
Research progress on the construction of synergistic electrocatalytic ORR/OER self-supporting cathodes for zinc–air batteries
Qi
Liu
a,
Lei
Wang
*b and
Honggang
Fu
 *ab
*ab
aKey Laboratory of Superlight Materials and Surface Technology of Ministry of Education, College of Materials Science and Chemical Engineering, Harbin Engineering University, Harbin 150001, P. R. China
bKey Laboratory of Functional Inorganic Material Chemistry, Ministry of Education of the People's Republic of China, Heilongjiang University, Harbin 150080, China. E-mail: wanglei0525@hlju.edu.cn; fuhg@hlju.edu.cn; fuhg@vip.sina.com
First published on 30th January 2023
Abstract
Zn–air batteries (ZABs) have developed rapidly as novel energy storage devices to replace metal-ion batteries. However, the kinetics of the air-cathode is slow, and thus it is necessary to develop highly efficient and stable bifunctional catalysts. Generally, self-supporting electrodes can avoid the use of binder; moreover, the growth of the catalyst in situ on the substrate can improve the performance of ZABs. In this review, we systematically summarize the application of non-precious metal-based self-supporting electrodes in ZABs according to the recent research progress. Firstly, the basic structure and principle of ZABs, including Zn anodes, air-cathodes and electrolytes, are proposed. Subsequently, a reasonable electrode preparation method is discussed by combining the reaction mechanism of ZABs during the charging and discharging processes. The reaction mechanism is further discussed by combining operando techniques and theoretical calculations for a deep understanding of the real active sites, which lays the foundation for the directional synthesis of highly efficient self-supporting electrodes. Then, the application progress of self-supporting electrodes in ZABs is discussed, and it is proposed that the construction of hybrid cells is an effective means to promote battery efficiency. Finally, the challenges associated with ZABs and their future directions are presented and have a guiding significance for the rapid development of related fields.
1. Introduction
To realize the proposal of “carbon peak, carbon neutral”, it is necessary to develop clean and sustainable energy conversion/storage devices.1,2 Accordingly, rechargeable batteries can effectively meet the reversible conversion of electrical energy to chemical energy, such as lithium-ion batteries, fuel cells and metal–air batteries.3,4 Among them, Zn–air batteries (ZABs) show relatively higher power density (≈1084 W h kg−1), stable discharge voltage, and produce water as a pollution-free product, which is consistent with sustainable development and environmental friendliness.5–7 The oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are the discharging and charging reactions of the air-cathode, respectively.8–10 However, these two reactions involve multiple electron transfer processes with slow reaction kinetics, and thus it is necessary to develop efficient catalysts to accelerate their kinetic processes.11–13 To date, although various electrocatalysts have been reported for the ORR and OER, the development of efficient bifunctional electrocatalysts remains a challenging task.14–16 The ORR and OER are a pair of reversible reactions, but the active sites are different in multiple processes of both reactions.17–19 It is critical to control the strength of O adsorption and the d-band center, and thus the internal charge–discharge mechanism is crucial for the rapid development of ZABs.20–22The complex electrochemical reactions always occur on the gas–liquid–solid three-phase interface, where the complex electron transfer process increases the diffusion resistance of electrons.23,24 The charge–discharge efficiency is often lower than 65%.25,26 Currently, powder catalysts are the main catalysts being explored as bifunctional catalysts.27 However, there are several drawbacks when ZABs are assembled with powder catalysts: (i) a binder is required to attach the powder catalyst to the conductive substrate for the air-cathode, which affects the conductivity of the electrode.28 Simultaneously, this not only makes the preparation process tedious but also reduces the energy density of the battery because of the increase in the overall weight of the electrode.29,30 (ii) The polymer binder will undergo severe degradation when the electrode is exposed to strong alkaline solution at a high oxidation potential for a long time, leading to an unsatisfactory lifetime of the battery.31,32 (iii) Moreover, the use of binder will reduce the effective active area of the catalyst, thus blocking the mass transfer channel and inhibiting the proton/electron transfer during the bifunctional oxygen reaction.33,34 The increase in the electrochemical polarization resistance, ohmic polarization resistance and diffusion polarization resistance of the electrocatalytic process can decrease the catalytic efficiency of the catalyst.35–37 In contrast, in self-supporting electrodes, the catalyst is chemically grown directly on the conductive substrate, and it is attached firmly, which effectively increases the contact area and eliminates the degradation of the catalyst during the long cycling of the battery.38,39 Significantly, the construction of self-supporting electrodes can enhance the stability and lifetime of ZABs, which can effectively solve the above-mentioned problems of powder catalysts.40,41
In recent years, significant research work has been devoted to self-supporting electrodes due to their special advantages. In this review, we elucidate the working principle and problems of ZABs, followed by a detailed discussion on the construction of self-supporting electrodes, such as wet chemical self-assembly, electrochemical deposition, and electrospinning. Finally, the charge–discharge reaction mechanism of ZABs is further clarified by combining in situ technology and theoretical calculation. This review will be beneficial for further research on self-supporting electrodes for ZABs.
2. Basic structure and principle of ZABs
ZABs consist of four parts, including a Zn anode, air-cathode, electrolyte and separator, assembled in the order of Zn anode-separator–catalyst–foam metal-waterproof breathable membrane.42 It should be noted that the size of the self-supporting electrode should be smaller than that of the separator to enable the electrode to fully function. Nickel foam is the typical metal foam employed in self-supporting electrodes, which has excellent conductivity and low reduction potential, and also avoids zinc self-corrosion. The area of the waterproof breathable film should be larger than that of the partition, which can effectively prevent the leakage of the electrolyte and does not affect the entry of O2 molecules, thus improving the stability of the ZAB.| Zn + 4OH− → Zn(OH)42− + 2e− (E = −1.25 V vs. SHE) | (1) |
| Zn(OH)42− → ZnO + H2O + 2OH− | (2) |
| O2 + 2H2O + 4e− → 4OH− (E = 0.4 V vs. SHE) | (3) |
| 2Zn + O2 → 2ZnO (E = 1.65 V vs. SHE) | (4) |
Zn flakes or Zn wire are generally used as the anode of ZABs.43,44 During the charging process, metal Zn is electrochemically coupled with the air-cathode, where the Zn anode loses electrons to become zinc ions. The lost electrons are transported to the air-cathode through the internal circulation of the battery, thus reducing the diffused oxygen to hydroxide ions at the gas–solid–liquid three-phase interface. The hydroxide ions return to the Zn anode through the recycling system in the battery, and further react with Zn2+ to form Zn(OH)42− (eqn (1)). The concentration of Zn(OH)42− increases with the progress of the reaction. When its concentration reaches a critical value, it will decompose into ZnO (eqn (2)). At the air-cathode, a reversible oxygen reaction occurs (eqn (3)). The overall reaction is shown as eqn (4) with the standard reaction potential of 1.65 V. Actually, even if a catalyst with excellent catalytic performance is used, the charging potential is about 1.2 V and the discharging potential is about 2.0 V at the current density of 10 mA cm−2, and thus the efficiency is only around 60%.45,46 In addition to the electron resistance affecting the overall efficiency during the reaction, the Zn anodes can also hinder the discharge process during long periods of charging/discharging processes.47 This problem is more pronounced in the case of large current discharge and deep discharging.11 The three main factors that affect the performance of Zn anodes are Zn dendrites, Zn passivation and HER reaction.48,49 The generation of these three factors and the current solutions will be described in detail below.
2.1 Zn anodes
The issue of Zn dendrites can be effectively solved by the lattice matching strategy. For example, Zheng et al. coated graphene nanosheets on a Zn anode via an electrochemical deposition method.53 Due to the high lattice matching between Zn and the graphene nanosheets, Zn achieved regular stripping/deposition in the form of epitaxial growth. The results showed that the reversibility of Zn deposition/dissolution on the electrode was 99.9%, and the cycle life of the electrode was 100 times that of the traditional Zn anode. In addition, other highly conductive materials with high lattice matching with zinc can also be investigated. Yi et al. developed a dendrite regulation strategy based on crystal face matching, that is, Zn(002)/Cu(111) crystal face matching was used to induce and regulate the formation and growth of Zn dendrites, thus achieving reversible galvanizing/Zn stripping at an ultra-high charge–discharge rate.54 Theoretically, a single large dendrite will significantly affect the distribution of the surrounding electric field, resulting in a high electric field intensity at the dendrite tip and uneven metal deposition (Fig. 1a–c). When the number of dendrite formation sites and their distribution are sufficient and the starting capacity of dendrite formation is the same, the expected dendrite, which does not affect the surrounding electric field, can be formed. The atomic arrangement of the Cu(111) plane is similar to that of the Zn(002) plane and epitaxial deposition on the Cu(111) surface can form uniform and small-size Zn dendrites, thus eliminating the abnormal growth of irregular dendrites and effectively inhibiting the “self-amplification” growth effect. The assembled symmetrical cell achieved a highly stable and rapid plating/stripping cycle of up to 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles at 100.0 mA cm−2 with an area capacity of up to 30 A h cm−2. The strategy of epitaxial deposition to induce and regulate dendrite growth can also be applied to other metal anodes, such as Mg and Al.55,56
000 cycles at 100.0 mA cm−2 with an area capacity of up to 30 A h cm−2. The strategy of epitaxial deposition to induce and regulate dendrite growth can also be applied to other metal anodes, such as Mg and Al.55,56
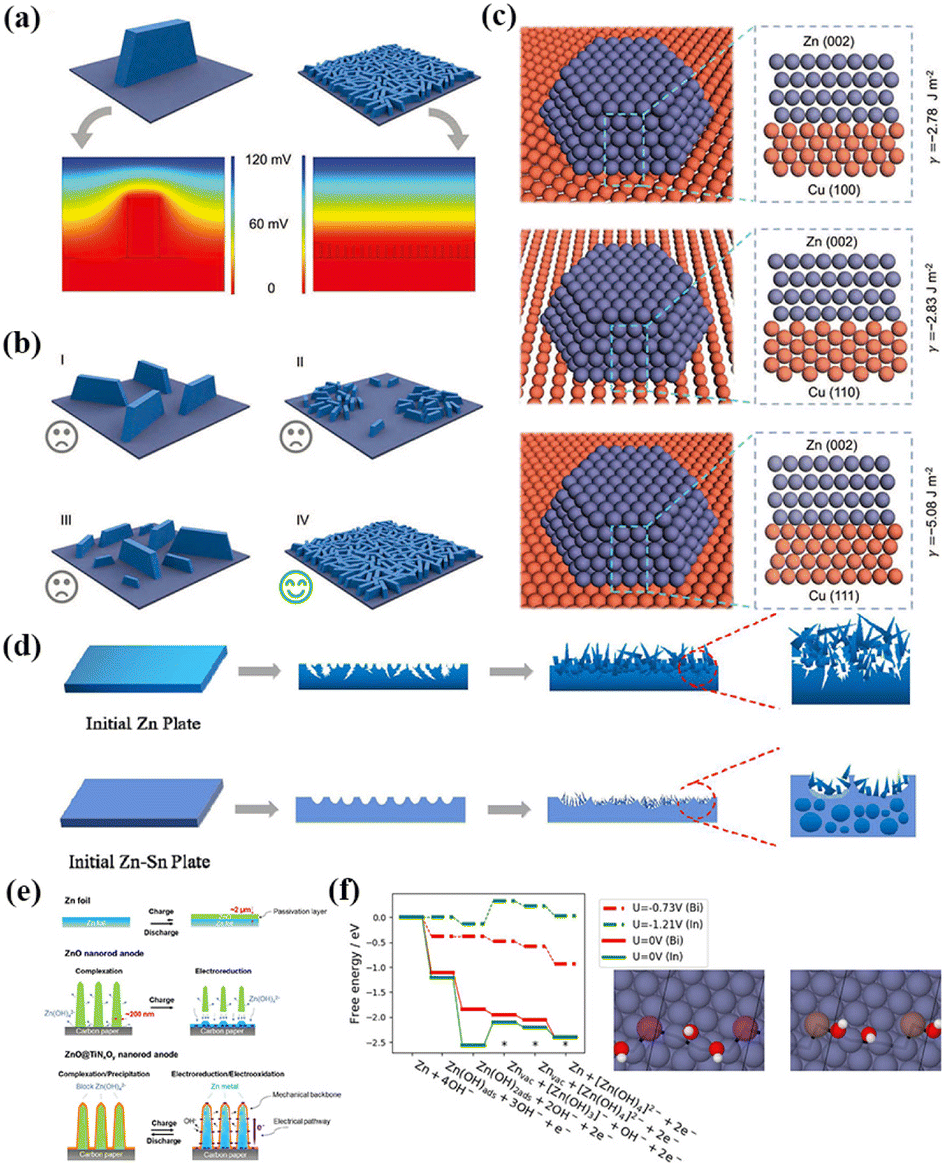 | ||
| Fig. 1 (a) Potential field distribution around a large dendrite and many small, homogeneously distributed, and identical dendrites. (b) Schematic of typical dendrite formation modes. Three unfavorable modes: (I) large and isolated dendrites, (II) irregularly distributed dendrites, (III) dendrites with different sizes; and one favorable mode: (IV) small, homogeneously distributed, and identical dendrites. (c) Interface structures and interfacial energies of (top) Cu(100)/Zn(002), (middle) Cu(110)/Zn(002), and (bottom) Cu(111)/Zn(002). Reproduced with permission from ref. 54, Copyright (2022) Wiley. (d) Schematic illustration of Zn plating on bare Zn and Zn–Sn alloy. Reproduced with permission from ref. 63, Copyright (2022) Elsevier. (e) Schematic of morphological changes of zinc electrode with Zn foil, ZnO nanorod and ZnO@TiNxOy nanorod as anode during electrochemical cycling. Reproduced with permission from ref. 71, Copyright (2018) Elsevier. (f) Free energy diagram of the electrochemical steps of Zn dissolution on In- and Bi-doped Zn surfaces. Reproduced with permission from ref. 75, Copyright (2018) Wiley. | ||
The deposition rate of zinc in the electrolyte can be controlled by adding organic additives.57–59 In the battery charging process, the organic solvent in the electrolyte preferentially accumulates in the protrusive part of the polar surface, thus inhibiting dendrite growth. In the discharging process, the aggregated organic molecules can prevent the diffusion of dissolved organic molecules in the electrolyte and slow the structural changes of the electrode. Huang et al. studied the scavenging ability of ethylene-diamine-tetraacetic-acid (EDTA), polysorbate 20 and tartaric acid for Zn dendrites.57 The three types of organic reagents had a good inhibitory effect on the formation of dendrites, among which EDTA showed the most obvious inhibitory effect. Lin et al. also effectively inhibited the self-corrosion process of the zinc anode by adding polyethylenimine (PEI) to the alkaline electrolyte of the ZAB, and consequently the battery had good cycle stability.60
Zn metal alloying can also solve dendrites to a certain extent. Theoretically, metal ions with a higher reduction potential are more likely to bind to Zn ions in the electrolyte and have more rapid and uniform deposition/stripping behavior, thus inhibiting dendrite growth.61,62 Peng et al. designed a Zn–Sn alloy anode, which facilitated the uniform deposition of zinc during discharge (Fig. 1d).63 Theoretical calculations indicated that the formation of alloys can regulate the electron density and charge distribution to promote the reversibility of Zn stripping/deposition non-dendrite formation. Zeng et al. developed a 3D multifunctional material (Cu NBs@NCFs) composed of Cu nanoboxes embedded in N-doped carbon nanofibers, which effectively reduced the nucleation overpotential of Zn. In the process of Zn deposition, a Cu–Zn solid solution is formed, and both Cu and Cu–Zn solid solution were zincophiles and had strong interaction with Zn ions, which could reduce the nuclear barrier of Zn and regulate the uniform deposition of Zn ions.64 Other metals such as Al, In and Ag can also form alloy structures with Zn electrodes to alleviate dendrite growth.
In this case, the addition of surfactants to the electrolyte can alleviate the Zn passivation problem. The formation of ZnO depends on the electrolyte to a large extent, and thus adjusting the composition of the electrolyte can effectively improve the dense passivation layer. The addition of a surfactant to the electrolyte can prevent the deposition of ZnO on the surface of active Zn and reduce the barrier of the passivation layer to the diffusion of Zn ions generated by the discharge reaction to the electrolyte.67 Yang et al. reported that the addition of sodium dodecyl benzene sulfonate (SDBS) to the alkaline electrolyte could effectively inhibit the passivation of the Zn anode during the electrochemical dissolution process and improve the discharge capacity of Zn.68 The additive adsorbed on the surface of Zn to form a porous passivation film, which promoted the diffusion and migration of the discharged products and solution reactants, thus improving the utilization rate of the Zn anode. Different types of surfactants will change the size of ZnO nanoparticles according to their hydrophobicity. Liu et al. added dodecyl trimethyl ammonium bromide (DTAB) to the electrolyte.69 The absorbability and good moisture retention of DTAB are conducive to forming a uniform and fluffy protective layer on the surface of the Zn anode. Thus, the modification effectively improved the diffusion rate of Zn ions, preventing the passivation of the Zn anode.
Chemical coating of the Zn anode is also an effective strategy to prevent passivation. To improve the reversibility of the electrochemical conversion process between Zn and ZnO, Sun et al. proposed the coating of the Zn anode with a chemical buffer layer to evenly disperse ZnO nanorods in GO (CBL@Zn).70 Due to the synergistic effect of the nuclear ZnO nanorods and GO adsorption affinity, the electrochemical deposition/dissolution process of ZnO could be effectively regulated. Zn (OH)42− was effectively confined in the chemical buffer layer, thus effectively eliminating zinc passivation. Zhang et al. reported the preparation of a ZnO@TiNxOy core/shell nanorod structure.71 As shown in Fig. 1e, the TiNxOy coating reduced the dissolution of Zn in the alkaline electrolyte, effectively delaying the passivation of ZnO. Finally, the ZnO@TiNxOy nanorod anode displayed excellent specific capacity and cyclic lifetime. Additionally, the passivation phenomenon can be alleviated by using Zn anode materials with a large specific surface area. Parker et al. used a Zn sponge as a ZAB anode.72 The porous structure with large specific surface area could accelerate the rapid transfer of zinc electrons, reduce the formation of passive ZnO, and make the utilization rate of zinc anode close to 90%. Thus, the addition of surfactants, chemical coating and using Zn anode with a large specific surface area are the most effective strategies to inhibit Zn passivation.
The hydrogen evolution reaction (HER) of the Zn anode is the main factor leading to the self-discharge behavior of the battery. In alkaline electrolyte, the hydrogen evolution reaction (HER) occurs as a side reaction when Zn is deposited (eqn (5)). This reduces the capacity of the battery by consuming active Zn, while the Zn(OH)2 produced increases the internal resistance of the battery. In addition, the H2 produced will cause the battery to expand, seriously affecting its lifetime and performance.73
| Zn + 2H2O → Zn(OH)2 + H2 | (5) |
At present, there are several methods to avoid the HER side reaction in ZABs. Zn and other metal alloys as electrode materials can significantly inhibit the HER of the Zn anode, resulting in faster and more uniform deposition/stripping behavior.74 Lysgaard et al. combined differential electrochemical mass spectrometry (DEM) and density functional theory (DFT) calculations to investigate the effects of In, Bi and Ag metal additives on Zn dissolution and HER reaction. It was found that co-doping of In and Bi significantly reduced the HER, where In could reduce the overpotential dissolution of Zn and Bi mainly stabilized In on the surface of Zn (Fig. 1f). Although Ag inhibited the adsorption of OH−, it did not hinder the HER.75 Chemical coatings of metal oxides and polymers can also inhibit the occurrence of the HER side reactions. Wongrujipairoj et al. directly coated Al2O3 on zinc nanoparticles via the sol–gel method, effectively alleviating the redox of Zn nanoparticles and inhibiting the occurrence of the HER.76 Jo et al. coated a Zn anode with polyaniline (PANI) to improve the corrosion inhibition and capacity retention of the zinc metal.77 Deyab et al. further added nano-zinc phthalocyanine (ZnPc) to the PANI coating to form a nano-PANI@ZnPc composite. This material has a higher Zn release rate (97.7%) than PANI alone (74.8%), resulting in a much higher capacity for ZABs.78
Although methods to solve the problems associated with zinc anodes have been reported to date, there is still a lack of methods to solve all these problems simultaneously. Also, there is no failure mechanism for standardized batteries and a lack of unified evaluation on the electrochemical performance of ZABs from the perspective of commercialization. Thus, it is still necessary to find solutions to these problems at the zinc anode level.
2.2 Air-cathodes
Four-electron reaction process:
| * + O2 + H2O + e− → *OOH + OH− |
| *OOH + e− → *O + OH− |
| *O + H2O + e− → *OH + OH− |
| *OH + e− → * + OH− |
Two-electron reaction process:
| * + O2 + H2O + e− → *OOH + OH− |
| *OOH + H2O + e− → H2O2 + OH− + * |
Four-electron catalytic reactions are mainly divided into three steps, i.e., reaction species adsorption, redox reactions (including charge transfer, proton transfer, bond cleavage and formation), and desorption process.80 Initially, oxygen is adsorbed on the surface of the catalyst through the gas diffusion layer to produce oxygen-active substances, and then reduced to OOH by combining with the transferred electrons from the anode. Finally, the reduction product OH− is desorbed from the catalyst surface. Generally, it is believed that the stronger O2 adsorption capacity of the catalyst will result in better ORR activity. In fact, the intermediate desorption process after O2 adsorption also plays an important role towards ORR activity. The four-electron ORR reaction occurs between O2 and OH, which can be used as a standard to evaluate the ORR activity of catalysts.81
In contrast, the two-electron ORR process produces H2O2, which is widely used in industrial H2O2 production. As shown in Fig. 2a and b, the best single-metal catalysts for the ORR and OER are precious metal-based Pt and IrO2, respectively.80 However, their high cost and unsatisfactory stability hinder the large-scale production of ZABs.82 Nørskov analyzed the relationship between the oxygen binding energy and the theoretical activity of the four-electron transfer processes towards the ORR and OER. It can be found that the oxygen binding energy at the peak activity of the ORR and OER is not the same, which makes it impossible for single-metal catalysts to achieve the optimal activity for the ORR and OER simultaneously.83
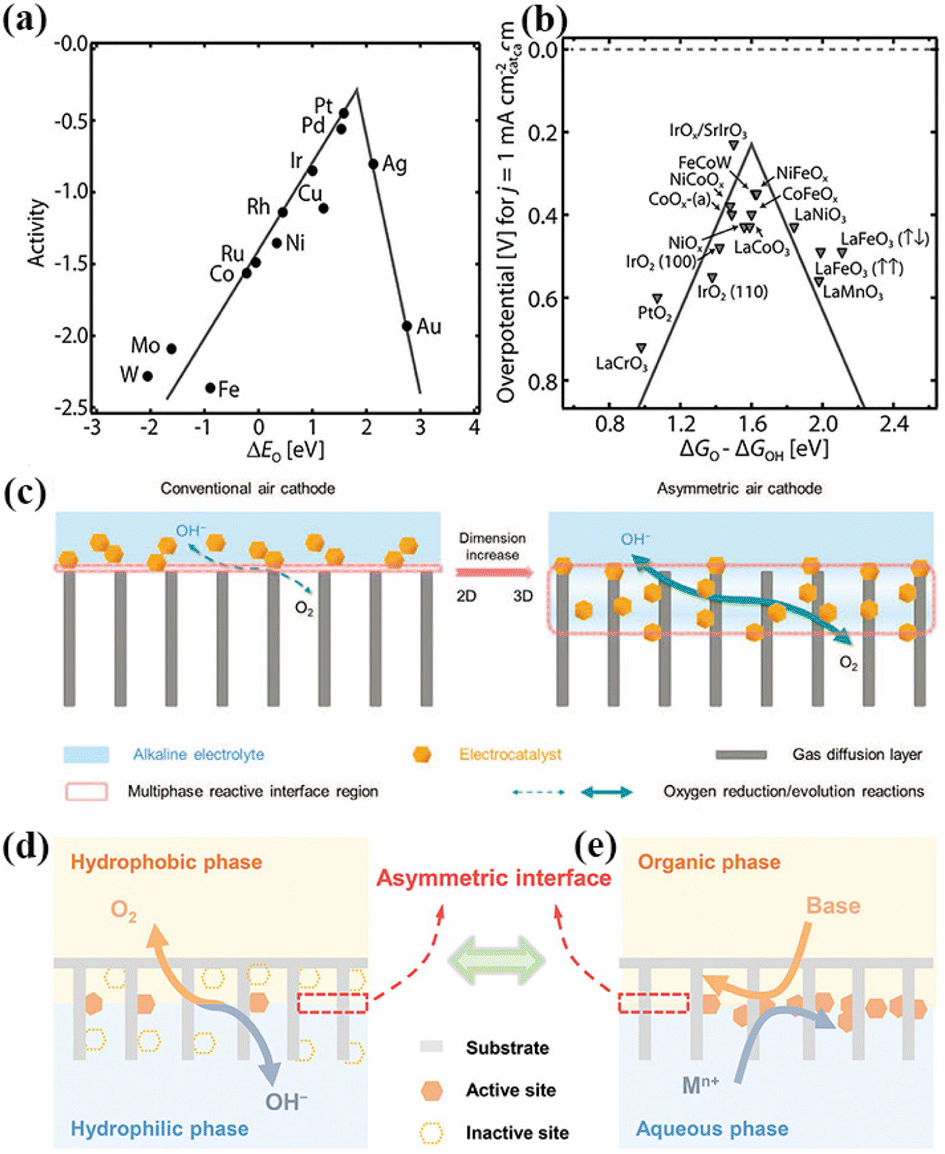 | ||
| Fig. 2 (a) ORR volcano plot for metals. (b) OER volcano plot for metal oxides. Reproduced with permission from ref. 80, Copyright (2019) Wiley. (c) Schematic illustrations of conventional and asymmetric air cathodes, which possess 2D and 3D multiphase reactive interfaces, respectively. The asymmetric air cathode exhibits an increased amount of active sites, accelerated mass transfer, and dynamically stabilized reactive interface. Reproduced with permission from ref. 94, Copyright (2020) Wiley. (d) Oxygen redox reactions undergo multiphase transformation processes between the aqueous electrolyte and the ambient atmosphere. (e) Asymmetric interface is preconstructed by introducing immiscible organic–aqueous diphases in the air cathode, at which the electrocatalysts are in situ formed to realize precise electrocatalyst loading at the asymmetric interface. Reproduced with permission from ref. 95, Copyright (2022) Wiley. | ||
The OER is the inverse reaction of the ORR, which also has a significant impact on the performance of ZABs. The reaction process equation of the OER is as follows:
| OH− + * → OH + e− |
| OH + OH− → O + e− + H2O |
| 2O2 → O2 + 2* |
There is almost no intersection for OER and ORR catalysts according to the volcano diagram, which is the fundamental reason why ORR catalysts with excellent activity are usually not suitable for the OER. Specifically, catalysts with a good ORR performance have strong O2 binding energy and relatively weak binding with OH and O.84 However, this is detrimental to the OER process because it is difficult for the reactants to adsorb on the electrode surface, while it is difficult for O2 to be released. Therefore, by adjusting the composition of the catalyst and its surface electronic structure to balance the electrocatalytic activity between the ORR and OER, the design and synthesis of bifunctional catalysts can be realized.85
The development of bifunctional ORR/OER catalysts is the most important aspect to improve the performance of ZABs at the air-cathode level.86 At present, the development of powder catalysts is relatively mature, but the required use of conductive agent and binder for assembling ZABs will lead to a series of problems, such increased resistance, easy detachment of the catalyst and occupation of the active sites.87 Therefore, self-supporting electrodes with simple preparation process and excellent catalytic performance have great potential for direct application in the air-cathode of ZABs. Simultaneously, it is very important to build efficient and stable non-precious metal catalysts.88 In Section 3, the strategy for the construction of non-precious metal-based self-supporting electrodes will be described in detail.
The ORR/OER bifunctional activity of catalysts is determined by the specific position of their metal component in the volcano diagram. It is necessary to select metals with high intrinsic activity as the starting point for the synthesis of catalysts. The relationship between the adsorption energy of oxygen-containing intermediates and the electronic structure of catalysts requires that the adsorption energy of the catalyst intermediates not be too large, which is conducive to the rapid ORR/OER when organic conductors and binders are used during the assembly of ZABs. However, the use of conductive agents and binders can flood the active sites of the catalysts, and thus reduce their ORR/OER activity and long-term cycle stability. Alternatively, a sufficiently large active surface area can significantly improve the electron reaction rate and ORR/OER activity.
Self-supporting electrodes possess a special 3D structure, which is beneficial for the adsorption/desorption of reactants on the active sites. A conductive self-supporting substrate can promote the electron transfer. The reaction rate directly affects the electrochemical reaction performance of the battery. In this case, the 3D structure and high conductivity of self-supporting electrodes are conducive to electron transfer and can improve the intrinsic activity and reaction rate of the ORR/OER.91 Heterogeneous catalytic reactions mainly occur on the surface/interface of the catalyst, and thus a well-designed and stable surface/interface can greatly promote the adsorption/activation of active species on the catalyst surface. Stability has an important influence on the catalytic performance for the ORR/OER. Generally, self-supporting electrodes are constructed via in situ growth on the conductive substrate and the catalyst is stably grown on the conductive substrate. Even in highly oxidized alkaline system, the electrochemical reaction can still occur stably without causing the shedding of the active species.92
In conventional air-cathodes, the 2D multiphase interface on the surface of the GDL inevitably leads to an insufficient number of active centers and poor interfacial contact, resulting in slow reaction kinetics.93 Thus, to solve this problem, Yu et al. designed an asymmetric air-cathode of NiFe layered double hydroxide to increase the active centers, accelerate mass transfer, and generate a dynamically stable reaction interface (Fig. 2c). It showed a small charge–discharge voltage gap, high power density, and stable cycling performance. The 3D reaction interface can significantly increase the number of active sites and accelerate the mass transfer due to its extended and interconnected interface structure.94 Recently, Liu et al. proposed an asymmetric interfacial pre-construction strategy to fabricate air-cathodes for high-performance ZABs (Fig. 2d and e). An asymmetric hydrophilic/hydrophobic configuration was achieved by constructing an immiscible organic–water asymmetric interface in the air-cathode and in situ generating the electrocatalyst. The asymmetric air-cathode significantly improved the utilization rate of the catalyst, and thus improved the performance of the ZAB.95 The integrated air-cathode can guarantee the maximum available active sites, fast electron and mass transport, and good long-term operation stability. Wang et al. constructed an integrated air-cathode using an in situ-grown Co3O4 nanosheet catalyst layer and GDL. By adjusting the pore size and the aperture of the GDL through efficient oxygen transfer channels and more three-phase interfaces, the bifunctional OER/ORR activity was significantly improved, opening a new way for the design of efficient integrated air electrodes for ZABs.96
2.3 Electrolytes
| 2KOH + CO2 → K2CO3 + H2O | (6) |
To solve the problem associated with alkaline electrolyte, Sun et al. proposed the use of a new non-alkaline hydrophobic zinc trifluoromesylate (Zn(OTf)2) electrolyte in ZABs.99 The efficient aprotic two-electron transfer process was realized by constructing a double-layer Zn-ion enrichment structure on the surface of the air-cathode. Compared with the traditional alkaline ZAB, the utilization of the Zn anode in the non-alkaline ZAB using Zn(OTf)2 electrolyte increased from 8.1% to 83.1% and the electrochemical stability was significantly improved (Fig. 3a). Non-alkaline ZABs exhibit consistent long-term stability in both air and pure O2 conditions (Fig. 3b). Sun et al. assembled a battery with good zinc dissolution/deposition negative electrode reversibility by employing weakly acidic zinc acetate (Zn(CH3CO2)2) aqueous solution as the electrolyte. The discharged product zinc glycolate dihydrate (Zn5(OH)8(CH3CO2)2·2H2O) had a high degree of reversible formation/solubility, and the constructed non-alkaline ZAB could run stably in air for nearly 600 h (Fig. 3c and d).100 Thus, it can be seen that the exploitation of non-alkaline electrolytes is an effective strategy to solve the problems faced by ZABs.
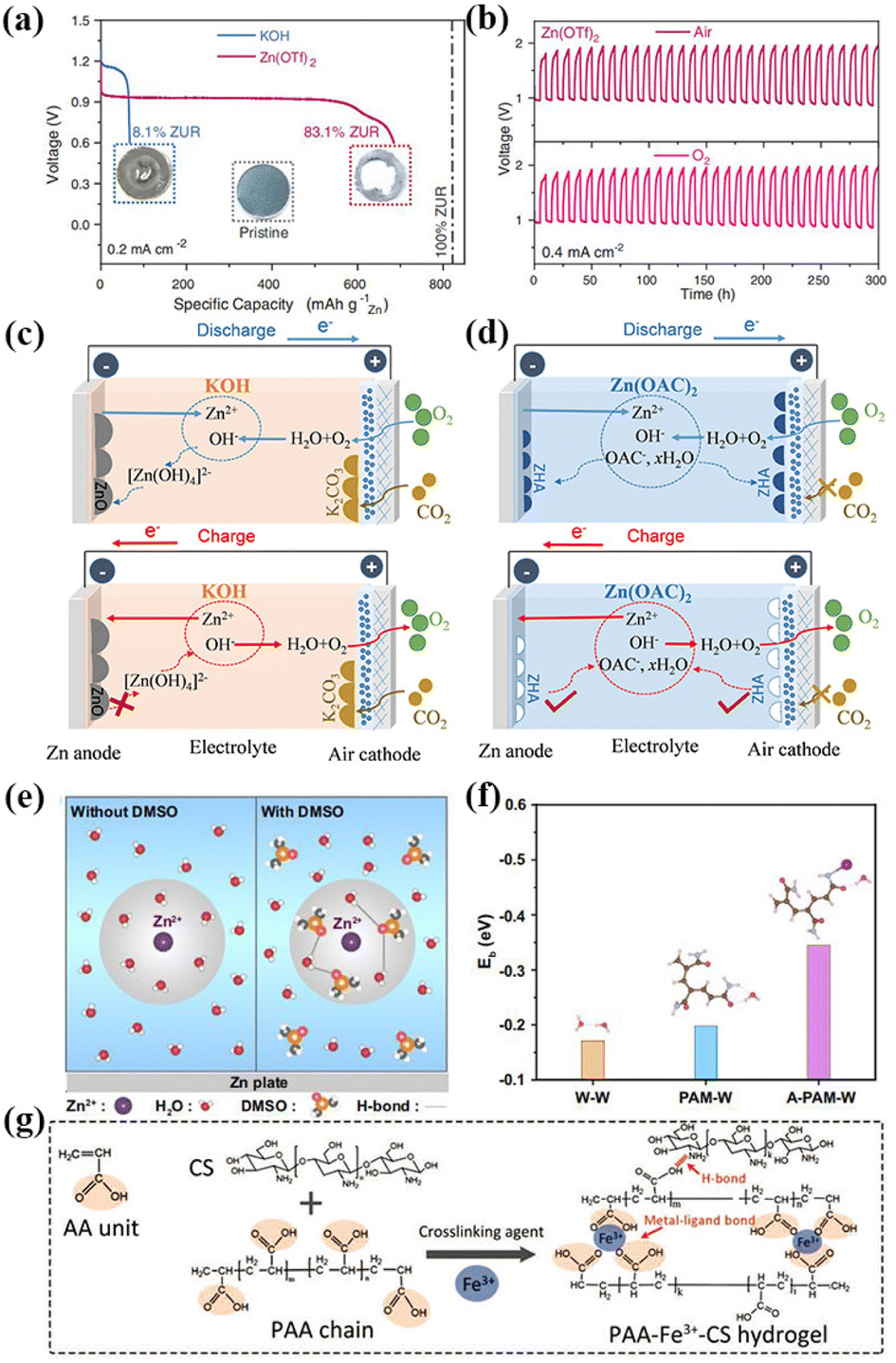 | ||
| Fig. 3 (a) Galvanostatic discharge profiles of ZABs in KOH (blue) and Zn(OTf)2 (red) electrolyte at 0.2 mA cm−2 (cutoff voltage: 0.6 V). The corresponding Zn utilization ratio (ZUR) was indexed for comparison. Insets are photographs of the pristine Zn anode (middle), the Zn anode after discharge in KOH (left), and Zn(OTf)2 (right) electrolytes. (b) Galvanostatic discharge and charge profiles of ZABs in Zn(OTf)2 electrolytes in a capacity fixed mode (fixed capacity: 2 mA h cm−2) at 0.4 mA cm−2 under an air and O2 atmosphere, respectively. Reproduced with permission from ref. 99, Copyright (2021) Science. Overview of electrically rechargeable ZABs using (c) alkaline KOH and (d) non-alkaline Zn(OAC)2 electrolytes, respectively. ZHA, zinc hydroxyacetate dihydrate (Zn5(OH)8(CH3CO2)2·2H2O). Reproduced with permission from ref. 100, Copyright (2022) Wiley. (e) Zn2+ solvation structure and formed H-bond between DMSO and H2O molecules. The gray circles correspond to the solvation sheath structure of Zn2+. (f) Eb of W-W, PAM-W, and A-PAM-W. Reproduced with permission from ref. 106, Copyright (2022) Springer Nature. (g) Schematic depiction of the mechanism of PAA-Fe3+-CS hydrogel entry into tough bonding to both the surfaces of air cathode and ZMA under the action of the Hofmeister effect. Reproduced with permission from ref. 107, Copyright (2022) Wiley. | ||
Based on the above analysis, some strategies need to be used to improve the low-temperature resistance and water retention of GPEs. Accordingly, the addition of organic reagents with a large number of hydrogen bond receptors to the electrolyte is considered to be one of the effective strategies. These additives have a high binding energy and can form hydrogen bonds with free water molecules, thus effectively reducing the freezing point of water. Simultaneously, the addition of organic reagents also promotes the rapid stripping and deposition of Zn2+ and reduces the formation of zinc dendrites. Recently, Wang et al. proposed a quasi-solid ZAB assembled from a polyacrylamide organic hydrogel electrolyte (A-PAM-W) with dimethyl sulfoxide modified in a hydrogen bond network anchored by atomically dispersed cobalt sites to nitrogen-doped graphene as an air-cathode. The battery could be stably cycled for 100 h at a high current of 100 mA cm−2. Even at the ultra-low temperature of −60 °C, and more than 90% of the capacity was maintained for more than 300 h at the charging–discharging current density of 0.5 mA cm−2. This GPE has a wide-range temperature adaptability (−60 to 60 °C). In principle, dimethyl sulfoxide has abundant hydrogen bond receptors and can form stable hydrogen bonds with free water molecules, thus effectively reducing the freezing point of water molecules. It can maintain certain conductivity of GPEs even at −60 °C (Fig. 3e). Simultaneously, the binding energy between A-PAM-W and water molecules is higher than that between PAM-W, which makes A-PAM-W have higher water retention. GPEs can still work stably without being destroyed by water evaporation for long-term cycles, (Fig. 3f).106 In addition to dimethyl sulfoxide, ethylene glycol, glycerol and other organic reagents can be used as additives to improve the water retention and reduce the freezing point of the electrolyte.
The construction of double hydrophilic network hydrogel electrolytes is another effective strategy. Compared with single network hydrogels, double network hydrogel electrolytes are composed of two interlaced cross-linked networks, which have a more hydrophilic backbone and can enhance the interaction between water molecules, thus effectively increasing the water retention of GPEs. Simultaneously, the interspersed skeleton structure can improve the polarity of terminal groups and increase the water binding capacity of GPEs, reducing the freezing point. Tang et al. synthesized polyacrylic acid-Fe3+-chitosan (PAA-Fe3+-CS) double-mesh hydrogels to enhance the chemical/mechanical durability of the electrode/electrolyte interface.107 When NH4Cl is absorbed by the Hofmeister effect near the neutral electrolyte, the chitosan molecular bundle introduced in the PAA-Fe3+-CS/NH4Cl hydrogel skeleton will precipitate and fold, effectively enhancing the adhesion strength of the PAA-Fe3+-CS/NH4Cl hydrogel electrolyte to the surface of the air positive electrode and zinc anode. This allows for efficient mechanical binding and effective reduction of the interface charge transfer resistance (Fig. 3g). The assembled flexible solid ZAB had good resistance to repeated mechanical deformation during the charging and discharging processes.
Recently, Gu et al. constructed a small molecule-based supramolecular polymer dual network (SP-DN) by introducing the non-covalently self-assembled guanosine supramolecular network in the covalently cross-linked polymer network. It was successfully applied in ZABs with high elasticity, waterproof property and wide temperature range. The assembled ZABs exhibited 1000% device-level elongation and excellent water resistance, capable of normal and stable operation at −50 °C to 100 °C. This study provides a new idea for the design of hydrogel electrolytes for high tensile and environmentally adaptable energy supply devices.108 Solid electrolytes play an important role in the practical application of solid ZABs. However, improving the water retention performance of the polymer is still a challenging topic.
In the mechanism of organic additives controlling the Zn deposition rate in ZABs, organic reagents preferentially gather on the surface of the zinc anode, limiting the two-dimensional diffusion of Zn2+ on the electrode surface, effectively preventing a large amount of Zn2+ from gathering on a certain position on the electrode surface, and thus controlling the zinc deposition rate and inhibiting the formation of large dendrites. In this case, organic reagents such as tartaric acid and citric acid are generally used. In the mechanism of surfactant alleviating the issue of zinc passivation, the adsorption of surfactant molecules on the zinc anode can prevent the contact between water and the anode, thus effectively limiting the formation of passivated ZnO. Generally, surfactants are classed as cationic surfactants, anionic surfactants and non-ionic surfactants. It is a requirement that the organic additives do not react during the charging and discharging processes of the ZAB, while the reaction substances are basic groups. Organic additives in the solution act as insulators and impurities, which can increase the impedance and impair the performance of the battery. Therefore, organic reagents only play a role in dendrite inhibition and passivation with any reaction.
2.4 Separator
As an essential component of ZABs, the separator plays an important role in their performance. The separator can prevent the short circuit of the battery and reasonably adjust the concentration of cations, reduce the uneven deposition of Zn, selectively permeate ions, and stabilize the battery performance. Suitable separators for ZABs must have the following characteristics: (i) Insulation: the separator must be insulating to isolate the anode and cathode to prevent short circuit of the battery. (ii) Stability: the electrolyte of ZABs is a strong alkaline solution, and thus the separator must have sufficient alkali resistance to ensure the stability of the battery. (iii) Toughness: the separator must have certain toughness to avoid short circuit caused by Zn dendrite puncture. (iv) Selective permeability: it is necessary to ensure the permeability of OH− and inhibit the passage of Zn(OH)42− through a selective separator.109,110Hydrogel polymer electrolytes are suitable for solid ZABs. With a certain tensile strain capacity, they provide the flexibility required in solid-state batteries, paving the way for the practical application of zinc air batteries. The polymers need to contain abundant hydrophilic groups (−OH, –COOH, –SO3 and –NH2) to effectively absorb lye and endows solid zinc–air batteries with long cycle capacity. The polymer maintains the unique characteristics of the diaphragm, namely, ionic permeability and flexibility, avoiding short circuit of the battery. The most widely used ZAB separators are polyolefin-based microporous membranes. A series of typical commercial battery separators is produced by Celgard (Polypore) in the United States, such as Celgard 5550, Celgard 3501, Celgard 3401, which are suitable for use in ZABs. At present, much effort has been also devoted to studying the effect of changing the separator on the performance of ZABs. Lee et al. used electrospinning to prepare a nanofiber felt composite separator with enhanced penetrative-selectivity (ERC separator). This separator was prepared by impregnating polyvinyl alcohol (PVA) in electrospun polyetherimide (PEI) nanofiber felt. The PEI nanofiber felt as a flexible frame could enhance the stability and mechanical strength of the separator. The results showed that the PVA substrate provided ion selectivity after being expanded by the electrolyte solution, and significantly inhibited the permeability of Zn(OH)42− without damaging the permeability of OH−, thus effectively improving the cycling performance of ZABs.111 Kim et al. prepared a novel (PVA/PVP) polymer electrolyte separator by electrospinning. The polymer phase acted as a selective ion transport channel, which facilitated the performance of electrochemical rechargeable ZABs. This new diaphragm was much more stable than the Celgard 3501 separator. The development direction of separators is to improve the ion selectivity, increase the permeability of OH− to reduce the permeability of Zn(OH)42−, and improve its toughness to promote the long-term cyclic stability of ZABs.112
Although the current development of ZABs is rapid, there are still some problems to be solved. Among them, Zn dendrites, Zn passivation and the HER on the Zn anode, and the exploration of bifunctional non-precious metal-based electrocatalysts for air-cathodes are the most important factors affecting the performance of ZABs. In the following section, we focus on the construction of non-noble metal-based self-supporting electrodes.
3. Construction strategies of self-supporting electrodes
In self-supported electrodes, the catalyst is grown directly on a conductive substrate, such as carbon cloth, carbon paper, foam metal and other materials, which can be directly used as a working electrode without using additional polymer binder and conductive agent.3.1 Wet chemical self-assembly method
The metal nitrogen bond is crucial for the bifunctional ORR/OER reaction, which has been proven to be the active center of the ORR.113 In this case, there are many methods to introduce N in self-supporting electrodes, such as calcination in NH3 atmosphere, carbonization after encapsulation with conductive polymer, and construction of metal–organic skeleton structure.114In a self-supporting electrode, the simplest approach is to take advantage of the unique coordination between a metal and organic solution by introducing N-containing ligands to form MOF (metal organic framework) structures by wet chemistry. MOFs can evolve into different types of catalysts via different treatment methods, such as metal nitride, phosphate, monatomic and monatomic clusters. ZIF-67 is a type of MOF with the coordination of Co metal, which is conducive to rapid charge transfer. Hou et al. synthesized two-dimensional Co-MOF (Co-MNS) nanosheets containing an N-group on the treated nickel foam. After heat treatment, the limited domain structure of the CoP nanoparticles anchored on carbon nanosheets, denoted as CoPx@CNS, exhibited three functional catalytic activities of OER, HER and ORR (Fig. 4a and b). The assembled ZAB could be stably cycled for 130 h under the charge discharge current density of 5 mA cm−2.115 Co metal salt solutions can form ZIF with different types of 2-methylimidazole solutions. For instance, ZIF-67 derived from methanol solution usually has a dodecahedral structure, whereas ZIF-67 derived from aqueous solution and ethanol solution usually displays a dendritic morphology. Zhong et al. innovatively constructed MOF on MOF structures by utilizing the different growth orientations of Co ZIF in 2-methylimidazole solution in water and methanol (Fig. 4c and d), resulting in hybrid 2D and 3D array structures. This layered structural design ensures that the air-cathode has a higher catalyst loading and improves the reaction kinetics. The liquid ZAB assembled with ZIF-D-Co3O4 as an independent cathode exhibited a high open circuit voltage of 1.66 V and could be stably cycled for 384 h at a charge discharge voltage interval of 0.83 V and charge discharge current density of 5 mA cm−2. However, it is difficult to regulate single-component catalysts show bifunctional ORR/OER activity.116
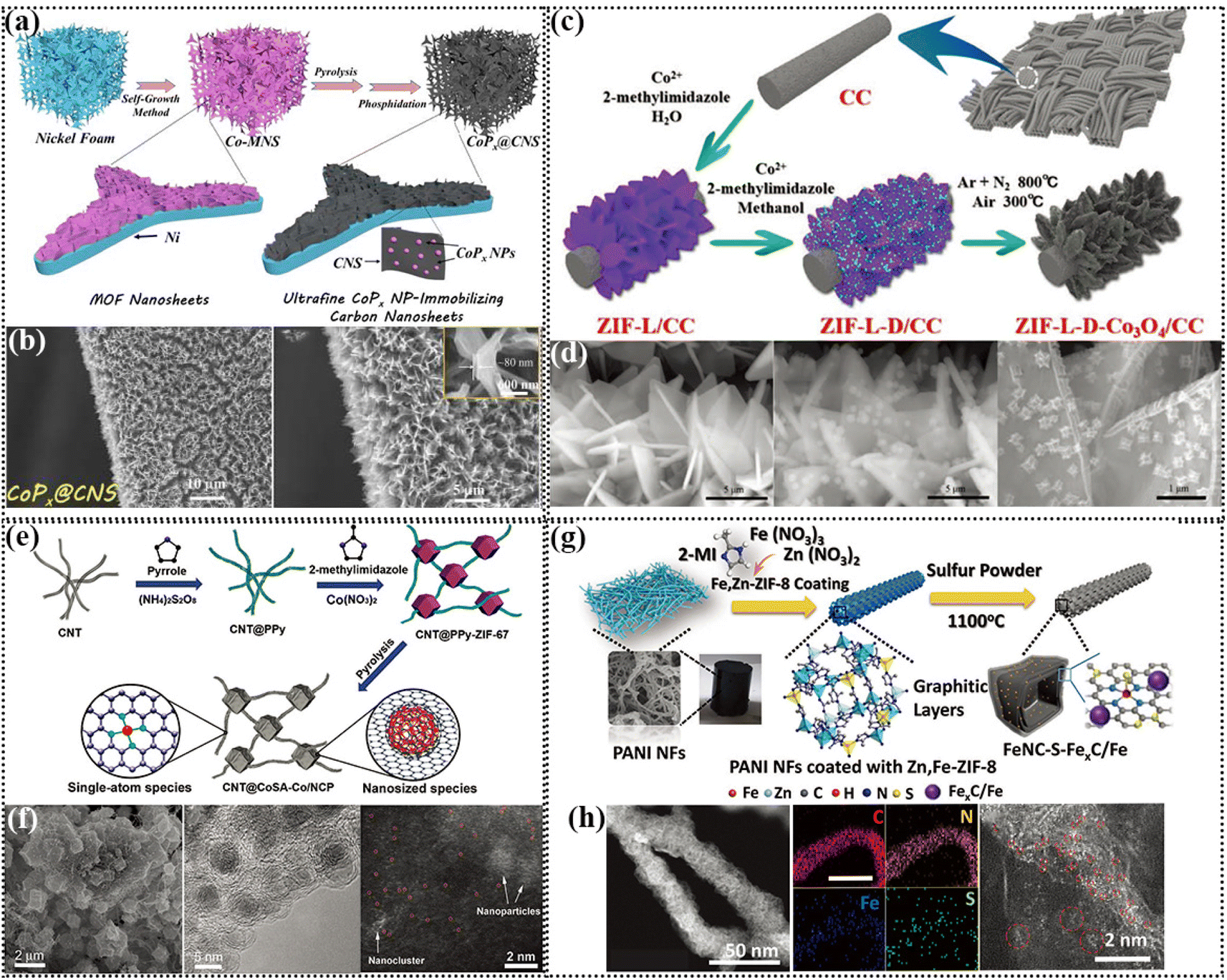 | ||
| Fig. 4 (a) Schematic illustration of the construction of a porous 3D nickel foam-templated superstructure of carbon nanosheets decorated with ultrafine cobalt phosphide nanoparticles. (b) SEM images (inset: thickness of CoPx@CNS). Reproduced with permission from ref. 115, Copyright (2020) Wiley. (c) Schematic illustration of the formation process of ZIF-L-D-Co3O4/CC. (d) SEM images of ZIF-L/CC, ZIF-L-D/CC, and ZIF-L-D-Co3O4/CC. Reproduced with permission from ref. 116, Copyright (2019) Wiley. (e) Schematic showing the synthetic process of CNT@SAC-Co/NC. (f) SEM, TEM and atomic-resolution TEM images of CNT@SAC-Co/NCP. (g) Synthesis scheme of the FeNC-S-FexC/Fe catalyst. Reproduced with permission from ref. 117, Copyright (2021) Wiley. (h) HAADF-STEM image, EDS elemental mappings with a scale bar of 100 nm and atomically scaled HAADF-STEM image of FeNC-S-FexC/Fe catalyst. Reproduced with permission from ref. 120, Copyright (2018) Wiley. | ||
Carbon-anchored single transition metal atoms have been shown to be the most efficient ORR active sites and transition metal species have excellent OER activity. Recently, Li et al. prepared a carbon-anchored metal SA and metal nanoparticle composite, namely “biphasic carbon” building strategy. Co SAs loaded on carbon nanotubes (single-atom phase) and encapsulated in a ZIF-derived carbon polyhedron (nanophase) were linked by carbon nanotube bridges. Due to the presence of Co in the ZIF during the synthetic process, a small part of it from the self-assembly process is adsorbed into the CNT@PPy system, and then calcined into SAs. The atomic-resolution TEM images showed the coexistence of SAs and nanoparticles (Fig. 4e and f). It was found through electrochemical performance tests that CNT@SAC-Co/NCP has excellent ORR/OER bifunction activity. The most impressive thing about the ZAB assembled is that its initial charge discharge efficiency was 71.88%, which remained at 67.77% even after 100 cycles, which proves that this catalyst has excellent stability. Although MOF structures have made great progress in electrocatalysis, there are still great challenges to achieve the controlled growth of MOF-based materials on flexible substrates such as carbon cloth (CC) and to apply them in flexible/wearable energy devices.117 Li et al. designed a self-assembly strategy based on MOFs, which assembled pre-synthesized colloidal MOF particles on the substrate surface through electrostatic self-assembly. The biggest advantage of this technique is that the MOF was prefabricated as required, and then self-assembled on the substrate surface.118
When bimetals form MOFs with a ligand, if one of the metals is volatile at high temperatures, the other metal can form an M-N-C catalyst. Wang et al. used Co and Zn coordinated with 2-methylimidazole to form a ZIF, which was pyrolyzed to produce Co–N with ultrahigh catalytic activity for both the ORR and OER. The solid-state ZAB assembled with Co–N-CNT as the catalyst and PVA/PEO as GPEs exhibited an open circuit voltage of 1.365 V, could be stably cycled for more than 15 h and used as a flexible device to drive small bulbs to emit light.119 Qiao et al. vulcanized heterogeneous FexC and Fe subatomic clusters to form Fe–S bonds, greatly increasing the availability of Fe atoms, and thereby enhancing the ORR activity (Fig. 4g and h). The larger atomic radius of S led to the formation of defects on the carbon carrier and the lower electronegativity of S altered the electronic structure of the Fe–N active center. DFT theoretical calculation showed that the significant increase in Fe–S–FexC/Fe activity was due to the synergistic effect of the Fe–Nx active sites with FexC/Fe and S-containing species. FeNC–S–FexC/Fe showed only a difference of ≈0.68 V between the ORR E1/2 and OER E10, which is a much smaller difference than that observed for Pt/C. The solid-state ZAB assembled with FeNC–S–FexC/Fe was found to have a stable cycle of 380 cycles with a charge discharge current density of 2 mA cm−2.120
The application of the wet chemical growth of metal MOFs for ZABs is as follows: (i) introducing a metal source and organic ligand on a substrate to obtain metal–N at high temperature, which is the active center of the oxygen reaction. (ii) The construction of functional catalysts is very important. The carbon sheet generated by the organic ligands at high temperature acts as a barrier, contributing to the formation of small-size nanoparticles or SA structures, and thus improving the kinetic rate of the ORR/OER. (iii) MOF-based materials can also be controlled on flexible substrates, and the catalysts can be directionally synthesized according to the performance requirements. (iv) The metal components in nickel foam, cobalt foam, iron foam, etc. are in situ used for coordination with organic ligands to form unique MOFs structures, which is also the development direction of ZAB air-cathodes.
3.2 Electrochemical deposition strategy
The electrochemical deposition strategy refers to the process of depositing thin films or deposits that meet the requirements on the surface of conductive substrates by electrochemical technologies. The synthetic conditions of electrodeposition are simple, environmentally friendly and low-cost, which can meet the current requirements for environmental protection and pollution free. Furthermore, the synthetic products have good uniformity and plentiful active sites, benefiting the electrocatalytic reaction. Thus, it is an effective method to prepare self-supporting electrodes for various energy storage and conversion devices. Below we describe various electrodeposition techniques for the preparation of self-supporting electrodes for ZABs. The size and morphology of the products can be adjusted by optimizing various electrodeposition parameters, such as deposition time, current, potential, additives, pH value and temperature.121The earliest application of potentiostatic electrodeposition was for metal oxides because they are located high on the volcanic map between 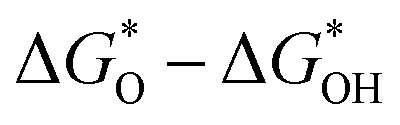 , attracting interest from researchers.122 Metal oxides mainly have the intrinsic activity for the OER, and nitrogen-containing species can be introduced to enhance the ORR activity through the use of nitrogen-containing substances via immersion and subsequent calcination. Fu's group used a three-electrode system with Co(NO3)2 solution as the electrolyte to obtain the Co3O4/CC-600 catalyst through constant potential electrochemical deposition of constant −1 V for 600 s and subsequent air oxidation (Fig. 5a). Then, the NC-Co3O4/CC-600 catalyst was obtained by immersion in dicyandiamide as the nitrogen source and calcination in a nitrogen atmosphere. The lamellar structure grew vertically on the surface of the carbon cloth, which were cross-linked, preventing the agglomeration of the structures. This catalyst possessed the largest contact area with the matrix, which was conducive to electronic transmission. The subsequent ZAB assembled with NC-Co3O4/CC-600 as an independent cathode had a charge discharge voltage interval of only 0.87 V under a current density of 10 mA cm−2. Three NC-Co3O4/CC-600-based ZABs in series could drive the 2.7 V LED lamp (Fig. 5b).123
, attracting interest from researchers.122 Metal oxides mainly have the intrinsic activity for the OER, and nitrogen-containing species can be introduced to enhance the ORR activity through the use of nitrogen-containing substances via immersion and subsequent calcination. Fu's group used a three-electrode system with Co(NO3)2 solution as the electrolyte to obtain the Co3O4/CC-600 catalyst through constant potential electrochemical deposition of constant −1 V for 600 s and subsequent air oxidation (Fig. 5a). Then, the NC-Co3O4/CC-600 catalyst was obtained by immersion in dicyandiamide as the nitrogen source and calcination in a nitrogen atmosphere. The lamellar structure grew vertically on the surface of the carbon cloth, which were cross-linked, preventing the agglomeration of the structures. This catalyst possessed the largest contact area with the matrix, which was conducive to electronic transmission. The subsequent ZAB assembled with NC-Co3O4/CC-600 as an independent cathode had a charge discharge voltage interval of only 0.87 V under a current density of 10 mA cm−2. Three NC-Co3O4/CC-600-based ZABs in series could drive the 2.7 V LED lamp (Fig. 5b).123
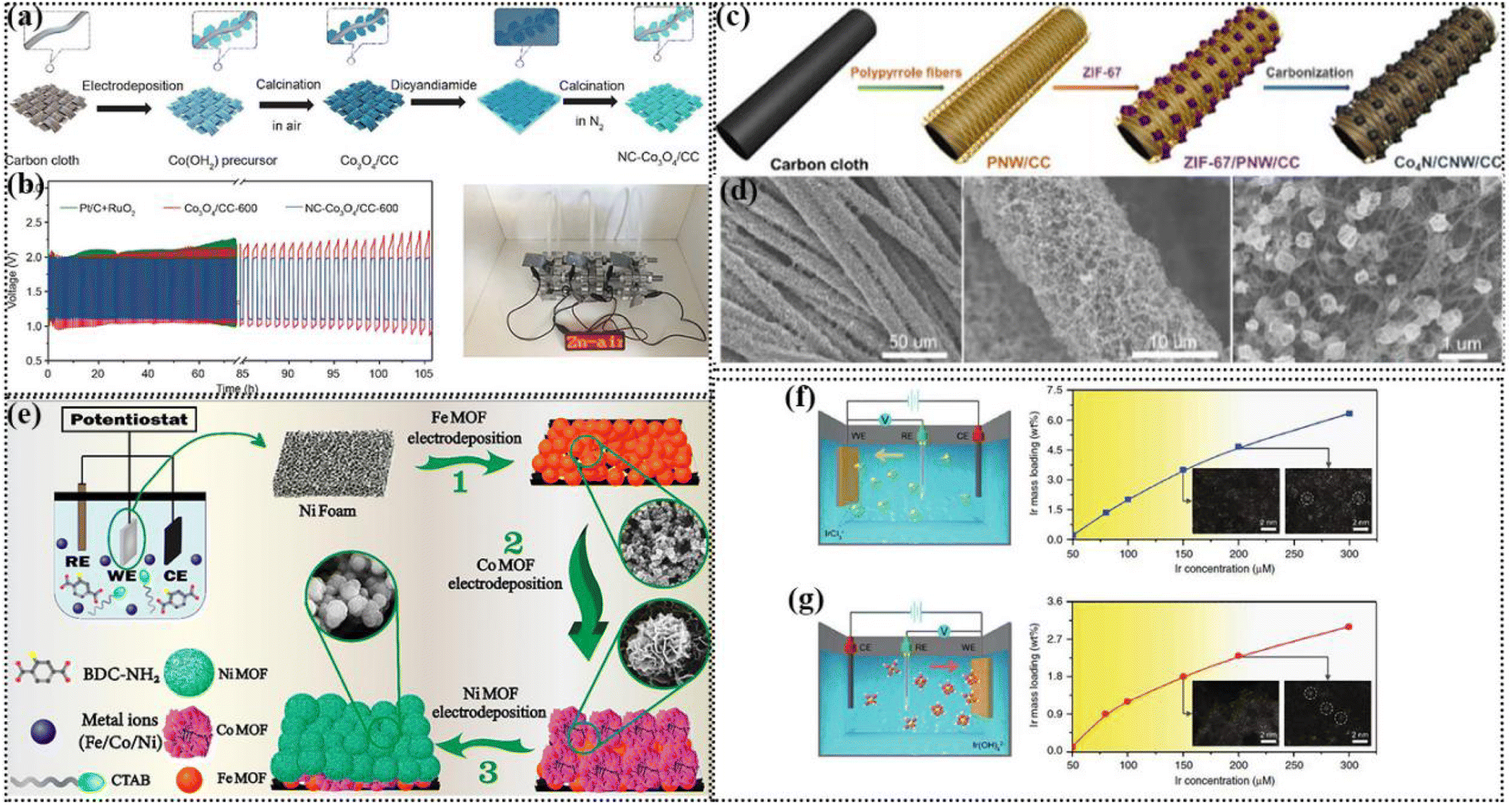 | ||
| Fig. 5 (a) Synthetic scheme of NC-Co3O4/CC. (b) Galvanostatic discharge–charge cycling curves at 10 mA cm−2 of rechargeable ZABs with NC-Co3O4/CC-600, Co3O4/CC-600 and mixture of Pt/C + RuO2 as air-cathodes, respectively, and photograph of a red LED indicator powered by three rechargeable ZABs with NC-Co3O4/CC-600 as the air-cathode in series. Reproduced with permission from ref. 123, Copyright (2018) Springer Nature. (c) Scheme of the synthesis of Co4N/CNW/CC. (d) Low and high-magnification SEM images of Co4N/CNW/CC. Reproduced with permission from ref. 133, Copyright (2016) ACS. (e) Three-electrode electrochemical cell comprised of an Ag/AgCl (KClO4) reference electrode, Ni foam working electrode, and Pt plate counter electrode immersed in a DMF-water mixed solvent containing BDC-NH2 linker, CTAB surfactant, and nitrate salts of Fe, Co, and Ni metal precursors. Electrochemical layer-by-layer assembly of (1) Fe MOF, (2) Co MOF, and (3) Ni MOF to prepare a trilayer and trimetallic Fe–Co–Ni MOF. Reproduced with permission from ref. 131, Copyright (2022) ACS. Schematic of cathodic (f) and anodic (g) deposition of Ir species. Reproduced with permission from ref. 135, Copyright (2020) Springer Nature. | ||
Different metal bonds in single metal catalysts can be used as active sites of the OER/ORR, respectively. Yu et al. synthesized a Co/Co–N–C catalyst on carbon felt via potentiostatic electrodeposition. The Co nanosheet structure obtained by electrochemical deposition could be used as an in situ template to build a Co nanoisland rooted in the Co–N–C nanosheet structure. The special structure exhibited good contact between the Co nanoisland and Co–N–C nanosheet. The coexistence of Co0+ and Co2+ was conducive to the bifunctional ORR/OER electrocatalytic performance. Under effective control, different components of monometallic compounds could also be used as active sites of the ORR/OER.124 The interesting phenomenon is that different products can be obtained by using different conductive substrates and carbon cloth and carbon felt cloth, respectively, under the same electrolyte conditions, which shows that the substrate has an essential influence on the electrodeposition products. When the substrate changes, due to the different wettability and roughness of the electrolyte, the degree of attachment of ions to atoms during deposition and the degree of combination of O will be different, and different products will be obtained. Heteroatom-doped single-metal compounds can also have good performance in ZABs. Marcus et al. reported a bottom-up and top-down method to produce an NiSx flexible porous film (FHF) through a combination of electrodeposition, electrochemical etching and vulcanization. The results showed that the NiSx FHF catalyst with independent structure was a bifunctional, additive-free catalyst with a layer of NiSx/Ni inside to enhance the electron transfer capability. The components of the single metal could synergistically promote the OER/ORR activity through different bond and coordination states in the single metal.125
Bimetallic catalysts have an intrinsic synergistic effect and more active sites than their single-metal counterparts. Also, integrated heterogeneous catalysts can better improve the electrocatalytic activity.126 Kim et al. reported a simple strategy mediated by agarose gel to selectively electrodeposit MnO2 and Co3O4 on 3D porous foam nickel to synthesize MnO2–Co3O4 electrodes. The prepared agarose water gel with metal precursor ions allowed electrochemical deposition only on the contact area between the gel and the matrix. This material showed excellent bifunctional catalytic activity.127 Non-noble metal layered double hydroxides (LDHs) are promising two-dimensional layered materials because of their low cost, abundant reserves and abundant electrochemical active sites. Due to its unique layered structure, which is conducive to the diffusion of water molecules and the rapid release of gas products, LDH-based materials have been studied as efficient OER catalysts. The electrochemical potential of transition metals such as Co, Fe, Mn and Ni is similar. Therefore, the LDH structure of bimetals can be obtained by mixing two metal solutions and applying a certain potential.128 Wan et al. successfully prepared porous shell NiCo2O4@FeNi LDH nanoarrays. Due to the high conductivity of the NiCo2O4 core, the adjustable electronic structure and the high specific surface area with FeNi LDH, their layered core shell structure greatly promoted the mass transfer, exposed a large number of catalytic sites, and significantly enhanced the bifunctional activity of the catalyst.129 Liu et al. used electrodeposition to grow CoFe layered double hydroxides (LDH) on CC, and then wrapped the polypyrrole coating. After pyrolysis, CoFe alloy consisting of uniform carbon-coated CoFe alloy nanoparticles was obtained (CoFe@NC/CC). Subsequently, it was confirmed by X-ray absorption spectroscopy and theoretical calculation that the active sites of Co species and metal O bond catalyzed the OER, while the active sites of Fe species and metal N bond catalyzed the ORR, achieving an excellent bifunctional OER/ORR performance. Therefore, the construction of bifunctional catalysts is more conducive to bifunctional catalytic reactions.130
Metal catalysts can simultaneously have the three functional electrocatalytic activities of OER/ORR/HER and used for ZAB and overall water splitting. Various materials with excellent electrochemical properties can be derived from MOF materials as precursors, which are generally synthesized via liquid-phase reactions, while there are few reports on their preparation via electrochemical deposition. Recently, Farahani et al. prepared a layer-by-layer self-assembled ternary metal Fe Co Ni MOF using the reductive electrochemical synthesis method, in which the metal ions between each layer or adjacent layers are connected by 2-amino-1,4-phenylenedicarboxylic acid through layer-by-layer assembly (Fig. 5e). The catalyst showed excellent triple electrocatalytic activity for the HER, OER and ORR. Through density functional theory of dispersion correction, it was found that the reason why this layer-by-layer MOF had excellent HER, OER, ORR multifunctional electrocatalytic activity is that the adsorption of water molecules on the metal nodes is an energy reduction process and the adsorption of H2O molecules leads to an increase in the bond length of the O–H chemical bonds.131
Polymeric conductive polymers, such as polypyrrole, polythiophene and polydopamine, as heterocyclic conjugated polymer materials, have unique super conductivity, which can significantly improve the conductivity of the substrate when attached to a conductive substrate and have potential to be used in electrochemical reactions.132 As shown in Fig. 5c and d, Meng et al. used the potentiostatic positive electrodeposition method to grow polypyrrole on CC to enhance the conductivity of the carbon cloth. Due to the special morphology of the polypyrrole carbon tubes, during the subsequent loading of ZIF-67, the loading area and loading amount of ZIF-67 effectively increased, maximizing the exposure of the active oxygen reaction center, and the excellent conductivity also significantly improved the overall activity of the ZAB.133 The conductive polymer not only improved the conductivity of the catalyst, but also be used as a special N source doped in the catalyst due to its N–C cross-linked ring structure compared with the N source introduced by other methods. Electrochemical deposition can completely wrap the polymer on the surface of the catalyst, which is uniform and stable, unifying the local and the whole of the catalyst, and significantly improving the stability of metal species. Not only polypyrrole, polyaniline, and polythiophene can be introduced as materials or N ligands to improve the conductivity.
Single atomic catalysts (SACs) as a new type of catalytic materials have attracted extensive attention in scientific research and industrial applications due to their remarkable activity. In addition, other catalytic properties of single metal atoms, including stability and selectivity, can be further improved by adjusting the electronic/geometric structure of single metal atoms and regulating the interaction between the metal carriers. Generally, SACs are composed of dispersed atoms and appropriate support materials, which are used to anchor, limit and coordinate isolated metal atoms. Therefore, the properties of single metal sites can achieve a maximum atom utilization rate of nearly 100%, effectively reducing the use of metal materials, which is beneficial to reduce the production cost of the catalyst. Also, this is of great significance for non-precious metal-based catalysts.134 Zhang et al. developed a universal method for the preparation of monatomic catalysts via cyclic voltammetric electrochemical deposition. By applying two different cyclic potentials, both processes generate metal SAs, but their valence states are quite different. The authors analyzed the reasons for this change and found that there were different electrodeposition processes for cathodic deposition and anodic deposition under these two potentials (Fig. 5f). During cathodic deposition, the applied potential is 0.1–0.4 V. Under the effect of external charges, metal ions are driven to the cathode and deposited on the carrier. The valence of single atoms is low. Most of the electrodeposition processes are cathodic deposition processes (Fig. 5g). During anodic deposition, the applied potential is 1.1–1.8 V. The metal ions are pushed to the anode under a large current and combined with the oxidation process of anions. The metal ions are in the oxidation state, resulting in high-valence single atoms. Later, it was also confirmed that different potentials, concentration of metal solution, and number of cyclic voltammetry segments can affect whether SAs are formed and the loading amount of metal SAs, indicating that the electrodeposition process is not static. After applying different potentials, new deposition processes will occur and different products will be obtained. This is a process of new crystal nucleus generation and regulation, which requires fine adjustment. Simultaneously, because bimetallic SA catalysts have different adaptive voltages, step potential deposition can be used to grow different monometals at different voltage points to effectively synthesize bimetallic SA catalysts.135
The ORR activity of electrodeposited single metal catalysts can be significantly improved by subsequently anchoring N ligands. Bimetallic LDH materials supported with catalysts with excellent ORR performance can be employed to construct heterogeneous interfaces or bimetallic LDH materials as precursors, and subsequently wrapped with nitrogen-containing species and carbonized-derived alloys containing metal–N bonds, which have excellent bifunctional catalytic activity. Reducing electrochemical synthesis can be used to prepare MOF materials with three functional activities. The conductive polymer synthesized by potentiostatic electrodeposition can be used as an independent material to improve the conductivity or as a nitrogen-containing ligand to wrap the surface of the catalyst. SAC catalysts can be synthesized via cyclic voltammetric electrochemical deposition.
3.3 Electrospinning
Electrospinning was first proposed in 1934,136 which is a manufacturing method that uses electrostatic action to pull the charged threads of high polymer solution or melt towards the average diameter of microfibers. Also, the fiber shape and structure are controllable.137 This method can be adapted to prepare a variety of polymer fibers, such as polyvinyl alcohol (PVA), polypropylene (PP), and polyurethane (PU) fibers.138 Polymer fibers have lower electrical conductivity and superior flexibility compared to metal fibers, which have important applications in energy devices.139 Some polymer fibers are stretchable, such as PU and SBS fibers, which can be used to construct flexible electronic devices with stretchable fibers.140 The nanofibers prepared by electrospinning have the properties of controllable chemical composition, high porosity, large specific surface area and volume ratio, and ideal mechanical strength, and thus they are considered ideal electrode substrates.141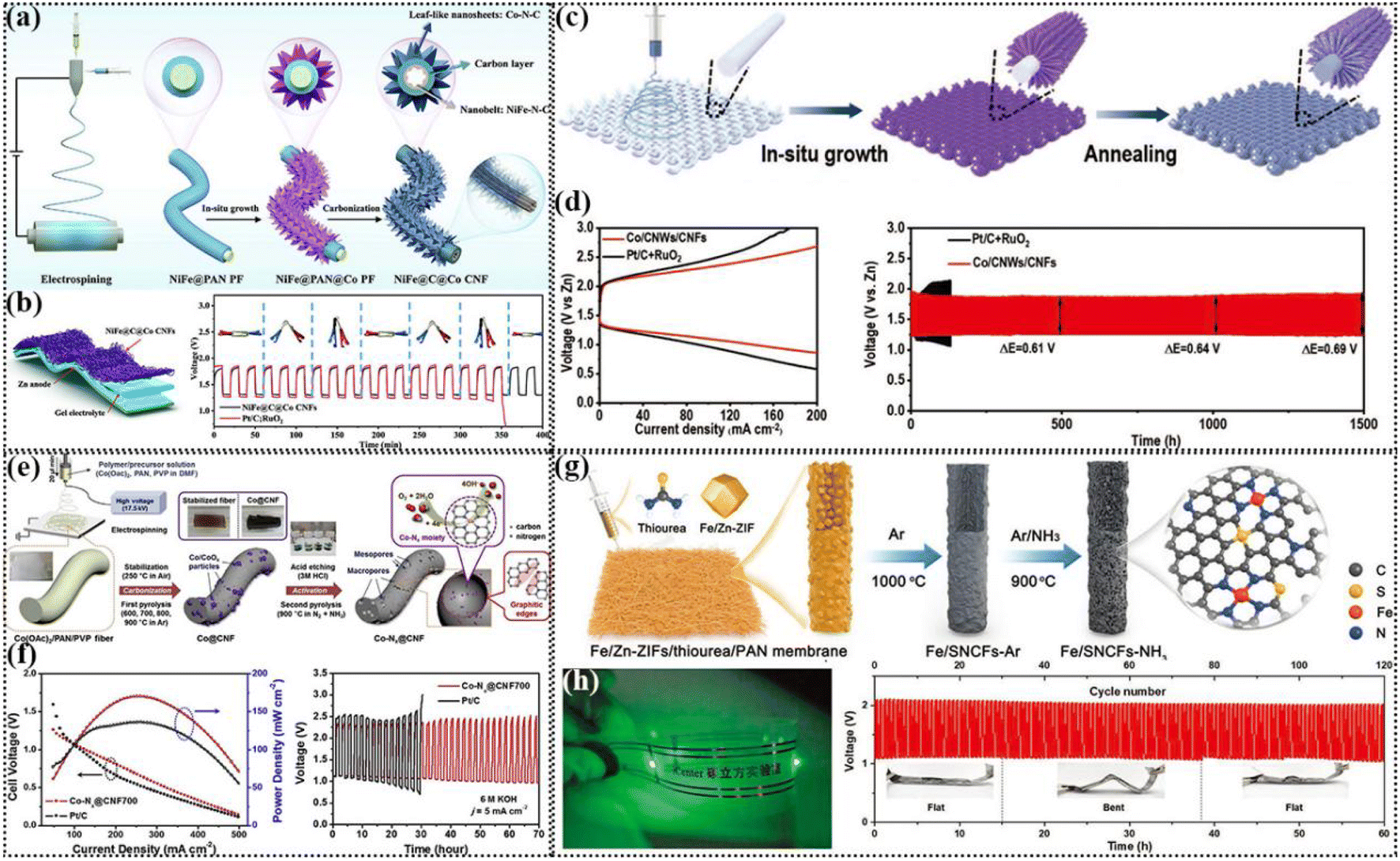 | ||
| Fig. 6 (a) Schematic illustration of the process for the synthesis of NiFe@C@Co CNFs. (b) Schematic illustration of a flexible solid-state ZAB and galvanostatic charge–discharge cycling curves at 5 mA cm−2 (insert: photo of the solid battery with different bending angles). Reproduced with permission from ref. 142, Copyright (2022) Wiley. (c) Schematic preparation of Co/CNWs/CNFs. (d) Discharge/charge polarization curves and cycling tests of Co/CNWs/CNFs and Pt/C + RuO2-based ZAB at 5 mA cm−2. Reproduced with permission from ref. 143, Copyright (2021) Wiley. (e) Schematic illustration of the preparation procedure of Co-Nx@CNF catalysts for the ORR. (f) Polarization curves and corresponding power density plot of ZABs using Co-Nx@CNF700 and Pt/C as the cathode catalyst and charge–discharge cycle curves. Reproduced with permission from ref. 146, Copyright (2018) Elsevier. (g) Schematic illustration of the synthesis of S and N co-doped porous carbon fiber membrane embedded with atomic Fe–N4/C sites (Fe/SNCFs-NH3). (h) Photograph of a wristband with a series of LED lamps lit by two series-connected solid-state ZABs and the stability of the solid-state ZAB at 1 mA cm−2. Inset: photographs of the solid-state ZAB at various flat/bent/flat states. Reproduced with permission from ref. 148, Copyright (2021) Wiley. | ||
A porous carbon nanofiber structure can be formed to facilitate electron transport when the polymer is mixed and electrospun. At present, PAN/PVP composite membranes are the most studied materials. Metal alloy- and oxide-based catalysts can be prepared by adding different metal salts to the spinning solution. Li et al. prepared a PVP/PAN hybrid composite fiber membrane via electrospinning to obtain a mesoporous carbon fiber-based Fe/Co–N–C structure containing FeCo nanoparticles. The prepared hybrid nanofibers possessed a one-dimensional mesoporous structure, large surface area and uniform distribution of active centers. The coexistence of FeCo alloy with Fe/Co–N active center could simultaneously increase the catalytic activity for the ORR and OER. When the composite was used as an air-cathode, the assembled ZAB showed a low voltage gap and good charge–discharge cycle stability.145 Yoon et al. obtained Co-Nx@CNFs by electrospinning a mixed solution containing Co precursors, PVP and PAN, and then carbonization, and further activation, which exhibited good ORR/OER activity (Fig. 6e). This was attributed to the existence of the graphene nanosheet structure, which could improve the conductivity of the carbon nanofibers, and the pore structure in the carbon nanofibers increased the active specific surface area, thus facilitating electron and mass transfer. The Co-Nx@CNF-based ZAB showed a superior power density compared to Pt/C and can be stably cycled for 70 h at a charge discharge current density of 5 mA cm−2 (Fig. 6f).146
At present, it is challenging and scientifically significant to construct SA catalysts using electrospinning technology. Ji et al. prepared high-performance binder-free air electrodes for wearable ZABs by anchoring SA in carbon nanofibers using an “impregnation–carbonation–acidizing” strategy. A PAN nanofiber film was prepared by electrospinning, and the resulting PAN nanofiber film was directly immersed in a solution containing a certain proportion of 2-methylimidazole and cobalt nitrate. In the mixed aqueous solution, the blade-like ZIF was impregnated on the electrospun nanofibers and transformed into N-doped carbon nanosheets under the optimized pyrolysis conditions. The Co–N coordination unit in ZIF was reduced in situ to form atomically dispersed Co–N active sites, and some Co atoms aggregated to form Co clusters. Finally, the carbon nanofiber structure of nitrogen-doped carbon sheet array modified by nano Co was obtained. The electrode material did not deform even when bent or twisted, proving its ultrahigh flexibility, which can be directly used as flexible ZABs. Due to its low coordination and unsaturated chemical environment, SA can produce abundant active centers, and thus it has very high electrocatalytic performance.147 The SA structure is not limited to the structure of metal–N4. When doped with other atoms such as S and P, the electronic structure of the metal will be adjusted to promote the activity of bifunctional groups. Yang et al. synthesized a flexible self-supporting carbon fiber membrane-supported iron SA catalyst with a hierarchical pore structure by electrospinning. Subsequently, a bimetallic organic skeleton material containing Zn and Fe (Fe/Zn-ZIF) was prepared, and then thiourea and PAN were added, and a flexible self-supporting carbon film electrocatalyst was obtained by spinning, pre-oxidation, high temperature carbonization and NH3 activation. After activation, the specific surface area of the material increased by about 9.5 times, which is beneficial for the full exposure of the Fe–N4 sites for enhancing the electrocatalytic activity (Fig. 6g). Optimizing the pore structure of carbon fibers is very important to improve the gas diffusion, electrolyte penetration, electron conduction and catalytic reaction kinetics. The N and S doping could regulate the local coordination environment of the Fe–N4/C active sites, thus promoting the ORR and OER activities, respectively. In the subsequent ZAB test, two Fe/SNCF NH3-based solid-state ZABs in series could drive a series of LED lamps, and also undergo more than 120 cycles of stable charging and discharging process in various flat/bending/flat states.148
The flexible self-supporting carbon fiber membrane formed by electrospinning possesses a special layered nanopore interwoven fiber structure of polymer fibers, which promotes gas transport, electrolyte penetration and electron transfer, maximizes the exposure of oxygen counter center, and effectively exposes the active sites of the metal catalysts with a large specific surface area. Simultaneously, not only one type, but also mixed polymer fibers can be used to synthesize catalysts with specific functions. It is feasible to synthesize the popular monatomic catalysts by electrospinning, and heteroatoms can be introduced to effectively prepare the electronic state of the monatomic catalyst to improve the catalyst activity.
3.4 Hydrothermal methods
The hydrothermal method is one of the most common methods for the synthesis of materials for energy applications.149 Specifically, the catalyst is in situ grown on a conductive substrate to construct a self-supporting electrode, and thus the catalyst and the substrate are in direct contact, which can enhance the stability and lifetime of the electrode.150 Compared with other synthetic strategies, this method is easy to adjust the structure and electronic state of the catalyst, thus improving the electrocatalytic performance. The most effective method is to construct a heterojunction interface by the interface engineering strategy.151Heterogeneous interface construction can regulate the activity of catalysts. The original coordination bonds and the electronic structure of metal species can be tuned by introducing other compounds. Song et al. synthesized hybrid nanostructured arrays (HNA) with branched and aligned structures constructed by cobalt selenide (CoSe2) nanotube arrays vertically oriented on CC with CoNi layered double hydroxide (CoSe2@CoNi LDH HNA) using a hydrothermal-selenization-hybridization strategy (Fig. 7a). As shown in Fig. 7b, the CoNi LDH and CoSe2 formed a heterogeneous interface, and elemental mapping further proved the uniform distribution of several elements. After constructing the heterogeneous interface, the electrons on the catalyst surface shifted, as evidenced by XPS. The strong interaction at the interface could effectively adjust the electronic structure of the hybrid, thus improving the reaction kinetics (Fig. 7c).152 Yin et al. reported the preparation of a porous NiO/CoN nanowire array via hydrothermal synthesis, which exhibited improved electrocatalytic performance and stability for the OER and ORR. X-ray photoelectron spectroscopy (XPS), electron spin resonance (EPR) and high-resolution transmission electron microscopy (HRTEM) showed that the decrease in the Co coordination number at the heterojunction and the increase in oxygen vacancies at the NiO/CoN interface were the main reasons for the excellent bi-functional electrocatalytic performance.153 Liu et al. constructed two-dimensional Mo–NiCo2O4/Co5.47N nanosheet arrays via hydrothermal reaction. The high-valent metals were successfully doped in the heterojunction nanosheet array grown directly on the conductive substrate, and subsequently electrode structures with balanced electronic structures, highly exposed active sites and no binder were synthesized. XPS characterization showed that the electron transfer of Co and Ni occurred after Mo atom doping, which could optimize the electronic structure of the active center in NiCo2O4/Co5.47N/NF (Fig. 7d–f). This porous nanosheet array had highly exposed active sites, which could greatly accelerate the electron/mass transfer, promote the electrolyte penetration and improve the electrocatalytic activity.154
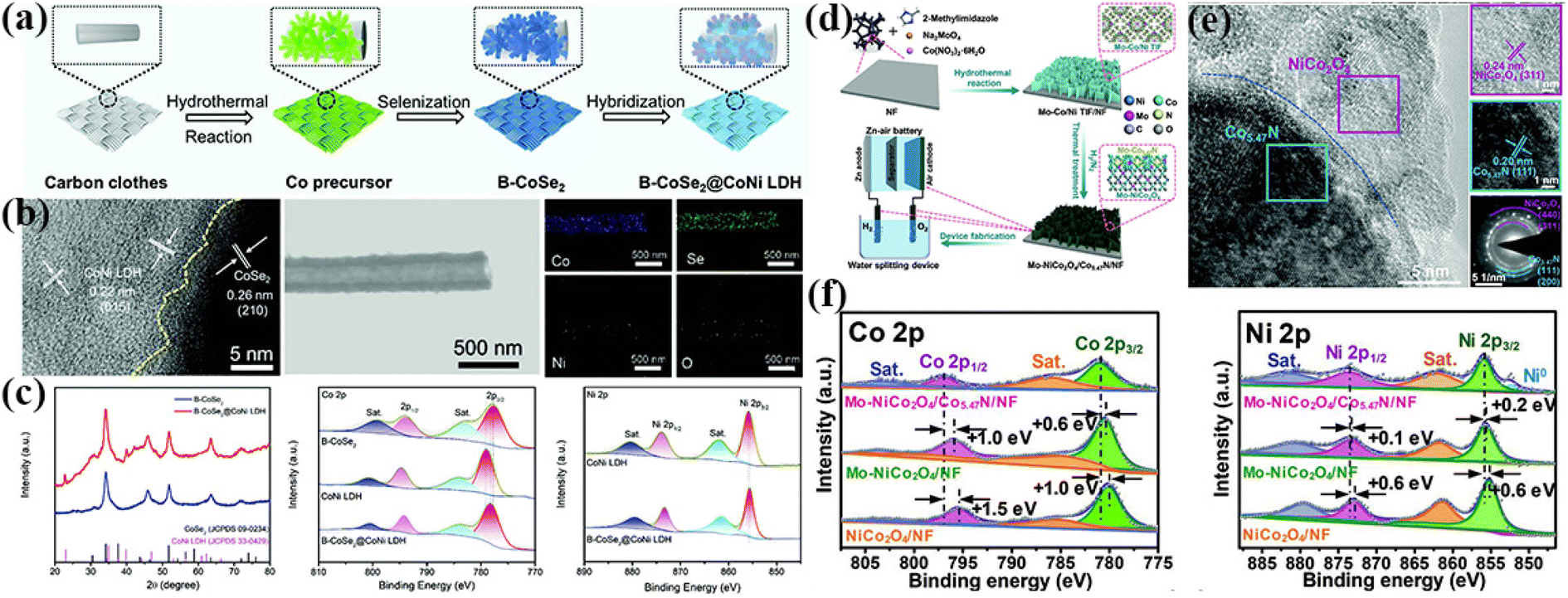 | ||
| Fig. 7 (a) Schematic illustration of the synthetic process of B-CoSe2@CoNi LDH HNA. (b) HRTEM image of B-CoSe2@CoNi LDH HNA highlighting the interface between CoSe2 and CoNi LDH and elemental mapping images of B-CoSe2@CoNi LDH HNA. (c) XRD spectra and XPS patterns for B-CoSe2, CoNi LDH, and B-CoSe2@CoNi LDH HNA. Reproduced with permission from ref. 152, Copyright (2022) Wiley. (d) Schematic illustration of the synthetic process of Mo-NiCo2O4/Co5.47N/NF. (e) HRTEM images and SAED pattern of Mo–NiCo2O4/Co5.47N/NF. (f) XPS spectra of Mo–NiCo2O4/Co5.47N/NF, Mo–NiCo2O4/NF, and NiCo2O4/NF. Reproduced with permission from ref. 154, Copyright (2020) Wiley. | ||
Given that the electrocatalytic process occurs on the surface of the catalyst, the heterogeneous interface constructed by the interface engineering strategy during the hydrothermal reaction can effectively adjust the electronic structure and enhance the activity for the ORR/OER. Simultaneously, the ORR/OER activity of the catalyst can also be adjusted by improving the electronic coordination microenvironment of the catalyst surface interface through heteroatom doping and manufacturing defects.
3.5 Modification strategies for improving self-supporting electrochemical performance
The modification of conductive substrates generally refers to carbon material substrates, such as carbon cloth, carbon paper and carbon felt cloth. Generally, the method for the treatment of carbon material substrates is the use of acetone or ethanol to remove impurities on the surface of carbon cloth. However, although the overall activity of the carbon substrate improves to a certain extent, its hydrophobicity makes it difficult to grow the catalyst and the poor wettability of the electrolyte affects the electrochemical activity of the self-supporting electrode. Subsequent experiments showed that the overall activity of the self-supporting electrode was significantly improved after the carbon material substrate was modified. At present, there are three main methods to modify carbon substrates: firstly, acid treatment, and then calcination in the air.155 The purpose of acid treatment is to remove the oxides and impurities on the surface of the carbon substrate. Uniform nanopores and super hydrophilic surface structures are generated on the surface of the carbon substrate due to the etching effect by the acid solution, which is conducive to the attachment of the catalyst and the contact of the electrolyte, and significantly improves the hydrophilicity of the carbon substrate. Air calcination can introduce oxygen-enriched functional group active species to improve the rate of oxygen reaction. Additionally, the carbon substrate can also be treated by the oxidation of KMnO4 and H2O2, thus effectively introducing a large number of oxygen-containing groups. Subsequently, high temperature reduction under an N2 atmosphere can introduce defects to manufacture active sites, thus improving the adsorption performance of carbon groups on the catalyst.156 Besides, carbon substrates are treated by plasma to make their surface defective, which is conducive for the growth of the catalyst and improves the electron transfer rate and electrocatalytic performance.157 In contrast, foam metal substrates (such as copper, iron, and nickel foam) cannot be modified due to the inherent characteristics of the metal substrate.The electrocatalytic process mainly occurs on the surface of the electrocatalyst. Therefore, the surface design and control of electrocatalysts are very important.158 The electrocatalytic performance of self-supported electrodes can be improved by adjusting the structure of the catalyst. In this case, heteroatom doping and construction of defects are the most common strategies for modifying self-supporting electrodes. Heteroatom doping can not only be used as the coordination site of metal atoms to achieve high stability, but also adjust the electron structure of metals by properly selecting heteroatoms with different atomic radii and electronegativity, thus reducing the activation barrier of the reaction and accelerating the electrochemical reaction rate.159 N atom doping is the most effective site for increasing ORR activity. A large number of N functional groups can provide abundant coordination sites for the bonding of metal atoms.160 S- and P-atom doping can significantly improve the OER activity of the catalyst, and the doping of heteroatoms can effectively improve the bi-functional ORR/OER activity of self-supporting electrodes.161 The defect structures can effectively adjust the electronic structure of the catalyst surface and make the electrons shift. The defects contain different types of anchor points and oxygen-rich vacancies, which can effectively improve the intrinsic activity of the catalyst and accelerate the electrochemical reaction rate.162 Accordingly, self-supporting electrodes can be modified by structural defects and heteroatomic doping.
4. Study the charging–discharging mechanisms of air-cathodes
At present, ZABs are developing rapidly, and thus it is still a research hotspot to explore ideal bifunctional electrocatalysts. In this case, it is necessary to explore the structure–activity relationship of materials for the construction of high-performance electrocatalysts.163 At present, many catalysts have been confirmed to undergo a phase change process during the charge and discharge process of ZABs and that the considered catalyst is not necessarily the active center of the reaction.164 Previously, due to the limited characterization, it was thought that the bulk catalyst was the active species of the reaction. However, with the rapid development of in situ characterization methods, the dynamic change process during the charge discharge process of ZABs can be visually observed.165 Various operando characterizations are the most effective means to reveal the black box process, such as operando X-ray diffraction (XRD), operando Raman, and operando X-ray absorption fine spectroscopy (XAFS).166 The following section presents a detailed description of the relevant research progress.4.1 Operando XRD
Operando XRD can be used to determine the crystal phase change of catalysts under different potentials in ZABs, which is applicable to the obvious crystal phase change trend of transition metal-based catalysts.167 Generally, it used to study the phase transition in the OER process and Zn–air hybrid battery. Shang et al. used copper oxide as an air-cathode material, creatively solving the problem that the traditional ZAB must rely on oxygen for operation, and developed a new ZAB that could freely switch to air and anaerobic conditions. When it operated in the ZAB mode, it displayed a voltage of 1.28 V, while the voltages of 1.13 V and 0.79 V when it operated in the Zn–Cu mode. In addition, the air-cathode also showed an ultra-fast self-charging performance when the battery was switched from an anaerobic environment to aerobic environment. Operando XRD spectroscopy was used to prove the reversible conversion process among Cu, Cu2O and CuO in the charging–discharging process (Fig. 8a).168 Zhang et al. also proved the reversible conversion process of CuO and CuO2 in different charging–discharging processes of a ZAB hybrid battery by operando XRD, which proved the presence of transition metal ion pairs (Fig. 8b).169 Li et al. synthesized a porous carbon nanofiber-supported CoS2/Cu2S heterostructure composite (CoS2/Cu2S-NF) as a high-efficient air-cathode for ZABs. The stability of the catalyst were investigated by operando XRD. As shown in Fig. 8c, the characteristic peaks of CoS2/Cu2S–NF slightly changed in the electrochemical charging–discharging processes. The individual peaks increased and decreased regularly, and finally returned to the original state.170 The results showed that the change in O2 adsorption and desorption on the catalyst surface was completely reversible. In addition, it also confirmed the stability of the electrode material under a wide working potential, which is an important condition to ensure the stability of the battery. Operando XRD can effectively explore the evolution process of catalysts with obvious crystal phase changes during the charge and discharge of ZABs and effectively clarify the real active sites and mechanisms of the charging–discharging processes. It also shows that the catalyst is unchanged in this process and that the bulk catalyst is the active center of the reaction, and thus the catalyst has excellent stability.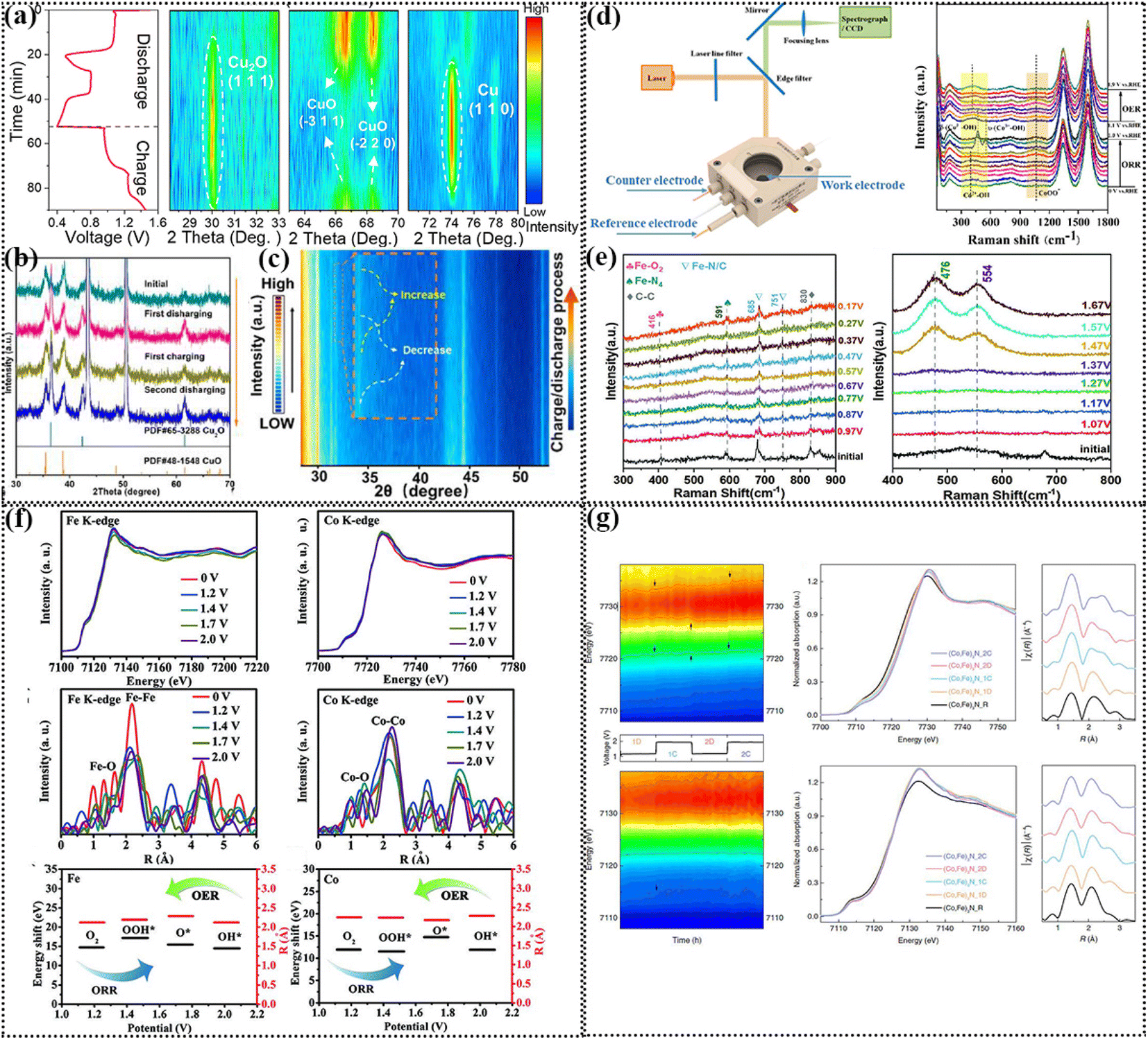 | ||
| Fig. 8 (a) Contour map of in situ XRD of the CuO electrode during the charging and discharging process. Reproduced with permission from ref. 168, Copyright (2022) Elsevier. (b) In situ XRD patterns of CuxO@NiFe-LDH electrode during charging and discharging processes. Reproduced with permission from ref. 169, Copyright (2022) ACS. (c) In situ XRD intensity map of ZAB with CoS2/Cu2S-NF electrodes during discharging and charging. Reproduced with permission from ref. 170, Copyright (2022) Elsevier. (d) Schematic diagram of in situ Raman spectroscopy equipment and in situ Raman spectra collected on Co@N-HPC-800 in 0.1 M KOH solution. Reproduced with permission from ref. 172, Copyright (2022) Elsevier. (e) In situ Raman spectra collected on FePc‖CNTs‖NiCo/CP for (a) ORR process and OER process in O2-saturated 0.1 M KOH solution. Reproduced with permission from ref. 173, Copyright (2022) Wiley. (f) In situ XANEs of the Fe K-edge and Co K-edge for CoFe/N-GCT at different potentials. Reproduced with permission from ref. 178, Copyright (2018) Wiley. (g) Operando X-ray absorption spectroscopic analysis of (Co, Fe)3N catalysts. Reproduced with permission from ref. 179, Copyright (2020) Springer Nature. | ||
4.2 Operando Raman
Operando Raman spectroscopy is also a very useful technique in exploring the origin of the activity of metallic species. By monitoring the change in metal valence and coordination bond during the reaction, the active center of the catalyst can be clarified.171Operando Raman spectroscopy can effectively elucidate the catalytic mechanism of the ORR/OER. Liu et al. reported a novel cobalt-ion-exchange and pyrolysis strategy with the well-designed bio-MOF-1 as a self-template to prepare ultra-small cobalt nanoparticles embedded in N-doped hierarchical porous carbon (Co@N-HPC-800) with excellent electrical conductivity, high surface area and abundant Co–N4 active sites. This catalyst showed excellent bifunctional ORR/OER catalytic activity under alkaline conditions, which was characterized by in situ Raman spectroscopy. They observed the continuous conversion of Co2+/Co3+ redox electric pairs in the ORR (0.6 V to 1.0 V) and OER processes (1.0 V to 1.9 V), which is the source of the catalytic active centers for the ORR/OER (Fig. 8d).172 Ding et al. also used in situ Raman spectroscopy to study the ORR/OER mechanism of the FePc‖CNTs‖NiCo/CP catalyst. As shown in Fig. 8e, the Fe–O2 vibration at 416 cm−1 indicated that O2 was adsorbed on the Fe active center, which disappeared after the ORR was potential applied. When the applied potential was 0.97 V, the three characteristic peaks at 591, 685 and 751 cm−1 related to the Fe active center became weaker. As the applied potential shifted negatively from 0.97 to 0.67 V, the three peaks continued to decay gradually. It was certified that O-containing intermediates (such as *OOH, *O and *OH) can be formed on the Fe active center, thus weakening the in-plane Fe–N4 and Fe–N/C vibrations. A further negative shift in the applied potential resulted in a relative enhancement in the signal, which may be due to the desorption of the *OH products. These results indicate that the ORR process may occur on the Fe active center via the O2 → *OOH → *O → *OH → OH− pathway. When the applied potential was 1.07–1.37 V, the OER could not occur and no peak was detected. However, as the applied potential increased to 1.47 V or more, two new peaks appeared at 476 and 554 cm−1, which were attributed to the metal (Ni, Co)–O vibration in (Ni, Co)–OOH. The results showed that the real active site for the OER was the corresponding hydroxide.173 Wu et al. constructed an NiFe LDH (d-NiFe-LDH) with defects. Operando Raman spectroscopy confirmed that cation defects were gradually formed with an increase in the applied potential (VM → VMOH → VMOOH), indicating the structural change sequence of crystalline Ni(OH)x→disordered Ni(OH)x → NiOOH, which revealed the real electrocatalytic active sites.174
Operando Raman spectroscopy can be employed to reasonably judge the catalytic sites of the reaction, which plays a role in the process through the appearance and disappearance of peaks and the weakening and strengthening of the peaks in the ORR/OER potential process and completely reveal the ORR/OER reaction mechanism.
4.3 Operando XAFS
Operando XAFS can be used to explore the chemical state and coordination structure during the electrocatalytic reaction based on the change in metal bonds under different electrochemical potentials. It can reveal the dynamic evolution and the real active sites in the electrocatalytic processes. It can be concluded that the coexistence of different coordination forms of metal species is the key to bifunctional activity for monometallic catalysts.175,176The synergistic effect between polymetallic species has a certain catalytic advantage, and thus it is important to reveal the related bifunctional electrocatalytic mechanism. Peng et al. investigated the dynamic structure change in catalysts during the OER by operando Raman and XAFS. The results showed that the co-doping of Fe and Ni accelerated the self-repair of the surface structure of Fe and Ni–CoS2 catalysts, which was conducive to the oxidation of Co and Fe to form hydroxyl oxide and prevent the oxidation of Ni to form Ni(OH)2, thus improving the activity and stability of the catalyst.177 Liu et al. constructed a graphene-entangled carbon nanotube-wrapped CoFe alloy composite (CoFe/N-GCT) based on Prussian blue precursor and studied the corresponding ORR/OER electrocatalytic mechanism by operando XAFS. The FT-EXAFS spectra showed that the Fe–Fe bond length first increased, and then decreased with an increase in potential, indicating that the ORR process at low potential mainly occurred on the surface of the iron species. The corresponding length of the Co–Co bond decreases first, and then increases with an increase in potential, indicating that the OER process mainly occurs on the surface of the cobalt species at high potential. In addition, the changes in the length of the Fe–Fe and Co–Co bonds were observed during the formation of *OOH and *O, further revealing that the ORR and OER active sites mainly existed on the surface of the Fe and Co species, respectively. Therefore, the synergistic effect of Fe and Co endowed CoFe/N-GCT with good bifunctional electrocatalytic activity (Fig. 8f).178
Deng et al. also innovatively proposed the concept of “dynamic catalyst” to describe the electrochemical behavior of metal-based materials in ZABs. Taking (Co, Fe)3N bimetallic nitrides as an example, a relatively obvious activation stage of the catalyst was found at the initial reaction stage of the ZAB (Fig. 8g). Operando XAFS certified that the cobalt species was the active center and becomes tetravalent and bivalent during the charging and discharging processes, respectively, while iron was relatively inert and always remained trivalent. During the electrochemical reaction, the recombination of the initial components between cobalt and iron improved the electrocatalytic activity. In the reaction process of ZABs, the metal species in catalyst will change dynamically, and the electrochemically active sites will evolve periodically during the charging and discharging potentials. A thorough understanding of the mechanism and nature of electrocatalysts has important guiding significance for the construction of high-performance catalysts for ZABs.179Operando characterizations can be used to clarify the crystal phase change, functional group evolution and coordination bond length change in the catalyst during the charge–discharge process of ZABs. Consequently, the microscopic change process affecting the catalytic activity can be explored in detail, which is crucial to understanding the source of the catalytic activity of the catalyst. These effective in situ methods are very important for the in-depth understanding of the electrocatalytic reaction mechanisms.
4.4 Theoretical calculations
Density functional theory (DFT) calculation plays an important role in understanding the catalytic active center and reaction mechanism. The change in electron density in the active center of the catalyst during the reaction can explain the reason for the excellent activity of the catalyst. Simultaneously, the structure of the catalyst can be explored according to the coordination theory, and the source of the active component explained. Theoretical calculation can strongly support the experimental data to reveal the electrocatalytic reaction mechanism.180The first is a catalyst with a crystalline phase. Tian et al. successfully fixed multiphase CoS/CoO nanocrystals on N-doped graphene (CoS/CoO@NGNs) in space and synthesized a bifunctional electrocatalyst. The ZAB assembled with this catalyst as the air-cathode showed a power density of 137.8 mW cm−2 and specific capacity of 723.9 mA h g−1. Then, theoretical calculations were performed to explain the interaction of bimetallic heterostructures by building a CoS/CoO@NGN model. Firstly, the typical diffraction peaks related to CoS and CoO were observed by XRD. Specifically, the diffraction peaks at 30.5°, 35.1°, 47.2° and 54.3° can be assigned to the (100), (002), (101) and (110) planes of CoS, respectively (JCPDS No. 75-0605). The peaks at 36.5°, 42.5°, 61.8° and 73.6° can be indexed to the (111), (200), (220) and (311) planes of CoO, respectively (JCPDS No. 71-1178) (Fig. 9a). In the high-resolution TEM (HRTEM) image, the identified lattice distances of 0.292 and 0.246 nm match well with the (100) plane of CoS and (111) plane of CoO, respectively (Fig. 9b). Then, the bimetal heterostructure was obtained by reasonably constructing a theoretical model based on the exposed crystal planes (Fig. 9c).181
 | ||
| Fig. 9 (a) XRD pattern of CoS/CoO@NGNs. (b) HRTEM images of CoS/CoO@NGNs. (c) Schematic of the CoS (100)/CoO (111) heterostructure model and contour plots of differential charge density of the CoS (100)/CoO (111) model. The yellow and cyan regions represent the charge accumulation and charge depletion, respectively. The iso-surface level was set to be 0.008 eÅ−1. Reproduced with permission from ref. 181, Copyright (2022) Springer Nature. (d) Operando Co K-edge XAFS spectra of Co9S8@Fe3O4 at different applied potentials. Co K-edge XAFS spectrum of CoOOH@Fe3O4 is shown as a reference and the corresponding FT curves of the Co K-edge extended XAFS spectra. (e) CoOOH@Fe3O4 model. Color codes for elements: Co, blue; O, red; H, pink; and Fe, yellow. Reproduced with permission from ref. 174, Copyright (2022) ACS. (f) FT-EXAFS spectra of Fe/Zn-NC-850, Fe/Zn-NC-950, Fe-NC, Fe foil, FeO, Fe2O3 and FT-EXAFS fitting curves of Fe K-edge for Fe/Zn-NC-950 (inset: structural model of Fe-N3C). (g) Bond order of Fe-N3C. Reproduced with permission from ref. 184, Copyright (2022) Wiley. | ||
The second is a catalyst with phase change during the OER process. Many catalysts have a certain phase conversion process in the OER process, that is, new metal phases will be generated. At this time, it is necessary to consider whether the new substance is the catalytic active species of the reaction. Lv et al. found that Co9S8 was reconstructed by successively transforming into CoOH and CoOOH in the Co9S8@Fe3O4 heterojunction during the OER process by operando XAFS. The resulting CoOOH was considered as the active species, while the existence of Fe3O4 could inhibit the aggregation of CoOOH. The theoretical calculation model was constructed based on the experimental results, clarifying that the real catalytic active site was CoOOH (Fig. 9d and e).182
The third is transition metal-based monatomic catalysts. Given that the loading of monatomic catalysts is less than 7%, XRD cannot characterize their crystal phase separately, and thus it is necessary to use X-ray absorption spectrum to fit the coordination number of monatomic catalysts, and reasonably build a theoretical calculation model in combination with the specific coordination environment.183 Tong et al. controlled the synthesis of Fe–N4 and Fe–N3C atomic dispersion structure by adjusting the pyrolysis temperature of the Fe/ZIF-8 precursor from 850 °C to 950 °C. Simultaneously, in situ C in ZIF-8 and high-temperature cracking can be used to obtain a Fe/Zn-NC-950 catalyst for the synthesis of Zn–N4. The Fe/Zn-NC-950 catalyst with Fe–N3C configuration showed better ORR activity and excellent durability. Given that XRD and TEM cannot characterize the specific structure of Fe, XAFS showed that the configuration of Fe is Fe–N3C, and subsequently a theoretical calculation model was built based on this structure (Fig. 9f and g). It was found through DFT that the valence state of iron in asymmetric Fe–N3C is higher than that in symmetric Fe–N4 because the electronegativity difference between the coordinated C atom and N atom results in the stronger electron delocalization of Fe–N3C. In addition, the residual Zn atom from ZIF-8 will transfer its d-orbital electron to Fe, and thus the synergistic effect between Fe–N3C and Zn–N4 will enhance the adsorption capacity of the oxygen-containing intermediates, which is conducive to the stretching and splitting of O–O on the Fe active center and reduce the formation energy of the rate-controlling step of OOH*.184
Liu et al. used theoretical calculations to demonstrate that heteroatom-doping could improve the electrocatalytic performance. Fig. 10a shows the free energies of various oxygen-containing intermediates of the ORR and OER at CoO@PWC and P-CoO@PWC-2. The adsorption free energy of the ORR significantly increased after P-doping because the P atom provided electrons to CoO, thus weakening the adsorption of OH*. The density of states results implied that the density of electron states near the Fermi level increased significantly after doping P, indicating the higher carrier density and more favorable charge transfer during the electrocatalytic reaction (Fig. 10b). The fundamental reason for the improvement in the electrocatalytic performance of the heteroatom-doped catalyst was proven by Gibbs free energy and density of state calculations.185
 | ||
| Fig. 10 (a) Free energy diagrams for the ORR and OER at different electrode potentials on CoO@PWC and P-CoO@PWC-2 through an oxygen associative mechanism. The reaction pathways for the ORR listed in this panel are Step 1: O2 (g) + * + H2O(l) + e− ↔OOH* + OH−, Step 2: OOH* + e− ↔ O* + OH−, Step 3: O* + H2O (l) + e− ↔OH* + OH−, and Step 4: OH* + e− ↔* + OH−; the OER occurs in reverse from Step 4 to Step 1. (b) Density of states of CoO@PWC and P–CoO@PWC-2. Reproduced with permission from ref. 185, Copyright (2021) Wiley. (c) Free-energy diagrams of Co-Nx [x = 4, 3, 2 (cis and trans), 1] and the theoretical ideal diagram for the ORR and OER. Reproduced with permission from ref. 186, Copyright (2022) Elsevier. (d) Proposed ORR mechanism on the Co4NC-2 model. (e) Projected density of states (PDOS) plots of Co4NC-1 and Co4NC-2 reaction process. (f) Calculated free–energy profiles for the ORR and OER pathways on the Co4NC-2 model with different electrode potentials. Reproduced with permission from ref. 188, Copyright (2022) Wiley. | ||
Moreover, theoretical calculations can also be used to illustrate the electrocatalytic reaction mechanism of SAs. Xu et al. investigated the ORR mechanism of Co–N4 single atom by theoretical calculation. The results showed that the Gibbs free energy of each step for the Co–N4 model during the ORR and OER was lower than that of Co–N1, Co–N2 and Co–N3, indicating that the structure of Co–N4 had better ORR/OER activity (Fig. 10c).186 It is more important to elucidate the catalytic mechanism of bimetallic SAs based on the synergistic effect on electrocatalytic activity. Fu's group prepared bimetallic SA catalysts containing Cu–N4 and Zn–N4, which showed high activity in ORR catalysis. The density of state calculation showed that the Cu metal center had an obvious negative shift in the D-band center of Cu–N4/Zn–N4, and the overlap area is larger compared with Cu–N4, further implying that the bimetallic SA has a stronger adsorption force on O2. The Gibbs free energy calculations further showed that the synergistic and electron transfer effects of bimetallic SAs can effectively reduce the reaction energy barrier and increase the reaction rate of the OOH* rate-determining step compared to single-metal SAs.187 Shu et al. synthesized bifunctional oxygen reaction electrocatalysts (CoCNTs/PNAs) via a simple self-assembly pyrolysis method. The effect of strong interaction between cobalt sites and nitrogen-doped carbon substrates on improving the ORR/OER catalytic activity was also studied through density functional theory calculations. Theoretical calculation further demonstrated that the electronic interaction between the Co sites and N-doped carbon substrate can activate oxygen molecules, significantly regulate the electronic d-band structure, and reduce the critical step reaction energy barrier, thus achieving efficient electrocatalytic ORR/OER (Fig. 10d–f).188
 | ||
| Fig. 11 Adsorption energies (Eads) on the (200) surfaces of CoFe and the optimized geometries of (a) CoFe@NC, (b) CoFe@Co and (c) CoFe@Fe systems. Reproduced with permission from ref. 130, Copyright (2020) RSC. Calculated charge density differences for (d) Zn–N4, (e) Cu–N4 and (f) Cu–N4/Zn–N4 (blue and red areas represent charge density increase and decrease, respectively, where larger values represent increased electron density). Reproduced with permission from ref. 187, Copyright (2021) Wiley. Differential charge density of (g) Fe-NPSA@NC and Fe-ACSA@NC (colors yellow and blue represent the increase and decrease in charge density, respectively). (h) ORR elemental steps in alkaline media and OH* adsorption energy as a function of Bader charge, ORR half-wave potential as a function of average Fe. Reproduced with permission from ref. 189, Copyright (2022) Wiley. | ||
5. Zn–air hybrid battery
At present, the low power density caused by the discharge voltage of ZABs makes it difficult for them to achieve 65% efficiency.190 Moreover, they cannot work in oxygen-free conditions, such as water, sealed and vacuum conditions, greatly hindering the practical application of ZABs.191 Zn–air hybrid batteries are a new type of battery derived from ZABs, which perfectly combine the advantages of ZABs and Zn-ion batteries. Compared with ZABs, Zn–air hybrid batteries have the following advantages (Fig. 12a): (i) Faraday redox reaction (transition metal redox reaction M–O–OH ↔ M–O, M = Co, Ni, Mn, etc.) can provide high working voltage and high energy density. (ii) The hybrid battery contains the redox process of metal ion pairs, which will increase the integral area of the discharge curve and reduce the integral area of the charging curve, and thus the efficiency of the ZAB will break through the bottleneck of 65%. (iii) Under oxygen-free conditions, the hybrid battery can work independently as a Zn-ion battery without damage to the battery. The utility model overcomes the problem that ZABs cannot work under the harsh high altitude conditions.192,193 To construct hybrid ZABs, it is necessary to consider the variation in the charging and discharging potential range by metal-based catalysts with Faraday redox electron pairs. Generally, metal-based catalysts, such as Co, Cu, Mn, and Ni, can undergo redox reactions in this potential range. The Faraday redox reaction should be driven by the adaptive potential of electron pair conversion, and the ORR/OER occurs while the reaction is proceeding.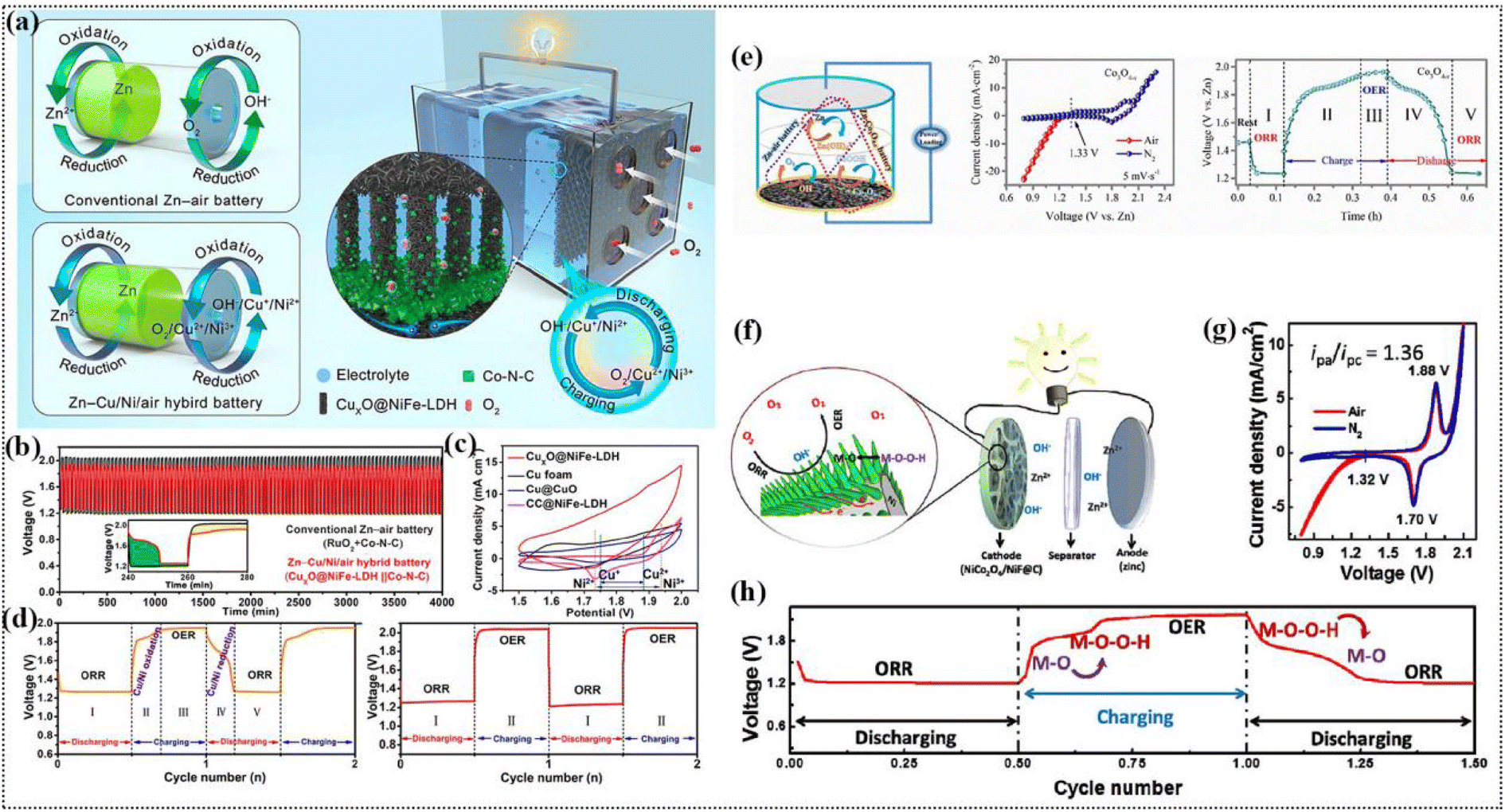 | ||
| Fig. 12 (a) Comparison of electrochemical mechanisms between hybrid battery and conventional ZAB. (b) Galvanostatic charging–discharging plots of Zn–Cu/Ni/air hybrid battery and conventional ZAB at 10 mA cm−2 (40 min per cycle). (c) CV curves of different air-cathode-assembled batteries in air ambient. The initial two cycles of galvanostatic charging–discharging voltage profiles for (d) Zn–Cu/Ni/air hybrid battery and conventional ZAB. Reproduced with permission from ref. 169, Copyright (2022) ACS. (e) Schematic illustration of Zn–Co3O4-x/Zn–air hybrid battery, CV curves of the hybrid battery with Zn plate as anode and Co3O4−x as cathode in air and N2 atmosphere and galvanostatic discharge–charge–discharge profile of the cycling measurement (at 5 mA cm−2, segment (I) 5 min discharge; segment (II) charge to 1.95 V; segment (III) 5 min charge; segment (IV) discharging to 1.25 V; segment (V) 5 min discharge), elaborating the transformation of the battery from a ZAB to a Zn–Co3O4−x/Zn–air hybrid battery. Reproduced with permission from ref. 194, Copyright (2018) ACS. (f) Schematic illustration. (g) CV curves of ZnAMB in ambient air and N2 atmosphere. (h) Voltage profile of the initial charge–discharge of the cycling test (at 5 mA cm−2, 15 min discharge followed by 15 min charge in each cycle), elaborating the transformation of the cell from a ZAB to ZnAMB. Reproduced with permission from ref. 195, Copyright (2017) ACS. | ||
Recently, Zhang et al. constructed a Zn/Cu/Ni/air hybrid battery system based on multi-pair coupling by using the Cu+/Cu2+ electric pair generated by the foam copper collector. The hybrid battery exhibited a charging–discharging mode different from the traditional ZAB. Different from the two processes of the ORR and OER of traditional ZABs, the operation process of the Zn Cu/Ni/air hybrid battery is as follows. Process I is the first discharge of hybrid battery on the 1.27 V voltage platform, mainly originating from the ORR process of the Co–N–C catalyst. Process II is the conversion process from Cu–O–Cu to Cu–O and from Ni–O to Ni–O–O–H, providing a charging voltage platform of 1.8–1.9 V. The 1.95 V charging platform of Process III benefits from the CuxO@NiFe-LDH OER process. Process IV is a reversible process of II, providing a discharge voltage platform of about 1.7 V for the conversion of Cu–O to Cu–O–Cu and Ni–O–O–H to Ni–O. The reversible transformation of Cu species and Ni species synergistically reduces the integral area of the charge curve and increases the integral area of the discharge curve, thus endowing the hybrid battery with ultra-high energy efficiency (Fig. 12b–d). Operando XRD and XPS were used to monitor the structural changes in the hybrid air cathode during charging and discharging. The results showed that the peak intensity ratio of Cu2O and CuO was significantly higher than that in the initial state after the first discharging process, indicating that Cu–O was accompanied by the transformation of Cu–O–Cu. After the first charging process, the peak intensity ratio of Cu2O and CuO decreased significantly, implying the transformation from Cu–O–Cu to Cu–O. In addition, XPS showed that the peak area of Cu+/Ni2+ increased and the peak area of Cu2+/Ni3+ decreased after the first discharge, which further confirmed that Cu2+/Ni3+ was transformed into Cu+/Ni2+ during the discharge process. After the charging process, the peak area of Cu2+/Ni3+ increased and the peak area of Cu+/Ni2+ decreased, which proves that a transformation from Cu+/Ni2+ to Cu2+/Ni3+ occurred during the charging process. The highest charge discharge efficiency of this hybrid battery could reach 79.6%, breaking through the 65% efficiency of traditional ZABs.169
Ma et al. synthesized a cobalt oxide rich in oxygen vacancies by plasma etching and constructed a new type of zinc–cobalt oxide and zinc–air hybrid battery. (Zinc–Co3O4−x) Co3O4 was the cathode material of zinc-ion battery. The Faraday redox reaction of Co–O ↔ Co–O–OH occurred during the charging and discharging processes of battery. The OER and ORR electrocatalytic performance could be effectively improved by introducing oxygen vacancy on the surface, thus improving its electrochemical performance as a cathode material for ZABs. CV showed that the redox peak at the 1.95/2.05 V potential corresponded to the change in the Faraday redox: Co–O ↔ Co–O–OH. The new hybrid zinc-based power battery exhibited a power density of 3200 W kg−1 and an energy density of 1060 W h kg−1, respectively, and it also exhibited good stability after 440 h and 1500 cycles (Fig. 12e). More importantly, the solid-state hybrid battery had good safety, excellent water resistance and washable properties after the gel electrolyte was used to assemble the complete battery. It could still maintain nearly 90% electrochemical performance after 20 h in water or 1 h after washing, which could solve the problem that traditional ZABs cannot work normally under harsh conditions.194
Zn–NiCo2O4 hybrid batteries were constructed by Li et al. The binary transition metal oxide was grown on carbon-coated nickel foam (NiCo2O4/NIF@C). The CV curves measured in an O2 atmosphere showed that the peak ORR/OER reaction of conventional ZABs is in the potential range of less than 1.3 V for the ORR and greater than 1.9 V for the OER, while the appearance of electron pairs at 1.7 V/1.86 V is attributed to changes in the redox reaction between metal–O and metal–O–OH (Fig. 12f and g). The assembled battery assembly displayed that the charge–discharge curve process was ORR-metal redox reaction-OER-metal redox reaction, and the reaction had cyclic reversibility. Compared with the traditional ZAB, the Zn–NiCo2O4 hybrid battery exhibited a higher discharge voltage and a smaller charging–discharging potential difference (Fig. 12h).195 Lee et al. also synthesized an NiO/Ni(OH)2 heterojunction for the assembly of hybrid batteries, which showed the electrochemical performance of Ni–Zn batteries and ZABs. The rapid kinetics of the reactive nickel Faraday redox reaction resulted in a high specific capacity and high discharge voltage platform. The hybrid battery had a high power and energy density, and dynamic charging and discharging from high current to low current proved the practical feasibility of the hybrid battery in electric and hybrid electric vehicles. Under a high current density, the Faraday redox reaction and oxygen reaction simultaneously discharged, which synergistically increased the battery power density.196 At a low current density, the ORR maintained the voltage and increased the energy density of the battery. Hence, the hybrid battery could be charged 10-times faster than it could be discharged without loss in its capacity.
The above-mentioned results show that the redox reaction of the metal species in the electrode occurring in the range of charging and discharging voltage is a necessary condition for the construction of novel Zn–air hybrid batteries. The combination of ZABs and metal-ion batteries can solve the bottleneck of traditional ZABs. In the future, related research on transition metal-based hybrid batteries should be vigorously carried out.
6. Outlook
At present, the self-supporting electrodes of ZABs have developed rapidly, but there are still some challenges that limit their development. The mechanism of metal-support interaction of self-supporting electrodes and catalytic mechanism of catalysts are not well understood. Thus, it is necessary to summarize a reasonable system to explain the reasons for the excellent performance of self-supporting electrodes and provide reference for the subsequent synthesis of high-performance self-supporting electrodes from the perspective of morphology, structure and intermediates in the catalytic process. The main substrates used for self-supporting electrodes are carbon base and foam metal base. In alkaline electrolyte, carbon-based materials corrode, while metal materials significantly reduce the energy density of the battery due to their weight. Therefore, developing new lightweight corrosion-resistant conductive substrate materials is a challenge. The test current density range of zinc–air batteries with self-supporting electrodes is generally below 20 mA cm−2, but in practical application, the standard of current density is generally > 50 mA cm−2, and thus it is necessary to explore self-supporting electrodes with high current density.Self-supporting electrodes have been used as the air-cathodes of ZABs, and their related development is very fast. It is necessary to explore the ORR/OER mechanism with advanced operando techniques and theoretical calculation for the directional synthesis of air-cathodes. In future research, the following problems should be solved for ZABs (Fig. 13).
 | ||
| Fig. 13 Scheme showing an outlook on the development of ZABs. | ||
(i) The rational design and synthesis of highly efficient and stable self-supporting electrodes are very important for the development of ZABs. The self-supporting electrode structure can guarantee the high stability of ZABs. The bifunctional oxygen reaction activity can be enhanced by adjusting the electronic state of the metal centers through heteroatom-doping, bimetal coordination and interface engineering. The growth of zinc dendrites can also be effectively controlled by adjusting the electrode structures and the composition of the electrolyte.
(ii) The in-depth study of the electrocatalytic reaction mechanism is of guiding significance for the directional synthesis of catalysts. Operando characterization can used to reveal the “black box” of the ZAB charging–discharging process and fully understand the electrocatalytic mechanism. It plays a key role in constructing theoretical models based on in situ characterization results and revealing the mechanism of the ORR/OER through DFT calculation.
(iii) Solid-state ZABs have the characteristics of portability, good flexibility, etc., which have great practical application value. The biggest problem of solid-state batteries is the water retention of solid electrolyte and the difficulty of working under extreme conditions. Therefore, much more effective solid electrolytes need to be developed and simultaneously functionalized to improve their water retention and stability.
Based on the practical application demand of ZABs, it is of practical significance to reasonably develop various novel batteries combined with other metal-ion batteries. Zinc–air hybrid batteries combine the ORR/OER and Faraday redox reaction of metal ions, which have the advantages of high discharging voltage, high power density and high efficiency. Importantly, they can overcome the problem that ZABs cannot be applied in harsh environments. Therefore, much more efforts are needed to develop novel hybrid batteries. We believe that the problems existing in ZABs will be gradually solved, which are expected to replace lithium-ion batteries in the future. It is hoped that this review will accelerate the development of ZABs.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors gratefully acknowledge the support of this research by the National Natural Science Foundation of China (U20A20250, 22179034).Notes and references
- I. E. L. Stephens, J. Rossmelsl and I. Chorkendorff, Science, 2016, 354, 1378–1379 CrossRef CAS PubMed.
- F. Duffner, N. Kronemeyer, J. Tübke, J. Leker, M. Winter and R. Schmuch, Nat. Energy, 2021, 6, 123–134 CrossRef CAS.
- G. Zhang, X. Liu, P. Yu, D. Shen, B. Liu, Q. Pan, L. Wang and H. Fu, Chin. Chem. Lett., 2022, 33, 3903–3908 CrossRef CAS.
- N. Shang, K. Wang, M. Wei, Y. Zuo, P. Zhang, H. Wang, Z. Chen and P. Pei, J. Mater. Chem. A, 2022, 10, 16369–16389 RSC.
- C.-X. Zhao, J.-N. Liu, J. Wang, D. Ren, J. Yu, X. Chen, B.-Q. Li and Q. Zhang, Adv. Mater., 2021, 33, 2008606 CrossRef CAS PubMed.
- M. Katsaiti, E. Papadogiannis, V. Dracopoulos, A. Keramidas and P. Lianos, J. Power Sources, 2023, 555, 232384 CrossRef CAS.
- X. Chen, B. Liu, C. Zhong, Z. Liu, J. Liu, L. Ma, Y. Deng, X. Han, T. Wu, W. Hu and J. Lu, Adv. Energy Mater., 2017, 7, 1700779 CrossRef.
- X. Guo, T. Zheng, G. Ji, N. Hu, C. Xu and Y. Zhang, J. Mater. Chem. A, 2018, 6, 10243–10252 RSC.
- H. Liu, F. Yu, K. Wu, G. Xu, C. Wu, H.-K. Liu and S.-X. Dou, Small, 2022, 18, 2106635 CrossRef CAS PubMed.
- Z. Guo, F. Wang, Y. Xia, J. Li, A. G. Tamirat, Y. Liu, L. Wang, Y. Wang and Y. Xia, J. Mater. Chem. A, 2018, 6, 1443–1453 RSC.
- X. Liu, G. Zhang, L. Wang and H. Fu, Small, 2021, 17, 2006766 CrossRef CAS PubMed.
- E. Davari and D. G. Ivey, Sustainable Energy Fuels, 2018, 2, 39–67 RSC.
- Y. Li and H. Dai, Chem. Soc. Rev., 2014, 43, 5257–5275 RSC.
- Y. Zhang, J. Wang, M. Alfred, P. Lv, F. Huang, Y. Cai, H. Qiao and Q. Wei, Energy Storage Mater., 2022, 51, 181–211 CrossRef.
- J. Zhu, M. Xiao, Y. Zhang, Z. Jin, Z. Peng, C. Liu, S. Chen, J. Ge and W. Xing, ACS Catal., 2016, 6, 6335–6342 CrossRef CAS.
- H. Cheng, M.-L. Li, C.-Y. Su, N. Li and Z.-Q. Liu, Adv. Funct. Mater., 2017, 27, 1701833 CrossRef.
- Z.-C. Yao, T. Tang, J.-S. Hu and L.-J. Wan, Energy Fuels, 2021, 35, 6380–6401 CrossRef CAS.
- T. Yang, Y. Chen, Y. Liu, X. Liu and X. Gao, Chin. Chem. Lett., 2022, 33, 2171–2177 CrossRef CAS.
- S. Wang, J. Qin, T. Meng and M. Cao, Nano Energy, 2017, 39, 626–638 CrossRef CAS.
- C.-X. Zhao, J.-N. Liu, J. Wang, D. Ren, B.-Q. Li and Q. Zhang, Chem. Soc. Rev., 2021, 50, 7745–7778 RSC.
- J. Wang, C.-X. Zhao, J.-N. Liu, Y.-W. Song, J.-Q. Huang and B.-Q. Li, Nano Energy, 2022, 104, 107927 CrossRef CAS.
- O. Y. Bisen, A. K. Yadav, B. Pavithra and K. K. Nanda, Chem. Eng. J., 2022, 449, 137705 CrossRef.
- J. Zhu, M. Xiao, G. Li, S. Li, J. Zhang, G. Liu, L. Ma, T. Wu, J. Lu, A. Yu, D. Su, H. Jin, S. Wang and Z. Chen, Adv. Energy Mater., 2019, 10, 1903003 CrossRef.
- C. Du, Y. Gao, J. Wang and W. Chen, J. Mater. Chem. A, 2020, 8, 9981–9990 RSC.
- W. Shang, W. Yu, P. Tan, B. Chen, Z. Wu, H. Xu and M. Ni, J. Mater. Chem. A, 2019, 7, 15564–15574 RSC.
- N. Liu, H. Hua, X. Xua and Q. Wang, J. Chem. Eng. Jpn., 2020, 49, 375–383 Search PubMed.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- Q. Liu, Y. Wang, L. Dai and J. Yao, Adv. Mater., 2016, 28, 3000–3006 CrossRef CAS PubMed.
- Y. Wang, Q. Cao, C. Guan and C. Cheng, Small, 2020, 16, 2002902 CrossRef CAS PubMed.
- Q. Cheng, C. Hu, G. Wang, Z. Zou, H. Yang and L. Dai, J. Am. Chem. Soc., 2020, 142, 5594–5961 CrossRef CAS PubMed.
- Z. Song, X. Han, Y. Deng, N. Zhao, W. Hu and C. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 22694–22703 CrossRef CAS PubMed.
- J. Pan, Y. Y. Xu, H. Yang, Z. Dong, H. Liu and B. Y. Xia, Adv. Sci., 2018, 5, 1700691 CrossRef PubMed.
- Z. Song, Z. Li, Y. Liu, L. Chen, J. Zhang and Z. Zheng, CrystEngComm, 2022, 24, 6980–6986 RSC.
- C. Lai, H. Li, Y. Sheng, M. Zhou, W. Wang, M. Gong, K. Wang and K. Jiang, Adv. Sci., 2022, 9, 2105925 CrossRef CAS PubMed.
- L. Chen, Z. Song, Z. Li, Z. Yang, J. Zhang, H. Li and Z. Zheng, J. Alloys Compd., 2022, 894, 162456 CrossRef CAS.
- Y. Yu and S. Hu, Chin. Chem. Lett., 2021, 32, 3277–3287 CrossRef CAS.
- X. Liu, L. Wang, G. Zhang, F. Sun, G. Xing, C. Tian and H. Fu, Chem. Eng. J., 2021, 414, 127569 CrossRef CAS.
- Y. Zhang, H. Sun, Y. Qiu, X. Ji, T. Ma, F. Gao, Z. Ma, B. Zhang and P. A. Hu, Carbon, 2019, 144, 370–381 CrossRef CAS.
- Q. Lu, X. Zou, K. Liao, R. Ran, W. Zhou, M. Ni and Z. Shao, Carbon Energy, 2020, 2, 461–471 CrossRef CAS.
- A. Sumboja, M. Lübke, Y. Wang, T. An, Y. Zong and Z. Liu, Adv. Energy Mater., 2017, 7, 1700927 CrossRef.
- S. S. Shinde, J.-Y. Yu, J.-W. Song, Y.-H. Nam, D.-H. Kim and J.-H. Lee, Nanoscale Horiz., 2017, 2, 333–341 RSC.
- X. Yan, Y. Ha and R. Wu, Small Methods, 2021, 5, 2000827 CrossRef CAS PubMed.
- H. Ma, C. S. Li, Y. Su and J. Chen, J. Mater. Chem., 2007, 17, 684 RSC.
- J.-F. Drillet, M. Adam, S. Barg, A. Herter, D. Koch, V. M. Schmidt and M. Wilhelm, ECS Trans., 2010, 28, 13–24 CrossRef CAS.
- G. Fu, Y. Chen, Z. Cui, Y. Li, W. Zhou, S. Xin, Y. Tang and J. B. Goodenough, Nano Lett., 2016, 16, 6516–6522 CrossRef CAS PubMed.
- P. Tan, B. Chen, H. Xu, W. Cai, W. He, M. Chen and M. Ni, J. Electrochem. Soc., 2019, 166, A616–A622 CrossRef CAS.
- J. Lee, B. Hwanga, M.-S. Park and K. Kim, Electrochim. Acta, 2016, 199, 164–171 CrossRef CAS.
- J. Hao, X. Li, X. Zeng, D. Li, J. Mao and Z. Guo, Energy Environ. Sci., 2020, 13, 3917–3949 RSC.
- J. Hao, X. Li, S. Zhang, F. Yang, X. Zeng, S. Zhang, G. Bo, C. Wang and Z. Guo, Adv. Funct. Mater., 2020, 30, 2001263 CrossRef CAS.
- M. T. Tsehaye, F. Alloin, C. Iojoiu, R. A. Tufa, D. Aili, P. Fischer and S. Velizarov, J. Power Sources, 2020, 475, 228689 CrossRef CAS.
- H. He and J. Liu, J. Mater. Chem. A, 2020, 8, 22100–22110 RSC.
- P. Liang, Q. Li, L. Chen, Z. Tang, Z. Li, Y. Wang, Y. Tang, C. Han, Z. Lan, C. Zhi and H. Li, J. Mater. Chem. A, 2022, 10, 11971–11979 RSC.
- J. Zheng, Q. Zhao, T. Tang, J. Yin, C. D. Quilty, G. D. Renderos, X. Liu, Y. Deng, L. Wang, D. C. Bock, C. Jaye, D. Zhang, E. S. Takeuchi, K. J. Takeuchi, A. C. Marschilok and L. A. Archer, Science, 2019, 366, 645–648 CrossRef CAS PubMed.
- Z. Yi, J. Liu, S. Tan, Z. Sang, J. Mao, L. Yin, X. Liu, L. Wang, F. Hou, S. X. Dou, H.-M. Cheng and J. Liang, Adv. Mater., 2022, 34, 2203835 CrossRef CAS PubMed.
- K. Zhang, Z. Yan and J. Chen, Joule, 2020, 4, 10–20 CrossRef CAS.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- M.-C. Huang, S.-H. Huang, S.-C. Chiu, K.-L. Hsueh, W.-S. Chang, C.-C. Yang, C.-C. Wu and J.-C. Lin, J. Chin. Chem. Soc., 2018, 65, 1239–1244 CrossRef CAS.
- K. Qiu, D. Trudgeon, X. Li, V. Yufit, B. Chakrabarti, N. Brandon and A. Shah, Batteries, 2022, 8, 106 CrossRef CAS.
- W. Shang, W. Yu, Y. Liu, R. Li, Y. Dai, C. Cheng, P. Tan and M. Ni, Energy Storage Mater., 2020, 31, 44–57 CrossRef.
- M.-H. Lin, C.-J. Huang, P.-H. Cheng, J.-H. Cheng and C.-C. Wang, J. Mater. Chem. A, 2020, 8, 20637–20649 RSC.
- Y.-J. Kim and K.-S. Ryu, Appl. Surf. Sci., 2019, 480, 912–922 CrossRef CAS.
- T. Nagy, L. Nagy, Z. Erdélyi, E. Baradács, G. Deák, M. Zsuga and S. Kéki, Batteries, 2022, 8, 212 CrossRef.
- Y. Peng, C. Lai, M. Zhang, X. Liu, Y. Yin, Y. Li and Z. Wu, J. Power Sources, 2022, 526, 231173 CrossRef CAS.
- Y. Zeng, P. X. Sun, Z. Pei, Q. Jin, X. Zhang, L. Yu and X. W. Lou, Adv. Mater., 2022, 34, 2200342 CrossRef CAS PubMed.
- H.-L. Kim, E.-J. Kim, S.-J. Kim and H.-C. Shin, J. Appl. Electrochem., 2015, 45, 335–342 CrossRef CAS.
- S. Jin, P.-Y. Chen, Y. Qiu, Z. Zhang, S. Hong, Y. L. Joo, R. Yang and L. A. Archer, J. Am. Chem. Soc., 2022, 144, 19344–19352 CrossRef CAS PubMed.
- M. Shimizu, K. Hiraharaa and S. Arai, Phys. Chem. Chem. Phys., 2019, 21, 7045–7052 RSC.
- H. Yang, Y. Cao, X. Ai and L. Xiao, J. Power Sources, 2004, 128, 97–101 CrossRef CAS.
- K. Liu, P. He, H. Bai, J. Chen, F. Dong, S. Wang, M. He and S. Yuan, Mater. Chem. Phys., 2017, 199, 73–78 CrossRef CAS.
- W. Sun, M. Ma, M. Zhu, K. Xu, T. Xu, Y. Zhu and Y. Qian, Small, 2022, 18, 2106604 CrossRef CAS PubMed.
- Y. Zhang, Y. Wu, H. Ding, Y. Yan, Z. Zhou, Y. Ding and N. Liu, Nano Energy, 2018, 53, 666–674 CrossRef CAS.
- J. F. Parker, C. N. Chervin, E. S. Nelson, D. R. Rolison and J. W. Long, Energy Environ. Sci., 2014, 7, 1117–1124 RSC.
- Z. Zhao, X. Fan, J. Ding, W. Hu, C. Zhong and J. Lu, ACS Energy Lett., 2019, 4, 2259–2270 CrossRef CAS.
- M. Naja, N. Penazzi, G. Farnia and G. Sandonà, Electrochim. Acta, 1993, 38, 1453–1459 CrossRef.
- S. Lysgaard, M. K. Christensen, H. A. Hansen, J. M. G. Lastra, P. Norby and T. Vegge, ChemSusChem, 2018, 11, 1933–1941 CrossRef CAS PubMed.
- K. Wongrujipairoj, L. Poolnapol, A. Arpornwichanop, S. Suren and S. Kheawhom, Phys. Status Solidi B, 2017, 254, 1600442 CrossRef.
- Y. N. Jo, S. H. Kang, K. Prasanna, S. W. Eom and C. W. Lee, Appl. Surf. Sci., 2017, 422, 406–412 CrossRef CAS.
- M. A. Deyab and G. mele, J. Power Sources, 2019, 443, 227264 CrossRef CAS.
- X. Xu, S. Ci, Y. Ding, G. Wang and Z. Wen, J. Mater. Chem. A, 2019, 7, 8006–8029 RSC.
- S. Jiang, J. Li, J. Fang and X. Wang, Small, 2019, 17, 1903760 CrossRef PubMed.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- Y. Xiao, J. Hong, X. Wang, T. Chen, T. Hyeon and W. Xu, J. Am. Chem. Soc., 2020, 142, 13201–13209 CrossRef CAS PubMed.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997–4008 CrossRef PubMed.
- J. Song, C. Wei, Z.-F. Huang, C. Liu, L. Zeng, X. Wang and Z. J. Xu, Chem. Soc. Rev., 2020, 49, 2196–2214 RSC.
- J. Yu, B.-Q. Li, C.-X. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241–11280 RSC.
- Y. Niu, X. Teng, S. Gong and Z. Chen, J. Mater. Chem. A, 2020, 8, 13725–13734 RSC.
- A. Pendashteh, J. S. Sanchez, J. Palma, M. Anderson and R. Marcilla, Energy Storage Mater., 2019, 20, 216–224 CrossRef.
- J. Yang, W. Liu, M. Xu, X. Liu, H. Qi, L. Zhang, X. Yang, S. Niu, D. Zhou, Y. Liu, Y. Su, J.-F. Li, Z.-Q. Tian, W. Zhou, A. Wang and T. Zhang, J. Am. Chem. Soc., 2021, 143(36), 14530–14539 CrossRef CAS PubMed.
- C. Lai, H. Li, Y. Sheng, M. Zhou, W. Wang, M. Gong, K. Wang and K. Jiang, Adv. Sci., 2022, 9, 2105925 CrossRef CAS PubMed.
- C. Tang, H. F. Wang and Q. Zhang, Acc. Chem. Res., 2018, 51, 881–889 CrossRef CAS PubMed.
- H. Xu, J. Yang, R. Ge, J. Zhang, Y. Li, M. Zhu, L. Dai, S. Li and W. Li, J. Energy Chem., 2022, 71, 234–265 CrossRef CAS.
- Y. Wang, X. Xu, J. Chen and Q. Wang, J. Power Sources, 2019, 430, 201–209 CrossRef CAS.
- J. Yu, B.-Q. Li, C.-C. Zhao, J.-N. Liu and Q. Zhang, Adv. Mater., 2020, 32, 1908488 CrossRef CAS PubMed.
- J.-N. Liu, C.-X. Zhao, D. Ren, J. Wang, R. Zhang, S.-H. Wang, C. Zhao, B.-Q. Li and Q. Zhang, Adv. Mater., 2022, 34, 2109407 CrossRef CAS PubMed.
- P. Wang, L. Wan, Y. Lin and B. Wang, Electrochim. Acta, 2019, 320, 134564 CrossRef CAS.
- M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433–11456 CrossRef CAS PubMed.
- G. Bieker, M. Winter and P. Bieker, Phys. Chem. Chem. Phys., 2015, 17, 8670–8679 RSC.
- W. Sun, F. Wang, B. Zhang, M. Zhang, V. Küpers, X. Ji, C. Theile, P. Bieker, K. Xu, C. Wang and M. Winter, Science, 2021, 371, 46–51 CrossRef CAS PubMed.
- W. Sun, V. Küpers, F. Wang, P. Bieker and M. Winter, Angew. Chem., Int. Ed., 2022, 61, e202207353 CAS.
- J. Fu, J. Zhang, X. Song, H. Zarrin, X. Tian, J. Qiao, L. Rasen, K. Li and Z. Chen, Energy Environ. Sci., 2016, 9, 663–670 RSC.
- Y. T. Wei, Y. C. Shi, Y. Chen, C. H. Xiao and S. J. Ding, J. Mater. Chem. A, 2021, 9, 4415–4453 RSC.
- X. Yang, X. Zheng, H. Li, B. Luo, Y. He, Y. Yao, H. Zhou, Z. Yan, Y. Kuang and Z. Huang, Adv. Funct. Mater., 2022, 32, 2200397 CrossRef CAS.
- S. Chen, L. Zhao, J. Ma, Y. Wang, L. Dai and J. Zhang, Nano energy, 2019, 60, 536–544 CrossRef CAS.
- X. F. Zhang, X. F. Ma and T. Hou, Angew. Chem., Int. Ed., 2019, 58, 7366–7370 CrossRef CAS PubMed.
- Q. Wang, Q. Feng, Y. Lei, S. Tang, L. Xu, Y. Xiong, G. Fang, Y. Wang, P. Yang, J. Liu, W. Liu and X. Xiong, Nat. Commun., 2022, 13, 3689 CrossRef CAS PubMed.
- K. Tang, J. Fu, M. Wu, T. Hua, J. Liu, L. Song and H. Hu, Small Methods, 2022, 6, 2101276 CrossRef CAS PubMed.
- C. Gu, X.-Q. Xie, Y. Liang, J. Li, H. Wang, K. Wang, J. Liu, M. Wang, Y. Zhang, M. Li, H. Kong and C.-S. Liu, Energy Environ. Sci., 2021, 14, 4451–4462 RSC.
- H.-J. Li, J.-M. Lim, H.-W. Kim, S.-H. Jeong, S.-W. Eom, Y. T. Hong and S.-Y. Li, J. Membr. Sci., 2016, 499, 526–537 CrossRef.
- C. C. Yang, J. M. Yang and C. Y. Wu, J. Power Sources, 2009, 191, 669–677 CrossRef CAS.
- H.-J. Lee, J.-M. Lim, H.-W. Kim, S.-H. Jeong, S.-W. Eom, Y. T. Hong and S.-Y. Lee, J. Membr. Sci., 2016, 499, 526–537 CrossRef CAS.
- H.-W. Kim, J.-M. Lim, H.-J. Lee, S.-W. Eom, Y. T. Hong and S.-Y. Lee, J. Mater. Chem. A, 2016, 4, 3711–3720 RSC.
- Y. Song, X. Zhang, X. Cui and J. Shi, J. Catal., 2019, 372, 174–181 CrossRef CAS.
- C. Han and Z. Chen, Appl. Surf. Sci., 2020, 511, 145382 CrossRef CAS.
- C.-C. Hou, L. Zou, Y. Wang and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 21360–21366 CrossRef CAS PubMed.
- Y. Zhong, Z. Pan, X. Wang, J. Yang, Y. Qiu, S. Xu, Y. Lu, Q. Huang and W. Li, Adv. Sci., 2019, 6, 1802243 CrossRef PubMed.
- J.-C. Li, Y. Meng, L. Zhang, G. Li, Z. Shi, P.-X. Hou, C. Liu, H.-M. Cheng and M. Shao, Adv. Funct. Mater., 2021, 31, 2103360 CrossRef CAS.
- Z. Li, J. Yang, X. Ge, Y.-P. Deng, G. Jiang, H. Li, G. Sun, W. Liu, Y. Zheng, H. Dou, H. Jiao, J. Zhu, N. Li, Y. Hu, M. Feng and Z. Chen, Nano Energy, 2021, 89, 106314 CrossRef CAS.
- T. Wang, Z. Kou, S. Mu, J. Liu, D. He, I. Saana Amiinu, W. Meng, K. Zhou, Z. Luo, S. Chaemchuen and F. Verpoort, Adv. Funct. Mater., 2017, 28, 1705048 CrossRef.
- Y. Qiao, P. Yuan, Y. Hu, J. Zhang, S. Mu, J. Zhou, H. Li, H. Xia, J. He and Q. Xu, Adv. Mater., 2018, 28, 1804504 CrossRef PubMed.
- X. Zhang, K. Wan, P. Subramanian, J. Luo and J. Fransaer, J. Mater. Chem. A, 2020, 8, 7569–7587 RSC.
- Y. Wang, W. Qiu, E. Song, F. Gu, Z. Zheng, X. Zhao, Y. Zhao, J. Liu and W. Zhang, Natl. Sci. Rev., 2018, 5, 327–341 CrossRef CAS.
- Q. Liu, L. Wang, X. Liu, P. Yu, C. Tian and H. Fu, Sci. China Mater., 2019, 62, 624–632 CrossRef CAS.
- P. Yu, L. Wang, F. Sun, Y. Xie, X. Liu, J. Ma, X. Wang, C. Tian, J. Li and H. Fu, Adv. Mater., 2019, 31, 1901666 CrossRef PubMed.
- K. Marcus, K. Liang, W. Niu and Y. Yang, J. Phys. Chem. Lett., 2018, 9, 2746–2750 CrossRef CAS PubMed.
- Y. Wang, Z. Li, P. Zhang, Y. Pan, Y. Zhang, Q. Cai, S. R. P. Silva, J. Liu, G. Zhang, X. Sun and Z. Yan, Nano energy, 2021, 87, 106147 CrossRef CAS.
- G.-P. Kim, H.-H. Sun and A. Manthiram, Nano energy, 2016, 30, 130–137 CrossRef CAS.
- L. Yu, H. Zhou, J. Sun, F. Qin, F. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2017, 10, 1820–1827 RSC.
- L. Wan, Z. Zhao, X. Chen, P.-F. Liu, P. Wang, Z. Xu, Y. Lin and B. Wang, ACS Sustainable Chem. Eng., 2020, 8, 11079–11087 CrossRef CAS.
- Q. Liu, X. Liu, Y. Xie, F. Sun, Z. Liang, L. Wang and H. Fu, J. Mater. Chem. A, 2020, 8, 21189–21198 RSC.
- F. S. Farahani, M. S. Rahmanifar, A. Noori, M. F. El-Kady, N. Hassani, M. N-Amal, R. B. Kaner and M. F. Mousavi, J. Am. Chem. Soc., 2022, 144, 3411–3428 CrossRef PubMed.
- D.-D. Sun, J.-J. Han, H. Ma, N. Zhang, T. Han and J.-N. Cheng, Ionics, 2019, 25, 3761–3768 CrossRef CAS.
- F. Meng, H. Zhong, D. Bao, J. Yan and X. Zhang, J. Am. Chem. Soc., 2016, 138, 10226–10231 CrossRef CAS PubMed.
- G. Xing, M. Tong, P. Yu, L. Wang, G. Zhang, C. Tian and H. Fu, Angew. Chem., Int. Ed., 2022, 61, e202211098 CAS.
- Z. Z. C. Feng, C. Liu, M. Zuo, L. Qin, X. Yan, Y. Xing, H. Li, R. Si, S. Zhou and J. Zeng, Nat. Commun., 2020, 11, 1215 CrossRef PubMed.
- A. Valizadeh and S. M. Farkhani, IET Nanobiotechnol., 2014, 8, 83–92 CrossRef CAS PubMed.
- I. Shepa, E. Mudra and J. Dusza, Mater. Today Chem., 2021, 21, 100543 CrossRef CAS.
- M. Tebyetekerwa and S. Ramakrishna, Matter, 2020, 2, 279–283 CrossRef CAS.
- M. Shneider, X. M. Sui, I. Greenfeld and H. D. Wagner, Polymer, 2021, 235, 124307 CrossRef CAS.
- M. Lech, J. M. Popławska and J. Laska, Materials, 2021, 14, 2459 CrossRef CAS PubMed.
- C.-L. Zhang and S.-H. Yu, Mater. Horiz., 2016, 3, 266–269 RSC.
- X. Chen, J. Pu, X. Hu, Y. Yao, Y. Dou, J. Jiang and W. Zhang, Small, 2022, 18, 2200578 CrossRef CAS PubMed.
- C. Xia, L. Huang, D. Yan, A. I. Douka, W. Guo, K. Qi and B. Y. Xia, Adv. Funct. Mater., 2021, 31, 2105021 CrossRef CAS.
- X. Wang, Y. Li, T. Jin, J. Meng, L. Jiao, M. Zhu and J. Chen, Nano Lett., 2017, 17, 7989–7994 CrossRef CAS PubMed.
- C. Li, M. Wu and R. Liu, Appl. Catal., B, 2019, 244, 150–158 CrossRef CAS.
- K. R. Yoon, J. Choi, S.-H. Cho, J.-W. Jung, C. Kim and J. Y. Cheong, J. Power Sources, 2018, 380, 174–184 CrossRef CAS.
- D. Ji, L. Fan, L. Li, S. Peng, D. Yu, J. Song, S. Ramakrishna and S. Guo, Adv. Mater., 2019, 31, 1808267 CrossRef PubMed.
- L. Yang, X. Zhang, L. Yu, J. Hou, Z. Zhou and R. Lv, Adv. Mater., 2022, 34, 2105410 CrossRef CAS PubMed.
- A. Sumboja, J. Chen, Y. Zong, P. S. Lee and Z. Liu, Nanoscale, 2017, 9, 774–780 RSC.
- A. Pendashteh, J. Palma, M. Anderson, J. J. Vilatela and R. Marcilla, ACS Appl. Energy Mater., 2018, 1, 2434–2439 CrossRef CAS.
- Z. Zhu, J. Zhang, X. Peng, Y. Liu, T. Cen, Z. Ye and D. Yuan, Energy Fuels, 2021, 35, 4550–4558 CrossRef CAS.
- J. Song, Y. Chen, H. Huang, J. Wang, S.-C. Huang, Y.-F. Liao, A. E. Fetohi, F. Hu, H.-Y. Chen, L. Li, X. Han, K. M. El-Khatib and S. Peng, Adv. Sci., 2022, 9, 2104522 CrossRef CAS PubMed.
- J. Yin, Y. Li, F. Lv, Q. Fan, Y.-Q. Zhao, Q. Zhang, W. Wang, F. Cheng, P. Xi and S. Guo, ACS Nano, 2017, 11, 2275–2283 CrossRef CAS PubMed.
- W. Liu, L. Yu, R. Yin, X. Xu, J. Feng, X. Jiang, D. Zheng, X. Gao, X. Gao, W. Que, P. Ruan, F. Wu, W. Shi and X. Cao, Small, 2020, 16, 1906775 CrossRef CAS PubMed.
- K. Kordek, L. Jiang, K. Fan, Z. Zhu, L. Xu, M. Al- Mamun, Y. Dou, S. Chen, P. Liu, H. Yin, P. Rutkowski and H. Zhao, Adv. Energy Mater., 2019, 9, 1802936 CrossRef.
- L. Ma, S. Chen, H. Li, Z. Ruan, Z. Tang, Z. Liu, Z. Wang, Y. Huang, Z. Pei, J. A. Zapien and C. Zhi, Energy Environ. Sci., 2018, 11, 2521–2530 RSC.
- Z. Liu, Z. Zhao, Y. Wang, S. Dou, D. Yan, D. Liu, Z. Xia and S. Wang, Adv. Mater., 2017, 29, 1606207 CrossRef PubMed.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- M. Fan, J. Cui, J. Wu, R. Vajtai, D. Sun and P. M. Ajayan, Small, 2020, 16, 1906782 CrossRef CAS PubMed.
- Q. Yang, Z. Xiao, D. Kong, T. Zhang, X. Duan, S. Zhou, Y. Niu, Y. Shen, H. Sun, W. Shaobin and Z. Linjie, Nano Energy, 2019, 66, 104096 CrossRef CAS.
- K. Ren, Z. Liu, T. Wei and Z. Fan, Nano-Micro Lett., 2021, 13, 129 CrossRef CAS PubMed.
- W. Li, D. Wang, Y. Zhang, L. Tao, T. Wang, Y. Zou, Y. Wang, R. Chen and S. Wang, Adv. Mater., 2020, 32, 1907879 CrossRef CAS PubMed.
- S. Li, L. Liu, Y. Wang, Y. Lu and X. Yan, Chin. Chem. Lett., 2017, 28, 2159–2163 CrossRef CAS.
- L.-A. Stern, L. Feng, F. Song and X. Hu, Energy Environ. Sci., 2015, 8, 2347–2351 RSC.
- T. Wu, S. Sun, J. Song, S. Xi, Y. Du, B. Chen, W. A. Sasangka, H. Liao, C. L. Gan, G. G. Scherer, L. Zeng, H. Wang, H. Li, A. Grimaud and Z. J. Xu, Nat. Catal., 2019, 2, 763–772 CrossRef CAS.
- Z. Wu, Y. Yu, G. Zhang, Y. Zhang, R. Guo, L. Li, Y. Zhao, Z. Wang, Y. Shen and G. Shao, Adv. Sci., 2022, 9, 2200614 CrossRef CAS PubMed.
- D. Stock, S. Dongmo, K. Miyazaki, T. Abe, J. Janek and D. Schröder, J. Power Sources, 2018, 395, 195–204 CrossRef CAS.
- W. Shang, H. Wang, W. Yu, Y. He, Y. Ma, R. Li, Z. Wu and P. Tan, Cell Rep. Phys. Sci., 2022, 3, 100904 CrossRef CAS.
- G. Zhang, X. Liu, L. Wang, G. Xing, C. Tian and H. Fu, ACS Nano, 2022, 16, 17139–17148 CrossRef CAS PubMed.
- W. Li, L. Wu, X. Wu, C. Shi, Y. Li, L. Zhang, H. Mi, Q. Zhang, C. He and X. Ren, Appl. Catal., B, 2022, 303, 120849 CrossRef CAS.
- K. Ding, J. Hu, J. Luo, W. Jin, L. Zhao, L. Zheng, W. Yan, B. Weng, H. Hou and X. Ji, Nano energy, 2022, 91, 106675 CrossRef CAS.
- Y. Liu, Z. Chen, N. Zhao, G. Tong, Z. Li, B. Wang, Y. Du, Q. Pan, Z. Li, Y. Xie and Y. Yang, Chem. Eng. J., 2022, 433, 134469 CrossRef CAS.
- S. Ding, L. He, L. Fang, Y. Zhu, T. Li, Z. Lyu, D. Du, Y. Lin and J.-C. Li, Adv. Energy Mater., 2022, 32, 2202984 CrossRef.
- Y.-J. Wu, J. Yang, T.-X. Tu, W.-Q. Li, P.-F. Zhang, Y. Zhou, J.-F. Li, J.-T. Li and S.-G. Sun, Angew. Chem., Int. Ed., 2021, 60, 26829–26836 CrossRef CAS PubMed.
- N. Ishiguro and M. Tada, Catal. Lett., 2018, 148, 1597–1609 CrossRef CAS.
- S. Kunze, P. Grosse, M. B. Lopez, I. Sinev, I. Zegkinoglou, H. Mistry, J. Timoshenko, M. Y. Hu, J. Zhao, E. E. Alp, S. W. Chee and B. R. Cuenya, Angew. Chem., Int. Ed., 2020, 59, 22667–22674 CrossRef CAS PubMed.
- W. Peng, A. Deshmukh, N. Chen, Z. Lv, S. Zhao, J. Li, B. Yan, X. Gao, L. Shang, Y. Gong, L. Wu, M. Chen, T. Zhang and H. Gou, ACS Catal., 2022, 12, 3743–3751 CrossRef CAS.
- X. Liu, L. Wang, P. Yu, C. Tian, F. Sun, J. Ma, W. Li and H. Fu, Angew. Chem., Int. Ed., 2018, 57, 16166–16170 CrossRef CAS PubMed.
- Y.-P. Deng, Y. Jiang, R. Liang, S.-J. Zhang, D. Luo, Y. Hu, X. Wang, J.-T. Li, A. Yu and Z. Chen, Nat. Commun., 2020, 11, 1952 CrossRef CAS PubMed.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2014, 16, 13800–13806 RSC.
- Y. Tian, L. Xu, M. Li, D. Yuan, X. Liu, J. Qian, Y. Dou, J. Qiu and S. Zhang, Nano-Micro Lett., 2020, 13, 3 CrossRef PubMed.
- L. Lv, C. Wang, F. Hu, W. Zhang, L. Cai and W. Yan, ACS Catal., 2022, 12, 4318–4326 CrossRef.
- W. Wan, X. Duan and Y. Huang, Adv. Energy Mater., 2020, 10, 1903815 CrossRef.
- M. Tong, P. Yu, Y. Xie, L. Wang, Y. Wang and H. Fu, Small, 2022, 18, 2201255 CrossRef CAS PubMed.
- H. Liu, Y. Liu, S. Mehdi, X. Wu, T. Liu, B. Zhou, P. Zhang, J. Jiang and B. Li, Adv. Sci., 2021, 8, 2101314 CrossRef CAS PubMed.
- Z. Xu, J. Zhu, J. Shao, Y. Xia, J. Tseng, C. Jiao, G. Ren, P. Liu, G. Li, R. Chen, S. Chen, F. Huang and H.-L. Wang, Energy Storage Mater., 2022, 47, 365–375 CrossRef.
- M. Tong, F. Sun, Y. Xie, Y. Wang, Y. Yang, C. Tian, L. Wang and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 14005–14012 CrossRef CAS PubMed.
- X. Shu, Q. Chen, M. Yang, M. Liu, J. Ma and J. Zhang, Adv. Energy Mater., 2022, 2202871 Search PubMed.
- H. Huang, D. Yu, F. Hu, S.-C. Huang, J. Song, H.-Y. Chen, L. L. Li and S. Peng, Angew. Chem., Int. Ed., 2022, 61, e202116068 CAS.
- M. Xu, D. G. Ivey, Z. Xie and W. Qu, J. Power Sources, 2015, 283, 358–371 CrossRef CAS.
- X. Chen, Z. Zhou, H. E. Karahan, Q. Shao, L. Wei and Y. Chen, Small, 2018, 14, 1801929 CrossRef PubMed.
- T. Zhang, S. Zhang, S. Cao, Q. Yao and J. Y. Lee, Energy Storage Mater., 2020, 33, 181–187 CrossRef.
- P. Tan, B. Chen, H. Xu, W. Cai, W. He, H. Zhang, M. Liu, Z. Shao and M. Ni, ACS Appl. Mater. Interfaces, 2018, 10, 36873–36881 CrossRef CAS PubMed.
- L. Ma, S. Chen, Z. Pei, H. Li, Z. Wang, Z. Liu, Z. Tang, J. A. Zapien and C. Zhi, ACS Nano, 2018, 12, 8597–8605 CrossRef CAS PubMed.
- B. Li, J. Quan, A. Loh, J. Chai, Y. Chen, C. Tan, X. Ge, T. S. A. Hor, Z. Liu, H. Zhang and Y. Zong, Nano Lett., 2017, 17, 156–163 CrossRef CAS PubMed.
- D. U. Lee, J. Fu, M. G. Park, H. Liu, A. G. Kashkooli and Z. Chen, Nano Lett., 2016, 16, 1794–1802 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |