
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Trojan horse approach for efficient drug delivery in photodynamic therapy: focus on taxanes
Vladimíra Svobodová
Pavlíčková
 ,
Jan
Škubník
,
Tomáš
Ruml
and
Silvie
Rimpelová
*
,
Jan
Škubník
,
Tomáš
Ruml
and
Silvie
Rimpelová
*
Department of Biochemistry and Microbiology, University of Chemistry and Technology, Prague, Technická 3, 166 28 Prague 6, Czech Republic. E-mail: vladimira.svobodova.pavlickova@vscht.cz; jan.skubnik@vscht.cz; tomas.ruml@vscht.cz; silvie.rimpelova@vscht.cz
First published on 16th March 2023
Abstract
Photodynamic therapy is an effective method for the treatment of several types of cancerous and noncancerous diseases. The key to the success of this treatment method is effective drug delivery to the site of action, for instance, a tumor. This ensures not only the high effectiveness of the therapy but also the suppression of side effects. But how to achieve effective targeted delivery? Lately, much attention has been paid to systems based on the so-called Trojan horse model, which is gaining increasing popularity. The principle of this model is that the effective drug is hidden in the internal structure of a nanoparticle, liposome, or nanoemulsion and is released only at the site of action. In this review article, we focus on drugs from the group of mitotic poisons, taxanes, and their use with photosensitizers in combined therapy. Here, we discuss the possibilities of how to improve the paclitaxel and docetaxel bioavailability, as well as their specific targeting for use in combined photo- and chemotherapy. Moreover, we also present the state of the art multifunctional drugs based on cabazitaxel which, owing to a suitable combination with photosensitizers, can be used besides photodynamic therapy and also in photoacoustic imaging or sonodynamic therapy.
1. Introduction
Photodynamic therapy (PDT) is a method with great potential for the treatment of both cancerous and noncancerous diseases. It is based on the simultaneous presence of three factors: light, oxygen, and a photosensitive drug (PS). Each component alone should not affect cell proliferation, however, when combined, they eliminate the diseased tissue. Due to the light absorption and energy transfer, a PS is excited, i.e., the transition of an electron of a PS from the ground state (S0) to a higher energy state (S1) occurs without changing its spin. This singlet excited state S1 of the PS is characterized by a short lifetime in the order of nanoseconds. The PS in the S1 state can radiate the obtained energy in the form of fluorescence emission or heat dissipation thereby returning to the ground state S0. However, due to the intersystem crossing, in which the spin changes occur, it may result in the transition of a PS from the S1 state to a more stable and long-lived triplet state (T1), which is very important from the point of view of PDT. Then, the PS relaxes from the T1 state back to its ground state S0 by radiation of the received energy in the form of phosphorescence, or by energy transfer to other molecules in the vicinity. This energy transfer can generate various types of radicals such as ˙OH, ˙HO2, and ˙OOR, collectively referred to as reactive oxygen species (ROS). When the energy is transferred directly to the molecular oxygen (3O2), highly reactive singlet oxygen (1O2) is formed. Reactive oxygen species together with singlet oxygen are responsible for the oxidation of other molecules present in the cell; this oxidative stress then leads to cell structure damage followed by cell death,1–3 for schematic depiction, see Fig. 1.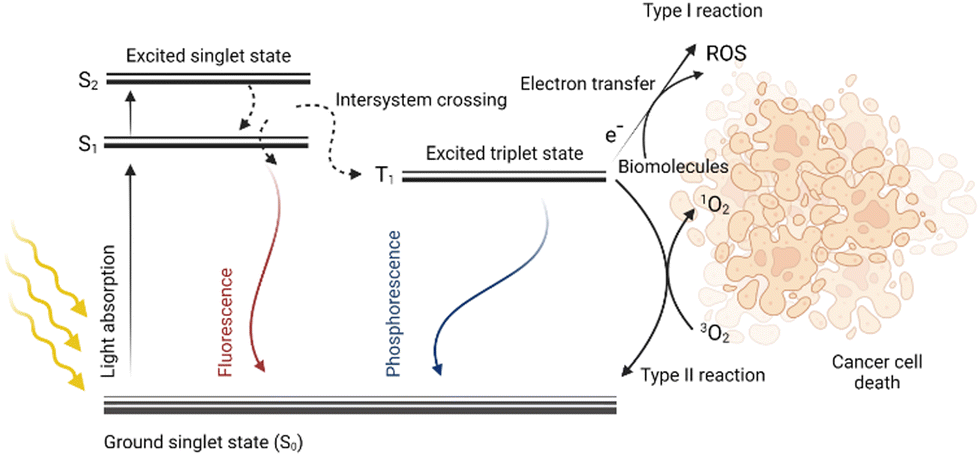 | ||
| Fig. 1 A simplified scheme of the Jablonski diagram extended by the mechanism of photodynamic therapy. After the light of the respective wavelength is absorbed by a photoactive molecule, it is excited and transitions from the ground state (S0) to the higher singlet excited states (S1, S2) occur. From these excited states, the molecule can return to the ground state S0 while releasing energy in the form of fluorescence emission. Via intersystem crossing, the molecule can transfer into a more stable triplet state (T1), and its energy can be radiated in the form of phosphorescence by the transition to the ground state S0. However, if a molecule in T1 reacts with biomolecules (type I reaction), electron transfer can occur to form reactive oxygen species (ROS). The molecule in T1 can also react directly with the molecular oxygen to form singlet oxygen (type II reaction). Created by BioRender.com. | ||
The PDT method has been successfully used already for many years for the treatment of several diseases such as age-related macular degeneration,4,5 head and neck cancer,6–9 bladder cancer,10–12 non-small lung cell carcinoma,13 actinic keratoses,14–16 cutaneous T-cell lymphoma,17,18 or other types of dermatological carcinomas.19 In clinical and pre-clinical trials, PDT and photodynamic diagnostics are also being evaluated for the treatment of brain cancer,20 prostate cancer,21–24 and gynecological diseases.25–27 However, the broader use of PDT for the treatment of other cancer types is still limited. One of the reasons is the relatively shallow (2–3 mm) tissue penetration of light with a wavelength of 630 nm.28 As a result, only cells with accumulated PS that are directly illuminated by the light source are effectively eliminated. This problem can be partially solved by the use of a PS absorbing in the near-infrared region of the electromagnetic spectra, which is characteristic of deeper tissue penetration. Another approach for PS photoactivation is the application of optical fibers, which can be introduced directly into the tumor lesion instead of light sources working only on the surface.29 However, there are still other limitations that hamper broader PDT utilization. These are insufficient capabilities in specific drug delivery to the diseased tissue caused either by low PS solubility, high aggregation, or poor PS selectivity for tumor cells.
Therefore, a more recent approach combining PDT with other treatment methods, for instance, chemotherapy, has emerged. Such a combination has been proven very effective, especially in cancer treatment. The combination of these approaches results in a highly effective reduction in the number of cancer cells, minimization of side effects, and also reduction of the risk of cancer cell resistance development.30–33 When a PS and an antitumor drug are applied as a mixture (in one injection), their simultaneous delivery to the target tissue and, thus, their synergistic effect is not guaranteed. These facts have led many research groups to the development of PSs that combine a PS and a chemotherapeutic agent in one moiety, often encapsulated in nanoparticles (NPs). The use of NPs is one of the possibilities for drug delivery based on the “Trojan horse strategy”, i.e., “hiding” the drug in the internal structure of NPs based on which the hidden drug can be more efficiently and in a more facile manner transported to the target tissue. Such an approach provides an advantage in crossing the cell membrane and in some cases also the blood-brain barrier. In addition, specific targeting can ensure that the drug is delivered to a specific tissue, thus eliminating or minimizing side effects on the whole organism. The advantage of the “Trojan horse approach” is also the possibility of encapsulating several active substances with different mechanisms of action in one system at the same time, which increases the probability of success in the fight against the disease.
Among the widely used chemotherapeutics, there belong drugs based on mitotic poisons, for instance, paclitaxel, colchicine, and vincristine. These compounds block the polymerization or depolymerization of microtubule fibers. The use of mitotic poisons has its limitations, especially, it is the low water solubility and high systemic toxicity of these compounds. However, due to their great clinical potential, they are of frequent choice for use in drug combinations, in which their side effects can be effectively eliminated.34 The encapsulation of multiple agents with therapeutic potential into a single particle provides an opportunity for accurate and controlled delivery of multiple drugs to a target area that enhances the synergistic effect of both therapeutics.
2. Taxanes in combined phototherapy and chemotherapy
Medicines based on clinically approved taxanes represent great potential in combined anticancer therapy. Taxanes belong among plant alkaloids that are responsible for the inhibition of tubulin depolymerization in cells. The mechanism of action of taxanes is based on their ability to stabilize microtubule fibers by binding to the β-tubulin subunit of the tubulin dimer. The exact effect of taxanes is dependent on the amount administered; for example, low doses of paclitaxel (PTX, 5–10 nM) lead only to cell cycle arrest but the cells survive. While at high doses of PTX (10–500 nM), there is a permanent cessation of mitosis, the formation of the atypical mitotic spindle, and abnormal microtubule aster.35 The formation of these structures prevents the further course of mitosis and leads to cell apoptosis. Therefore, PTX has been widely used in medicine as a chemotherapeutic for the treatment of breast, ovarian, and non-small cell lung carcinoma. However, the low solubility of PTX has led scientists to the development of novel formulations and derivatives, in which case, drug combinations have come to the forefront of interest. A detailed overview of the various drug combinations involving PTX is given in Škubník et al.,36 which summarizes not only the co-administration of two different separate drugs but also two drugs joined in one molecule. The progress in the development of nano-formulated taxanes via the use of novel biomaterials and NP drug delivery systems was summarized by Zhang et al.37 In this comprehensive review, they discussed the importance of nano-formulated taxanes in synergistic therapy strategies (e.g., combination with surgery, chemo- and radiotherapy) and nano-formulated taxanes in targeted drug delivery in preclinical and clinical use.37 Here, we focus mainly taxanes, paclitaxel, docetaxel, and cabazitaxel, used as chemotherapeutics in combination with photosensitizers adminitered together in one particle/delivery agent.2.1. Paclitaxel-based prodrugs
One of the possibilities in the development of novel drugs is the preparation of so-called prodrugs, i.e., inactive forms of drugs that are converted into active substances only after their administration, or rather after their delivery to the target tissue. Prodrugs can be prepared by modifying the original chemical structure of a drug, but also by conjugation with another molecule or cleavable linker (Fig. 2).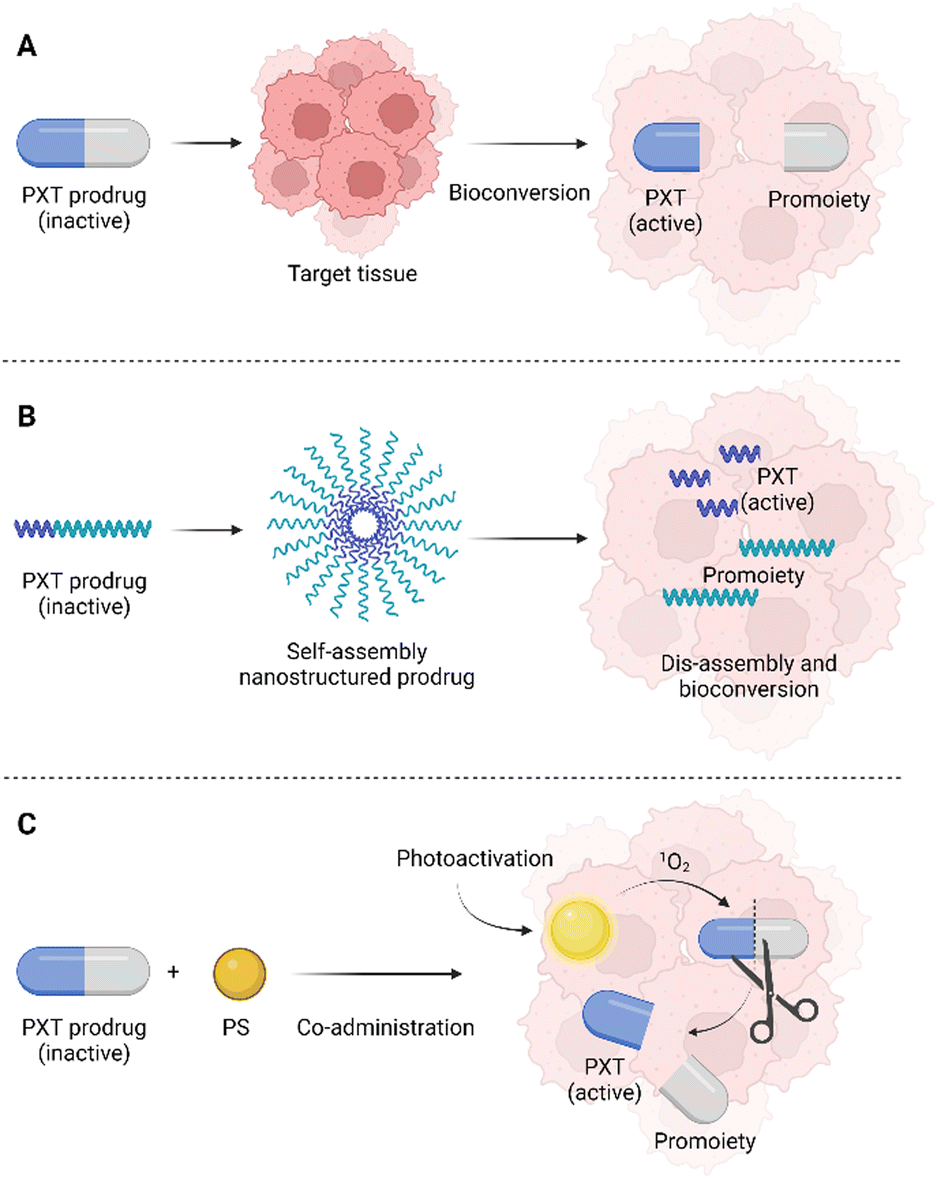 | ||
| Fig. 2 (A) An illustration of paclitaxel modification to inactive PTX prodrug which is converted to an active PTX form in the target tissue; (B) an amphiphilic PTX prodrug can create self-assembling nanostructures, which are disassembled in the target tissue and an accessible form of PTX is released; (C) a combination of singlet oxygen-cleavable PTX prodrug with a photosensitizer as a multimodal approach in cancer treatment. Created by BioRender.com. | ||
The prodrug approach is a strategy chosen also by Bio et al.,38 who prepared a series of three singlets oxygen-activable PTX prodrugs targeted to the mitochondria of cancer cells. These newly prepared prodrugs consisted of PTX covalently linked to one of three mitochondriotropic cations: rhodamine (RH), 4-carboxy-1-methylpyridinium chloride (CAT), or triphenylphosphonium (TPP), which were connected to a singlet oxygen-cleavable amino acrylate linker. The second molecule used in the system, Bio et al. presented, is hexyl-5-aminolevulinic acid (hALA), resp. protoporphyrin IX (PpIX), which after photoactivation generates singlet oxygen. hALA has been approved by the U.S. Food and drug administration for the diagnosis of non-muscle-invasive bladder cancer. In the body, hALA, alike 5-aminolevulinic acid, serves as a precursor in the biosynthesis of the PpIX photosensitizer in high yields.39,40 In the study of Bio et al.,38 the prodrug chemotherapeutics RH-L-PTX, TPP-L-PTX, or CAT-L-PTX were co-administered (a concentration range of 0.25–1.25 μM) with hALA (0.5 mM) to the rat cells derived from bladder carcinoma (AY-27). After 2 h, the PpIX internalized in the cells was photoactivated (530 nm, 10 mW cm−2) which induced the formation of singlet oxygen. The increased amount of singlet oxygen leads to fast cleavage of the prodrug molecule and PTX release. This photoinduced chemotherapy resulted in 84, 82, and 80% elimination of AY-27 cells after treatment with 0.25 μM concentration of RH-L-PTX, TPP-L-PTX, and CAT-L-PTX, respectively. PDT alone (hALA/PpIX + photoactivation) induced the elimination of only 40% of the AY-27 cells. Without hALA, the prodrugs did not induce significant phototoxicity or cytotoxicity. In addition, the intracellular localization of PpIX was detected in the mitochondria of AY-27 cells. The same localization site was determined also for the fluorescent RH-L-PTX prodrug. This result suggests that the prepared prodrugs effectively target the AY-27 cell mitochondria, which is an attractive compartment for effective photo-/chemotherapy.38 However, the presented study does not report on any other studies of the prepared prodrugs, for instance, using 3D cell models of spheroids, or in vivo tests using experimental animals. It is therefore a question of whether the high efficiency would be preserved in more complex systems as well. In this case, what could be a limitation is the different delivery efficiency/rate of prodrugs and hALA, which were reported to be administered separately.
The second example of a PTX-based prodrug is the direct conjugation of PTX to a PS, thus creating one molecule.41 In this study, PTX was conjugated to a PS (phthalocyanine) together with a folate receptor-targeting group attached via a polyethylene glycol (PEG) linker of various lengths (1, 2, 3.5, and 5k) which was compared to direct conjugation of both moieties (without the PEG linker). The optimal length of the PEG linker for efficient cell uptake of the prodrug was determined as 1–3.5k. In the case of direct PTX-PS conjugation, a formation of prodrug aggregates occurred which resulted in a reduction of prodrug transport into cells. In contrast, for the longest PEG linker (5k), there was a steric hindrance of the prodrug binding to the folate receptor, and due to the high hydrophilicity of the conjugate, free transport of the drug across the plasma membrane was limited. The prodrug phototoxicity was determined 72 h after laser irradiation (690 nm, 10 J cm−2) when internalized in human cells derived from ovarian carcinoma (SKOV-3, folate receptor positive), in which the half-maximal inhibitory concentration (IC50) values of the prodrugs with medium lengths of the PEG linker (1–3.5k) reached to ca. three times lower values than that of the directly conjugated prodrug and prodrug with the longest PEG linker (5k). Without photoactivation, only negligible prodrug cytotoxicity was detected at 500 nM concentration of all conjugates with more than 90% of cells surviving after 72 h treatment.40 The anticancer activity of the prodrug with 2k PEG linker was further verified in BALB/c mice with implanted mouse cells derived from colon carcinoma (Colon-26). The study focused on the optimization of the so-called drug-light interval (DLI), i.e., the time at which tumor photoactivation occurs counted by the drug administration. DLI appears to be a very important parameter of anticancer therapy utilizing PDT. This study evaluated three DLIs, 0.5, 9, and 48 h, for 2k PEG (PTX-phthalocyanine). In all three times, the colon-26 tumor progression in BALB/c mice slowed down or completely stopped. In addition, in the case of DLI of 9 h, the tumor was fully eliminated without remission within 90 days after photoactivation. With shorter or, on the contrary, longer DLIs, the tumor growth was initially suppressed after 3–7 days from treatment, however, then, the tumor growth continued, which shows the great importance of the correct timing of the photoactivation after drug administration.42
2.2. Nanoparticles and nanoemulsions with paclitaxel
Along with the rapid development in the field of nanotechnologies, nanomaterials, and biomaterial research, there has been an increasing effort to use them in the preparation of novel (bio)medicinal products. The most common delivery systems that enhance PTX solubility and improve its pharmacological properties like liposomes, NPs, micelles, prodrugs, emulsions, implants, and cosolvent methods were summarized by Haddad et al. (Fig. 3). The review focuses especially on nanocrystal formulations of PTX.43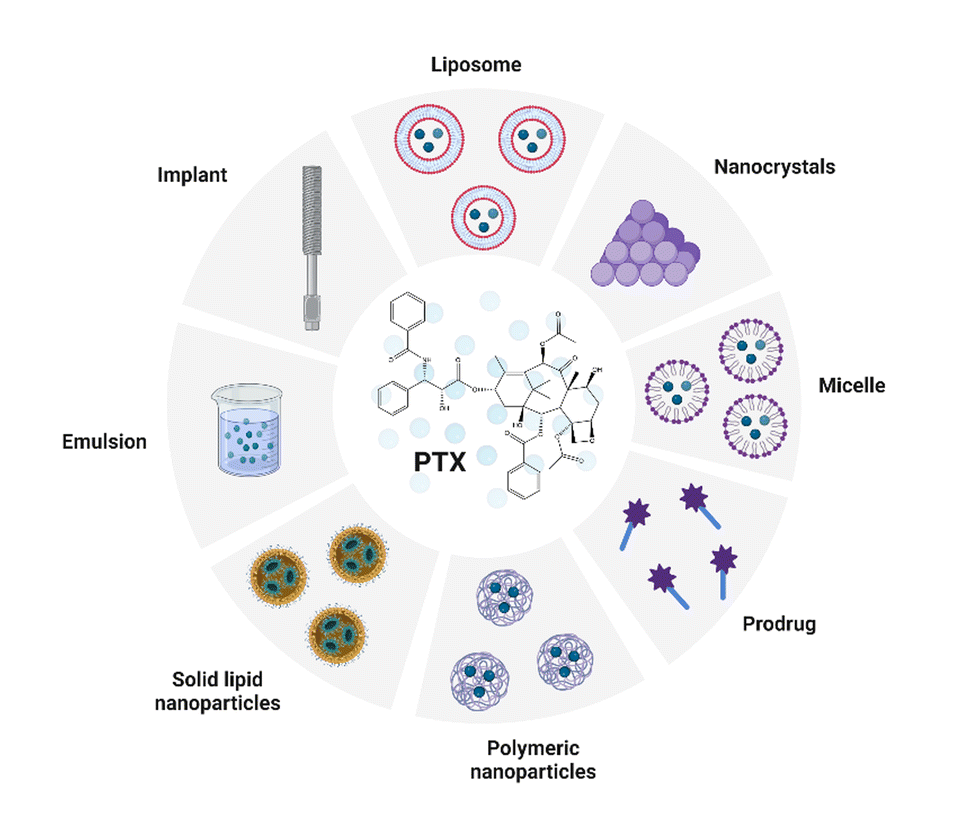 | ||
| Fig. 3 Common delivery systems of paclitaxel (PTX). Taken and edited from Haddad et al. 2022.43 Created by BioRender.com. | ||
However, the preparation of NPs that leads to improved drug bioavailability, prolonged drug circulation in the bloodstream, and specifically targeted drug accumulation at the site of a tumor has remained an immense challenge, especially for multimodal drugs. Moreover, researchers have also increasingly pointed out that relatively large NPs are retained around the tumor's blood supply and do not penetrate the tumor tissue. In contrast, small NPs penetrate tumor tissue easily but are also more rapidly eliminated from the site of action.44,45 In the case of PDT, the situation is all worse since many of the used carriers increase the aggregation of PSs, thereby affecting their photochemical properties and limiting their use in PDT.46 Yu et al.47 tried to solve these problems by preparation of several size-reducible NPs (Fig. 4). The NP core is composed of small gold nanoclusters (CAuNCs) into which ROS-responsive cinnamaldehyde- and thioacetal-based PTX dimers (PXTK) are encapsulated together with a PS pheophorbide a. The NP core is coated with a hyaluronic acid (HA) layer which determines the NP size. The amounts of 0.67, 1, and 2 mg mL−1 of HA corresponding to a 150, 200, and 300 nm NP diameter, respectively, were evaluated. Moreover, the HA layers also served as a size-reducing factor for the NPs given the possibility of hyaluronidase cleavage. The NP surface was further coated with a red blood cell membrane (RBC) to ensure longer systemic circulation and improved NP delivery to the site of the tumor. To enhance the immunotherapeutic effect, the particles were supplemented with a hydrolysis-resistant D-peptide antagonist (dPPA) targeting the programmed cell death receptor-1/programmed cell death receptor–ligand 1 pathway (PD-1/PD-L1). The use of the RBC membrane coating was found to reduce NP cytotoxicity in mouse macrophages (RAW 264.7). In addition, the cytotoxicity was based on the size of the NPs, for which larger particles exhibited increased toxicity compared to the smaller ones; the IC50s were equal to 1.09, 0.45, and 0.86 μg mL−1 for 150, 200, and 300 nm NP diameters, respectively. Moreover, based on measurements of fluorescence emission intensity, significantly lower uptake of RBC-treated NPs by RAW 264.7 cells was detected after 2–4 h of treatment than for uncoated NPs. Contrary to that, in cells derived from mouse mammary carcinoma (4T1), the RBC-coated NPs induced higher cytotoxicity than the uncoated ones. However, at the same time, no statistically significant relationship between the NP size and in vitro cytotoxicity was found for 4T1 cells, for which the IC50s of 0.092, 0.26, and 0.10 μg mL−1 were determined for 150, 200, and 300 nm NP diameters, respectively. To further verify the anticancer activity of the prepared NPs, Yu et al.47 performed an in vivo experiment in 4T1 tumor-bearing mice, using 150 nm NPs as the most suitable ones for mammary tumor elimination. The efficacy of 84.2% inhibition of tumor growth was reached upon treatment with RBC-coated NPs containing PXTK, pheophorbide a, and dPPA with laser photoactivation, i.e., photo-, chemo-, and immunotherapy were combined. In the case, in which only photo- and chemotherapy (NPs without dPPA) were combined, the tumor growth was inhibited to a lesser extent, i.e., 74.3%. In the case of PDT monotherapy (photoactivated NPs containing only pheophorbide a) or combined chemo-/immunotherapy (no photoactivation), the tumor growth inhibition ranged between 65–66%, when compared to the control group. In addition, the prepared NPs also showed significant antimetastatic potential in combination with photo-/chemo-/immunotherapy,47 which appears very promising.
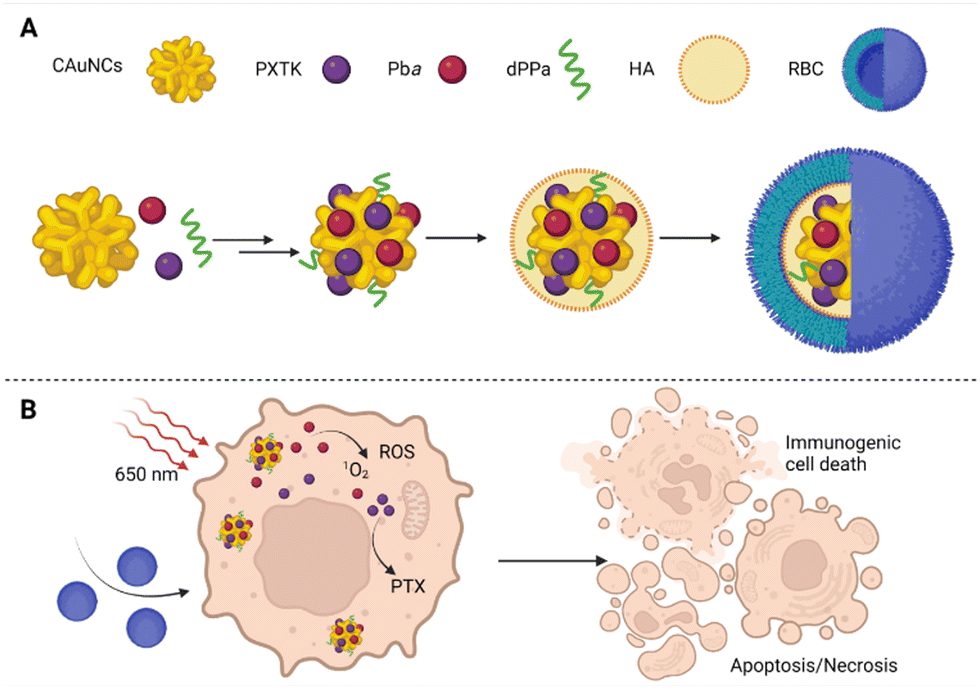 | ||
| Fig. 4 (A) Size-reducible nanoparticle composition: the core of nanoparticles composed of small gold nanoclusters (CAuNCs), into which ROS-responsive cinnamaldehyde- and thioacetal-based PTX dimers (PXTK) were encapsulated together with a PS pheophorbide a (Pba) and, for increased immunotherapeutic effect, the particles were supplemented with hydrolysis-resistant D-peptide antagonist (dPPA) targeting the programmed cell death receptor-1/programmed cell death receptor–ligand 1 pathway (PD-1/PD-L1). The core of the nanoparticles was covered with a layer of hyaluronic acid (HA), which determined their diameter, and the surface was formed by a red blood cell membrane (RBC). (B) A schematic representation of the therapeutic effect of the prepared nanoparticles in 4T1 cells after photoactivation at 650 nm. Taken and adapted from Yu et al. 2019.47 Created with BioRender.com. | ||
A different approach for the development of combined photo-/chemotherapy was chosen by Luo et al.,48 who prepared a thioether bond-bridged heterotypic dimer (PPa-S-PTX) of PTX and pyropheophorbide a (PPa) for multimodal synergistic cancer therapy. Owing to the ROS-sensitive thioether bond, the cleavage of dimers into two separately active parts of the PS (PPa) and the chemotherapeutic moiety (PTX) was assured. As a control, they synthesized a ROS-insensitive dimer, in which the thioether bond was replaced by a carbon chain (PPa-C-PTX). For the formation of NPs, a one-step nanoprecipitation method was used during which the dimers (PPa-S-PTX or PPa-C-PTX) were dispersed in water and self-packaged. The NP surface was subsequently modified by PEGylation with 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] to increase the stability of the NPs. When the prepared PPa-S-PTX NPs were exposed to H2O2 or photoactivated by laser radiation (660 nm, 10 min), there occurred a release of more than 90 and 40% of PTX, respectively. Contrary to that, in the case of a control PPa-C-PTX, more than 90% of the NPs remained intact. The efficacy of PPa-S-PTX treatment (a concentration range of 1.562–100 nM) was evaluated in human epidermoid carcinoma (KB), human lung adenocarcinoma (A549), and 4T1 cells, for which a synergistic effect of the phototherapeutic and chemotherapeutic moieties was detected after 4 h upon laser photoactivation (660 nm, 58 mW cm−2, 6 min). These in vitro experiments were followed by in vivo tests in two animal models with implanted KB and 4T1 tumor cell xenografts. In these models, a clear reduction in tumor growth occurred after five doses of PPa-S-PTX NPs administered in intravenous injections (the total dose of NPs was 9.4 μmol kg−1 from which PTX was 8 mg kg−1 and PPa of 5 mg kg−1 for PPa) followed by laser irradiation (660 nm, 200 mW cm−2, 6 min), compared to both nonphotoactivated sole PTX (commercially available Taxol®), and PPa co-administered with unconjugated Taxol® (+laser irradiation).48 A similar approach was chosen by Sun et al.,49 who prepared a light-activatable porphyrin–PEG polymer-coated prodrug. The NP was based on a self-assembled hydrophobic core composed of PTX and oleic acid (OA) linked by a ROS-cleavable thioether bond (PTX-S-OA). The surface of the NPs was composed of a PS, PPa, conjugated to PEG (PPa-PEG2k). Also, these NPs (PTX-S-OA/PPa-PEG2k) exhibited good stability in phosphate buffer solution and efficient release of PTX (>90%) in the presence of 2 mM H2O2 in combination with 2 min laser photoactivation at 660 nm (58 mW cm−2, 6 min). Experiments in KB tumor-bearing nude mice revealed good antitumor activity of these NPs in vivo. Significant tumor reduction was detected after repeated treatment of the mice with PTX-S-OA/PPa-PEG2k (5 injections; the dose corresponds to 8 mg kg−1 for PTX, 1 mg kg−1 for PPa) followed by laser photoactivation (660 nm, 200 mW cm−2, 6 min), in comparison to sole Taxol® treatment or Taxol®/PPa/laser and PTX-S-OA/PPa-PEG2k without photoactivation.49
Yang et al.50 synthesized a new amphiphilic polymer that was composed of chondroitin sulfate (ChS) linked to hydrophobic blocks of ROS-responsive poly(propylene sulfide) [PPS]. This polymer (ChS-g-PPS) formed the basis of NPs into which a PS chlorine e6 (Ce6) was encapsulated together with the PTX chemotherapeutic. The prepared NPs named CP/ChS-g-PPS NPs (a diameter of ∼200 nm) exhibited a high rate of drug loading efficiency with a Ce6 and PTX content of 14.93% and 24.31%, respectively, as well as a higher rate of drug retention in tumor foci. A synergistic photo-/chemotherapeutic effect was detected after treatment of human breast cancer cells (MCF-7) with CP/ChS-g-PPS followed by photoactivation (100 mW cm−2, 1 min), upon which cancer cell elimination was effective to a much higher extent than just monochemotherapy with CP/ChS-g-PPS (not photoactivated) or monophototherapy using Ce6/ChS-g-PPS (photoactivated). At the same time, the photo-/chemotherapy using CP/ChS-g-PPS (dose corresponding to 5 μg mL−1 PTX, 2.625 μg mL−1 Ce6) also induced the highest percentage of apoptotic cells MCF-7 compared to the monotherapies. Moreover, interestingly, the toxicity of non-photoactivated CP/ChS-g-PPS was reduced compared to free PTX (dissolved in dimethyl sulfoxide) and Taxol®. The anticancer activity of CP/ChS-g-PPS NPs was examined in vivo in two animal models a 4T1 tumor-bearing mouse and MCF-7 xenografts in nude BALB/c mice. After 14 days, 92.76% resp. 88.57% inhibition of the tumor size was observed in 4T1 and MCF-7 xenografts in mice (in this order) treated with CP/ChS-g-PPS (dosage: 16 mg kg−1 PTX, 8.5 mg kg−1 Ce6) with photoactivation (660 nm, 200 mW cm−2, 15 min) compared to untreated mice. This was the best result achieved even in comparison with sole Taxol® or non-photoactivated CP/ChS-g-PPS treatment. Histochemical analysis of selected tissues showed no significant organ damage compared to the control group.50
Different ChS-based NPs, this time targeting multi-drug resistant tumors with increased P-glycoprotein (P-gp) expression, were prepared by Shi et al.51 A PS Ce6 and the P-gp inhibitor quercetin (Q) were attached to the ChS backbone using a redox-responsive cystamine linker (C) to obtain ChS-C-Q/Ce6 (CQE) conjugate. The thus prepared conjugate spontaneously assembled into NPs in an aqueous medium. With the addition of PTX, PTX/CQE NPs with 120 nm in diameter were prepared; they contained three molecules with different mechanisms of action. The NP's in vitro cytotoxicity was determined using a multi-drug-resistant breast cancer cell line MCF-7/ADR and wild-type MCF-7 cells. The measurements clearly show that combination therapy, whether in the form of co-administration of free PTX + Q (IC50 of 7.12 ± 1.94 μg mL−1) or NPs with PTX/CQE (IC50 of 3.71 ± 0.44 μg mL−1) has in the case of MCF-7/ADR cells significantly higher efficiency after 24 h than PTX alone (IC50 of 36.37 ± 13.26 μg mL−1). The highest decrease in the number of MCF-7/ADR cells was observed after administration of PTX/CQE NPs in combination with photoactivation (600 nm, 100 mW cm−2, 1 min). In this case, also the highest proportion of apoptotic cells MCF-7/ADR (62.08%) was recorded. In vivo experiments with 4T1 tumor-bearing mice showed efficient accumulation of CQE NPs in the tumor tissue but also in the liver, in which free Ce6 accumulated significantly lesser than the NPs. The authors explain the accumulation of CQE NPs in the liver by the NP size, since it is known that NPs with a diameter of 50–250 nm can be taken up by liver sinusoidal endothelial cells. The second explanation for the liver uptake is the NP composition, which, due to the content of ChS, can bind to the cluster of differentiation 44 (CD44) expressed not only by tumor cells but also by liver cells. The liver of experimental mice accumulated PTX in high concentrations also in the aforementioned study by Yang et al.50 In the same experimental model (4T1 tumor-bearing mice), the number of lung metastases was significantly reduced after two-week treatment by PTX/CQE NPs followed by photoactivation, compared to the Taxol®-only treated group. The therapeutic effect of PTX/CQE NPs was verified in mice bearing MCF-7/ADR tumor xenograft, in which 84.28% suppression of tumor growth occurred after administration of the PTX/CQE NPs (intravenous injection every 4 days 3 times at 10 mg kg−1 of PTX equivalent) in combination with photoactivation (600 nm, 100 mW cm−2, 15 min).51
Another possible approach for improving drug hydrophilicity and specific targeting is the creation of nanoemulsions, which are popularly used for drug delivery. Nanoemulsions are dispersions of two immiscible liquids, usually oil-in-water or water-in-oil, which are dispersed in each other in the form of submicron droplets. Surfactants are often used to increase the stability of nanoemulsions, however, they have also certain limitations, since they can lead to adverse drug effects when administered systemically.52–54 Currently, the use of nanoemulsions is investigated in five active clinical trials for treating basal cell carcinoma by PDT (NCT02367547),55 for joint pain reduction caused by treatment with aromatase inhibitors in breast cancer survivors (NCT03865992),56 against pandemic flu (NCT05397119),57 plaque psoriasis (NCT04971239)58 and dry eye disease (NCT05245604).59 More than 15 other clinical studies of different nanoemulsion applications were completed in the recent four years which indicates the great potential of this approach.60
However, nanoemulsions can be also used in combination therapies with PDT, photothermal therapy, and chemotherapy, as shown in Hou et al.61 They prepared a surfactant-free nanoemulsion composed of an amphiphilic porphyrin salt shell (pyropheophorbide a and mono-sodium salt) and an oil core consisting of glyceryl trioctanoate (PyroNewPS). The addition of hydrophobic drugs such as the chemotherapeutic PTX to the particle core then created a multifunctional nanoplatform (PyroNewPS@PTX) that can be used in combination phototherapy. The absorption spectrum of the PyroNewPS showed a redshift of 44 nm (671–715 nm) compared to the disrupted nanoemulsion, which was probably due to the presence of J-aggregates and π-interaction systems The PyroNewPS showed a stable photoacoustic signal at 715 nm. At the same time, an increase in temperature (>52 °C) was observed after laser photoactivation (660 nm) of the nanoemulsion. After some time, the nanoemulsion disintegrated in the cells and the photoacoustic signal and photothermal effect were suppressed. On the contrary, an increase in the fluorescence emission intensity and an augmented amount of singlet oxygen production were recorded, which indicates the presence of a PS in a monomeric form. The same phenomenon was observed also in vivo after PyroNewPS administration to mice bearing subcutaneous KB tumors. After 2 h from the administration, a strong photoacoustic signal was observed at 715 nm, after another 24 h, a decrease in this signal and, conversely, an increase of the fluorescence emission intensity at the tumor site was recorded. When PTX was added to the nanoemulsion oil core (PyroNewPS@PTX), the IC50 in KB, A549, and NCI-H460 cells was reduced 94, 85, and 24 times, respectively, when compared to Taxol®. The in vivo experiments showed a high potential of the nanoemulsions for efficient PTX delivery, with a significant decrease in tumor size observed in mice bearing KB tumors treated with PyroNewPS@PTX (7.2 mg kg−1 of PTX, two doses) compared to mice treated only with Taxol® (7.2 mg kg−1) or PyroNewPS. Unfortunately, the study does not provide any data on combination PDT/PA/chemotherapy that could enhance drug efficacy.61 A similar approach was used by Chang et al.,60 who used porphyrin lipids as the basic building blocks for the preparation of a combined PDT/chemotherapeutic drug. The porphyrin–lipid nanoemulsion containing PTX embedded in the oil core of the particle (PLNE-PTX) was stabilized by the addition of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino (polyethylene glycol)-2000] (DSPE–PEG 2000), which also prolonged the circulation time of PLNE-PTX in the blood plasma of experimental animals (BALB/c mice). Viability tests performed using KB cancer cells showed an increase in the PLNE-PTX cytotoxicity when photoactivated by a laser (671 nm, 10 J cm−2, 500 s), compared to Taxol® monotherapy, PLNE-PTX without light, or PLNE with photoactivation (no PTX). Interestingly, PLNE-PTX particles alone, even without photoactivation, led to a more significant reduction in KB cell viability (24.22 ± 0.74% of viable cells) than after using Taxol® alone at the same dose (65.77 ± 1.86% of viable cells). This could probably be caused by the more efficient delivery of PTX into cells. Even lower viability of only 8.75 ± 1.1% was measured for PLNE-PTX combined laser irradiation. In vivo tests in a KB-xenograft mouse model showed effective accumulation of PLNE-PTX in the tumor tissue (∼5.4 of injected dose (ID %) per gram of tissue) and 80% inhibition of tumor growth using the combination therapy. Neither the total weight of the animals nor other biochemical parameters were significantly changed after PLNE-PTX treatment.62
2.3. Docetaxel in combination chemo-/phototherapy
Another well-known and broadly used taxane chemotherapeutic is docetaxel (DTX). It is a semi-synthetic derivative of PTX, which is used as a cytostatic in the treatment of breast, lung, prostate, stomach, head, and neck cancer. It has been also used with good results in combination therapy with doxorubicin, trastuzumab, prednisone, or cisplatin.36 Due to its low bioavailability, caused by its limited solubility in water, new directions for its targeted delivery are being sought nowadays. Owing to its high potential in cancer treatment, DTX is a frequent choice for the preparation of multifunctional nanoplatforms with a combined photo-/chemotherapeutic effect, as, for instance, in the case of double-coated NPs prepared by Maiolino et al.63 These NPs consist of a negatively charged core composed of poly(lactide-co-glycolide) with DTX, followed by a layer of polyethyleneimine containing tetrasodium-meso-tetra(4-sulfonatophenyl) porphyrin (TPPS4) and an upper layer of HA for targeted delivery to cells with increased CD44 expression. Using human breast cancer cells MDA-MD-131 and MCF-7 which differ in the CD44 expression levels, higher uptake of DTX/TPP4–dcNPs was detected for MDA-MD-131 exhibiting pronounced CD44 expression. At the same time, they confirmed the importance of particle-based drug delivery, when the elimination of MDA-MD-131 cells was observed already at very low concentrations (0.01 μg mL−1) of DTX-dcNPs, compared to a similar amount of free DTX which does not affect the growth of MDA-MD-131 cells. A similar trend was observed in the case of DTX/TPP4–dcNPs-treated MDA-MD-131 cells, in which the IC50 was 0.008 μg mL−1 24 h after photoactivation (8 J cm−2, the light of 390–460 nm). When the drugs were co-administered in the free forms, the IC50 significantly augmented to a value of 0.16 μg mL−1 and, thus, lower cytotoxicity was detected 24 h after photoactivation.63 Various drug combinations with DTX are also discussed in studies by Gaio et al.64–66 They have proposed several new types of polymeric NPs for combined DTX photo-/chemotherapy. One of them is based on large 200 nm bilayer NPs, the hydrophobic core of which consists of poly(lactide-co-glycolide) (PLGA) with DTX. Around the NPs core, there is a layer of polyethyleneimine (PEI) into which, there is, based on electrostatic interactions, anchored a negatively charged PS meso-tetraphenyl chlorine disulfonate (TPCS2a, Fimaporfin). It is used as a light-triggered cytosolic delivery of endocytosed drugs that are unable to cross the cell plasma membrane. The top layer of the particles is formed by hyaluronan to target tumor cells with increased CD44 expression. The active substance content in the particles is in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 for DTX and TPCS2a, respectively, which roughly corresponds to the calculated optimal drug ratio (1:30) for the combined treatment of HeLa and MDA-MB-231 cells. In vitro cytotoxicity experiments showed that the DTX/TPCS2a-NPs lead to an increase in the therapeutic efficacy in comparison to DTX and TPCS2a in their free forms or simultaneous administration of DTX-NPs and TPCS2a-NPs. Higher cytotoxicity of the tested system was determined in CD44-positive MDA-MB231 cells, which are less sensitive to DTX treatment than HeLa cells. Moreover, fluorescence microscopy showed intracellular localization of TPCS2a-NPs in lysosomes of MDA-MB231 cells, suggesting preferential particle transport via CD44-mediated endocytosis. At the same time, the influence of free DTX, which led to a reduced entry of TPCS2a into cells, was co-administered with both drugs in the free form. No effect was observed when both drugs were co-administered in an NP form (DTX-NPs, TPCS2a-NPs). For both cell lines (HeLa, MDA-MB-231) co-delivery of DTX with TPCS2a loaded in separate NPs brings an advantage in cell toxicity concerning the co-administration of free forms of drugs. The highest inhibition of cancer cell growth was observed after treatment with DTX/TPCS2a-NPs, especially regarding MDA-MB-231. The potential DTX/TPCS2a-NPs in the treatment of multidrug-resistant cancer was evaluated using the prepared DTX-resistant HeLa cell line, the high toxicity of DTX/TPCS2a-NPs was detected as well as a significant increase in the synergistic effect of both drugs, compared to other conditions: free DTX with TPCS2a, DTX-NP with TPCS2a-NPs.64 Another vivid approach consists of NPs prepared by Gaio et al.,65 which are based on natural polymer keratin. They were designed for co-delivery of DTX with a Ce6 photosensitizer (DTX/Ce6-KNPs). The biological activity of these 133 nm-sized NPs was verified using DTX-sensitive HeLa cells as well as the derived DTX-resistant HeLa cell line (a cell monolayer and 3D spheroids). At the same time, NPs containing only the photosensitizer (Ce6-KNPs) and only the drug (DTX-KNPs) were prepared. Cell in vitro tests showed that when DTX is administered in the NP form, there is a significantly bigger decrease in HeLa cell viability compared to cells treated only with free DTX; the IC50s after 24 h treatment were 0.024 and 0.007 μg mL−1 for DTX and DTX-KNPs, respectively. In contrast, the phototoxicity (photoactivation conditions: 15 J cm−2, 600–800 nm) of free Ce6 does not significantly differ from Ce6-KNPs, the IC50s are 1.03 and 1.204 μg mL−1, respectively. Without photoactivation, Ce6 and Ce6-KNPs do not show a toxic effect. Likewise, empty KNPs and NPs do not affect HeLa cell viability under the same conditions tested. The use of DTX/Ce6-KNPs in combined photo-/chemotherapy has led to a reduction in cell viability and a reduction in cancer cell spheroid volume in both DTX-sensitive and DTX-resistant HeLa cells.65 The third type of NPs from the study of Gaio et al.66 are polymeric NPs (D,L-lactide-co-glycolide, PLGA) with DTX content in the core combined with a photosensitizer TPCS2a and hyaluronic acid surface treatment (HA-NPs). This type of NPs was developed to target breast cancer cells with increased CD44 expression, both in differentiated and cancer stem cell (CSC) forms. These are significant factors in the development of tumor metastases, their progression, and the resumption of disease after tumor treatment. Targeting and effective elimination of CSC thus becomes an important part of anticancer therapy. However, the removal of CSCs is very difficult due to their slow proliferation and the large number of efflux pumps they express. Gaio et al.66 described a promising approach for breast CSC eradication by combined photo-/chemotherapy using DTX/TPCS2a HA-NPs, which could be a breakthrough in this field. Breast tumor cell lines MCF-7 and MDA-MB-231 were selected for this study. These cell lines differ in the expression profile of CSC breast tumor markers CD44 and CD24, depending on whether they grow in a 2D monolayer or as 3D mammospheres. For MCF-7 cells growing in the 2D monolayer, only 1.7% of cells with breast CSC characteristics (CD44high/CD24low) were identified, in the case of first-generation mammospheres, 6% of the cells exhibited the given characteristics, in the case of second-generation mammospheres, it was 29% of MCF-7 cells. In contrast, the MDA-MB-231 cell line showed a 93% breast CSC-like phenotype already in the 2D monolayer and, thus, a high stemness potential. Based on the mammosphere forming efficacy of MDA-MB-231, a significant reduction in their ability to form mammospheres was observed after treatment of the cell monolayer with combination therapy using prepared DTX/TPCS2aHA-NPs (red light 600–800 nm, 15 mW cm−2) than in the case of chemotherapy (DTX/HA-NPs) or PDT (TPCS2a/HA-NPs, red light 600–800 nm, 15 mW cm−2) alone. No significant difference in the ability to form mammospheres was detected for adherently growing MCF-7 cells after combination therapy (DTX/TPCS2aHA-NPs) or monotherapy with DTX/HA-NPs. After treatment of the first generation of mammospheres by combination therapy (DTX/TPCS2aHA-NPs; the light of 600–800 nm, 15 mW cm−2), a significantly lower ability to produce a second generation of mammospheres was detected for both MCF-7 and MDA-MB-231 cells, compared to monotherapy (DTX/HA-NPs or TPCS2a/HA-NPs + red light 600–800 nm, 15 mW cm−2). Interestingly, DTX/HA-NPs monotherapy resulted in only a minimal reduction in second-generation MDA-MB-231 cell mammosphere formation, compared to combination therapy or PDT monotherapy. Using fluorescence microscopy, a significantly higher fluorescence emission intensity was observed 2 h after treatment with/TPCS2aHA-NPs, indicating a higher accumulation of NPs in the MDA-MB-231 mammospheres compared to MCF-7, probably caused by the efficient binding of NPs to CD44 and their rapid transport into cells. Prolonged incubation (24 h) of mammosphere with TPCS2aHA-NPs led to an increase in the TPCS2a fluorescence emission intensity even in the case of MCF-7 cells into which the NPs were probably nonspecifically endocytosed. The results published by Gaio et al. indicate the great potential of combined
photo-/chemotherapy not only for breast cancer but also for breast CSC elimination.66
:35 for DTX and TPCS2a, respectively, which roughly corresponds to the calculated optimal drug ratio (1:30) for the combined treatment of HeLa and MDA-MB-231 cells. In vitro cytotoxicity experiments showed that the DTX/TPCS2a-NPs lead to an increase in the therapeutic efficacy in comparison to DTX and TPCS2a in their free forms or simultaneous administration of DTX-NPs and TPCS2a-NPs. Higher cytotoxicity of the tested system was determined in CD44-positive MDA-MB231 cells, which are less sensitive to DTX treatment than HeLa cells. Moreover, fluorescence microscopy showed intracellular localization of TPCS2a-NPs in lysosomes of MDA-MB231 cells, suggesting preferential particle transport via CD44-mediated endocytosis. At the same time, the influence of free DTX, which led to a reduced entry of TPCS2a into cells, was co-administered with both drugs in the free form. No effect was observed when both drugs were co-administered in an NP form (DTX-NPs, TPCS2a-NPs). For both cell lines (HeLa, MDA-MB-231) co-delivery of DTX with TPCS2a loaded in separate NPs brings an advantage in cell toxicity concerning the co-administration of free forms of drugs. The highest inhibition of cancer cell growth was observed after treatment with DTX/TPCS2a-NPs, especially regarding MDA-MB-231. The potential DTX/TPCS2a-NPs in the treatment of multidrug-resistant cancer was evaluated using the prepared DTX-resistant HeLa cell line, the high toxicity of DTX/TPCS2a-NPs was detected as well as a significant increase in the synergistic effect of both drugs, compared to other conditions: free DTX with TPCS2a, DTX-NP with TPCS2a-NPs.64 Another vivid approach consists of NPs prepared by Gaio et al.,65 which are based on natural polymer keratin. They were designed for co-delivery of DTX with a Ce6 photosensitizer (DTX/Ce6-KNPs). The biological activity of these 133 nm-sized NPs was verified using DTX-sensitive HeLa cells as well as the derived DTX-resistant HeLa cell line (a cell monolayer and 3D spheroids). At the same time, NPs containing only the photosensitizer (Ce6-KNPs) and only the drug (DTX-KNPs) were prepared. Cell in vitro tests showed that when DTX is administered in the NP form, there is a significantly bigger decrease in HeLa cell viability compared to cells treated only with free DTX; the IC50s after 24 h treatment were 0.024 and 0.007 μg mL−1 for DTX and DTX-KNPs, respectively. In contrast, the phototoxicity (photoactivation conditions: 15 J cm−2, 600–800 nm) of free Ce6 does not significantly differ from Ce6-KNPs, the IC50s are 1.03 and 1.204 μg mL−1, respectively. Without photoactivation, Ce6 and Ce6-KNPs do not show a toxic effect. Likewise, empty KNPs and NPs do not affect HeLa cell viability under the same conditions tested. The use of DTX/Ce6-KNPs in combined photo-/chemotherapy has led to a reduction in cell viability and a reduction in cancer cell spheroid volume in both DTX-sensitive and DTX-resistant HeLa cells.65 The third type of NPs from the study of Gaio et al.66 are polymeric NPs (D,L-lactide-co-glycolide, PLGA) with DTX content in the core combined with a photosensitizer TPCS2a and hyaluronic acid surface treatment (HA-NPs). This type of NPs was developed to target breast cancer cells with increased CD44 expression, both in differentiated and cancer stem cell (CSC) forms. These are significant factors in the development of tumor metastases, their progression, and the resumption of disease after tumor treatment. Targeting and effective elimination of CSC thus becomes an important part of anticancer therapy. However, the removal of CSCs is very difficult due to their slow proliferation and the large number of efflux pumps they express. Gaio et al.66 described a promising approach for breast CSC eradication by combined photo-/chemotherapy using DTX/TPCS2a HA-NPs, which could be a breakthrough in this field. Breast tumor cell lines MCF-7 and MDA-MB-231 were selected for this study. These cell lines differ in the expression profile of CSC breast tumor markers CD44 and CD24, depending on whether they grow in a 2D monolayer or as 3D mammospheres. For MCF-7 cells growing in the 2D monolayer, only 1.7% of cells with breast CSC characteristics (CD44high/CD24low) were identified, in the case of first-generation mammospheres, 6% of the cells exhibited the given characteristics, in the case of second-generation mammospheres, it was 29% of MCF-7 cells. In contrast, the MDA-MB-231 cell line showed a 93% breast CSC-like phenotype already in the 2D monolayer and, thus, a high stemness potential. Based on the mammosphere forming efficacy of MDA-MB-231, a significant reduction in their ability to form mammospheres was observed after treatment of the cell monolayer with combination therapy using prepared DTX/TPCS2aHA-NPs (red light 600–800 nm, 15 mW cm−2) than in the case of chemotherapy (DTX/HA-NPs) or PDT (TPCS2a/HA-NPs, red light 600–800 nm, 15 mW cm−2) alone. No significant difference in the ability to form mammospheres was detected for adherently growing MCF-7 cells after combination therapy (DTX/TPCS2aHA-NPs) or monotherapy with DTX/HA-NPs. After treatment of the first generation of mammospheres by combination therapy (DTX/TPCS2aHA-NPs; the light of 600–800 nm, 15 mW cm−2), a significantly lower ability to produce a second generation of mammospheres was detected for both MCF-7 and MDA-MB-231 cells, compared to monotherapy (DTX/HA-NPs or TPCS2a/HA-NPs + red light 600–800 nm, 15 mW cm−2). Interestingly, DTX/HA-NPs monotherapy resulted in only a minimal reduction in second-generation MDA-MB-231 cell mammosphere formation, compared to combination therapy or PDT monotherapy. Using fluorescence microscopy, a significantly higher fluorescence emission intensity was observed 2 h after treatment with/TPCS2aHA-NPs, indicating a higher accumulation of NPs in the MDA-MB-231 mammospheres compared to MCF-7, probably caused by the efficient binding of NPs to CD44 and their rapid transport into cells. Prolonged incubation (24 h) of mammosphere with TPCS2aHA-NPs led to an increase in the TPCS2a fluorescence emission intensity even in the case of MCF-7 cells into which the NPs were probably nonspecifically endocytosed. The results published by Gaio et al. indicate the great potential of combined
photo-/chemotherapy not only for breast cancer but also for breast CSC elimination.66
Another combination of the DTX chemotherapeutic, a near-infrared emitting photosensitizer IR780, was chosen for the treatment of breast cancer by Yang et al.67 The surface of the NPs was coated with a red blood cell membrane to form NPs 150 nm in diameter (IR780/DTX PCEC@RBC). The NPs were designed not only for combined photo-/chemotherapy but also for the photochemical properties of IR780, photothermal therapy, fluorescence, and photoacoustic imaging. Cytotoxicity assays performed using the MCF-7 cells did not show a significant difference in toxicity of DTX PCEC, IR780/DTX PCEC@RBC, IR780/DTX PCEC (without RBC membrane) without laser irradiation. In contrast, a significant reduction in MCF-7 cell viability was detected when the cells were treated with IR780 PCEC, IR780/DTX PCEC@RBC, or IR780/DTX PCEC and simultaneously illuminated with a laser (808 nm, 1.5 W cm−2, 5min), especially in the case of IR780/DTX PCEC@RBC and IR780/DTX PCEC. Tests in healthy BALB/c mice, which followed, showed the prolonged circulation of IR780/DTX PCEC@RBCs in the bloodstream compared to IR780 PCEC and DTX PCEC alone. In mice with implanted MCF-7 xenografts, based on fluorescence emission intensity, the highest accumulation of IR780/DTX PCEC@RBC was detected in the tumor tissue. At the same time, the highest photoacoustic signal was detected in the tumor tissue after IR780/DTX PCEC@RBC treatment, and in the case of laser treatment, the temperature at the tumor site increased from 34 to 50 °C, thus indicating a high PTT potential of the drug. The high therapeutic effect of IR780/DTX PCEC@RBC (DTX and IR780 amount corresponding to 10 and 1.67 mg kg−1) was also noted; in combination with laser treatment, complete tumor elimination occurred 42 days after the treatment. Without photoactivation, tumor regrowth was observed 24 days after IR780/DTX PCEC@RBC administration, and a 45% reduction in tumor size compared to the control group (saline treatment) size was assessed 42 days after treatment. At the same time, no negative impact on total mouse weight was detected, indicating low systemic toxicity and good biocompatibility of IR780/DTX PCEC@RBC.67
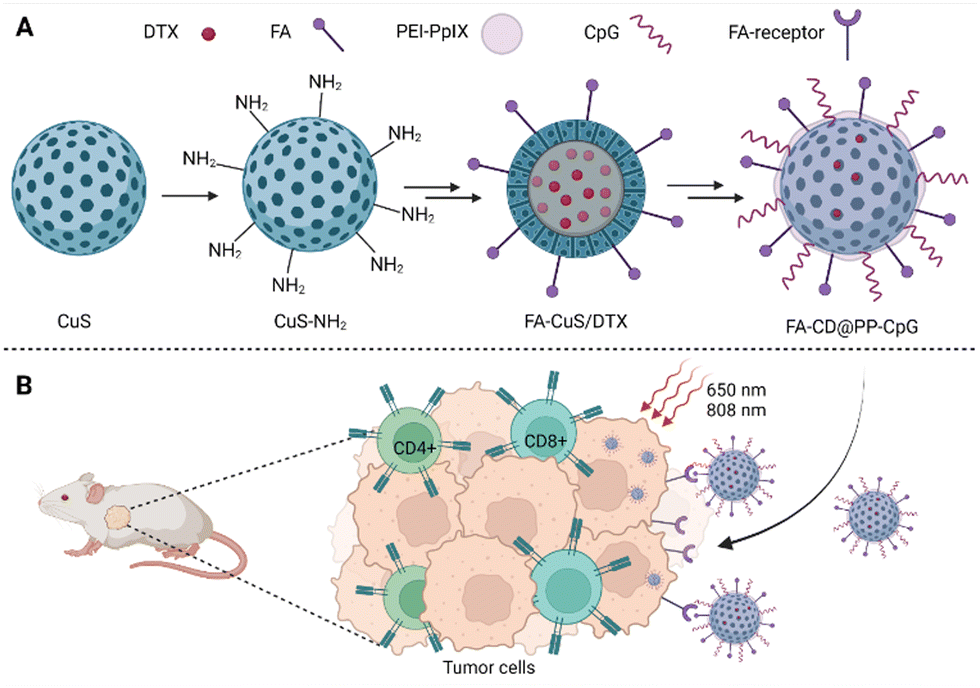 | ||
| Fig. 5 (A) Composition of FA-CD@PP-CpG nanoparticles: the basis of nanoparticles is CuS, docetaxel (DTX) is incorporated into the core of the nanoparticles, polyethyleneimine–protoporphyrin IX (PEI–PpIX) was attached to the nanoparticle's surface together with folic acid (FA). Cytosine–phosphate–guanine oligonucleotides (CpG), specifically binding toll-like receptor 9 antigen-presenting cells, were used as an immunoadjuvant. (B) Schematic representation of the action of FA-CD@PP-CpG. Taken and edited from Chen et al., 2019.68 Created with BioRender.com. | ||
Another type of multifunctional platform, this time targeting castration-resistant prostate cancer, was introduced by Tan et al.69 They presented 120 nm large NPs based on gold nanostars, which served as DTX and PS IR820 carriers (the loading efficacy was 11.5% and 9.7%, respectively). The surface of the gold nanostars was coated with PEG to increase the particle solubility in water and also to bind the anti-CD133 antibody to ensure their specific targeting. Thanks to the gold nanostars and IR820, the possibility of monitoring the distribution of NPs using NIR fluorescence as well as photoacoustic imaging (PAI) was ensured. Using human prostate cancer cells PC-3, successful transport and intracellular accumulation of the NPs were verified already 1 h after treatment based on fluorescence emission measurements; the fluorescence intensity raised with prolonged incubation time. When the PC-3 cells were treated with NPs conjugated to an anti-CD133 antibody, higher fluorescence emission intensity was detected. Without photoactivation, the NPs had only a negligible impact on PC-3 cell viability (the concentration of the IR820 active substance in the NPs did not exceed 5 μg mL−1). When PC-3 cells were treated with NPs and photoactivated at the same time (808 nm, 1 min), there was a significant decrease in their viability, and a higher number of apoptotic cells was also detected. The anticancer effect of the NPs was determined also in vivo using BALB/c athymic nude mice with subcutaneously implanted PC-3 cells. Fluorescence emission at the tumor site was detected already 1 h after intravenous administration of the NPs; with prolonged time, a consistent increase in the fluorescence emission intensity was observed until 24 h when the NP distribution completely predominated in the tumor tissue. A similar result was obtained also for PAI imaging, for which, the highest fluorescence emission intensity was also determined 24 h after treatment. When evaluating the anticancer efficacy of the NPs, it was found that after photoactivation (808 nm, 0.8 W cm−2, 6 min for 1 min after every 3 min), there is a significant increase in the temperature at the tumor site (44.6 °C), which indicates a good PTT activity. The most effective tumor growth inhibition was achieved 21 days after initiation of the treatment with the prepared NPs + light. At the same time, no significant changes in animal weight or damage to other organs were measured in this group. The prepared NPs thus seem to be a suitable platform for combined PDT/PTT/CH therapy and F/PA imaging.69
For combined CH/PTT/immunotherapy, NPs were prepared based on DTX, which was encapsulated together with polysaccharides serving as immunomodulators (CSP) in protein NPs formed by zein (the main storage protein of corn seeds). The surface of the NPs was modified with a thin layer of green tea polyphenol (GTP) with FeIII, which served as a photothermal agent inducing the conversion of NIR laser radiation into heat and facilitating pH-responsive drug release. It was found that the prepared DTX-loaded Zein/CSP-GTP/FeIII NPs induced a concentration-dependent decrease in the viability of Lewis lung carcinoma (LLC) cells and, at the same time, only moderate cytotoxicity to the mammary epithelial cell line (HC11) was detected. The cytotoxicity of DTX-loaded Zein/CSP-GTP/FeIII NPs against LLC was further increased after photoactivation using a laser (808 nm, 1.2 W cm−2, 5 min), after which the DTX-loaded Zein/CSP-GTP/FeIII NPs reduced the viability of LLC cells at the highest rate in the comparison of DTX-loaded Zein NPs and DTX-loaded Zein/CSP NPs. At the same time, it was found that DTX-loaded Zein/CSP-GTP/FeIII NPs induce apoptosis and immunogenic cell death in LLC cells. In vivo tests with mice-bearing LLC cell xenografts showed that three doses of intravenous administration of DTX-loaded Zein/CSP-GTP/FeIII NPs in combination with photoactivation by a laser (808 nm, 2.0 W cm−2, 5 min, 24 h postinjection) led to complete elimination of the primary tumor without further regrowth 30 days after the first treatment. Interestingly, complete tumor elimination occurred even after treatment with Zein-GTP/FeIII NPs (without DTX). At the same time, the thermal camera revealed an increase in the temperature of the tumor tissue of mice treated with DTX-loaded Zein/CSP-GTP/FeIII NPs from 39.9 to 50.2 °C after only 3 min of photoactivation. After treating animals with photoactivated DTX-loaded Zein/CSP-GTP/FeIII NPs, a strong antitumor immune response and the ability to eliminate distant tumor lesions were also observed. A more than 80% reduction in the tumor size, compared to the control group, occurred after the administration of photoactivated DTX-loaded Zein/CSP-GTP/FeIII NPs also in the case of the second animal model 4T1 tumor-bearing mice. The results showed that the combined CH/I/PTT therapy mediated by DTX-loaded Zein/CSP-GTP/FeIII NPs (+laser) can promote intratumoral infiltration of T lymphocytes, regulate the release of antitumor immune cytokines, and generate a strong antitumor immune response.70
2.4. Cabazitaxel in combined chemo-/PDT a PTT
Another semisynthetic derivative belonging to taxanes is cabazitaxel (CBZ). It was approved in combination with prednisone for the treatment of metastatic castration-resistant prostate cancer in patients who relapsed after treatment with DTX.71 In a clinical trial, CBZ was also tested for the treatment of breast,72 ovary,72 head and neck cancer,72,73 and other types of cancer. Its advantage is in particular that it also acts on tumors previously resistant to taxane treatment. However, the wider use of CBZ is limited by several side effects due to high toxicity. This could be solved by modifying the structure of the CBZ, or by “hiding” the drug in another non-toxic structure. This was attempted by Pandya et al.,74 who presented NPs for combined photo-/chemotherapy consisting of the biodegradable polysaccharide chitosan (CS) conjugated to PS tetraphenyl chlorin (TPC) and CBZ. The prepared polymer (TPC–CS) forms self-assembled NPs in an aqueous medium, into which it was possible to incorporate the desired drug CBZ. Stability and high loading capacity were ensured due to π–π interactions of PS aromatic groups with the lipophilic drug. The TPC–CS polymer can also be used in combination with other drugs such as a chemotherapeutic mertansine. Tests with MDA-MB-231 and MDA-MB-468 breast cancer cells showed that both CBZ alone and CBZ–TPC–CS NPs inhibited cell growth by 50–60% without photoactivation. In addition, when cells treated with CBZ–TPC–CS NPs were photoactivated, their growth was completely suppressed. A similar result was achieved at the highest concentrations used in the case of TPC–CS NPs (without CBZ), which indicates a high PDT potential. Determination at lower concentrations indicates a synergistic effect of both active substances.74 Another possibility of applying CBZ was presented by Tian et al.,75 who prepared self-assembled polymer NPs made of a biocompatible and at the same time thermoresponsive polymer, intended for programmed thermo-/chemotherapy. The basis of the NPs was formed from amphiphilic copolymer blocks: PEG, acrylonitrile, and acrylamide [PEG-b-P(AAm-co-AN)], into which a heptamethinium dye IR780 and the chemotherapeutic CBZ were co-encapsulated (Fig. 6). After laser illumination of the thus prepared NPs (808 nm, 500 mW cm−2, 5 min), an increase in the temperature of the solution (up to 60 °C) was detected, as well as a rapid release of CBZ into the solution (92% when the temperature reached 50 °C, 19% at 37 °C). The entry of NPs into 4T1 cells was observed as early as 4 h after treatment, the cells treated with NPs also exhibited higher fluorescence emission intensity than that treated with IR780. Without photoactivation, significant toxicity of free CBZ (24 g mL−1) and IR780 (20 μg mL−1) was detected, with only 16 and 18% of 4T1 cells remaining viable after 24 h of treatment. After administration of NPs (corresponding to 24 μg mL−1 CBZ and 20 μg mL−1 IR780 concentration), more than 90% viability of 4T1 cells was maintained after 24 h. In contrast, a significant decrease in 4T1 viability was determined after administration of NPs and photoactivation (808 nm, 500 mW cm−2, 5 min), and the rate of phototoxicity increased with augmenting concentration of NPs 24 h after photoactivation. In vivo tests in nude mice with 4T1 cell xenografts showed accumulation of NPs in the tumor tissue 12 h after administration. By monitoring the surface temperature of the tumor, it was found that the average temperature reached 50 °C after only 3 min of photoactivation (808 nm, 500 mW cm−2). By monitoring the tumor size, the highest decrease in tumor size was found after the application of NPs + laser 16 days after initiation of the treatment. Further, tumor shrinkage was also detected in groups, to which IR780 or CBZ were administered alone, however, the efficacy was lower. In contrast, in the group in which NPs were administered without photoactivation, no significant decrease in tumor size occurred compared to the control (PBS). The results, therefore, show a significant benefit of combined PTT/CH.75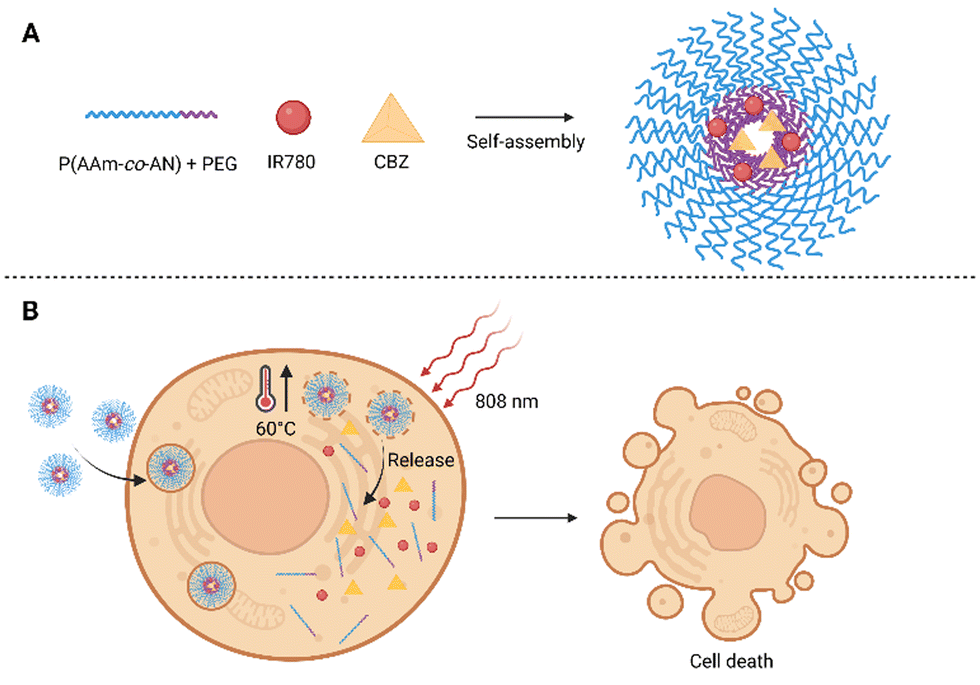 | ||
| Fig. 6 (A) Composition of polymeric self-assembled nanoparticles. The nanoparticles are based on blocks of amphiphilic co-polymers composed of acrylonitrile and acrylamide [P(AAm-co-AN)] and polyethylene glycol (PEG), the heptamethinium dye IR780 and cabazitaxel (CBZ) are encapsulated inside. (B) After photoactivation (808 nm) of cells treated with the nanoparticles, the temperature rises rapidly up to 60 °C, the nanoparticles disintegrate and the CBZ chemotherapeutic is released. Taken and edited from Tian et al., 2019.75 Created with BioRender.com. | ||
:Ce6, 1:1). Fluorescence microscopy using human melanoma cells A375 showed the localization of NPs in the lysosomes of the cells, suggesting their transport by endocytosis. NP-treated cells also showed a significantly higher fluorescence emission intensity than free Ce6, which indicates easier transport of NPs into the cells. Toxicity tests showed a significant decrease in A375 cell viability after treatment with NPs followed by light irradiation (660 nm, 300 mW cm−2, 5 min), compared to nonphotoactivated samples (IC50 dark = 84.0 ± 5.2 nM, IC50 light = 47.2 ± 1.6 nM). The cytotoxicity mechanism of NP action was subsequently confirmed by microscopic analysis. After photoactivation of the NPs localized in A375 cells, the thioketal bond is cleaved by ROS and free CBZ is released. This causes the polymerization of tubulin fibers and the formation of multinucleated cells, which can lead to cell apoptosis. Owing to the combination with PDT, the antitumor effect of the treatment is further enhanced. Acute toxicity tests in healthy mice showed good tolerance of NPs up to 50 mg kg−1, which is ten times more than the co-administration of free drugs (CBZ, Ce6). At the same time, no significant decrease in white blood cell count (neutropenia), which is a characteristic side effect of CBZ administration, was observed after the NP's application. The anticancer effects of the NPs were investigated using BALB/c mice with subcutaneously implanted tumor tissue collected from the patient's lymph nodes. In the case of NP treatment in conjunction with laser treatment (660 nm, 10 min, 600 mW cm−2), the highest decrease in tumor volume was recorded 21 days after initiation of the treatment (day 0 = 82.2 ± 41.4 mm3, day 21 = 21.5 ± 23.9 mm3). In addition, in three of the six mice, complete tumor elimination occurred. Immunohistological analysis showed tumor damage by apoptosis and necrosis, without concomitant damage to other tissues (liver, kidney). The prepared NPs thus combine the high potential of CBZ and Ce6 while suppressing side effects, the rapid release of free CBZ is supported by the cleavage of the thioketal bond between CBZ dimers.76 Another study presents a carrier-free system based on CBZ heterodimers connected by a thioketal bond using hydrogen bonds, hydrophobic and π–π interactions, formed with PPa NPs, the surface of which was modified by PEG. To reach an ideal synergistic effect, a CBZ:PPa ratio in NPs was determined 1:2. Using KB and mouse 4T1 cells, cytotoxicities of the free CBZ form and the prepared CBZ/PPa NPs were determined. Without photoactivation, the CBZ and PPa mixture showed comparable cytotoxicity to free CBZ. In contrast, CBZ/PPa NPs exhibited lower cytotoxicity than free CBZ (nonphotoactivated). On the contrary, after photoactivation (660 nm, 60 mW cm−2, 5 min), CBZ/PPa NPs and a mixture of free CBZ with PPa showed higher cytotoxicity compared to free CBZ after 48 and 72 h of treatment. The combined chemotherapeutic and PDT effect of NPs with CBZ and PPa was verified in vivo using 4T1 tumor-bearing BALB/c mice. Free CBZ and a mixture of CBZ with PPa (3 and 3.8 mg kg−1, respectively) without photoactivation showed comparable antitumor efficacy (tumor growth suppression compared to PBS-treated control) as free photoactivated PPa (660 nm, 100 mW cm−2, 5 min). The best result was achieved after the application of CBZ/PPa NPs with photoactivation (660 nm, 100 mW cm−2, 5 min), upon which the tumor growth was suppressed and its size decreased on day 15 of the treatment compared to the size before the start of treatment. During treatment with CBZ/PPa NPs, no significant change in animal weight or major organ damage was detected. Conversely, the tumor tissue showed significant damage with necrotic and apoptotic areas, thus indicating the high efficacy of CBZ/PPa NPs therapy combined with photoactivation.77
Different types of ROS-responsive linkers were used in other types of NPs. These self-assembled NPs contained a thioether or selenoether linker linking CBZ to oleic acid, as well as a PPa PS. To improve the solubility of the NPs in water and prolong their circulation in the bloodstream, the structure of the NPs was supplemented with 20% (w/w) 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000]. In total, there were prepared three types of NPs, which differed in the linkers, i.e., without a ROS-responsive linker, with a thioether linker, and with a selenoether one; the NPs were called PPa@CBZ-OA/DSPE-PEG2k, PPa@CBZ-S-OA/DSPE-PEG2k, and PPa@CBZ-Se-OA/DSPE-PEG2k, respectively. The binding capacity of the drug in all three types of NPs reached 50.1–60.3%. The ability of the NPs to cross the cell plasma membrane was determined microscopically; a higher fluorescence emission intensity was observed in 4T1 cells 2 h after NPs administration compared to free PPa administration. Furthermore, 4T1 cells treated with PPT@CBZ-Se-OA/DSPE-PEG2k and photoactivated exhibited higher ROS level production than cells treated with PPa@CBZ-S-OA/DSPE-PEG2k and photoactivated. In vitro tests then showed negligible cytotoxicity of nonphotoactivated NPs, and, conversely, a significant decrease in 4T1 cell viability after laser irradiation (660 nm, 60 mW cm−2, 8 min). The IC50 values of photoactivated PPa@CBZ-S-OA/DSPE-PEG2k and PPa@CBZ-Se-OA/DSPE-PEG2k reached 40.06 and 5.68 nM (48 h after photoactivation), respectively. This difference in IC50s was probably caused by elevated CBZ release from PPa@CBZ-Se-OA/DSPE-PEG2k due to higher ROS production. To study the photo-/chemotherapeutic effect of the NPs, they were intravenously administered (5 injections at a dose equivalent to 6 mg kg−1 of CBZ) into BALB/c mice with subcutaneously implanted 4T1 xenografts. At 12 h after the NP administration, a portion of the mice was exposed to laser radiation (660 nm, 0.1 W cm−2, 12 min), and the highest inhibition of tumor size was subsequently observed in this group, especially in mice treated with the redox-sensitive PPa@CBZ-S-OA/DSPE-PEG2k and PPa@CBZ-Se-OA/DSPE-PEG2k. To a lesser extent, a reduction in tumor size was measured after treatment with PPa@CBZ-S-OA/DSPE-PEG2k and PPa@CBZ-Se-OA/DSPE-PEG2k even without subsequent photoactivation. The tumor growth was not affected in the control groups (PBS, free PPa) and surprisingly, only slight tumor suppression was detected after administration of PPa@CBZ-OA/DSPE-PEG2k (with and without light), probably due to the strong ester bond between CBZ and OA. The results thus show a significant synergistic effect of endogenously and PDT-produced ROS.78
Another type of photoactivatable nanosystem combines two molecules, CBZ and PS Ce6 linked via a ROS-responsive thioketal linker (TkdC). The novel nanosystem (termed psTKdC) is highly stabilized by intermolecular interactions between Ce6 and TkdC (e.g., hydrogen bond, π–π stacking, or hydrophobic interactions). The stability of psTKdC was confirmed by no significant variations in particle sizes when stored in water at room temperature for one month. On the other hand, TKdC alone was stable for only a few hours under the same condition. High stability, efficient drug loading capacity (97.3 and 91.95% for CBZ and Ce6, respectively), and high ROS production predict psTKdC as a potent photo-/chemotherapeutic. The potential of psTKdC in cancer treatment was verified in vitro using a human melanoma cell line (A375). Fluorescence microscopy showed higher fluorescence emission intensity of psTKdC than of free Ce6, at the same concentration of 16 μmol L−1 (after 6 h) and its localization in the lysosomes of A375 cells. The synergistic photo-/chemotherapy was exhibited by a significant decrease in viability of A375 cells treated with psTKdC (0.15–1200 nmol L−1) followed by photoactivation (660 nm, 300 mW cm−2, 5 min). After 42 h treatment, the IC50 of photoactivated psTKdC was equal to 47.2 ± 1.6 nM, which was significantly lower than without photoactivation 84.0 ± 5.2 nM. In vivo tests in immunocompromised BALB/c nude mice implanted with patient-derived melanoma cells showed a preferential accumulation of psTKdC in the tumor tissue (amount of drug per gram of tissue 35.85 ± 10.37 μg g−1) 24 h post-administration and also a longer circulation time of the drug in the bloodstream than free CBZ. In addition, the authors evaluated also the therapeutic potential of the psTKdC by monitoring the changes in tumor size in different mice groups. The first group were mice administered with 7 mg kg−1 of psTKdC followed by photoactivation (660 nm, 300 mW cm−2, 10 min), in which a significant reduction in the tumor size, without a concomitant decrease in the animal weight, was measured. Unfortunately, a similar result was achieved also without photoactivation. A better outcome was accomplished after the administration of 14 mg kg−1 of psTKdC + photoactivation, which resulted in a remarkable reduction of the tumor volume from 82.2 ± 41.4 mm3 (first day of the treatment) to 21.5 ± 23.9 mm3 on day 21. In the same group, in three of the six mice, complete tumor elimination with no obvious damage to organs occurred. In comparison to the other drug combination with photoactivation, i.e., TKdC with free Ce6 or free CBZ with free Ce6, psTKdC was more effective in tumor size reduction (219.9 ± 130.0 mm3; 339.2 ± 93.5 mm3; 337.3 ± 198.9 mm3, respectively). The combination of free drugs (CBZ with Ce6) leads to systemic toxicity manifested by animal weight loss (20.2%) compared to other groups. These results show the good therapeutic potential of psTKdC, for melanoma therapy with the simultaneous suppression of side effects and the possibility of applying higher doses of the drug.79
2.5. Use of nanoparticles with taxanes in clinical practice
The use of NP-based drugs is not only part of preclinical research but also belongs to clinically approved methods for the treatment and diagnosis of numerous types of cancer. Among the first clinically approved chemotherapeutics based on NPs, there belong medicinal formulations containing taxanes as active ingredients. The first clinically approved nano-chemotherapeutic agent in this sense was the liposomal form of DOX (Doxil)80 used for the treatment of ovarian cancer, Kaposi's sarcoma, and melanomas. Next, for the treatment of metastatic breast cancer, pancreatic cancer, non-small cell lung carcinoma, and also other types of cancer, albumin-based NPs with bound PTX (Abraxane)81 have been approved. The great potential of nanomedicine has been also evidenced by the fact that several drugs formulated in NPs have been currently undergoing clinical evaluation for the treatment of different cancer types. An overview of clinical studies dealing with the use of NPs in anticancer therapy (chemotherapy, immunotherapy, radiotherapy, PDT, and PTT) was recently summarized in detail by Jing et al.82 To the best of our knowledge, only aminolevulinic acid nanoemulsion (BF-200 ALA) for the treatment of basilioma (NCT02367547)55 is currently in clinical evaluation for its use in PDT. None of the multifunctional NPs using combined CH/PDT/PTT strategies have yet entered clinical evaluation. Given that the number of scientific publications dealing with the use of NPs in anticancer PDT and PTT has increased more than eight times83 over the last ten years, we assume that the number of clinical studies will also raise.3. Prospective future of nanoparticles with taxanes
It is clear that the future of successful anticancer therapy depends not only on the development of completely new drugs but also on the search for novel, more suitable drug formulations and combinations of already known drugs. Chemotherapy combined with PDT and/or PTT, in the form of prodrugs or nanoformulations, has great therapeutic potential. Owing to the knowledge of the pharmacophores of PTX and other taxanes, it is possible to modify these molecules in a targeted manner in order to create prodrugs. In combined CH/PDT, the combination of two active molecules, i.e. a chemotherapeutic with a PS, into one molecule is highly advantageous. This ensures the transport of both active molecules into the cells at the same time, which is superior to the simultaneous administration of two drugs in their free forms. The drawback, in this case, can be the increased size of the resulting molecule and also changed steric properties, due to which the newly prepared drug can be transported into cells with higher difficulty. However, this can be avoided, for example, by optimization of the spacer length between the active parts of the molecule. And the use of a suitable spacer can also affect water solubility and other properties of the resulting molecule.42 What is an interesting option, here, is the use of cleavable linkers sensitive to changes in pH, the presence of singlet oxygen, and ROS inside cells or are activable by light. Owing to this possibility, the prepared prodrugs can be activated in cancer cells in a targeted manner, and thus, the side effects of the treatment can be reduced.41 What is questionable, though, is the stability of the drugs prepared in this way and the possibility of their long-term storage.Like the preparation of prodrugs, the use of nanotechnology is undoubtedly a vivid option for the preparation of novel drug forms. The increasing popularity of NPs has been mainly due to their relatively facile and economically feasible preparation, low cytotoxicity, and available surface modifiability. At the same time, however, NPs are highly reactive due to their large surface area to weight ratio, they can heavily bind to proteins present in plasma and can be efficiently eliminated from the body. However, this can be prevented by proper NP surface modification, which cannot only be a vital way to control their size but also affect their circulation time in the bloodstream, bioavailability, pharmacokinetics, and specific targeting. As for the NP surface modification, PEG is often being used, however, it can be also utilized as a spacer for binding of the functional groups,48,49,60,69,75,76,78 which increases the hydrophilic nature of the particles, augments their stability, and protects rapid removal of the used drugs from the body. Even though PEG is a clinically approved polymer that should be nontoxic, non-immunogenic, and non-antigenic, it has turned out that its long-term administration can lead to accumulation in healthy tissues and undesired side effects. The formation of specific anti-PEG antibodies has also been reported. Such antibodies can not only lead to reduced treatment efficacy but also cause undesired immune reactions. However, it always depends on the specific molecular weight of the PEG applied, its amount, and the method of its use. These aforementioned PEG-related issues were summarized in detail in several review articles by Ivens et al.,84 Baumann et al.,85 and others.86,87
Besides the PEG surface modification of NPs, what is also frequent is the use of HA.63,64,66 In addition to reduced NPs uptake by macrophages, HA also serves to selectively target the NPs to cells expressing the CD44 receptor. Thanks to this, conjugation of NPs with another targeting molecule is not necessary when HA is used.
The indisputable advantage of various types of polymeric as well as solid NPs is that the drugs used do not need to be modified before encapsulation and, thus, their properties do not change. At the same time, several active substances can be encapsulated in NPs at once, for instance, a chemotherapeutic and a PS, which makes them a suitable tool for multimodal therapy. Therefore, the use of multimodal NPs seems to be a vital way how to overcome the increasingly occurring potential drug resistance.64 The use of biologically degradable polymeric materials as a basis for NPs, as opposed to inorganic materials, reduces the risk of undesired accumulation of NPs in tissues after drug release and potential side effects. NPs based on inorganic materials, however, bring advantageous physicochemical properties that can be used in photothermal conversion,68,69 and photoacoustic imaging.69 Each of the mentioned systems has its advantages and disadvantages and deciding which types of NPs are the best option for the treatment of individual cancer diseases will be shown in the future after proper clinical evaluation.
4. Conclusions
Combination therapy undoubtedly represents a significant benefit in cancer treatment.88 However, the combination of just two treatment approaches, such as resection of the tumor tissue and further treatment, for instance, with chemotherapy, is often not sufficient to fully eliminate the disease. Therefore, approaches in which multiple therapies act simultaneously prove to be much more effective. One such approach is to apply drug combinations of two or more compounds with different mechanisms of action in one particle. In this review, we presented the currently studied drug combinations with mitotic poisons, taxanes, and photoactivable compounds designated for photodynamic therapy. These drugs are often prepared in prodrug forms, NPs, or nanoemulsions, which are transported through the bloodstream with higher efficacy than the parent drugs alone. By “hiding” taxanes in the internal structures of NPs, their relatively high systemic toxicity is reduced. Taxanes are released after photoactivation of the PS, which leads to ROS production and cleavage of ROS-responsive moieties, such as the thioether bond or poly(propylene sulfide) group. The particular linker ensuring the lower toxicity of taxanes before photoactivation greatly affects the success of anticancer therapy in different tumors. However, also the size of NPs and their shape, as well as the proper timing of photoactivation play important roles in the treatment efficacy. Interestingly, owing to the convenient photochemical properties of PSs, they have potential not only in PDT and PTT,89 but also in fluorescent and photoacoustic imaging, for which the prepared drugs could be also potent diagnostic tools. Such “all-in-one” NPs are gaining increasing attention in the field of anticancer therapies and diagnostics and will for sure be further developed in the coming era.Abbreviations
| 4T1 | Mouse mammary carcinoma-derived cells |
| A375 | Human melanoma cell line |
| A549 | Human cells from lung carcinoma |
| AY-27 | Rat bladder carcinoma cells |
| BALB/c | An albino, the laboratory-bred strain of the house mouse |
| C | Cystamine linker |
| CAT | 4-Carboxy-1-methylpyridinium chloride |
| CAT-L-PTX | 4-Carboxy-1-methylpyridinium chloride-paclitaxel prodrug |
| CAuNCs | Gold nanoclusters |
| CBZ | Cabazitaxel |
| CBZ/PPa NPs | Nanoparticles based on cabazitaxel dimers with pyropheophorbide a |
| CD44 | Cluster of differentiation 44 |
| Ce6 | Chlorine e6 |
| Ce6-KNPs | Nanoparticles based on keratin with encapsulated chlorine e6 |
| CH | Chemotherapy |
| ChS-g-PPS | Polymer block based on chondroitin sulfate and poly(propylene sulfide) |
| ChS | Chondroitin sulfate |
| Colon-26 | Mouse cells from colon carcinoma |
| CP/ChS-g-PPS NPs | Nanoparticles with encapsulated chlorine e6 and PTX |
| CpG | Cytosine–phosphate–guanine oligonucleotides |
| CQE | Conjugate based on chondroitin sulfate, quercetin, and chlorine e6 |
| CS | Polysaccharide chitosan |
| CSC | Cancer stem cells |
| CSP | Polysaccharide with immunomodulating effect |
| DLI | Drug-light interval |
| DMSO | Dimethyl sulfoxide |
| dPPA | Hydrolysis-resistant D-peptide antagonist |
| DSPE-PEG 2000 | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] |
| DTX | Docetaxel |
| DTX/Ce6-KNPs | Nanoparticles based on keratin with encapsulated docetaxel and chlorine e6 |
| DTX PCEC | Nanoparticles based on poly(caprolactone) loaded with docetaxel |
| DTX/HA NPs | Polymeric nanoparticles with docetaxel in the core and hyaluronic acid surface treatment |
| DTX-KNPs | Nanoparticles based on keratin with encapsulated docetaxel |
| DTX/TPCS2a HA-NPs | Polymeric nanoparticles with docetaxel and tetrasodium-meso-tetra (4-sulfonatophenyl) porphyrin content in the core and hyaluronic acid surface treatment |
| FDA | Food and drug administration |
| FA | Folic acid |
| FA-CD@PP-CpG NPs | Nanoparticles based on CuS, with incorporated docetaxel, the surface is modified with polyethyleneimine–protoporphyrin IX and folic acid, cytosine–phosphate–guanine oligonucleotides are used as an immuno-adjuvant |
| FR | Folate receptor |
| GTP | Green tea polyphenol |
| H460 | Non-small-cell lung cancer cells |
| HA | Hyaluronic acid |
| hALA | Hexyl-5-aminolevulinic acid |
| HBL-100 | Noncancerous cell line established from human breast milk |
| HC11 | Noncancerous mammary epithelial cell line |
| HeLa | Human cells from cervical carcinoma |
| IC50 | Half-maximal inhibitory concentration |
| IR780 | Heptamethin cyanine dye; 2-[(2E)-2-[2-chloro-3-[(E)-2-(3,3-dimethyl-1-propylindol-1-ium-2-yl)ethenyl]cyclohex-2-en-1-ylidene]ethylidene]-3,3-dimethyl-1-propylindole |
| IR780/DTX PCEC | Nanoparticles based on poly(caprolactone) co-loaded with heptamethinium dye and docetaxel |
| IR780/DTX PCEC@RBC | Nanoparticles based on poly(caprolactone) co-loaded with heptamethinium dye and docetaxel, surface coated with red blood cell membrane |
| IR780 PCEC | Nanoparticles based on poly(caprolactone) heptamethinium dye |
| KB | Human cells from epidermal carcinoma |
| KNPs | Nanoparticles based on keratin (empty) |
| LLC | Lewis lung carcinoma cells |
| MCF-7 | Human cells from breast carcinoma |
| MCF-7/ADR | Human multidrug-resistant breast cancer cells |
| MDA-MD-131 | Human breast cancer cells |
| MDA-MD-468 | Human breast cancer cell line |
| NCI-H460 | Human cells from lung carcinoma |
| NewPS | Nanoemulsion with porphyrin shell |
| NP | Nanoparticle |
| OA | Oleic acid |
| PAI | Photoacoustic imaging |
| PBS | Phosphate-buffered saline |
| Pba | Pheophorbide a |
| PD-1 | Programmed cell death receptor 1 |
| PD-L1 | Programmed cell death receptor ligand 1 pathway |
| PDD | Photodynamic diagnosis |
| PDT | Photodynamic therapy |
| PEG | Polyethylene glycol |
| PEG-b-P(AAm-co-AN) | Co-polymer PEG, acrylonitrile, and acrylamide |
| PEI | Polyethyleneimine |
| PEI–PpIX | Polyethyleneimine–protoporphyrin IX conjugate |
| P-gp | P-Glycoprotein 1 |
| PLNE-PTX | Porphyrin–lipid nanoemulsion with paclitaxel |
| PLGA | Poly(lactide-co-glycolide) |
| PPa | Pyropheophorbide a |
| PPa-C-PTX | ROS-insensitive dimer with a carbon chain |
| PPa@CBZ-OA/DSPE-PEG2k | Nano drug delivery system composed of pyropheophorbide a, and cabazitaxel linked with oleic acid by ROS non-responsive linker |
| PPa@CBZ-S-OA/DSPE-PEG2k | Nano drug delivery system composed of pyropheophorbide a, and cabazitaxel linked with oleic acid by thioether linker |
| PPa@CBZ-Se-OA/DSPE-PEG2k | Nano drug delivery system composed of pyropheophorbide a, and cabazitaxel linked with oleic acid by selenoether linker |
| PPa-PEG2k | Pyropheophorbide a conjugated to a PEG polymer |
| PPa-S-PTX | Thioether bond-bridged heterotypic dimer |
| PpIX | Proto-porphyrin IX |
| PPS | Poly(propylene sulfide) |
| PS | Photosensitizer |
| PTT | Photothermal therapy |
| PTX | Paclitaxel |
| PTX/CQE NPs | Nanoparticles with quercetin, chlorine e6, cysteamine, and paclitaxel |
| PTX-S-OA | Self-assembled hydrophobic nanoparticles composed of PTX and oleic acid, linked by a thioether bond |
| PXTK | ROS-responsive cinnamaldehyde and thioacetal-based PTX dimers |
| PyroNewPS | Nanoemulsion with porphyrin shell co-loaded with pyropheophorbide a |
| PyroNewPS@PTX | Nanoemulsion with porphyrin shell co-loaded with pyropheophorbide a and paclitaxel |
| Q | Quercetin |
| RAW 264.7 | Mouse macrophages |
| RBC | Red blood cell membrane |
| ROS | Reactive oxygen species |
| RH | Mitochondriotropic cation rhodamine |
| RH-L-PTX | Rhodamine-paclitaxel prodrug |
| ROS | Reactive oxygen species |
| SKOV-3 | Human cells from ovarian carcinoma |
| TPC | Tetraphenylchlorin |
| TPCS2a | meso-Tetraphenyl chlorine disulfonate |
| TPP | Mitochondriotropic cation triphenylphosphonium |
| TPP-L-PTX | Triphenylphosphonium-paclitaxel prodrug |
| TPPS4 | Tetrasodium-meso-tetra (4-sulfonatophenyl) porphyrin |
| TPPS4/HA NPs | Polymeric nanoparticles with tetrasodium-meso-tetra (4-sulfonatophenyl) porphyrin and hyaluronic acid surface treatment |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported from EuroNanoMed 3 program project TEIGER funded under national scheme TACR Episolon project number TH73020001; the grant of Specific university research – grant No. A2_FPBT_2022_049 and A1_FPBT_2022_001. Image by story set on Freepik.com.References
- B. W. Henderson and T. J. Dougherty, Photochem. Photobiol., 1992, 55, 145–157 CrossRef CAS PubMed.
- L. Benov, Med. Princ. Pract., 2015, 24(suppl 1), 14–28 CrossRef PubMed.
- T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, JNCI, J. Natl. Cancer Inst., 1998, 90, 889–905 CrossRef CAS PubMed.
- B. J. Fenner, D. S. W. Ting, A. C. S. Tan, K. Teo, C. M. Chan, R. Mathur, I. Y. S. Yeo, T. Y. Wong, E. Y. M. Wong and C. M. G. Cheung, Ophthalmol. Retina, 2020, 4, 403–414 CrossRef PubMed.
- A. F. Cruess, G. Zlateva, A. M. Pleil and B. Wirostko, Acta Ophthalmol., 2009, 87, 118–132 CrossRef CAS PubMed.
- B. Karakullukcu, K. van Oudenaarde, M. P. Copper, W. M. Klop, R. van Veen, M. Wildeman and I. Bing Tan, Eur. Arch. Oto-Rhino-Laryngol. Head Neck, 2011, 268, 281–288 CrossRef PubMed.
- M. A. Biel, Photochem. Photobiol., 2007, 83, 1063–1068 CrossRef CAS PubMed.
- A. Sieron, G. Namyslowski, M. Misiolek, M. Adamek and A. Kawczyk-Krupka, Eur. Arch. Oto-Rhino-Laryngol. Head Neck, 2001, 258, 349–352 CrossRef CAS PubMed.
- H. Ikeda, T. Tobita, S. Ohba, M. Uehara and I. Asahina, Photodiagn. Photodyn. Ther., 2013, 10, 229–235 CrossRef CAS PubMed.
- M. J. Bader, H. Stepp, W. Beyer, T. Pongratz, R. Sroka, M. Kriegmair, D. Zaak, M. Welschof, D. Tilki, C. G. Stief and R. Waidelich, Urol. Oncol.: Semin. Orig. Invest., 2013, 31, 1178–1183 CrossRef CAS PubMed.
- J. Y. Lee, R. R. Diaz, K. S. Cho, M. S. Lim, J. S. Chung, W. T. Kim, W. S. Ham and Y. D. Choi, J. Urol., 2013, 190, 1192–1199 CrossRef PubMed.
- K. Inoue, S. Anai, K. Fujimoto, Y. Hirao, H. Furuse, F. Kai, S. Ozono, T. Hara, H. Matsuyama, M. Oyama, M. Ueno, H. Fukuhara, M. Narukawa and T. Shuin, Photodiagn. Photodyn. Ther., 2015, 12, 193–200 CrossRef CAS PubMed.
- G. Shafirstein, A. Battoo, K. Harris, H. Baumann, S. O. Gollnick, J. Lindenmann and C. E. Nwogu, Ann. Am. Thorac. Soc., 2016, 13, 265–275 CrossRef PubMed.
- M. B. Ericson, A.-M. Wennberg and O. Larkö, Ther. Clin. Risk Manage., 2008, 4, 1–9 CrossRef CAS.
- B. J. Osiecka, P. Nockowski and J. C. Szepietowski, Acta Derm.-Venereol., 2018, 98, 689–693 CrossRef CAS PubMed.
- Y. Xiaoqin, H. Chan, W. Long, X. Yuting, U. Keyal, Z. Guolong, W. Peiru and W. Xiuli, Technol. Cancer Res. Treat., 2018, 17, 1533033818820091 Search PubMed.
- E. A. Coors and P. von den Driesch, J. Am. Acad. Dermatol., 2004, 50, 363–367 CrossRef PubMed.
- J. A. Leman, D. C. Dick and C. A. Morton, Clin. Exp. Dermatol., 2002, 27, 516–518 CrossRef CAS PubMed.
- X.-L. Wang, H.-W. Wang, M.-X. Guo and S.-Z. Xu, Photodiagn. Photodyn. Ther., 2008, 5, 127–133 CrossRef PubMed.
- D. Bechet, S. R. Mordon, F. Guillemin and M. A. Barberi-Heyob, Cancer Treat. Rev., 2014, 40, 229–241 CrossRef PubMed.
- C. M. Moore, A.-R. Azzouzi, E. Barret, A. Villers, G. H. Muir, N. J. Barber, S. Bott, J. Trachtenberg, N. Arumainayagam, B. Gaillac, C. Allen, A. Schertz and M. Emberton, BJU Int., 2015, 116, 888–896 CrossRef CAS PubMed.
- D. Zaak, R. Sroka, M. Höppner, W. Khoder, O. Reich, S. Tritschler, R. Muschter, R. Knüchel and A. Hofstetter, Medical Laser: Appl., 2003, 18, 91–95 CrossRef.
- C. Adam, G. Salomon, S. Walther, D. Zaak, W. Khoder, A. Becker, O. Reich, A. Blana, R. Ganzer, S. Denzinger, G. Popken, R. Sroka, R. Knüchel-Clarke, J. Köllermann, G. Sauter, A. Hartmann, S. Bertz, M. Graefen, H. Huland, W. Wieland and C. G. Stief, Eur. Urol., 2009, 55, 1281–1288 CrossRef CAS PubMed.
- M. Osuchowski, D. Bartusik-Aebisher, F. Osuchowski and D. Aebisher, Photodiagn. Photodyn. Ther., 2021, 33, 102158 CrossRef CAS PubMed.
- Y. Matoba, K. Banno, I. Kisu and D. Aoki, Photodiagn. Photodyn. Ther., 2018, 24, 52–57 CrossRef PubMed.
- A. Maździarz, Photodiagn. Photodyn. Ther., 2019, 28, 65–68 CrossRef PubMed.
- L. Gu, M. Cheng, Z. Hong, W. Di and L. Qiu, Photodiagn. Photodyn. Ther., 2021, 33, 102172 CrossRef CAS PubMed.
- M. M. Kim and A. Darafsheh, Photochem. Photobiol., 2020, 96, 280–294 CrossRef CAS PubMed.
- T. M. Baran and T. H. Foster, Med. Phys., 2014, 41, 022701 CrossRef PubMed.
- M. Zhang, E. Liu, Y. Cui and Y. Huang, Cancer Biol. Med., 2017, 14, 212–227 CrossRef CAS PubMed.
- R. Jadia, C. Scandore and P. Rai, Int. J. Nanotechnol. Nanomed., 2016, 1 Search PubMed.
- Y. Chen, Y. Gao, Y. Li, K. Wang and J. Zhu, J. Mater. Chem. B, 2019, 7, 460–468 RSC.
- L. Pinto da Silva, C. M. Magalhães, A. Núñez-Montenegro, P. J. O. Ferreira, D. Duarte, J. E. Rodríguez-Borges, N. Vale and J. C. G. Esteves da Silva, Biomolecules, 2019, 9, 384 CrossRef CAS PubMed.
- J. Škubník, M. Jurášek, T. Ruml and S. Rimpelová, Molecules, 2020, 25, 4632 CrossRef PubMed.
- A. L. Risinger, S. M. Riffle, M. Lopus, M. A. Jordan, L. Wilson and S. L. Mooberry, Molecular Cancer, 2014, 13, 41 CrossRef PubMed.
- J. Škubník, V. Pavlíčková, T. Ruml and S. Rimpelová, Plants, 2021, 10, 569 CrossRef PubMed.
- Y. Zhang, W. Zhang, Y. Wang, J. Zhu, M. Zhou, C. Peng, Z. He, J. Sun, Z. Li and S. Gui, Biomaterials, 2021, 272, 120790 CrossRef CAS PubMed.
- M. Bio, K. M. M. Rahman, I. Lim, P. Rajaputra, R. E. Hurst and Y. You, Bioorg. Med. Chem. Lett., 2019, 29, 1537–1540 CrossRef CAS PubMed.
- P. Uehlinger, M. Zellweger, G. Wagnières, L. Juillerat-Jeanneret, H. van den Bergh and N. Lange, J. Photochem. Photobiol., B, 2000, 54, 72–80 CrossRef CAS PubMed.
- I. Cosserat-Gerardin, L. Bezdetnaya, D. Notter, C. Vigneron and F. Guillemin, J. Photochem. Photobiol., B, 2000, 59, 72–79 CrossRef CAS PubMed.
- P. Thapa, M. Li, R. Karki, M. Bio, P. Rajaputra, G. Nkepang, S. Woo and Y. You, ACS Omega, 2017, 2, 6349–6360 CrossRef CAS PubMed.
- M. Li, L. Nguyen, B. Subramaniyan, M. Bio, C. J. Peer, J. Kindrick, W. D. Figg, S. Woo and Y. You, J. Controlled Release, 2019, 308, 86–97 CrossRef CAS PubMed.
- R. Haddad, N. Alrabadi, B. Altaani and T. Li, Polymers, 2022, 14, 658 CrossRef CAS PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344–15349 CrossRef CAS PubMed.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2011, 6, 815–823 CrossRef CAS PubMed.
- C. Ren, H. Wang, D. Mao, X. Zhang, Q. Fengzhao, Y. Shi, D. Ding, D. Kong, L. Wang and Z. Yang, Angew. Chem., Int. Ed., 2015, 54, 4823–4827 CrossRef CAS PubMed.
- W. Yu, X. He, Z. Yang, X. Yang, W. Xiao, R. Liu, R. Xie, L. Qin and H. Gao, Biomaterials, 2019, 217, 119309 CrossRef CAS PubMed.
- C. Luo, B. Sun, C. Wang, X. Zhang, Y. Chen, Q. Chen, H. Yu, H. Zhao, M. Sun, Z. Li, H. Zhang, Q. Kan, Y. Wang, Z. He and J. Sun, J. Controlled Release, 2019, 302, 79–89 CrossRef CAS PubMed.
- B. Sun, Y. Chen, H. Yu, C. Wang, X. Zhang, H. Zhao, Q. Chen, Z. He, C. Luo and J. Sun, Acta Biomater., 2019, 92, 219–228 CrossRef CAS PubMed.
- X. Yang, X. Shi, Y. Zhang, J. Xu, J. Ji, L. Ye, F. Yi and G. Zhai, J. Controlled Release, 2020, 323, 333–349 CrossRef CAS PubMed.
- X. Shi, X. Yang, M. Liu, R. Wang, N. Qiu, Y. Liu, H. Yang, J. Ji and G. Zhai, Carbohydr. Polym., 2021, 254, 117459 CrossRef CAS PubMed.
- S. Ganta, M. Talekar, A. Singh, T. P. Coleman and M. M. Amiji, AAPS PharmSciTech, 2014, 15, 694–708 CrossRef CAS PubMed.
- R. J. Wilson, Y. Li, G. Yang and C.-X. Zhao, Particuology, 2021, 64, 85–97 CrossRef.
- E. Sánchez-López, M. Guerra, J. Dias-Ferreira, A. Lopez-Machado, M. Ettcheto, A. Cano, M. Espina, A. Camins, M. L. Garcia and E. B. Souto, Nanomaterials, 2019, 9, 821 CrossRef PubMed.
- ClinicalTrials.gov, Superficial Basal Cell Cancer's Photodynamic Therapy: Comparing Three Photosensitizers: HAL and BF-200 ALA Versus MAL, https://clinicaltrials.gov/ct2/show/NCT02367547, accessed 20.9.2022.
- ClinicalTrials.gov, Curcumin in Reducing Joint Pain in Breast Cancer Survivors With Aromatase Inhibitor-Induced Joint Disease, https://clinicaltrials.gov/ct2/show/NCT03865992, accessed 20.9.2022.
- ClinicalTrials.gov, A Safety and Immunogenicity of Intranasal Nanoemulsion Adjuvanted Recombinant Pandemic Flu Vaccine in Healthy Adults (IN-NE-rH5), https://clinicaltrials.gov/ct2/show/NCT05397119, accessed 20.9.2022.
- ClinicalTrials.gov, Topical Methotrexate Microemulsion in the Treatment of Plaque Psoriasis, https://clinicaltrials.gov/ct2/show/NCT04971239, accessed 20.9.2022.
- ClinicalTrials.gov, Evaluating the Efficacy and Safety of TJO-087 in Moderate to Severe Dry Eye Disease Patients, https://clinicaltrials.gov/ct2/show/NCT05245604, (20.9.2022).
- ClinicalTrials.gov, Nanoemulsion, https://clinicaltrials.gov/ct2/results?cond=&term=Nanoemulsion&cntry=&state=&city=&dist=, accessed 20.9.2022.
- W. Hou, J. W. H. Lou, J. Bu, E. Chang, L. Ding, M. Valic, H. H. Jeon, D. M. Charron, C. Coolens, D. Cui, J. Chen and G. Zheng, Angew. Chem., Int. Ed., 2019, 58, 14974–14978 CrossRef CAS PubMed.
- E. Chang, J. Bu, L. Ding, J. W. H. Lou, M. S. Valic, M. H. Y. Cheng, V. Rosilio, J. Chen and G. Zheng, J. Nanobiotechnol., 2021, 19, 154 CrossRef CAS PubMed.
- S. Maiolino, F. Moret, C. Conte, A. Fraix, P. Tirino, F. Ungaro, S. Sortino, E. Reddi and F. Quaglia, Nanoscale, 2015, 7, 5643 RSC.
- E. Gaio, C. Conte, D. Esposito, G. Miotto, F. Quaglia, F. Moret and E. Reddi, Mol. Pharmaceutics, 2018, 15, 4599–4611 CrossRef CAS PubMed.
- E. Gaio, A. Guerrini, M. Ballestri, G. Varchi, C. Ferroni, E. Martella, M. Columbaro, F. Moret and E. Reddi, J. Photochem. Photobiol., B, 2019, 199, 111598 CrossRef CAS PubMed.
- E. Gaio, C. Conte, D. Esposito, E. Reddi, F. Quaglia and F. Moret, Cancers, 2020, 12, 278 CrossRef CAS PubMed.
- Q. Yang, Y. Xiao, Y. Yin, G. Li and J. Peng, Mol. Pharmaceutics, 2019, 16, 4086 CrossRef CAS PubMed.
- L. Chen, L. Zhou, C. Wang, Y. Han, Y. Lu, J. Liu, X. Hu, T. Yao, Y. Lin, S. Liang, S. Shi and C. Dong, Adv. Mater., 2019, 31, 1904997 CrossRef CAS PubMed.
- H. Tan, N. Hou, Y. Liu, B. Liu, W. Cao, D. Zheng, W. Li, Y. Liu, B. Xu, Z. Wang and D. Cui, Nanomedicine, 2020, 27, 102192 CrossRef CAS PubMed.
- X. Yu, N. Han, Z. Dong, Y. Dang, Q. Zhang, W. Hu, Ch Wang, S. Du and Y. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 42988–43009 CrossRef CAS PubMed.
- G. Nightingale and J. Ryu, Pharmacy Therapy, 2012, 37, 440–448 Search PubMed.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01693549?term=cabazitaxel&draw=3&rank=17, accessed 6.11.2021.
- ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01620242?term=cabazitaxel&draw=4&rank=21, accessed 6.11.2021.
- A. D. Pandya, A. Øverbye, P. Sahariah, V. S. Gaware, H. Høgset, M. Masson, A. Høgset, G. M. Mælandsmo, T. Skotland, K. Sandvig and T.-G. Iversen, Biomacromolecules, 2020, 21, 1489–1498 CrossRef CAS PubMed.
- J. Tian, B. Huang, H. Li, H. Cao and W. Zhang, Biomacromolecules, 2019, 20, 2338–2349 CrossRef CAS PubMed.
- L. Huang, X. Chen, Q. Bian, F. Zhang, H. Wu, H. Wang and J. Gao, J. Controlled Release, 2020, 328, 325–338 CrossRef CAS PubMed.
- S. Zhang, Z. Wang, Z. Kong, Y. Wang, X. Zhang, B. Sun, H. Zhang, Q. Kan, Z. He, C. Luo and J. Sun, Theranostics, 2021, 11, 6019–6032 CrossRef CAS PubMed.
- B. Yang, K. Wang, D. Zhang, B. Sun, B. Ji, L. Wei, Z. Li, M. Wang, X. Zhang, H. Zhang, Q. Kan, C. Luo, Y. Wang, Z. He and J. Sun, Biomater. Sci., 2018, 6, 2965–2975 RSC.
- L. Huang, J. Wan, H. Wu, X. Chen, Q. Bian, L. Shi, X. Jiang, A. Yuan, J. Gao and H. Wang, Nano Today, 2021, 36, 101030 CrossRef CAS.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- M. R. Green, G. M. Manikhas, S. Orlov, B. Afanasyev, A. M. Makhson, P. Bhar and M. J. Hawkins, Ann. Oncol., 2006, 17, 1263–1268 CrossRef CAS PubMed.
- Z. Jing, Q. Du, X. Zhang and Y. Zhang, Chem. Eng. J., 2022, 446, 137147 CrossRef CAS.
- T. P. Pivetta, C. E. A. Botteon, P. A. Ribeiro, P. D. Marcato and M. Raposo, Nanomaterials, 2021, 11, 3132 CrossRef CAS PubMed.
- I. A. Ivens, W. Achanzarm, A. Baumann, A. Brändli-Baiocco, J. Cavagnaro, M. Dempster, B. O. Depelchin, A. R. I. Rovira, L. Dill-Morton, J. H. Lane, B. M. Reipert, T. Salcedo, B. Schweighardt, L. S. Tsuruda, P. L. Turecek and J. Sims, Toxicol. Pathol., 2015, 43, 959–983 CrossRef CAS PubMed.
- A. Baumann, D. Tuerck, S. Prabhu, L. Dickmann and J. Sims, Drug Discovery Today, 2014, 19, 1623–1631 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhang, S. Song, L. Yin, D. Sun and J. Gu, J. Sep. Sci., 2020, 43, 1978–1997 CrossRef CAS PubMed.
- L. Shi, J. Zhang, M. Zhao, S. Tang, X. Cheng, W. Zhang, W. Li, X. Liu, H. Peng and Q. Wang, Nanoscale, 2021, 13, 10748–10764 RSC.
- J. Škubník, V. S. Pavlíčková, T. Ruml and S. Rimpelová, Biology, 2021, 10, 849 CrossRef PubMed.
- V. Pavlíčková, J. Škubník, M. Jurášek and S. Rimpelová, Appl. Sci., 2021, 11, 2254 CrossRef.
| This journal is © The Royal Society of Chemistry 2023 |