
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nickel(II)-based one-dimensional organic–inorganic halide perovskite ferroelectric with the highest Curie temperature†
Hao-Fei
Ni‡
a,
Lou-Kai
Ye‡
a,
Peng-Cheng
Zhuge
a,
Bo-Lan
Hu
a,
Jia-Rui
Lou
a,
Chang-Yuan
Su
 b,
Zhi-Xu
Zhang
b,
Li-Yan
Xie
*a,
Da-Wei
Fu
*ab and
Yi
Zhang
*ab
b,
Zhi-Xu
Zhang
b,
Li-Yan
Xie
*a,
Da-Wei
Fu
*ab and
Yi
Zhang
*ab
aInstitute for Science and Applications of Molecular Ferroelectrics, Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Normal University, Jinhua, 321004, China. E-mail: liyanxie@zjnu.edu.cn; dawei@zjnu.edu.cn; yizhang1980@seu.edu
bJiangsu Key Laboratory for Science and Applications of Molecular Ferroelectrics, Southeast University, Nanjing, 211189, China
First published on 19th January 2023
Abstract
Organic–inorganic halide perovskites (OIHPs) are very eye-catching due to their chemical tunability and rich physical properties such as ferroelectricity, magnetism, photovoltaic properties and photoluminescence. However, no nickel-based OIHP ferroelectrics have been reported so far. Here, we designed an ABX3 OIHP ferroelectric (3-pyrrolinium)NiCl3, where the 3-pyrrolinium cations are located on the voids surrounded by one-dimensional chains composed of NiCl6-face-sharing octahedra via hydrogen bonding interactions. Such a unique structure enables the (3-pyrrolinium)NiCl3 with a high spontaneous polarization (Ps) of 5.8 μC cm−2 and a high Curie temperature (Tc) of 428 K, realizing dramatic enhancement of 112 and 52 K compared to its isostructural (3-pyrrolinium)MCl3 (M = Cd, Mn). To our knowledge, remarkably, (3-pyrrolinium)NiCl3 should be the first case of nickel(II)-based OIHP ferroelectric to date, and its Tc of 428 K (35 K above that of BaTiO3) is the highest among all reported one-dimensional OIHP ferroelectrics. This work offers a new structural building block for enriching the family of OIHP structures and will inspire the further exploration of new nickel(II)-based OIHP ferroelectrics.
Introduction
Organic–inorganic halide perovskites (OIHPs) have caught phenomenal research attention in recent years for their promising application potential in photovoltaics,1 photoelectricity,2,3 light emitting diodes,4 ferroelectric memory,5,6 and electron spin devices.7,8 Among them, ferroelectrics feature switchable spontaneous polarization that acts as the essential core in nonvolatile memory, capacitors and sensors, etc.9–14 OIHPs have occupied an important position in the ferroelectric field by taking advantage of their chemical tunability, light weight, mechanical flexibility and so on, compared with their inorganic counterparts. Structurally, OIHPs with the typical ABX3-type (A = organic cation, B = metal cation, and X = halide anion) formula generally adopt two structural forms, either a three-dimensional BX6-corner-sharing cubic structure or a one-dimensional BX6-face-sharing hexagonal structure.15–17 The A-site organic cations are embedded in the voids surrounded by the inorganic framework of BX6 octahedra via weak interactions, endowing the OIHPs with great structural diversity and chemical variability.18–23 Such unique structural characteristics open a rich platform for design synthesis and performance optimization of ferroelectric materials.24–33 Nevertheless, hundreds of ABX3-type OIHPs have been discovered, while only a few cases show ferroelectricity. Exploring reliable design concepts to assemble OIHP structures for developing high-performance ferroelectrics is an imminent ongoing challenge.To be a ferroelectric, the crystal must have polar symmetry, whose polarization switching is inseparable from the reorientational arrangement of molecular dipoles in lattice. Accordingly, several design strategies involving chemical modifications on organic cations of OIHPs have been proposed to efficiently obtain molecular ferroelectrics.34–36 For example, by modifying some structurally flexible building blocks, like spherical [(CH3)4N]+, 1,4-diazabicyclo[2.2.2]octonium (Dabco) and quinuclidine, to be suitable A-site cations, a series of OIHP ferroelectrics have been designed with superior performance such as multiple polarization axes, large piezoelectric response and high Curie temperature (Tc).37–45 Emphatically, a high Tc is essential for ferroelectric applications because it directly determines the working temperature range in many areas. Besides the organic cation itself, the weak interactions between organic and inorganic components in OIHPs cannot be ignored, which play an important role in crystallographic engineering to induce desired physical properties.46–49 Previous studies have demonstrated the rational modifications on A-site organic cations to modulate weak interactions to realize the design and performance optimization of OIHP ferroelectrics such as (4,4-difluorohexahydroazepine)2PbI4,37 and (ethylammonium)2(EA)2Pb3Br10.50 In this stage, it has to be mentioned that, however, the BX6 inorganic components in OIHPs are equally important while they are always easily overlooked under the halo of organic components. BX6 octahedra in the reported ferroelectric OIHPs also have considerable diversity, with a variety of optional B-site metal ions such as Pb2+, Mn2+, Cd2+, Cr2+, Sn2+ and so on.51–58 Notably, Xiong et al. reported a special nickel(II) nitrite-based organic–inorganic hybrid perovskite ferroelectric, [N-fluoromethyl tropine][Ni(NO2)3], designed by rational chemical modification and structural assembly.59 Zhang et al. also reported several zero-dimensional packing phase transition materials with the Ni(NCS)6 framework.60 For nickel(II) halide crystals, a series of perovskite variants ABX3 (B = Ni, A = Gu, FA, MA, X = Cl, Br; B = Mn, A = MA, X = Br) have been demonstrated to show optical properties and high electronic capacity (CH3NH3NiCl3).61,62 However, no nickel(II) halide-based OIHP ferroelectrics have been reported so far.
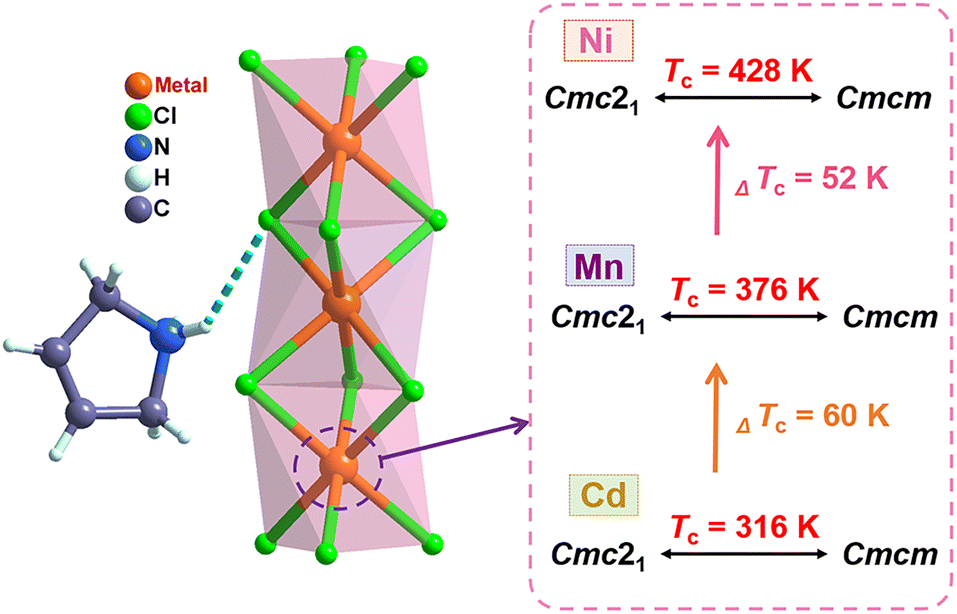
In the OIHP system, particularly, the 3-pyrrolinium cation has been shown to be a very promising candidate and an effective functional unit for building ferroelectrics, as reported in several previous studies such as (3-pyrrolinium)CdBr3 and (3-pyrrolinium)MCl3 (M = Cd, Mn).29,55,56 On the other hand, compared to Mn(II) and Cd(II), the six-coordinated Ni(II) with a smaller radius can form a smaller nickel(II) halide octahedron with a reduced octahedron volume, which may compress the space for organic cation movements and reduce the distance of hydrogen bonding interactions, thereby resulting in high phase transition temperature. Accordingly, through an elaborate structural assembly, we here present a new nickel(II)-based OIHP ferroelectric, (3-pyrrolinium)NiCl3, with a high Tc of 428 K, above that (about 393 K) of the inorganic perovskite ferroelectric BaTiO3.63 Systematic characterization reveals its remarkable ferroelectricity with a high saturated polarization (Ps) of 5.8 μC cm−2, and the order–disorder transition of the 3-pyrrolinium cations which leads to a ferroelectric phase transition. Compared to its isostructural compounds previously reported, (3-pyrrolinium)MCl3 (M = Cd, Mn) (Scheme 1),55,56 the variation of the inorganic framework of BX6 octahedra enables the Tc to get a significant increase of 112 and 52 K, due to the stronger hydrogen bonding interactions that act as a rope between the 3-pyrrolinium cations and [NiCl3]− anionic chains. Moreover, to our knowledge, (3-pyrrolinium)NiCl3 should be the first case of nickel(II)-based OIHP ferroelectric to date, which could provide a new structural paradigm for one-dimensional ABX3 OIHP ferroelectrics. More importantly, its Tc of 428 K is the highest among all reported one-dimensional OIHP ferroelectrics, making it competitive for ferroelectric-related devices with a wide operating temperature range.
 | ||
| Scheme 1 The design concept of ferroelectric (3-pyrrolinium)NiCl3 with the one-dimensional ABX3 OIHP structure. | ||
Results and discussion
(3-Pyrrolinium)NiCl3 was prepared by the reaction of 3-pyrrolinium chloride and nickel chloride in dilute hydrochloric acid solution, and its prism-like colorless crystals were easily obtained by a slow solution evaporating method. The preliminary differential scanning calorimetry (DSC) traces show a pair of endothermic/exothermic peaks in the heating/cooling runs (Fig. 1), indicating a reversible phase transition at about 428 K (Curie temperature, Tc), higher than those of the analogous (3-pyrrolinium)CdCl3 (Tc = 316 K) and (3-pyrrolinium)MnCl3 (Tc = 376 K).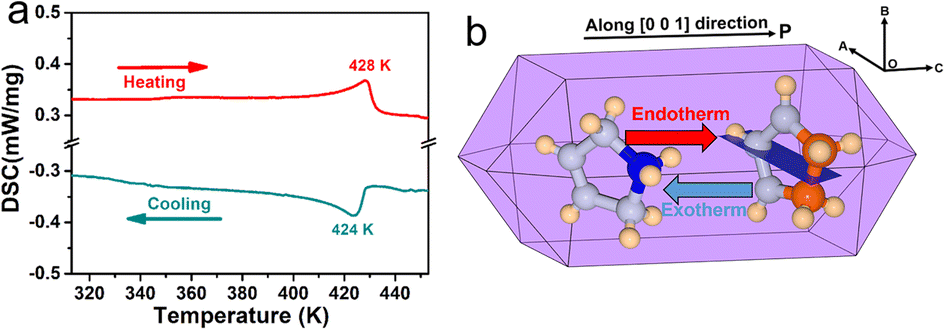 | ||
| Fig. 1 (a) DSC curves of (3-pyrrolinium)NiCl3 in heating/cooling runs. (b) Simulative single crystal shape of (3-pyrrolinium)NiCl3. | ||
To understand the phase transition mechanism and ferroelectric origin of (3-pyrrolinium)NiCl3, we determined its crystal structures and the resulting crystal data at various temperatures are summarized in Table S1.† (3-Pyrrolinium)NiCl3 adopts a one-dimensional hexagonal ABX3-type perovskite structure, where the infinite linear chains of the face-sharing NiCl6 octahedron along the [0 0 1] direction are separated by the A-site 3-pyrrolinium cations (Fig. 2). The anion chains and organic cations are loosely connected by N–H⋯Cl hydrogen bonding interactions. At room temperature in the phase below Tc (marked as LTP), the (3-pyrrolinium)NiCl3 crystallized in an orthorhombic space group Cmc21 (polar point group mm2) with the well-ordered 3-pyrrolinium cations and distorted NiCl6 octahedra. From the packing view of the crystal structure, all of the 3-pyrrolinium cations are aligned along the c-axis, which could result in a spontaneous polarization. On the basis of the point charge model for (3-pyrrolinium)NiCl3 in LTP (Fig. S1†), the estimated spontaneous polarization along the [0 0 1] direction is about 6.08 μC cm−2. In the phase above Tc (marked as HTP), the crystal symmetry of (3-pyrrolinium)NiCl3 transforms into the centrosymmetric space group Cmcm, which belongs to the point group mmm. Such crystallographic symmetry of mmm cancels out the molecular dipoles in each direction, leading to zero macroscopic polarization. In the HTP, the 3-pyrrolinium cations are located on the special symmetry site of 2mm in the lattice, and consequently the cations are required to show 2-fold orientational disorder, where the C and N atoms distribute equally in two positions by the mirror plane symmetry perpendicular to the c-axis. The anion chains of the NiCl6 octahedron experience little change except the octahedron become more regular. Therefore, the phase transition mechanism can be mainly attributed to the order–disorder transition of 3-pyrrolinium cations. From HTP to LTP, the symmetric elements are halved from eight (E, i, C2, 2C2′, σh, 2σv) in HTP to four (E, C2, 2σv) in LTP (Fig. S2†). The symmetry breaking occurs with the disappearance of the mirror element, leading to the orientational ordering of cations to generate spontaneous polarization, which means that the (3-pyrrolinium)NiCl3 is an mmmFmm2-type ferroelectric with two equivalent polarization directions.
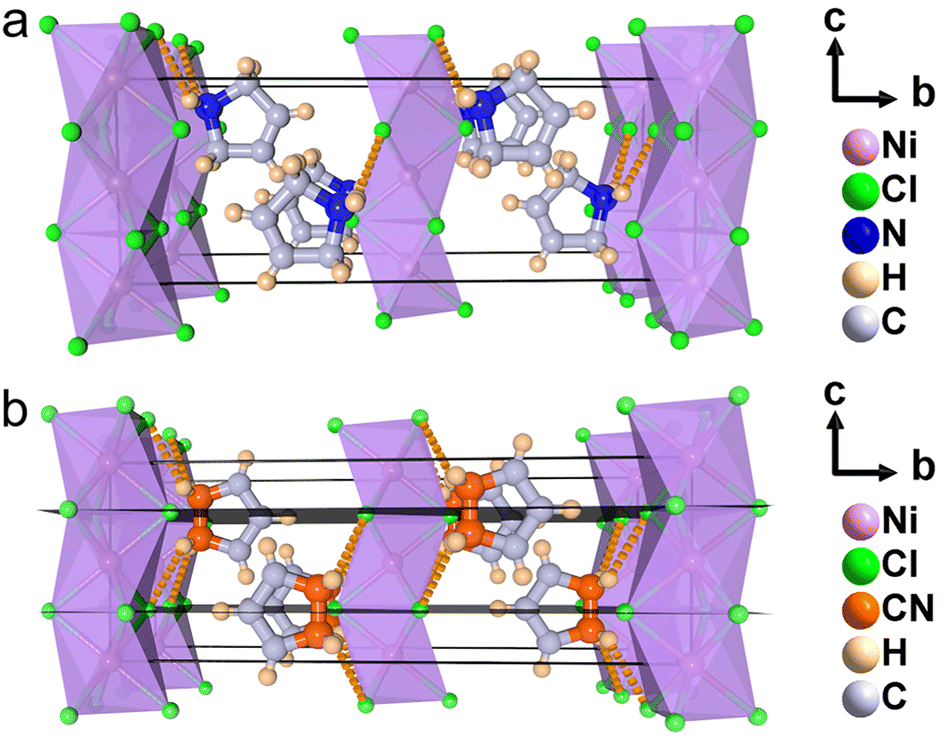 | ||
| Fig. 2 Packing view of crystal structures of (3-pyrrolinium)NiCl3 in (a) LTP and (b) HTP. The orange dotted lines represent N–H⋯Cl hydrogen bonding, and the black plane represents mirror symmetry elements. | ||
The stacking structure and phase transition mechanism of (3-pyrrolinium)NiCl3 are similar to those of (3-pyrrolinium)MCl3 (M = Cd, Mn), while the Tc has been greatly improved to up to 428 K. This would be closely associated with the changes of hydrogen-bonding interactions by different inorganic anion chains, and then affect the energy barrier of cationic orientational motions. As shown in Fig. 3a–c, for (3-pyrrolinium)MCl3 (M = Cd, Mn, Ni), the M–Cl bond distances undergo a decrease in sequence from Cd, Mn to Ni, with the range changes from 2.621–2.669 Å to 2.527–2.583 Å to 2.390–2.464 Å. Obviously, the MCl6 (M = Cd, Mn, Ni) octahedra gradually become smaller with the variable M–M distances in anion chains from 3.341 Å to 3.234 Å to 3.089 Å. The M–M distances between adjacent anion chains also experienced a gradual decrease with the changes of MCl6 octahedra from Cd, Mn to Ni (Fig. S3†). These changes make the void smaller, resulting in a confinement effect to increase the energy for cation rotation (Table S2†). Based on the Density Functional Theory (DFT) calculations, the energy barrier for the rotation of 3-pyrrolinium cations in (3-pyrrolinium)MCl3 (M = Cd, Mn, Ni) are 4.27, 4.51, and 4.97 eV respectively (Fig. S4–S7 and Table S3†), which indicates that (3-pyrrolinium)NiCl3 has a higher Tc, consistent with the results for structural analysis. In the OIHP system, besides the anionic component, hydrogen bonding interactions also have to be considered, which have been utilized as an indispensable tool in crystal engineering to design desired physical properties. The N–H⋯Cl hydrogen-bonding in (3-pyrrolinium)NiCl3 is stronger than the other two compounds, which can be clearly seen from the shortest distance between the donor (N) and acceptor (Cl) in (3-pyrrolinium)MCl3 (M = Cd, Mn, Ni) with 3.335, 3.306 and 3.302 Å respectively (Fig. S3†). The Hirshfeld dnorm surfaces and related 2D fingerprint plots of 3-pyrrolinium cations in (3-pyrrolinium)MCl3 (M = Cd, Mn, Ni) were further calculated to get the information about the environment of cations and intermolecular contacts (Fig. 3d–i).
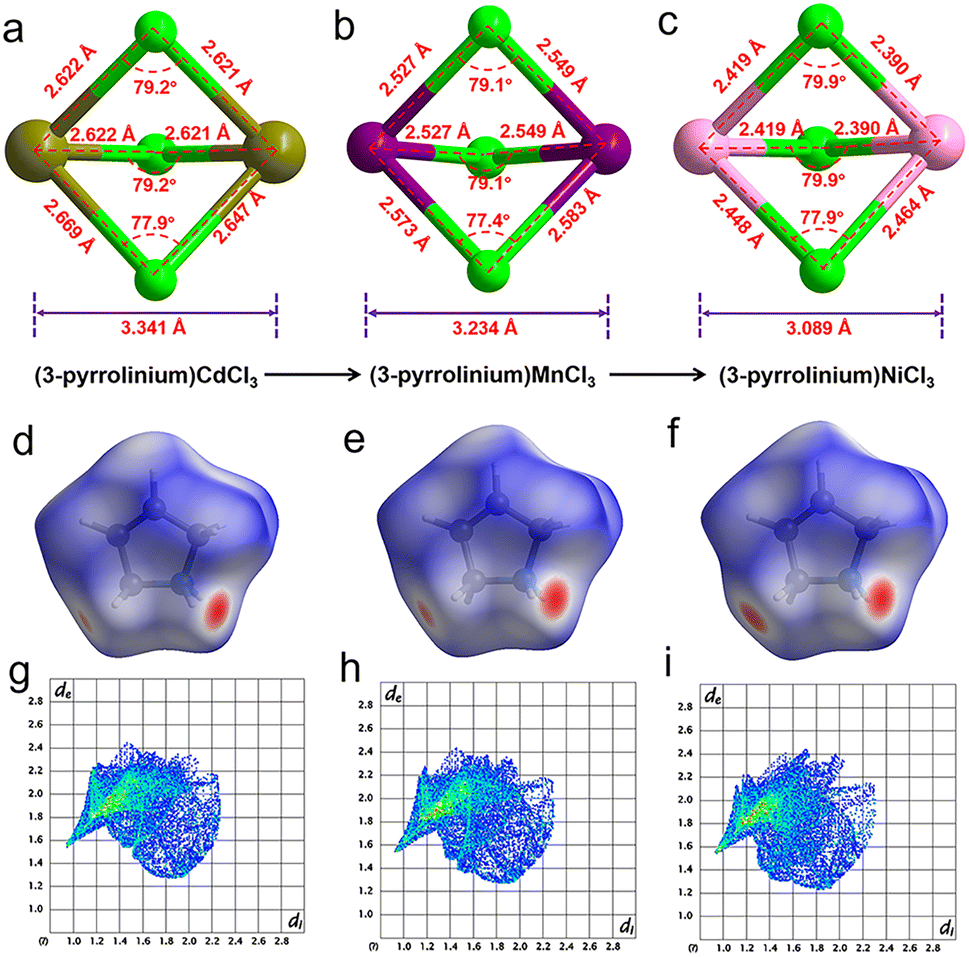 | ||
| Fig. 3 Comparison of anionic chains of (a) (3-pyrrolinium)CdCl3, (b) (3-pyrrolinium)MnCl3 and (c) (3-pyrrolinium)NiCl3. The Hirshfeld dnorm surfaces (d–f) and two-dimensional (2D) fingerprint plots (g–i) for the 3-pyrrolinium cations in (3-pyrrolinium)MCl3 (M = Cd, Mn, Ni). The di of the x-axis coordinate and the de of the y-axis represent the distance (Å) from the atoms inside the cation to the Hirshfeld surface and the external atoms to the Hirshfeld surface respectively. | ||
From the Hirshfeld dnorm surfaces, the short contacts (represented by the deep red circular depressions on the surfaces) between the cation and its surroundings for (3-pyrrolinium)NiCl3 are closer than those for (3-pyrrolinium)MCl3 (M = Cd, Mn), which are reflected in 2D fingerprints by quantifying the mean dnorm value for (3-pyrrolinium)NiCl3 (0.384), (3-pyrrolinium)MnCl3 (0.422) and (3-pyrrolinium)MnCl3 (0.424). The calculated results show that the total interactions in (3-pyrrolinium)NiCl3 are stronger than those in (3-pyrrolinium)MCl3 (M = Cd, Mn), which like a strong rope pull the cations, leading to a larger energy barrier required for the structural phase transition of (3-pyrrolinium)NiCl3.
Ferroelectric phase transitions are generally accompanied by significant anomalies of dielectric permittivity at around Tc. For (3-pyrrolinium)NiCl3, the real part ε′ of the complex dielectric constant exhibits a sharp λ-shaped peak-like response at about Tc = 428 K in each frequency (Fig. 4a). The peak values are several hundred to one thousand times larger than those in the stable state. We also carried out the second harmonic generation (SHG) measurements to study the symmetry changes of (3-pyrrolinium)NiCl3 during ferroelectric phase transition, by taking advantage of the high sensitivity of the SHG response to the breaking of the space-inversion symmetry. The SHG signal shows non-zero intensity in LTP (ferroelectric phase), consistent with the polar space group Cmc21, and experiences a change with temperature increasing until Tc (Fig. 4b). Above Tc, the SHG signal becomes inactive and maintains the zero intensity in HTP (paraelectric phase), corresponding to the centrosymmetric space group Cmcm.
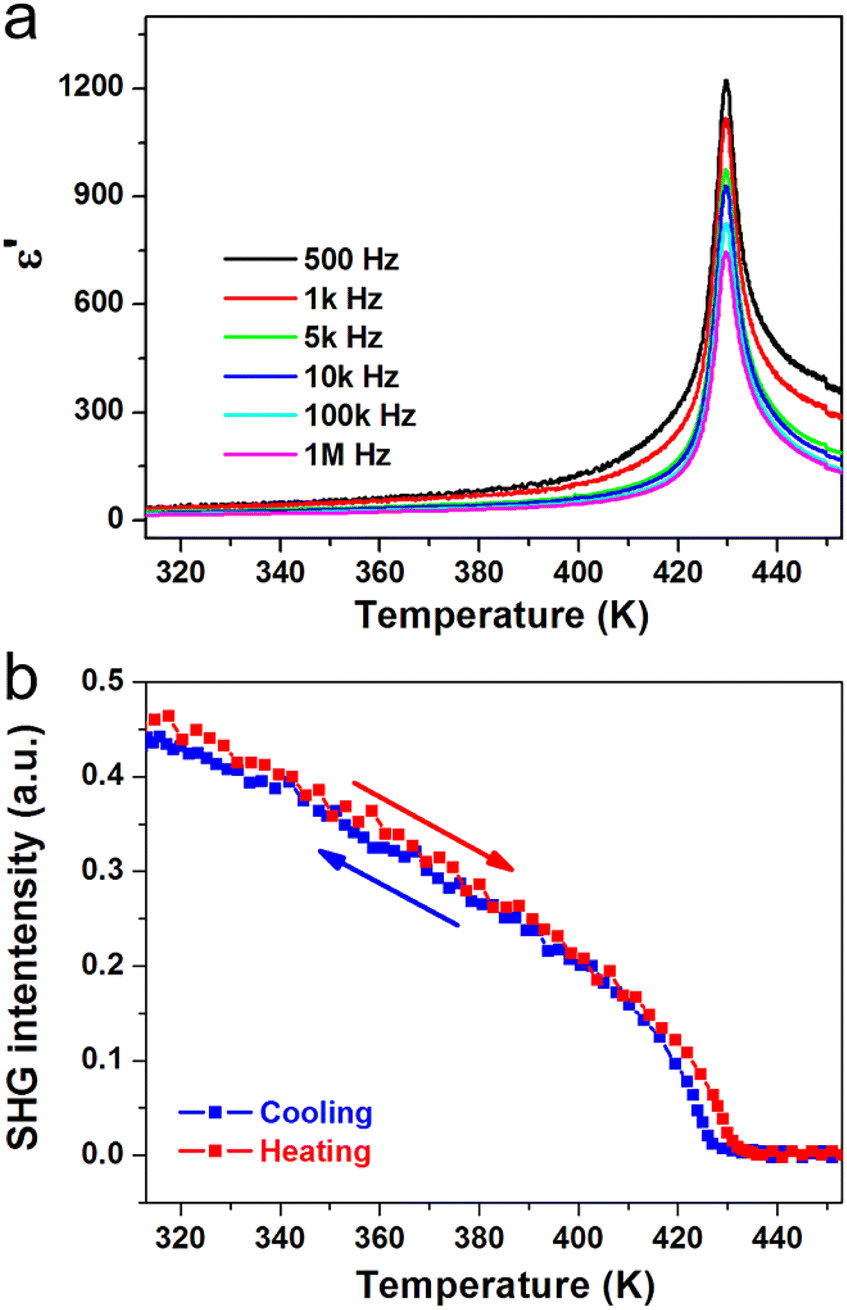 | ||
| Fig. 4 Temperature-dependent (a) real part of the complex dielectric constant and (b) SHG intensity of (3-pyrrolinium)NiCl3. | ||
The ferroelectricity with polarization reversal of (3-pyrrolinium)NiCl3 was directly verified by measuring polarization–electric field (P–E) hysteresis loops (Fig. 5). A linear P–E curve was obtained at 443 K above its Tc, indicating that it is currently in the paraelectric phase. On cooling to 428 K at about Tc, the result begins to show a compressed S-shaped curve. And then, the P–E curve opens to form a typical loop at 423 K below Tc, entering the ferroelectric phase. Upon further cooling, the ferroelectric loops expand and the Pr (remnant polarization) value increases gradually to reach about 5.8 μC cm−2 at 303 K, in accordance with the estimated value of 6.08 μC cm−2 from the crystal structure. The measured saturated polarization (Ps) and remnant polarization (Pr) values are close to each other. The polarization value of 5.8 μC cm−2 is higher than those of the ABX3-type OIHP ferroelectrics reported thus far (Table S4†), and larger than that of the reported nickel(II) nitrite-based hybrid perovskite ferroelectric [N-fluoromethyl tropine][Ni(NO2)3] (3.0 μC cm−2). Moreover, its Tc of 428 K should be the highest among all reported one-dimensional OIHP ferroelectrics (Table S4†), which combined with the small coercive field Ec in the range of 2.6–4.8 kV cm−1, makes (3-pyrrolinium)NiCl3 more competitive for relatively high-temperature, low operation voltage, low-power information storage devices.
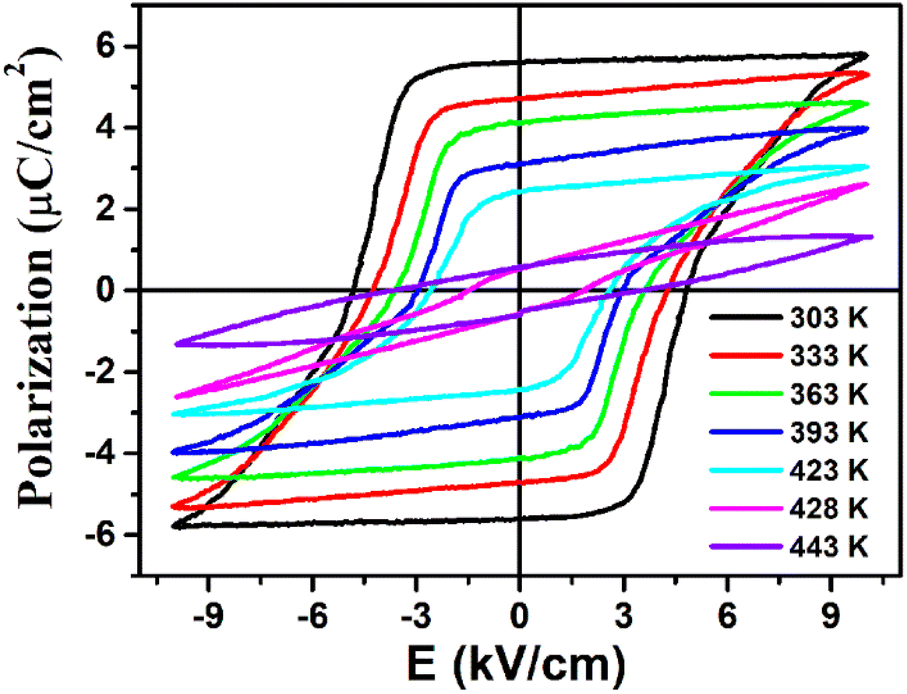 | ||
| Fig. 5 Ferroelectric hysteresis loops of (3-pyrrolinium)NiCl3 measured at different temperatures in the LTP. | ||
Conclusions
In summary, we successfully designed a new nickel(II)-based OIHP ferroelectric, (3-pyrrolinium)NiCl3, through an elaborate structural assembly of 3-pyrrolinium organic cations and one-dimensional [NiCl3]n− chains of NiCl6-face-sharing octahedra. It undergoes an mmmFmm2-type ferroelectric phase transition at 428 K, induced by the thermally driven transition of 3-pyrrolinium cations. The confined environment of 3-pyrrolinium cations caused by anion chains and relatively strong hydrogen bonding interactions give rise to a high energy barrier for phase transition, inducing ferroelectricity up to a high Curie temperature of 428 K. To our knowledge, such a Tc of 428 K should be the highest among all reported one-dimensional OIHP ferroelectrics, offering a wide operating temperature range for ferroelectric-related devices. More strikingly, (3-pyrrolinium)NiCl3 is the first nickel(II)-based OIHP ferroelectric, enriching the hybrid perovskite ferroelectric families. Considering the structural diversity and chemical tunability of OIHPs, one can expect more such nickel(II)-based OIHPs to be discovered with excellent ferroelectricity.Author contributions
H.-F. N. conceived the experiments and wrote the paper. L.-K. Y., P.-C. Z. G., B.-L. H. and J.-R. L. performed the experiments. C.-Y. S. and Z.-X. Z. assisted in the auxiliary analysis of data. L.-Y. X., D.-W. F. and Y. Z. guided and supervised this work.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant 21991141) and Zhejiang Normal University. The authors acknowledge the financial support from the Open Research Fund of the Key Laboratory of the Ministry of Education for Advanced Catalysis Materials and Zhejiang Key Laboratory.Notes and references
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036–3103 CrossRef CAS PubMed.
- H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC.
- Y. Zhao, C. Li and L. Shen, InfoMat, 2019, 1, 164–182 CrossRef CAS.
- L. N. Quan, B. P. Rand, R. H. Friend, S. G. Mhaisalkar, T.-W. Lee and E. H. Sargent, Chem. Rev., 2019, 119, 7444–7477 CrossRef CAS PubMed.
- W.-J. Xu, S. Kopyl, A. Kholkin and J. Rocha, Coord. Chem. Rev., 2019, 387, 398–414 CrossRef CAS.
- H.-Y. Zhang, X.-J. Song, X.-G. Chen, Z.-X. Zhang, Y.-M. You, Y.-Y. Tang and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 4925–4931 CrossRef CAS PubMed.
- B. Huang, J.-Y. Zhang, R.-K. Huang, M.-K. Chen, W. Xue, W.-X. Zhang, M.-H. Zeng and X.-M. Chen, Chem. Sci., 2018, 9, 7413–7418 RSC.
- K. Liao, X. Hu, Y. Cheng, Z. Yu, Y. Xue, Y. Chen and Q. Gong, Adv. Opt. Mater., 2019, 7, 1900350 CrossRef.
- X.-G. Chen, X.-J. Song, Z.-X. Zhang, P.-F. Li, J.-Z. Ge, Y.-Y. Tang, J.-X. Gao, W.-Y. Zhang, D.-W. Fu, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 1077–1082 CrossRef CAS PubMed.
- X. Li, F. Wu, Y. Yao, W. Wu, C. Ji, L. Li, Z. Sun, J. Luo and X. Liu, J. Am. Chem. Soc., 2022, 144, 14031–14036 CrossRef CAS PubMed.
- W.-Q. Liao, D. Zhao, Y.-Y. Tang, Y. Zhang, P.-F. Li, P.-P. Shi, X.-G. Chen, Y.-M. You and R.-G. Xiong, Science, 2019, 363, 1206–1210 CrossRef CAS PubMed.
- M. E. Lines and A. M. Glass, Principles and applications of ferroelectrics and related materials, Oxford University Press, 2001 Search PubMed.
- L. P. Miao, N. Ding, N. Wang, C. Shi, H. Y. Ye, L. Li, Y. F. Yao, S. Dong and Y. Zhang, Nat. Mater., 2022, 21, 1158–1164 CrossRef CAS PubMed.
- J. F. Scott, Science, 2007, 315, 954–959 CrossRef CAS PubMed.
- T. Chen, Y. Zhou, Z. Sun, S. Zhang, S. Zhao, Y. Tang, C. Ji and J. Luo, Inorg. Chem., 2015, 54, 7136–7138 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- K. Liu, Y. Jiang, Y. Jiang, Y. Guo, Y. Liu and E. Nakamura, J. Am. Chem. Soc., 2019, 141, 1406–1414 CrossRef CAS PubMed.
- P.-Z. Huang, H.-F. Ni, C.-Y. Su, M.-M. Lun, H.-F. Lu, D.-W. Fu and Q. Guo, CCS Chem., 2022, 1–10 Search PubMed.
- Q. Jia, T. Shao, L. Tong, C. Su, D. Fu and H. Lu, Chin. Chem. Lett., 2022 DOI:10.1016/j.cclet.2022.05.053.
- W. Li, Z. Wang, F. Deschler, S. Gao, R. H. Friend and A. K. Cheetham, Nat. Rev. Mater., 2017, 2, 16099 CrossRef.
- W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2018, 140, 3975–3980 CrossRef CAS PubMed.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CrossRef CAS PubMed.
- C.-Y. Su, Y. Yao, Z. Zhang, Y. Wang, M. Chen, P.-Z. Huang, Y. Zhang, W. Qiao and D.-W. Fu, Chem. Sci., 2022, 13, 4794–4800 RSC.
- Y. Ai, X.-G. Chen, P.-P. Shi, Y.-Y. Tang, P.-F. Li, W.-Q. Liao and R.-G. Xiong, J. Am. Chem. Soc., 2019, 141, 4474–4479 CrossRef CAS PubMed.
- H. L. B. Boström, M. S. Senn and A. L. Goodwin, Nat. Commun., 2018, 9, 2380 CrossRef PubMed.
- K. Gesi, J. Phys. Soc. Jpn., 1990, 59, 432–434 CrossRef CAS.
- Y. Hu, K. Parida, H. Zhang, X. Wang, Y. Li, X. Zhou, S. A. Morris, W. H. Liew, H. Wang, T. Li, F. Jiang, M. Yang, M. Alexe, Z. Du, C. L. Gan, K. Yao, B. Xu, P. S. Lee and H. J. Fan, Nat. Commun., 2022, 13, 5607 CrossRef CAS PubMed.
- Y. Hu, L. You, B. Xu, T. Li, S. A. Morris, Y. Li, Y. Zhang, X. Wang, P. S. Lee, H. J. Fan and J. Wang, Nat. Mater., 2021, 20, 612–617 CrossRef CAS PubMed.
- P.-F. Li, W.-Q. Liao, Y.-Y. Tang, H.-Y. Ye, Y. Zhang and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 8752–8757 CrossRef CAS PubMed.
- M.-M. Lun, T. Zhang, C.-Y. Su, J. Li, Z.-X. Zhang, D.-W. Fu and H.-F. Lu, Mater. Chem. Front., 2022, 6, 1929–1937 RSC.
- X. J. Song, T. Zhang, Z. X. Gu, Z. X. Zhang, D. W. Fu, X. G. Chen, H. Y. Zhang and R. G. Xiong, J. Am. Chem. Soc., 2021, 143, 5091–5098 CrossRef CAS PubMed.
- H. Wei, Y. Yang, S. Chen and H. J. Xiang, Nat. Commun., 2021, 12, 637 CrossRef CAS PubMed.
- Y. Zhang, H.-Y. Ye, W. Zhang and R.-G. Xiong, Inorg. Chem. Front., 2014, 1, 118–123 RSC.
- H.-Y. Zhang, Y.-Y. Tang, P.-P. Shi and R.-G. Xiong, Acc. Chem. Res., 2019, 52, 1928–1938 CrossRef CAS PubMed.
- T. Zhang, K. Xu, J. Li, L. He, D.-W. Fu, Q. Ye and R.-G. Xiong, Natl. Sci. Rev., 2022 DOI:10.1093/nsr/nwac240.
- Z.-X. Zhang, H.-Y. Zhang, W. Zhang, X.-G. Chen, H. Wang and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 17787–17794 CrossRef CAS PubMed.
- X.-G. Chen, X.-J. Song, Z.-X. Zhang, H.-Y. Zhang, Q. Pan, J. Yao, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2020, 142, 10212–10218 CrossRef CAS PubMed.
- D. W. Fu, J. X. Gao, W. H. He, X. Q. Huang, Y. H. Liu and Y. Ai, Angew. Chem., Int. Ed., 2020, 59, 17477–17481 CrossRef CAS PubMed.
- J.-X. Gao, W.-Y. Zhang, Z.-G. Wu, Y.-X. Zheng and D.-W. Fu, J. Am. Chem. Soc., 2020, 142, 4756–4761 CrossRef CAS PubMed.
- W.-Q. Liao, Y.-Y. Tang, P.-F. Li, Y.-M. You and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 18071–18077 CrossRef CAS PubMed.
- Y.-Y. Tang, Y. Xie, Y.-L. Zeng, J.-C. Liu, W.-H. He, X.-Q. Huang and R.-G. Xiong, Adv. Mater., 2020, 32, 2003530 CrossRef CAS PubMed.
- C.-F. Wang, H. Li, M.-G. Li, Y. Cui, X. Son, Q.-W. Wang, J.-Y. Jiang, M.-M. Hua, Q. Xu, K. Zhao, H.-Y. Ye and Y. Zhang, Adv. Funct. Mater., 2021, 31, 2009457 CrossRef CAS.
- Y.-M. You, W.-Q. Liao, D. Zhao, H.-Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P.-F. Li, D.-W. Fu, Z. Wang, S. Gao, K. Yang, J.-M. Liu, J. Li, Y. Yan and R.-G. Xiong, Science, 2017, 357, 306–309 CrossRef CAS PubMed.
- J. Ma, Q. Xu, L. Ye, Q. Wang, Z. Gong, C. Shi, H. Ye and Y. Zhang, J. Rare Earths, 2022, 40, 937–941 CrossRef CAS.
- C. F. Wang, C. Shi, A. Zheng, Y. Wu, L. Ye, N. Wang, H. Y. Ye, M. G. Ju, P. Duan, J. Wang and Y. Zhang, Mater. Horiz., 2022, 9, 2450–2459 RSC.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem.–Eur. J., 2006, 12, 8952–8960 CrossRef CAS PubMed.
- L. Zhang, X. Liu, J. Li and S. McKechnie, Sol. Energy Mater. Sol. Cells, 2018, 175, 1–19 CrossRef CAS.
- T. Zhang, K. Ding, J. Y. Li, G. W. Du, L. L. Chu, Y. Zhang and D. W. Fu, Chin. J. Chem., 2022, 40, 1559–1565 CrossRef CAS.
- Y. Zhao, P. Zhu, S. Huang, S. Tan, M. Wang, R. Wang, J. Xue, T.-H. Han, S.-J. Lee, A. Zhang, T. Huang, P. Cheng, D. Meng, J.-W. Lee, J. Marian, J. Zhu and Y. Yang, J. Am. Chem. Soc., 2020, 142, 20071–20079 CrossRef CAS PubMed.
- Y. Liu, S. Han, J. Wang, Y. Ma, W. Guo, X.-Y. Huang, J.-H. Luo, M. Hong and Z. Sun, J. Am. Chem. Soc., 2021, 143, 2130–2137 CrossRef CAS PubMed.
- Y. Ai, R. Sun, Y.-L. Zeng, J.-C. Liu, Y.-Y. Tang, B.-W. Wang, Z.-M. Wang, S. Gao and R.-G. Xiong, Chem. Sci., 2021, 12, 9742–9747 RSC.
- Q. Pan, Z.-B. Liu, Y.-Y. Tang, P.-F. Li, R.-W. Ma, R.-Y. Wei, Y. Zhang, Y.-M. You, H.-Y. Ye and R.-G. Xiong, J. Am. Chem. Soc., 2017, 139, 3954–3957 CrossRef CAS PubMed.
- C.-F. Wang, H. Li, Q. Ji, C. Ma, L. Liu, H.-Y. Ye, B. Cao, G. Yuan, H.-F. Lu, D.-W. Fu, M.-G. Ju, J. Wang, K. Zhao and Y. Zhang, Adv. Funct. Mater., 2022, 32, 2205918 CrossRef CAS.
- W.-J. Xu, C.-T. He, C.-M. Ji, S.-L. Chen, R.-K. Huang, R.-B. Lin, W. Xue, J.-H. Luo, W.-X. Zhang and X.-M. Chen, Adv. Mater., 2016, 28, 5886–5890 CrossRef CAS PubMed.
- H.-Y. Ye, Y. Zhang, D.-W. Fu and R.-G. Xiong, Angew. Chem., Int. Ed., 2014, 53, 11242–11247 CrossRef CAS PubMed.
- H.-Y. Ye, Q. Zhou, X. Niu, W.-Q. Liao, D.-W. Fu, Y. Zhang, Y.-M. You, J. Wang, Z.-N. Chen and R.-G. Xiong, J. Am. Chem. Soc., 2015, 137, 13148–13154 CrossRef CAS PubMed.
- H.-Y. Zhang, X.-G. Chen, Z.-X. Zhang, X.-J. Song, T. Zhang, Q. Pan, Y. Zhang and R.-G. Xiong, Adv. Mater., 2020, 32, 2005213 CrossRef CAS PubMed.
- Y. Zhang, W.-Q. Liao, D.-W. Fu, H.-Y. Ye, C.-M. Liu, Z.-N. Chen and R.-G. Xiong, Adv. Mater., 2015, 27, 3942–3946 CrossRef CAS PubMed.
- Y.-A. Xiong, T.-T. Sha, Q. Pan, X.-J. Song, S.-R. Miao, Z.-Y. Jing, Z.-J. Feng, Y.-M. You and R.-G. Xiong, Angew. Chem., Int. Ed., 2019, 58, 8857–8861 CrossRef CAS PubMed.
- Z.-H. Jia, J.-Y. Liu, D.-X. Liu, S.-Y. Zhang, Z.-Y. Du, C.-T. He, W.-X. Zhang and X.-M. Chen, J. Mater. Chem. C, 2021, 9, 8076–8082 RSC.
- M. Daub, I. Ketterer and H. Hillebrecht, Z. Anorg. Allg. Chem., 2018, 644, 280–287 CrossRef CAS.
- L. T. Lopez, D. Ramirez, F. Jaramillo and J. A. Calderon, Electrochim. Acta, 2020, 357, 136882 CrossRef CAS.
- G. H. Haertling, J. Am. Chem. Soc., 1999, 82, 797–818 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S7, Table S1–S4. CCDC 2214383 and 2214384. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05857j |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |