A165) gave worse results, i.e. the clotrimazole transported in it was also toxic to non-cancer HMEC1 cells, and therefore the very clear selectivity of the system was blurred.
Nonetheless, almost all the systems effectively transported clotrimazole into the cells more effectively than the free drug. Moreover, clotrimazole encapsulated in the unimolecular micelles was rather less toxic to non-cancer cells (HMEC1) than to cancer cells (HeLa). To better illustrate the difference between the tested systems and the free drug, as well as their toxicity to both cell lines, the IC50 parameter was determined, which indicates the concentration of the tested compound that causes the death of half of the cells (Table 3).
| HeLa [μM] | HMEC1 [μM] | |||
|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | |
| Clotrimazole | 127.42 ± 7.11 | 97.33 ± 6.46 | >1000 | >1000 |
| En 170 | 30.41 ± 2.56 | 20.48 ± 4.25 | 766.80 ± 3.48 | 76.14 ± 1.90 |
| En 171 | 27.27 ± 6.39 | 21.40 ± 7.48 | 681.49 ± 3.37 | 127.84 ± 2.67 |
| En 172 | 26.37 ± 1.91 | 21.41 ± 0.61 | >1000 | 310.23 ± 3.06 |
| En 30 | 103.39 ± 2.59 | 61.88 ± 2.51 | 154.45 ± 3.70 | 201.00 ± 4.84 |
| En 156 | 19.57 ± 2.38 | 22.20 ± 4.07 | 87.42 ± 5.86 | 27.95 ± 3.61 |
| En 157 | 99.50 ± 2.41 | 129.68 ± 3.37 | 330.47 ± 4.36 | 147.90 ± 2.17 |
| En 163 | 449.79 ± 5.67 | 378.20 ± 4.53 | >1000 | 781.87 ± 2.39 |
| En 143 | 305.91 ± 7.09 | 105.28 ± 6.28 | >1000 | 736.28 ± 4.29 |
| En 144 | 319.48 ± 5.11 | 231.22 ± 2.89 | >1000 | >1000 |
| En 145 | 63.54 ± 3.98 | 65.78 ± 4.12 | 172.24 ± 3.72 | 106.28 ± 4.16 |
To identify systems with clotrimazole that are (1) more toxic than the free drug, (2) more potent against cancer than non-cancerous cells, and (3) more potent after 48 h than after 24 h incubation with the drug, we created the following list:
HeLa – 24 h
En 156 < En 172 < En 171 < En 170 < En 145 < En 157 < En 30 < clotrimazole ≈ En143 < En 144 < En 163.HeLa – 48 h
En 170 < En 171 < En 172 < En 156 < En30 < En 145 < clotrimazole < En 143 < En 157 < En 144.HMEC1 – 24 h
En 156 < En 30 < En 145 < En 157 < En 171 < En 170 < clotrimazole ≈ En 172, En 163, En 143, En 144.HMEC1 – 48 h
En 156 < En 170 < En 145 < En 171 < En 157 < En 30 < En 172 < En 143 < En 163 < clotrimazole ≈ En 144.Based on the list, we selected, in our opinion, three of the best systems, i.e. the En170, En171, and En172 (A86) systems, and three other systems that also met our requirements: En30 (A92), En156 (A165), and En145 (A71). Since they were more toxic than the free drug (1), it would be possible to use clotrimazole at a lower concentration, which would positively affect the reduction of undesirable side effects. Also, their selectivity and stronger effect on cervical cancer cells than on non-cancerous endothelial cells (2) can avoid side effects when the drug is applied locally in the form of a gel. Selected systems provided prolonged and maintained selective toxicity (3), which is important in the treatment of cervical cancer, where the cells double rapidly and the effect of the drug decreases with each cell division.
Observing the selective and long-term delivery of clotrimazole to cervical cancer cells, the question arises whether the carrier used affects the mechanism of action of the drug. Apart from the postulated mechanism of clotrimazole action, involving the strong effect of the drug on the viability of mammalian cancer cells by affecting the glycolytic enzymes related to the cytoskeleton and inhibiting cell glycolysis and ATP production,23,24 it is considered that clotrimazole can cause cell cycle arrest at the G0/G1 phase and the induction of cellular apoptosis.47
Therefore, to answer the posted question, we investigated the effect of clotrimazole treatment on cell cycle and apoptosis. We found that both the free and encapsulated clotrimazole blocked the progression of the HeLa cycle in the G0/G1 phase and induced apoptosis, observed as a sub-G1 population. This result was consistent with previous studies on glioblastoma and lung cancer,48,49 with the difference that in our study encapsulated clotrimazole had a stronger anticancer/proapoptotic effect compared to the free drug. Fig. 6 shows the results of the flow cytometric analyses for cell cycle progression with the apoptotic cell population shown as a sub-G1 peak for the control cells, pure clotrimazole, and encapsulated in the most efficient and selective system, i.e. En170. Table 4 shows the apoptotic cell ratio calculated from the flow cytometric sub-G1 populations and G0/G1 phase.
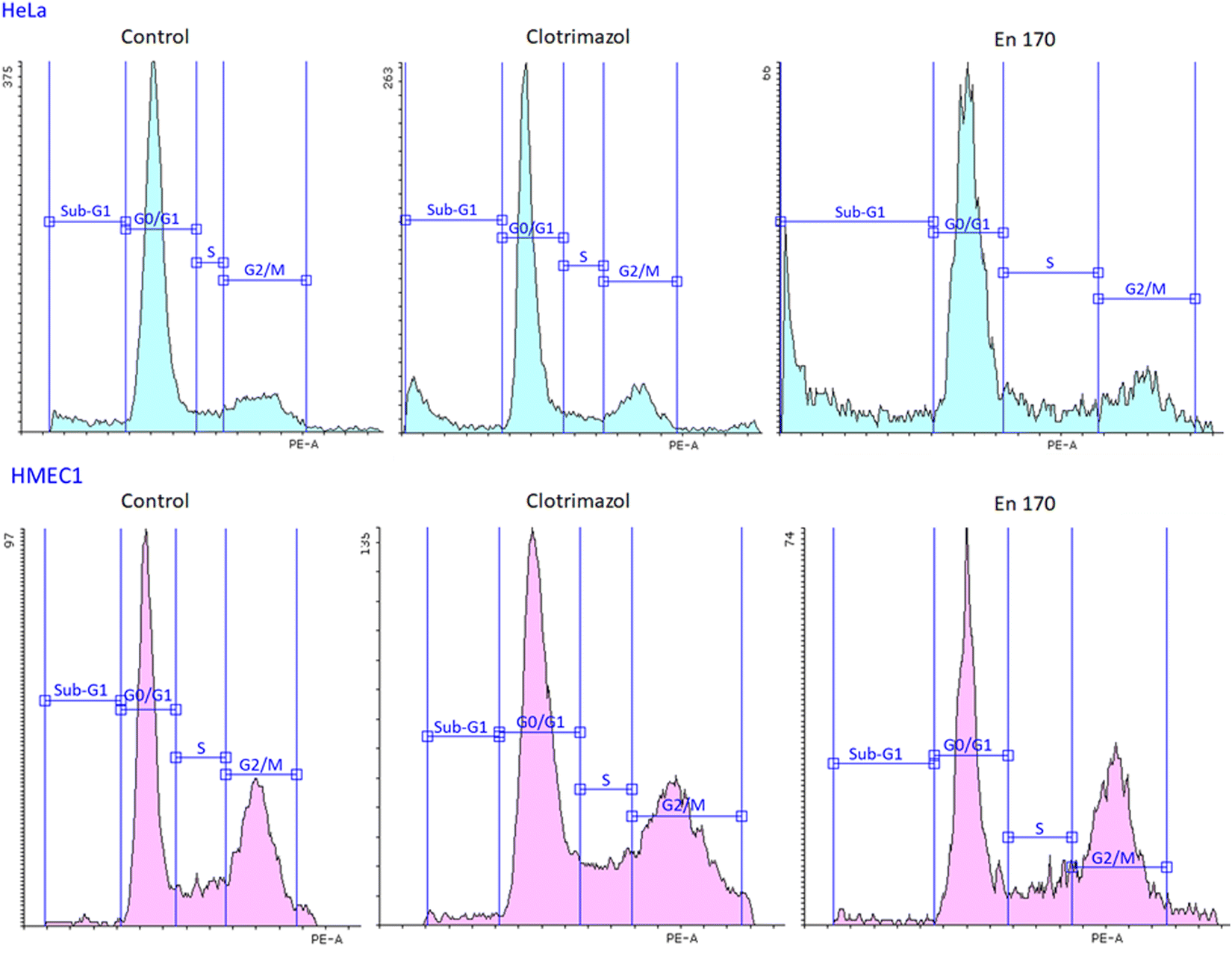 | ||
| Fig. 6 Flow cytometric analysis of the cell cycle phase distribution for cervical cancer endothelial HeLa cells (upper panel) and dermal microvascular endothelium HMEC1 cells (lower panel). The apoptotic cell population is shown as the Sub-G1 population. | ||
| HeLa [%] | HMEC1 [%] | |||
|---|---|---|---|---|
| 24 h Sub-G1 | 24 h G0/G1 | 24 h Sub-G1 | 24 h G0/G1 | |
| Control | 3.38 ± 1.44 | 69.12 ± 1.54 | 1.57 ± 0.07 | 47.26 ± 0.62 |
| Clotrimazole | 12.48 ± 2.79 | 51.73 ± 0.71 | 5.66 ± 0.51 | 43.87 ± 0.44 |
| En 170 | 7.66 ± 0.88 | 66.26 ± 9.50 | 5.08 ± 0.96 | 44.55 ± 0.87 |
| En 171 | 8.59 ± 0.19 | 66.64 ± 6.38 | 4.77 ± 1.25 | 41.73 ± 3.93 |
| En 172 | 13.84 ± 4.21 | 54.91 ± 6.57 | 3.71 ± 1.20 | 43.44 ± 0.16 |
| En 30 | 9.59 ± 0.19 | 72.70 ± 0.20 | 3.79 ± 0.97 | 46.28 ± 1.93 |
| En 145 | 12.16 ± 1.00 | 67.63 ± 2.89 | 6.19 ± 1.22 | 46.86 ± 1.60 |
| En 156 | 12.71 ± 0.71 | 67.83 ± 2.60 | 5.30 ± 2.52 | 47.80 ± 2.91 |
| HeLa [%] | HMEC1 [%] | |||
|---|---|---|---|---|
| 48 h Sub-G1 | 48 h G0/G1 | 48 h Sub-G1 | 48 h G0/G1 | |
| Clotrimazole | 6.22 ± 0.04 | 67.56 ± 0.41 | 12.55 ± 3.21 | 33.11 ± 3.79 |
| En 170 | 18.14 ± 7.39 | 55.44 ± 1.14 | 8.70 ± 1.63 | 48.37 ± 2.67 |
| En 171 | 14.19 ± 6.46 | 59.77 ± 8.87 | 7.03 ± 1.00 | 48.35 ± 1.57 |
| En 172 | 11.50 ± 0.26 | 67.78 ± 4.64 | 8.19 ± 0.66 | 46.90 ± 2.10 |
| En 30 | 11.70 ± 3.88 | 66.19 ± 0.97 | 10.51 ± 1.44 | 44.91 ± 8.34 |
| En 145 | 12.78 ± 0.69 | 60.16 ± 0.71 | 7.41 ± 0.78 | 49.85 ± 5.79 |
| En 156 | 13.95 ± 2.38 | 65.32 ± 0.58 | 8.00 ± 0.13 | 55.80 ± 0.22 |
Based on the data of the apoptotic cell ratio of the sub-G1 and G0/G1 cell cycle phase, it could be stated that all the analyzed compounds showed similar cell cycle progression and resulted in a significant accumulation of the sub-G1 cell population in HeLa cells, which indicated apoptosis. Considering that clotrimazole's main mechanism of action was also G0/G1 cycle arrest, we expected the same from our encapsulated drug systems and we indeed got this result.
This means that the transferred clotrimazole, even at such a low concentration of 10 μM, could selectively block the cell cycle of cervical cancer cells and lead them to the controlled death path, i.e. apoptosis, and at the same time did not affect the metabolism of the non-cancer endothelial cells. The obtained results were consistent with the data on the cytotoxicity of the free drug and the drug encapsulated in the system of the star-hyperbranched copolymers. The analyzed carriers did not weaken the effect of the hydrophobic drug but enabled its transport inside the cell and prolonged its release, thus increasing its anticancer efficacy.
Formation of hydrogel systems based on amphiphilic star-hyperbranched copolymers: rheological properties
The prolonged action of a drug in the afflicted area is crucial to assure the efficiency of anticancer therapy via the intravaginal route. The usage of the suspension of unimolecular micelles led to uncontrolled leakage due to its unsuitable rheological properties. Thus the construction of unimolecular micelles based on a star-shaped hydrophobic core and hyperbranched polyglycidol shell that could be easily built in the hydrogel structure would be beneficial. In view of the intravaginal therapy, the hydrogel, however, has to be constructed on reversible cross-links to assure the formation of a continuous layer on the covered surface, and its eventual removal after the fulfilment of the function. Such hydrogels are generally determined to be dynamic because the network integrity is controlled by the equilibrium of the reaction between the product (crosslink) and the substrate. To obtain this type of hydrogel, the terminal 1,2-diol units present in the hyperbranched polyglycidol shell of the amphiphilic constructs were employed for cross-linking with an acrylamide copolymer of 2-acrylamidephenyl boronic acid, poly(acrylamide-2-acrylamidephenyl boronic acid), and poly(AM-APBA) with 20 mol% fraction of 2-APBA units (ESI,† Fig. S24).The rheological experiments were carried out at a physiological temperature (37 °C) to investigate the behaviour of the hydrogels under the administration conditions. The frequency sweep experiments demonstrated that all the synthesized star-hyperbranched constructs formed viscoelastic networks in water (Fig. 7) In the higher frequency range, i.e. shorter time scales, the storage modulus exceeded the value of the loss modulus (G′ > G′′). This resulted from the fact that the lifetime of the boronic ester cross-links was longer in comparison to the applied strain rate. The decrease in frequency resulted in the inversion of G′ and G′′ at the crossover frequency (ωc), which corresponded to the gelation point (gel–liquid transition) as an effect of the cross-links reshuffling. Here, ωc denotes the onset of the macroscopic chain displacement, while at frequencies below ωc (longer time scales), the material begins to flow (G′ < G′′) due to the dominant contribution of the liquid-like behaviour. The crossover frequency for all the prepared hydrogels was similar, ranging from 3.2 to 4.1 rad s−1, indicating that differences in the core structure of the synthesized polymers only had a minor effect on the macromolecules dynamics in the reversibly cross-linked hydrogels. This is especially important in the view of the self-healing properties of such hydrogels.
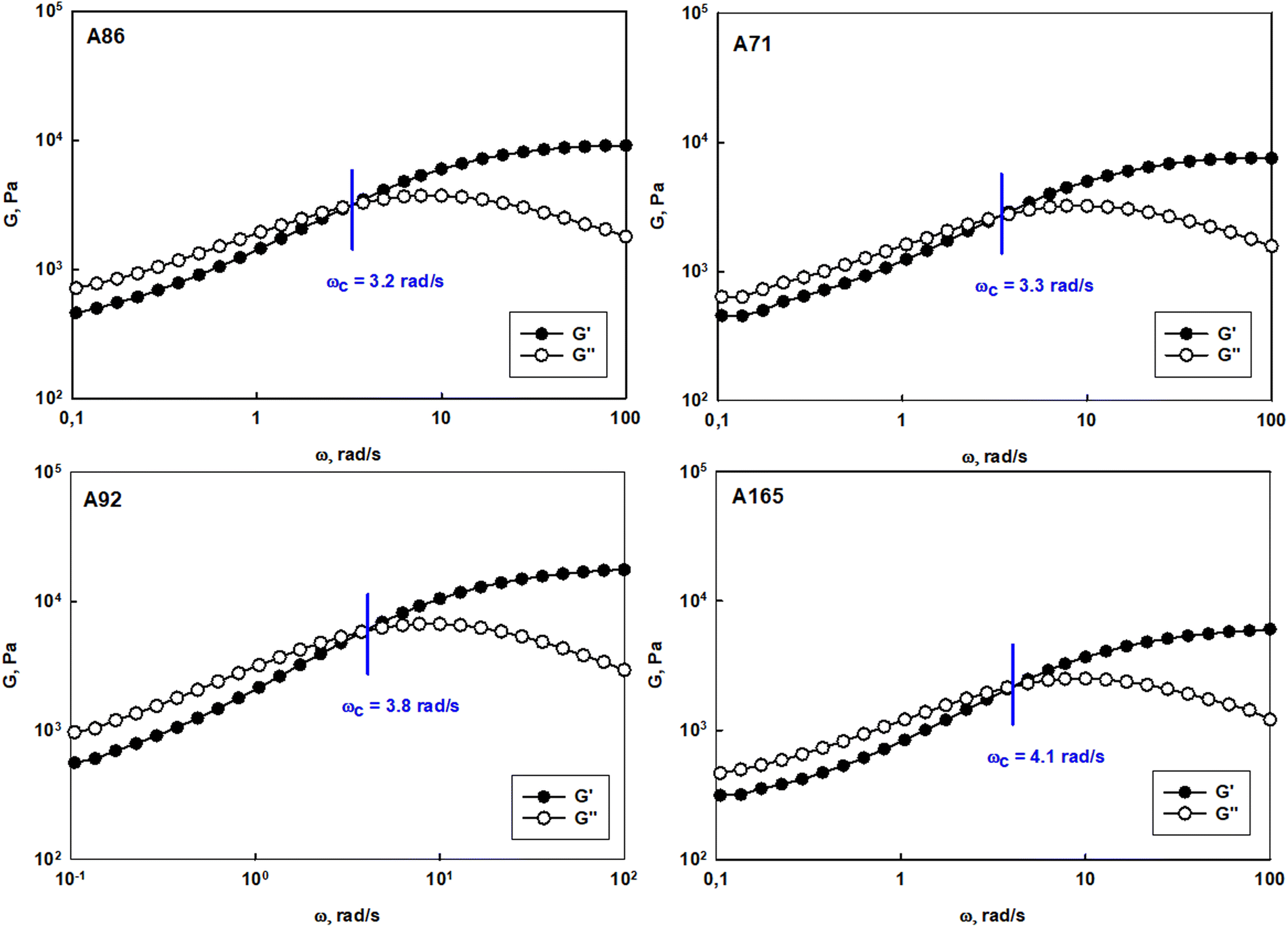 | ||
| Fig. 7 Frequency sweep tests performed for hydrogel systems constructed of polyether-based star-hyperbranched unimolecular micelles at 37 °C. | ||
Contrary to previously published reports on hydrogels constructed on unimolecular micelles made of hyperbranched polyglycidol hydrophobized directly underneath the terminal units by modification of the monohydroxyl groups,15 the star-hyperbranched amphiphiles in this study formed both injectable (ESI,† Video film) and self-healable hydrogels (Fig. 8). These properties can assure the convenient administration of the therapeutic platform to the target place and homogeneous hydrogel distribution at the afflicted area. The reported approach for the design of unimolecular micelles assures that the hydrophobic core would be well-protected with a hydrophilic shell, while the macromolecules are well-soluble in water, and the continuous reorganization of the dynamic boronic ester cross-links would be assured.
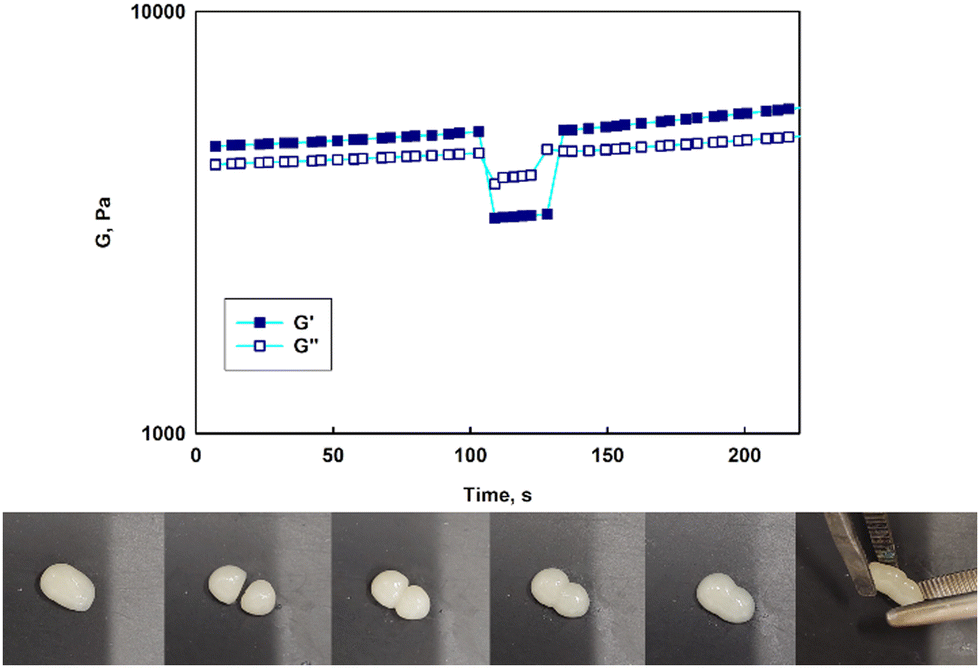 | ||
| Fig. 8 Time-dependent self-healing of the hydrogel constructed of poly(1,2-epoxybutane-b-hyperbranched polyglycidol) at room temperature (at the top) and visual demonstration of the hydrogel reforming from two distinct pieces (at the bottom). The hydrogel sample was divided into two pieces. The fracture gradually vanished thanks to the self-healable properties of the 2-AAPBA-based boronic ester cross-links leading to its complete decay. The damaged hydrogel was completely repaired by regaining its initial mechanical properties. | ||
Strain sweep tests revealed that the stability of the hydrogel networks constructed of various star-hyperbranched polyether-based constructs was comparable (Fig. S25, ESI†). At the low amplitude range, both the storage and loss moduli exhibited a plateau, characteristic for the linear viscoelastic region, which was followed by a decrease in both moduli at the amplitude characteristic for each hydrogel. The boundary amplitude values of the linear viscoelasticity region for all the prepared hydrogels were comparable and ranged from 63% to 88%. After exceeding the critical strain, at which point the network was disrupted, the inversion of G′ and G′′ was observed, and G′′ became larger than G′. The crossover of both moduli corresponded to a transition from a solid (hydrogel) to a liquid state and the material then exhibited viscous flow. The calculated yield stress for the prepared hydrogels was approximately 6 kPa.
To verify whether the hydrogels prepared with synthesized micelles were self-healable, experiments showing the response to the drastic applied strain were carried out (Fig. 8, at the top). Upon increasing the strain (γ = 300%), a significant decrease in both moduli was observed along with the inversion of G′ and G′′, which indicated the network disruption. A subsequent reduction of the strain to 1% resulted in a rapid regeneration of the hydrogel structure after the strain-induced failure, as evidenced by the return of the moduli to their initial values (Fig. 8, at the top). The experiments confirmed that the hydrogels could undergo structural recovery as a result of the fast cross-links reshuffling. The self-healing of the hydrogels was also visualized in a simple experiment in which a piece of the gel was cut into two pieces, which then reassembled into one piece after about 10 min. (Fig. 8, at the bottom). Due to the dynamic character of the boronic ester cross-links, the hydrogels composed of the star-hyperbranched copolymer unimolecular micelles could be easily shaped and formed a continuous layer on the covered surface. These features are very important in biomedical applications because they facilitate hydrogel deposition at the target site and ensure proper contact with the tissue, thus providing control over local drug delivery.
Experimental section
Materials
1,1,1-Tris(hydroxymethyl)propane was purchased from Sigma-Aldrich. It was dissolved under reflux in acetone, precipitated with ethyl ether, and then dried before usage. NaH (60 wt%) in mineral oil was purchased from Merck. The oil was removed by washing with dry tetrahydrofuran, and then by drying under reduced pressure. 1,2-Epoxybutane,1,2-epoxyhexane (purchased from TCI) and ethylene oxide (Sigma-Aldrich) were dried over CaH2 and distilled before use. THF (Sigma-Aldrich) was dried over Na/K alloy. DMSO (Sigma-Aldrich) was purified and dried according to the procedure described in ref. 50, and stored over 4 Å molecular sieves. Ethoxyethyl glycidyl ether (EEGE) was synthesized by the reaction of glycidol (Sigma-Aldrich) and ethyl vinyl ether (Acros Organics) according to the procedure described by Fitton51 and Wurm.52 Clotrimazole was purchased from Thermo Scientific, and used without further purification.Synthesis of poly(1,2-epoxybutane)/poly(1,2-epoxyhexane)
1,1,1-Tris(hydroxymethyl)propane (THMP) was weighed into a Schlenk flask and dried with benzene under argon, followed by its removal by distillation at 100 °C. Next, 10 mol% of the OH groups of THMP were converted into alcoholate groups with sodium hydrate in dry THF at 40 °C under a vacuum overnight.Next, THF was removed under reduced pressure and the dry initiator was dissolved in dry DMSO, followed by the addition of 1,2-epoxybutane or 1,2-epoxyhexane. The polymerization was conducted at 50 °C under a vacuum for 48 h. Then the polymer was extracted from the reaction mixture with hexane. The collected hexane fractions were dried and analyzed by GPC and 1H NMR spectroscopy.
Synthesis of poly(1,2-epoxybutane-b-glycidol) and poly(1,2-epoxyhexane-b-glycidol)
A sample of poly(1,2-epoxybutane) or poly(1,2-epoxyhexane) was weighed into a Schlenk flask and dried with benzene under argon at 100 °C. Next, 10 mol% of the end OH groups of a polymer were converted into alcoholate groups with sodium hydrate in dry THF at 40 °C under a vacuum overnight. Subsequently, ethoxyethyl glycidyl ether, EEGE was distilled into a Schlenk flask, and the copolymerization was carried out at 40 °C for 48 h. The structures of the synthesized copolymers were characterized by GPC and 1H NMR spectroscopy. Then, to deprotect the monohydroxyl groups in the EEGE units, the polymer was dissolved in methanol, and a 0.1 M aqueous solution of HCl was added. The polymer solution was stirred at 37 °C for 6 h, and dialyzed against deionized water using the dialysis tube with MWCO = 1 kDa. The copolymer was characterized by 1H NMR spectroscopy.Synthesis of poly(1,2-epoxybutane)-b-hyperbranched polyglycidol/poly(1,2-epoxyhexane)-b-hyperbranched polyglycidol
A sample of poly(1,2-epoxybutane-b-glycidol) or poly(1,2-epoxyhexane-b-glycidol) was weighed into a Schlenk flask and dried with benzene under argon at 100 °C. Next, 10 mol% of the end OH groups of a polymer were converted into alcoholate groups with sodium hydrate in dry THF at 40 °C under a vacuum overnight. Then, THF was removed and glycidol was added to the reaction mixture dropwise via a syringe pump with an injection speed of 0.5 mL h−1. The reaction was held under argon for 24 h at 95 °C. After that time, the reaction was terminated by exposing it to air. The reaction mixture was dissolved in MeOH and precipitated into acetone. The crude product was isolated, dissolved in DMSO, and dialyzed against DMSO for 48 h using the dialysis tubes with MWCO = 3.5 kDa. The copolymer structure was characterized by 1H and 13C INVGATE spectroscopy and 1H DOSY NMR spectroscopy.Synthesis of poly(1,2-epoxybutane-b-ethylene oxide)-b-hyperbranched polyglycidol
A sample of poly(1,2-epoxybutane) was weighed into a Schlenk flask and dried with benzene under argon at 100 °C. Next, 10 mol% of the end OH groups of a polymer were converted into alcoholate groups with sodium hydrate in dry THF at 40 °C under a vacuum overnight. Subsequently, ethylene oxide was distilled into a Schlenk flask, and the copolymerization was conducted at 70 °C for 48 h. The copolymer was dialyzed against water using the dialysis tube with MWCO = 1 kDa. The structure of a synthesized copolymer was characterized by 1H and 13C INVGATE spectroscopy and 1H DOSY NMR spectroscopy.Next, 10 mol% of the end OH groups of poly(1,2-epoxybutane-b-ethylene oxide) were converted into alcoholate groups with sodium hydrate in dry THF at 40 °C under a vacuum overnight. Then, THF was removed and glycidol was added to the reaction mixture dropwise via a syringe pump with an injection speed of 0.5 mL h−1. The reaction was held under argon for 24 h. After that time, the reaction was terminated by exposing it to air. The reaction mixture was dissolved in MeOH and precipitated in acetone. The crude product was isolated, redissolved in DMSO, and dialyzed against DMSO for 48 h using the dialysis tubes with MWCO = 3.5 kDa. The copolymer structure was characterized by 1H and 13C INVGATE spectroscopy and 1H DOSY NMR spectroscopy.
Synthesis of poly(acrylamide-ran-2-acrylamidephenylboronic acid), poly(AM-APBA)
The copolymer was prepared by conventional radical polymerization initiated with AIBN by applying acrylamide (2 g; 28.10 mmoL) and 2-acrylamidophenylboronic acid pinacol ester (1.226 g; 4.48 mmoL). The polymerization was carried out in 15 mL of a DMF/dioxane mixture (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) at 70 °C. The synthesis was conducted for 16 h. The polymerization mixture was diluted in water, and the copolymer was precipitated into acetone and then dried. Next, the copolymer was dissolved in an alkaline solution of NaOH (1 wt%) and dialyzed using a 1000 MW cut-off dialysis membrane, at first against the alkaline aqueous solution and then against water, which was changed several times to reach the neutral pH. Dialysis was necessary to hydrolyze pinacol boronic esters and remove released pinacol. The copolymer was characterized using 1H NMR spectroscopy and GPC. The molar fraction of 2-AAPBA units in the copolymer was equal to 20 moll%, whereas Mn was 63000 (Mw/Mn = 1.86).
:1 v/v) at 70 °C. The synthesis was conducted for 16 h. The polymerization mixture was diluted in water, and the copolymer was precipitated into acetone and then dried. Next, the copolymer was dissolved in an alkaline solution of NaOH (1 wt%) and dialyzed using a 1000 MW cut-off dialysis membrane, at first against the alkaline aqueous solution and then against water, which was changed several times to reach the neutral pH. Dialysis was necessary to hydrolyze pinacol boronic esters and remove released pinacol. The copolymer was characterized using 1H NMR spectroscopy and GPC. The molar fraction of 2-AAPBA units in the copolymer was equal to 20 moll%, whereas Mn was 63000 (Mw/Mn = 1.86).
Solubilization of clotrimazole within unimolecular micelles based on the star-hyperbranched amphiphilic polyethers
A stock solution of clotrimazole (9 mg mL−1) in methanol was prepared. A total of 75 mg of each copolymer was dissolved in 1 mL of methanol. Next, 3 mL of clotrimazole solution was added to the copolymer solution. The mixture was stirred for 6 h. Subsequently, methanol was allowed to evaporate at 37 °C overnight. The dry polymer–drug content was suspended in 10 ml of deionized water The suspension was filtered using an 0.8 μm SFCA filter, and lyophilized. The amount of drug loaded into the HbPGL-based micelles was determined by 1H NMR spectroscopy.Drug-release profile
A sample of a copolymer containing 1.3 mg of clotrimazole was dissolved in 9 mL PBS at pH 5.6 and transferred into a regenerated cellulose dialysis membrane (MWCO = 3500) with a magnetic stirrer inside. The dialysis tube was then immersed in 250 mL of PBS at pH 5.6 with 1% of Tween 80 (v/v). At given time points, 20 mL of the solution was collected and replaced with 20 mL of fresh PBS/Tween 80 solution. Subsequently, 3 × 50 mL of dichloromethane was added to each of the collected samples to extract clotrimazole from the aqueous phase. The organic phases were dried over MgSO4 for 30 min while stirring and then liberated from dichloromethane by evaporation under reduced pressure. The dry product was dissolved in 4 mL of acetonitrile and filtered using a 0.2 μm PTFE filter. The quantification of the released clotrimazole was determined by an ACQUITY UPLC I-Class chromatography system equipped with a binary solvent pump and a photodiode array detector (Waters Corp., Milford, MA, USA). The separation of the analyte was achieved using an ACQUITY UPLC™ BEH C18 column (100 × 2.1 mm, 1.7 μm) maintained at 45 °C. The mobile phase was prepared by mixing 0.1% formic acid (A) and 0.1% formic acid in acetonitrile (B). The elution gradient was: 32% B (0–1.0 min), 32%–95% B (1.0–3.0 min), 95%–95% B (3.0–3.5 min), 95%–32% B (3.5%–3.52 min), and 32%–32% B (3.52–7.0 min). The flow rate was 0.45 mL min−1 and the injection volume was 4 μL. The optimal absorption wavelength for clotrimazole was determined and set at 195 nm. The initial stock calibration solution of standards was created with a concentration of 1 mg ml−1 in acetonitrile.The stock solution was serially diluted with acetonitrile to obtain working solutions at several concentration levels. The calibration curves were prepared at seven different concentrations of clotrimazole solutions and were linear over a concentration range from 0.78 to 50 μg ml−1 with a correlation coefficient of >0.999. The system was controlled using MassLynx software (Version 4.1) and data processing (peak area integration, construction of the calibration curve) was performed by the TargetLynxTM program.
Cell culture
Dermal microvascular endothelium cells (HMEC1) were grown in MCDB131 medium supplemented with hydrocortisone, L-glutamine, and epidermal growth factor. Human cervical cancer endothelial (HeLa) cells were grown in Dulbecco's modified Eagle's medium (DMEM). Next, 10% foetal bovine serum (FBS) and streptomycin (100 mg ml−1) were added to both cell culture media. The cells were grown in T-75 culture flasks at 310 K in an atmosphere containing 5% CO2. The cells were subcultured every 2 or 3 days, then harvested and used in experiments after obtaining 80–90% confluence. The number of viable cells was determined by the trypan blue exclusion assay with the use of a Countess Automated Cell Counter (Invitrogen, Carlsbad, CA, USA).Determination of cytotoxicity
The influence of the neat star-hyperbranched unimolecular micelles (A86, A92, A165, A71) and their drug-loaded constructs, i.e. En 30, En 143, En 144, En 145, En 156, En 157, En 163, En 170, En 171, and En 172, carrying different amounts of clotrimazole on the cell viability was determined with the use of the MTT assay. In addition, pure clotrimazole was used as a control and reference point.Briefly, to the 96-well plates containing cells at the density of 1.5 × 104 cells per well in medium, different concentrations of all the compounds were added, in the 0.1–100 μM concentration range. The cells were incubated with unimolecular micelles for 24 and 48 h in a 310 K humidified atmosphere containing 5% CO2. After the incubation period, the cells were washed with 50 μl of phosphate-buffered saline (PBS). Next, 50 μl of a 0.5 mg mL−1 solution of MTT in PBS was added to each well, and the cells were further incubated under normal culture conditions for 3 h. After incubation, the residue MTT solution was removed and the obtained formazan precipitate was dissolved in DMSO (100 μl per well). The conversion of the tetrazolium salt (MTT) to a coloured formazan by mitochondrial and cytosolic dehydrogenases is a marker of cell viability. Before the absorbance measurement, the plates were shaken for 1 min and the absorbance at 570 nm was measured on a PowerWave HT Microplate Spectrophotometer (BioTek, USA).
Cell cycle analysis
Both cell lines 1.5 × 105 cells per well were incubated with the analyzed compounds (10 μM final concentration) in 12-well cell culture plates for 24 and 48 h in a 310 K humidified atmosphere containing 5% CO2. Then, the cells were fixed with cold 70% ethanol and stained for total DNA content with RNase A and propidium iodide staining buffer (BD, San Diego, CA, USA) according to the manufacturer's instructions. A minimum of 10000 cells were analyzed for the propidium iodide (PE-A) fluorescence signal using a Becton Dickinson LSR II flow cytometer (BD Biosciences, USA). Data were plotted by using Flowing Software 2.5.1 (Turku Centre for Biotechnology, University of Turku, Finland).
Hydrogel formation
Hydrogels were prepared by mixing a 0.25 mL aqueous solution containing 0.05 g of poly(AM-ran-2-AAPBA) with 0.15 mL aqueous solution containing 0.055 g of each star-hyperbranched construct.NMR spectroscopy
1H and 13C INVGATED NMR spectra were recorded using a Bruker Avance NEO AV 400 MHz instrument. 1H DOSY measurements were carried out at 295 K on a Bruker Avance III 500 spectrometer equipped with a 5 mm BBI probe head with a z-gradients coil and GAB/2 gradient unit capable of producing B0 gradients with a maximum strength of 50 G cm−1. The BCU-05 cooling unit, managed by the BVT3300 variable temperature unit, was used for temperature control and stabilization. The spectrometer was controlled with a PC computer running under Windows 7 (64 bit) OS with the TopSpin 3.1 program.For the measurements, each sample was stabilized at 295 K for at least 10 min before data accumulation, and the 1H π/2 pulse length was checked and adjusted carefully for each sample. The standard Bruker pulse program dstebpgp3s was selected for the measurements using the double stimulated echo for convection compensation and LED (Longitudinal Eddy Current Delay) using bipolar gradient pulses for the diffusion and 3 spoil gradients. The shape of all gradient pulses was sinusoidal, while the gradient spoil pulse was 0.6 ms, the delay for gradient recovery was set at 0.2 ms, and the LED was set at 5 ms and kept constant in all the experiments. The gradient pulse (small delta; δ/p30) was kept constant throughout the entire series of temperature measurements, and the diffusion time (big delta; Δ/D20) was changed to achieve the desired signal attenuation at the maximum gradient strength. The DOSY experiments were run in pseudo 2D mode with the gradients varied exponentially from 5% up to 95%, typically in 16 steps, with 16 scans per step. The spectra were processed by TopSpin 3.1 software supplied by the spectrometer manufacturer. The 1 Hz line broadening Lorenzian function was applied and each row was phased and baseline-corrected before executing the Fourier transformation in the F2 dimension. Diffusion coefficient values, for the resolved 1H signals, were extracted from the T1/T2 analysis module of the TopSpin 3.1 program. The NMR spectra of the polymer solutions were recorded at deuterated solvents (CDCl3, DMSO-d6, MeOD-d1).
Gel permeation chromatography
The average molecular weight of HbPGL was determined by gel permeation chromatography (GPC) using a Shimadzu Pump LC-20AD system and a Shimadzu SIL-20A HT Autosampler. A refractometer RI-Optilab-T-rex-Wyatt and laser photometer DAWN 8+ (Wyatt Technology) were used as detectors. N,N′-Dimethylformamide was used as an eluent at a flow rate of 0.8 mL min−1 at 25 °C. Here, Mw = 7800, Mw/Mn = 1.70.Rheology
Gel formation was confirmed by oscillation frequency sweep tests carried out in the linear viscoelastic regime using a parallel plate–plate geometry of 8 mm diameter with a 0.3 mm gap on a Thermo Scientific HAAKE MARS 40 rheometer. Strain sweep measurements for the hydrogel samples were performed at a frequency of 1 Hz in the range of strain from 0.02% to 2000%. Frequency sweep tests were carried out at 10 °C, 25 °C, and 40 °C in the linear viscoelastic regime. Temperature sweep tests in the range from 10 °C to 60 °C were performed at a frequency of 1 Hz using the continuous heating program with a heating rate 5 °C min−1. The self-healing test was carried out in the 1 Hz oscillation time mode at 37 °C by monitoring both the storage and loss moduli. First, the sample was placed under a 1% strain for 180 s, and then it was destroyed with a 5 s 300% strain pulse, after which 1% was again applied for sample restoration.Conclusions
We successfully elaborated a synthetic route to prepare polyether-based amphiphilic constructs with a star-shaped hydrophobic core by applying poly(n-alkyl epoxides) and a shell composed of hyperbranched polyglycidol using a step-by-step ring-opening polymerization of epoxide comonomers. The constructs presented here displayed enhanced clotrimazole solubilization in the aqueous media. The in vitro release profiles demonstrated a sustained drug release from the unimolecular micelles after approximately 48 h. The construction of amphiphiles assured not only the lack of an initial burst release of the drug molecules but also enabled the formation of dynamic hydrogels displaying both injectable and self-healable properties, which are crucial in the administration of hydrogel-based drug carriers. This is the first report on the usage of polymer constructs to increase clotrimazole bioavailability towards the enhancement of anticancer activity.The in vitro study revealed that the encapsulation of clotrimazole in most of the synthesized amphiphilic constructs displayed prolonged and selective toxicity to cervical cancer cells, which allows for reducing the dose of the drug while maintaining the therapeutic effect.
From a preclinical and potential clinical perspective, the newly synthesized clotrimazole carrier systems described in this paper may be useful in the treatment of cervical cancer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland (Project Number: UMO-2018/30/E/ST5/00576). This article has been completed while Mr Piotr Ziemczonek was the Doctoral Candidate in the Interdisciplinary Doctoral School at the Lodz University of Technology, Poland.References
- A. M. Jhaveri and V. P. Torchilin, Front. Pharmacol., 2014, 5, 77 CrossRef PubMed.
- Y. Lu, E. S. Zhang, J. H. Yang and Z. Q. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef PubMed.
- G. L. Li, J. Y. Liu, Y. Pang, R. B. Wang, L. M. Mao, D. Y. Yan, X. Y. Zhu and J. Sun, Biomacromolecules, 2011, 12, 2016–2026 CrossRef CAS PubMed.
- S. Eetezadi, S. N. Ekdawi and C. Allen, Adv. Drug Delivery Rev., 2015, 91, 7–22 CrossRef CAS PubMed.
- X. R. Sun, G. W. Wang, H. Zhang, S. Q. Hu, X. Liu, J. B. Tang and Y. Q. Shen, ACS Nano, 2018, 12, 6179–6192 CrossRef CAS PubMed.
- H. L. Xu, Q. Yao, C. F. Cai, J. X. Gou, Y. Zhang, H. J. Zhong and X. Tang, J. Controlled Release, 2015, 199, 84–97 CrossRef CAS PubMed.
- C. E. Wang, P. S. Stayton, S. H. Pun and A. J. Convertine, J. Controlled Release, 2015, 219, 345–354 CrossRef CAS PubMed.
- A. V. Ambade, E. N. Savariar and S. Thayumanavan, Mol. Pharmaceutics, 2005, 2, 264–272 CrossRef CAS PubMed.
- Z. L. X. Fan and X. J. Loh, Polym. Chem., 2016, 7, 5898 RSC.
- X. L. X. Liang, X. Gao, Y. Zhang, W. Wei and X. Liu, Polymer, 2020, 202, 122716 CrossRef CAS.
- X. H. S. Luo, Y. Zhang, C. Ling, X. Liu and S. Chen, Polym. J., 2011, 43, 41–50 CrossRef.
- H. S. A. S. J. T. Rezaei, M. R. Nabid and H. Niknejad, J. Colloid Interface Sci., 2014, 425, 27–35 CrossRef PubMed.
- M. R. N. S. J. T. Rezaei, H. Niknejad and A. A. Entezami, Polymer, 2012, 53, 3485–3497 CrossRef.
- P. Ziemczonek, M. Gosecka, M. Gosecki, M. Marcinkowska, A. Janaszewska and B. Klajnert-Maculewicz, Int. J. Mol. Sci., 2021, 22, 8386 CrossRef CAS PubMed.
- M. Gosecka, D. Jaworska-Krych, M. Gosecki, E. Wielgus, M. Marcinkowska, A. Janaszewska and B. Klajnert-Maculewicz, Biomacromolecules, 2022, 23, 4203–4219 CrossRef CAS PubMed.
- S. Mantha, S. Pillai, P. Khayambashi, A. Upadhyay, Y. Zhang, O. Tao, H. M. Pham and S. D. Tran, Materials, 2019, 12, 3323 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS PubMed.
- H. Aktas, R. Fluckiger, J. A. Acosta, J. M. Savage, S. S. Palakurthi and J. A. Halperin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 8280–8285 CrossRef CAS PubMed.
- M. A. Burgess and G. P. Bodey, Antimicrob. Agents Chemother., 1972, 2, 423–426 CrossRef CAS PubMed.
- F. Saadatfar, A. Shayanfar, E. Rahimpour, M. Barzegar-Jalali, F. Martinez, M. Bolourtchian and A. Jouyban, J. Mol. Liq., 2018, 256, 527–532 CrossRef CAS.
- F. Kareem, A. M. Bhayo, M. Imran, M. R. Shah, K. M. Khan and M. I. Malik, J. Appl. Polym. Sci., 2019, 136, 47769 CrossRef.
- J. Penso and R. Beitner, Mol. Genet. Metab., 2002, 76, 181–188 CrossRef CAS PubMed.
- J. Penso and R. Beitner, Eur. J. Pharmacol., 2002, 451, 227–235 CrossRef CAS PubMed.
- L. Hegemann, S. M. Toso, K. I. Lahijani, G. F. Webster and J. Uitto, J. Invest. Dermatol., 1993, 100, 343–346 CrossRef CAS PubMed.
- M. M. Marinho-Carvalho, P. Zancan and M. Sola-Penna, Mol. Genet. Metab., 2006, 87, 253–261 CrossRef CAS PubMed.
- B. Fauvel and A. Yasri, MAbs, 2014, 6, 838–851 CrossRef PubMed.
- S. K. Parks, J. Chiche and J. Pouyssegur, Nat. Rev. Cancer, 2013, 13, 611–623 CrossRef CAS PubMed.
- O. Warburg, Science, 1956, 123, 309–314 CrossRef CAS PubMed.
- C. M. Furtado, M. C. Marcondes, M. Sola-Penna, M. L. S. de Souza and P. Zancan, PLoS One, 2012, 7, e30462 CrossRef CAS PubMed.
- B. Prabagar, B. K. Yoo, J. S. Woo, J. A. Kim, J. D. Rhee, M. G. Piao, H. G. Choi and C. S. Yong, Arch. Pharmacal Res., 2007, 30, 249–254 CrossRef CAS PubMed.
- G. O. R. Porras, J. C. Nogueda and A. P. Chacon, Rep. Pract. Oncol. Radiother., 2018, 23, 533–539 CrossRef PubMed.
- X. S. Fan, Z. B. A. Li and X. J. Loh, Polym. Chem., 2016, 7, 5898–5919 RSC.
- G. J. Chen, Y. Y. Wang, R. S. Xie and S. Q. Gong, Adv. Drug Delivery Rev., 2018, 130, 58–72 CrossRef CAS PubMed.
- W. Celentano, G. Neri, F. Distante, M. Li, P. Messa, C. Chirizzi, L. Chaabane, F. De Campo, P. Metrangolo, F. B. Bombelli and F. Cellesi, Polym. Chem., 2020, 11, 3951–3963 RSC.
- M. Gosecki, B. Zgardzinska and M. Gosecka, J. Phys. Chem. C, 2016, 120, 18323–18332 CrossRef CAS.
- M. Gosecki, S. Kazmierski and M. Gosecka, Biomacromolecules, 2017, 18, 3418–3431 CrossRef CAS PubMed.
- M. Jafari, S. S. Abolmaali, H. Najafi and A. M. Tamaddon, Int. J. Pharm., 2020, 576, 118959 CrossRef CAS PubMed.
- H. Ying, G. J. He, L. F. Zhang, Q. F. Lei, Y. S. Guo and W. J. Fang, J. Appl. Polym. Sci., 2016, 133, 42951 CrossRef.
- C. Z. Wu, C. Strehmel, K. Achazi, L. Chiapisi, J. Dernedde, M. C. Lensen, M. Gradzielski, M. B. Ansorge-Schumacher and R. Haag, Biomacromolecules, 2014, 15, 3881–3890 CrossRef CAS PubMed.
- I. Postnova, V. Silant'ev, M. H. Kim, G. Y. Song, I. Kim, C. S. Ha and Y. Shchipunov, Colloids Surf., B, 2013, 103, 31–37 CrossRef CAS PubMed.
- Haryanto, D. Singh, P. H. Huh and S. C. Kim, J. Biomed. Mater. Res., Part A, 2016, 104, 48–56 CrossRef CAS PubMed.
- M. Schomer, C. Schull and H. Frey, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 995–1019 CrossRef.
- D. Wilms, S. E. Stiriba and H. Frey, Acc. Chem. Res., 2010, 43, 129–141 CrossRef CAS PubMed.
- A. Sunder, R. Hanselmann, H. Frey and R. Mulhaupt, Macromolecules, 1999, 32, 4240–4246 CrossRef CAS.
- B. M. S. dos Santos, T. dos Santos and F. E. Antunes, Drug delivery systems: Advanced technologies potentially applicable in personalized treatments, Springer, 2013 Search PubMed.
- J. Wang, L. H. Jia, Z. R. Kuang, T. Wu, Y. Hong, X. B. Chen, W. K. Leung, J. Xia and B. Cheng, PLoS One, 2014, 9, e98885 CrossRef PubMed.
- M. H. Khalid, S. Shibata and T. Hiura, J. Neurosurg., 1999, 90, 918–927 CAS.
- M. Y. Cao, Y. Lee, N. P. Feng, R. A. Al-Qawasmeh, S. Viau, X. P. Gu, L. Lau, H. Jin, M. Wang, A. Vassilakos, J. A. Wright and A. H. Young, J. Pharmacol. Exp. Ther., 2004, 308, 538–546 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of laboratory chemicals, Elsevier/Butterworth-Heinemann, Amsterdam London, 7th edn, 2013 Search PubMed.
- A. O. Fitton, J. Hill, D. E. Jane and R. Millar, Synthesis, 1987, 1140–1142 CrossRef CAS.
- F. Wurm, J. Nieberle and H. Frey, Macromolecules, 2008, 41, 1909–1911 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 1H DOSY and 13C INVGATE NMR spectra of synthesized star-hyperbranched unimolecular micelles, strain sweep experiments performed for hydrogel platforms. See DOI: https://doi.org/10.1039/d2tb02629e |
| This journal is © The Royal Society of Chemistry 2023 |