
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the guest-induced spatiotemporal response of isostructural dynamic frameworks towards efficient gas separation and storage†
Kornel
Roztocki
 *a,
Szymon
Sobczak
ab,
Arkadiusz
Smaruj
ab,
Anna
Walczak
ab,
Mateusz
Gołdyn
a,
Volodymyr
Bon
c,
Stefan
Kaskel
c and
Artur R.
Stefankiewicz
*ab
*a,
Szymon
Sobczak
ab,
Arkadiusz
Smaruj
ab,
Anna
Walczak
ab,
Mateusz
Gołdyn
a,
Volodymyr
Bon
c,
Stefan
Kaskel
c and
Artur R.
Stefankiewicz
*ab
aFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, Poznań 61-614, Poland. E-mail: kornel.roztocki@amu.edu.pl
bCenter for Advanced Technology, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, Poznań 61-614, Poland. E-mail: ars@amu.edu.pl
cChair of Inorganic Chemistry I, Technische Universität Dresden, Bergstrasse 66, Dresden 01062, Germany
First published on 23rd August 2023
Abstract
Understanding and control of the spatiotemporal stimuli-responsiveness of flexible metal–organic frameworks are crucial for the development of novel adsorbents for gas storage and separation technologies. Herein, we report two isostructural pillared-layer dynamic frameworks differing only in one atom that bridge a benzenocarboxylate linker. Through a synthetic approach, we switch the stepwise CO2-induced transformation into a continuous one. Our findings are proved by equilibrium and time-resolved in situ powder X-ray diffraction collected during CO2 adsorption at 195 K. Finally, we use high-pressure single and multi-gas adsorption experiments to show the superiority of continuous breathing in CH4 storage and CH4/CO2 separation at 298 K. This report demonstrates that the desirable mechanism of flexible frameworks can be readily achieved through single-atom exchange enabling efficient gas separation and storage.
Flexible metal–organic frameworks (MOFs) are porous coordination polymers that adapt their structure in response to external stimuli1–5 such as pressure,6 temperature,7 electrical field8 and light.9 Their nano-elasticity is responsible for a plethora of novel macroscopic phenomena, which do not occur in rigid adsorbents, including the shape memory effect,10 negative gas adsorption,11 self-accelerating adsorption12 or swelling.13 Moreover, the spatiotemporal adaptability of the frameworks opens the door to various potential applications, inter alia, gas storage14,15 and separation,16,17 logical operation,18 proton conductivity,19 molecular recognition,20 catalysis,21 drug delivery22 and water isotopologue separation.23
Solvent removal from the nano-cavities of flexible MOFs transforms their porous structure (open pore phase – op) into a non-porous or less porous structure (close pore phase – cp) expected in some cases as the desolvation of hydrated MIL-53(Cr).24 Gas adsorption reverses these processes and the structural transformation occurs in a discontinuous (stepwise) manner. For example, ELM-11 when exposed to N2 or Ar exhibits a one-step transformation described as gating (cp → op).25 The same effect is observed in DUT-8(Ni); this pillared layer framework transforms from the non-porous cp phase into the op phase during the n-butane adsorption,26 while the N2 adsorption profile of CoBDP has several steps, which correspond to the different well-defined intermediate phases.27 On the other hand, MIL-53 breathes CO2 which is represented as two distinguished steps on the isotherm.25
Rosseinsky and others made a very intriguing comparison of the conformational energy landscape of a three-dimensional chiral MOF with flexible macromolecules – human hemoglobin.28 The authors determined nine different crystal structures and calculated their energetical minima using the DFT methodology. However, there are flexible porous materials that have a continuous spectrum of substructures29–31 represented by an infinite set of numbers. MIL-88(Fe) reported by Férey should be considered an important example of such swelling behavior.13,32 The cell volume of MIL-88 strongly depends on the type of solvent in its cavities. On the other hand, in 2017 Brammer and co-workers have reported SHF-61 which continuously changes the unit cell volume during the time-dependent desolvation.29 However, in most cases, the limited number of advanced structural characterization does not fully reveal the complete phase transition pathway.
A spatiotemporal response of MOFs to external stimuli is of paramount importance for most of the flexibility-related applications, however, at the moment, there is no clear understanding of all factors influencing the phase transition kinetics, e.g. repeatability, size effects, sample “history” etc.33 In a prospective review, Van Speybroeck and co-workers described the vision and pathways for in silico prediction of the spatiotemporal response and encouraged the community to use machine learning potential and coarse-grained model techniques in combination with enhanced sampling techniques in a finer phase space.34
Herein, using an example of two nearly identical flexible MOFs that differ only in one atom of the repeat unit, we show both continuous and discrete structural transformations. To shine light on the observed phenomena, we employed in situ PXRD measurements applied under equilibrium and out-of-equilibrium conditions in parallel to CO2 adsorption. The former involves simultaneous measurement of PXRD patterns at the defined points of the CO2 isotherm at 195 K. The latter consists of a kinetic study in which 100 PXRD patterns were collected per second upon the CO2 pressure jump from vacuum to 60 kPa at 195 K. The last part of our investigation shows that continuous transformation is superior to the discrete one in terms of CO2/CH4 separation and CH4 storage at 298 K.
Heating of the 4,4′-oxydibenzoic acid (H2oba) or 4,4′-thiodibenzoic acid (H2sba) in the presence of 2,5-di(pyridin-4-yl)thiazolo[5,4-d]thiazole (TzTz) and zinc(II) cations in N,N-dimethylformamide (DMF) yields two 3D isostructural moisture-stable metal–organic frameworks, UAM-1O and UAM-1S, respectively (Fig. 1 and S1;† UAM-1 = Uniwersytet Adama Mickiewicza material number 1). Single crystal X-ray diffraction analysis reveals that they crystallize in the monoclinic space group P21/n (Table S1†). Zn2+ cations form “paddlewheel” secondary building blocks linked in [Zn2(xba)2]n layers by the μ4-κ1κ1κ1κ1 bridging anions. The μ2-κ1κ1 TzTz linkers connect these layers to a three-dimensional non-interpenetrated network with the primitive cubic (pcu) topology (Fig. S2†). Both materials have a two-dimensional pore system occupied by DMF molecules and their calculated free void fraction (probe radius = 1.2 Å; Mercury software) and theoretical BET surface35 are approx. 34–36% of unit cell volume and ∼500 m2 g−1, respectively. Nevertheless, due to the different C–X–C bond angles (X = O 115.2°; X = S 101.4°), the pore geometry is slightly different, e.g., the maximum diameter and pore window size for UAM-1O are 6.08 Å and 3.71 Å, while for UAM-1S those values are 6.20 Å and 4.17 Å.
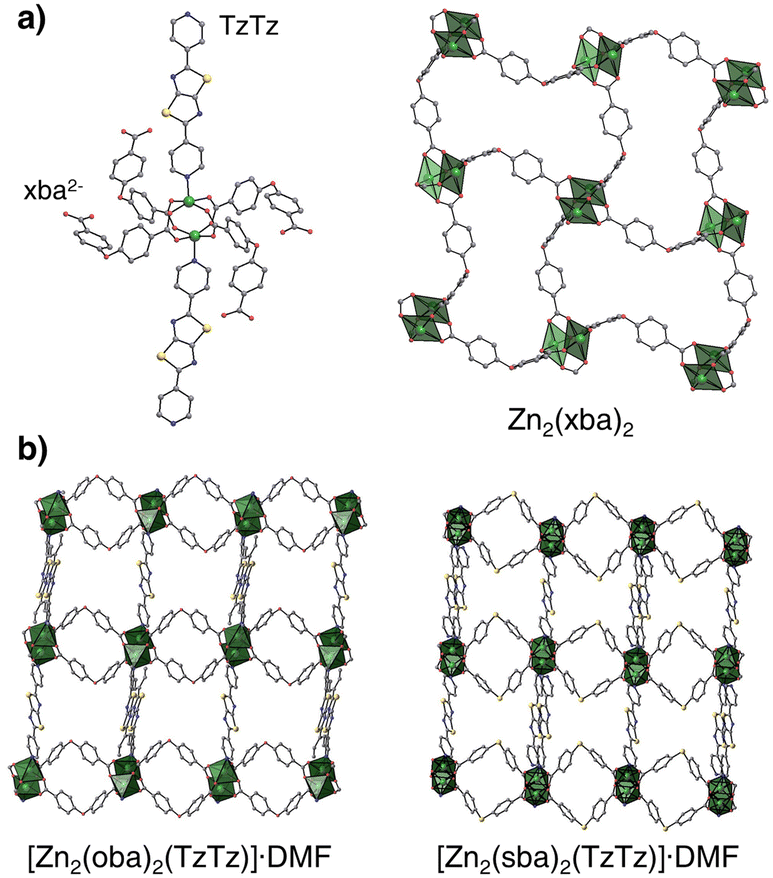 | ||
| Fig. 1 Structural features of as-synthesized UAM-1X. (a) Coordination environment of the di-zinc unit and representation of the 2D layers of Zn2(xba)2; for simplicity, shown only for UAM-1O. (b) Differences in pore geometry originates from different C–X–C angles: X = O or S; 115.2° and 101.4°, respectively. | ||
Before desolvation, the N,N′-dimethylformamide was exchanged for dichloromethane. Then both materials were activated under dynamic vacuum at 353 K. Comparison of the ex situ IR-ATR and PXRD of as-synthesized materials with the desolvated ones reveals considerable structural changes and indicates the flexible nature of both compounds (Fig. S3†), for example, signals at 2θ of 5.78 and 5.76° for UAM-1O and UAM-1S, respectively, disappear. Furthermore, the OCO stretching region of IR-ATR spectra proves the reorganization of secondary building blocks. Due to the fracture of the crystals, we were unable to determine the closed structure of desolvated MOFs, however, this will be subject of further studies. Exposure of the collapsed phases to gaseous carbon dioxide at 195 K causes its adsorption characterized by singularities15 on the recorded isotherms (Fig. 2). Firstly, both UAM-1X adsorb ∼20 cm3 g−1 which practically does not affect their structures. However, exceeding a pressure of 0.31 bar (UAM-1O) and 0.01 bar (UAM-1S) triggered different single-atom-dependent structural transformations which increased CO2 uptake to 155 cm3 g−1 and 152 cm3 g−1 at p = 0.99 bar, respectively. Based on these maximal uptakes and applying the Gurvich rule, we calculated that the experimental pore volume is equal to 0.27 cm3 g−1 (UAM-1O) and 0.28 cm3 g−1 (UAM-1S), which agrees with the theoretical values of 0.28 cm3 g−1 and 0.27 cm3 g−1.
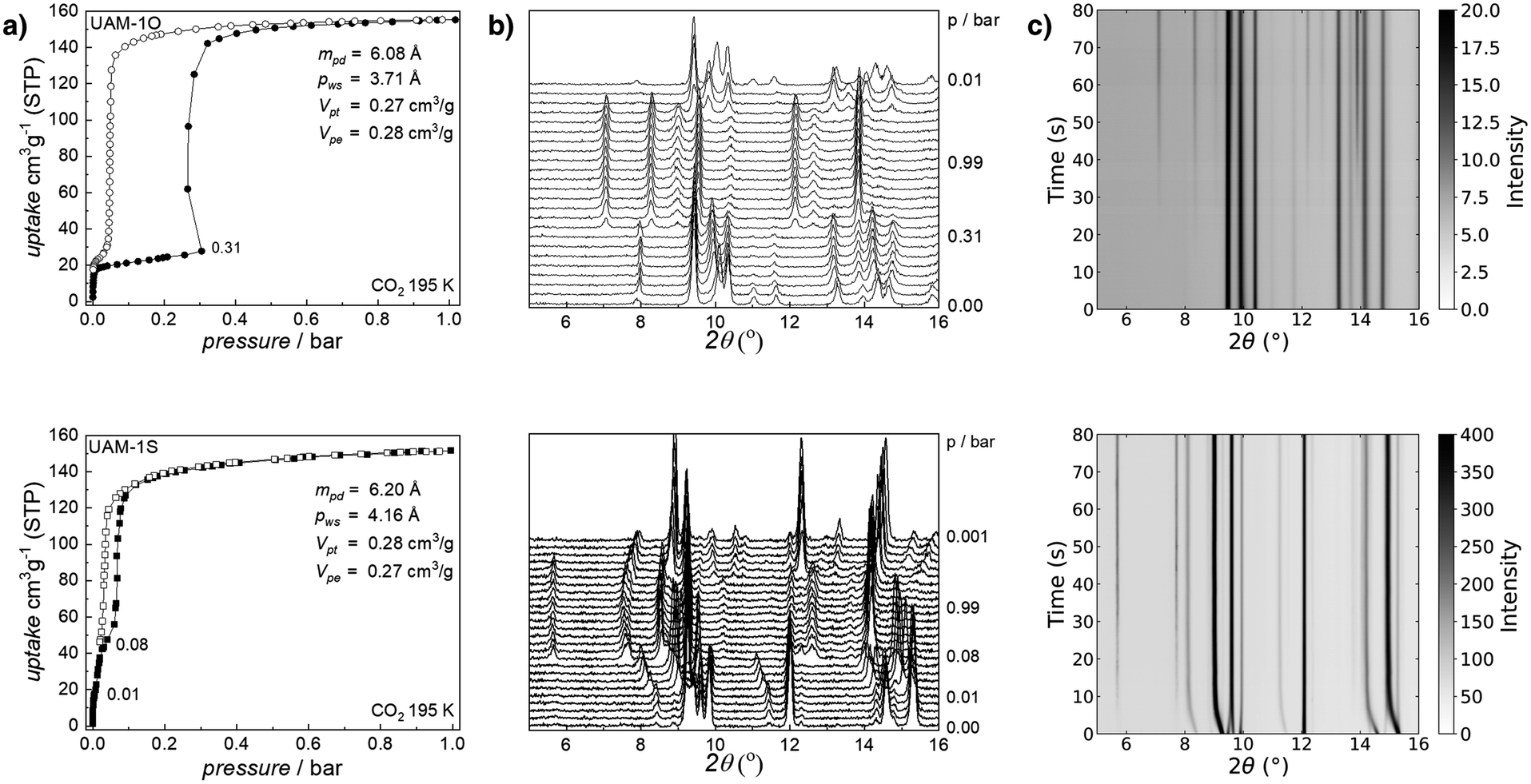 | ||
| Fig. 2 Mechanistic understanding of CO2-driven structural transformation in UAM-1X (O – top row; S – bottom row): (a) CO2 adsorption (full symbols) and desorption (open symbols) at 195 K juxtaposed with (b) corresponding in situ PXRD patterns collected at selected pressure (λ = 1.540599 Å); (c) in situ time-resolved PXRD during the CO2 adsorption at 195 K (see Fig. S5;†λ = 0.6199 Å, for consistency data were converted to λ = 1.540599 Å). To eliminate the influence of crystal size dependency, the samples underwent repeatable adsorption stress and subsequent SEM imaging prior to the in situ time-resolved measurements (Fig. S4†). mpd: maximum pore diameter; pws: pore window size; Vpt, theoretical pore volume; and Vpe, experimental pore volume for CO2 adsorption calculated at ∼0.99 bar according to the Gurvich's rule. | ||
Equilibrium in situ PXRD collected for UAM-1O above 0.31 bar shows evidence for only two states, thus the UAM-1O phase transition is described as 1st order and the MOF phases could be represented by a set of two numbers. The case of UAM-1S is more complicated since the fully CO2 saturated phase was preceded by many intermediate steps. This raises the fundamental question of whether the observed process is truly continuous or whether it is multistep adsorption as described previously by Long and co-workers.27 On the other hand, Carrington et al. have described29 nine H2O desorption steps monitored by PXRD as continuous breathing. Herein, we prove for the first time that the observed mechanism is continuous. For this purpose, we employed time-resolved synchrotron PXRD during CO2 adsorption at 195 K (Fig. 2). Importantly, before crucial experiments, we exclude the influence of crystal size dependency on adsorbate diffusion by subjecting desolvated phases to repeated adsorption–desorption stress. Scanning electron microscopy imaging confirms that both materials have comparable crystal sizes (Fig S4†).
For UAM-1O, analogous to the static conditions, we observed only two states, with the phase transformation (cp → op) beginning approximately 7 s after CO2 exposure. In contrast, CO2 injection into UAM-1S has an immediate effect on its structure, the transformation occurring as soon as the framework comes into contact with the gas but more importantly, an infinitive number of intermediate phases are detectable. This is shown in the continuous smooth change of reflection position from 2θ = 8.4° to 8.0° but also other reflections (Fig. 2c). This is clear evidence of a 2nd order transformation. As both frameworks are isostructural, the different transition mechanism is caused by the O or S atoms. In comparison to oxygen, sulfur has an additional electron shell thus is more easily polarizable and creates longer bonds. Crystal structures of UAM-1X reveal that the C–X bond length is 1.374 Å and 1.783 Å for UAM-1O and UAM-1S, respectively. These factors influence the accessible range of vibrational modes between benzenocarboxylate rings in both xba2− linkers. While in the case of UAM-1O, the C–O–C vibrational range has a narrower value, favoring the existence of two different states, the C–S–C range is higher. Therefore, a small increase in CO2 pressure triggers small changes in the unit cell parameters of UAM-1S, resulting in continuous breathing. In the case of UAM-1O, there was limited oba2− reorganization, restricting its ability to undergo gas-induced transformation.
Additionally, it has implications for the transformation pressure. Since UAM-1O exists only in closed or open states, the switching between these states requires overcoming a high energy barrier, reflected on the isotherm as a “gate opening” pressure of 0.31 bar at 195 K. On the other hand, multiple intermediate phases with low energy barriers between them can be observed in UAM-1S, resulting in a transition at a very low pressure of 0.01 bar at 195 K.
Furthermore, we assessed the influence of grinding on the adsorption properties of UAM-1O (Fig. S5†). We found that the uptake in saturation slightly increased for the ground sample, from 152 cm3 g−1 for the pristine material containing macrocrystals to 163 cm3 g−1 for the microcrystalline sample. However, the isotherm exhibited a slightly different shape, with a steady increase in uptake in the first stage, which ended with an even higher transformation pressure of 0.34 bar compared to the as-synthesized material. The larger uptake before the transformation pressure indicates that some grains open before others. All of this suggests the creation of unevenly distributed defects caused by mechanical force, which should be studied further, although it is beyond the scope of this investigation.
Understanding CO2 adsorption mechanisms poses a second fundamental question “does continuous breathing affect the two important properties of novel porous materials: gas separation and delivery?” To answer this question, we investigated high pressure single and multi-component adsorption at 298 K (Fig. 3 and 4).
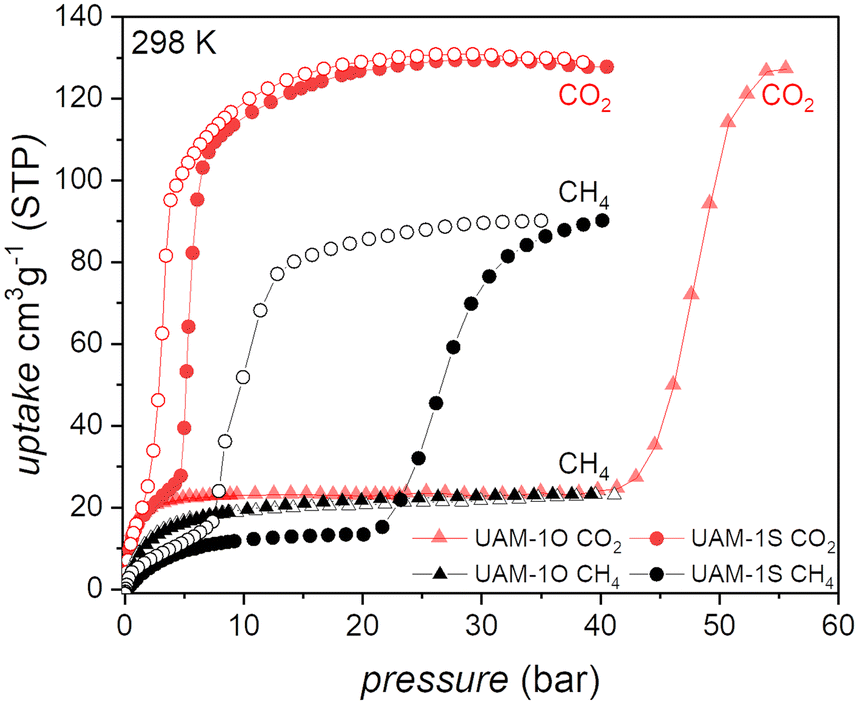 | ||
| Fig. 3 The first indication that continuous transformation is superior to the discrete one in CH4 separation and delivery processes: single-gas adsorption measurements for UAM-1X at 298 K; red CO2, black CH4, full symbols adsorption, open symbols desorption. Due to the remarkably high transformation pressure and the technical limitation that arose from it, no CO2 desorption branch for UAM-1O was recorded. | ||
Methane was not taken up by UAM-1O, while CO2 triggers the cp → op transformation at 45 bar, hindering the potential applicability of this material. CH4 induced the transformation of UAM-1S at 7.4 bar, the total gas uptake being 90 cm3 g−1 at 40 bar and the material released 80 cm3 g−1 at 5.8 bar, making UAM-1S a moderate candidate as a CH4 tank filler.36
At a CO2 transition pressure of 7.5 bar, considerably lower than CH4 (22.7 bar), UAM-1S adsorbs 109 cm3 g−1, while on desorption 98 cm3 g−1 is released at 0.5 bar. This adsorption–desorption window indicates the possibility of efficient CO2/CH4 separation, but is based on considering only the separate adsorption of CH4 and CO2. To answer the question of whether the excellent selectivity factor (Sfactor = 10; pCO2 = 7.5 bar and pCH4 = 7.5 bar, see eqn S(1)†) derived from single-adsorption isotherms would be maintained during co-adsorption, we conducted a series of co-adsorption measurements at various CO2/CH4 ratios at 298 K (Fig. 4).
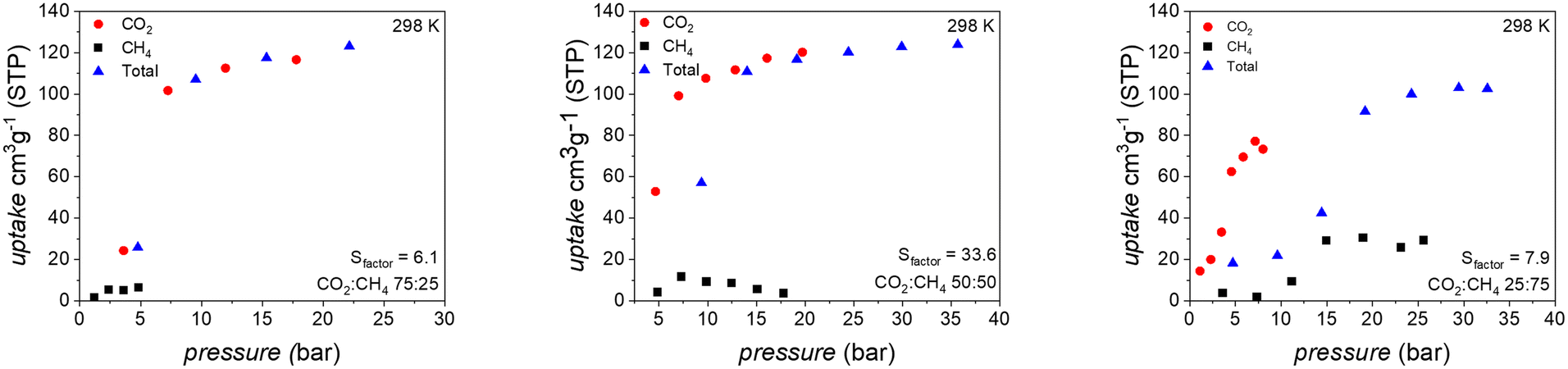 | ||
Fig. 4 Isothermal multicomponent adsorption experiments for different CO2/CH4 compositions (75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25; 50:50; 25:75 v/v) in UAM-1S at 298 K. Black squares and red circles – partial pressure of CH4 and CO2, respectively; blue triangles – total adsorbed volume. Sfactor was calculated based on the last point of each isotherm. :25; 50:50; 25:75 v/v) in UAM-1S at 298 K. Black squares and red circles – partial pressure of CH4 and CO2, respectively; blue triangles – total adsorbed volume. Sfactor was calculated based on the last point of each isotherm. | ||
In an atmosphere containing 75% v/v of CO2, the methane uptake reaches the maximum value of 6 cm3 g−1, while the maximum CO2 uptake is 117 cm3 g−1 at total pressure 22 bar. The calculated Sfactor for this point is 6.0. Enriching the composition of the gas mixture to an equimolar composition does not affect the ability of UAM-1S to exclusively adsorb CO2. Due to the considerable CO2 uptake of 120 cm3 g−1, the real Sfactor increases to 33.6 for the last point. A further increase of CH4 to 75% v/v caused a peculiar behavior of UAM-1S. At the two first points of the isotherm, the CH4 uptake is negligible, but then both gases compete for the adaptable nanoconfined space of UAM-1S. A clear manifestation of this is the desorption of 4 cm3 g−1 CO2 at the last point (ptotal 32.6 bar). In our opinion, this competitive adsorption accompanied by structural changes needs to be studied in detail by theory and experiment. It is important to answer the questions as to what is the environment of each gas in such a complex system and how does the spatial adaptivity of UAM-1S changes during the adsorption of one component from the mixture.
Methane co-adsorption at the ratio of 25:75 has a minor impact on selectivity, dropping, for example, for the total pressure of 23.1 bar, from Sfactor = 9.7 to 7.9 at the last point. These values proved here that continuous structural transformation was superior in comparison to the discrete one as well as that UAM-1S has a comparable Sfactor to benchmark materials (Tables S2 and S3†). We plan to assess next whether gradual changes can overcome the considerable problem of flexible adsorbents, the slipping off phenomenon.37
In summary, we have characterized two flexible thiazolothiazolate MOFs differing only by one atom in the linker, O or S. Upon desolvation, both materials transform into a closed phase, while the exposure of the cp phases to CO2 rebuilds their porosity. Static in situ PXRD collected at specific pressures of CO2 has shown that CO2 induced transformation of UAM-1O (cp → op) occurs in two steps, while the analogous structure, UAM-1S, exhibits multiple intermediate steps between the cp and op phases. By the utilization of time-resolved powder X-ray diffraction, we have shown that UAM-1S transformation is gradual, with a formally infinite number of intermediate states between the open and closed states. Furthermore, our findings show the superiority of continuous transformation when applied to CO2/CH4 separation and CH4 storage. We consider that the obtained results represent important findings in the chemistry of flexible MOFs, ergo the following aspects of both frameworks should be studied and compared: (i) thermal management, (ii) detailed adsorption mechanisms, and their (iii) compatibility with mixed-matrix membranes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support of the National Science Centre (NCN), Poland (Grant nos. 2020/36/C/ST4/00534 and 2018/30/E/ST5/00032) and DFG (FOR2433). We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at the P23 beamline of the PETRA III synchrotron. We would like to thank Dr A. Khadiev for support with in situ time-resolved PXRD measurements. Beamtime was allocated for proposal I-20211060. V. B. thanks the German Federal Ministry of Education and Research for financial support (BMBF Projects no. 05K22OD1). We thank Dr F. Formalik for help with Fig. 2 preparation.Notes and references
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 Search PubMed.
- S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695 CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 CAS.
- J. H. Lee, S. Jeoung, Y. G. Chung and H. R. Moon, Coord. Chem. Rev., 2019, 389, 161–188 CAS.
- S. Krause, N. Hosono and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 15325–15341 CAS.
- S. A. Moggach, T. D. Bennett and A. K. Cheetham, Angew. Chem., Int. Ed., 2009, 48, 7087–7089 CAS.
- Y. Liu, J.-H. Her, A. Dailly, A. J. Ramirez-Cuesta, D. A. Neumann and C. M. Brown, J. Am. Chem. Soc., 2008, 130, 11813–11818 CAS.
- A. Knebel, B. Geppert, K. Volgmann, D. I. Kolokolov, A. G. Stepanov, J. Twiefel, P. Heitjans, D. Volkmer and J. Caro, Science, 2017, 358, 347 CAS.
- R. Lyndon, K. Konstas, B. P. Ladewig, P. D. Southon, P. C. J. Kepert and M. R. Hill, Angew. Chem., Int. Ed., 2013, 52, 3695–3698 CAS.
- Y. Sakata, S. Furukawa, M. Kondo, K. Hirai, N. Horike, Y. Takashima, H. Uehara, N. Louvain, M. Meilikhov, T. Tsuruoka, S. Isoda, W. Kosaka, O. Sakata and S. Kitagawa, Science, 2013, 339, 193–196 CAS.
- S. Krause, V. Bon, I. Senkovska, U. Stoeck, D. Wallacher, D. M. Többens, S. Zander, R. S. Pillai, G. Maurin, F.-X. Coudert and S. Kaskel, Nature, 2016, 532, 348 CAS.
- H. Sato, W. Kosaka, R. Matsuda, A. Hori, Y. Hijikata, R. V. Belosludov, S. Sakaki, M. Takata and S. Kitagawa, Science, 2014, 343, 167 CAS.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CAS.
- J. A. Mason, J. Oktawiec, M. K. Taylor, M. R. Hudson, J. Rodriguez, J. E. Bachman, M. I. Gonzalez, A. Cervellino, A. Guagliardi, C. M. Brown, P. L. Llewellyn, N. Masciocchi and J. R. Long, Nature, 2015, 527, 357 CAS.
- Q.-Y. Yang, P. Lama, S. Sen, M. Lusi, K.-J. Chen, W.-Y. Gao, M. Shivanna, T. Pham, N. Hosono, S. Kusaka, J. J. Perry IV, S. Ma, B. Space, L. J. Barbour, S. Kitagawa and M. J. Zaworotko, Angew. Chem., Int. Ed., 2018, 57, 5684–5689 CAS.
- M. K. Taylor, T. Runčevski, J. Oktawiec, J. E. Bachman, R. L. Siegelman, H. Jiang, J. A. Mason, J. D. Tarver and J. R. Long, J. Am. Chem. Soc., 2018, 140, 10324–10331 CAS.
- J. Y. Kim, L. Zhang, R. Balderas-Xicohténcatl, J. Park, M. Hirscher, H. R. Moon and H. Oh, J. Am. Chem. Soc., 2017, 139, 17743–17746 CAS.
- K. Roztocki, V. Bon, I. Senkovska, D. Matoga and S. Kaskel, Chem. –Eur. J., 2022, 28, e202202255 CAS.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J.-R. Li and B. Chen, Nat. Energy, 2017, 2, 877–883 CAS.
- R. Haldar, R. Matsuda, S. Kitagawa, S. J. George and T. K. Maji, Angew. Chem., Int. Ed., 2014, 53, 11772–11777 CAS.
- S. Yuan, L. Zou, H. Li, Y.-P. Chen, J. Qin, Q. Zhang, W. Lu, M. B. Hall and H.-C. Zhou, Angew. Chem., Int. Ed., 2016, 55, 10776–10780 CAS.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CAS.
- Y. Su, K. Otake, J.-J. Zheng, S. Horike, S. Kitagawa and C. Gu, Nature, 2022, 611, 289–294 CAS.
- N. Guillou, F. Millange and R. I. Walton, Chem. Commun., 2011, 47, 713–715 CAS.
- D. Li and K. Kaneko, Chem. Phys. Lett., 2001, 335, 50–56 CAS.
- N. Klein, C. Herzog, M. Sabo, I. Senkovska, J. Getzschmann, S. Paasch, M. R. Lohe, E. Brunner and S. Kaskel, Phys. Chem. Chem. Phys., 2010, 12, 11778–11784 CAS.
- F. Salles, G. Maurin, C. Serre, P. L. Llewellyn, C. Knöfel, H. J. Choi, Y. Filinchuk, L. Oliviero, A. Vimont, J. R. Long and G. Férey, J. Am. Chem. Soc., 2010, 132, 13782–13788 CAS.
- A. P. Katsoulidis, D. Antypov, G. F. S. Whitehead, E. J. Carrington, D. J. Adams, N. G. Berry, G. R. Darling, M. S. Dyer and M. J. Rosseinsky, Nature, 2019, 565, 213–217 CAS.
- E. J. Carrington, C. A. McAnally, A. J. Fletcher, S. P. Thompson, M. Warren and L. Brammer, Nat. Chem., 2017, 9, 882 CAS.
- G. K. Angeli, E. Loukopoulos, K. Kouvidis, A. Bosveli, C. Tsangarakis, E. Tylianakis, G. Froudakis and P. N. Trikalitis, J. Am. Chem. Soc., 2021, 143, 10250–10260 CAS.
- E. J. Carrington, S. F. Dodsworth, S. van Meurs, M. R. Warren and L. Brammer, Angew. Chem., Int. Ed., 2021, 60, 17920–17924 CAS.
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284–286 Search PubMed.
- D. J. Cerasale, D. C. Ward and T. L. Easun, Nat. Rev. Chem., 2022, 6, 9–30 Search PubMed.
- V. Van Speybroeck, S. Vandenhaute, A. E. J. Hoffman and S. M. J. Rogge, Trends Chem., 2021, 3, 605–619 Search PubMed.
- T. F. Willems, C. H. Rycroft, M. Kazi, J. C. Meza and M. Haranczyk, Microporous Mesoporous Mater., 2012, 149, 134–141 CAS.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557–580 CAS.
- S. Hiraide, Y. Sakanaka, H. Kajiro, S. Kawaguchi, M. T. Miyahara and H. Tanaka, Nat. Commun., 2020, 11, 3867 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data and structural visualizations (PDF). CCDC 2247315 (UAM-1O) and 2247321 (UAM-1S). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ta02167j |
| This journal is © The Royal Society of Chemistry 2023 |