
Prediction of zwitterion hydration and ion association properties using machine learning†
Daniel
Christiansen
,
Gang
Cheng
 and
Shafigh
Mehraeen
*
and
Shafigh
Mehraeen
*
Department of Chemical Engineering, University of Illinois at Chicago, 929 West Taylor Street, Chicago, Illinois 60607, USA. E-mail: tranzabi@uic.edu
First published on 12th April 2023
Abstract
Molecular dynamics simulations were performed to study the hydration and ion association properties of a library of zwitterionic molecules with varying charged moieties and spacer chemistries in pure water and with Na+ and Cl− ions. The structure and dynamics of associations were calculated using the radial distribution and residence time correlation function. Resulting association properties are used as target variables for a machine learning model, with cheminformatic descriptors of the molecule subunits used as descriptors. Prediction of hydration properties revealed that steric and hydrogen bonding descriptors were of greatest importance and there was influence from the cationic moiety on the anionic moiety hydration properties. Ion association properties prediction performed poorly, which is attributed to the role of hydration layers in ion association dynamics. This study is the first to quantitatively describe the influence of subunit chemistry on hydration and ion association properties of zwitterions. These quantitative descriptions supplement prior studies of zwitterion association and previously described design principles.
Introduction
In recent years, zwitterions and zwitterionic polymers have become a frequently studied class of materials due to their strong antifouling,1,2 ion dissociation enhancement,3–6 and tunable properties.7–10 These characteristics arise from the electrostatic properties of the zwitterion's dual-charged moieties, enabling the molecules to form tight, highly ordered water layers,10 and simultaneously push or pull ions in solution.3Careful design of the zwitterion can enable it to become super-antifouling, resisting nonspecific protein adsorption and biofilm formation indefinitely,7 and restrict undesired ion motion,3 enabling more mobile counterions to move freely, enhancing ionic conductivity of the system. These properties have considerable market applications, ranging from coatings for implantable biomedical devices11–13 to materials for batteries.6,14,15 With such important potential applications, it is imperative that structure-design principles are found to enable the design of next-generation materials with improved performance.
Current zwitterion design principles have primarily been made from experimental3,6 and simulation10 studies by modifying the molecular chemistry or polymer structure, as well as changing system conditions, then observing the response in properties such as hydration,16–18 ionic conductivity,6 and mechanical strength,19,20 to name a few.
Among studies of zwitterion hydration and ion association structure–property relationships, Shao et al. used molecular dynamics (MD) simulations to thoroughly explore how modifying the cationic, anionic, and nonionic zwitterion subunits will affect hydration and ion association properties.10,17,18,21,22 In their works, they suggested the partial charge of the ionic moieties was a principal factor affecting the moieties ability to associate with water or ions. These studies laid a foundation for understanding the atomic-scale interactions, however their work lacked quantitative evidence of how features of the zwitterionic moieties, such as charge density, affected the properties of interest.
Interest has considerably grown in applying machine learning (ML) and other data science techniques to produce design principles and quantitative structure–property relationships of materials.23,24 In this approach, the collected properties are used as target variables and predictors typically consist of molecular cheminformatics or fingerprints. Presently, there have been few applications of ML for the prediction or understanding of zwitterionic materials properties due to limited quantity and lack of uniformity of existing data.25–28
Here, we present a ML study of the structure–property relationships of zwitterion hydration and ion association. A library of common zwitterion chemistries is generated using MD simulations to provide the training and test datasets of hydration and ion association properties, which can be predicted by cheminformatic descriptors to produce quantitative and understandable design principles.
Methods
Library of zwitterion chemistries
We consider a library of zwitterion chemistries, which consists of molecular subunits that frequently appear in the literature, as shown in Fig. 1. These subunits belong to one of three categories: anionic, cationic, and nonionic. A zwitterion must have anionic and cationic moieties, and some contain a nonionic intermediate separating these charged groups. It has been shown that the presence of an intermediate (spacer) group can considerably affect the hydration22,29 and ion association22 properties of zwitterions, though it can also lead to their self-association, which can weaken both properties.30 Methylene is a predominant spacer chemistry; however, methyl and hydroxyl functional groups are sometimes included to provide additional properties, such as mechanical stability,20 so these groups were also included for consideration. The quantity of spacer groups, also called the carbon spacer length (CSL), has been shown to control the strength of associations between the charged groups and water or ions, though the effect of the spacer groups diminished as the CSL increased to 3 and beyond.22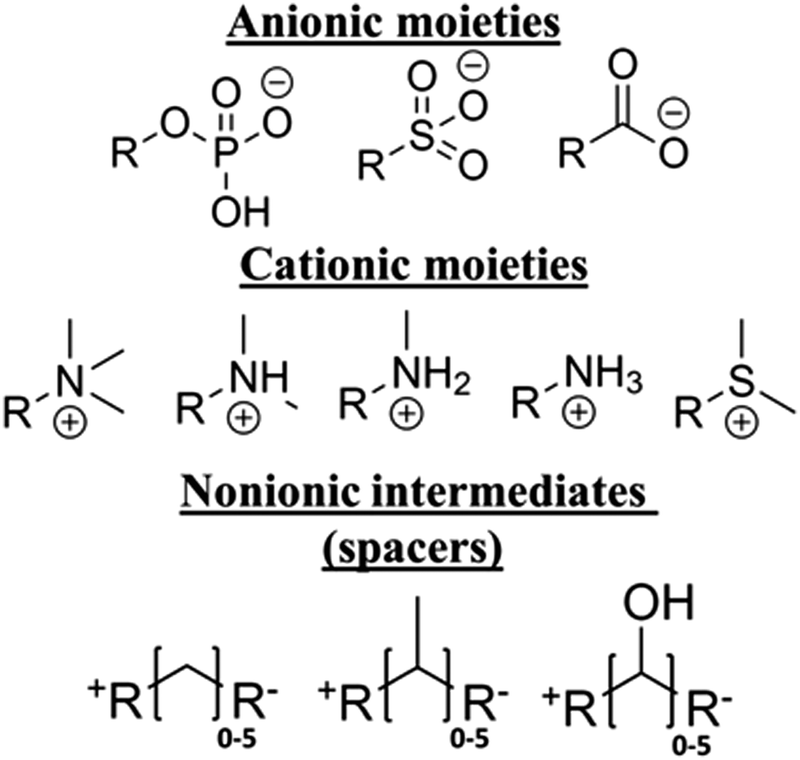 | ||
| Fig. 1 Library of subunits used to construct zwitterionic molecules, each having one cationic moiety, one anionic moiety, and between 0 and 5 nonionic intermediates. | ||
With these observations in mind, the library of zwitterion subunit chemistries was arranged to produce 240 unique molecules, each having one of the three anionic moieties, one of the five cationic moieties, and either no spacer or one of the spacer groups with CSL between 1 and 5. For example, a molecule containing carboxyl anion, trimethylamine cation, and a methylene spacer with CSL equal to 2 would have the following chemical formula, N(CH)3(CH2)2CO2. Despite the previous observation showing a maximum practical CSL of 3,22 the maximum here is 5 to account for the unseen properties, which may have greater sensitivity to the CSL.
Computational simulations
To model the atomic-scale hydration and ion association to the individual atoms of the charged moieties of the zwitterions, MD simulations were performed in GROMACS.31 The All-Atom Optimized Potentials for Liquid Simulations (OPLS-AA) forcefield32 was chosen to describe the intra- and interatomic potentials between atoms during simulation, due to its historic quality in simulating small organic molecules.Molecular topologies
LigParGen33 was used to produce a molecular structure and topology file describing the atoms, bonds, angles, etc. that is compatible with OPLS-AA. This software uses SMILES strings as input, so the subunit chemistries were translated into SMILES, then assembled in the above description to create the molecular SMILES. When executing LigPargen, the molecules were not structurally optimized, and the 1.14*CM1A-LBCC34 charge model was used. Further structural optimization and charge calculation are described in the following section. Table S1 (ESI†) presents the 240 molecular SMILES strings used for input to LigParGen.Quantum chemical simulations
Shao et al.22 previously demonstrated the importance of an accurate charge model when studying the hydration of zwitterions. Therefore, we applied additional structure optimization and partial charge calculation to the structures output from LigParGen. To do so, quantum chemical simulations were performed in Gaussian1635 at the B3LYP level with the 6-31G(d,p) basis set for optimization, and the CHELP algorithm36 was used to assign atomic partial charges from electrostatic potentials. The optimized structure and corrected charges replaced those produced by LigParGen. All other interactions remained the same. A sample structure and topology are included in Tables S2 and S3 (ESI†), respectively.Molecular dynamics simulations
Using the molecule structure and topology files created as mentioned above, we performed MD simulations to model the motion of zwitterions, waters, and ions. Next, we will present the overall workflow used for simulations. Specific simulation parameters can be found in Tables S4–S6 (ESI†). Each zwitterion is simulated with or without 0.2 M salt concentration and the workflow is repeated 5 times.For each zwitterion, a simulation box is generated with edge-length of 4 nm, containing 3 of the zwitterion molecules separated by at least 1.2 nm (see NPT/NVT simulation parameters in Tables S5 and S6, ESI†). Extended simple point charge (SPC/E) water model37 is used to solvate the system. Following solvation, energy minimization (see parameters in Table S4, ESI†) is performed to relax the system, before a 500 ps NPT equilibration step at 298 K and 1 bar (Table S5, ESI†). A stability analysis of the molecules was determined by root mean-squared displacement (RMSD) of the zwitterions during NVT simulation to ensure the length of NVT simulation is sufficient for the molecules to be stable. Samples of RMSD for five selected zwitterions are shown in Fig. S1 (ESI†). Before production simulation, half of the trials have 0.2 M Na+ and Cl− ions inserted by randomly replacing water molecules. Ion topologies are described by Aqvist's parameters.38 Finally, 10 ns NVT simulations (see parameters in Table S6, ESI†) are used for data-collection.
Target properties
To examine the structure and dynamics of hydration and ion association to the charged moieties of the zwitterions, the radial distribution function (RDF) and residence time correlation function,![[C with combining macron]](https://www.rsc.org/images/entities/i_char_0043_0304.gif) (t), of water and ions (Na+ and Cl−) with respect to the position of the moieties are calculated during NVT simulations. From the RDF and residence time correlation function, the coordination shell radius (rshell), coordination number (N), and effective residence time (τ) are calculated. Fig. 2 shows a schematic of the associations and measured properties. These three properties are calculated for four kinds of interactions: (i) cationic moiety-water, (ii) anionic moiety-water, (iii) cationic moiety-Cl−, and (iv) anionic moiety-Na+. These three properties will be used as the target variables for training and evaluating the ML algorithm.
(t), of water and ions (Na+ and Cl−) with respect to the position of the moieties are calculated during NVT simulations. From the RDF and residence time correlation function, the coordination shell radius (rshell), coordination number (N), and effective residence time (τ) are calculated. Fig. 2 shows a schematic of the associations and measured properties. These three properties are calculated for four kinds of interactions: (i) cationic moiety-water, (ii) anionic moiety-water, (iii) cationic moiety-Cl−, and (iv) anionic moiety-Na+. These three properties will be used as the target variables for training and evaluating the ML algorithm.
 | ||
| Fig. 2 (a) Graphic of water and ions associating to the charged moieties of a zwitterion and forming structured coordination shells. (b) Example RDF with rshell and N indicated by the red vertical dashed line and hatched area, respectively. (c) Example of residence time correlation function, (t), expressing an exponentially decaying relationship with time, t. | ||
Coordination shell radius
The RDF is calculated to study the change in density of water molecules or ions from the reference atoms on the zwitterion moieties. All RDF calculations were averaged over 5 trials. For interactions involving zwitterion cationic moieties, the carbon of the cationic methyl group(s) is used as the reference except when no methyl group is present (i.e., NH3), where the hydrogen is used for the reference. This was decided because of the observed overlap between the hydration shells, where 99–100% of all water molecules are shared between the coordination shells of the carbon(s) and hydrogen(s). Nitrogen or sulfur atoms were not selected, as the observed coordination shells were significantly influenced by the anionic moiety. For interactions with anionic moieties, the oxygens of the anionic moiety are used for reference. All interactions with water use the water's oxygen atom as the reference due to its position in the molecule approximately representing the center-of-mass.17Coordination number
With rshell found, it is possible to calculate the coordination number, N, which is the average number of a species (oxygen of water or ion) within a distance r = rshell from the reference atom in the zwitterionic moiety during the entire simulation.Effective residence time
The effective residence time, τ, is the averaged time during which a species stays within the coordination shell around a charged moiety before leaving. To calculate τ, the residence time correlation function,(t), is calculated for each interaction, which is then fitted to an exponential function of the form f(t) = A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) exp(−t/τ). (t) is found by normalizing the autocorrelation function, C(t), of water or ions within their respective coordination shell with radius rshell, to its initial value, C(0).39 The autocorrelation function is described by
exp(−t/τ). (t) is found by normalizing the autocorrelation function, C(t), of water or ions within their respective coordination shell with radius rshell, to its initial value, C(0).39 The autocorrelation function is described by  , where tk is the k th time interval and vi(t) is 1 when the ith water molecule or ion is within the coordination shell radius at time t and 0 otherwise.
, where tk is the k th time interval and vi(t) is 1 when the ith water molecule or ion is within the coordination shell radius at time t and 0 otherwise.
The following fitting process requires selecting an appropriate maximum lag time. Fig. S2 (ESI†) shows a sample of (t) for four possible interactions for a selected zwitterion, where the lag time varies from 0 to 300 ps to clearly represent the effective residence time.
Cheminformatic descriptors
Prior studies have identified electrostatic interactions and hydrogen bonding as dominating forces in the hydration and ion association of zwitterions.17,22,30,40,41 To describe these properties of the zwitterions, cheminformatics describing the electrostatic, hydrogen bonding, and geometric characteristics of the cationic and anionic moieties, and spacer groups were calculated as shown in Table 1, and will be used as inputs for the ML algorithm. Further explanation of descriptor calculations can be found in the ESI.†| Descriptor | Description |
|---|---|
| Volume* | Subunit van der Waals volume in nm3 |
| Surface area* | Subunit van der Waals surface area in nm2 |
| Charge† | Sum of subunit partial charges in Coulomb |
| HBondAcceptors | Number of hydrogen-bond acceptor sites |
| HBondDonors | Number of hydrogen-bond donor sites |
Results and discussion
Role of molecular design in hydration and ion association
Both graphical and statistical methods are used to understand how chemical design affects hydration and ion association properties.Distribution of association properties by chemistry in the presence of salt
Fig. 3a–c show the distribution of rshell, N, and τ, respectively, color coded for five different zwitterion cation (ZwCation) moieties and water interaction in the presence of salt. Fig. 3d–f illustrate the same but for three different zwitterion anion (ZwAnion) moieties and water interaction. The distribution of rshell, N, and τ for interactions of other ZwCations and ZwAnions with water and salt are provided in Fig. S3 in ESI.† The most obvious trends from these Figures occur when the changing subunit chemistry agrees with the association of interest (e.g., changing cation for ZwCation–water interactions, or changing anion for ZwAnion–water interactions). Additionally, Table 2 shows the mean and standard deviation of the distributions for each ZwCation and ZwAnion illustrated in Fig. 3.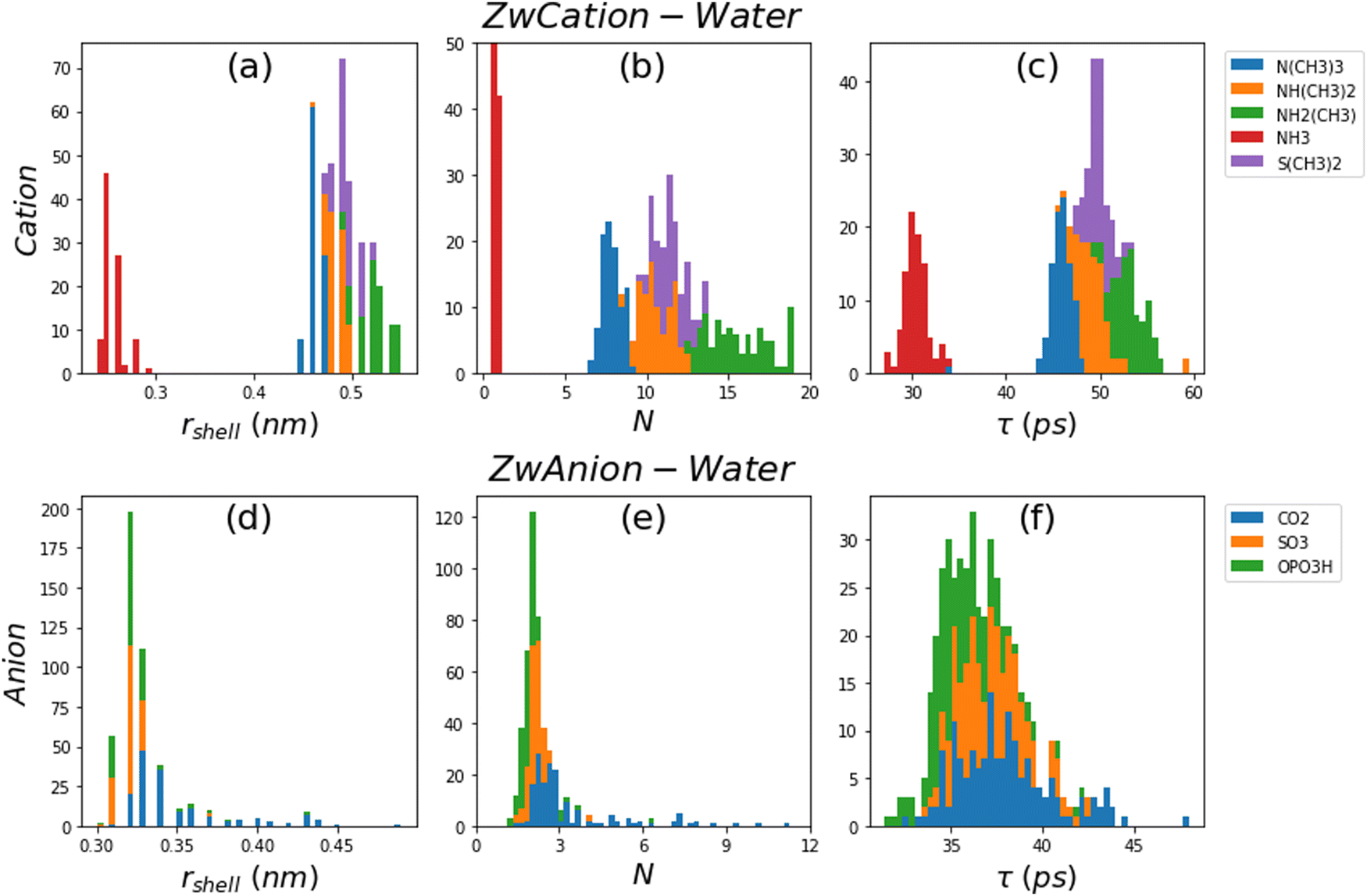 | ||
| Fig. 3 Distribution of rshell (a and d), N (b and e), and τ (c and f) with changing subunit chemistries indicated in the legend for ZwCation–water (a–c) and ZwAnion–water (d–f) associations. | ||
| Subunit | r shell (nm) | N | τ (ps) | |
|---|---|---|---|---|
| ZwCation | N(CH3)3 | 0.46 ± 0.01 | 7.86 ± 0.62 | 45.80 ± 1.58 |
| NH(CH3)2 | 0.48 ± 0.01 | 10.63 ± 0.95 | 49.20 ± 1.99 | |
| NH2(CH3) | 0.52 ± 0.02 | 15.57 ± 1.86 | 53.07 ± 1.64 | |
| NH3 | 0.26 ± 0.01 | 0.83 ± 0.15 | 30.36 ± 1.26 | |
| S(CH3)2 | 0.50 ± 0.01 | 11.53 ± 0.95 | 49.84 ± 1.11 | |
| ZwAnion | CO2 | 0.35 ± 0.03 | 3.35 ± 1.77 | 37.92 ± 2.65 |
| SO3 | 0.32 ± 0.01 | 2.17 ± 0.32 | 37.24 ± 1.85 | |
| OPO3H | 0.32 ± 0.02 | 2.01 ± 0.61 | 35.53 ± 1.83 |
Fig. 3a–c, showing ZwCation–water interactions with changing cation group, are highly bimodal with notable peaks on the left-hand side of rshell, N, and τ distribution corresponding to the NH3 cationic moiety chemistry. These lesser property values are likely caused by the considerably smaller volume of NH3 compared to the other cations where the methyl group(s) can sterically hinder water and ion associations. Beyond the left peak belonging to NH3, it appears there is an inverse relationship between the distribution of the properties and the number of methyl groups in the cationic moieties, following N(CH3)3 < NH(CH3)2 ∼ S(CH3)2 < NH2(CH3). This trend is also supported by the mean property values shown in Table 2. Additionally, Pearson correlations between descriptors and subunit association properties show the cation group association properties are highly correlated with the steric and hydrogen bonding descriptors, which will be discussed later in this section. Prior literature has also demonstrated this trend.42
Fig. 3d–f, show ZwAnion–water interaction properties with changing zwitterion anionic chemistry. Fig. 3d–f and Table 2 suggest patterns for all properties following OPO3H ∼ SO3 < CO2, though the effect is less pronounced than the trend for ZwCation–water interactions. Property correlation data from Fig. 4 would suggest steric (volume and surface area) and hydrogen bonding effects (number of hydrogen bond donating and accepting sites) again influence the anionic association properties. However, there is also a strong correlation between these properties and the charge of the cationic moiety. This is indicative of a strong electrostatic influence from the cationic moiety affecting the hydration and ion association of the anionic moiety. This has been previously observed, where the hydration shells of some cationic moieties can be overlapping those of anionic moieties.42
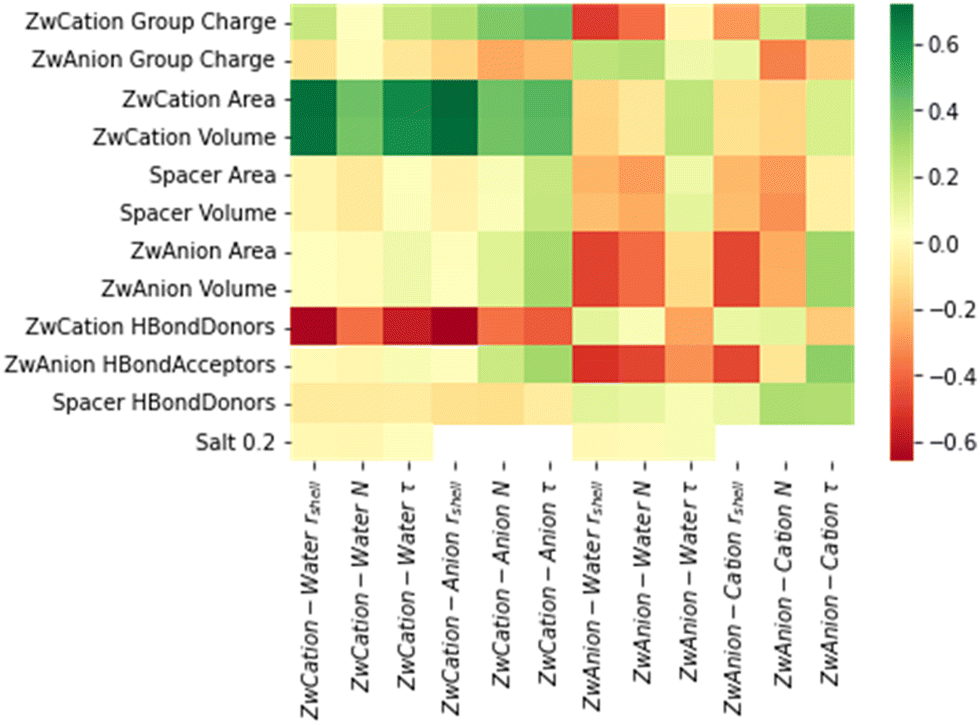 | ||
| Fig. 4 Heatmap of Pearson correlation between descriptors (y-axis) and association properties (x-axis). Color indicates feature's Pearson correlation value to the corresponding target variable. | ||
Above trends are far less pronounced for interactions with ions as shown in Fig. S3c and d (ESI†). There remains a notable peak in Fig. S3c (top row) (ESI†) corresponding to the NH3 cationic moiety interaction with Cl−; however, this peak is far less pronounced for N and τ variables in ZwCation–Cl− interactions than for those in ZwCation–water interactions in Fig. S3a (top row) (ESI†). Furthermore, results in Fig. 4 suggest that there is a strong correlation between the steric and hydrogen bonding properties and ZwCation–Cl− association, but it is less obvious what descriptors affect the anion association properties.
Multivariate analysis of variance
To gain more detailed understanding of how zwitterion design affects hydration (rshell, N, and τ for interaction with water) and ion association (rshell, N, and τ for interaction with Na+ and Cl−) properties before ML is applied, multivariate analysis of variance (MANOVA) is performed. In this analysis, there are 5 design factors (cationic/anionic/spacer groups, CSL, presence of salt) and 3 dependent variables (rshell, N, τ) for 4 interactions (ZwCation with water or Cl−, ZwAnion with water or Na+). MANOVA is performed to investigate the effects of the abovementioned factors on the dependent variables while also considering the correlation between the dependent variables.MANOVA is applied with Pillai's trace with the formula for each interaction following rshell + N + τ as a dependent variable, and cation, anion, spacer, CSL, and salt as independent variables, except where the interaction is with respect to ions, where salt is removed as a factor. MANOVA results are presented in Table S7 (ESI†). All factor p-values were less than α-level of 0.05 for all interactions, suggesting the factors are statistically significant to the combined dependent variable.
Cheminformatic descriptor–property correlation
The Pearson correlation between descriptors and target values is shown in Fig. 4, which reveals the nature of the descriptor–property relationships. In Fig. 4, there are clusters of similarly shaded correlation properties corresponding to the type of the association. In particular, the surface area and volume of the zwitterion cationic moiety have similar correlations with rshell, N, and τ of ZwCation–water and ZwCation–anion (Cl−) interactions. Other notable features are the inverse correlations between the number of hydrogen bond donors of cationic moieties and rshell, N, and τ of ZwCation–water and ZwCation–anion interactions. Similar inverse correlations are observed between the number of hydrogen bond acceptors and the rshell, N, and τ of ZwAnion–water and ZwAnion–cation interactions except for τ of ZwAnion–cation interaction. On its own, this data generally prescribes design principles for controlling the association properties of the zwitterionic moieties. However, once this data is combined with feature importances from the ML algorithm, it will give more clear definitions for how important a descriptor is to the rshell, N, and τ of water and ion association, and whether it will positively or negatively influence these association properties.Machine learning structure–property relationships
Before applying ML algorithms to reveal the desired relationship between structure and property (rshell, N, and τ) for aforementioned zwitterionic molecules, it is important to discuss the limitations of the produced data. The controlled factors, cation, anion, spacer groups, CSL, and presence of salt, are a small representation of the complex chemistries and systems which could be studied for zwitterions. For this reason, all analysis to follow is performed with the expectation that the information revealed may not be extended to other materials and systems.Preprocessing and model workflow
To prepare for training and evaluating the ML algorithm, several preprocessing steps are required. Firstly, cheminformatics describing the spacer groups are NaN-valued for all cases where CSL is equal to zero, making the data unusable for training the ML algorithm. To correct this, the missing data is zero-imputed, which is reasonable given the cheminformatics at-hand are all strictly positive and additive. The salt value is one-hot encoded with a drop-first scheme to convert the continuous salt concentration into a binary-valued variable. The predictors are CSL, salt, and cheminformatics as indicated in Table 1, and predicted variables are rshell, N, and τ. Predictor variables are min–max scaled. Finally, some predictor variables have constant values or are correlated; thus, feature pruning is necessary. Pruning is manually performed by removing any constant-valued descriptors, as well as descriptors which are explicitly used in the calculation of other descriptors (e.g., subunit volume is used to calculate ovality).Following preprocessing, the data is supplied to train and evaluate a scikit-learn XGBoostRegressor algorithm43 with 5-fold cross-validation with shuffles and grid search hyperparameter tuning, exploring number of iterations (i.e., number of trees) between 50 and 500 in intervals of 50, with all other hyperparameters left as default. The XGBoostRegressor algorithm is selected as it is considered a highly interpretable model and is capable of training model parameters on both one-hot encoded (presence of salt) and continuous inputs (all other variables) with no need for feature scaling. Additionally, as will be further discussed later, it is possible to extract feature importances from the models, which quantitatively describe the impact a feature has on a model's predictions. These feature importances are useful independently to describe how a model acts, but they may also be compared to one another to demonstrate which features are more or less impactful. The performance of the above-mentioned algorithm is scored by R2.
Model performance
As mentioned, there is a bimodal distribution of rshell, N, and τ caused by the NH3 cation moiety, which makes up approximately 20% of all data. To determine the impact of such outliers on model performance, the ML algorithm was trained with and without data containing this cation moiety, as well as a model trained with this subset alone to evaluate the extensibility of the model. It was hoped that the cheminformatic descriptors would sufficiently describe the trends, and thus be capable of describing outlier data.Table 3 shows training and testing model performance as determined by the coefficient of determination, R2. R2 values for models not explicitly using NH3 data as the test set are the mean cross-validated scores from the best-performing hyperparameter-tuned model. Otherwise, R2 is the performance on models using the NH3 in the test set after hyperparameter tuning. Across all subsets, the models performed considerably better on the training set than the test set, indicating overfitting, as could be expected for such a small dataset.
| R 2 Train/R2Test | |||||
|---|---|---|---|---|---|
| Property | Train and test with NH3 | Train and test without NH3 | Train without, and test with NH3 | ||
| ZwCation | Water | r shell | 1.00/0.99 | 0.96/0.83 | 0.96/−0.09 |
| N | 1.00/0.97 | 0.99/0.91 | 0.98/0.50 | ||
| τ | 1.00/0.97 | 1.00/0.80 | 0.93/0.02 | ||
| Cl− | r shell | 0.99/0.95 | 0.88/0.36 | 0.87/−0.08 | |
| N | 0.98/0.78 | 0.93/0.47 | 0.93/0.08 | ||
| τ | 0.91/0.60 | 0.90/0.30 | 0.96/0.17 | ||
| ZwAnion | Water | r shell | 0.99/0.71 | 1.00/0.68 | 0.93/0.42 |
| N | 0.99/0.65 | 0.99/0.62 | 0.95/0.21 | ||
| τ | 1.00/0.72 | 1.00/0.71 | 0.99/0.33 | ||
| Na+ | r shell | 0.79/0.21 | 0.85/0.29 | 0.85/0.00 | |
| N | 0.90/0.57 | 0.91/0.42 | 0.90/0.30 | ||
| τ | 0.79/0.11 | 0.83/0.14 | 0.82/0.07 | ||
Comparing R2 values between the subsets with and without NH3 data, we find that the inclusion of the data improved model performance. However, when the NH3 subset is not used in the training, but used in the test data, the model lacks any meaningful predictive capabilities and cannot generalize to this unseen data. For this reason, the subset excluding the NH3 from the training data is chosen for further analysis.
Focusing solely on the model without NH3 data, reveals interactions with the cationic moieties generally performed better than those with the anionic moieties. This may be due to the greater variation of cation-specific descriptors from 4 cationic moieties (excluding NH3) versus 3 for the anions and corresponding diversity of descriptor values.
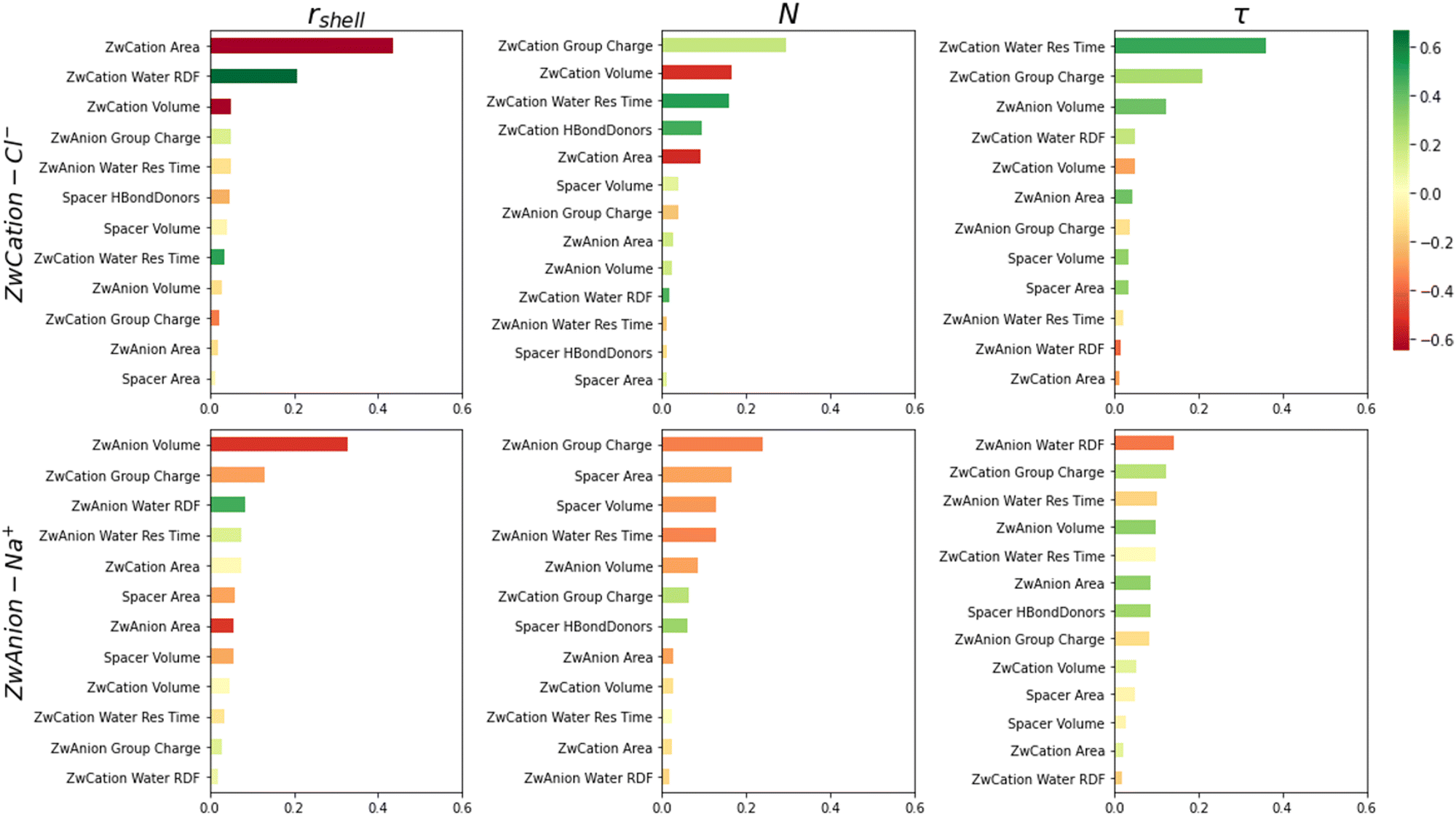
Feature importances are extracted from the ML algorithm and are shown in Fig. 5, ranking the descriptors by their utility in model predictions and colored by their correlation coefficients with respect to target variables. ZwCation interactions have descriptors predominantly related to steric properties (area and volume), though electrostatics (charge) and hydrogen bonding descriptors are frequently present. This result suggests that the methyl groups of the cationic moiety are primarily affecting the hydration layer and hydrogen bonding is a secondary influence. Surprisingly, electrostatic properties are not among the highest importances for interactions with water. However, these properties do rise in importance for ZwCation–Cl− models.
 | ||
| Fig. 5 Feature importances extracted from XGBoostRegressor algorithm. Features with an importance value greater than 0.01 are only included for clarity. Color bar indicates feature's Pearson correlation value to the corresponding target variable. | ||
Top feature importances for ZwAnion interactions are mostly steric and electrostatic descriptors. The ZwCation charge is frequently one of the top descriptors for these interactions. As mentioned, this result is likely due to the cationic moiety hydration layer and charge cloud overlapping the anionic moiety.42
Considering the correlation of important features, there is a general positive correlation between steric features (e.g., area, volume) and target variables for interactions between cationic moieties and water or ions. Moiety area and volume depend upon several lower-order features; thus, the meaning of this correlation cannot be exactly determined here. The number of hydrogen bond donors has relatively high importance for all water-association models and a negative correlation, indicating the number of hydrogen bond donors would be antithetical to hydration. ZwAnion interactions depend more heavily on electrostatics and have predominately negative correlations. Generally, if cationic moiety feature properties increase, there will be increases in hydration and ion association properties, and the opposite will be true for anionic moieties.
These results are consistent with Table 3, which also indicates that predictions of rshell, N, and τ for ion (Cl− and Na+) interactions are not as good as those for water interactions. From prior studies, it is known that the hydration layers impact the association abilities of ions.44 This result suggests it is necessary to include hydration properties in predicting ion association. Additionally, there are overlap effects demonstrated by the presence of ZwCation descriptors in the top feature importances of ZwAnion–water and ZwAnion–Na+ interactions. The proximity of the oppositely charged moieties, dictated by the spacer volume and area, is also an important feature, which is sometimes seen in ZwAnion–water and ZwAnion–Na+ interactions in Fig. 5.
Hydration-inclusive prediction of ion association
To explore the influence of the hydration layer on the ion association abilities of the zwitterionic moieties, hydration rshell and τ were included in the descriptors of the ML algorithm. Since there is a strong correlation between rshell and N for interactions with water as shown in Fig. S4 (ESI†), only one of the two properties is required to be included. We also included hydration τ as an approximate description of the thermodynamics of hydration. Correlation analysis of features and target properties is performed, as before, and results are presented in Fig. 6. There are strong correlations, both negative and positive, across the feature-target matrix for both ZwCation–Anion and ZwAnion–Cation associations. Among each interaction kind, there are mixed correlations for feature-target relationships, such as the ZwCation Area feature having strongly negative correlation with ZwCation–Anion and rshell, while ZwCation–Water and rshell has a strong positive correlation. | ||
| Fig. 6 Heatmap of Pearson correlation between descriptors (y-axis) and ion association properties (x-axis). Color indicates feature's Pearson correlation value to the corresponding target variable. | ||
Table 4 and Fig. 7 show resulting model performance and feature importances, respectively. There was no notable improvement to model performance, though the hydration descriptors are present as important features, suggesting hydration is important to ion association. It should be noted that generally the model quality is not sufficient to fully trust importances, though this is true for ZwAnion–Cation association. Taking model accuracy for what it's worth, examining the high-importance features and strength of correlation, a few conclusions may be suggested. Interestingly, each of the target properties, rshell, N, and τ appear to have similar feature-kinds despite the target ion. For rshell, there are primarily steric features, such as the area or volume of like-charged features. N sees the electrostatic properties become dominant, though steric and hydration residence time influences still carry significant importance. τ deviates with less concise description of its important features, likely due to its significantly lower model accuracies, leaving the only interesting feature that the ZwCation–Anion residence time has water residence time and group charge as its most important features.
| Property | R 2 Train/R2Test | |
|---|---|---|
| ZwCation–Cl− | r shell | 0.89/0.44 |
| N | 0.95/0.54 | |
| τ | 0.93/0.39 | |
| ZwAnion–Na+ | r shell | 0.86/0.24 |
| N | 0.92/0.40 | |
| τ | 0.84/0.09 |
 | ||
| Fig. 7 Feature importances including hydration rshell and τ in the descriptors to predict the ion association properties, rshell, N, and τ. Color bars indicate value of feature's Pearson correlation to the corresponding target variable. | ||
Conclusions
MD simulations and a ML algorithm have been used to examine the hydration and ion association properties of a library of zwitterionic molecules with varying cationic, anionic, alkane spacer chemistries, and number of spacer groups. Structural and dynamic properties of hydration and ion association were calculated from simulations. Cheminformatics describing molecule's steric, electrostatic, and hydrogen bonding properties were used as descriptors for the ML prediction of association properties.The coordination shell radius, coordination number, and effective residence time of waters and ions associating to cationic and anionic moieties were calculated from simulations. Results showed fully protonated amines had hydration layers, which were distinct from all other cationic moiety chemistries. This finding resulted in lower quality ML predictions and it was decided to remove molecules with NH3 cationic moiety from the final ML predictions. Correlation between descriptors and target values revealed that the number of hydrogen bonding sites and steric effects (areas and volumes) significantly affect hydration shell radius, coordination number, and effective residence time of waters around the cationic moieties. We found similar correlations between the three hydration properties of anionic moieties and hydrogen bonding and steric effects, although there was also meaningful electrostatic influence from the cationic groups. Ion association had less immediately recognizable trends, likely due to the dependence of ion association on the hydration abilities of the molecules.
XGBoostRegressor algorithm was trained using the cheminformatic descriptors to predict three hydration and ion association properties. The resulting model feature importances were used to rank the impact of the descriptors on the target variables. The three hydration properties prediction outperformed the three ion association properties prediction. Furthermore, prediction of cationic association properties consistently outperformed that of anionic association properties. The former was due to the influence of the zwitterion hydration layers in ion association while the latter is expected to be due to the cationic hydration layer overlapping the anionic moiety, and significantly influencing the anionic association properties, making distinct association property prediction of the anionic moiety less feasible.
Feature importances generally concurred with correlation data, where steric and hydrogen bonding properties were frequently among the most important values in predicting hydration properties. Surprisingly, electrostatics was not among the top descriptors for ZwCation–water properties, though it was important in predicting all other association properties. It was found that the cationic moieties affect the association properties of the anionic moieties, as reported prior. The ML algorithm performed poorly when predicting ion association properties. This result was hypothesized to be due to the role of hydration in ion association, which was not included in the descriptors. When hydration properties (rshell and τ) were included in the descriptors, there was no notable improvement to prediction accuracy; however, the hydration properties became some of the important descriptors in the ML algorithm, indicating the hypothesis was correct.
The findings presented here quantitatively demonstrate the influence of molecular design on the hydration and ion association abilities of zwitterions. Descriptor correlation and feature importances concur with prior studies, identifying the hydrogen bonding, steric, and electrostatic influences are important to the association properties. It was found that there is a more notable impact from steric effects on cationic group hydration properties than what has been discussed in prior literature. This result leaves room for further exploration of the role of the methyl groups in the hydration properties of cationic moieties.
Finally, there remain additional features, which are expected to influence hydration and ion association, such as the mobility of cationic methyl groups, hydration free energy of the charged moieties, and self-association of the zwitterions. There also remains the challenge of accurately defining the associations of the cationic moiety, where there are heterogeneous associations due to the methyl and hydrogen groups.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. Christiansen and S. Mehraeen gratefully acknowledge seed funding from the College of Engineering at University of Illinois, Chicago.References
- M. He, K. Gao, L. Zhou, Z. Jiao, M. Wu and J. Cao, et al., Zwitterionic materials for antifouling membrane surface construction, Acta Biomater., 2016, 40, 142–152, DOI:10.1016/j.actbio.2016.03.038.
- K. Wang, H. Seol, X. Liu, H. Wang, G. Cheng and S. Kim, Ultralow-fouling zwitterionic polyurethane-modified membranes for rapid separation of plasma from whole blood, Langmuir, 2021, 37(33), 10115–10125, DOI:10.1021/acs.langmuir.1c01477.
- C. Tiyapiboonchaiya, J. M. Pringle, J. Sun, N. Byrne, P. C. Howlett and D. R. MacFarlane, et al., The zwitterion effect in high-conductivity polyelectrolyte materials, Nat. Mater., 2004, 3(1), 29–32, DOI:10.1038/nmat1044.
- N. Byrne, P. C. Howlett, D. R. MacFarlane and M. Forsyth, The zwitterion effect in ionic liquids: Towards practical rechargeable lithium-metal batteries, Adv. Mater., 2005, 17(20), 2497–2501, DOI:10.1002/adma.200500595.
- L.-H. Tseng, P.-H. Wang, W.-C. Li, C.-H. Lin and T.-C. Wen, Enhancing the ionic conductivity and mechanical properties of zwitterionic polymer electrolytes by betaine-functionalized graphene oxide for high-performance and flexible supercapacitors, J. Power Sources, 2021, 516(230624), 230624, DOI:10.1016/j.jpowsour.2021.230624.
- C.-J. Lee, H. Wu, Y. Hu, M. Young, H. Wang and D. Lynch, et al., Ionic conductivity of polyelectrolyte hydrogels, ACS Appl. Mater. Interfaces, 2018, 10(6), 5845–5852, DOI:10.1021/acsami.7b15934 Available from.
- H. Wang, X. Liu, D. E. Christiansen, S. Fattahpour, K. Wang and H. Song, et al., Thermoplastic polyurethane with controllable degradation and critical anti-fouling properties, Biomater. Sci., 2021, 9(4), 1381–1396, 10.1039/d0bm01967d.
- B. Cao, L. Li, Q. Tang and G. Cheng, The impact of structure on elasticity, switchability, stability and functionality of an all-in-one carboxybetaine elastomer, Biomaterials, 2013, 34(31), 7592–7600, DOI:10.1016/j.biomaterials.2013.06.063.
- G. Cheng, H. Xue, Z. Zhang, S. Chen and S. Jiang, A switchable biocompatible polymer surface with self-sterilizing and nonfouling capabilities, Angew. Chem., Int. Ed., 2008, 47(46), 8831, DOI:10.1002/anie.200803570.
- Q. Shao and S. Jiang, Molecular understanding and design of zwitterionic materials, Adv. Mater., 2015, 27(1), 15–26, DOI:10.1002/adma.201404059.
- S. Paschke and K. Lienkamp, Polyzwitterions: From surface properties and bioactivity profiles to biomedical applications, ACS Appl. Polym. Mater., 2020, 2(2), 129, DOI:10.1021/acsapm.9b00897.
- W. Peng, P. Liu, X. Zhang, J. Peng, Y. Gu and X. Dong, et al., Multi-functional zwitterionic coating for silicone-based biomedical devices, Chem. Eng. J., 2020, 398(125663), 125663, DOI:10.1016/j.cej.2020.125663.
- C.-H. Hsu, A. Venault, H. Zheng, C.-T. Lo, C.-C. Yang and Y. Chang, Failure of sulfobetaine methacrylate as antifouling material for steam-sterilized membranes and a potential alternative, J. Membr. Sci., 2021, 620(118929), 118929, DOI:10.1016/j.memsci.2020.118929.
- F. Lu, X. Gao, A. Wu, N. Sun, L. Shi and L. Zheng, Lithium-containing zwitterionic poly(ionic liquid)s as polymer electrolytes for lithium-ion batteries, J. Phys. Chem. C, 2017, 121(33), 17756, DOI:10.1021/acs.jpcc.7b06242.
- M. T. Nguyen and Q. Shao, Effects of zwitterionic molecules on ionic association in ethylene oxide-based electrolytes, Fluid Phase Equilib., 2020, 515(112572), 112572, DOI:10.1016/j.fluid.2020.112572.
- S. Chen, L. Li, C. Zhao and J. Zheng, Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials, Polymer, 2010, 51(23), 5283–5293, DOI:10.1016/j.polymer.2010.08.022.
- Q. Shao, Y. He, A. D. White and S. Jiang, Difference in hydration between carboxybetaine and sulfobetaine, J. Phys. Chem. B, 2010, 114(49), 16625, DOI:10.1021/jp107272n.
- Q. Shao and S. Jiang, Influence of charged groups on the properties of zwitterionic moieties: A molecular simulation study, J. Phys. Chem. B, 2014, 118(27), 7630–7637, DOI:10.1021/jp5027114.
- D. Dong, C. Tsao, H.-C. Hung, F. Yao, C. Tang and L. Niu, et al., High-strength and fibrous capsule-resistant zwitterionic elastomers, Sci. Adv., 2021, 7(1), eabc5442, DOI:10.1126/sciadv.abc5442.
- Y. He, H.-K. Tsao and S. Jiang, Improved mechanical properties of zwitterionic hydrogels with hydroxyl groups, J. Phys. Chem. B, 2012, 116(19), 5766, DOI:10.1021/jp300205m.
- Q. Shao, Y. He and S. Jiang, Molecular dynamics simulation study of ion interactions with zwitterions, J. Phys. Chem. B, 2011, 115(25), 8358, DOI:10.1021/jp204046f.
- Q. Shao and S. Jiang, Effect of carbon spacer length on zwitterionic carboxybetaines, J. Phys. Chem. B, 2013, 117(5), 1357, DOI:10.1021/jp3094534.
- T. D. Huan, A. Mannodi-Kanakkithodi, C. Kim, V. Sharma, G. Pilania and R. Ramprasad, A polymer dataset for accelerated property prediction and design, Sci. Data, 2016, 3(1), 160012, DOI:10.1038/sdata.2016.12.
- A. Tkatchenko, Machine learning for chemical discovery, Nat. Commun., 2020, 11(1), 4125, DOI:10.1038/s41467-020-17844-8.
- T. C. Le, M. Penna, D. A. Winkler and I. Yarovsky, Quantitative design rules for protein-resistant surface coatings using machine learning, Sci. Rep., 2019, 9(1), 265, DOI:10.1038/s41598-018-36597-5.
- Y. Liu, D. Zhang, Y. Tang, Y. Zhang, Y. Chang and J. Zheng, Machine learning-enabled design and prediction of protein resistance on self-assembled monolayers and beyond, ACS Appl. Mater. Interfaces, 2021, 13(9), 11306–11319, DOI:10.1021/acsami.1c00642.
- Y. Liu, D. Zhang, Y. Tang, Y. Zhang, X. Gong and S. Xie, et al., Machine learning-enabled repurposing and design of antifouling polymer brushes, Chem. Eng. J., 2021, 420(129872), 129872, DOI:10.1016/j.cej.2021.129872.
- A. P. Sgouros, S. Knippenberg, M. Guillaume and D. N. Theodorou, Multiscale simulations of polyzwitterions in aqueous bulk solutions and brush array configurations, Soft Matter, 2021, 17(48), 10873–10890, 10.1039/d1sm01255j.
- Y. Higaki, Y. Inutsuka, T. Sakamaki, Y. Terayama, A. Takenaka and K. Higaki, et al., Effect of charged group spacer length on hydration state in zwitterionic poly(sulfobetaine) brushes, Langmuir, 2017, 33(34), 8404–8412, DOI:10.1021/acs.langmuir.7b01935.
- Q. Shao, L. Mi, X. Han, T. Bai, S. Liu and Y. Li, et al., Differences in cationic and anionic charge densities dictate zwitterionic associations and stimuli responses, J. Phys. Chem. B, 2014, 118(24), 6956, DOI:10.1021/jp503473u.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith and B. Hess, et al., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25, DOI:10.1016/j.softx.2015.06.001.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236, DOI:10.1021/ja9621760.
- L. S. Dodda, I. Cabeza de Vaca, J. Tirado-Rives and W. L. Jorgensen, LigParGen web server: an automatic OPLS-AA parameter generator for organic ligands, Nucleic Acids Res., 2017, 45(W1), W331, DOI:10.1093/nar/gkx312.
- L. S. Dodda, J. Z. Vilseck, J. Tirado-Rives and W. L. Jorgensen, 14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations, J. Phys. Chem. B, 2017, 1, 3864–3870 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, et al., Revision B.01, Vol. 16, Wallingford CT, 2016 Search PubMed.
- L. E. Chirlian and M. M. Francl, Atomic charges derived from electrostatic potentials: A detailed study, J. Comput. Chem., 1987, 8(6), 894–905, DOI:10.1002/jcc.540080616.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The missing term in effective pair potentials, J. Phys. Chem., 1987, 91(24), 6269–6271, DOI:10.1021/j100308a038.
- J. Aqvist, Ion-water interaction potentials derived from free energy perturbation simulations, J. Phys. Chem., 1990, 94(21), 8021–8024, DOI:10.1021/j100384a009.
- R. M. Brunne, E. Liepinsh, G. Otting, K. Wüthrich and W. F. Van Gunsteren, Hydration of proteins: A comparison of experimental residence times of water molecules solvating the bovine pancreatic trypsin inhibitor with theoretical model calculations, J. Mol. Biol., 1993, 231, 1040–1048 CrossRef CAS PubMed.
- H. Huang, C. Zhang, R. Crisci, T. Lu, H.-C. Hung and M. S. J. Sajib, et al., Strong surface hydration and salt resistant mechanism of a new nonfouling zwitterionic polymer based on protein stabilizer TMAO, J. Am. Chem. Soc., 2021, 143(40), 16786–16795, DOI:10.1021/jacs.1c08280.
- A. White and S. Jiang, Local and bulk hydration of zwitterionic glycine and its analogues through molecular simulations, J. Phys. Chem. B, 2011, 115(4), 660–667, DOI:10.1021/jp1067654.
- C. Leng, S. Sun, K. Zhang, S. Jiang and Z. Chen, Molecular level studies on interfacial hydration of zwitterionic and other antifouling polymers in situ, Acta Biomater., 2016, 40, 6–15, DOI:10.1016/j.actbio.2016.02.030.
- C. L. Ritt, M. Liu, T. A. Pham, R. Epsztein, H. J. Kulik and M. Elimelech, Machine learning reveals key ion selectivity mechanisms in polymeric membranes with subnanometer pores, Sci. Adv., 2022, 8(2), eabl5771, DOI:10.1126/sciadv.abl5771.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion and O. Grisel, et al. Scikit-learn: Machine Learning in Python. arXiv. 2012. http://arxiv.org/abs/1201.0490.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00062a |
| This journal is © The Royal Society of Chemistry 2023 |