
High-areal-capacity anode-free all-solid-state lithium batteries enabled by interconnected carbon-reinforced ionic-electronic composites†
Wen-Ze
Huang
 a,
Ze-Yu
Liu
b,
Pan
Xu
a,
Wei-Jin
Kong
a,
Xue-Yan
Huang
a,
Peng
Shi
a,
Peng
Wu
c,
Chen-Zi
Zhao
*a,
Hong
Yuan
d,
Jia-Qi
Huang
d and
Qiang
Zhang
*a
a,
Ze-Yu
Liu
b,
Pan
Xu
a,
Wei-Jin
Kong
a,
Xue-Yan
Huang
a,
Peng
Shi
a,
Peng
Wu
c,
Chen-Zi
Zhao
*a,
Hong
Yuan
d,
Jia-Qi
Huang
d and
Qiang
Zhang
*a
aDepartment of Chemical Engineering, Beijing Key Laboratory of Green Chemical Reaction Engineering and Technology, Tsinghua University, Beijing 100084, China. E-mail: zcz@mail.tsinghua.edu.cn; zhang-qiang@mails.tsinghua.edu.cn
bZhili College, Tsinghua University, Beijing 100084, China
cResearch & Development Center BMW, BMW China Services Ltd, Beijing 101318, China
dSchool of Materials Science and Engineering, Advanced Research Institute of Multidisciplinary Science, Beijing Institute of Technology, Beijing 100081, China
First published on 10th February 2023
Abstract
Taking energy density and safety into account, the anode-free all-solid-state lithium battery is a strong candidate to surpass the capabilities of routine lithium-ion batteries. However, achieving uniform stable lithium metal plating under high areal capacity is a grand challenge facing practical applications of lithium metal batteries. We report a high-performance anode-free all-solid-state lithium battery with a current collector consisting of carbon-reinforced ionic-electronic composites. When an interconnected carbon paper is compounded with a solid electrolyte, a three-dimensional ionic-electronic conduction network can be achieved, affording a large number of sites and scalable spaces for the nucleation and growth of lithium metal. The composite layer can achieve a long cycle life (>5000 cycles), stable lithium metal plating with a high areal capacity (>8 mAh cm−2), which is significantly better than that of the copper current collectors for routine anode-free configurations. The application of high-areal-capacity (4 mAh cm−2) pouch cell provides an efficient and effective strategy for practical anode-free all-solid-state lithium batteries.
10th anniversary statementCongratulations on the 10th anniversary of the Journal of Materials Chemistry A. The development of advanced energy materials is a core issue if we want to achieve a decarbonized society. The fundamental science to address this issue is the primary interest of Journal of Materials Chemistry A, which has contributed substantially to the development of emerging materials in this field. We are honored and delighted for the opportunities to contribute to the journal, and we hope and believe that Journal of Materials Chemistry A will continue to lead the materials for energy and sustainability in the next decade and beyond. |
1. Introduction
All-solid-state batteries (ASSBs) are superstars and game changers in the current energy sector due to their better safety characteristics than existing lithium-ion batteries.1,2 ASSBs can be used in a cell with bipolar stacked electrodes to achieve a series connection of cells, which greatly reduces the weight and size of the package and increases the energy density of rechargeable cells. Solid-state electrolytes (SEs) are also promising to enhance the battery power density due to the lithium transmission number (tLi+) being close to 1.3 The increasing ionic conductivity of SEs enables their successful application in solid-state batteries. Some inorganic SEs, such as sulfides, have a high ionic conductivity of over 10 mS cm−1 at ambient temperature.2,3 The recent advances in terms of reduced costs,4 improved preparation methods,5 and superior mechanical properties6 of SEs have also paved the way for large-scale production. With the widespread acceptance of ASSBs, the feasibility of using energy-dense lithium metal as an anode is reviving.7–14 However, current lithium batteries often employ an excessive amount of lithium as the anode, which hinders the increase in battery energy density.15 Besides, the preparation and handling of ultra-thin lithium metal pose significant challenges in terms of manufacturing processes, cost, and safety.16The anode-free all-solid-state lithium metal battery (AFSSLB) is introduced to achieve the maximum energy density for any given lithiated cathode system without compromising on safety.17,18 The initial lithium anode is formed by extracting lithium ions from the lithiated cathode during the first charge. Nevertheless, the implementation of AFSSLBs faces several challenges of (1) accommodating the large volume change of the electrodes during repeated cycling and (2) the growth of lithium dendrites due to the uneven flux of lithium ions.17,19 The synergy of the above factors often leads to short-circuit (Fig. 1a). Besides, copper (Cu) foil is one of the most unfavorable substrates for lithium metal plating as it is not conducive to lithium metal dissolution,20 while substrates with smaller nucleation overpotential and weaker surface diffusion barriers enable stable growth of deposited lithium leading to enhanced electrochemical performance.21
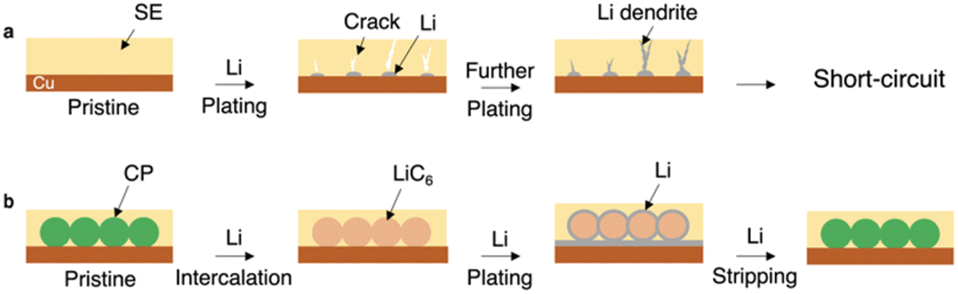 | ||
| Fig. 1 Schematic diagrams of different current collectors for AFSSLBs. (a) Lithium nuclei are formed on the surface of Cu. Volumetric changes during the Li plating process can easily break the SE, especially at high areal capacity. This behavior leads to the growth of cracks and subsequent Li dendrites, which results in short-circuit. (b) The modification of the Cu substrate with an interconnected carbon paper creates a host with large plating sites for stabilizing the lithium intercalation and the subsequent plating. The volumetric change of the Li plating is accommodated by the flexible interconnected carbon paper. | ||
Some strategies have been proposed to address these issues, including the manipulation of current collectors,22–25 the rational design of solid electrolytes,26,27 and the adjustment of cycling protocols.28 Cells designed based on these strategies pioneered the feasibility of AFSSLBs, despite their limited areal-capacity and moderate cycle life. Recently, regulating lithium metal plating behaviors by the modification of the working surfaces of current collectors has attracted much attention.29–31 The implementation of silver-carbon composites affords impressive high areal capacity and long cycle life.32 However, their preparation processes are often complex and the mechanisms remain elusive. This raises a fundamental question of how the structural and physicochemical properties of a current collector affect the deposition properties of lithium metal. Compared with a conventional planar Cu current collector, interconnected three-dimensional (3D) hosts with lithiophilic sites are promising to decrease local current density and modulate lithium nucleation for a homogeneous lithium deposition,33–39 relieving stresses arising from volume expansion. The effectiveness of such hosts is apparent in liquid systems and through theoretical simulation. Therefore, it would be very interesting and important to construct such hosts in AFSSLBs to understand their roles in regulating lithium deposition behaviors and to evaluate them under relevant structural or physicochemical parameters.
In this contribution, an AFSSLB with a high areal capacity greater than 8 mAh cm−2 is reported, achieved by using a 3D interconnected carbon paper (CP) as current collector (Fig. 1b). A uniform and dense LiC6 interfacial layer forms on the surface of the CP host during the initial lithium intercalation, which facilitates the wettability of lithium on the CP and reduces the lithium metal nucleation overpotential (∼1.5 mV). Lithium metal can be further deposited on the lithiophilic LiC6 surface (ESI, Fig. S1†), inducing stable plating of lithium and adapting to volume changes through the 3D scaffolds, ultimately improving the cycle stability (>5000 cycles).
2. Results and discussion
2.1. 3D interconnected carbon layer
The interconnected interfaces are constructed through CP with 3D woven carbon fibers (CFs) (Fig. 2a). The abundance of macropores between the fibers provides enough space to accommodate the subsequent plating of lithium metal. The diameter of a single CF bundle is approximately 6.5 μm, which is made up of many fine CFs (Fig. 2b). The total thickness of the CP layer is 50 μm without pressing (cross section shown in Fig. 2d). The large porosity of the fibers in the direction perpendicular to the CP mitigates the stress caused by volumetric changes during lithium metal plating. | ||
| Fig. 2 The 3D interconnected CP. SEM images of (a) CP, which is composed of bundles of CFs, and (b) a bundle of CFs with a diameter of 6.5 μm. (c) Digital camera image of CP. (d) Cross-sectional SEM image of CP with a thickness of 50 μm. (e) HRTEM image of CP showing d(002) plane of graphite with an interplanar spacing of 0.34 nm. (f) SEM image of CP at the corner of a bend. Red dashed line: the trace of bending curvature. | ||
Transmission electron microscopy (TEM) and X-ray diffraction patterns exhibit that the CFs are composed of well-crystalline graphite with a (002) layer spacing of 0.34 nm (Fig. 2e, ESI, Figs. S2 and S3†). The LiC6 compound is formed during the first lithium intercalation. In turn, the excellent ionic conductivity and lithiophilic nature of the LiC6 layer promote close contacts between the lithium metal and the CP host, which facilitates dendrite-free plating/stripping behaviors over many cycles.34 Digital camera images (Fig. 2c) and scanning electron microscopy (SEM) images (Fig. 2f) exhibit that the CP films have excellent bending properties, ensuring that the CP does not fracture under large volume changes, rendering their application in flexible and wearable energy storage devices feasible.40
2.2. Ionic-electronic conduction network
Both CP and SE were assembled into a 3D carbon-reinforced ionic-electronic composite (CRIEC) as current collector by one-step pressing. X-ray computed tomography (XCT) was carried out to probe the fine structure of the composite layer. The tight fit of the CP to the SE is observed on the cross sections of the CRIEC (Fig. 3a). The yellow dashed line points out the undulating interfaces between them. Apparently, the CP is subjected to an overall compression in the vertical direction, which is approximately 40 μm at the highest point and only a few micrometers at the lowest position (ESI, Fig. S4†). In the meantime, the electrolyte is embedded into the pores of the CP to form linked ion channels. | ||
| Fig. 3 Phase-contrast XCT of CRIEC showing a 3D ionic-electronic conduction network. (a) Cross-sectional image slice at the edge of the current collectors revealing the tight contact interfaces between the CP and the electrolyte (marked with a yellow dashed line). (b) Virtual image slice taken along a plane parallel to the CP. The dark gray indicates the CP substrate and the light gray indicates the SE, which act as conductors of electrons and lithium ions, respectively. (c) Virtual image slices taken along planes parallel to the CP from the surface of CP to the SE layer (positions indicated by the red and blue arrows in (a)). A magnified image of (c) (iv) is shown in (b) for detail. | ||
To further elucidate the 3D structure of the CRIEC, XCT image slices were acquired along a plane parallel to the CRIEC, starting from the red dashed line and ending at the blue dashed line (Fig. 3a). The positions of the image slices in Fig. 3c are indicated by the red and blue arrows on the virtual cross-sectional image slice in Fig. 3a. Fig. 3b exhibits the details in the image of Fig. 3c(iv). The interconnected CP layer (black) acts as a substrate, with the SE (grey) dispersed within, forming a tightly bound interfacial layer with the CP (yellow dashed line). Herein the CP layer is an excellent electronic conductor, while the SE layer is a particularly good diffusion channel of lithium ions. They nest with each other to afford a 3D structure for the nucleation and growth of the lithium metal anode. In the direction of the longitudinal section (indicated by the black arrow in Fig. 3a), Fig. 3c(i) exhibits a surface consisting entirely of CP layer (where the red arrow points in Fig. 3a). With further longitudinal sectioning towards Fig. 3c(ii) and (iii), a small portion of electrolyte and its interfaces with the CP appears (yellow dashed line); while in Fig. 3c(iv) and (v) it can be seen that the percentage of electrolyte is expanding while the percentage of CP is reducing; in Fig. 3c(vi) and (vii), as one progresses further in depth, the electrolyte dominates while the CP layer has only a few small isolated patches; and finally, the layer is entirely composed of electrolyte at the position of the blue line (where the blue arrow points in Fig. 3a). These results clearly demonstrate that the CP and SE form a 3D interconnected, tightly bound composite structure with a well-established electron and ion conduction network, providing abundant sites and buffer intervals for subsequent plating of lithium metal.
2.3. Lithium plating/stripping behaviors in the CRIEC
To evaluate the performance of the CRIEC, it was subjected to lithium metal plating and compared to the performance of Super P (SP) modified and unmodified Cu foils. In the CRIEC as well as in the SP modified foil, the lithium intercalation reactions occurred prior to forming lithiophilic Li–C composite to the observed voltage plateau for lithium metal plating (ESI, Fig. S5†). The intercalation of Li atoms into carbon is thermodynamically preceded by the plating of lithium metal due to the negative Gibbs free energy of the reaction (Li + 6C → LiC6, −10.59 kJ mol−1).41 For objectivity of comparison, only the voltage profile at the start of lithium metal plating is recorded in Fig. 4a. The bare Cu foil exhibited a large nucleation overpotential (8.4 mV) and was short-circuited after deposition of very little lithium metal (∼0.015 mAh cm−2). SE is easily damaged due to the non-uniform growth of lithium, which increases the probability of short circuiting. In contrast, the SP modified foil exhibited a smaller nucleation overpotential, suggesting that the lithiophilic Li–C composite reduces the nucleation energy of lithium metal, which induces a more uniform lithium plating. However, the voltage profile converges to zero gradually during plating, suggesting a micro-short-circuit,42 where the plated lithium metal is penetrating the SE and the distance between the electrodes is reducing. The short circuit occurs after plating a lithium metal capacity of ∼0.25 mAh cm−2. This indicates that a routine two-dimensional carbon film is insufficient to match the high areal capacity. Notably, lithium metal nucleation in the CRIEC exhibits a smooth voltage dip and the overpotential is extremely small (only 1.5 mV), demonstrating that the lithiophilic graphitic Li–C composite can effectively regulate the nucleation behavior of lithium metal anode. In addition, the subsequent plating plateau is ultra-smooth, indicating that the abundance of 3D macropores and large porosity between the fibers in the CRIEC to accommodate the volumetric change promise the stability of the composite electrode for high areal capacity. Fig. 4b summarizes and compares the nucleation overpotential and the maximum lithium deposited areal capacity for the three different current collectors. The coulombic efficiency of the lithium plating/stripping in the CRIEC is >95% with an areal capacity of 3.0 mAh cm−2 (including the capacity of lithium intercalation, Fig. 4c). The CRIEC can substantially reduce the nucleation overpotential and regulate the subsequent lithium plating/stripping behavior, enabling stable cycling with high areal capacity.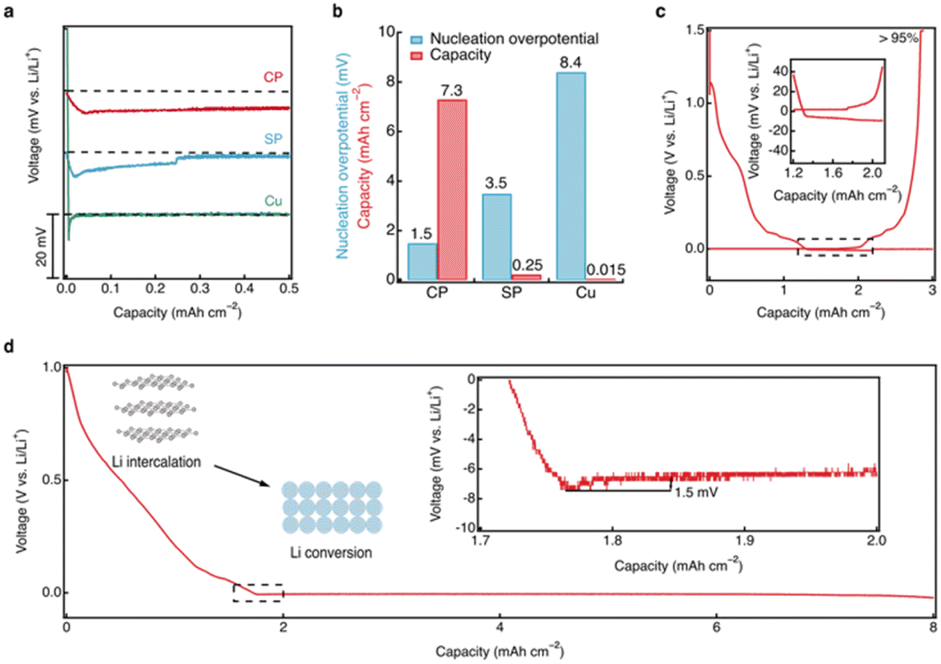 | ||
| Fig. 4 Lithium plating/stripping behaviors in the CRIEC. (a) Voltage profiles of AFSSLBs with CP, SP, and bare Cu. To enhance the comparison, the curves are shifted horizontally according to the onset of lithium nucleation and vertically with a constant shift of 20 mV. (b) The nucleation overpotential and the maximum areal capacity of lithium plating with different interfacial layers. (c) coulombic efficiency of the plating/stripping of the CRIEC after depositing an areal capacity of 3.0 mAh cm−2 (including the capacity of the lithium intercalation). (d) Stable lithium plating in CRIEC with an areal capacity of 8 mAh cm−2 and a nucleation overpotential of 1.5 mV. The current density is 0.05 mA cm−2 at ambient temperature. | ||
The ultimate plating capacity of the CRIEC was further probed (Fig. 4d). The CRIEC exhibited an exceptionally high plating capacity, remaining stable at a capacity of 8 mAh cm−2. After depositing a capacity of more than 9 mAh cm−2, a short-circuit behavior occurred (ESI, Fig. S6†). The increased overpotential is due to the contact loss caused by the over-discharge of lithium metal counter-electrode, which eventually triggers the growth of lithium dendrites. When pairing with a more stable counter electrode than lithium metal, better plating performance can be expected. The potential of the CRIEC was further probed under various extreme conditions. A relatively high value of 0.3 mA cm−2 can be applied at 333 K with an areal capacity of over 3.5 mAh cm−2 (ESI, Fig. S7†). Besides, the critical current density of lithium stripping reaches 0.4 mA cm−2 at ambient temperature when a pressure of 3.5 MPa is applied (ESI, Fig. S8†).
2.4. Electrochemical performance of the AFSSLBs
Motivated by the exceptional abilities of the CRIEC to induce lithium metal nucleation and to accommodate the large volumetric changes associated with lithium metal plating/stripping, we performed cycling tests on both mold cells and pouch cells with the CRIEC. In the mold cell, representative single-crystal NCM-622 and NCM-811 ternary cathodes were selected with an areal loading of 8.9 mg cm−2. Even at ambient temperature, there is an excellent charging capacity of 172 and 201 mAh g−1 on single-crystal NCM-622 and NCM-811 ternary cathodes, respectively (Fig. 5a and b). In contrast, for the control current collector Cu matches, there is a sudden voltage drop (ESI, Fig. S9†) and the inability to reach the cut-off voltage results in a continuous overcharge, which is attributed to the occurrence of a soft short-circuit.43 During the discharge process, the NCM-622 and NCM-811 mold cells showed a capacity loss of 0.82 and 0.87 mAh cm−2, respectively, presumably due to the generation of the SEI and CEI in the initial cycle.44,45 This implies that a further rise in areal capacity can improve the discharge capacity and coulombic efficiency of a working cell. Pouch cells were operated with an areal capacity up to 4.0 mAh cm−2 to emphasize the electrochemical performance of the CRIEC, exhibiting an excellent discharge capacity of 2.2 mAh cm−2.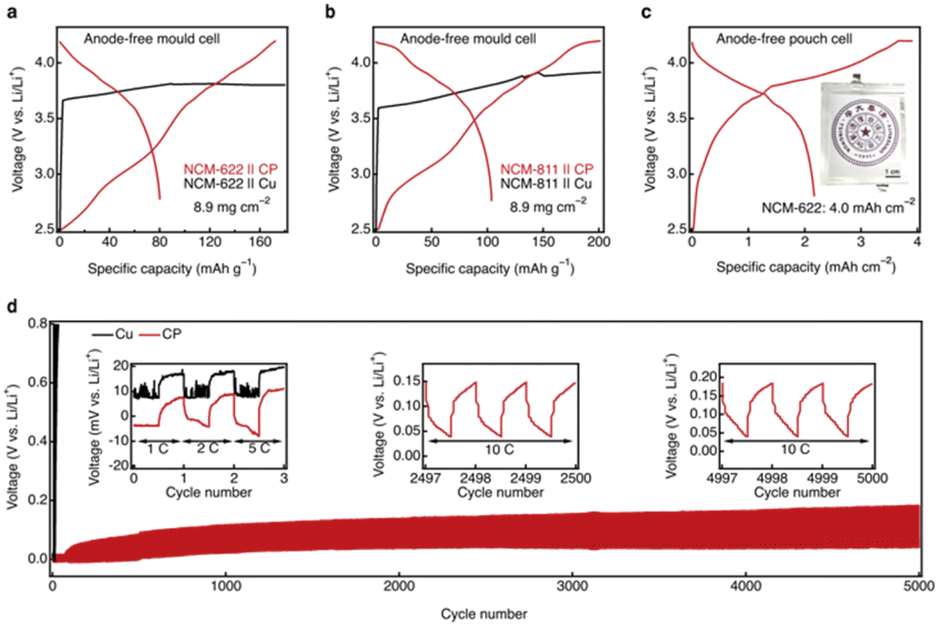 | ||
| Fig. 5 Electrochemical performance of AFSSLBs with the CRIEC. (a and b) Comparison of charge–discharge performances of the CRIEC and bare Cu with LiNbO3-coated single crystal (a) NCM-622 and (b) NCM-811 as cathode active materials. The areal loading of active materials is 8.9 mg cm−2. (c) Charge–discharge profiles of anode-free pouch cell with an areal capacity of 4.0 mAh cm−2. The inset shows a digital camera image of a pouch cell. Scale bars, 1 cm. (d) Comparison of the cycling performance of LiIn‖CP (marked in red) and LiIn‖Cu (marked in black) cells at various current densities (1C = 0.05 mA cm−2). Insets: magnified voltage profiles for different cycles. | ||
Inspired by the outstanding charge–discharge performance of the CRIEC, long-time cycling tests were carried out (Fig. 5d). In the first few cycles, the cell with bare Cu showed unstable voltage profiles, which was ascribed to the non-uniform growth of lithium leading to soft short-circuit. In stark contrast, the cell with the CRIEC exhibited stable cycling with an extremely low polarization of 10 mV at 5C (1C = 0.05 mA cm−2). This low polarization is attributed to the conformal interface between the CP and the SE layer, which can substantially reduce the interfacial resistance and induce a uniform flux of lithium ions. Notably, the cell with bare Cu showed rapidly deteriorating overpotential indicating the contact loss between the bare Cu and the SE at a high current density of 10C in the subsequent cycles, while the cell with the CRIEC exhibited ultrastable voltage profiles even after 5000 cycles. The distinct cycling stability again verifies that the CRIEC layer can form a conformal interface with a 3D ionic-electronic conduction network to induce a uniform flux of lithium ions and adapt to the volumetric change during cycling.
3. Conclusions
In summary, AFSSLBs are constructed using interconnected CP as 3D current collector, a SE, and a high-nickel NCM cathode. The 3D ionic-electronic conduction network of the CRIEC affords abundant lithiophilic sites and scalable spaces for lithium nucleation and inhibits dendritic growth of metallic lithium. The cells with CRIEC layer exhibit superior cycle life (>5000 cycles) with an extremely low nucleation overpotential (∼1.5 mV). The CRIEC enables an ultrahigh areal capacity (>8 mAh cm−2) demonstrating its great potential to stabilize the lithium metal plating. The successful application of high-areal-capacity pouch cells (4 mAh cm−2) paves the way for the design of practical AFSSLBs. This work sheds fresh light on the optimization of anode-free all-solid-state batteries with much-enhanced energy density and safety.Author contributions
Q. Z., C. Z., and W. H. conceived the ideas and designed the experiments. Z. L., P. X., W. K., X. H., and P. S. were involved with the methods and characterizations of materials. W. H., C. Z., Q. Z., P. W., H. Y., and J. H. analysed the data and wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Key Research and Development Program (2021YFB2500300), National Natural Science Foundation of China (22108151, 22109084, 22075029, U1910202, and 21825501), Key Research and Development Program of Yunnan Province (202103AA080019), S&T Program of Hebei (22344402D), International Postdoctoral Exchange Fellowship Program (YJ20210125), and the ‘Shuimu Tsinghua Scholar’ Program of Tsinghua University.Notes and references
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Nat. Res. Mater., 2020, 5, 229–252 CrossRef CAS.
- N. Kamaya, K. Homma, Y. Yamakawa, M. Hirayama, R. Kanno, M. Yonemura, T. Kamiyama, Y. Kato, S. Hama, K. Kawamoto and A. Mitsui, Nat. Mater., 2011, 10, 682–686 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- W. Huang, N. Matsui, S. Hori, K. Suzuki, M. Hirayama, M. Yonemura, T. Saito, T. Kamiyama, Y. Sasaki, Y. Yoon, S. Kim and R. Kanno, J. Am. Chem. Soc., 2022, 144, 4989–4994 CrossRef CAS.
- S. Yubuchi, S. Teragawa, K. Aso, K. Tadanaga, A. Hayashi and M. Tatsumisago, J. Power Sources, 2015, 293, 941–945 CrossRef CAS.
- G. L. Zhu, C. Z. Zhao, H. J. Peng, H. Yuan, J. K. Hu, H. X. Nan, Y. Lu, X. Y. Liu, J. Q. Huang, C. A. X. He, J. Zhang and Q. Zhang, Adv. Funct. Mater., 2021, 31, 2101985 CrossRef CAS.
- X. B. Cheng, R. Zhang, C. Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS.
- S. Y. Yuan, T. Y. Kong, Y. Y. Zhang, P. Dong, Y. J. Zhang, X. L. Dong, Y. G. Wang and Y. Y. Xia, Angew. Chem., Int. Ed., 2021, 60, 25624–25638 CrossRef CAS.
- X. Shen, X. Q. Zhang, F. Ding, J. Q. Huang, R. Xu, X. Chen, C. Yan, F. Y. Su, C. M. Chen, X. J. Liu and Q. Zhang, Energy Mater. Adv., 2021, 2021, 1205324 Search PubMed.
- X.-B. Cheng, H. Liu, H. Yuan, H.-J. Peng, C. Tang, J.-Q. Huang and Q. Zhang, SusMat, 2021, 1, 38–50 CrossRef CAS.
- J. F. Ding, R. Xu, C. Yan, Y. Xiao, Y. R. Liang, H. Yuan and J. Q. Huang, Chin. Chem. Lett., 2020, 31, 2339–2342 CrossRef CAS.
- Q. J. Yu, K. C. Jiang, C. L. Yu, X. J. Chen, C. J. Zhang, Y. Yao, B. Jiang and H. J. Long, Chin. Chem. Lett., 2021, 32, 2659–2678 CrossRef CAS.
- B. L. Zhao, L. X. Ma, K. Wu, M. X. Cao, M. G. Xu, X. X. Zhang, W. Liu and J. T. Chen, Chin. Chem. Lett., 2021, 32, 125–131 CrossRef CAS.
- Y. Na, Z. Chen, Z. K. Xu, Q. An, X. Zhang, X. H. Sun, S. Cai and C. M. Zheng, Chin. Chem. Lett., 2022, 33, 4037–4042 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- H. Chen, Y. F. Yang, D. T. Boyle, Y. K. Jeong, R. Xu, L. S. de Vasconcelos, Z. J. Huang, H. S. Wang, H. X. Wang, W. X. Huang, H. Q. Li, J. Y. Wang, H. K. Gu, R. Matsumoto, K. Motohashi, Y. Nakayama, K. J. Zhao and Y. Cui, Nat. Energy, 2021, 6, 790–798 CrossRef CAS.
- W. Z. Huang, C. Z. Zhao, P. Wu, H. Yuan, W. E. Feng, Z. Y. Liu, Y. Lu, S. Sun, Z. H. Fu, J. K. Hu, S. J. Yang, J. Q. Huang and Q. Zhang, Adv. Energy Mater., 2022, 12, 2201044 CrossRef CAS.
- C. Heubner, S. Maletti, H. Auer, J. Huttl, K. Voigt, O. Lohrberg, K. Nikolowski, M. Partsch and A. Michaelis, Adv. Funct. Mater., 2021, 31, 2106608 CrossRef CAS.
- E. Kazyak, M. J. Wang, K. W. Lee, S. Yadavalli, A. J. Sanchez, M. D. Thouless, J. Sakamoto and N. P. Dasgupta, Matter, 2022, 5, 3912–3934 CrossRef CAS.
- K. Yan, Z. Lu, H.-W. Lee, F. Xiong, P.-C. Hsu, Y. Li, J. Zhao, S. Chu and Y. Cui, Nat. Energy, 2016, 1, 16010 CrossRef CAS.
- V. Pande and V. Viswanathan, ACS Energy Lett., 2019, 4, 2952–2959 CrossRef CAS.
- X. Xing, Y. J. Li, S. Wang, H. D. Liu, Z. H. Wu, S. C. Yu, J. Holoubek, H. Y. Zhou and P. Liu, ACS Energy Lett., 2021, 6, 1831–1838 CrossRef CAS.
- O. Garcia-Calvo, A. Gutierrez-Pardo, I. Combarro, A. Orue, P. Lopez-Aranguren, I. Urdampilleta and A. Kvasha, Front. Chem., 2022, 10, 934365 CrossRef CAS.
- D. Gu, H. Kim, J. H. Lee and S. Park, J. Energy Chem., 2022, 70, 248–257 CrossRef CAS.
- S. H. Park, D. Y. Jun, G. H. Lee, S. G. Lee, J. E. Jung, K. Y. Bae, S. Son and Y. J. Lee, Adv. Sci., 2022, 9, 2203130 CrossRef CAS.
- M. J. Wang, E. Carmona, A. Gupta, P. Albertus and J. Sakamoto, Nat. Commun., 2020, 11, 5201 CrossRef CAS.
- B. J. Neudecker, N. J. Dudney and J. B. Bates, J. Electrochem. Soc., 2000, 147, 517–523 CrossRef CAS.
- T. Fuchs, J. Becker, C. G. Haslam, C. Lerch, J. Sakamoto, F. H. Richter and J. Janek, Adv. Energy Mater., 2023, 13, 2203174 CrossRef CAS.
- J. Oh, S. H. Choi, B. Chang, J. Lee, T. Lee, N. Lee, H. Kim, Y. Kim, G. Im, S. Lee and J. W. Choi, ACS Energy Lett., 2022, 7, 1374–1382 CrossRef CAS.
- J. Lee, S. H. Choi, G. Im, K.-J. Lee, T. Lee, J. Oh, N. Lee, H. Kim, Y. Kim, S. Lee and J. W. Choi, Adv. Mater., 2022, 34, 2203580 CrossRef CAS.
- S. H. Park, D. Jun, J. E. Jung, S. G. Lee, G. H. Lee and Y. J. Lee, J. Mater. Chem. A, 2022, 10, 21995–22006 RSC.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- R. Zhang, X. Chen, X. Shen, X.-Q. Zhang, X.-R. Chen, X.-B. Cheng, C. Yan, C.-Z. Zhao and Q. Zhang, Joule, 2018, 2, 764–777 CrossRef CAS.
- P. Shi, T. Li, R. Zhang, X. Shen, X. B. Cheng, R. Xu, J. Q. Huang, X. R. Chen, H. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1807131 CrossRef.
- H. Kwon, J. H. Lee, Y. Roh, J. Baek, D. J. Shin, J. K. Yoon, H. J. Ha, J. Y. Kim and H. T. Kim, Nat. Commun., 2021, 12, 5537 CrossRef CAS.
- S. Cho, D. Y. Kim, J. I. Lee, J. Kang, H. Lee, G. Kim, D. H. Seo and S. Park, Adv. Funct. Mater., 2022, 32, 2208629 CrossRef CAS.
- J. Yang, T. T. Feng, J. M. Hou, X. R. Li, B. Y. Chen, C. Chen, Z. Chen, Y. C. Song and M. Q. Wu, J. Energy Chem., 2022, 65, 583–591 CrossRef CAS.
- W. Cao, Q. Li, X. Yu and H. Li, eScience, 2022, 2, 47–78 CrossRef.
- R. Zhang, X. Shen, H. T. Ju, J. D. Zhang, Y. T. Zhang and J. Q. Huang, J. Energy Chem., 2022, 73, 285–291 CrossRef CAS.
- K. Liu, B. Kong, W. Liu, Y. Sun, M.-S. Song, J. Chen, Y. Liu, D. Lin, A. Pei and Y. Cui, Joule, 2018, 2, 1857–1865 CrossRef CAS.
- Y. Shao, H. Wang, Z. Gong, D. Wang, B. Zheng, J. Zhu, Y. Lu, Y.-S. Hu, X. Guo, H. Li, X. Huang, Y. Yang, C.-W. Nan and L. Chen, ACS Energy Lett., 2018, 3, 1212–1218 CrossRef CAS.
- L. C. Dejonghe, L. Feldman and A. Beuchele, J. Mater. Sci., 1981, 16, 780–786 CrossRef CAS.
- W. Ping, C. Wang, Z. Lin, E. Hitz, C. Yang, H. Wang and L. Hu, Adv. Energy Mater., 2020, 10, 2000702 CrossRef CAS.
- X.-R. Chen, C. Yan, J.-F. Ding, H.-J. Peng and Q. Zhang, J. Energy Chem., 2021, 62, 289–294 CrossRef CAS.
- F.-N. Jiang, S.-J. Yang, H. Liu, X.-B. Cheng, L. Liu, R. Xiang, Q. Zhang, S. Kaskel and J.-Q. Huang, SusMat, 2021, 1, 506–536 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ta00121k |
| This journal is © The Royal Society of Chemistry 2023 |