
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in synthesis of water-stable metal halide perovskites and photocatalytic applications†
He
Zhao
 a,
Krisztian
Kordas
*b and
Satu
Ojala
*a
a,
Krisztian
Kordas
*b and
Satu
Ojala
*a
aEnvironmental and Chemical Engineering Research Unit, Faculty of Technology, University of Oulu, PO Box 4300, FI-90014 Oulu, Finland. E-mail: satu.ojala@oulu.fi
bMicroelectronics Research Unit, Faculty of Information Technology and Electrical Engineering, University of Oulu, P. O. Box 4500, FI-90014 Oulu, Finland. E-mail: krisztian.kordas@oulu.fi
First published on 9th October 2023
Abstract
Solar-driven photocatalytic reactions have attracted wide interest as a viable method to generate green energy and alleviate environmental challenges posed by fossil fuels. Although, various classes of photocatalysts have been explored during the past decades, the pursuit towards even more efficient ones is still ongoing. Metal halide perovskites (MHPs) have been recently proposed as novel photocatalysts owing to their wide light absorption range and excellent optoelectronic properties. However, the instability of MHPs in water is the main obstacle that impedes their applications in practice and prompts stabilization strategies to be developed. This review focuses on the recent approaches for stabilizing MHPs in water, including surface engineering, common-ion effect, and intrinsic water stability. The photocatalytic applications of water-stable MHPs are summarized and an outlook with perspectives over the current challenges are provided.
 He Zhao | He Zhao is a doctoral researcher in the Faculty of Technology at the University of Oulu. He received his BSc degree in Materials Chemistry at Zhengzhou University in 2014 and his MSc degree in Materials Physics and Chemistry from Xinjiang Technical Institute of Physics & Chemistry, University of Chinese Academy of Sciences in 2017. His current research interests include lead-free halide perovskites, semiconductor photocatalysis, 2D materials and photocatalytic hydrogen production. |
 Krisztian Kordas | Krisztian Kordas, MSc Physics and Chemistry (1998, Univ. Szeged), Dr Tech. in Microelectronics, Docent of Nanotechnology, Professor of micro- and nanoelectronic materials and components for ICT applications (2002, 2005, 2016, Univ. Oulu). He has led or participated in more than 20 national and international research projects, published 190+ papers, co-authored 6 book chapters, and supervised 11 doctoral students to graduation. His team's research is focused on synthesis, characterization, and implementation of nanostructured materials for electronics, sensors as well as for energy and environmental applications. |
 Satu Ojala | Satu Ojala gained her MSc in Chemical Engineering in 2000 and DSc (Tech) in Environmental Engineering in 2005 with Distinction from the University of Oulu, Finland. She worked as a postdoc at the University of Poitiers, France between 2006 and 2007, and at VTT Technology Research Center between 2008 and 2013. She gained her adjunct professorship in 2012 in the field of Environmental Catalysis from the University of Oulu. Her main field of research is Environmental Engineering, especially heterogeneous catalytic processes in gas and liquid phases. She has a special interest in in situ and operando spectroscopy applied in catalysis research. |
1 Introduction
During recent years, the exploration of greener energy sources has become of utmost importance to alleviate the inordinate reliance on non-renewable fossil fuel reserves and the consequent serious energy shortage and environmental pollution.1 Specifically, solar radiation, as an abundant, virtually endless, and green source of energy, has been seen as a promising option for the replacement of fossil fuels.2 In this context, the question of how to convert solar energy into fuels and value-added chemicals in an efficient and economic manner is a pivotal issue. So far, diverse solar-to-chemical energy conversion systems, including conventional heterogeneous photocatalysis, photoelectrochemical and combined photovoltaic-electrocatalytic systems have been proven to meet the requirements and developed for practical use.3 Compared with other reported systems, photocatalytic routes are probably the simplest ones and easiest to implement. For example, Domen's group has already designed a 100 m2 array of panel reactors to demonstrate hydrogen evolution from water over several months safely, with a maximum solar-to-hydrogen (STH) energy conversion efficiency of 0.76%.4 In a recent work of Zhou et al., the efficiency has been improved significantly to 9.2%,5 which is close to the proposed target for commercialization according to techno-economic analysis.6 However, compared with the latest 47.6% solar-to-electricity conversion efficiency of the state-of-the-art four-junction solar cells,7 there is still a long road ahead to achieve such values in photocatalytic systems.Exploring efficient, stable photocatalysts is thus one vital research field. In photocatalysis, the key component of the process is the photocatalyst (Fig. 1), whose role is to transform solar energy to charge carriers (electrons and holes) after light absorption, which then drift to the surface of the photocatalyst and participate in reduction/oxidation reactions.8 To date, several different photocatalysts either homogenous or heterogeneous have been proposed,9 including e.g., transition metal complexes,10 organic photoredox catalysts,9 titanium dioxide,11,12 carbon nitride,13,14 and perovskites.15 As most of the so far explored photocatalytic materials suffer from (i) limited photostability, high cost and potential toxicity,16 (ii) narrow utilizable solar spectral absorption range,17 (iii) sluggish charge carrier transport from the bulk of a photocatalyst to the surface and/or (iv) fast charge carrier recombination,11 there is a room to improve conversion efficiencies.18,19
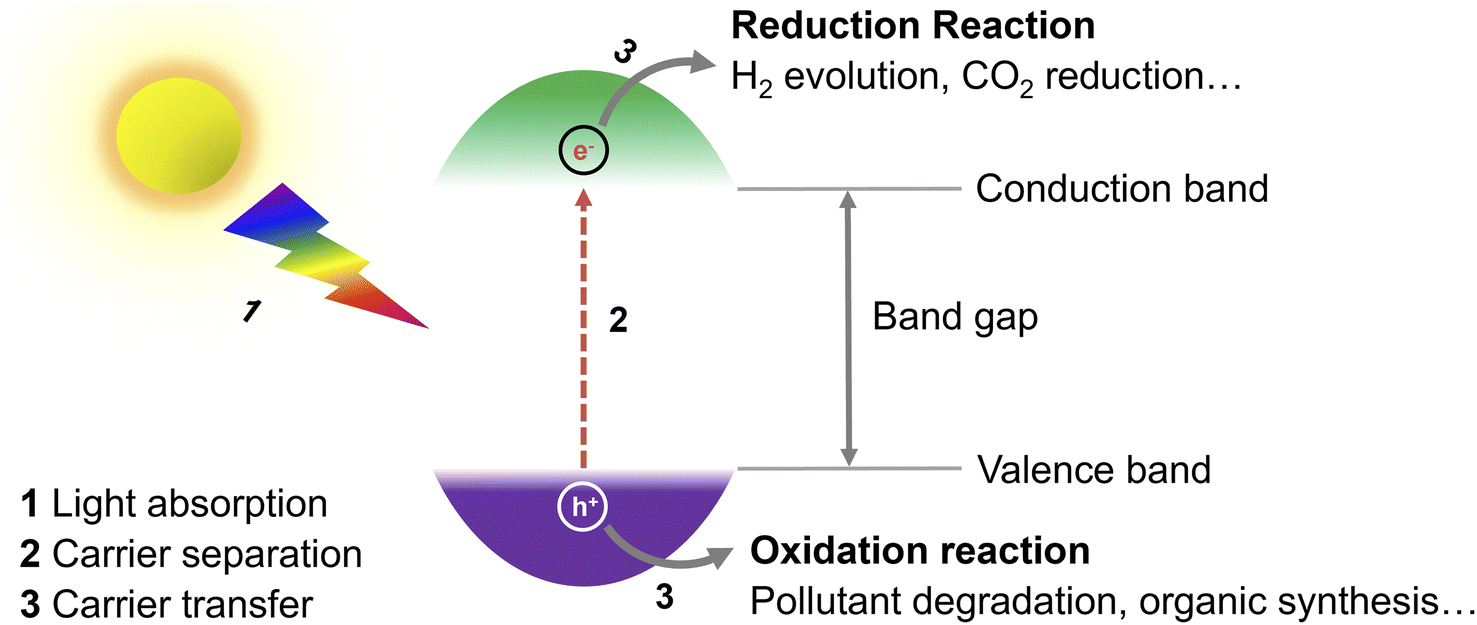 | ||
| Fig. 1 Basic mechanism of heterogeneous photocatalysis. | ||
The family of metal halide perovskites (MHPs) appears to offer features that tackle the aforementioned issues simultaneously. Organolead trihalide perovskites (e.g., CH3NH3PbI3) have emerged as novel high-performance photoactive absorbers with rocketing power conversion efficiency from 3.8% to certified 25.5% in just a few years.20,21 In general, the remarkable photovoltaic performance of MHPs originates from their high absorption coefficients,22,23 wide absorption window,20 long electron–hole diffusion lengths22,24 and high carrier mobility.25 All these excellent optical and charge-transport characteristics predestinate MHPs as ideal candidates for photocatalytic processes.26 However, every coin has two sides, and unfortunately MHPs are not exceptions either. The ionic nature of MHPs imparts their vulnerability toward moisture and polar solvents (especially water), which severely restricts their photocatalytic applications,27 and remains to be resolved. In 2016, Park et al. reported the pioneering work concerning the stabilization of CH3NH3PbI3 (MAPbI3) in aqueous HI solution by exploiting the common-ion effect; however, this happens under harsh conditions, at pH < −0.5 and −log[I−] < −0.4.28 Nevertheless, environmental concerns, costs, and the demand for practical conditions necessitate photocatalytic processes to be performed preferably in aqueous systems having less extreme chemistries.29 While MHP-based photocatalysts have been reviewed from different perspectives, such as photoredox organic synthesis,30–33 CO2 reduction,34–39 and H2 generation,40–47 strategies to synthesize water stable MHPs or their photocatalytic applications are not yet adequately summarized. Therefore, in this review, we survey and overview the recent progress in stabilizing MHPs against water, and discuss the encountered challenges and potential limitations of their applications, with the goal of providing new ideas and inspiration to advance the design new generations of efficient MHP-based photocatalytic systems.
2 Stabilization methods
Conventional lead-based MHPs are prone to decompose when exposed to moisture. It is reported that the perovskite solar cells lose more than 80% of their initial efficiency after one-day storage in ambient air, and only 5% is retained after 6 days.48 Encapsulation of the MHP films using materials having diffusion barrier properties have been proposed to protect the MHP solar cells from moisture hence maintaining more than 90% of their initial performance for almost 2 months.49 This is still far from the commercial long-term stability expected (25 years is typical for silicon solar cells),50 thus the challenge in the MHP development remains to be solved.To improve the water stability of MHPs, the role of water in the degradation of MHPs needs to be elucidated. The published results indicate that the MHPs adsorb water molecules quickly with a time scale of seconds51 and a single water molecule can accelerate the degradation of perovskite via acid–base reaction.52 What is even more troublesome, is that the water intake is not limited only to surface adsorption, but the water molecules also diffuse and penetrate the bulk and even infiltrate the MAPbI3 unit cells (Fig. 2). However, some studies claim that MHPs have a minor tolerance to water. For example, MAPbI3 was reported to remain intact when exposed to below 2 × 1010 Langmuir of H2O (one Langmuir equals to an exposure of about 1.33 × 10−4 Pa for one second).53 Only ∼1% volume expansion of the crystal structure is observed although water incorporated in the perovskite.54 These results are consistent with previous reports of a reversible process between the hydration and dehydration stages of MAPbI3.55,56 To figure out the detailed degradation mechanism under moist conditions, the dependence of spatially resolved external quantum efficiency (EQE) under various humidity exposure conditions has been investigated by using laser beam induced current mapping, and a four-stage degradation process has been proposed.57 When a tiny amount of water (1.6% H2O in N2) is introduced and kept for a short time (6 min), EQE increases marginally and reaches a maximum, which is caused by the solvation of CH3NH3+ (MA+) and I− ions,58 that heals some defects, reduces the trap density, and thus improves the uniformity of perovskite films (Stage 1). As the time increases, a slow drop in EQE is observed due to the change of the electronic structure and carrier mobility of hole-transporting materials (Stage 2). In the next stage (Stage 3), a sharp decrease of EQE indicates the breakdown of 3D structure, while monohydrated 1-dimensional (1D) chains of CH3NH3PbI3·H2O or 0-dimensional (0D) dots of dihydrates (CH3NH3)4PbI6·2H2O are formed (Stage 3). Finally, the degradation of MHPs results in the formation of CH3NH3I (MAI), PbI2 and water (Stage 4). In general, the decomposition starts at the surface, especially at the MAI-terminations.54 The loss of MA+ results in an open inorganic framework to form vacancies inside the crystal lattice that finally leads to a rapid deterioration or decomposition of the perovskite structure.59 The dissolution of I− ions in water is easier than that of MA+ ions since the hydrophobicity of –CH3 group in MA+ requires higher dissolution energy.60 These studies indicate that water molecules penetrate easily into the bulk of MHPs and the removal of ions (especially the I− ions due to the lower energy barrier) is the main cause of the degradation. Based on the observations above, thus, the main strategies to obtain water stable MHPs are: (i) preventing the structure from the contact of water, (ii) compensating for the depletion of ions from the surrounding, and (iii) reducing the solubility of organic cations. In these efforts, three different strategies have been proposed, including surface engineering, utilization of the common-ion effect, and enhancement of the intrinsic stability of perovskites.
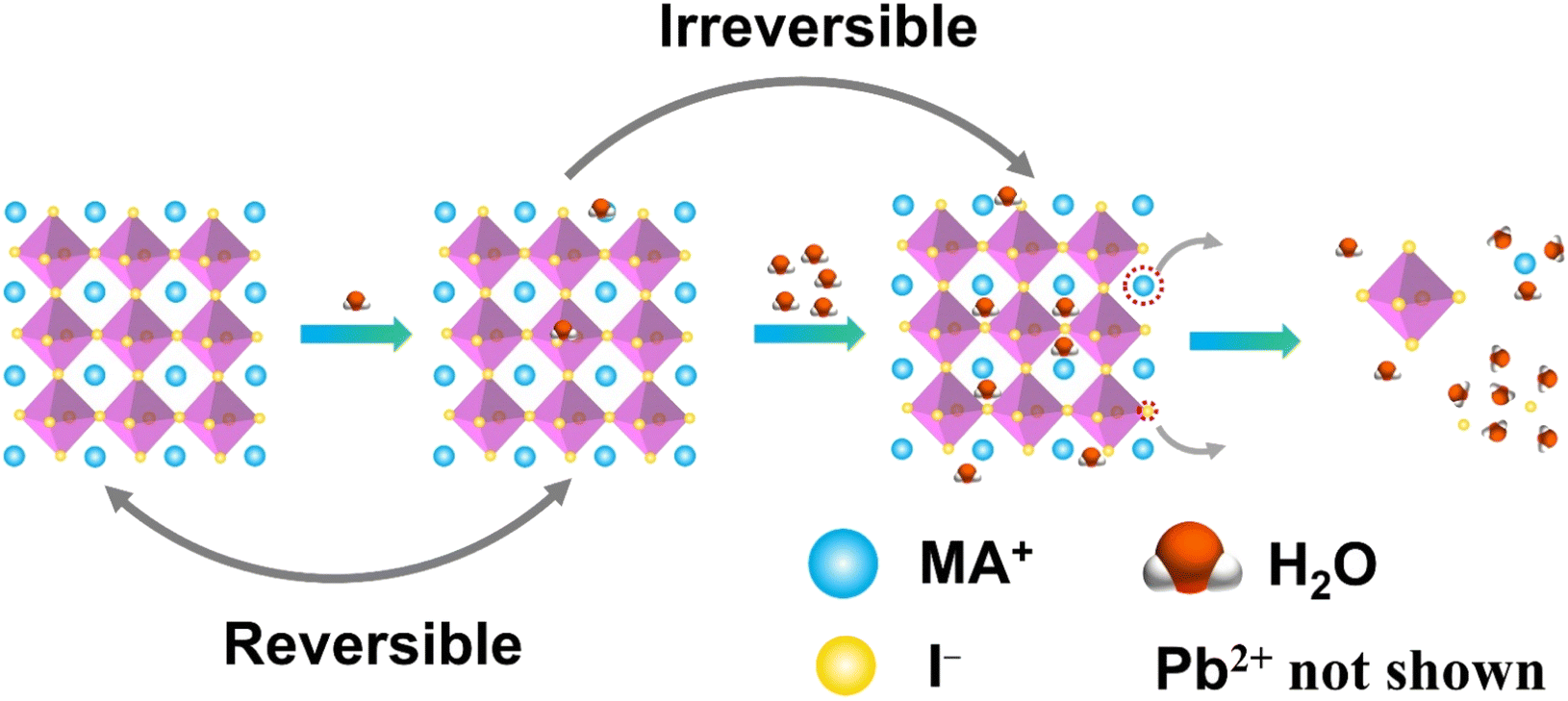 | ||
| Fig. 2 Schematic illustration of MAPbI3 decomposition in water. | ||
2.1 Surface engineering
Surface engineering has been explored to generate protecting layers on the MHPs, thus avoiding the direct contact of water with MHPs. In this section, six surface passivation methods are discussed: (1) organic ligands, (2) organic polymers, (3) inorganic materials, (4) metal–organic frameworks (MOFs), (5) phase engineering, and (6) water-assisted engineering.The introduction of hydrophobic ligands on the surface of MHPs is one of the most efficient and convenient approaches to stabilize MHPs. A post-surface functionalization with hydrophobic cations via simple ligand exchange process can substitute surface methylammonium (MA+) ions and enhance the stability of MHPs. Many studies have indicated that the presence of hydrophobic quaternary ammonium cations, such as tetra-methyl ammonium, (CH3)4N+;65–67 tetra-ethyl ammonium, (C2H5)4N+;68 tetra-butyl ammonium, (C4H9)4N+;69,70 and tetra-hexyl ammonium, (C6H13)4N+;71 have a vital influence on the moisture-stability of MHPs. These quaternary ammonium cations adsorb chemically on the surface of MHPs, and inhibit the water intake of the lattice thus keeping the perovskite films stable under 90 ± 5% relative humidity (RH) for more than 30 days without a photovoltaic loss. This happens because the bulky organic cations shift the surface Pb5C–I1c (I1c represents the surface I atom coordinated with one Pb atom) bonds owing to the steric effects and impede the water adsorption on the five-coordinated surface Pb atoms (Pb5c).71 In addition, these molecules can also suppress the iodide migration,72 evidenced by the shortened Pb–I bond (from 3.17 Å to 3.07 Å, Fig. 3a). Besides these factors, the formation of water-stable quaternary ammonium lead iodide shell may also contribute to the enhanced stability.66,73,74 For example, with (C4H9)4NI post-treatment over CsPbI3, the (C4H9)4N+ cations can intercalate the inorganic framework of MHP and exchange Cs+ ions, forming a one-dimensional (C4H9)4NPbI3 layer75 exhibiting intrinsic water stability (Fig. 3b).76
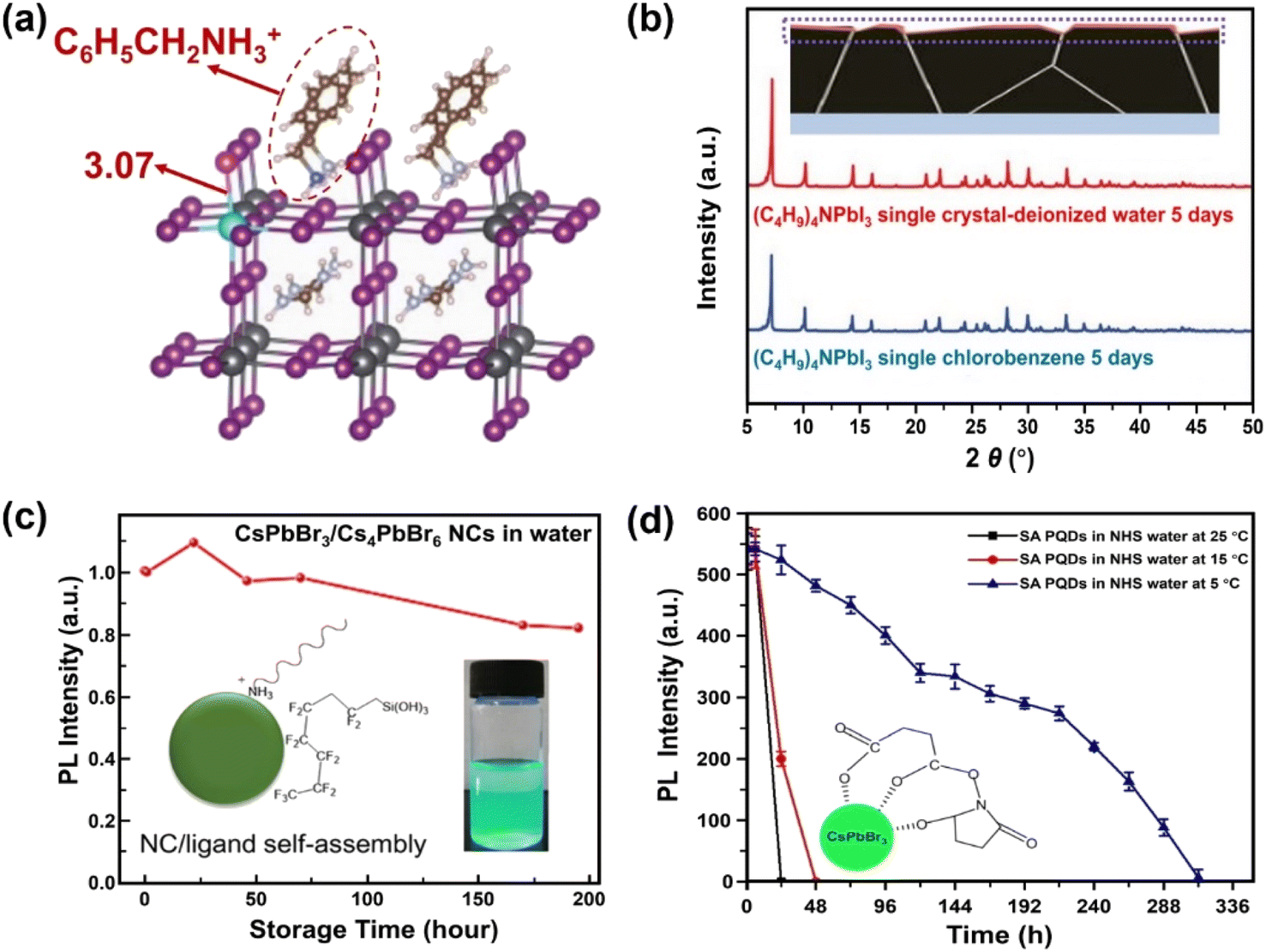 | ||
| Fig. 3 Ligand engineering for water-stable MHPs. (a) Length of Pb–I bond over FA0.83Cs0.17PbI3 after adding phenylmethylammonium ions. Adapted from ref. 72. (b) Formation of (C4H9)4NPbI3 layers over MHP from a (C4H9)4NI post-treatment and XRD patterns of (C4H9)4NPbI3 single crystals after 5 days immersion in water and toluene. Adapted from ref. 76. Copyright 2021 John Wiley and Sons. (c) Variations of PL intensity over fluorocarbon-coating CsPbBr3/Cs4PbBr6 NCs in water. Adapted from ref. 86. Copyright 2018 American Chemical Society. (d) PL intensity evolution of succinic acid-capping CsPbBr3 QDs in N-hydroxy succinimide water and the formation of tridentate ligands over QDs. Adapted from ref. 93. Copyright 2021 Elsevier. | ||
Sufficient interactions between MHPs and ligands should also be considered because the instability MHP NCs partly originates from the easy detachment of ligands as described above.77 Clearly, introducing reactive groups in the capping ligands and forming a covalent or ionic bonding between ligands and MHPs are deemed to improve the water stability of MHPs. For example, polyhedral oligomeric silsesquioxane (POSS) having a mercaptopropyl anchor group attaches to the surface of MHP NCs and forms a cage-like structure. Such POSS-protected CsPbX3 (X = Br and/or I) NCs were shown to maintain the original green light emission and stability in water for 10 weeks.78 Unlike the physical encapsulation strategy with hydrocarbons,79,80 the impressive enhancement of water stability results from the strong metal–thiol interactions between POSS and MHPs.81,82
Compared with metal–thiol interaction, the fluorine in fluorocarbons is expected to form a stronger interaction with MHPs owing to its high electronegativity.83 The fluorocarbon agents (FCAs) have a low surface energy, featuring superior hydrophobicity compared to the corresponding hydrocarbons.84 In addition, the abundance of –CF2– and –CF3 groups and the amphiphilic nature trigger the self-assembly in aqueous solution.85 As an example, after coating with FCAs ((C6F13CH2CH2Si(OCH2CH3)3, C6F13CH2CH2OOCCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, C6F6H7Si(OCH2CH3)3 or C3F6CH2CH2OOCCHCH2)), the CsPbBr3/Cs4PbBr6 NCs exhibited a high absolute photoluminescence quantum yield (PLQY) of ∼80% in water for weeks as expected (Fig. 3c). Because of their excellent performance, short fluorocarbon chains (down to C4) of FACs are also able to stabilize the perovskite NCs in water. After additional coating with BaSO4, the CsPbBr3/Cs4PbBr6/BaSO4 NCs retain 90% of the initial fluorescence intensity after 1 day storage at room temperature in saline solution, which broadens the potential applications in biology.86 Among the above FCAs, C3F6CH2CH2OOCCHCH2 can be further transformed to amphiphilic hexafluorobutanol (HFBO) with a polar C–OH head in the presence of water resulting in good water solubility and long-term stability in aqueous solution for more than 100 hours.87 The concentrated hydroxyl ligands may also serve as a water-proof layer to avoid further water attack.88
CH2, C6F6H7Si(OCH2CH3)3 or C3F6CH2CH2OOCCHCH2)), the CsPbBr3/Cs4PbBr6 NCs exhibited a high absolute photoluminescence quantum yield (PLQY) of ∼80% in water for weeks as expected (Fig. 3c). Because of their excellent performance, short fluorocarbon chains (down to C4) of FACs are also able to stabilize the perovskite NCs in water. After additional coating with BaSO4, the CsPbBr3/Cs4PbBr6/BaSO4 NCs retain 90% of the initial fluorescence intensity after 1 day storage at room temperature in saline solution, which broadens the potential applications in biology.86 Among the above FCAs, C3F6CH2CH2OOCCHCH2 can be further transformed to amphiphilic hexafluorobutanol (HFBO) with a polar C–OH head in the presence of water resulting in good water solubility and long-term stability in aqueous solution for more than 100 hours.87 The concentrated hydroxyl ligands may also serve as a water-proof layer to avoid further water attack.88
Efforts to explore ligands with dual functions, namely, hydrophobicity and bonding property, have been also extended to metal stearates (e.g., AlSt3, ZnSt2, NaSt). The long carbon chains protect MHPs from the surrounding environment, meanwhile, the coordinate bonding of St–Pb,89 St–Cs90 or ionic bonding of Al3+–Br− also provides a stronger interaction than the van der Waals bonds between OA (or OAm) and MHPs. The CsPbBr3-AlSt3 nanocomposite suspension exhibited bright photoluminescence (PL) after 25 min sonication in water or overnight storage91 and excellent stability in water for more than 60 days.92
However, introducing long chain ligands on the surface of MHPs usually deteriorates the charge transfer from MHPs to the coating and the adsorption of reactants, which is unfavorable to the photocatalytic activity.95–100 It is found that the employment of short chain ligands over NCs or QDs can enhance charge transfer rate101,102 and subsequently improve the photocatalytic activity of CsPbBr3 NCs.103 These findings indicate that utilizing short chain ligands is an exciting means for improving the stabilization of the MHPs without a compromised charge mobility. Up to now, a series of short chain multidentate ligands have been explored, which can be classified as acids (succinic acid,93 2,2′-iminodibenzoic acid,104 4-mercaptobenzoic acid,105 octadecanedioic acid106,107), amino acids (aminocaproic acid108), pyridines (2,2′-bipyridine, 2,2′:6′,2′′-terpyridine,109 2-mercaptopyridine107,110,111), salts (potassium dichloroacetate,112 N1,N2-didodecyl-N1,N1,N2,N2-tetramethylethane-1,2-diaminium bromide113), and zwitterions.114 One example is succinic acid, which has two carboxylic acid groups. After coupling with CsPbBr3 QDs, the composites exhibit enhanced water-stability because of the improved crystallinity and stronger bonding with CsPbBr3 QDs.93 Further activating the carboxyl groups with N-hydroxy succinimide (NHS) in water would form a tridentate ligand over CsPbBr3 QDs as depicted in Fig. 3d. The light emission over the composites lasts for 24 h and 48 h at 25 °C and 15 °C, respectively, and expands to two weeks at 5 °C. The reviewed various ligand strategies and their water stabilities are summarized in Table 1.
| Water-stable MHPs | Stability | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Materials | Method of preparation | Medium | Characterizations | Retained PL intensity | PLQY before (after) | Observed durability | |
| (C4H9)4NPbI3 | Solvent evaporation | Water (immersion) | PXRD | — | — | 5 days | 76 |
| NBCAnPbI3/CH3NH3PbI3 | Precipitation | Water (immersion) | UV-vis absorption spectroscopy | — | — | 30 min | 73 |
| PXRD | |||||||
| Paraffin-CsPbX3 | Physical blending | Water (immersion) | PL | 78% | 20 days | 79 | |
| Paraffin-CsPb0.7Sn0.3Br2I QDs | Physical blending | Water (immersion) | PL | — | — | 20 days | 80 |
| POSS-CsPbBr3 | Physical blending | Water (suspension) | PL | — | 62% (—) | 10 weeks | 78 |
| POSS-CsPb(Br/I)3 | Physical blending | Water (immersion) | PL | — | 45% (—) | 10 weeks | 78 |
| PFOTHS-CsPbBr3/Cs4PbBr6 NCs | Aqueous synthesis | Water (suspension) | PL | >80% | 79.2% (—) | 200 h | 86 |
| PFOTHS-CsPbBr3/Cs4PbBr6/BaSO4 | Aqueous synthesis | Saline solution (suspension) | PL | 90% | — | 1 day | 86 |
| Hexafluorobutyl methacrylate-coated Co2%@CsPbBr3/Cs4PbBr6 NCs | Physical blending | Water (suspension) | PL | >90% | ∼80% (—) | 100 h | 87 |
| CsPbBr3-AlSt3 NCs | Physical blending | Toluene/water (floating) | PL | — | — | Overnight | 91 |
| CsPbBr3-AlSt3 QDs | Co-precipitation | Water (suspension) | PL | ∼100% | — | 60 days | 92 |
| Succinic acid-capped CsPbBr3 QDs | Ligand exchange | Toluene/water (suspension) | PL | 85% | — | — | 93 |
| Succinic acid-capped CsPbBr3 QDs in NHS water | Ligand exchange and physical blending | Toluene/water 5 °C (suspension) | PL | — | — | 312 h | 93 |
| SLNs-CsPbBr3 | Sonication-assisted melt homogenization | Water (suspension) | PL | — | 13% (—) | 2 months | 94 |
| SLNs-CsPb(Br0.2I0.8)3 | Sonication-assisted melt homogenization | Water (suspension) | PL | — | 9% (—) | 2 months | 94 |
Photopolymers, such as Norland Optical Adhesive (NOA) 63,121 NOA 61,7 and Ergo® optical adhesive 8500 (Ergo),122 have been exploited firstly to stabilize MHPs because of their popularity in optoelectronic devices. They are polyurethane-related polymers, which can form crosslinked networks quickly upon UV curing. For instance, the Ergo-coated CsPbBr0.6I2.4 film maintained 91% PL intensity after soaking in water for 24 hours with only 6% loss of PLQY. Although the photocuring process improves the robustness of MHPs,123 this physical blending strategy may cause heterogeneous phase aggregation and can lead to instability.124 This is because of the specific surface area and volume effects of MHP NCs125 as well as the large difference in polarity of perovskites and polymers.126
In contrast to ligand modifications occurred on the surface of MHPs, most polymer–MHP NC composites have been produced by swelling–deswelling process of polymers. During the dynamic process,127 the dissolved solutes (mainly MHP's precursors) can penetrate into the polymer and the MHPs then grow inside the 3D network of polymer, after which the polymer shrinks during solvent evaporation or the addition of theta solvent. Therefore, MHPs become confined within the polymer thus limiting their contact with the surrounding environment. One important parameter to be optimized for such composites is the ratio of polymer and MHP constituents. Too small amounts of the polymer phase lead to insufficient protection and consequent degradation, whereas the other extreme results in turbid mixtures. As one example, the ratio of poly(isobutylene-alt-maleic anhydride)-graft-dodecyl (PMA) and MHP NCs showed a significant effect on the final water stability of the composites. It is found that the polymer-to-nanoparticle ratio (Rpol/area, number of monomers of polymer per nanoparticle area [nm2]) should be controlled in the range of 1000–3000. CsPbBr3 NCs-PMA composites with a Rpol/area of 1500 show good water stability for at least 8 months.128 The ratio also seems to influence the PL intensity of the composites. Too high MHP NC content will reduce the PL intensity due to the fluorescence self-quenching of NCs.124
Besides the molar ratio, the chemical structure of polymer also dominates the stability. Many polymers have been examined as hosts, including polystyrene (PS), polycarbonate (PC), acrylonitrile butadiene styrene (ABS), cellulose acetate (CA), polyvinyl chloride (PVC), poly(methyl methacrylate) (PMMA) and poly(styrene/acrylamide).129 Among those, PS, PC, PVC and ABS can protect MHP NCs from being attacked by water for periods of months, while the CA loses stability after two days, and PMMA-protected MHP NCs fail instantly (Fig. 4a). The failure of CA originates from its hydrophilic character and thus its high-water permeability, whereas the reason of PMMA is likely stemming from its low swelling ratio of PMMA in DMF solvent and weak bonding interaction with MHPs. Specifically, the MAPbBr3–PS and MAPbBr3–PC composite films can survive in boiling water for 30 min with less than 15% and 7% of decay in PLQY, respectively.129 The protecting effect of crosslinked PS is much better than other uncross-linked polymers because it cannot swell in polar solvents. For instance, the CsPbBr3@PS composites maintain 65.7% of the absolute PLQYs after stirring in water for 3 days, and still exhibit strong green luminescence after 9 months storage.130 Note that the composites have a good stability in acid/alkali aqueous solution, and even biologic buffers, showing advantages in monitoring pH, urea, and urease.131
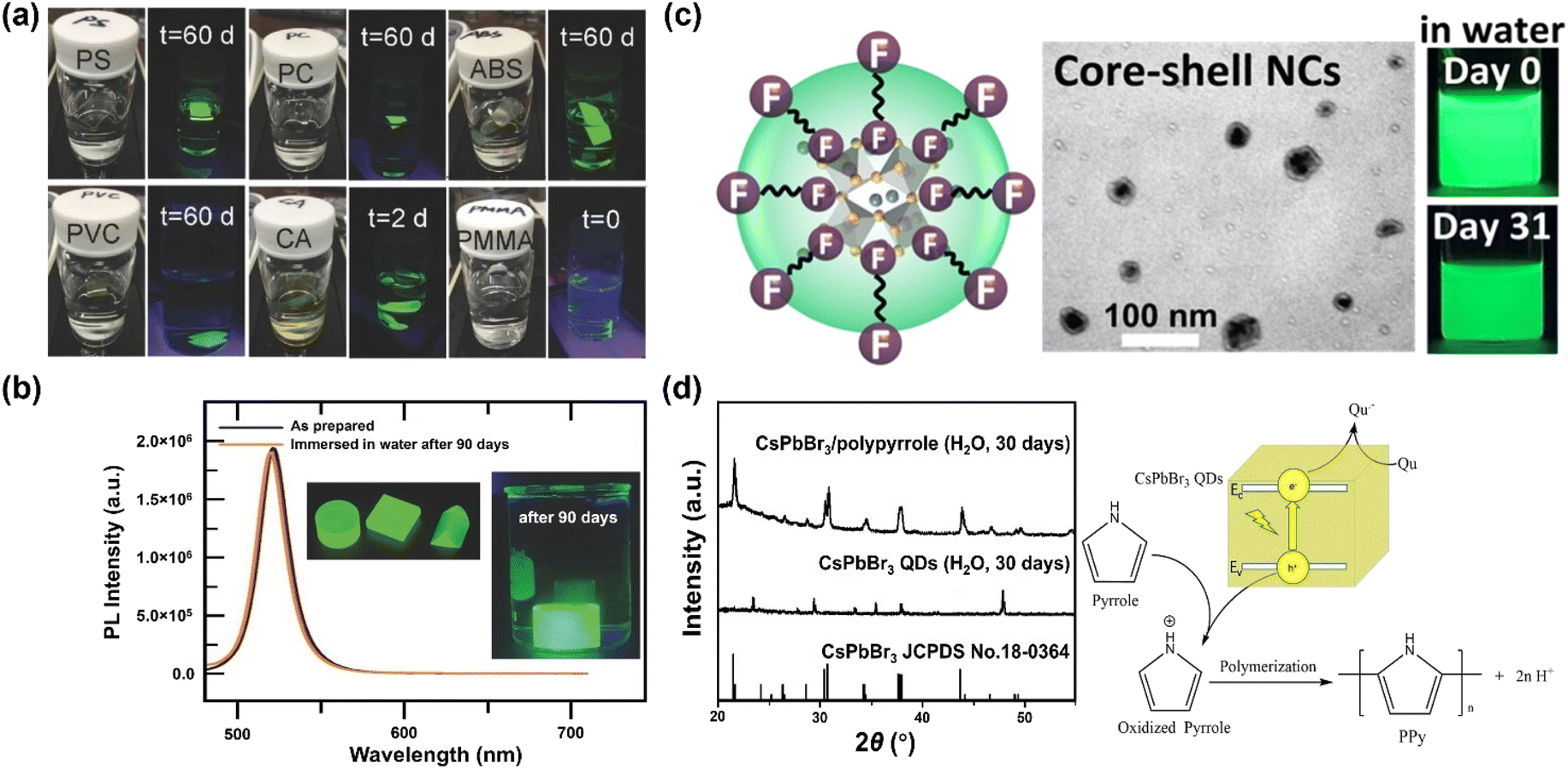 | ||
| Fig. 4 Polymer encapsulation over MHPs. (a) Fluorescent photos of polystyrene, polycarbonate, acrylonitrile butadiene styrene, cellulose acetate and poly(methyl methacrylate)–MAPbBr3 composites at different periods. Adapted from ref. 129. Copyright 2016 John Wiley and Sons. (b) Comparison of PL spectra over MAPbBr3 NC–V18 composites before and after a 90 days immersion in water. Adapted from ref. 133. Copyright 2017 John Wiley and Sons. (c) TEM image and photos of CsPbBr3/Cs4PbBr6 NCs protected by fluoropolymer shell. Adapted from ref. 138. Copyright 2022 American Chemical Society. (d) Visible light photopolymerization synthesis of CsPbBr3/polypyrrole composites and XRD patterns after 30 days water immersion. Adapted from ref. 145. Copyright 2020 Elsevier. | ||
Usually, the dissolution or swelling of polymers is time-consuming, especially for those of high-molecular-weight. Hence, the MHPs need to be prepared prior to coupling; however, serious degradation or aggregation may occur during the storage of MHP NCs or QDs. Through adding crosslinkable monomers into the MHP's precursor solution, polymer–MHPs composites can be achieved in a one-pot reaction assisted by heating or UV irradiation.132 For example, a crosslinked 4-vinylbenzyl-dimethyloctadecylammonium chloride (V18)-MAPbBr3 NC composite has been prepared via in situ polymerization under 90 °C for 30 min by employing azobisisobutyronitrile (AIBN) as an initiator.133 The crosslinked MAPbBr3 NC–V18 retains about 85% of PL intensity after immersion in water for 120 min. After copolymerization with methyl methacrylate, the water-resistance extends to 90 days without the change of its PL (Fig. 4b). However, the decomposition products of remaining photo-initiator may have an unintended consequence on the property of polymers and MHPs.147 The alternative approach is to utilize polymers (for example, copolymer micelles) as nanoreactors and grow the MHPs inside the polymers. In contrast to random growth of MHPs during swelling–deswelling process, block copolymer micelles are highly ordered structures, featuring superior size uniformity and high stability of individual micellar building blocks, which are often employed as templates and scaffolds for producing arrays and ordered structures of nanomaterials.134,148,149 One good example is employing polystyrene-poly(2-vinlypyridine) (PS-b-P2VP) diblock copolymer micelles as nanoreactors to prepare monodisperse polymer–MHP NCs. The resultant composites display strong stability against water over 75 days.134
Similar to the strategies with ligands, improving the interface interactions between MHPs and polymers by introducing anchoring groups, such as –COOH groups139,150 and –CF2 groups,136 can enhance the stability of polymer–MHPs.151 For example, PMMA has been widely used as passivation to protect solar cells from oxygen and moisture.152–154 On the other hand, it fails to stabilize MHPs in water owing to the too weak interaction between those. Jiang et al. found that partially hydrolyzing PMMA (below 10%, h-PMMA) would form methacrylic acid anchors in PMMA chain.137,150 These anchors work as ligands and enhance the PMMA–MHP interaction, thus improving the water stability. Thus, a good interaction between the MHPs and polymers is a prerequisite to the success of good stability. For example, a fluoropolymer (Hyflon) was used to stabilize CsPbBr3/Cs4PbBr6 NCs and it was found that the CF2 groups from Hyflon are strongly bonded on the surface of MHPs (Fig. 4c), forming a ligand shell over CsPbBr3/Cs4PbBr6 NCs core having good stability for at least one month.138
The strength of interaction at the polymer–MHP interface is also influenced by the compatibility of surface organic ligands on MHPs and hydrophobic polymers.155 For instance, OA and OAm ligands capped MHP particles can be stabilized by polymers having long alkyl chain ligands because the similar alkyl chains has a good compatibility and thus create a better MHP–polymer interface.143 Introducing interfacial layers (e.g., polyvinyl pyrrolidone, PVP) can not only serve as an additional barrier, but also improve the compatibility of MHP with polymer matrix. For PVP, it is a widely used coupling agent. Owing to the amphiphilic characteristics, PVP can be adsorbed on different surfaces, including metals, metal oxides, polymers (e.g., PS, cellulose).156 Inspired by this, PVP has been employed serving as an effective interfacial layer between MHP NCs and polymer for the design of water-stable MHPs.140,141 Similar to the multidentate ligands, block copolymers have inherently different domains and provide more functional features. As an example, polystyrene-block-poly(ethylene-ran-butylene)-block-polystyrene (PS-PEB-PS) and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEG-PPG-PEG) have been applied to encapsulate the CsPbBr3 QDs. The obtained nanocomposites display luminescence in water for 8 days.142 The results suggested that the PS blocks interact with the hydrophobic parts (surface ligands) of QDs; the PEG moiety, as Lewis base, would have a strong interaction with Lewis acid (PbBr2) and acts as water protection barrier.182
In addition to the interfacial interaction, the thickness of coating also has a significant effect on water-resistance.183 While thicker coatings provide better stability, those can seriously compromise charge transport (tunneling), when the coating thickness exceeds a few nanometers.184,185 Thus, tailoring the thickness to balance the water-resistance and charge transportation is necessary. By chemical crosslinking the polymer chains, a thinner dense covalent-bonded 3D polymer network can be formed. Therefore, molecules having multiple sites for crosslinking, such as, 2-((acryloyloxy)methyl)-2-(((12-guanidinododecanoyl)oxy)methyl)propane-1,3-diyl diacrylate (PETA-G) can be polymerized efficiently to provide a sufficiently thin shell around MHPs without compromising the charge transport property.144
Based on the above analysis, polymers offer a good strategy to form protecting layers on MHPs for stabilization. However, in most cases, their limited electrical transport properties impede the extraction and transfer of charge, and consequently can compromise their applications in photocatalysis and photoelectrocatalysis. To overcome such limitations, polymers having conjugated electronic structure (e.g., polypyrrole and polyaniline) and hence reasonable charge transport properties (typically via hopping mechanisms) have been applied.186 As shown in Fig. 4d, the composites exhibit dramatic enhancement of water stability as well as improved charge transport behavior.145,146 Unfortunately, these conducting polymers also suffer from limited stability especially in O2 and water, caused by doping effects and also by the electroactive nature of such polymers.187,188
Accordingly, to build a successful strategy to produce water-stable polymer coated MHPs (Table 2), simultaneously, several aspects of materials selection and synthetic routes shall be considered (Fig. 5).
| Water-stable MHPs | Stability | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Materials | Methods | Medium | Characterizations | Retained PL intensity | PLQY before (after) | Observed durability | |
| a NOA61: Norland Optical Adhesives 61; Ergo: Ergo® optical adhesive 8500; PMSR: phenyl methyl silicon resin; SSDC: Silicone Sealant Dow Corning® 937; PMA: poly(isobutylene-alt-maleic anhydride)-graft-dodecyl; Rpol/area: the number of monomers of polymer per NP area [nm2]; Hyflo: Hyflon AD 60; DFTHS: dodecafluoroheptylpropyl-trihydroxysilane; V18: 4-vinylbenzyl-dimethyloctadecylammonium chloride. | |||||||
| NOA61/CH3NH3PbBr3/glass | Photocurring | Water (washing) | PL | ∼100% | — | Four cycles | 7 |
| CsPbBr0.6I2.4 QD/Ergo films | Photocurring | Water (immersion) | PL | 91% | 43% (—) | 24 h | 122 |
| MAPbBr3/PMSR (1.13 wt%) | Physical blending | Water (immersion) | PL | 93% | 53% (—) | 576 h | 124 |
| MAPbBr3/PMSR (1.13 wt%) | Physical blending | Water (immersion, 70 °C) | PL | 91% | 53% (—) | 50 min | 124 |
| MAPbBr3/PMSR (1.13 wt%) | Physical blending | Water (immersion, 100 °C) | PL | 77% | 53% (—) | 20 min | 124 |
| MAPbBr3/SSDC (1.13 wt%) | Physical blending | Water | PL | 77% | 62% (—) | 36 h | 124 |
| MAPbBr3/SSDC (1.13 wt%) | Physical blending | Water (immersion, 70 °C) | PL | 52% | 62% (—) | 10 min | 124 |
| MAPbBr3/SSDC (1.13 wt%) | Physical blending | Water (immersion, 100 °C) | PL | 44% | 62% (—) | 10 min | 124 |
| CsPbBr3 NCs-PMA (Rpol/area of 1500) | Physical blending | Water (immersion) | PL | — | — | >8 months | 128 |
| MAPbBr3–polystyrene | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 34% (32%) | 2 months | 129 |
| MAPbBr3–polycarbonate | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 31% (31%) | 2 months | 129 |
| MAPbBr3–acrylonitrile butadiene | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 48% (45%) | 2 months | 129 |
| MAPbBr3–polyvinyl chloride | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 16% (15%) | 2 months | 129 |
| MAPbBr3–cellulose acetate | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 47% (—) | 2 months | 129 |
| MAPbBr3–cellulose acetate | Swelling–deswelling microencapsulation | Water (immersion) | PL | 5% | — | 48 h | 129 |
| MAPbBr3–poly(methyl methacrylate) | Swelling–deswelling microencapsulation | Water (immersion) | PL | — | 14% (—) | 2 months | 129 |
| MAPbBr3–polystyrene | Swelling–deswelling microencapsulation | Boiling water (immersion) | PL | — | 34% (29%) | 30 min | 129 |
| MAPbBr3–polyvinyl chloride | Swelling–deswelling microencapsulation | Boiling water (immersion) | PL | — | 31% (29%) | 30 min | 129 |
| CsPbBr3 QDs@polystyrene | Swelling–shrinking | Water (immersion, stirring) | PL | — | 68% (64.7%) | 3 days | 130 |
| CsPbBr3 QDs@polystyrene | Swelling–shrinking | Water (immersion, stirring) | PL | 20–30% | 68% (—) | 30 days | 130 |
| CsPbBr3–poly(methyl methacrylate) | One-pot thermal and UV polymerization | Water (immersion) | PL | 54% | 54.6% (—) | 48 h (30 days) | 132 |
| CsPbBr3–poly(butyl methacrylate) | One-pot thermal and UV polymerization | Water (immersion) | PL | 56% | 62.2% (—) | 48 h (30 days) | 132 |
| MAPbX3 NCs-polystyrene-poly(2-vinlypyridine) (PS-b-P2VP) | In situ growth | Water (immersion) | PL | — | — | 75 days | 134 |
| Polyimide-coated CsPbBr3 NCs | In situ growth | Water (immersion) | PL | ∼80% | 88.1% (—) | 60 min | 135 |
| MAPbBr3 (8 wt%) NCs/polyvinylidene fluoride | In situ growth | Water (immersion) | PL | — | 94.6 ± 1% (68.1 ± 1%) | 400 h | 136 |
| Hydrolyzed poly(methyl methacrylate)-coated CH3NH3PbBr3 | Mechanical grinding | Water (suspension) | PL | ∼80% | — | 40 days | 137 |
| CsPbBr3/Cs4PbBr6 NCs-Hyflon-DFTHS/OLA | Physical blending | Water (suspension) | PL | 68% | 73% (—) | 31 days | 138 |
| CsPbBr3/octylamine-modified polyacrylic acid + OAm NCs | Ligand engineering | Water (suspension) | PL | 80.13% | — | 15 days | 139 |
| PVP-capped CsPbX3 NCs@polystyrene microhemispheres | Self-assembly | Water (washing) | PL | — | — | 3 times | 140 |
| Silicone resin/PVP-CsPbBr3 nanofibrous membranes | One-step electrospun | Water (immersion) | PL | — | — | Several hours | 141 |
| PS-PEB-PS and PEG-PPG-PEG coated CsPbBr3 QDs | Physical blending | Water (immersion) | PL | 60% | 88% (86%) | 1 month | 142 |
| CsPbBr3 QDs–poly(styrene-ethylene-butylene-styrene) films | Physical blending | Water (immersion) | PL | — | — | 122 days | 143 |
| V18–MAPbBr3 NCs | Thermal polymerization | Water (immersion) | PL | 85% | — | 120 min | 133 |
| V18-co-MMA-MAPbBr3 NCs | Thermal polymerization | Water (immersion) | PL | — | — | 90 days | 133 |
| Crosslinked PETA-G/FA0.92MA0.08PbI3 films | Spin-coating and thermal polymerization | Water (immersion) | Photograph evolution | — | — | 420 s | 144 |
| CsPbBr3/polypyrrole | Visible light polymerization | Water (immersion) | PXRD | — | — | 30 days | 145 |
| TEM | |||||||
| CsPbBr3/polyaniline | Visible light polymerization | Water (immersion) | PL | ∼93% | — | 4 weeks | 146 |
| PXRD | |||||||
| TEM | |||||||
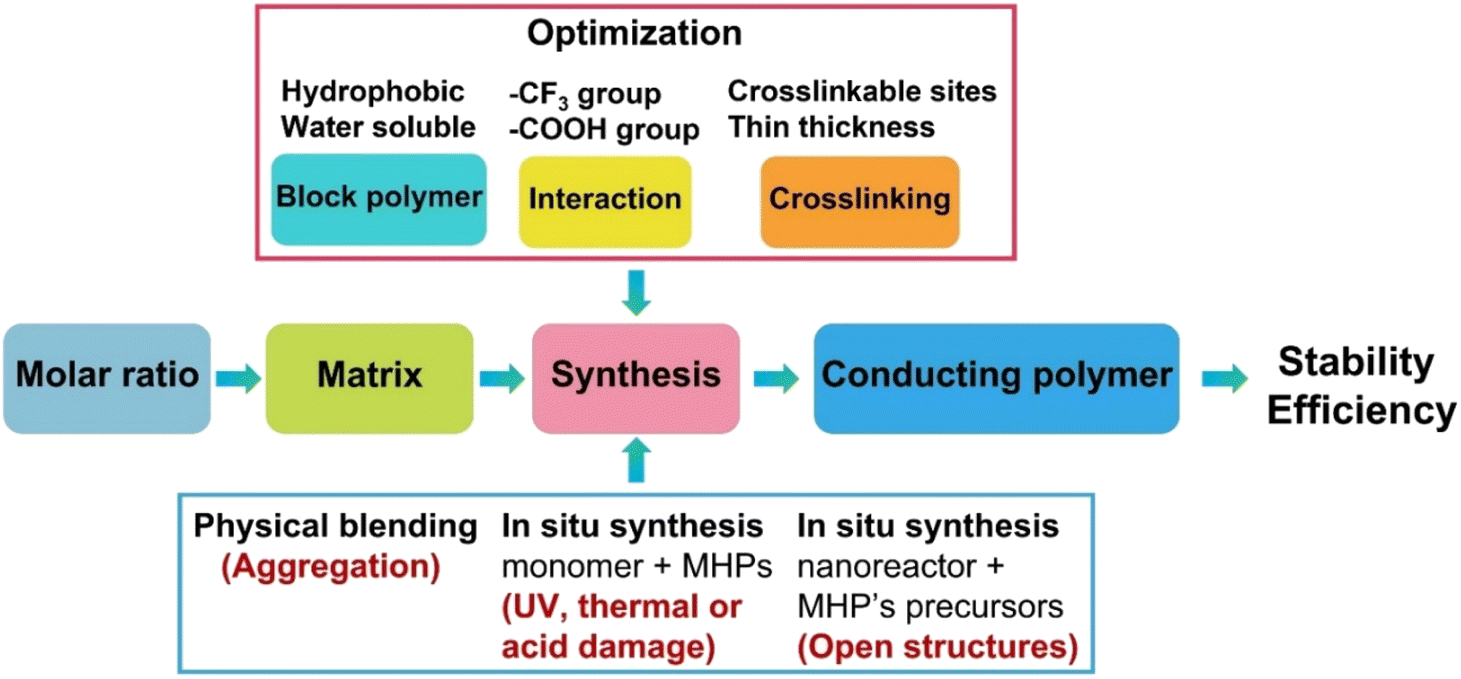 | ||
| Fig. 5 Scheme of suggested strategies for the synthesis of polymer–MHPs composites. | ||
For coatings with a thin Al2O3 layer, trimethylaluminum (TMA) vapor-based atomic layer deposition (ALD) has been reported to form covalent bonds with MHP NCs.190 The ALD is emerging as a useful method for depositing thin films with excellent conformality, uniformity, precise thickness control and high quality. Considering that it can be carried out at moderate temperatures,191 the technique is particularly attractive to coat MHPs. Amorphous alumina (AlOx) encapsulated CsPbX3 QDs have shown improved stability for at least 1 hour.157 The short stability in water originates from the intrinsic instability of this amorphous overcoat. The stability can be further improved (up to one month) by increasing the growth temperature and hence the crystallinity of alumina on CsPbBr3 nanoplates, as shown in Fig. 6a.158
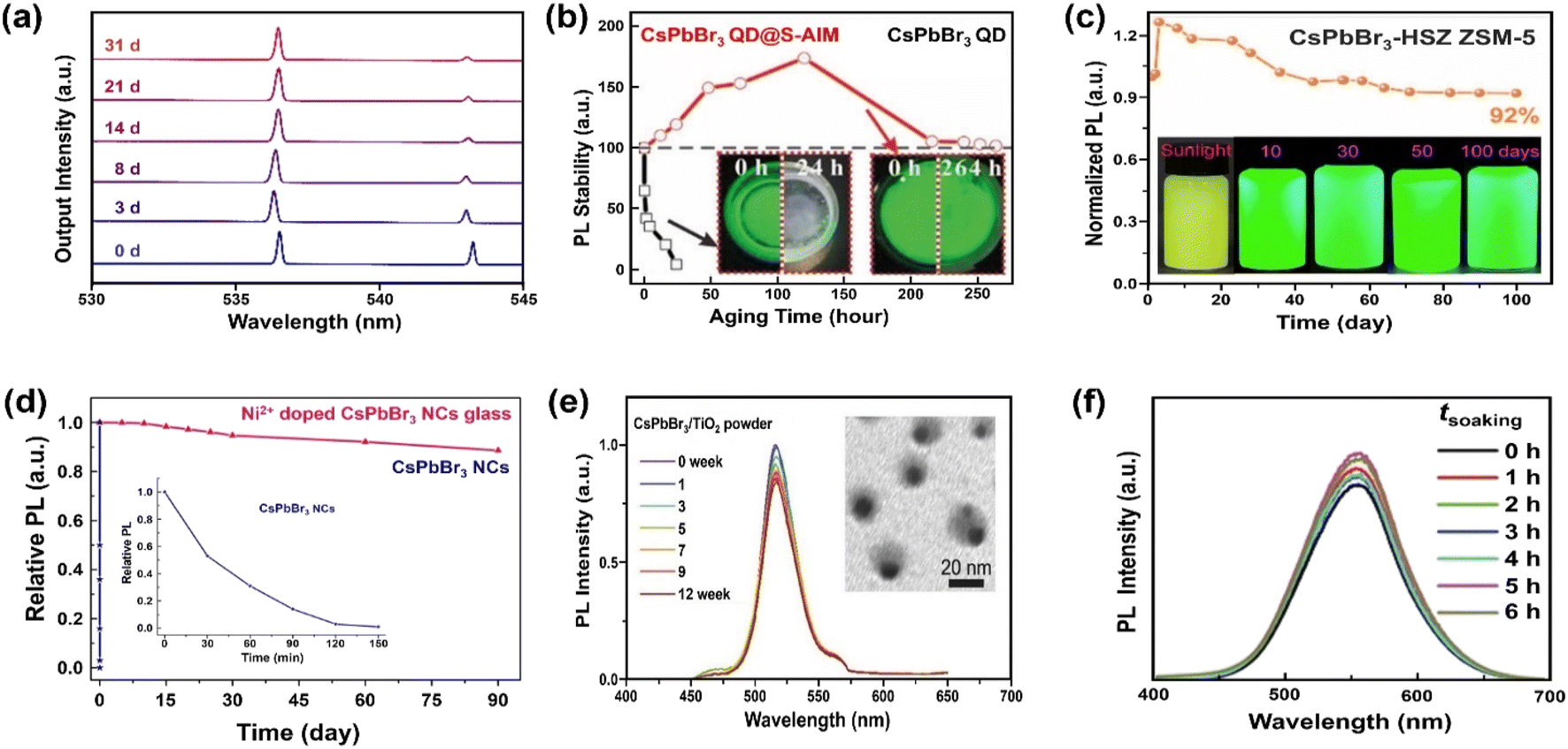 | ||
| Fig. 6 Stabilizing MHPs by using inorganic materials. (a) Time-dependent normalized lasing spectra of the ALD Al2O3 coated CsPbBr3 nanoplate after being immersed in water for 1 month. Adapted from ref. 158. Copyright 2020 American Chemical Society. (b) PL intensity evolution over CsPbBr3 QDs and CsPbBr3 QDs@superhydrophobic aerogel inorganic matrix (S-AIM) completely immersed in water with time. Adapted from ref. 163. Copyright 2020 John Wiley and Sons. (c) PL stability of CsPbBr3-HSZ ZSM-5-700 composites when exposed in water and luminescent photographs of CsPbBr3-HSZ ZSM-5-700 composite immersed in water for various time (5 mg mL−1). Adapted from ref. 175. Copyright 2022 Elsevier. (d) The change in relative PL of Ni2+ doped CsPbBr3 NCs glass and CsPbBr3 NCs in water (inset: enlarged the change of PL with CsPbBr3 NCs in water). Adapted from ref. 177. Copyright 2019 Elsevier. (e) The relative PL intensity of CsPbBr3/TiO2 NCs after immersing in Milli-Q water (0–12 weeks), inset shows a TEM image of CsPbBr3/TiO2 NCs after immersing in Milli-Q water for 12 weeks. Adapted from ref. 181. Copyright 2017 John Wiley and Sons. (f) Normalized PL spectra of Cs2Sn0.89Te0.11Cl6versus different soaking time. Adapted from ref. 180. Copyright 2020 John Wiley and Sons. | ||
Silicon oxide (SiO2) is another attractive coating material, featuring good chemical stability, blocking of moisture and oxygen, and excellent transparency. It has been selected as protecting layers for the stabilization of carbon dots,192 and metal nanoparticles193 among others. Silica-based mesoporous aerogels (AGs) have been developed as matrix owing to high porosity, large specific surface area, low density and thermal conductivity.194 The hydrophobic property can be tailored by grafting hydrophobic functional groups over SiO2 or directly using the silica precursor holding hydrophobic groups. Diverse types of pores (micropore, mesopore and macropore) can host the CsPbBr3 QDs and supply enhanced water stability for more than two weeks in hydrophobic AG.162 However, the polydisperse pores also bring the potential aggregation of MHP QDs. A fine control over the pore size of mesoporous silica will ease the uniform distribution of MHP QDs in random-distributed AGs because of the compatibility of pore size and QDs' mean size195 and excellent adsorption of QDs on the hydrophobic surface. For example, Li et al. have encapsulated the QDs into superhydrophobic aerogel inorganic matrix (S-AIM) with open structures.163 The composites preserved their initial PL intensity after 11 days of soaking in water (Fig. 6b) and achieved a relatively high PLQY stability (50.5%) after soaking for 3.5 months. This good stability without the sealing of open structure may originate from the matched size between the pore and the particle, the super-hydrophobicity from rough surface,196 and the hydrophobic functional groups from the AGs.163
However, the open shell after impregnation is still a threat to long-term stability. Thus, a second coating is normally needed. For this purpose, ALD grown compact AlOx,168 polymers169,170 and biomedical phospholipids171,172 have been reported. The mSiO2–CsPbBr3@AlOx obtained with 100 ALD cycles keeps up to 95% of PL intensity after 8 h in water dispersion under stirring, and the stability extends to 90 days under static conditions.168 He et al. embedded Mn-doped CsPbCl3 QDs into SiO2/Al2O3 monolith through a facile sol–gel process, followed by a physical grinding. The obtained Mn-doped CsPbCl3 QDs-SiO2/Al2O3 monolith sample maintained around 92% of the initial PL intensity after 7 days under accelerated aging condition (85 °C and 85% RH).173 For polymers, this strategy can trigger the formation of micro/nano structured SiO2 surface, which is one essential feature needed for super-hydrophobicity.197,198 The coating of biomedical phospholipid can also further improve the stability over SiO2 coated MHPs and broaden their applications in bioimaging and biosensing.171,172
Another issue is the aggregation during the synthesis, which may cause quenching and instability.124,199 In conjunction with polymer-type coatings, most of the studies focus on multiple MHP NCs. However, the synthesis of monodisperse MHP NCs at atomic-level is more desirable in LEDs, bio- or catalysis-related fields. Through a fast hydrolysis of highly reactive silica precursor (for example, tetramethoxysilane, TMOS) with a small amount of water, the MHP NCs or QDs can be wrapped by silica at the interface of water/nonpolar solvent via a simple sol–gel method to synthesize monodisperse CsPbX3/SiO2 nanoparticles.159 The CsPbBr3/SiO2 Janus NCs suspension showed bright PL and remained 80% of the PL intensity after placing in water 7 days.159 This method can be extended to other oxides, such as Ta2O5, ZrO2.160 However, the Janus structure means that part of the surfaces of MHPs are exposed to the environment simultaneously and would suffer from degradation. Subsequent work by Zhang's group has achieved monodisperse CsPbBr3@SiO2 core–shell nanoparticles with the aid of ammonia via a modified supersaturated recrystallization method. They also found that ammonia is not mandatory for hydrolysis, but influences the morphology, size of the products and the formation rates of SiO2. Under a harsh condition (ultrasound irradiation in water for 40 min), CsPbBr3@SiO2 core–shell nanoparticles still exhibited bright emission.161
SiO2 is usually synthesized by the hydrolysis of Si precursor via a sol–gel method. Note that the choice of precursor and synthetic methods will also impact the water stability of MHP–SiO2 composites. Owing to the fast hydrolysis rate of TMOS, it is widely used in synthesis of highly stable MHP–SiO2 composites.159,161 However, it is reported that the MHP–SiO2 composites synthesized via a modified ligand-assisted reprecipitation (LARP) method showed a lower stability in water, probably caused by the amorphous and porous structure.164 Generally, tetraethoxysilane (TEOS) has been regarded as an inefficient precursor to envelop MHP NCs owing to its slower hydrolysis rate, which would form separate MHP NCs or big aggregates. However, monodisperse CsPbBr3@SiO2 NCs has been reported by using TEOS via one-pot hot-injection strategy.165 By introducing the hydrophobic and multibranched trioctylphosphine oxide (TOPO) as ligands, the TOPO would anchor on the surface of NCs, and effectively tune the hydrolysis rate of TEOS and surface property of NCs resulting in the formation of core@shell NCs at a nanoscale-particle level. The CsPbBr3@SiO2 NCs retained more than 70% of their initial fluorescence intensity within 8 days immersion in hexane–water (1/1.5, v/v) mixture. Accordingly, based on these results, we may conclude that the quality of SiO2 and hence its barrier properties strongly depend on the synthetic routes applied.
Besides the choice of suitable precursors, the interaction of SiO2 and MHPs would also influence the final water stability. It is found that coatings produced from phenyltriethoxysilane (phTEOS) could not envelope the MHP NCs because of the weak adsorption of intermediate silsesquioxanes over MHPs.200 Only combining TMOS with phTEOS (especially equimolar amount), the MHPs can be covered by 3D silica grafting with hydrophobic phenyl group, showing an improved water-resistance (at least for 24 hours).164 The interaction can be strengthened by the addition of interfacial ligands (such as PVP166) or the introduction of bonding interactions.167 For example, Li et al. have introduced a Pb–S bonding by directly using (3-mercaptopropyl)trimethoxysilane (C6H15O3Si-SH, MPTMS) as SiO2 precursor.167 The strong bonding makes the perovskite@silica nanodots stable in water for over six weeks.
Zeolites represent a large family of inorganic porous crystalline materials, in which the anions of [AlO4]5− and [SiO4]4− tetrahedra are linked with oxo-bridges forming networks of 3D cages or channels.201 The pore structure makes them vastly used in catalysis, separation, gas adsorption and ion-exchange.202,203 Cation exchange property of zeolite can activate the introduction of A site ions from MHPs, and the CsPbBr3 QDs can grow into the zeolite structure via in situ crystallization. Such composites exhibit excellent photostability204 and water resistance.174 Contrary to the commonly recognized CsPbBr3 QDs, new species of [Na4Cs6PbBr4]8+ QDs have been confirmed in the zeolite's super cage, in which the tetrahedral PbBr42− ions are surrounded by Na+ and Cs+ ions. A recent study has shown that the high water-resistance (for 100 days) of CsPbBr3 QDs-HSZ ZSM-5 (Fig. 6c) is attributed to the interconnected micro–mesoporous network in hierarchically structured zeolite (HSZ). The micropores act as a shielding wall to isolate MHPs from the external environment and mesopores promote the diffusivity of precursors towards the successful space-confinement of CsPbX3 (X = Cl and Br) QDs.175
As an alternative, inorganic glass matrices (silicate, soda-lime, lead-oxide etc.) have been proven as another effective way to stabilize MHPs. The glass matrixes feature excellent mechanical, thermal and chemical stability. Up to now, mostly CsPbX3 NCs have been successfully incorporated into glass matrices, demonstrating dramatic enhancement of water stability, up to 90 days (Fig. 6d).176,177 This solution needs high processing temperature, which would unavoidable to engender the volatilization of halides precursor (e.g., CsBr and PbBr2).205 It has been reported that embedding CsPbBr3 QDs into TeO2-based inorganic glass can lower the temperature (from 1100 °C to 630 °C). However, the water stability decreases to 120 hours correspondingly.178 However, the glass matrix is only studied in all-inorganic MHPs because of their higher thermal stability and structure integrity during the formation process of glass.
Although the encapsulation with AlOx, SiO2, zeolites and glassy materials can solve the instability issue of MHPs, similar to the non-conductive organic polymers or ligands, these insulating materials restrain charge extraction across the coating shell and thus greatly disable applications in catalysis. Therefore, instead, application of semiconducting inorganic coating materials such as SnO2 and TiO2 can be a more viable option. Apart from enabled carrier transport, another benefit of semiconducting coatings is a possibility to have electron–hole rectification across the interfacial junctions (depending on the band structures of the core and shell materials), which can eventually inhibit/delay recombination.206 Stable CsPbBr3/TiO2 core/shell NCs have been reported by calcination of CsPbBr3 NCs and titanium butoxide at 300 °C for 2 h.181 The tight TiO2 shell over CsPbBr3 core ensures the stability of PL peak position and intensity (≈80%) for over three months (Fig. 6e). To obtain a robust protecting layer, crystalline TiO2 is favored over an amorphous phase.207 However, the poor thermal stability of MHPs (mostly blow 300 °C for hybrid ones208) limits the thermal budgets of processes. In contrast to the instability of amorphous TiO2 shell, amorphous SnO2 obtained by hydrothermal route was found to be able to stabilize Cs2Sn1−xTexCl6 (x = 0.11)180 and Cs2PtxSn1−xCl6 (0 ≤ x ≤ 1)209 solid solutions in water (Fig. 6f). Thus, it appears that not only the choice of the inorganic materials coating but also the chemistry plays a role in water stability of the core (Table 3). However, it is worth noting that both TiO2 and SnO2 absorb only in the UV region, which only accounts for less than 4% in solar spectrum.
| Water-stable MHPs | Stability | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Materials | Methods | Medium | Characterizations | Retained PL intensity | PLQY before (after) | Observed durability | |
| CsPbX3 QDs/AlOx films | ALD | Water (immersion) | PL | 100% | — | >1 h | 157 |
| Al2O3-coated CsPbBr3 nanoplate | ALD | Water (immersion) | Lasing spectra | — | — | 31 days | 158 |
| CsPbBr3/SiO2 | Water-triggered transformation and sol–gel method | Hexane/water (floating) | PL | 80% | 80% (—) | 7 days | 159 |
| CsPbBr3/ZrO2-10 | Water-triggered transformation and sol–gel method | Hexane/water (floating) | PL | 80% | 90% (—) | 8 days | 160 |
| CsPbBr3@SiO2 NPs | Modified supersaturated recrystallization | Water (ultrasonication, suspension) | PL | — | — | 40 min | 161 |
| CsPbBr3 QDs/mesoporous silica AGs | Physical blending | Water (immersion) | PL | 50% | — | 14 days | 162 |
| Green-S-AIM/CsPbBr3 QDs | Postadsorption | Water (immersion) | PL | 100% | 75.6% (50.5%) | 11 days | 163 |
| Green-S-AIM/CsPbBr3 QDs | Postadsorption | Hexane/water (floating) | PL | — | 75.6% (77.6%) | 4.5 months | 163 |
| phTEOS-TMOS@CsPbBr3 NCs | Hydrolysis-condensation | Water (immersion) | PL | — | — | 24 h | 164 |
| PXRD | |||||||
| FTIR | |||||||
| CsPbBr3 QDs@SiO2 | Nonpolar solvent | Hexane/water (floating) | PL | 70% | ∼87% (—) | 8 days | 165 |
| PVP–CsPbBr3 QD@SiO2-octadecyl trimethoxysilane-lecithin core–shell nanoparticles | Physical blending, hydrolysis-condensation and self-assembly | Water (immersion) | PL | 98% | 41.6% (—) | 10 days | 166 |
| CsPbBr3 QDs-Pb-S-SiO2-SH nanodots | Hydrolysis-condensation | Water (suspension) | PL | 50% | 78% (—) | 20 days | 167 |
| CsPbBr3 QDs-Pb-S-SiO2-SH nanodots | Hydrolysis-condensation | Water (suspension) | PL | — | — | 6 weeks | 167 |
| Mesoporous SiO2–CsPbBr3@AlOx | Modified template-assisted formation and ALD | Water (suspension) | PL | 95% | — | 8 h | 168 |
| Mesoporous SiO2–CsPbBr3@AlOx | Modified template-assisted formation and ALD | Water (suspension) | PL | 20% | — | 90 days | 168 |
| MAPbBr3@SiO2/PVDF nanoparticles | Impregnation and physical blending | Water (floating) | PL | — | 85.5% (—) | 1 month | 169 |
| MAPbBr3@SiO2/PVDF films | Impregnation and C | Water (immersion) | PL | 83% | 85.5% (—) | 20 min | 169 |
| MAPbBr3@SiO2/PVDF films | Impregnation and physical blending | Water (immersion) | PL | 55% | 85.5% (—) | 2 h | 169 |
| CsPbI3@polystyrene@SiO2 | Confined condensation | Water (boiling) | PL | 97.8% | 86% (—) | 48 h | 170 |
| CsPbBr3/SiO2/PEGylated phospholipid | Hydrolysis-condensation and physical blending | Water (suspension) | PL | 80% | — | 2 weeks | 171 and 172 |
| Mn-doped CsPbCl3 QDs-SiO2/Al2O3 monolith | One-pot synthesis | 85 °C and 85% RH | PL | ∼92% | — | 7 days | 173 |
| [Na4Cs6PbBr4]8+-zeolite | Ion-exchange and in situ growth | Water (immersion) | PL | 100% | 35% (—) | 40 days | 174 |
| PXRD | |||||||
| XPS | |||||||
| CsPbBr3 QDs-HSZ ZSM-5 | Grinding-calcination | Water (immersion) | PL | 92% | 62% (—) | 100 days | 175 |
| CsPbBr1.2/I1.8 NCs@P-Si-Zn glass | Melt-quenching and subsequent heat-treatment | Water (immersion) | PL | ∼90% | — | 40 days | 176 |
| Ni2+-doped CsPbBr3 NCs@B-Si-Zn glass | Melt-quenching | Water (immersion) | PL | 88.2% | 84.3% (—) | 90 days | 177 |
| CsPbBr3 QDs@TeO2-based glass | In situ nanocrystallization | Water (immersion) | PL | ∼90% | 70% (—) | 120 h | 178 |
| CsPbBr3 QDs@TeO2-based glass | In situ nanocrystallization | Water (immersion) | PL | ∼60% | 70% (—) | 45 days | 178 |
| CsPbBr3@ZnO nanoparticles | Physical blending | Water (ultrasonication) | PL | — | — | 30 min | 179 |
| CsPbBr3@NaYF4 nanoparticles | Physical blending | Water (ultrasonication) | PL | — | — | 30 min | 179 |
| Cs2Sn0.89Te0.11Cl6 | Hydrothermal method | Water (immersion) | PL | 100% | 95.42% (—) | 360 min | 180 |
| PXRD | |||||||
| FTIR | |||||||
| XPS | |||||||
| CsPbBr3/TiOx | Hydrolysis-drying | Water (immersion) | PL | — | — | 1 week | 181 |
| CsPbBr3/TiO2 core/shell NCs | Hydrolysis-calcination | Water (immersion) | PL | ∼85% | — | 3 months | 181 |
| TEM | |||||||
| PXRD | |||||||
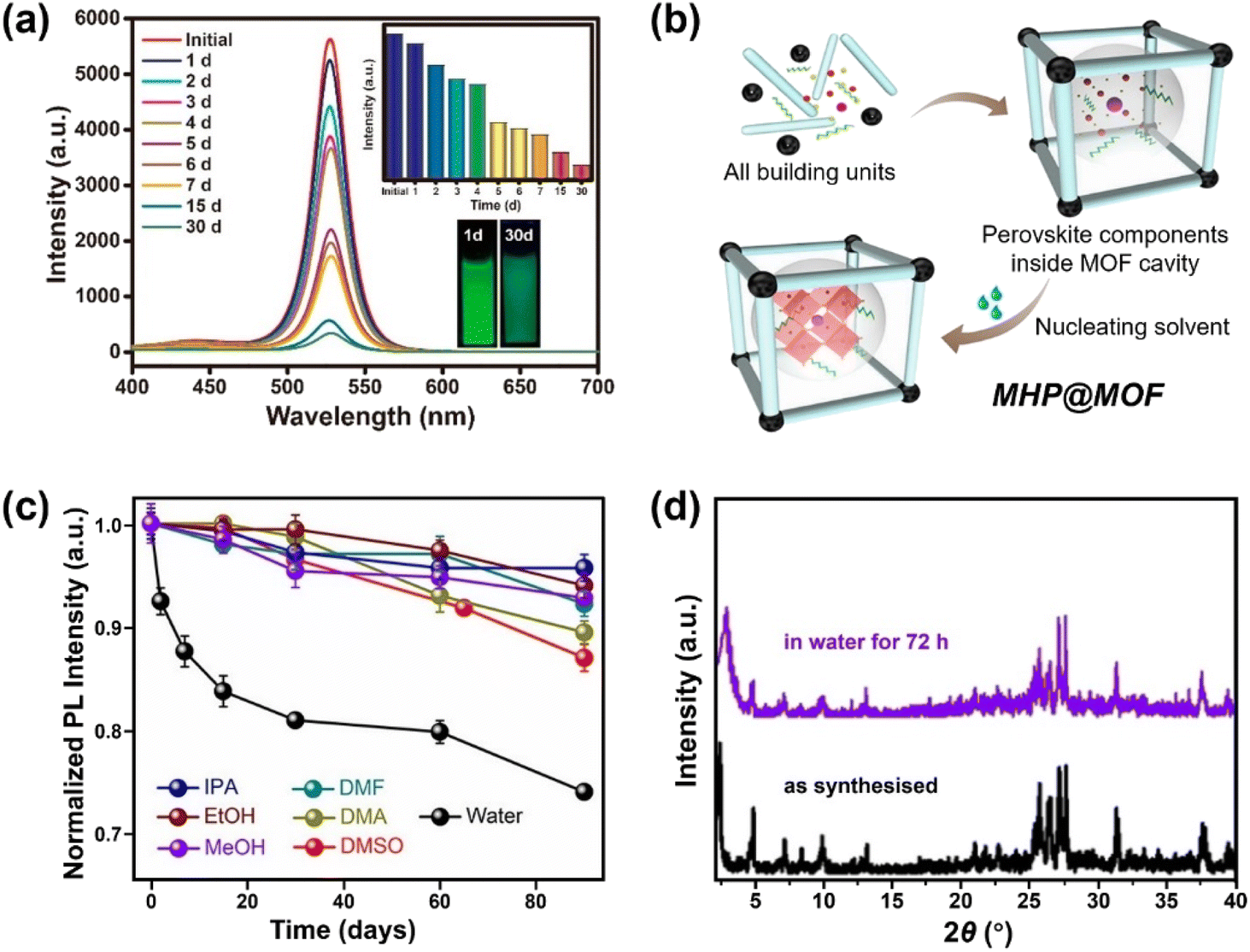 | ||
| Fig. 7 MHPs encapsulated by MOF. (a) Time-dependent PL spectra of CH3NH3PbBr3@MOF-5 composites in water for different days (inset: the evolution of PL intensity and the images of CH3NH3PbBr3@MOF-5 composites under 365 nm light after 1 and 30 days). Reproduced with permission from ref. 213. Copyright 2018 American Chemical Society. (b) Scheme for preparing MHP@MOF composites and (c) normalized PL intensity as a function of time in different polar solvents over a period of 90 days. Adapted from ref. 215. Copyright 2019 American Chemical Society. (d) XRD patterns of CsPbI3@PCN-222(20%) before and after immersion in water for 72 h. Adapted from ref. 218. Copyright 2022 John Wiley and Sons. | ||
Apart from the removal of surface MHPs, the stability and the pore size (cavity diameter and opening aperture size) of MOF matrix also dominate the water stability of MHP@MOF. The MOF should be stable during the synthesis of composites, and the pore size should be well-tailored, which can accommodate monodispersed MHP nanoparticles in the cage, but also suppress the MHP leaching through the opening apertures. For instance, to confine the monodispersed CsPbBr3 nanocrystals (4–5 nm) into MOF, mesopores (pore size >4 nm) instead of common micropores (e.g., ZIF-8, pore size <4 nm)216 are desirable, since the MHP NCs can grow freely inside the cage and would not damage the porous structure of MOFs. To this end, Yu and coauthors utilized the high-valent metal-based MOF (PCN-333(Fe)) as host and constructed a CsPbBr3@PCN-333(Fe) composite material, working as a stable photocathode in Li–O2 battery for at least 200 hours.217 In addition, the long-channel of MOF can also serve as a template for the growth of MHP nanowires. Xia et al. reported that the CsPbI3 nanowires can be grown and encapsulated into Zr-based MOF (PCN-222). The CsPbI3@PCN-222 maintained structure integrity (Fig. 7d) and morphology after water immersion for 72 h due to the excellent protection of the MOF walls.218
Another breakthrough over MOF has been reported by Hou and coworkers. They have successfully prepared (CsPbI3)0.25(agZIF-62)0.75 composites at 350 °C by liquid-phase sintering (ZIF-62, {Zn[(Im)1.95(bIm)0.05]}; Im, imidazolate; bIm, benzimidazolate). The composites extended water stability up to over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours.219 Compared to the silica-based glasses mentioned earlier, the ZIF glasses can be prepared at a lower temperature220 because of the metal–imidazolate–metal linkages and large voids.221
000 hours.219 Compared to the silica-based glasses mentioned earlier, the ZIF glasses can be prepared at a lower temperature220 because of the metal–imidazolate–metal linkages and large voids.221
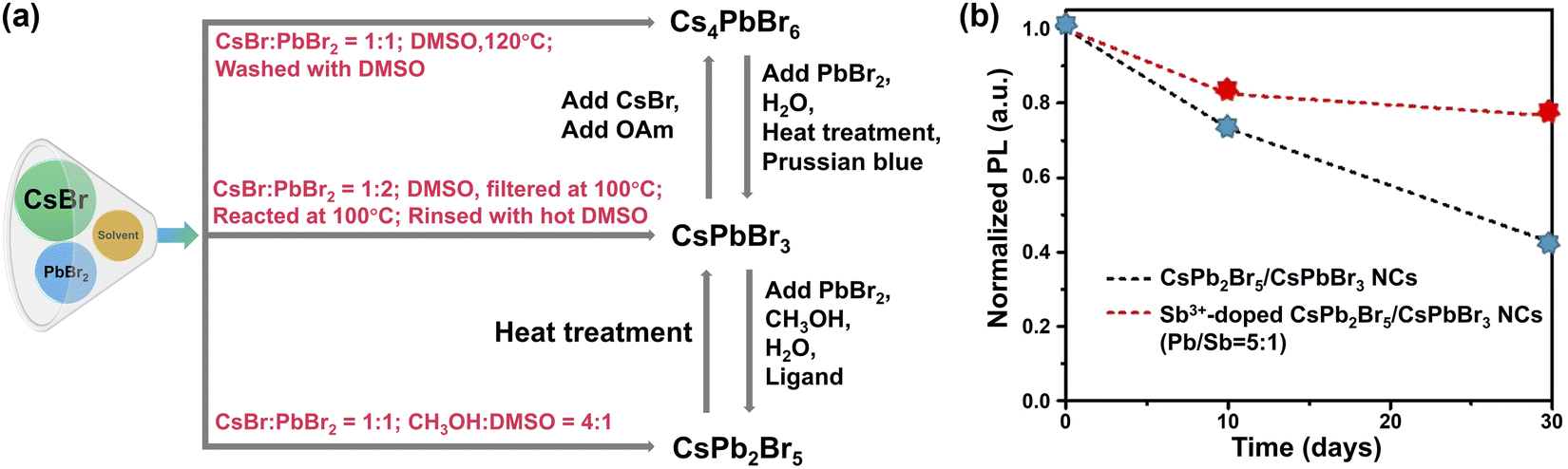 | ||
| Fig. 8 Phase engineering. (a) Schematic diagram illustrating the synthesis of pure CsXPbYBrZ-based MHPs and their potential transformations. (b) PL intensity of CsPbBr3/CsPb2Br5 NCs and Sb3+-doped dual phase CsPbBr3/CsPb2Br5 NCs (Pb/Sb = 5:1) as a function of time. Adapted from ref. 237. Copyright 2021 Elsevier. | ||
Dual phases such as CsPbBr3–CsPb2Br5 NCs232 and CsPbBr3/Cs4PbBr6 NCs233 seem to show enhanced stability. With excess PbBr2, CsPbBr3/CsPb2Br5 core–shell NCs have been synthesized via a modified non-stoichiometric solution-phase method.234 Layered 2D CsPb2Br5 nanosheets are coated on the surface of CsPbBr3 nanocubes. The CsPbBr3/CsPb2Br5 core–shell NCs remained luminous after ultrasonication in water for 2 h. The elevated stability originates from the unique sandwiched structure of 2D CsPb2Br5 in which the Cs+ ions are inserted into the compact-bound inorganic (Pb2Br5)− layers via strong electrostatic interactions.235,236 Further improvement of water stability was realized with Sb3+-doped dual-phase CsPb2Br5/CsPbBr3 NCs (preserving 80% of the original PL value up to 30 days, Fig. 8b).237 This is because the replacement of Pb2+ (1.19 Å) ions with smaller Sb3+ ions (0.92 Å) leads to increased lattice energy.238 Meanwhile, rapid hydrolysis of Sb3+ ions in water may also play a role.239,240 The presence of hydrophobic antinomy oxychloride in Sb-doped MHPs is also considered to be the reason for improved water stability.238,241,242
The reasons for the enhanced stability are manifold. As depicted in Fig. 9a, one is the dissolution of the defective surface and thus the formation of near to ideal stoichiometry (CsPbBr3) having high stability.247,248 Benefiting from improved crystal quality of MHP NCs and the immiscibility of hexane with water, further dissolution of CsPbBr3 NCs would not happen temporarily. Another explanation is the concurrent passivation and the formation of halide-rich surface upon CsX-stripping.249 The origin of this passivation results from the dissolution of surface layers of MHP NCs during the CsX-stripping process and subsequently the formation of non-stoichiometric MHP surface (Fig. 9b).250,251 For example, with Cs4PbBr6 NCs as starting materials, the following water treatment engenders a phase transformation into CsPbBr3, accompanied by a passivation effect from the CsBr salt in water. The resultant CsPbBr3 NCs display ultra-stability (over 200 days with ∼20% decrease in the initial PL value, Fig. 9d).250 A third potential mechanism is based on the attached isomorphic hydroxyl (OH) ligands over CsPbBr3 nanocrystals that are assumed to prevent MHP NCs from further water attack (Fig. 9c).88 The formation of hydroxyl might have originated from the self-ionization of water. It is reported that polar solvent (for example, isopropanol, C3H7OH) will ionize itself to produce C3H7O− and C3H7OH2+ and replace OA− and OAm+ respectively, acting as shorter and more reactive ligands and inducing the oriented growth of MHP NCs.252 Similar ionization is expected in the case of water, which triggers the formation of OH ligands on MHP NCs.253 Additionally, the hydroxyl groups might provide a passivation effect by forming hydrogen bonding interaction with halide ions in MHPs.254 Besides these possibilities, the formation of atomically thin quasi-2D CsPbBr3 nanosheets (NSs) also favors the stability,245 because the (quasi-)2D structures features improved stability255 and suppressed ion migration256 than their 3D counterparts.
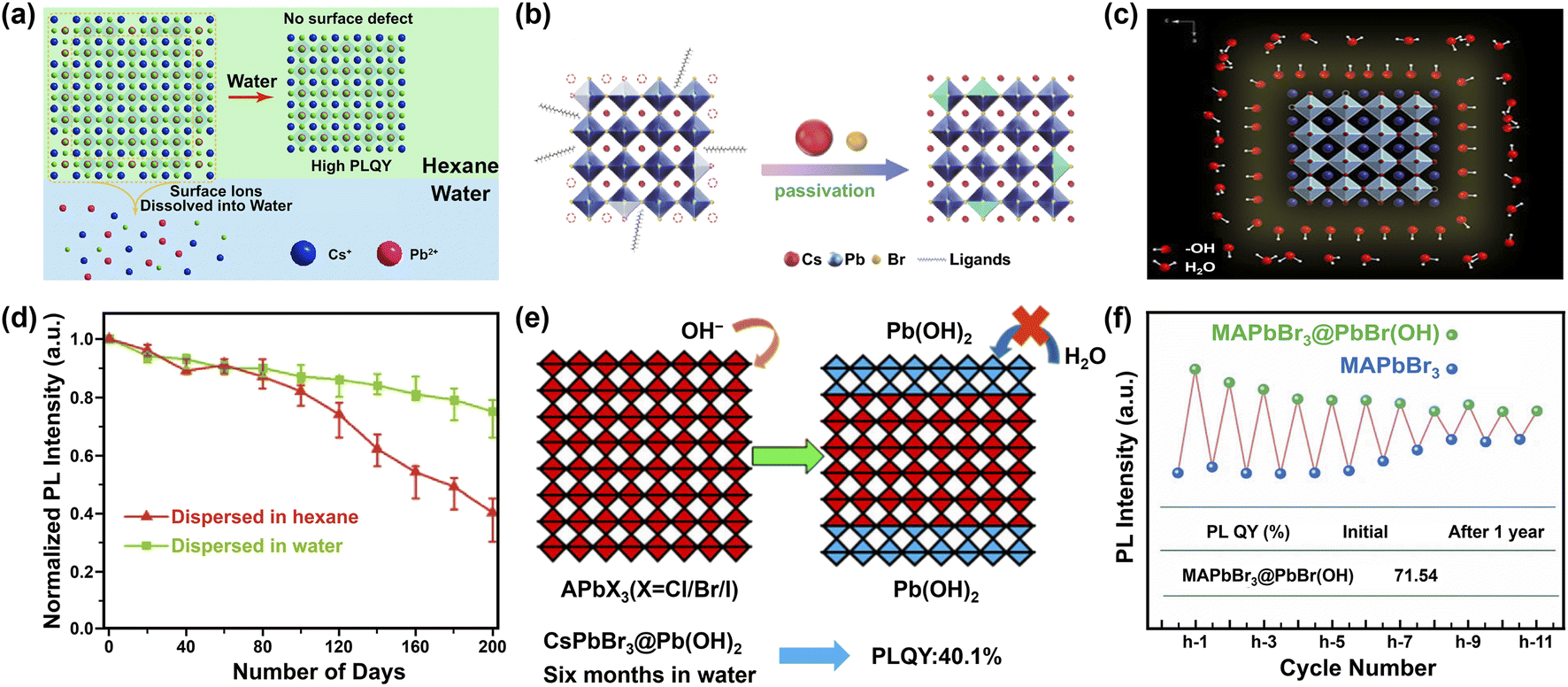 | ||
| Fig. 9 Water-assisted engineering for preparing water-stable MHPs. Three possible mechanisms (a–c) for the synthesis of water-stable MHPs with water-assisted engineering strategy: (a) illustration of forming CsPbX3 NCs in hexane with perfect unit cell after water-treatment. Adapted from ref. 247. Copyright 2018 American Chemical Society. (b) Schematic diagrams of CsPbBr3 NCs passivated by CsBr. Reproduced with permission from ref. 250. (c) Illustration of CsPbBr3 NCs stabilized by hydroxyl (OH) ligands. Reproduced from ref. 88. (d) Normalized PL intensity's evolution of CsPbBr3 NCs dispersed in water and hexane respectively. Adapted from ref. 250. (e) Formation mechanism of Pb(OH)2 by Lewis base vapor diffusion (LBVD) method. Adapted from ref. 259. Copyright 2018 American Chemical Society. (f) The variant PL intensity of MAPbBr3@PbBr(OH) and MAPbBr3 during cycling. Adapted from ref. 261. | ||
Efforts have been devoted to decoding the underlying mechanism with density-functional theory (DFT) calculations. Recent studies indicate that Cs-rich precursor favors the formation of CsBr-terminated surface, whereas low Cs concentration results in PbBr2 terminations. Compared with PbBr2-terminated surface, the former case is more stable even after the adsorption of water molecules according to the DFT results.257 Yoo et al. proposed that a ligand transition from anionic ligands to cationic ligands in metal halide medium also contributes the improved water stability.258
In situ grown Pb(OH)2via a Lewis base vapor diffusion (LBVD) method has been proposed as another strategy to stabilize the MHPs (Fig. 9e).259 When excess methylamine is diffused into the solution of MHPs, a basic solution (pH > 12) is formed. Then highly nucleophilic OH− ions react with the peripherical layer of [PbX6]4− on MHPs, forming a dense Pb(OH)2 layer. Notably, the as-obtained Pb(OH)2-coated perovskites maintained structural stability in water for more than 6 months and retained the fluorescence property in water even after grinding or sonication. The Pb(OH)2-coated MAPbX3 perovskites can be also obtained via organic cation exchange between formamidinium (FA+) and MA+ in water, which can further extend the stability up to one year.260 Instead of the time-consuming LBVD method, to form surface hydroxides may be obtained by simply adjusting the pH of metal halide precursor solution with ammonium hydroxide.261,262 Such MAPbBr3@PbBr(OH) retained 89.9% of the initial PL value (i.e., 64.28%) after being immersed in water for 1 year (Fig. 9f). The PbBr(OH) layer can be also extended to all-inorganic MHPs. For example, by modulating the water content, water-stable CsPbBr3/CsPb2Br5@PbBr(OH) and CsPbBr3@PbBr(OH) nano/micro-spheres have been obtained respectively, in which CsPbBr3/CsPb2Br5@PbBr(OH) showed excellent water stability and maintained 91% of initial PL intensity after 18 months of storage in water.263 Dong et al. also found that rod-like CsPb2Br5-embedded Pb(OH)Br obtained 92.2% of initial PL intensity after soaking in water for 165 days, indicating a good stability.264 PbBrF matrix also shows good protecting ability in CsPbBr3/PbBrF composites having no decrease of PLQY after 30 days in water.265 DFT calculations indicated that the improved stability originates from the increased decomposition enthalpy after introducing insoluble PbBr(OH) compared with that of bare MAPbBr3 (ref. 261 and 263) or the positive energy cost for water entering the lattice of PbBr(OH).266
Even though water-assisted engineering strategy can dramatically improve MHPs stability in water, it should be mentioned that no photocatalytic applications over this class of water-stable MHPs have been reported yet.
2.2 Common-ion effect
It is well-known that a salt can be precipitated by adding other soluble salts having common ions to the solution. Inspired by this, Nam's group have proposed the idea of common-ion effect to prepare water-stable MHPs in aqueous solution.28 Although the MHPs are stabilized, it is a difficult-to-implement strategy in the practice due to the highly corrosive nature of the solution (pH < −0.5 and [I−] > 2.5 mol L−1). Clearly, a more practical medium is needed. Later, Geng et al. found that CH3NH3PbX3 (X = Br or Cl/Br) nanocrystals could be synthesized in aqueous solution when the pH value was in the range of 0–5.267 In this aqueous solution, the [PbX6]4− ions would adsorb on NCs, facilitating the formation of halide-rich surface of NCs, thus preventing the dissolution of MHP NCs in water. However, they would decompose in neutral solution.To realize the water stability of MHPs, our group explored to employ the bismuth halide perovskites as photocatalysts owing to their better stability in solar cells and photocatalysis.268–270 We found that the tri(dimethylammonium) hexaiodobismuthate (DA3BiI6) could be stabilized in aqueous solution using dimethylammonium iodide (DAI) without addition of acids.271 We noted that a stepwise transformation of BiI3 → [BiI4]− → [BiI6]3− as the increase of DAI concentration and only [BiI6]3− ions existed when the concentration of DAI was higher than 0.15 M (Fig. 10a and b). The structural integrity of DA3BiI6 could be preserved for more than two weeks (Fig. 10c and d). Similarly, a series of 2D MHPs, including C6H5CH2NH3PbI4, C6H5(CH2)2NH3PbI4 and C6H5(CH2)3NH3Pb2I7 have been stabilized in iodide salt aqueous solutions.272
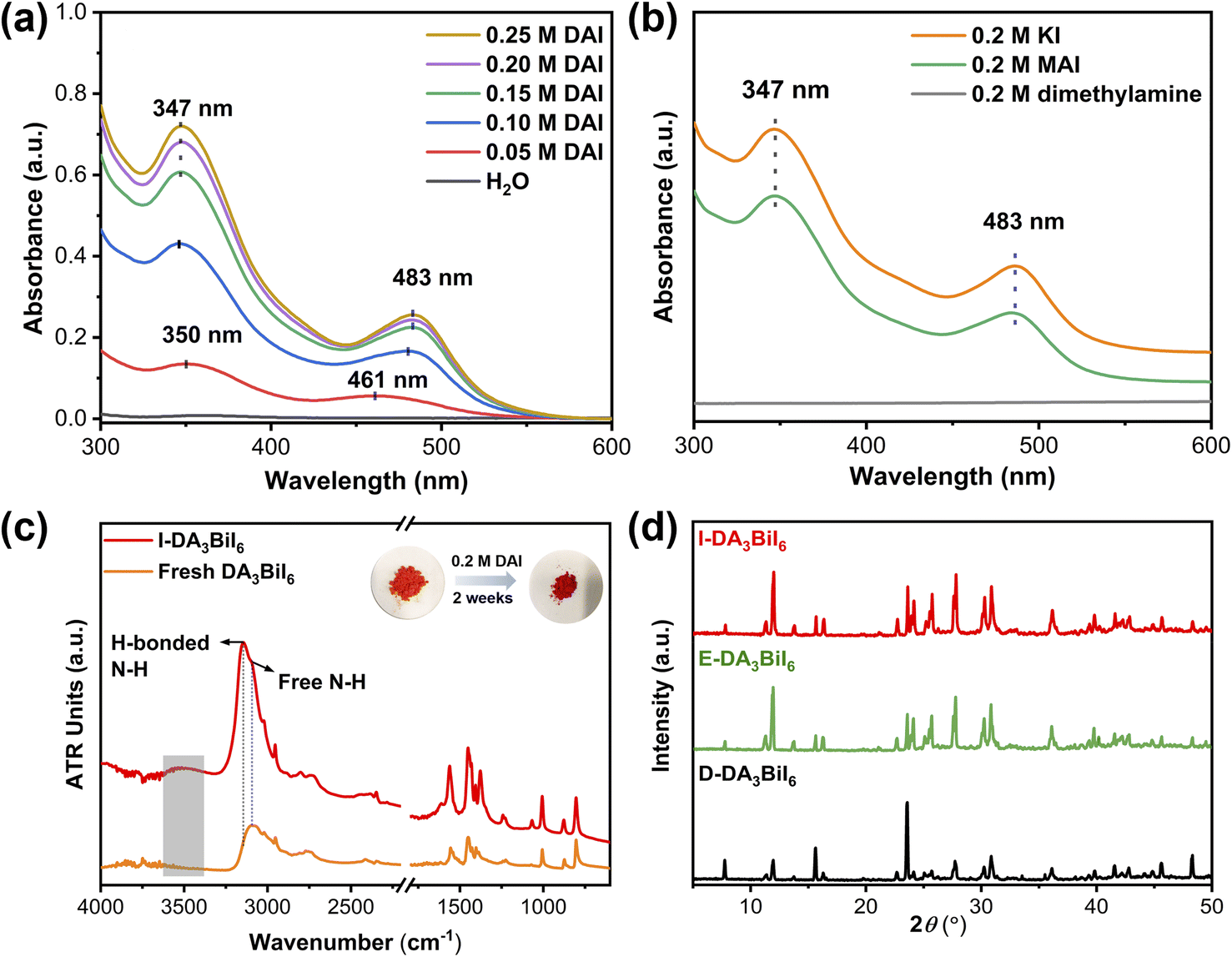 | ||
| Fig. 10 Common-ion effect. (a) UV-vis absorption spectra of DAI aqueous solutions after immersing DA3BiI6 for 1 day. (b) UV-vis absorption spectra of 0.2 M aqueous dimethylamine, KI, and MAI solutions after immersing DA3BiI6 for 5 min. (c) ATR-FTIR spectra and color change of DA3BiI6 and immersed-DA3BiI6 powder. (d) XRD patterns of DA3BiI6 after immersion in water, ethanol and DA3BiI6 synthesized from DAI. Reproduced from ref. 271. | ||
2.3 Intrinsic water stability
Besides the strategies developed based on surface engineering and common-ion effect discussed above, exploring intrinsically water-stable MHPs are potentially advantageous because they can bypass the obstacles of water instability in aqueous media.A few 3D MHPs have exhibited an intrinsic stability in water. As far as we know, the first reported intrinsically water-stable MHPs were hydroxyl ammonium lead iodo chloride (OHNH3PbI2Cl) and hydroxyl ammonium lead chloride (OHNH3PbCl3).273 After stirring in deionized water for 1 h, no leaching of Pb2+ was detected. In addition, no color change of the solids was observed after a 45 days immersion in water, suggesting outstanding water-stability. It is speculated that strong hydrogen bonding interactions among MHPs contribute to the water stability. However, it is worth noting that their crystal structures are still under dabate.274,275 Also, C6H4NH2CuBr2I with ABX3 structure has exhibited no structure change after water immersion for 4 h.276,277 Currently, another widely studied water-stable MHPs are dimethylammonium tin halide perovskites,278,279 which feature no decomposition after 20 h in deionized water (see Fig. 11a). Based on DFT calculations (Fig. 11b), the intrinsic water-stability originates from higher water surface adsorption energy, higher water osmotic energy barrier, and smaller intralayer spacing inside DMASnI3 structure compared with the Pb-based counterpart.279
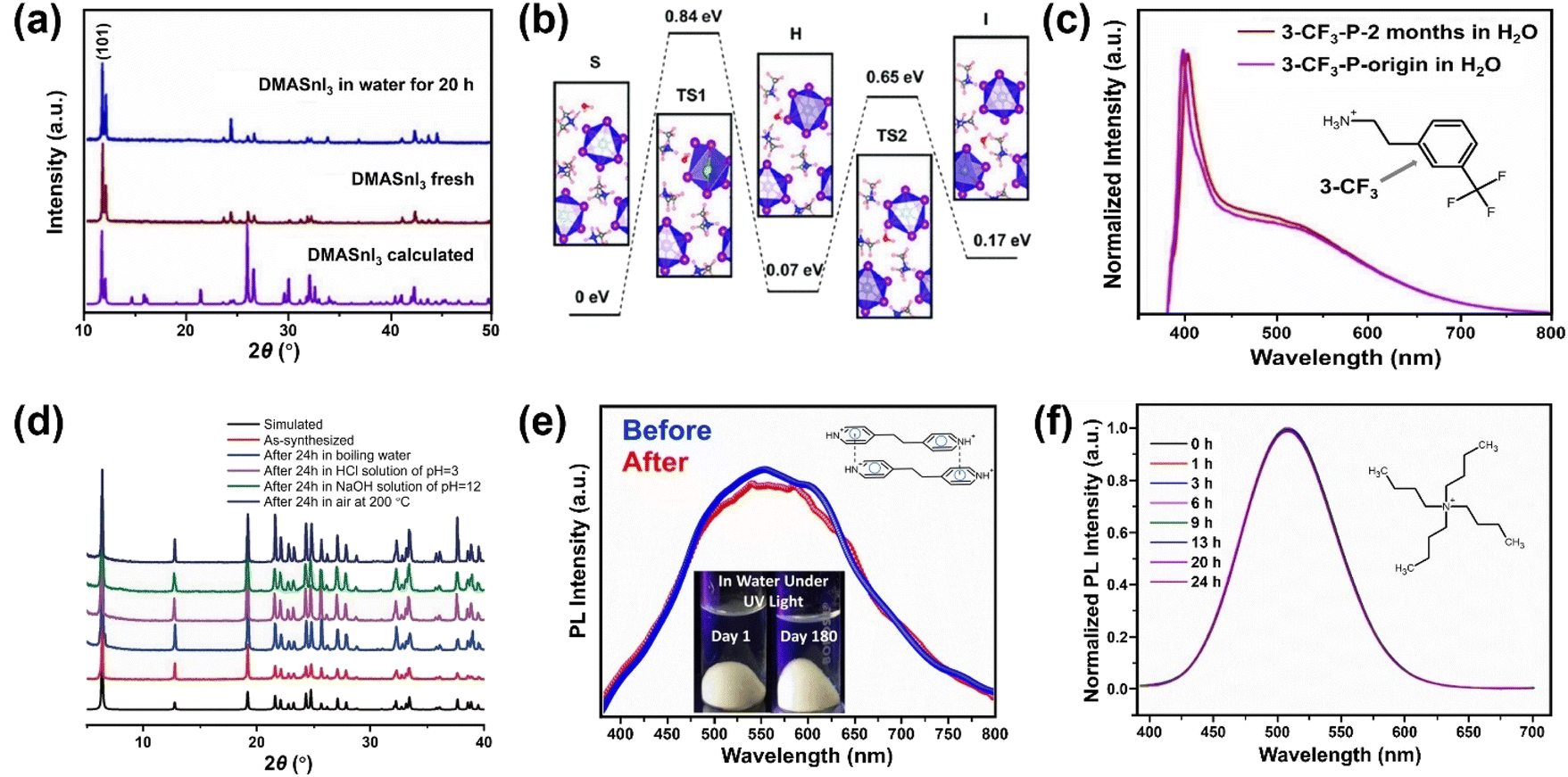 | ||
| Fig. 11 Intrinsically water-stable MHPs. (a) The powder XRD (PXRD) patterns of DMASnI3 before and after water treatment. Adapted from ref. 279. Copyright 2020 John Wiley and Sons. (b) Relative energy profile for water infiltration into (101) surface of DMASnI3 crystal. Adapted from ref. 279. Copyright 2020 John Wiley and Sons. (c) PL spectra of 3-CF3-MHP before and after being kept in water for 2 months. Adapted from ref. 285. Copyright 2022 American Chemical Society. (d) PXRD patterns of [Pb2Cl2]2+ [−O2C(CH)2CO2−] before and after chemical treatment for 24 h. Adapted from ref. 288. Copyright 2017 John Wiley and Sons. (e) PL spectra of fresh and immersed (4,4′-EDP)Pb2Br6 for 180 days. Adapted from ref. 294. Copyright 2021 John Wiley and Sons. (f) Normalized PL spectra evolution of (C4H9)4NCuCl2 after water immersion for different periods. Adapted from ref. 296. Copyright 2021 American Chemical Society. | ||
In contrast to 3D MHPs, low-dimensional (2D, 1D and 0D) perovskites have shown better environmental stability.280–282 A typical 2D perovskite can be regarded as interdigitating bulky organic bilayers intercalated by inorganic layers. The replacement of MA+ with bulkier alkylammonium cations results in enhanced stability.255 Inspired by this, several (quasi-) 2D perovskites having a bulky organic cation, e.g., phenylethylammonium (C8H9NH3+, PEA+),283–285 1-hexadecylammonium (CH3(CH2)15NH3+, HDA+),286 have been developed (Fig. 11c). Among them, PEA-based MHPs have been widely investigated and proven that increased van der Waal's interactions255 and reduced water adsorption energy are the key factors for the improved stability.283 Another possibility is to have a better interaction between the A site (such as cysteamine,287 α,ω-alkanedicarboxylates [−O2C(CH2)4CO2−],288,289 bipyridine290) and inorganic framework (Fig. 11d).288 As supported by DFT calculations, the strong coordination between Pb atoms and adipate dianions increases the energy cost of surface hydrolysis and limits the penetration of water molecules.289
Meanwhile, a series of 1D and 0D MHPs with intrinsic water stability have been reported including 1D [N-methyldabconium]PbI3,291 [(AD)Pb2Cl5],292 (DAO)Sn2I6 (DAO = 1,8-octyldiammonium),293 (4,4′-TMDP)Pb2Br6 (TMDP = trimethylenedipyridinium),294 (4,4′-EDP)Pb2Br6 (EDP = ethylenedipyridinium),294 and 0D (3-ethylbenzo[d]thiazol-3-ium)4Bi2I10,295 (C4H9)4NCuCl2.296 The cations are summarized in Fig. 12, and the details of stability are listed in Table 4. The molecular design strategies aim to increase the ionization energy,291 introduce cation–π interaction (Fig. 11e),294 enhance steric hindrance effect (Fig. 11f),292,296 or utilize hydrogen-bond-free A sites.295 Among them, one report introduced the concept of long-range intermolecular cation–π interactions among A-site cations (4,4′-TMDP or 4,4′-EDP) of hybrid perovskites and facilitate the formation of polymer-like network, imparting water stability up to 180 days (Fig. 11e). Cation–π interaction is originated from the noncovalent interaction between the π face of aromatic ring and cation (such as alkali cations, ammonium ions),297 which is stronger than the cation–water interactions during the decomposition of MHPs.298 Undoubtedly, the development of intrinsic stability of MHPs in water may provide new directions and opportunities to advance the photocatalytic applications.
 | ||
| Fig. 12 Chemical structures of organic cations used for fabricating water-stable MHPs. | ||
| Water-stable MHPs | Stability | Ref. | |||||
|---|---|---|---|---|---|---|---|
| Materials | Methods | Medium | Characterizations | Retained PL intensity | PLQY before (after) | Observed durability | |
| a NBCAnPbI3: 4-[(N-3-butyne)carboxyamido]anilinium lead(ll) iodide; ICP-OES: inductively coupled plasma optical emission spectrometry. | |||||||
| (C4H9)4NPbI3 | Solvent evaporation | Water (immersion) | PXRD | — | — | 5 days | 76 |
| NBCAnPbI3{CH3NH3PbI3} | In situ synthesis | Water (immersion) | UV-vis absorption spectroscopy | — | — | 30 min | 73 |
| PXRD | |||||||
| Rb0.05Cs2.95Bi2I9 single crystals | Temperature lowering method | Water (immersion) | PL, PXRD, XPS and UV-vis absorption spectroscopy | ∼100% | 17.63% (—) | 24 h | 308 |
| OHNH3PbI2Cl crystals | Solution method | Water (stirring) | UV-vis absorption spectroscopy | — | — | 45 days | 273 |
| OHNH3PbCl3 crystals | Solution method | Water (stirring) | UV-vis absorption spectroscopy | — | — | 45 days | 273 |
| C6H4NH2CuBr2I thin film | Grinding and spin-coating | Water (immersion) | PXRD | — | — | 4 h | 276 |
| C6H4NH2CuBr2I thin film | — | Water (immersion) | PXRD | — | — | 2 h | 277 |
| UV-vis absorption spectroscopy | |||||||
| (CH3)2NH2SnI3 (DMASnI3) single crystals | Temperature lowering method | Water (immersion) | PXRD | — | — | 16 h | 278 |
| DMASnIXBr3−X crystals | Temperature lowering method | Water (immersion) | PXRD | — | — | 20 h | 279 |
| XPS | |||||||
| FTIR | |||||||
| UV-vis diffuse reflectance spectroscopy | |||||||
| Mn-doped (PEA)2PbBr4 crystals | Lewis base-assisted precipitated method | Water (immersion) | High-power XRD | — | >45% (—) | 45 days | 283 |
| PEA2SnBr4 | Wet-chemistry (solvent evaporation) | Water (stirring) | PXRD | — | — | 4 h | 284 |
| ICP-OES | |||||||
| UV-vis absorption spectroscopy | |||||||
| XPS | |||||||
| Trifluoromethyl-modified PEA2PbBr4 | Temperature lowering method | Water (immersion) | PL | ∼100% | 18.11% (—) | 74 days | 285 |
| (HDA)2PbI4 (HDA+ = CH3(CH2)15NH3+) | Modified ligand-assisted reprecipitation | Water (immersion) | PXRD | — | — | 30 min | 286 |
| PL | |||||||
| (HCya)2PbI4 (Cya = HS(CH2)2NH2) crystals | Temperature lowering method | 50% isopropanol-water (immersion) | — | — | — | >30 s | 287 |
| [Pb2X22+] [−O2C(CH2)4CO2−] crystals | Hydrothermal method | Boiling water, HCl solution (pH 3), and NaOH solution (pH 12) | PXRD | — | — | 24 h | 288 |
| [Pb2X22+] [−O2C(CH2)4CO2−] crystals | Hydrothermal method | HCl solution (pH 3–6), pure water (pH 7) | PXRD | — | — | 24 h | 289 |
| NaOH solution (pH 8–11) and boiling water | |||||||
| APbX2 (A = bipyridine) crystals | Ligand-assisted reprecipitation | Water (immersion) | PXRD | — | — | 24 h | 290 |
| XPS | |||||||
| SEM | |||||||
| [N-Methyldabconium]PbI3 crystals | Solvent evaporation | Water (immersion) | PXRD | — | — | 15 h | 291 |
| Dielectric permittivity | |||||||
| [(AD)Pb2Cl5] (AD = acridine) micro-belts | Precipitation | Water (immersion) | PL | ∼100% | 7.45% (58.79%) | 60 days | 292 |
| PXRD | |||||||
| SEM | |||||||
| (DAO)Sn2I6 (DAO = 1,8-octyldiammonium) crystals | Temperature lowering method | Water (immersion) | XPS | — | 20.3% (—) | >15 h | 293 |
| UV-vis absorption and Raman spectra, PXRD | |||||||
| 4,4′-Trimethylenedipyridinium lead bromide crystals [(4,4′-TMDP)Pb2Br6] | Temperature lowering method | Water (immersion) | PL | ∼100% | 3.7% (—) | 180 days | 294 |
| UV-vis absorption, PXRD | |||||||
| 4,4′-Ethylenedipyridinium lead bromide crystals [(4,4′-EDP)Pb2Br6] | Temperature lowering method | Water (immersion) | PL | ∼100% | 4% (—) | 180 days | 294 |
| UV-vis absorption, PXRD | |||||||
| (3-Ethylbenzo[d]thiazol-3-ium)4Bi2I10 | Solvothermal method | Water (immersion) | PXRD | — | — | 7 days | 295 |
| (EtbtBi2I10) single crystals | |||||||
| (C4H9)4NCuCl2 single crystals | Solvent evaporation | Water (immersion) | PL, XPS | 98.6% | 82% (—) | 24 h | 296 |
| PXRD | |||||||
| Cs2PtI6 | Hydrothermal method | Water (immersion) | PXRD | — | — | 4 h | 302 |
| CH3NH3Pb(SCN)2I | Solvent evaporation | 95% RH | UV-vis absorption spectra | — | — | 4 h | 309 |
Besides the selection of A cations, substitution of the B sites with smaller divalent metals could reduce the lattice parameter and increase the cohesive energy, which has been reported to improve the MHP stability.299–301 Substitution of Pb2+ with smaller divalent metals would also favor the formation of vacancy-ordered A2BX6 double perovskites with improved stability. Hamdan et al. synthesized a vacancy ordered halide perovskite Cs2PtI6 exhibiting extraordinary stability up to 1 year under ambient condition and showing stability under high temperature (350 °C), extremely acidic (pH 1) and basic (pH 13) solutions.302 DFT calculations suggest that the improved stability is due to the strong covalent interaction of the B–X bonds in the isolated the [BX6]2− clusters.303
Halogen anion substitution is another approach. A partial replacement of iodide with bromide was found to enhance stability and water tolerance in mixed iodide-bromide MHP compositions.304 This is related to the suppression of oxygen incorporation and the presence of stronger hydrogen bonds between the MA+ cation and Br− ions as the bromide content increases305 as well as the weaker interaction of water with bromide.306 Apart from halogen ions, pseudohalides (cyanide, cyanate, thiocyanate, selenocyanate, azide, BF4−, PF6−, BH4−, N3− and HCOO−) have shown significant stability enhancement of MHPs.307 For instance, thiocyanate (SCN)-substituted CH3NH3Pb(SCN)2I was shown to be more stable than pristine CH3NH3PbI3 (for 4 h vs. <1.5 h) under 95% humidity.309
3 Photocatalytic applications
In the past seven years, water-stable MHPs have attracted much attention for numerous potential applications (Table S1 and Fig. S1†) owing to their enhanced stability and improved performance. Very recently, it has been exploited as potential photocatalysts in different reactions. In this section, we will provide an overview of these photocatalytic applications and will show lots of nice and encouraging studies for pollutant degradation, H2 generation, CO2 reduction and organic synthesis over water-stable MHP photocatalysts.3.1 Pollutant degradation
Photocatalytic degradation is an attractive way to remove pollutants from wastewater and thus a great amount of work has been devoted to exploring effective photocatalysts. Aamir et al. firstly reported on water-stable OHNH3PbI2Cl and OHNH3PbCl3 for photocatalytic dye degradation of Direct Yellow 27 dye under sunlight with a degree of degradation of 93.98% within 20 min and almost 100% after 55 min, respectively.273 However, both catalysts are only UV-responsive. On the other hand, Ghosh and co-workers reported that the water-stable MAPbBr3@ZIF-8 composites could degrade different pollutants (methyl orange, methyl red and nitrofurazone) under visible light (60 W LED lamp, λmax ∼ 530 nm) or sunlight. The composite exhibited a higher degradation for methyl orange and could degrade methyl orange up to 90% in 100 min (degradation rate constant: 0.02723 min−1), shown in Fig. 13a.215 Although the degradation rate is not high enough compared to the other materials, it can be improved by using photoactive MOFs.310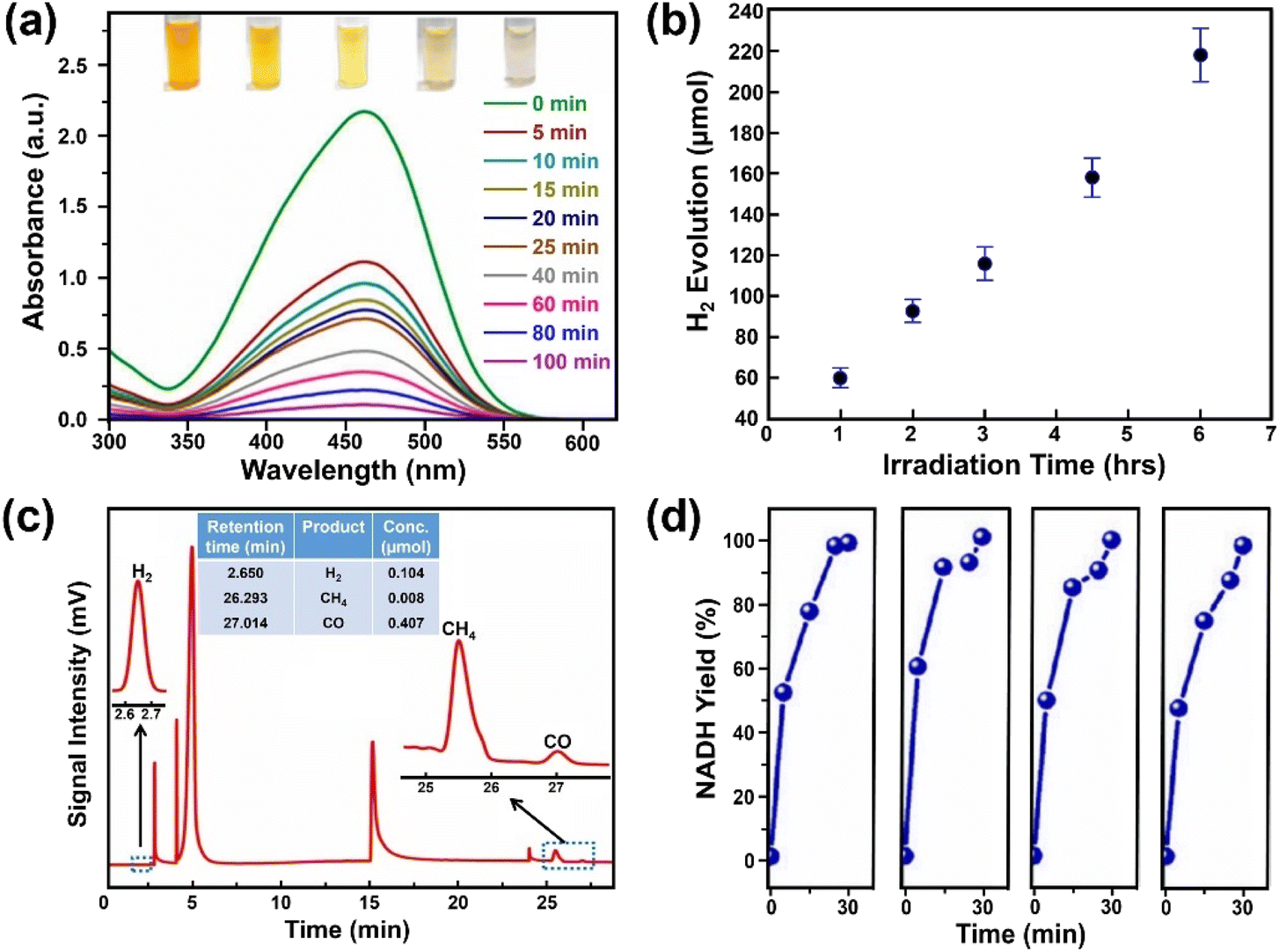 | ||
| Fig. 13 Photocatalytic applications over water-stable MHPs. (a) Absorption spectra evolution of methyl orange solution degraded by MAPbBr3@ZIF-8 under visible light irradiation. Adapted from ref. 215. Copyright 2019 American Chemical Society. (b) Hydrogen evolution over 33 wt% DMASnBr3@g-C3N4 composite (1 g L−1, 10% v/v triethanolamine, 3 wt% Pt) under simulated solar light. Adapted from ref. 313. Copyright 2020 John Wiley and Sons. (c) CO2 reduction products over CsPbBr3@g-C3N4 composite under AM1.5 G simulated sunlight. Adapted from ref. 317. Copyright 2022 Elsevier. (d) Nicotinamide adenine dinucleotide (NADH) yields over DMASnI3 irradiated by blue LED lamp (wavelength of 450 nm) with four independent tests. Adapted from ref. 279. Copyright 2020 John Wiley and Sons. | ||
To alleviate the toxicity associated with Pb, some lead-free MHPs have been explored. Wu et al. prepared a C6H4NH2CuCl2I film to degrade rhodamine B (RhB) under visible light irradiation. Degradation of 18% of RhB in 60 min was achieved (degradation rate constant: 0.003 min−1). They found that the sluggish transfer rate of holes restrains photocatalytic performance of this catalyst. After coupling the photocatalyst with a hole-transporting material (CuO), the charge separation was enhanced, resulting in a degradation rate of 0.005 min−1.311 As also suggested by the study, one may enhance the overall photocatalytic activity by constructing heterojunctions that rectify electron–hole transport at the interface of two semiconductors to facilitate better charge separation and to limit photocharge recombination. A good example is a nanocomposite of PEA2SnBr4/g-C3N4 prepared via ball milling of the constituents. Compared with pristine g-C3N4, the heterojunction decreased the degradation time of methylene blue (10−5 M) from 90 min to 45 min (degradation rate constant: 0.078 min−1) under solar light.284 Among the so far reported photocatalysts, DMASnI3 seems to show the best photocatalytic performance with a complete conversion of methyl orange (100 mg L−1) in 12–15 min (degradation constant rate: ∼0.13 min−1) under visible light.279
3.2 H2 evolution reaction (HER)
MHPs have also been regarded as a promising family of materials for photocatalytic HER. However, due to the constraint of water stability, previous works in MHP photocatalysts mainly focused on hydrogen generation from concentrated halide acids as summarized before.40–47 Here, the results related to MHP-based photocatalysts for H2 production from water are presented. The first results of H2 evolution from water with MHPs was reported by Tao's group278 using DMASnI3, H2 evolution from water under 300 W Xe-lamp was explored and H2 evolution rate of 3.2 μmol g−1 h−1 was obtained. One year later, Malavasi's group observed that DMASnBr3 is also highly air-resistant.312 They proved that bare DMASnBr3 photocatalyst exhibited a HER rate of 6 μmol g−1 h−1 (50 mW cm−2, 1500 W Xenon lamp with UV filter), which was further improved to 11 μmol g−1 h−1 after introducing triethanolamine (TEOA) as sacrificial agent and 1 wt% Pt as co-catalyst. Later, g-C3N4 was introduced to further improve the HER performance by constructing heterojunctions. As shown in Fig. 13b, the DMASnBr3@g-C3N4 (33 wt% DMASnBr3) exhibited stable HER activities and reached an impressive H2 production of 1730 μmol g−1 h−1 with an apparent quantum yield (AQY) of 6.6% (50 mW cm−2, 1500 W Xenon lamp with UV filter).313 The enhancement of photocatalytic activity is closely tied to the suitable bandgap and matched band alignment. Same strategy was extended to PEA2SnBr4/g-C3N4 and Cs3Bi2Br9/g-C3N4 systems and the maximum HER rates of 1600 μmol g−1 h−1 (50 mW cm−2, 1500 W Xenon lamp with UV filter) and 1050 μmol g−1 h−1 (50 mW cm−2, 1500 W Xenon lamp with UV filter) were achieved, respectively.284,314 Apart from hydrid MHPs, inorganic MHPs have also been employed as photocatalysts for HER due to their better stability in humid air.315 Yin et al. prepared a series of inorganic Cs2PtxSn1−xCl6 (0 ≤ x ≤ 1) crystals via a hydrothermal method. The Cs2Pt0.05Sn0.95Cl6 catalyst exhibited good phase stability in water for at least 4 hours and obtained a rate of 16.11 μmol g−1 h−1 (300 W Xe lamp) for hydrogen evolution from water using TEOA as the sacrificial agent.209 Encouragingly, Fei's group reported the first organolead iodide layered crystalline material [Pb8I8(H2O)3]8+[−O2C(CH2)4CO2−]4 (TJU-16) with overall photocatalytic water splitting characteristics few years ago. Combing Rh as co-catalysts, the TJU-16-Rh0.22 exhibited a hydrogen evolution rate of 31 μmol g−1 h−1 (300 W Xenon lamp with an AM1.5 G filter) with AQY of 0.13% at 320 nm.2893.3 CO2 reduction
To alleviate environmental issues related to ever-increasing levels of CO2 concentration in the atmosphere, photocatalytic valorization of CO2 to useful compounds such as CO, methane, formic acid and alike have been placed in the focus of research for decades. Recently, Chen et al. combined water-stable perovskite-like organolead iodide [Pb8I8(H2O)3]8+[−O2C(CH2)4CO2−]4 (TJU-16) with Au co-catalyst (Au0.19/TJU-16) for photocatalytic CO2 reduction in aqueous solution.316 Without using any sacrificial agent, the Au0.19/TJU-16 with loading of 0.19 wt% of Au nanoparticles showed the highest photocatalytic CO production rate of 2.5 μmol g−1 h−1 and CH4 production rate of 10.1 μmol g−1 h−1 in water under AM 1.5 G simulated irradiation, achieving a solar-to-fuel conversion efficiency of 0.034%. In addition, the consumption of electrons calculated from CO and CH4 of Au0.19/TJU-16 is 2.2-fold of individual TJU-16. It was concluded that the improvement in the activity originated from the spatial charge accumulation and enhanced interfacial charge transfer when applying the Au co-catalyst. In another study, photocatalyst film of CsPbBr3 nanoparticles coated with monolayered C3N4 (CsPbBr3@g-C3N4) was found to be capable of transforming CO2 to CO, CH4 and H2 (0.407, 0.104 and 0.008 μmol cmcat−2, respectively) in the presence of water vapor under AM1.5 G solar without using scavengers.317 Nanoparticles of the individual phases (i.e., g-C3N4 or CsPbBr3) alone did not produce any reduction species of CO2 (Fig. 13c). Isotopic labeling experiments confirmed the origin of products from CO2 reduction. The improved performance was ascribed to the enhanced stability of CsPbBr3 coated with the g-C3N4 shell as well as by the promoted separation of photocarriers at the heterojunction interface.Doping of the photocatalyst with ionic impurities offers a further strategy to improve their optical absorption, catalytic behavior as well as carrier transport properties.87,251 For example, Co-doped CsPbBr3/Cs4PbBr6 NCs was demonstrated to be superior compared to its pristine counterpart for water-assisted CO2 photoreduction with a conversion of 247 μmol g−1 under visible light irradiation (with CO and CH4 as the main products). The Co-dopant is assumed to provide not only additional active sites, but it is also believed to be responsible for altering the adsorption energies of reactants and intermediates thus influencing the reaction pathways.
3.4 Organic synthesis
Photocatalytic synthesis of organic compounds is another fascinating field to offer environmentally benign and cost-effective routes. Although numerous works have been done with MHPs for organic synthesis, such as C–X (X = C, N, O, P) formation, carbon–carbon cleavage and carbon–hydrogen activation, all these photocatalytic reactions are limited to nonpolar solvent systems.30–33 To broaden their applications in aqueous system, a prerequisite factor is to utilize water-stable MHPs. Ju et al. firstly extended the organic synthesis to aqueous solution with DMASnIxBr3−x for photoenzyme catalysis.279 The nicotinamide adenine dinucleotide (NADH) yield was nearly 100% within only 30 min and exhibited stable reproducibility in four tests under blue LED irradiation (Fig. 13d). 320 μM formic acid can be obtained over a period of 60 min in the photoenzymatic reaction, suggesting an efficient photocatalytic process in artificial photosynthesis.4 Challenges and prospects
In this review, we summarize the significant advances in the development and applications of water-stable MHPs. To date, the water stability of MHPs has been achieved by surface engineering, common-ion effect and intrinsically stable MHPs. As an emerging class of photocatalysts, the photocatalytic study of water-stable MHPs is still in their infancy. Several challenges and future research directions related to water-stable MHPs are identified as follows:(1) Although various strategies have been proposed to obtain water stability of MHPs, to realize their photocatalytic applications, studies should be further focused on the charge carrier transport properties by exploring novel MHPs with intrinsic water compatibility (quasi-3D and 2D MHPs) and/or employing surface passivation that can protect MHPs from water while preserving good carrier transport features.318–323 For example, constructing a MHP@shell core–shell heterostructure is a highly promising way to hit two birds with one stone. By bringing MHPs with semiconducting/conductive shell, not only new catalytically active sites324 or protective coatings325 can be introduced by the second phase, but more importantly rectifying interfaces may be obtained.
(2) While some water-stable MHP based photocatalytic systems have shown good photocatalytic activity in the degradation of organic dyes, more challenging and highly relevant fields such as hydrogen evolution and activation/valorization of CO2 are still in their early stages of development, and catalysts exhibit lower photocatalytic performance compared to traditional photocatalysts.326 One possibility for the inferior activity might lie in the severe surface charge recombination.327 In this regard, constructing a close–contact interface among MHP-based composites, which benefits the sufficient transfer and rectification of photogenerated charges, is highly desirable.328–330 It has been proven that chemical modification or in situ synthesis can result in an intimate heterointerface.151,325,330,331 Therefore, in the future, it is plausible to expect new avenues in this direction as well.
(3) Another vital direction of research is towards an in-depth understanding of reaction mechanisms over MHP-based photocatalysts, as the contemporary results are not entirely coherent and conclusive. Taking CO2 reduction reaction as an example, there are debates over the CO2 adsorption sites on bismuth-based A3Bi2I9 (A = Rb+, Cs+ or MA+)332–335 and the origin of products (photocatalysis317 or photolysis336,337). The photocatalytic HER reaction mechanism over DMASnBr3 is also not exhaustive. It is reported that the binding energy of electron polarons over DMASnBr3 surface dominates the HER activity, but the specific role of electron polarons, as driving force or trapping charges, needs further investigation.338 In this regard, combining analytics, such as in situ Fourier-transform infrared spectroscopy, in situ Raman, etc., with theoretical investigations, will provide new insights in understanding the mechanisms involved and thereby to the design of water-stable and efficient MHPs.
Author contributions
H. Z.: conceptualization, formal analysis, investigation, writing – original draft; K. K.: supervision, funding acquisition, resources, writing – review & editing; S. O.: supervision, funding acquisition, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Kvantum Institute Emerging Project (Charge carrier recombination dynamics in semiconductor materials) at the University of Oulu. H. Z. acknowledges the financial support of Tauno Tönning Foundation (grant no. 20210036) and University of Oulu Scholarship Fund (grant no. 20220010).Notes and references
- L. Gibson, E. N. Wilman and W. F. Laurance, Trends Ecol. Evol., 2017, 32, 922–935 CrossRef PubMed.
- N. Kannan and D. Vakeesan, Renewable Sustainable Energy Rev., 2016, 62, 1092–1105 CrossRef.
- J. H. Kim, D. Hansora, P. Sharma, J.-W. Jang and J. S. Lee, Chem. Soc. Rev., 2019, 48, 1908–1971 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef CAS PubMed.
- P. Zhou, I. A. Navid, Y. Ma, Y. Xiao, P. Wang, Z. Ye, B. Zhou, K. Sun and Z. Mi, Nature, 2023, 613, 66–70 CrossRef CAS PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983 RSC.
- Y.-H. Song, J. S. Yoo, E. K. Ji, C. W. Lee, G. S. Han, H. S. Jung and D.-H. Yoon, Chem. Eng. J., 2016, 306, 791–795 CrossRef CAS.
- S. Chen, T. Takata and K. Domen, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS.
- J. Schneider, M. Matsuoka, M. Takeuchi, J. Zhang, Y. Horiuchi, M. Anpo and D. W. Bahnemann, Chem. Rev., 2014, 114, 9919–9986 CrossRef CAS PubMed.
- Y. Ma, X. Wang, Y. Jia, X. Chen, H. Han and C. Li, Chem. Rev., 2014, 114, 9987–10043 CrossRef CAS PubMed.
- G. Liao, Y. Gong, L. Zhang, H. Gao, G.-J. Yang and B. Fang, Energy Environ. Sci., 2019, 12, 2080–2147 RSC.
- A. Kumar Singh, C. Das and A. Indra, Coord. Chem. Rev., 2022, 465, 214516 CrossRef CAS.
- V.-H. Nguyen, H. H. Do, T. Van Nguyen, P. Singh, P. Raizada, A. Sharma, S. S. Sana, A. N. Grace, M. Shokouhimehr, S. H. Ahn, C. Xia, S. Y. Kim and Q. Van Le, Sol. Energy, 2020, 211, 584–599 CrossRef CAS.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- M. Rahman, H. Tian and T. Edvinsson, Angew. Chem., Int. Ed., 2020, 59, 16278–16293 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- H. Min, D. Y. Lee, J. Kim, G. Kim, K. S. Lee, J. Kim, M. J. Paik, Y. K. Kim, K. S. Kim, M. G. Kim, T. J. Shin and S. Il Seok, Nature, 2021, 598, 444–450 CrossRef CAS PubMed.
- G. Xing, N. Mathews, S. Sun, S. S. Lim, Y. M. Lam, M. Graẗzel, S. Mhaisalkar and T. C. Sum, Science, 2013, 342, 344–347 CrossRef CAS PubMed.
- M. A. Green, A. Ho-Baillie and H. J. Snaith, Nat. Photonics, 2014, 8, 506–514 CrossRef CAS.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967–970 CrossRef CAS PubMed.
- C. Wehrenfennig, G. E. Eperon, M. B. Johnston, H. J. Snaith and L. M. Herz, Adv. Mater., 2014, 26, 1584–1589 CrossRef CAS PubMed.
- P. Chen, W. Ong, Z. Shi, X. Zhao and N. Li, Adv. Funct. Mater., 2020, 30, 1909667 CrossRef CAS.
- Y. Zhou and Y. Zhao, Energy Environ. Sci., 2019, 12, 1495–1511 RSC.
- S. Park, W. J. Chang, C. W. Lee, S. Park, H.-Y. Ahn and K. T. Nam, Nat. Energy, 2017, 2, 16185 CrossRef CAS.
- P. C. K. Vesborg, Nat. Energy, 2017, 2, 16205 CrossRef.
- Y. Lin, J. Guo, J. San Martin, C. Han, R. Martinez and Y. Yan, Chem. –Eur. J., 2020, 26, 13118–13136 CrossRef CAS PubMed.
- M. Corti, S. Bonomi, R. Chiara, L. Romani, P. Quadrelli and L. Malavasi, Inorganics, 2021, 9, 56 CrossRef CAS.
- M. Zhang, W. Sun, H. Lv and Z.-H. Zhang, Curr. Opin. Green Sustainable Chem., 2021, 27, 100390 CrossRef CAS.
- V. Murugesh and S. P. Singh, Chem. Rec., 2020, 20, 1181–1197 CrossRef CAS PubMed.
- S. Shyamal and N. Pradhan, J. Phys. Chem. Lett., 2020, 11, 6921–6934 CrossRef CAS PubMed.
- S. Trivedi, D. Prochowicz, A. Kalam, M. M. Tavakoli and P. Yadav, Renewable Sustainable Energy Rev., 2021, 145, 111047 CrossRef CAS.
- M. A. Raza, F. Li, M. Que, L. Zhu and X. Chen, Mater. Adv., 2021, 2, 7187–7209 RSC.
- X. Zhang, R. Tang, F. Li, R. Zheng and J. Huang, Sol. RRL, 2022, 6, 2101058 CrossRef CAS.
- J. Wang, Y. Shi, Y. Wang and Z. Li, ACS Energy Lett., 2022, 7, 2043–2059 CrossRef CAS.
- C. B. Hiragond, N. S. Powar and S.-I. In, Nanomaterials, 2020, 10, 2569 CrossRef CAS PubMed.
- L. Romani and L. Malavasi, ACS Omega, 2020, 5, 25511–25519 CrossRef CAS PubMed.
- S. Purohit, K. L. Yadav and S. Satapathi, Adv. Mater. Interfaces, 2022, 9, 2200058 CrossRef CAS.
- V. Armenise, S. Colella, F. Fracassi and A. Listorti, Nanomaterials, 2021, 11, 433 CrossRef CAS PubMed.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- J. Chen, C. Dong, H. Idriss, O. F. Mohammed and O. M. Bakr, Adv. Energy Mater., 2020, 10, 1902433 CrossRef CAS.
- Y. Xu, M. Cao and S. Huang, Nano Res., 2021, 14, 3773–3794 CrossRef CAS.
- J. Luo, W. Zhang, H. Yang, Q. Fan, F. Xiong, S. Liu, D. Li and B. Liu, EcoMat, 2021, 3, eom2.12079 CrossRef.
- S. Pan, J. Li, Z. Wen, R. Lu, Q. Zhang, H. Jin, L. Zhang, Y. Chen and S. Wang, Adv. Energy Mater., 2022, 12, 2004002 CrossRef CAS.
- H. Zhou, Q. Chen, G. Li, S. Luo, T. B. Song, H. S. Duan, Z. Hong, J. You, Y. Liu and Y. Yang, Science, 2014, 345, 542–546 CrossRef CAS PubMed.
- N. Tripathi, M. Yanagida, Y. Shirai, T. Masuda, L. Han and K. Miyano, J. Mater. Chem. A, 2015, 3, 12081–12088 RSC.
- W. Oh, S. Bae, S. Kim, N. Park, S.-I. Chan, H. Choi, H. Hwang and D. Kim, Microelectron. Reliab., 2019, 100–101, 113392 CrossRef CAS.
- C. Müller, T. Glaser, M. Plogmeyer, M. Sendner, S. Döring, A. A. Bakulin, C. Brzuska, R. Scheer, M. S. Pshenichnikov, W. Kowalsky, A. Pucci and R. Lovrinčić, Chem. Mater., 2015, 27, 7835–7841 CrossRef.
- J. M. Frost, K. T. Butler, F. Brivio, C. H. Hendon, M. van Schilfgaarde and A. Walsh, Nano Lett., 2014, 14, 2584–2590 CrossRef CAS PubMed.
- Y. Li, X. Xu, C. Wang, C. Wang, F. Xie, J. Yang and Y. Gao, J. Phys. Chem. C, 2015, 119, 23996–24002 CrossRef CAS.
- E. Mosconi, J. M. Azpiroz and F. De Angelis, Chem. Mater., 2015, 27, 4885–4892 CrossRef CAS.
- A. M. A. Leguy, Y. Hu, M. Campoy-Quiles, M. I. Alonso, O. J. Weber, P. Azarhoosh, M. van Schilfgaarde, M. T. Weller, T. Bein, J. Nelson, P. Docampo and P. R. F. Barnes, Chem. Mater., 2015, 27, 3397–3407 CrossRef CAS.
- J. Yang, B. D. Siempelkamp, D. Liu and T. L. Kelly, ACS Nano, 2015, 9, 1955–1963 CrossRef CAS PubMed.
- Z. Song, A. Abate, S. C. Watthage, G. K. Liyanage, A. B. Phillips, U. Steiner, M. Graetzel and M. J. Heben, Adv. Energy Mater., 2016, 6, 1600846 CrossRef.
- G. E. Eperon, S. N. Habisreutinger, T. Leijtens, B. J. Bruijnaers, J. J. van Franeker, D. W. DeQuilettes, S. Pathak, R. J. Sutton, G. Grancini, D. S. Ginger, R. A. J. Janssen, A. Petrozza and H. J. Snaith, ACS Nano, 2015, 9, 9380–9393 CrossRef CAS PubMed.
- G. Grancini, V. D'Innocenzo, E. R. Dohner, N. Martino, A. R. Srimath Kandada, E. Mosconi, F. De Angelis, H. I. Karunadasa, E. T. Hoke and A. Petrozza, Chem. Sci., 2015, 6, 7305–7310 RSC.
- C. Zheng and O. Rubel, J. Phys. Chem. C, 2019, 123, 19385–19394 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- H. Zhang, M. K. Nazeeruddin and W. C. H. Choy, Adv. Mater., 2019, 31, 1805702 CrossRef PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- Y. Wei, Z. Cheng and J. Lin, Chem. Soc. Rev., 2019, 48, 310–350 RSC.
- X. Du, R. Qiu, T. Zou, X. Chen, H. Chen and H. Zhou, Adv. Mater. Interfaces, 2019, 6, 1900413 CrossRef.
- A. Ciccioli, R. Panetta, A. Luongo, B. Brunetti, S. Vecchio Ciprioti, M. L. Mele and A. Latini, Phys. Chem. Chem. Phys., 2019, 21, 24768–24777 RSC.
- B. Parida, I. S. Jin and J. W. Jung, Chem. Mater., 2021, 33, 5850–5858 CrossRef CAS.
- Y. Miao, M. Zheng, H. Wang, C. Chen, X. Ding, C. Wu, B. Wang, M. Zhai, X. Yang and M. Cheng, J. Power Sources, 2021, 492, 229621 CrossRef CAS.
- I. Poli, S. Eslava and P. Cameron, J. Mater. Chem. A, 2017, 5, 22325–22333 RSC.
- Z. Xu, R. Chen, Y. Wu, R. He, J. Yin, W. Lin, B. Wu, J. Li and N. Zheng, J. Mater. Chem. A, 2019, 7, 26849–26857 RSC.
- S. Yang, Y. Wang, P. Liu, Y.-B. Cheng, H. J. Zhao and H. G. Yang, Nat. Energy, 2016, 1, 15016 CrossRef CAS.
- Y. Guo, S. Apergi, N. Li, M. Chen, C. Yin, Z. Yuan, F. Gao, F. Xie, G. Brocks, S. Tao and N. Zhao, Nat. Commun., 2021, 12, 644 CrossRef CAS PubMed.
- S. Sasmal, S. Valiyaveettil, A. P. Upadhyay, R. G. S. Pala, S. Sivakumar, C. S. Sundar and D. Sornadurai, MRS Commun., 2018, 8, 289–296 CrossRef CAS.
- M. Aamir, A. F. Butt, M. D. Khan, M. Sher, A. Iqbal, M. A. Malik, U. Jabeen and J. Akhtar, Optik, 2020, 207, 163828 CrossRef CAS.
- X. Liu, X. Wang, T. Zhang, Y. Miao, Z. Qin, Y. Chen and Y. Zhao, Angew. Chem., Int. Ed., 2021, 60, 12351–12355 CrossRef CAS PubMed.
- H. Wang, Z. Zhang, J. V. Milić, L. Tan, Z. Wang, R. Chen, X. Jing, C. Yi, Y. Ding, Y. Li, Y. Zhao, X. Zhang, A. Hagfeldt, M. Grätzel and J. Luo, Adv. Energy Mater., 2021, 11, 2101082 CrossRef CAS.
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 394–405 CrossRef CAS PubMed.
- H. Huang, B. Chen, Z. Wang, T. F. Hung, A. S. Susha, H. Zhong and A. L. Rogach, Chem. Sci., 2016, 7, 5699–5703 RSC.
- H. Wu, S. Lin, R. Wang, X. You and Y. Chi, Nanoscale, 2019, 11, 5557–5563 RSC.
- S. Liu, L. Yuan, Y. Zhao, Y. Chen, W. Xiang and X. Liang, J. Alloys Compd., 2019, 806, 1022–1028 CrossRef CAS.
- C.-H. Lu, S.-W. Kuo, C.-F. Huang and F.-C. Chang, J. Phys. Chem. C, 2009, 113, 3517–3524 CrossRef CAS.
- B. P. Kennedy and A. B. P. Lever, Can. J. Chem., 1972, 50, 3488–3507 CrossRef CAS.
- H. Plenio, ChemBioChem, 2004, 5, 650–655 CrossRef CAS PubMed.
- C. J. Drummond, G. Georgaklis and D. Y. C. Chan, Langmuir, 1996, 12, 2617–2621 CrossRef CAS.
- K. C. Hoang and S. Mecozzi, Langmuir, 2004, 20, 7347–7350 CrossRef CAS PubMed.
- Z. Li, Q. Hu, Z. Tan, Y. Yang, M. Leng, X. Liu, C. Ge, G. Niu and J. Tang, ACS Appl. Mater. Interfaces, 2018, 10, 43915–43922 CrossRef CAS PubMed.
- Y. Mu, W. Zhang, X. Guo, G. Dong, M. Zhang and T. Lu, ChemSusChem, 2019, 12, 4769–4774 CrossRef CAS PubMed.
- L. Yang, T. Wang, Q. Min, B. Liu, Z. Liu, X. Fan, J. Qiu, X. Xu, J. Yu and X. Yu, ACS Omega, 2019, 4, 6084–6091 CrossRef CAS.
- A. Gericke and H. Hühnerfuss, Thin Solid Films, 1994, 245, 74–82 CrossRef CAS.
- T. Lu, Y. Zhu, Y. Kang, J. Xu and A. Wang, Int. J. Biol. Macromol., 2021, 193, 1676–1684 CrossRef CAS PubMed.
- Y. Chang, Y. J. Yoon, G. Li, E. Xu, S. Yu, C.-H. Lu, Z. Wang, Y. He, C. H. Lin, B. K. Wagner, V. V. Tsukruk, Z. Kang, N. Thadhani, Y. Jiang and Z. Lin, ACS Appl. Mater. Interfaces, 2018, 10, 37267–37276 CrossRef CAS PubMed.
- B. Shu, Y. Chang, L. Dong, L. Chen, H. Wang, S. Yang, J. Zhang, X. Cheng and D. Yu, J. Lumin., 2021, 234, 117962 CrossRef CAS.
- C. G. Sanjayan, M. S. Jyothi, M. Sakar and R. G. Balakrishna, J. Colloid Interface Sci., 2021, 603, 758–770 CrossRef CAS PubMed.
- L. Gomez, C. de Weerd, J. L. Hueso and T. Gregorkiewicz, Nanoscale, 2017, 9, 631–636 RSC.
- A. Jana, K. N. Lawrence, M. B. Teunis, M. Mandal, A. Kumbhar and R. Sardar, Chem. Mater., 2016, 28, 1107–1120 CrossRef CAS.
- S. Biswas, S. Akhil, N. Kumar, M. Palabathuni, R. Singh, V. G. V. Dutt and N. Mishra, J. Phys. Chem. Lett., 2023, 14, 1910–1917 CrossRef CAS PubMed.
- Z. Chen, Y. Hu, J. Wang, Q. Shen, Y. Zhang, C. Ding, Y. Bai, G. Jiang, Z. Li and N. Gaponik, Chem. Mater., 2020, 32, 1517–1525 CrossRef CAS.
- J.-C. Wang, N. Li, A. M. Idris, J. Wang, X. Du, Z. Pan and Z. Li, Sol. RRL, 2021, 5, 1–8 Search PubMed.
- J. G. Smith and P. K. Jain, J. Am. Chem. Soc., 2016, 138, 6765–6773 CrossRef CAS PubMed.
- Y. Yuan, H. Zhu, K. Hills-Kimball, T. Cai, W. Shi, Z. Wei, H. Yang, Y. Candler, P. Wang, J. He and O. Chen, Angew. Chem., Int. Ed., 2020, 59, 22563–22569 CrossRef CAS PubMed.
- M. Zhao, D. Ding, F. Yang, D. Wang, J. Lv, W. Hu, C. Lu and Z. Tang, Nano Res., 2017, 10, 1249–1257 CrossRef CAS.
- J. Chang, Y. Ogomi, C. Ding, Y. H. Zhang, T. Toyoda, S. Hayase, K. Katayama and Q. Shen, Phys. Chem. Chem. Phys., 2017, 19, 6358–6367 RSC.
- H. Lu, X. Zhu, C. Miller, J. San Martin, X. Chen, E. M. Miller, Y. Yan and M. C. Beard, J. Chem. Phys., 2019, 151, 204305 CrossRef PubMed.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- S. Han, H. Zhang, R. Wang and Q. He, Mater. Sci. Semicond. Process., 2021, 131, 105847 CrossRef CAS.
- C. Lin, J. Li, N. She, S. Huang, C. Huang, I. Wang, F. Tsai, C. Wei, T. Lee, D. Wang, C. Wen, S. Li, A. Yabushita, C. Luo, C. Chen and C. Chen, Small, 2022, 18, 2107881 CrossRef CAS PubMed.
- C. Gao, F. Zhang, X. Gu, J. Huang, K. Wang, S. Zhang, S. Liu and Q. Tian, ACS Appl. Energy Mater., 2021, 4, 4021–4028 CrossRef CAS.
- C. Zhao, Z. He, P. Wangyang, J. Tan, C. Shi, A. Pan, L. He and Y. Liu, ACS Appl. Nano Mater., 2022, 5, 13737–13744 CrossRef CAS.
- J. Chen, S.-G. Kim, X. Ren, H. S. Jung and N.-G. Park, J. Mater. Chem. A, 2019, 7, 4977–4987 RSC.
- W. Zhang, Q. Li and Z. Li, Adv. Mater. Interfaces, 2022, 9, 2101881 CrossRef CAS.
- H. Zhang, Y. Wu, C. Shen, E. Li, C. Yan, W. Zhang, H. Tian, L. Han and W. Zhu, Adv. Energy Mater., 2019, 9, 1803573 CrossRef.
- Y. Yang, J. Liang, Z. Zhang, C. Tian, X. Wu, Y. Zheng, Y. Huang, J. Wang, Z. Zhou, M. He, Z. Chen and C. Chen, ChemSusChem, 2022, 15, e202102474 CrossRef CAS PubMed.
- Y. Li, M. Cai, M. Shen, Y. Cai and R.-J. Xie, J. Mater. Chem. C, 2022, 10, 8356–8363 RSC.
- Q. Chen, X. Yang, Y. Zhou and B. Song, New J. Chem., 2021, 45, 15118–15130 RSC.
- H. Wu, S. Wang, F. Cao, J. Zhou, Q. Wu, H. Wang, X. Li, L. Yin and X. Yang, Chem. Mater., 2019, 31, 1936–1940 CrossRef CAS.
- A. F. Carrizo, G. K. Belmonte, F. S. Santos, C. W. Backes, G. B. Strapasson, L. C. Schmidt, F. S. Rodembusch and D. E. Weibel, ACS Appl. Mater. Interfaces, 2021, 13, 59252–59262 CrossRef CAS PubMed.
- B. Yu, S. Liang, F. Zhang, Z. Li, B. Liu and X. Ding, Photonics Res., 2021, 9, 1559 CrossRef.
- S. Wang, Z. Zhang, Z. Tang, C. Su, W. Huang, Y. Li and G. Xing, Nano Energy, 2021, 82, 105712 CrossRef CAS.
- Y. Yang and H. Zhao, Appl. Surf. Sci., 2022, 577, 151895 CrossRef CAS.
- J. Han, M. Sharipov, S. Hwang, Y. Lee, B. T. Huy and Y.-I. Lee, Sci. Rep., 2022, 12, 3147 CrossRef CAS PubMed.
- H. C. Yoon, H. Kang, S. Lee, J. H. Oh, H. Yang and Y. R. Do, ACS Appl. Mater. Interfaces, 2016, 8, 18189–18200 CrossRef CAS PubMed.
- J. Ren, X. Dong, G. Zhang, T. Li and Y. Wang, New J. Chem., 2017, 41, 13961–13967 RSC.
- Y. Zhang, B. Zhang, Y. Fu, Y. Han, T. Zhang, L. Zhang, J. Guo and X. Zhang, J. Mater. Chem. C, 2022, 10, 8609–8616 RSC.
- H. Wang, H. Lin, X. Piao, P. Tian, M. Fang, X. An, C. Luo, R. Qi, Y. Chen and H. Peng, J. Mater. Chem. C, 2017, 5, 12044–12049 RSC.
- S. Kango, S. Kalia, A. Celli, J. Njuguna, Y. Habibi and R. Kumar, Prog. Polym. Sci., 2013, 38, 1232–1261 CrossRef CAS.
- S. Masi, A. Rizzo, F. Aiello, F. Balzano, G. Uccello-Barretta, A. Listorti, G. Gigli and S. Colella, Nanoscale, 2015, 7, 18956–18963 RSC.
- B. Erman and P. J. Flory, Macromolecules, 1986, 19, 2342–2353 CrossRef CAS.
- C. Carrillo-Carrión, P. del Pino and B. Pelaz, Appl. Mater. Today, 2019, 15, 562–569 CrossRef.
- Y. Wang, J. He, H. Chen, J. Chen, R. Zhu, P. Ma, A. Towers, Y. Lin, A. J. Gesquiere, S. Wu and Y. Dong, Adv. Mater., 2016, 28, 10710–10717 CrossRef CAS PubMed.
- Y. Wei, X. Deng, Z. Xie, X. Cai, S. Liang, P. Ma, Z. Hou, Z. Cheng and J. Lin, Adv. Funct. Mater., 2017, 27, 1703535 CrossRef.
- J. An, M. Chen, G. Liu, Y. Hu, R. Chen, Y. Lyu, S. Sharma and Y. Liu, Anal. Bioanal. Chem., 2021, 413, 1739–1747 CrossRef CAS PubMed.
- Y. Xin, H. Zhao and J. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 4971–4980 CrossRef CAS PubMed.
- H. Sun, Z. Yang, M. Wei, W. Sun, X. Li, S. Ye, Y. Zhao, H. Tan, E. L. Kynaston, T. B. Schon, H. Yan, Z.-H. Lu, G. A. Ozin, E. H. Sargent and D. S. Seferos, Adv. Mater., 2017, 29, 1701153 CrossRef PubMed.
- V. A. Hintermayr, C. Lampe, M. Löw, J. Roemer, W. Vanderlinden, M. Gramlich, A. X. Böhm, C. Sattler, B. Nickel, T. Lohmüller and A. S. Urban, Nano Lett., 2019, 19, 4928–4933 CrossRef CAS PubMed.
- J. Zhang, P. Jiang, Y. Wang, X. Liu, J. Ma and G. Tu, ACS Appl. Mater. Interfaces, 2020, 12, 3080–3085 CrossRef CAS PubMed.
- Q. Zhou, Z. Bai, W. Lu, Y. Wang, B. Zou and H. Zhong, Adv. Mater., 2016, 28, 9163–9168 CrossRef CAS PubMed.
- G. Jiang, C. Guhrenz, A. Kirch, L. Sonntag, C. Bauer, X. Fan, J. Wang, S. Reineke, N. Gaponik and A. Eychmüller, ACS Nano, 2019, 13, 10386–10396 CrossRef CAS PubMed.
- Z. Lu, Y. Li, Y. Xue, W. Zhou, S. Bayer, I. D. Williams, A. L. Rogach and S. Nagl, ACS Appl. Nano Mater., 2022, 5, 5025–5034 CrossRef CAS.
- Y. Shu, Y. Wang, J. Guan, Z. Ji, Q. Xu and X. Hu, Anal. Chem., 2022, 94, 5415–5424 CrossRef CAS PubMed.
- H. Zhang, X. Wang, Q. Liao, Z. Xu, H. Li, L. Zheng and H. Fu, Adv. Funct. Mater., 2017, 27, 1604382 CrossRef.
- J. Hai, H. Li, Y. Zhao, F. Chen, Y. Peng and B. Wang, Chem. Commun., 2017, 53, 5400–5403 RSC.
- A. Pramanik, K. Gates, S. Patibandla, D. Davis, S. Begum, R. Iftekhar, S. Alamgir, S. Paige, M. M. Porter and P. C. Ray, ACS Appl. Bio Mater., 2019, 2, 5872–5879 CrossRef CAS PubMed.
- S. N. Raja, Y. Bekenstein, M. A. Koc, S. Fischer, D. Zhang, L. Lin, R. O. Ritchie, P. Yang and A. P. Alivisatos, ACS Appl. Mater. Interfaces, 2016, 8, 35523–35533 CrossRef CAS PubMed.
- J. Xi, Y. Wu, W. Chen, Q. Li, J. Li, Y. Shen, H. Chen, G. Xu, H. Yang, Z. Chen, N. Li, J. Zhu, Y. Li and Y. Li, Nano Energy, 2022, 93, 106846 CrossRef CAS.
- Z. Zhang, L. Liu, H. Huang, L. Li, Y. Wang, J. Xu and J. Xu, Appl. Surf. Sci., 2020, 526, 146735 CrossRef CAS.
- Z. Zhang, L. Li, L. Liu, X. Xiao, H. Huang and J. Xu, J. Phys. Chem. C, 2020, 124, 22228–22234 CrossRef CAS.
- F.-J. Kahle, C. Saller, A. Köhler and P. Strohriegl, Adv. Energy Mater., 2017, 7, 1700306 CrossRef.
- T. Xu, J. Stevens, J. A. Villa, J. T. Goldbach, K. W. Guarini, C. T. Black, C. J. Hawker and T. P. Russell, Adv. Funct. Mater., 2003, 13, 698–702 CrossRef CAS.
- Y. Liu, Z. Wang, S. Liang, Z. Li, M. Zhang, H. Li and Z. Lin, Nano Lett., 2019, 19, 9019–9028 CrossRef CAS PubMed.
- K. Manokruang and E. Manias, Mater. Lett., 2009, 63, 1144–1147 CrossRef CAS.
- W. Hu, Z. Wen, X. Yu, P. Qian, W. Lian, X. Li, Y. Shang, X. Wu, T. Chen, Y. Lu, M. Wang and S. Yang, Adv. Sci., 2021, 8, 2004662 CrossRef CAS PubMed.
- F. Wang, M. Endo, S. Mouri, Y. Miyauchi, Y. Ohno, A. Wakamiya, Y. Murata and K. Matsuda, Nanoscale, 2016, 8, 11882–11888 RSC.
- S. Kundu and T. L. Kelly, Mater. Chem. Front., 2018, 2, 81–89 RSC.
- C. Wu, K. Wang, Y. Yan, D. Yang, Y. Jiang, B. Chi, J. Liu, A. R. Esker, J. Rowe, A. J. Morris, M. Sanghadasa and S. Priya, Adv. Funct. Mater., 2019, 29, 1804419 CrossRef.
- M. Meyns, M. Perálvarez, A. Heuer-Jungemann, W. Hertog, M. Ibáñez, R. Nafria, A. Genç, J. Arbiol, M. V. Kovalenko, J. Carreras, A. Cabot and A. G. Kanaras, ACS Appl. Mater. Interfaces, 2016, 8, 19579–19586 CrossRef CAS PubMed.
- A. F. Demirörs, A. van Blaaderen and A. Imhof, Langmuir, 2010, 26, 9297–9303 CrossRef PubMed.
- A. Loiudice, S. Saris, E. Oveisi, D. T. L. Alexander and R. Buonsanti, Angew. Chem., Int. Ed., 2017, 56, 10696–10701 CrossRef CAS PubMed.
- H. Yu, X. Xu, H. Liu, Y. Wan, X. Cheng, J. Chen, Y. Ye and L. Dai, ACS Nano, 2020, 14, 552–558 CrossRef CAS PubMed.
- H. Hu, L. Wu, Y. Tan, Q. Zhong, M. Chen, Y. Qiu, D. Yang, B. Sun, Q. Zhang and Y. Yin, J. Am. Chem. Soc., 2018, 140, 406–412 CrossRef CAS PubMed.
- H. Liu, Y. Tan, M. Cao, H. Hu, L. Wu, X. Yu, L. Wang, B. Sun and Q. Zhang, ACS Nano, 2019, 13, 5366–5374 CrossRef CAS PubMed.
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed.
- Y.-T. Hsieh, Y.-F. Lin and W.-R. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 58049–58059 CrossRef CAS PubMed.
- Z. Li, C. Song, J. Li, G. Liang, L. Rao, S. Yu, X. Ding, Y. Tang, B. Yu, J. Ou, U. Lemmer and G. Gomard, Adv. Mater. Technol., 2020, 5, 1900941 CrossRef CAS.
- P. M. Talianov, O. O. Peltek, M. Masharin, S. Khubezhov, M. A. Baranov, A. Drabavičius, A. S. Timin, L. E. Zelenkov, A. P. Pushkarev, S. V. Makarov and M. V. Zyuzin, J. Phys. Chem. Lett., 2021, 12, 8991–8998 CrossRef CAS PubMed.
- F. Gao, W. Yang, X. Liu, Y. Li, W. Liu, H. Xu and Y. Liu, Chem. Eng. J., 2021, 407, 128001 CrossRef CAS.
- H. Wu, Y. Chen, W. Zhang, M. S. Khan and Y. Chi, ACS Appl. Nano Mater., 2021, 4, 11791–11800 CrossRef CAS.
- S. Li, D. Lei, W. Ren, X. Guo, S. Wu, Y. Zhu, A. L. Rogach, M. Chhowalla and A. K. Y. Jen, Nat. Commun., 2020, 11, 1192 CrossRef CAS PubMed.
- C.-Y. You, F.-M. Li, L.-H. Lin, J.-S. Lin, Q.-Q. Chen, P. M. Radjenovic, Z.-Q. Tian and J.-F. Li, Nano Energy, 2020, 71, 104554 CrossRef CAS.
- Y. Huang, F. Li, L. Qiu, F. Lin, Z. Lai, S. Wang, L. Lin, Y. Zhu, Y. Wang, Y. Jiang and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 26384–26391 CrossRef CAS PubMed.
- Z. Zheng, L. Liu, F. Yi and J. Zhao, J. Lumin., 2019, 216, 116722 CrossRef CAS.
- K. K. Chan, D. Giovanni, H. He, T. C. Sum and K.-T. Yong, ACS Appl. Nano Mater., 2021, 4, 9022–9033 CrossRef CAS.
- K. K. Chan, S. H. K. Yap, D. Giovanni, T. C. Sum and K.-T. Yong, Microchem. J., 2022, 180, 107624 CrossRef CAS.
- M. He, Y. Cheng, L. Shen, C. Shen, H. Zhang, W. Xiang and X. Liang, Appl. Surf. Sci., 2018, 448, 400–406 CrossRef CAS.
- J. Y. Kim, K. I. Shim, J. W. Han, J. Joo, N. H. Heo and K. Seff, Adv. Mater., 2020, 32, 2001868 CrossRef CAS PubMed.
- Y. Zhang, L. Han, B. Li and Y. Xu, Chem. Eng. J., 2022, 437, 135290 CrossRef CAS.
- J. Jiang, G. Shao, Z. Zhang, L. Ding, H. Zhang, J. Liu, Z. Chen, W. Xiang and X. Liang, Chem. Commun., 2018, 54, 12302–12305 RSC.
- C. Shen, Y. Zhao, L. Yuan, L. Ding, Y. Chen, H. Yang, S. Liu, J. Nie, W. Xiang and X. Liang, Chem. Eng. J., 2020, 382, 122868 CrossRef CAS.
- S. Yuan, D. Chen, X. Li, J. Zhong and X. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 18918–18926 CrossRef CAS PubMed.
- P. Song, B. Qiao, D. Song, J. Cao, Z. Shen, G. Zhang, Z. Xu, S. Zhao, S. Wageh and A. Al-Ghamdi, J. Mater. Sci., 2020, 55, 9739–9747 CrossRef CAS.
- Z. Tan, Y. Chu, J. Chen, J. Li, G. Ji, G. Niu, L. Gao, Z. Xiao and J. Tang, Adv. Mater., 2020, 32, 2002443 CrossRef CAS PubMed.
- Z. Li, E. Hofman, J. Li, A. H. Davis, C.-H. Tung, L.-Z. Wu and W. Zheng, Adv. Funct. Mater., 2018, 28, 1704288 CrossRef.
- Y. You, W. Tian, M. Wang, F. Cao, H. Sun and L. Li, Adv. Mater. Interfaces, 2020, 7, 2000537 CrossRef CAS.
- G. Kang, H. Lee, J. Moon, H.-S. Jang, D.-H. Cho and D. Byun, ACS Appl. Nano Mater., 2022, 5, 6726–6735 CrossRef CAS.
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed.
- J. Zeng, C. Meng, X. Li, Y. Wu, S. Liu, H. Zhou, H. Wang and H. Zeng, Adv. Funct. Mater., 2019, 29, 1904461 CrossRef CAS.
- A. Pron and P. Rannou, Prog. Polym. Sci., 2002, 27, 135–190 CrossRef CAS.
- Y. Li and R. Qian, Synth. Met., 1993, 53, 149–154 CrossRef CAS.
- R. Ansari, E-J. Chem., 2006, 3, 186–201 CrossRef CAS.
- M. D. Groner, S. M. George, R. S. McLean and P. F. Carcia, Appl. Phys. Lett., 2006, 88, 051907 CrossRef.
- G. Li, F. W. R. Rivarola, N. J. L. K. Davis, S. Bai, T. C. Jellicoe, F. de la Peña, S. Hou, C. Ducati, F. Gao, R. H. Friend, N. C. Greenham and Z.-K. Tan, Adv. Mater., 2016, 28, 3528–3534 CrossRef CAS PubMed.
- R. W. Johnson, A. Hultqvist and S. F. Bent, Mater. Today, 2014, 17, 236–246 CrossRef CAS.
- C.-C. Shih, P.-C. Chen, G.-L. Lin, C.-W. Wang and H.-T. Chang, ACS Nano, 2015, 9, 312–319 CrossRef CAS PubMed.
- S. Liu and M.-Y. Han, Chem.–Asian J., 2009, 5, 36–45 CrossRef PubMed.
- A. Soleimani Dorcheh and M. H. Abbasi, J. Mater. Process. Technol., 2008, 199, 10–26 CrossRef CAS.
- H.-C. Wang, S.-Y. Lin, A.-C. Tang, B. P. Singh, H.-C. Tong, C.-Y. Chen, Y.-C. Lee, T.-L. Tsai and R.-S. Liu, Angew. Chem., Int. Ed., 2016, 55, 7924–7929 CrossRef CAS PubMed.
- M. Liu, S. Wang and L. Jiang, Nat. Rev. Mater., 2017, 2, 17036 CrossRef CAS.
- S. Wang, H. Wang, D. Zhang, Y. Dou, W. Li, F. Cao, L. Yin, L. Wang, Z.-J. Zhang, J. Zhang and X. Yang, Chem. Eng. J., 2022, 437, 135303 CrossRef CAS.
- X. Tian, T. Verho and R. H. A. Ras, Science, 2016, 352, 142–143 CrossRef CAS PubMed.
- D. K. Sharma, S. Hirata and M. Vacha, Nat. Commun., 2019, 10, 4499 CrossRef PubMed.
- R. H. Baney, M. Itoh, A. Sakakibara and T. Suzuki, Chem. Rev., 1995, 95, 1409–1430 CrossRef CAS.
- J. M. Newsam, Science, 1986, 231, 1093–1099 CrossRef CAS PubMed.
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268–7316 CrossRef CAS PubMed.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. A. Catlow, Chem. Soc. Rev., 2015, 44, 7044–7111 RSC.
- J.-Y. Sun, F. T. Rabouw, X.-F. Yang, X.-Y. Huang, X.-P. Jing, S. Ye and Q.-Y. Zhang, Adv. Funct. Mater., 2017, 27, 1704371 CrossRef.
- G. Tong, W. Song, L. K. Ono and Y. Qi, Appl. Phys. Lett., 2022, 120, 161604 CrossRef CAS.
- J.-F. Liao, Y.-F. Xu, X.-D. Wang, H.-Y. Chen and D.-B. Kuang, ACS Appl. Mater. Interfaces, 2018, 10, 42301–42309 CrossRef CAS PubMed.
- Y. Xu, X. Wang, J. Liao, B. Chen, H. Chen and D. Kuang, Adv. Mater. Interfaces, 2018, 5, 1801015 CrossRef.
- Y. Liu, Z. Yang, D. Cui, X. Ren, J. Sun, X. Liu, J. Zhang, Q. Wei, H. Fan, F. Yu, X. Zhang, C. Zhao and S. F. Liu, Adv. Mater., 2015, 27, 5176–5183 CrossRef CAS PubMed.
- H. Yin, J. Chen, P. Guan, D. Zheng, Q. Kong, S. Yang, P. Zhou, B. Yang, T. Pullerits and K. Han, Angew. Chem., Int. Ed., 2021, 60, 22693–22699 CrossRef CAS PubMed.
- S. Wang and X. Wang, Small, 2015, 11, 3097–3112 CrossRef CAS PubMed.
- W. Lu, Z. Wei, Z.-Y. Gu, T.-F. Liu, J. Park, J. Park, J. Tian, M. Zhang, Q. Zhang, T. Gentle III, M. Bosch and H.-C. Zhou, Chem. Soc. Rev., 2014, 43, 5561–5593 RSC.
- W. Nie and H. Tsai, J. Mater. Chem. A, 2022, 10, 19518–19533 RSC.
- D. Zhang, Y. Xu, Q. Liu and Z. Xia, Inorg. Chem., 2018, 57, 4613–4619 CrossRef CAS PubMed.
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310–316 CrossRef CAS PubMed.
- S. Mollick, T. N. Mandal, A. Jana, S. Fajal, A. V. Desai and S. K. Ghosh, ACS Appl. Nano Mater., 2019, 2, 1333–1340 CrossRef CAS.
- S. Ahmed, S. Lahkar, S. Doley, D. Mohanta and S. Kumar Dolui, J. Photochem. Photobiol., A, 2023, 443, 114821 CrossRef CAS.
- G.-Y. Qiao, D. Guan, S. Yuan, H. Rao, X. Chen, J.-A. Wang, J.-S. Qin, J.-J. Xu and J. Yu, J. Am. Chem. Soc., 2021, 143, 14253–14260 CrossRef CAS PubMed.
- Z. Xia, B. Shi, W. Zhu, Y. Xiao and C. Lü, Adv. Funct. Mater., 2022, 32, 2207655 CrossRef CAS.
- J. Hou, P. Chen, A. Shukla, A. Krajnc, T. Wang, X. Li, R. Doasa, L. H. G. Tizei, B. Chan, D. N. Johnstone, R. Lin, T. U. Schülli, I. Martens, D. Appadoo, M. S. Ari, Z. Wang, T. Wei, S. Lo, M. Lu, S. Li, E. B. Namdas, G. Mali, A. K. Cheetham, S. M. Collins, V. Chen, L. Wang and T. D. Bennett, Science, 2021, 374, 621–625 CrossRef CAS PubMed.
- T. D. Bennett, Y. Yue, P. Li, A. Qiao, H. Tao, N. G. Greaves, T. Richards, G. I. Lampronti, S. A. T. Redfern, F. Blanc, O. K. Farha, J. T. Hupp, A. K. Cheetham and D. A. Keen, J. Am. Chem. Soc., 2016, 138, 3484–3492 CrossRef CAS PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed.
- Y. Chen, Z. Lai, X. Zhang, Z. Fan, Q. He, C. Tan and H. Zhang, Nat. Rev. Chem, 2020, 4, 243–256 CrossRef CAS PubMed.
- R. Kappera, D. Voiry, S. E. Yalcin, B. Branch, G. Gupta, A. D. Mohite and M. Chhowalla, Nat. Mater., 2014, 13, 1128–1134 CrossRef CAS PubMed.
- D. Voiry, A. Mohite and M. Chhowalla, Chem. Soc. Rev., 2015, 44, 2702–2712 RSC.
- S. Caicedo-Dávila, H. Funk, R. Lovrinčić, C. Müller, M. Sendner, O. Cojocaru-Mirédin, F. Lehmann, R. Gunder, A. Franz, S. Levcenco, A. V. Cohen, L. Kronik, B. Haas, C. T. Koch and D. Abou-Ras, J. Phys. Chem. C, 2019, 123, 17666–17677 CrossRef.
- J. Hui, Y. Jiang, Ö. Ö. Gökçinar, J. Tang, Q. Yu, M. Zhang and K. Yu, Chem. Mater., 2020, 32, 4574–4583 CrossRef CAS.
- M. I. Saidaminov, M. A. Haque, J. Almutlaq, S. Sarmah, X.-H. Miao, R. Begum, A. A. Zhumekenov, I. Dursun, N. Cho, B. Murali, O. F. Mohammed, T. Wu and O. M. Bakr, Adv. Opt. Mater., 2017, 5, 1600704 CrossRef.
- C. de Weerd, J. Lin, L. Gomez, Y. Fujiwara, K. Suenaga and T. Gregorkiewicz, J. Phys. Chem. C, 2017, 121, 19490–19496 CrossRef PubMed.
- I. Dursun, M. De Bastiani, B. Turedi, B. Alamer, A. Shkurenko, J. Yin, A. M. El-Zohry, I. Gereige, A. AlSaggaf, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, ChemSusChem, 2017, 10, 3746–3749 CrossRef CAS PubMed.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- Q. A. Akkerman, T. P. T. Nguyen, S. C. Boehme, F. Montanarella, D. N. Dirin, P. Wechsler, F. Beiglböck, G. Rainò, R. Erni, C. Katan, J. Even and M. V. Kovalenko, Science, 2022, 3616, 1–13 Search PubMed.
- X. Zhang, B. Xu, J. Zhang, Y. Gao, Y. Zheng, K. Wang and X. W. Sun, Adv. Funct. Mater., 2016, 26, 4595–4600 CrossRef CAS.
- J. Xu, W. Huang, P. Li, D. R. Onken, C. Dun, Y. Guo, K. B. Ucer, C. Lu, H. Wang, S. M. Geyer, R. T. Williams and D. L. Carroll, Adv. Mater., 2017, 29, 1703703 CrossRef PubMed.
- B. Qiao, P. Song, J. Cao, S. Zhao, Z. Shen, D. Gao, Z. Liang, Z. Xu, D. Song and X. Xu, Nanotechnology, 2017, 28, 445602 CrossRef PubMed.
- X. Tang, Z. Hu, W. Yuan, W. Hu, H. Shao, D. Han, J. Zheng, J. Hao, Z. Zang, J. Du, Y. Leng, L. Fang and M. Zhou, Adv. Opt. Mater., 2017, 5, 1600788 CrossRef.
- Z. Ma, F. Li, G. Qi, L. Wang, C. Liu, K. Wang, G. Xiao and B. Zou, Nanoscale, 2019, 11, 820–825 RSC.
- J. Deng, J. Xun, W. Shen, M. Li and R. He, Mater. Res. Bull., 2021, 140, 111296 CrossRef CAS.
- X. Zhang, H. Wang, Y. Hu, Y. Pei, S. Wang, Z. Shi, V. L. Colvin, S. Wang, Y. Zhang and W. W. Yu, J. Phys. Chem. Lett., 2019, 10, 1750–1756 CrossRef CAS PubMed.
- J. P. McKaveney and M. D. Buck, Anal. Chem., 1974, 46, 650–654 CrossRef CAS.
- B. Zhou, Z. Liu, H. Li, S. Fang, F. Fang, Y. Wang, F. Chen and Y. Shi, Adv. Photonics Res., 2021, 2, 2100143 CrossRef CAS.
- J. Li, Z. Tan, M. Hu, C. Chen, J. Luo, S. Li, L. Gao, Z. Xiao, G. Niu and J. Tang, Front. Optoelectron., 2019, 12, 352–364 CrossRef.
- Q. Ba, J. Kim, H. Im, S. Lin and A. Jana, J. Colloid Interface Sci., 2022, 606, 808–816 CrossRef CAS PubMed.
- J. You, Y. Yang, Z. Hong, T.-B. Song, L. Meng, Y. Liu, C. Jiang, H. Zhou, W.-H. Chang, G. Li and Y. Yang, Appl. Phys. Lett., 2014, 105, 183902 CrossRef.
- L. Wu, H. Hu, Y. Xu, S. Jiang, M. Chen, Q. Zhong, D. Yang, Q. Liu, Y. Zhao, B. Sun, Q. Zhang and Y. Yin, Nano Lett., 2017, 17, 5799–5804 CrossRef CAS PubMed.
- M. Xie, H. Liu, F. Chun, W. Deng, C. Luo, Z. Zhu, M. Yang, Y. Li, W. Li, W. Yan and W. Yang, Small, 2019, 15, 1901994 CrossRef PubMed.
- A. Pramanik, S. Patibandla, Y. Gao, K. Gates and P. C. Ray, JACS Au, 2021, 1, 53–65 CrossRef CAS PubMed.
- Y. Liu, F. Li, Q. Liu and Z. Xia, Chem. Mater., 2018, 30, 6922–6929 CrossRef CAS.
- S. Mamgain, V. Kunnathodi and A. Yella, Energy Technol., 2020, 8, 1900890 CrossRef CAS.
- Q. Jing, M. Zhang, X. Huang, X. Ren, P. Wang and Z. Lu, Nanoscale, 2017, 9, 7391–7396 RSC.
- K. Chen, K. Qi, T. Zhou, T. Yang, Y. Zhang, Z. Guo, C.-K. Lim, J. Zhang, I. Žutic, H. Zhang and P. N. Prasad, Nano–Micro Lett., 2021, 13, 172 CrossRef CAS PubMed.
- J. Zhu, Y. Zhu, J. Huang, L. Hou, J. Shen and C. Li, Nanoscale, 2020, 12, 11842–11846 RSC.
- F. Fang, W. Chen, Y. Li, H. Liu, M. Mei, R. Zhang, J. Hao, M. Mikita, W. Cao, R. Pan, K. Wang and X. W. Sun, Adv. Funct. Mater., 2018, 28, 1706000 CrossRef.
- X. Zhang, X. Bai, H. Wu, X. Zhang, C. Sun, Y. Zhang, W. Zhang, W. Zheng, W. W. Yu and A. L. Rogach, Angew. Chem., Int. Ed., 2018, 57, 3337–3342 CrossRef CAS PubMed.
- H. Jiang, Z. Yan, H. Zhao, S. Yuan, Z. Yang, J. Li, B. Liu, T. Niu, J. Feng, Q. Wang, D. Wang, H. Yang, Z. Liu and S. F. Liu, ACS Appl. Energy Mater., 2018, 1, 900–909 CrossRef CAS.
- L. N. Quan, M. Yuan, R. Comin, O. Voznyy, E. M. Beauregard, S. Hoogland, A. Buin, A. R. Kirmani, K. Zhao, A. Amassian, D. H. Kim and E. H. Sargent, J. Am. Chem. Soc., 2016, 138, 2649–2655 CrossRef CAS PubMed.
- Y. Lin, Y. Bai, Y. Fang, Q. Wang, Y. Deng and J. Huang, ACS Energy Lett., 2017, 2, 1571–1572 CrossRef CAS.
- Y. Yang, C. Hou and T.-X. Liang, Phys. Chem. Chem. Phys., 2021, 23, 7145–7152 RSC.
- D. Yoo, J. Y. Woo, Y. Kim, S. W. Kim, S.-H. Wei, S. Jeong and Y.-H. Kim, J. Phys. Chem. Lett., 2020, 11, 652–658 CrossRef CAS PubMed.
- A. Jana and K. S. Kim, ACS Energy Lett., 2018, 3, 2120–2126 CrossRef CAS.
- A. Jana and K. S. Kim, ACS Appl. Energy Mater., 2019, 2, 4496–4503 CrossRef CAS.
- K.-K. Liu, Q. Liu, D.-W. Yang, Y.-C. Liang, L.-Z. Sui, J.-Y. Wei, G.-W. Xue, W.-B. Zhao, X.-Y. Wu, L. Dong and C.-X. Shan, Light: Sci. Appl., 2020, 9, 44 CrossRef CAS PubMed.
- L. Jia, Z. Xu, L. Zhang, Y. Li, T. Zhao and J. Xu, Appl. Surf. Sci., 2022, 592, 153170 CrossRef CAS.
- K. Du, L. He, S. Song, J. Feng, Y. Li, M. Zhang, H. Li, C. Li and H. Zhang, Adv. Funct. Mater., 2021, 31, 2103275 CrossRef CAS.
- H. Dong, S. Kareem, X. Gong, J. Ruan, P. Gao, X. Zhou, X. Liu, X. Zhao and Y. Xie, ACS Appl. Mater. Interfaces, 2021, 13, 23960–23969 CrossRef CAS PubMed.
- S. Lou, S. Si, L. Huang, W. Gan, B. Lan, J. Zhang, M. Li, T. Xuan and J. Wang, Chem. Eng. J., 2022, 430, 132680 CrossRef CAS.
- H. Lin, X. Zhang, L. Cai, J. Lao, R. Qi, C. Luo, S. Chen, H. Peng, R. Huang and C. Duan, J. Mater. Chem. C, 2020, 8, 5594–5599 RSC.
- C. Geng, S. Xu, H. Zhong, A. L. Rogach and W. Bi, Angew. Chem., Int. Ed., 2018, 57, 9650–9654 CrossRef CAS.
- P. Cheng, K. Han and J. Chen, ACS Mater. Lett., 2022, 60–78 Search PubMed.
- X. Chen, M. Jia, W. Xu, G. Pan, J. Zhu, Y. Tian, D. Wu, X. Li and Z. Shi, Adv. Opt. Mater., 2023, 11, 2202153 CrossRef CAS.
- H. Zhao, Y. Li, B. Zhang, T. Xu and C. Wang, Nano Energy, 2018, 50, 665–674 CrossRef CAS.
- H. Zhao, K. Chordiya, P. Leukkunen, A. Popov, M. Upadhyay Kahaly, K. Kordas and S. Ojala, Nano Res., 2021, 14, 1116–1125 CrossRef CAS.
- H. Wang, H. Zhang, J. Wang, Y. Gao, F. Fan, K. Wu, X. Zong and C. Li, Angew. Chem., Int. Ed., 2021, 60, 7376–7381 CrossRef CAS PubMed.
- M. Aamir, Z. H. Shah, M. Sher, A. Iqbal, N. Revaprasadu, M. A. Malik and J. Akhtar, Mater. Sci. Semicond. Process., 2017, 63, 6–11 CrossRef CAS.
- M. Becker and M. Wark, J. Phys. Chem. C, 2018, 122, 3548–3557 CrossRef CAS.
- A. D'Annibale, R. Panetta, O. Tarquini, M. Colapietro, S. Quaranta, A. Cassetta, L. Barba, G. Chita and A. Latini, Dalton Trans., 2019, 48, 5397–5407 RSC.
- X. Li, X. Zhong, Y. Hu, B. Li, Y. Sheng, Y. Zhang, C. Weng, M. Feng, H. Han and J. Wang, J. Phys. Chem. Lett., 2017, 8, 1804–1809 CrossRef CAS PubMed.
- K. Ahmad and S. M. Mobin, Energy Technol., 2020, 8, 1901185 CrossRef CAS.
- D. Ju, X. Zheng, J. Liu, Y. Chen, J. Zhang, B. Cao, H. Xiao, O. F. Mohammed, O. M. Bakr and X. Tao, Angew. Chem., Int. Ed., 2018, 57, 14868–14872 CrossRef CAS PubMed.
- D. Ju, G. Lin, H. Xiao, Y. Zhang, S. Su and J. Liu, Sol. RRL, 2020, 4, 2000559 CrossRef CAS.
- M. I. Saidaminov, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2017, 2, 889–896 CrossRef CAS.
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2018, 3, 54–62 CrossRef CAS.
- T. Zhu and X. Gong, InfoMat, 2021, 3, 1039–1069 CrossRef CAS.
- Q. Ba, A. Jana, L. Wang and K. S. Kim, Adv. Funct. Mater., 2019, 29, 1904768 CrossRef CAS.
- L. Romani, A. Bala, V. Kumar, A. Speltini, A. Milella, F. Fracassi, A. Listorti, A. Profumo and L. Malavasi, J. Mater. Chem. C, 2020, 8, 9189–9194 RSC.
- W. Bi, Z. Wang, H. Li, Y. Song, X. Liu, Y. Wang, C. Ge, A. Wang, Y. Kang, Y. Yang, B. Li and Q. Dong, J. Phys. Chem. Lett., 2022, 13, 6792–6799 CrossRef CAS PubMed.
- Z. Hong, W. K. Chong, A. Y. R. Ng, M. Li, R. Ganguly, T. C. Sum and H. Sen Soo, Angew. Chem., Int. Ed., 2019, 58, 3456–3460 CrossRef CAS PubMed.
- S.-K. Yu, Z.-R. Zhang, Z.-H. Ren, H.-L. Zhai, Q.-Y. Zhu and J. Dai, Inorg. Chem., 2021, 60, 9132–9140 CrossRef CAS PubMed.
- Z. Zhuang, C. Peng, G. Zhang, H. Yang, J. Yin and H. Fei, Angew. Chem., Int. Ed., 2017, 56, 14411–14416 CrossRef CAS PubMed.
- X. Song, G. Wei, J. Sun, C. Peng, J. Yin, X. Zhang, Y. Jiang and H. Fei, Nat. Catal., 2020, 3, 1027–1033 CrossRef CAS.
- D. Ju, G. Lin, M. Zhou, Y. Hua, X. Li, H. Li and J. Liu, J. Mater. Chem. A, 2022, 10, 17752–17759 RSC.
- C. Xue, Z.-Y. Yao, J. Zhang, W.-L. Liu, J.-L. Liu and X.-M. Ren, Chem. Commun., 2018, 54, 4321–4324 RSC.
- X. Yang, L.-F. Ma and D. Yan, Chem. Sci., 2019, 10, 4567–4572 RSC.
- I. Spanopoulos, I. Hadar, W. Ke, P. Guo, S. Sidhik, M. Kepenekian, J. Even, A. D. Mohite, R. D. Schaller and M. G. Kanatzidis, J. Am. Chem. Soc., 2020, 142, 9028–9038 CrossRef CAS PubMed.
- T. Sheikh, S. Maqbool, P. Mandal and A. Nag, Angew. Chem., Int. Ed., 2021, 60, 18265–18271 CrossRef CAS PubMed.
- G.-N. Liu, R.-Y. Zhao, B. Xu, Y. Sun, X.-M. Jiang, X. Hu and C. Li, ACS Appl. Mater. Interfaces, 2020, 12, 54694–54702 CrossRef CAS PubMed.
- H. Peng, X. Wang, Y. Tian, T. Dong, Y. Xiao, T. Huang, Y. Guo, J. Wang and B. Zou, J. Phys. Chem. Lett., 2021, 12, 6639–6647 CrossRef CAS PubMed.
- D. A. Dougherty, Acc. Chem. Res., 2013, 46, 885–893 CrossRef CAS PubMed.
- O. M. Cabarcos, C. J. Weinheimer and J. M. Lisy, J. Chem. Phys., 1998, 108, 5151–5154 CrossRef CAS.
- Q. A. Akkerman, D. Meggiolaro, Z. Dang, F. De Angelis and L. Manna, ACS Energy Lett., 2017, 2, 2183–2186 CrossRef CAS PubMed.
- H. Liu, Z. Wu, J. Shao, D. Yao, H. Gao, Y. Liu, W. Yu, H. Zhang and B. Yang, ACS Nano, 2017, 11, 2239–2247 CrossRef CAS PubMed.
- M. Lu, X. Zhang, Y. Zhang, J. Guo, X. Shen, W. W. Yu and A. L. Rogach, Adv. Mater., 2018, 30, 1804691 CrossRef PubMed.
- M. Hamdan and A. K. Chandiran, Angew. Chem., Int. Ed., 2020, 59, 16033–16038 CrossRef CAS PubMed.
- Z. Xiao, H. Lei, X. Zhang, Y. Zhou, H. Hosono and T. Kamiya, Bull. Chem. Soc. Jpn., 2015, 88, 1250–1255 CrossRef CAS.
- S. Pont, D. Bryant, C.-T. Lin, N. Aristidou, S. Wheeler, X. Ma, R. Godin, S. A. Haque and J. R. Durrant, J. Mater. Chem. A, 2017, 5, 9553–9560 RSC.
- A. Aziz, N. Aristidou, X. Bu, R. J. E. Westbrook, S. A. Haque and M. S. Islam, Chem. Mater., 2020, 32, 400–409 CrossRef CAS.
- Q. Li, Z. Chen, I. Tranca, S. Gaastra-Nedea, D. Smeulders and S. Tao, Appl. Surf. Sci., 2021, 538, 148058 CrossRef CAS.
- P. Lin, A. Loganathan, I. Raifuku, M. Li, Y. Chiu, S. Chang, A. Fakharuddin, C. Lin, T. Guo, L. Schmidt-Mende and P. Chen, Adv. Energy Mater., 2021, 11, 2100818 CrossRef CAS.
- R. Babu, S. Bhandary, D. Chopra and S. P. Singh, Chem. –Eur. J., 2020, 26, 10519–10527 CrossRef CAS PubMed.
- Q. Jiang, D. Rebollar, J. Gong, E. L. Piacentino, C. Zheng and T. Xu, Angew. Chem., Int. Ed., 2015, 54, 7617–7620 CrossRef CAS PubMed.
- C.-C. Wang, J.-R. Li, X.-L. Lv, Y.-Q. Zhang and G. Guo, Energy Environ. Sci., 2014, 7, 2831–2867 RSC.
- F. Wu, R. Pathak, J. Liu, R. Jian, T. Zhang and Q. Qiao, ACS Appl. Mater. Interfaces, 2021, 13, 44274–44283 CrossRef CAS PubMed.
- A. Pisanu, A. Speltini, P. Quadrelli, G. Drera, L. Sangaletti and L. Malavasi, J. Mater. Chem. C, 2019, 7, 7020–7026 RSC.
- L. Romani, A. Speltini, F. Ambrosio, E. Mosconi, A. Profumo, M. Marelli, S. Margadonna, A. Milella, F. Fracassi, A. Listorti, F. De Angelis and L. Malavasi, Angew. Chem., Int. Ed., 2021, 60, 3611–3618 CrossRef CAS PubMed.
- L. Romani, A. Speltini, C. N. Dibenedetto, A. Listorti, F. Ambrosio, E. Mosconi, A. Simbula, M. Saba, A. Profumo, P. Quadrelli, F. De Angelis and L. Malavasi, Adv. Funct. Mater., 2021, 31, 2104428 CrossRef CAS.
- J. Liang, C. Wang, Y. Wang, Z. Xu, Z. Lu, Y. Ma, H. Zhu, Y. Hu, C. Xiao, X. Yi, G. Zhu, H. Lv, L. Ma, T. Chen, Z. Tie, Z. Jin and J. Liu, J. Am. Chem. Soc., 2016, 138, 15829–15832 CrossRef CAS PubMed.
- R. Chen, G. Gao and J. Luo, Nano Res., 2022, 1–6 Search PubMed.
- D. Laishram, S. Zeng, K. M. Alam, A. P. Kalra, K. Cui, P. Kumar, R. K. Sharma and K. Shankar, Appl. Surf. Sci., 2022, 592, 153276 CrossRef CAS.
- M. V. Kovalenko, M. Scheele and D. V. Talapin, Science, 2009, 324, 1417–1420 CrossRef CAS PubMed.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- E. A. Muljarov, S. G. Tikhodeev, N. A. Gippius and T. Ishihara, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 14370–14378 CrossRef CAS PubMed.
- H. Tsai, W. Nie, J.-C. Blancon, C. C. Stoumpos, R. Asadpour, B. Harutyunyan, A. J. Neukirch, R. Verduzco, J. J. Crochet, S. Tretiak, L. Pedesseau, J. Even, M. A. Alam, G. Gupta, J. Lou, P. M. Ajayan, M. J. Bedzyk, M. G. Kanatzidis and A. D. Mohite, Nature, 2016, 536, 312–316 CrossRef CAS PubMed.
- I. C. Smith, E. T. Hoke, D. Solis-Ibarra, M. D. McGehee and H. I. Karunadasa, Angew. Chem., Int. Ed., 2014, 53, 11232–11235 CrossRef CAS PubMed.
- D. A. Hines and P. V. Kamat, J. Phys. Chem. C, 2013, 117, 14418–14426 CrossRef CAS.
- W. Guan, Y. Li, Q. Zhong, H. Liu, J. Chen, H. Hu, K. Lv, J. Gong, Y. Xu, Z. Kang, M. Cao and Q. Zhang, Nano Lett., 2021, 21, 597–604 CrossRef CAS PubMed.
- Y. Wang, T. Wu, J. Barbaud, W. Kong, D. Cui, H. Chen, X. Yang and L. Han, Science, 2019, 365, 687–691 CrossRef CAS PubMed.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Y. Yang, M. Yang, D. T. Moore, Y. Yan, E. M. Miller, K. Zhu and M. C. Beard, Nat. Energy, 2017, 2, 16207 CrossRef CAS.
- D. W. Wakerley, M. F. Kuehnel, K. L. Orchard, K. H. Ly, T. E. Rosser and E. Reisner, Nat. Energy, 2017, 2, 17021 CrossRef CAS.
- S. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037–5040 CrossRef CAS PubMed.
- X.-D. Wang, Y.-H. Huang, J.-F. Liao, Y. Jiang, L. Zhou, X.-Y. Zhang, H.-Y. Chen and D.-B. Kuang, J. Am. Chem. Soc., 2019, 141, 13434–13441 CrossRef CAS PubMed.
- Y. Jiang, H. Chen, J. Li, J. Liao, H. Zhang, X. Wang and D. Kuang, Adv. Funct. Mater., 2020, 30, 2004293 CrossRef CAS.
- S. S. Bhosale, A. K. Kharade, E. Jokar, A. Fathi, S. Chang and E. W.-G. Diau, J. Am. Chem. Soc., 2019, 141, 20434–20442 CrossRef CAS PubMed.
- J. Sheng, Y. He, J. Li, C. Yuan, H. Huang, S. Wang, Y. Sun, Z. Wang and F. Dong, ACS Nano, 2020, 14, 13103–13114 CrossRef CAS PubMed.
- J. Sheng, Y. He, M. Huang, C. Yuan, S. Wang and F. Dong, ACS Catal., 2022, 12, 2915–2926 CrossRef CAS.
- N. Li, X. Chen, J. Wang, X. Liang, L. Ma, X. Jing, D.-L. Chen and Z. Li, ACS Nano, 2022, 16, 3332–3340 CrossRef CAS PubMed.
- R. Das, S. Chakraborty and S. C. Peter, ACS Energy Lett., 2021, 6, 3270–3274 CrossRef CAS.
- J. San Martin, N. Dang, E. Raulerson, M. C. Beard, J. Hartenberger and Y. Yan, Angew. Chem., Int. Ed., 2022, 61, e202205572 CrossRef CAS PubMed.
- D. Ricciarelli, W. Kaiser, E. Mosconi, J. Wiktor, M. W. Ashraf, L. Malavasi, F. Ambrosio and F. De Angelis, ACS Energy Lett., 2022, 7, 1308–1315 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta04994a |
| This journal is © The Royal Society of Chemistry 2023 |