
Cu-Catalysed sustainable synthesis of formamide with glycerol derivatives as a carbonyl source via a radical-relay mechanism†
Xingchao
Dai‡
 *b,
Xinzhi
Wang‡
a,
Carsten
Kreyenschulte
b,
Hangkong
Yuan
a,
Angelika
Brückner
b,
Feng
Shi
*a and
Jabor
Rabeah
*b
*b,
Xinzhi
Wang‡
a,
Carsten
Kreyenschulte
b,
Hangkong
Yuan
a,
Angelika
Brückner
b,
Feng
Shi
*a and
Jabor
Rabeah
*b
aState Key Laboratory for Oxo Synthesis and Selective Oxidation, Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, No. 18, Tianshui Middle Road, Lanzhou, 730000, China. E-mail: fshi@licp.cas.cn
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Xingchao.Dai@catalysis.de; Jabor.Rabeah@catalysis.de
First published on 23rd January 2023
Abstract
Value-added formamides were sustainably synthesized by the N-formylation of amines with glycerol derivatives (1,3-dihydroxyacetone, glyceraldehyde and glycolic acid) as the carbonyl source via in situ C–C bond cleavage catalyzed over zeolite 5A supported Cu-based catalysts. Introducing a second metal ZrIV ions can greatly increase the activity of the Cu/5A catalyst. Catalyst characterization results revealed that the introduction of ZrIV ions decreases the amount of surface acidic sites, in particular, medium–strong acidic sites and promotes the formation of ˙OOH radicals. Isotopic tracing experiments confirmed that all the carbon atoms in the carbonyl group of formamide products are indeed from glycerol derivatives. Combined electron paramagnetic resonance (EPR) spin-trapping, operando attenuated total reflection (ATR)-FTIR spectroscopy and control experiments revealed that the reaction of aniline and DHA with formanilide involves GA and HCOOH intermediates and ˙NHPh radicals via a ˙OH–˙OOH radical-relay mechanism.
Introduction
With the increasing production of biodiesel, a surplus of glycerol is obtained as a by-product,1–3 for which a suitable method must be found to turn waste into treasure. The highly functionalized molecular structure of glycerol provides a chance to synthesize valuable chemicals and liquid fuels by different transformation processes such as oxidation, hydrogenolysis, carboxylation, etherification, cyclization and so on.4–14 Among them, the selective oxidation of glycerol to formic acid via oxidative C–C bond cleavage has revealed the great potential of glycerol as a carbonyl source for carbonyl-containing chemical production. As one of the important glycerol derivatives, 1,3-dihydroxyacetone (DHA) can be obtained via the catalytic oxidation of glycerol,5 and its value-added utilization can in turn valorise glycerol to some extent. Moreover, compared to the C–C bond in an alkane (83–90 kcal mol−1), the Cα–C bond in DHA is relatively weak (around 80 kcal mol−1 for acetone).15 Therefore, it should be easier to split the C–C bond of DHA than that of glycerol to in situ generate formic acid for the synthesis of carbonyl-containing molecules.As one of the important nitrogen-containing chemicals, formamides have widespread applications in organic synthesis and industrial production, and various methods have been developed for their production.16–28N-Formylation of amines with formylation reagents is a widely used method, and the common formylation reagents include CO,27 CO2,17,18,29–34 formic acid,23,27,35 formaldehyde36–43 and methanol.43–46 Although some excellent results have been obtained, obvious problems such as high toxicity and cost, requirement of harsh equipment and reaction conditions, poor product selectivity and so on have limited their large-scale application. From a sustainability point of view, it would be attractive to directly use bio-based feedstocks as formylation reagents to synthesize formamides by the reaction with amines.
In continuation of our efforts towards the synthesis of nitrogen-containing chemicals from renewable feedstocks,47–51 we recently achieved the N-formylation of amines to formamides with glycolic acid (GA) as the carbonyl source using Cu-containing zeolite 5A as the catalyst and H2O2 as the oxidant. In this case, the C–C bond in GA was split off to form carbonyl-containing intermediates in situ.52 Since GA must first be formed by the oxidation of DHA, it would be much more attractive to use DHA right away as a carbonyl source for formamide synthesis. Indeed, we could show that with a supported Cu/Al2O3 catalyst, DHA and various amines can be converted to the corresponding formamides in high yields even at room temperature.47 Unfortunately, the efficiency of the carbon atom in these early attempts was still insufficient. This has been markedly improved recently using a Cu/5A zeolite catalyst, which enabled for the first time the complete utilization of all carbon atoms in DHA for the formation of formamides from the corresponding amines.52 However, satisfactory formamide yields could only be achieved at 70 °C. Moreover, excess H2O2 was necessary since the latter partly decomposed at this temperature.
The reaction of DHA with an amine to form formamide should involve three main steps (Scheme 1): the cleavage of one C–C bond of DHA to GA and a C1 molecule (I) followed by the cleavage of the C–C bond of GA to C1 molecules (II) and finally the C–N bond formation between a C1 molecule and the amine (III). It has been shown that both conversion of DHA to GA (step I)47,53,54 and N-formylation of amines with C1 molecules (step III)21,55–58 can be achieved with H2O2 as the oxidant at room temperature. This suggests that the splitting of GA to C1 molecules (step II) might be the rate-limiting step, which could be the reason why the reaction of amines and GA to formamides requires a higher reaction temperature.52 It has been shown that ˙OH radicals play a key role in the cleavage of DHA to GA,47,53 while ˙OOH radicals are important to split the C–C bond in GA to C1 molecules and to activate the amine.52 This suggests that the reaction of DHA and amines to form formamides is co-mediated by both ˙OH and ˙OOH radicals, whereby an optimum ratio of both radicals might be required to maximize the formation of formamides and to prevent undesired deep degradation of reactants and target products by excess of highly reactive ˙OH radicals. It is known that H2O2 is split by Cu-based catalysts in a Fenton-like reaction into ˙OH and ˙OOH radicals.47,52,59 Usually ˙OH radicals dominate but the specific radical ratio depends on the electronic properties of the Cu centres since high electron density promotes the formation of ˙OH radicals (eqn (1) and (2)).59
| CuII + H2O2 → CuI + ˙OOH + H+ | (1) |
| CuI + H2O2 → CuII + ˙OH + OH− | (2) |
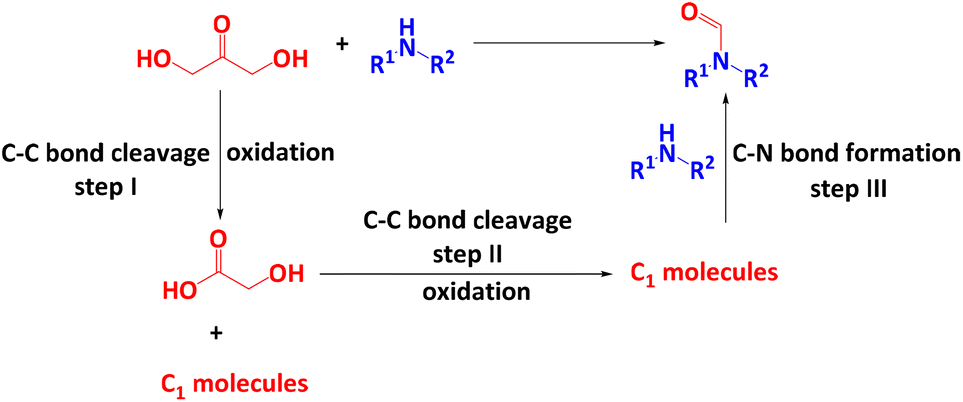 | ||
| Scheme 1 Synthesis of formamides with DHA as the carbonyl source. | ||
Thus, the above-mentioned optimum ˙OH/˙OOH ratio required for the highly selective conversion of DHA and amines to formamides (Scheme 1) might require (I) active Cu centres of tailored redox behaviour and electron density and (II) acidic surface sites of optimum strength to enable appropriate adsorption/desorption of reactants and products. To achieve requirements (I) and (II), we have explored bimetallic CuM/5A zeolite catalysts (M = Zr, Ni, Ag, Pd, Rh) using a series of metals with electronic properties different from the active Cu component whereby reaction temperatures as low as possible are desired to prevent the detrimental decomposition of H2O2. As a result, a highly active bimetallic catalyst CuZr/5A was developed for the N-formylation of amines with three glycerol derivatives such as DHA, GLA and GA as the carbonyl sources. Compared to our previous study,52 this catalytic system can lower the reaction temperature to 50 °C and reduce the amount of catalyst to 5 mg and H2O2 to 4 mmol, respectively. Moreover, it showed a good amine substrate scope, and 24 examples of different amines including aromatic, benzylic, aliphatic and secondary amines can be converted into the corresponding formamide product with good to excellent yields. Besides, for the first time a ˙OH–˙OOH radical-relay reaction mechanism was revealed for the N-formylation of amines with DHA as the carbonyl source.
Results and discussion
Catalytic test
Besides monometallic Cu/5A, various bimetallic CuM/5A catalysts (M = Ni, Zr, Ag, Pd and Rh) were prepared by ion exchange and tested in the N-formylation of aniline with DHA as the carbonyl source and H2O2 as the oxidant at 50 °C in 1,4-dioxane (Fig. 1a). 74% product yield was obtained with Cu/5A while the yield increased to 78% and 88%, respectively, when the second metal Ag or Zr was introduced. In contrast, lower yields were obtained with Ni, Pd and Rh, leaving Zr as the best second metal for this reaction. Therefore, CuZr/5A was used to optimize the amounts of aniline, H2O2 and catalysts. The product yield decreased from 88% to 61% when reducing the aniline amount to 2 mmol (Table 1, entry 1). The amount of H2O2 has a great effect on the product yields and much lower yields were obtained when <6 mmol H2O2 were used (Table 1, entries 2–5). When the catalyst dosage was changed from 0 to 100 mg, the highest product yield of 89% was obtained with 10 mg and an almost same product yield (88%) was obtained with 5 mg (Fig. 1b). Decreasing the content of the second metal Zr while keeping the Cu content unchanged lowered the product yield to 72% (Table 1, entry 6). Only 49% product yields were obtained with the bare support 5A, indicating that both Cu and Zr are essential (Table 1, entry 7). No formamide product was formed in the absence of DHA, implying that aniline does not react with the solvent 1,4-dioxane to generate the formamide product (Table 1, entry 8).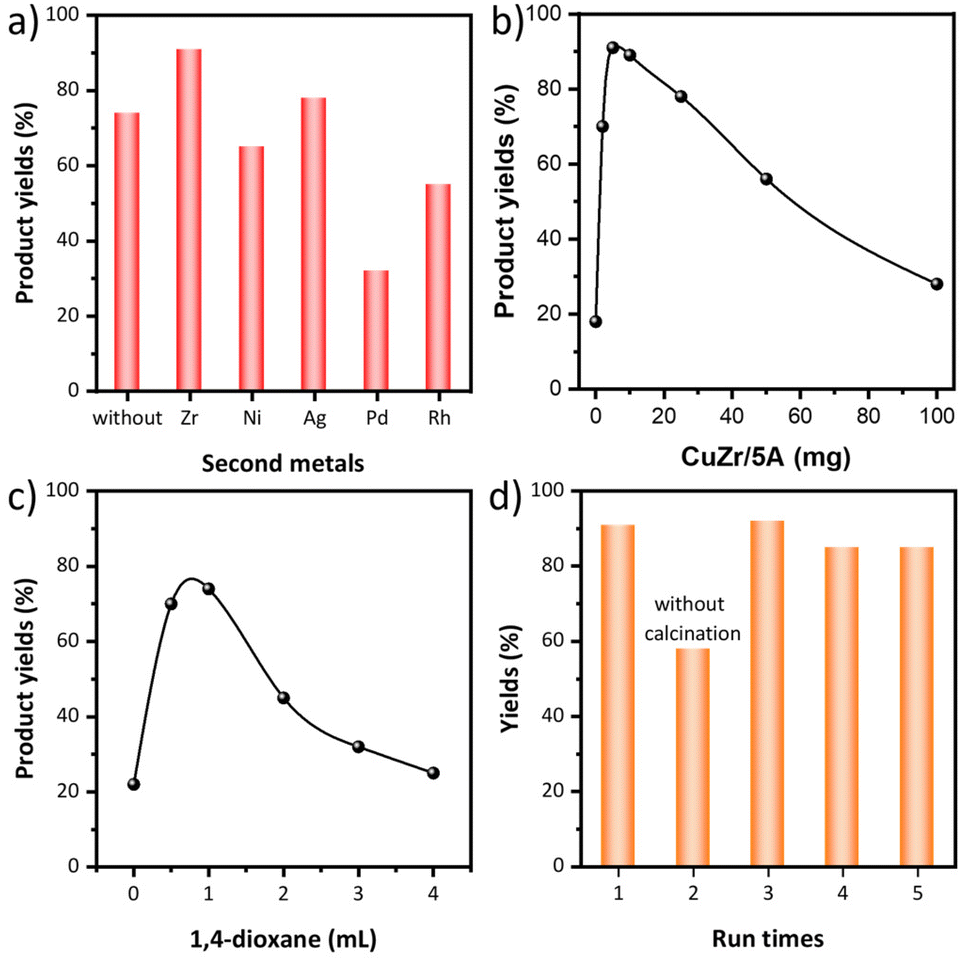 | ||
| Fig. 1 (a) Activity test of different catalysts. Reaction conditions: 0.33 mmol DHA (1 mmol carbon source), 3 mmol aniline, 6 mmol H2O2 (35 wt%), 5 mg catalysts, 2 mL 1,4-dioxane, 50 °C, 24 h. The effect of (b) catalyst (reaction conditions: 0.33 mmol DHA, 3 mmol aniline, 6 mmol H2O2 (35 wt%), 0–100 mg CuZr/5A, 2 mL 1,4-dioxane, 50 °C, 24 h) and (c) solvent amount (reaction conditions: 0.33 mmol DHA, 3 mmol aniline, 4 mmol H2O2 (35 wt%), 5 mg CuZr/5A, 0–4 mL 1,4-dioxane, 50 °C, 12 h) on the catalytic performance. (d) Recyclability of the active catalyst CuZr/5A (reaction conditions: 0.33 mmol DHA, 3 mmol aniline, 6 mmol H2O2 (35 wt%), 5 mg CuZr/5A, 2 mL 1,4-dioxane, 50 °C, 24 h). | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalysts | Aniline/mmol | DHA/mmol | H2O2/mmol | Yieldsb/% | UECAc |
a Reaction conditions: catalysts 5 mg, aniline 2–3 mmol, DHA 0.33 mmol (1 mmol carbon), H2O2 (35 wt%) 2–6 mmol, 1,4-dioxane 2 mL, 50 °C, 24 h.
b Yields were determined by GC-FID with biphenyl as the external standard and calculated based on the carbon atoms in DHA.
c Utilization efficiency of carbon atoms (UECA) in DHA = 100% × (nformamide product yields with DHA − nformamide product yields without DHA)/ncarbon atoms in DHA (mol/mol).
d Cu/Zr = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5 (mol/mol).
e 1 mL 1,4-dioxane.
f 12 h. :0.5 (mol/mol).
e 1 mL 1,4-dioxane.
f 12 h.
|
||||||
| 1 | CuZr/5A | 2 | 0.33 | 6 | 61 | 61 |
| 2 | CuZr/5A | 3 | 0.33 | 5 | 46 | 46 |
| 3 | CuZr/5A | 3 | 0.33 | 4 | 45 | 45 |
| 4 | CuZr/5A | 3 | 0.33 | 3 | 28 | 28 |
| 5 | CuZr/5A | 3 | 0.33 | 2 | 22 | 22 |
| 6d | CuZr/5A | 3 | 0.33 | 6 | 72 | 72 |
| 7 | 5A | 3 | 0.33 | 6 | 49 | 49 |
| 8 | CuZr/5A | 3 | — | 6 | 0 | 0 |
| 9e | CuZr/5A | 3 | 0.33 | 4 | 90 | 90 |
| 10f | CuZr/5A | 3 | 0.33 | 6 | 43 | 43 |

Considering the similar results with 4 mmol and 5 mmol H2O2, further catalyst screening and reaction parameter optimization were performed with 10 mg catalyst and 4 mmol H2O2. The catalytic test results showed the same trend as before and CuZr/5A is still the most active catalyst (Fig. S1a†). The Zr/5A catalyst delivered lower product yields than Cu/5A, suggesting Cu as the main active component. The control experiments without DHA showed that no formamide products were formed in the case of all catalysts when DHA was removed from the reaction system (Table S1,† entries 1–8), implying that all the prepared catalysts can't catalyze the reaction of aniline with dioxane to form the desired formanilide and DHA should be the carbonyl source for formamide formation. Thus, the yield of the formamide product, which was calculated based on the carbon atoms of DHA, is directly equal to the utilization efficiency of the carbon atom (Fig. S1a†). The highest product yields of 78% was obtained with 10 mg catalyst when changing the catalyst dosage from 0–100 mg, and both reduced and increased catalyst dosages lowered the product yields (Fig. S1b†). A test of the solvent amount effect revealed that 1 mL 1,4-dioxane was optimal, providing 93% product yield (Fig. S1c†). Reducing the catalyst dosage to 5 mg slightly decreased the product yield (Fig. 1c and Table 1, entry 9). The product yields continuously decreased when the reaction temperature was gradually lowered from 50 to 25 °C (Table S1,† entries 9 and 10).
Recyclability tests revealed a drop in the product yield after the first run; however, activity has been restored by re-calcination (Fig. 1d). After this, the catalyst could be used for another 3 runs without any obvious activity loss. To test whether the reaction is indeed heterogeneously catalyzed, the catalyst was removed by hot filtration after a 12 h reaction (Table 1, entry 10). With the filtered solution the reaction was continued for another 12 h, but no more products were formed, indicating that the supported metal species are the real active species. In order to investigate the activity and durability of the CuZr/5A catalyst for practical applications, we further tested its catalytic performance using a continuous reactor. Within the 78 h continuous test, the catalytic activity stabilized after 24 h with slightly decreased formamide product yields of 76–78% due to the dilution of H2O2 required for safe operation (Fig. S1d†).
With the optimized reaction conditions, the scope of the substrates and their tolerance towards functional groups were evaluated (Table 2). 85–99% product yields were obtained when aniline and its derivatives with the groups of –CH3 and –Cl were used as the substrates, but only a moderate yield was obtained with aniline containing the strong electron-withdrawing group –F. Aminoindane could be N-formylated with 95% product yield and aliphatic primary amines and secondary amines were also well tolerated. Benzylamine and its derivatives with –CH3, –OCH3, –Cl and –F substituents also gave excellent product yields although higher amounts of amines were required. In comparison with benzylic amines, aliphatic primary amines showed slightly better reactivity and excellent product yields were obtained in some cases even with only 3 mmol of amine. Aromatic and aliphatic secondary amines, i.e. N-methylaniline and morpholine, gave 72% and 93% product yields, respectively.
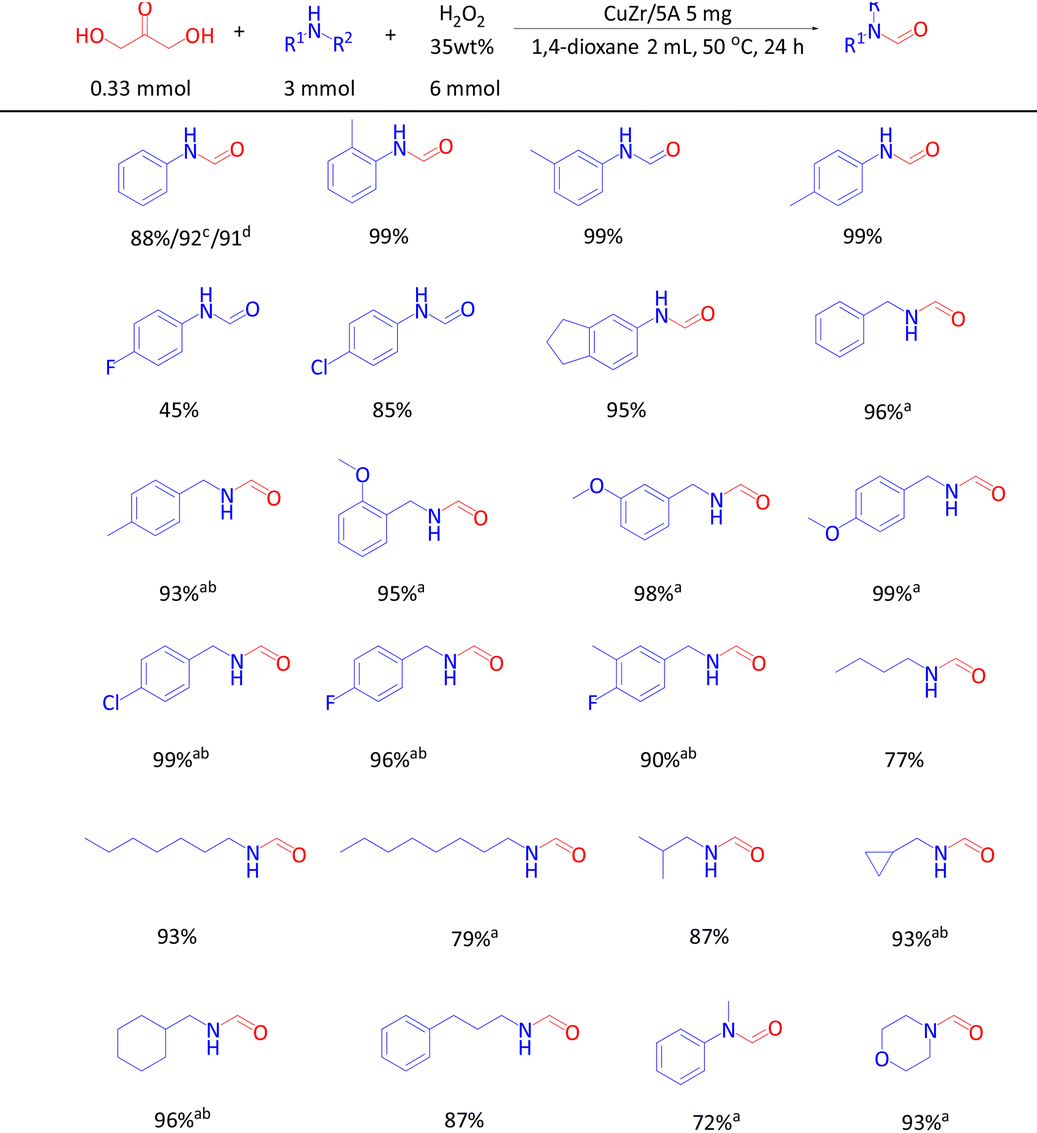
Inspired by the successful synthesis of various formamides by direct N-formylation with DHA, other glycerol derivatives were also explored as carbonyl sources in the reaction with aniline to formanilide. Thus, 92% and 91% product yields were obtained, respectively, when GLA and GA were used as the carbonyl sources.
Catalyst characterization
To gain insight into the correlation between the structure and activity, the catalysts were comprehensively characterized by inductively coupled plasma optical emission spectrometry (ICP-OES), N2-adsorption–desorption analysis, X-ray powder diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), EPR and NH3-temperature programmed desorption (TPD). ICP-OES revealed a similar Cu content (0.51–0.56 wt%) in all catalysts (Table S2†). N2-Adsorption–desorption analysis showed that the introduction of the second metal lowered the BET surface area and total pore volume (Table S2 and Fig. S2†), suggesting that a part of pores might be blocked. The N2 adsorption–desorption isotherm and pore size distribution patterns showed that all the catalysts have similar pore structures with pore sizes of 3.4–4.2 nm and pore volumes of 0.362–0.512 cm3 g−1 (Table S2 and Fig. S2†). The most active catalyst CuZr/5A has a medium pore size of 3.6 nm and a pore volume of 0.441 cm3 g−1. Although there is no obvious linear correlation between the activity and pore structure, some speculations can be made on the effect of the pore structure on the reaction: (1) the pore size of >4 nm may be adverse for the reaction because CuPd/5A with the lowest activity is the only catalyst with a pore size >4 nm; (2) the pore size of 3.6 nm may be more favorable to the reaction because both CuZr/5A and CuAg/5A catalysts with the same pore size of 3.6 nm exhibited better activity than Cu/5A. XRD patterns show the typical reflections of zeolite 5A with an LTA-type crystal structure60 and these do not change after introducing the metals, indicating that the zeolite structure remains stable (Fig. S3†). No reflections of Cu or second metal species were detected, as expected, given the low metal content.The XPS spectra of all catalysts show a Cu 2p3/2 peak centered at around 934 eV and a weak satellite peak at around 943 eV (Fig. S4†), suggesting the presence of surface CuII species.61,62 All second metals are present as high valent ions, i.e. NiII (Ni 2p3/2 at 856.61 eV),63 and 3d5/2 peaks of ZrIV (182.36 eV),64 AgI (368.23 eV),65 PdII (337.06 eV)66 and RhIII (309.40 eV)67 (Fig. S4†). Only in the case of Rh some metallic Rh0 species are also present (3d5/2 peak at 306.50 eV).68
The HAADF-STEM images of the CuZr/5A catalyst show some tube-like particles (Fig. 2a) typical of the support. While Cu seems to be homogeneously distributed throughout the whole sample (Fig. 2b and c), Zr is enriched in particles of different shapes and sizes (Fig. 2c and e) and is also present on the surface in general as indicated in the spectrum of the central part (Fig. 2b and f). On the one hand, Zr forms larger particles attached to the support in general (Fig. 2e). On the other hand, a thin layer of very small clusters seems to cover the surface of the tube-like particles (Fig. 2f). In any case, Cu species are highly dispersed and no distinct Cu clusters were observed.
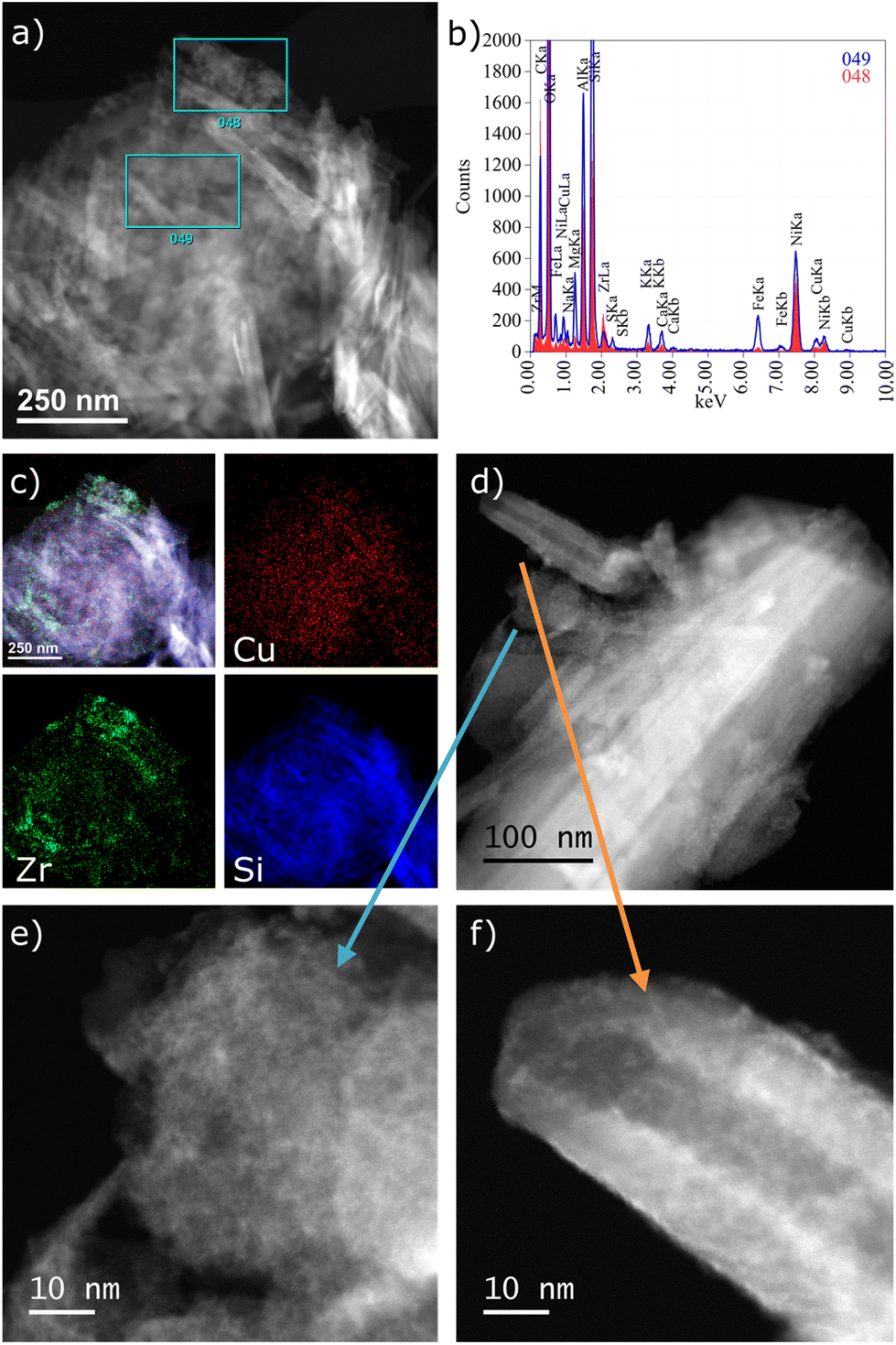 | ||
| Fig. 2 (a) Overview HAADF-STEM image, (b) EDX spectra from regions marked in (a), (c) EDX elemental maps of CuZr/5A, (d) more detailed HAADF-STEM image with Zr containing regions highlighted in (e), showing a rather large amorphous particle, and (f), showing bright smaller clusters adhering to the support surface. | ||
To gain specific insight into the structure of the CuII centres, the EPR spectra of the catalysts were recorded at 95 K and 298 K, respectively (Fig. 3a and S5†). The EPR spectrum of Cu/5A shows an axial signal at g⊥ = 2.06 and g∥ = 2.36 with a hyperfine structure (hfs) A∥ = 139 G due to the coupling of the single electron spin of CuII (d9, S = 1/2) with the nuclear spin of 65,63CuII (I = 3/2), indicating the presence of isolated CuII ions.69,70 Additionally, a broad structureless singlet is superimposed on the hfs signal, suggesting the presence of magnetically interacting CuII ions.71 A relative measure of the strength and nature of such interaction is the temperature dependence of the EPR signal intensity. For pure paramagnetic CuII species without any mutual magnetic interactions, the intensity of the EPR signal is inversely proportional to temperature. For monometallic Cu/5A, the intensity ratio I95 K/I298 K = 2.31 (Fig. S5†) is lower than that expected for pure paramagnetic behavior (I95/I298 = 3.13). This indicates that a part of the CuII sites interacts antiferromagnetically. However, since TEM micrographs did not show any Cu-containing particles, such antiferromagnetic CuII moieties might be very small and/or not connected by direct Cu–O–Cu bridges.
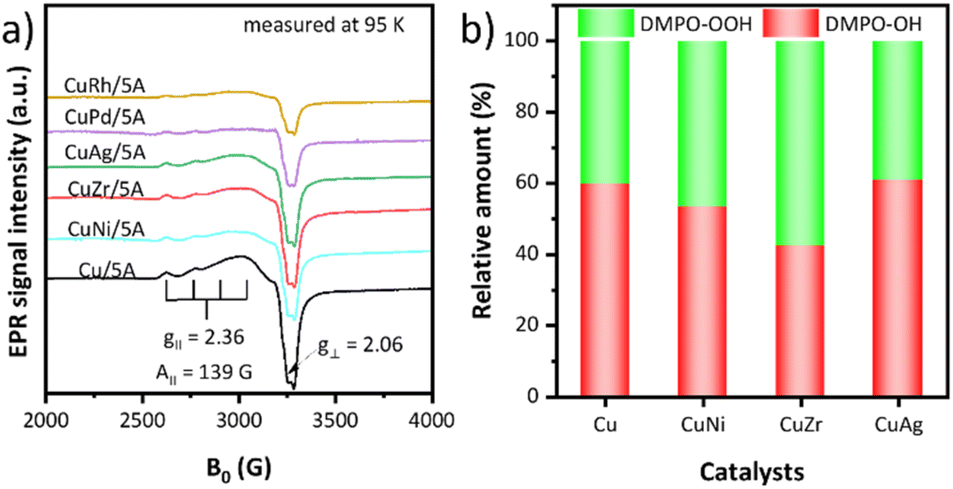 | ||
| Fig. 3 (a) EPR spectra of the prepared catalysts recorded at 95 K. (b) H2O2 activation abilities of catalysts. | ||
Introduction of a second metal led to EPR spectra with a similar shape but a lower intensity, despite the almost same Cu content (Table S2†). This suggests that antiferromagnetic coupling between the CuII ions is strengthened and/or the size of antiferromagnetic CuxOy moieties increased, whereby the latter might still be below the detection limit of TEM. The I95 K/I298 K EPR intensity ratios decrease in the order Cu/5A > CuAg/5A > CuNi/5A = CuZr/5A > CuRh/5A > CuPd/5A (Fig. S5†), suggesting that the second metals promote clustering of Cu in the same order.
To reveal the reason for the regeneration of catalytic activity after calcination treatment, the EPR spectra of the used CuZr/5A catalyst before and after calcination treatment were recorded at 105 K (Fig. S5†). Clearly, compared with the fresh CuZr/5A catalyst, the EPR signal intensity of the used CuZr/5A catalyst decreased significantly, suggesting that some CuII species were reduced to CuI during the reaction. The signal intensity increased again after high temperature calcination treatment, indicating that the reduced CuI can be re-oxidized to CuII by the high temperature calcination process.
The deconvolution results of NH3–TPD patterns revealed four desorption peaks at different temperatures in all catalysts, suggesting the presence of four different kinds of acidic sites (Fig. S6†). Two strong peaks centered at around 220 and 290 °C can be attributed to the weak acidic sites while the other two weak peaks centered at around 520 and 620 °C correspond to the medium and strong acidic sites, respectively. Quantification of the acidic sites showed that the exchange of protons by Cu ions reduced the number of acidic sites, as expected, and this trend continued by introducing a second metal (Table 3). The most active catalyst CuZr/5A has the lowest amount of medium–strong acidic sites, which can be attributed to the fact that the introduction of higher valence ZrIV ions replaced more H+ in zeolite 5A. This suggests that those sites are detrimental for high catalytic performance since they may lead to strong adsorption of aniline and/or hinder product desorption.
| Catalysts | Total acidic sites/mmol g−1 | Acidic sites/mmol g−1 | |
|---|---|---|---|
| Weak | Medium–strong | ||
| 5A | 2.30 | 1.65 | 0.65 |
| Cu/5A | 1.92 | 1.33 | 0.59 |
| CuNi/5A | 1.62 | 1.22 | 0.40 |
| CuZr/5A | 1.54 | 1.25 | 0.29 |
| CuAg/5A | 1.79 | 1.38 | 0.41 |
| CuPd/5A | 1.69 | 1.30 | 0.39 |
| CuRh/5A | 1.78 | 1.30 | 0.48 |
The ability of the catalysts to activate H2O2 was evaluated by EPR analyzing the kind and relative amount of radicals generated from H2O2 after trapping with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a spin trap (Fig. 3b).72 To exclude the effect of H2O2 decomposition resulting from the reaction temperature, the experiments were performed at room temperature and a detailed description of these experiments is provided in the ESI (SI5).† After adding DMPO to a suspension of the catalyst in H2O2, ˙OH and ˙OOH radicals were detected in all cases. The relative percentage of both radicals was calculated and the ˙OH/˙OOH ratio is 59.7/40.3 of Cu/5A, 53.4/46.6 of CuNi/5A, 42.3/57.7 of CuZr/5A, and 60.7/39.3 of CuAg/5A, respectively. The detection in the case of CuPd/5A and CuRh/5A failed due to the formation of intense gas bubbles caused from fast H2O2 decomposition, probably explaining their poor catalytic performance.73 Clearly, the most active CuZr/5A catalyst produces the highest proportion of ˙OOH radicals, suggesting that Zr can promote the selective formation of the latter (Fig. 3b) and that the higher percentage of ˙OOH should be favorable for the reaction. However, no linear relationship was observed between the proportion of ˙OOH radicals and the catalytic activity. Thus, it can be concluded that the ˙OH/˙OOH ratio is an important factor rather than the only one to determine the activity of catalysts and the ˙OH/˙OOH ratio of 42.3/57.7 proved to be the optimal among our screened results.
To uncover the effect of Zr on the Cu species, the EPR spectra of Cu/5A and CuZr/5A before and after interaction with H2O2 and aniline were compared (Fig. S7†). An obvious decrease of the EPR signal was observed for Cu/5A after contact with H2O2 while the spectrum of CuZr/5A remained almost unchanged. This suggests that H2O2 can be decomposed faster over Cu/5A than CuZr/5A, which is probably due to a higher amount of H+ sites in Cu/5A than in CuZr/5A, as the introduction of ZrIV ions replaced more H+ in zeolite 5A. According to the Fenton-like reaction of CuII/I species with H2O2, the presence of H+ favored the decomposition of H2O2 to ˙OH radicals.59 Also, the EPR signals of Cu/5A decreased much more upon contact with aniline but not in the case of CuZr/5A, implying that compared to CuZr/5A, CuII species in Cu/5A are more easily reduced to CuI, which is favorable for the formation of ˙OH radicals (eqn (2)). Based on these results, the fact that Cu/5A has a higher proportion of ˙OH radicals than CuZr/5A can be well understood.
Study of the reaction mechanism
To gain deep mechanistic insight, the reaction of aniline and DHA was investigated for the CuZr/5A catalyst by EPR spin trapping experiments with DMPO and operando ATR-IR. As mentioned above, the signals of DMPO-OH and DMPO-OOH spin adducts appeared after suspending the catalyst in aqueous H2O2, indicating the formation of ˙OH and ˙OOH radicals (Fig. 4). Subsequent addition of the DHA substrate dissolved in 1,4-dioxane results in the appearance of a new EPR signal, suggesting the formation of a new radical (Fig. 4a middle). Spectral simulation revealed the formation of organic carbon-centered radicals ˙R accompanied by a decrease of the relative amount of ˙OH from 51.7% to 18.8% (Fig. 4b, B). This might be due to the attack of DHA by ˙OH to produce ˙C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)CH2OH and ˙CH2OH radicals.47 After adding aniline, a new EPR signal appeared (Fig. 4a top) which must be assigned to the DMPO–˙N spin adduct as evident from the spectral fit.74 Most probably a ˙NHPh radical intermediate was formed (42.2%, Fig. 4b, C). This intermediate might stem from the attack of aniline by ˙OOH radicals52 since the relative amount of DMPO-OOH decreased from 63.6% to 25.6% immediately after adding aniline while the amount of DMPO-OH declined only slightly (Fig. 4b, C). After reaction for 1 h, the concentration of both ˙NHPh and ˙R radicals decreased, probably due to the formation of non-radical formamide products (Fig. 4b, D). Interestingly, the ˙NHPh radicals disappeared much faster than the ˙R radicals, suggesting that ˙NHPh radicals are not only involved in the synthesis of formamide products but also in the formation of by-products such as azobenzenes and azoxybenzenes, identified by GC-MS. After 1 h, the relative amount of ˙OOH radicals increased again due to the interaction of the catalyst and excess H2O2.
O)CH2OH and ˙CH2OH radicals.47 After adding aniline, a new EPR signal appeared (Fig. 4a top) which must be assigned to the DMPO–˙N spin adduct as evident from the spectral fit.74 Most probably a ˙NHPh radical intermediate was formed (42.2%, Fig. 4b, C). This intermediate might stem from the attack of aniline by ˙OOH radicals52 since the relative amount of DMPO-OOH decreased from 63.6% to 25.6% immediately after adding aniline while the amount of DMPO-OH declined only slightly (Fig. 4b, C). After reaction for 1 h, the concentration of both ˙NHPh and ˙R radicals decreased, probably due to the formation of non-radical formamide products (Fig. 4b, D). Interestingly, the ˙NHPh radicals disappeared much faster than the ˙R radicals, suggesting that ˙NHPh radicals are not only involved in the synthesis of formamide products but also in the formation of by-products such as azobenzenes and azoxybenzenes, identified by GC-MS. After 1 h, the relative amount of ˙OOH radicals increased again due to the interaction of the catalyst and excess H2O2.
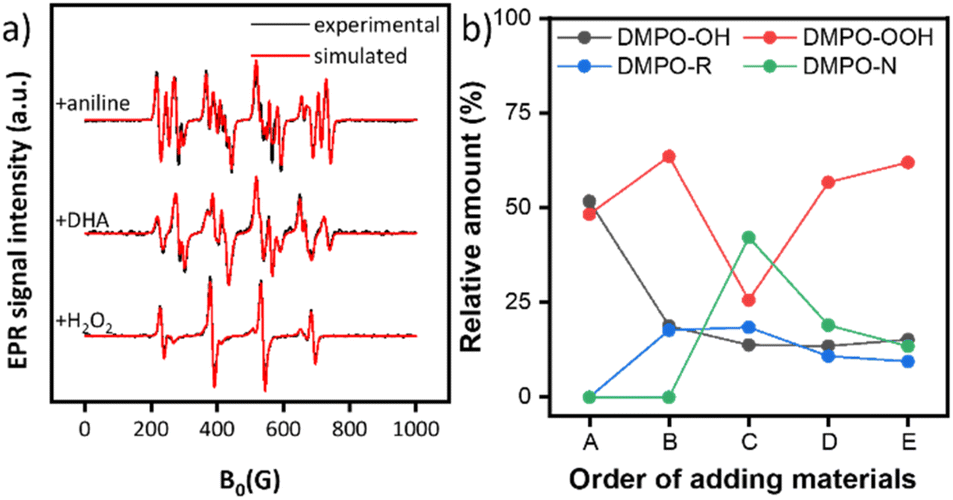 | ||
| Fig. 4 (a) Experimental (black) and fitted EPR spectra (red) of DMPO spin adducts. For fitting, the following hfs parameters were used:74AN = 14.3, AH = 12.3 G for ˙OH, AN = 13.3, AβH = 11.1, AγH = 1.2 G for ˙OOH, AN = 15.1, AH = 20.9 G for ˙R, and AN1 = 14.9, AH = 16.3 G, AN2 = 2.7 for ˙NHPh. (b) Relative amount of the DMPO-X (X = OH, OOH, R, NHPh) spin adducts. A: CuZr/5A + H2O2, B: +DHA in 1,4-dioxane, C: +aniline, and D: +1 h, E: +4 h. | ||
In addition, the reaction of aniline and DHA was tracked by operando ATR-FTIR (Fig. 5a). Two bands at 1697 and 1542 cm−1 increased gradually with increasing reaction time and they can be respectively assigned to the ν(CO) and ν(C–N) vibrations of the formamide product by comparison with a reference sample of formanilide in dioxane (Fig. S8†). Due to the interference of the broad background signal of water (formed during the reaction from H2O2), bands in the carbonyl region above the ν(CO) band of formamide are hardly discernable. Therefore, the 2nd derivative of the IR spectra was used to better visualize the changes in this region (Fig. 5b), in which two new IR bands at 1710 and 1728 cm−1 appeared besides the one of formanilide at 1697 cm−1 once H2O2 was added. They can be assigned to the ν(CO) vibration of the reaction intermediates formic acid (FA) and glycolic acid (GA), respectively (Fig. 5b).47 As confirmed by control experiments, these intermediates are formed by the reaction of DHA with H2O2 (Scheme 2b) and react further with aniline to formanilide (Scheme 2c and d). They were also identified by 1H-NMR (Fig. S9†) although the observed signal peak of formic acid is very weak, which may be due to the oxidative decomposition of most formic acid to CO2 caused by excess H2O2. The quantitative analysis revealed the formation of 0.05 mmol FA, so the FA yields were 15% based on the added DHA (0.33 mmol).
 | ||
| Fig. 5 (a) Operando ATR-FTIR spectra. (b) 2nd derivative of the ATR-FTIR spectra. | ||
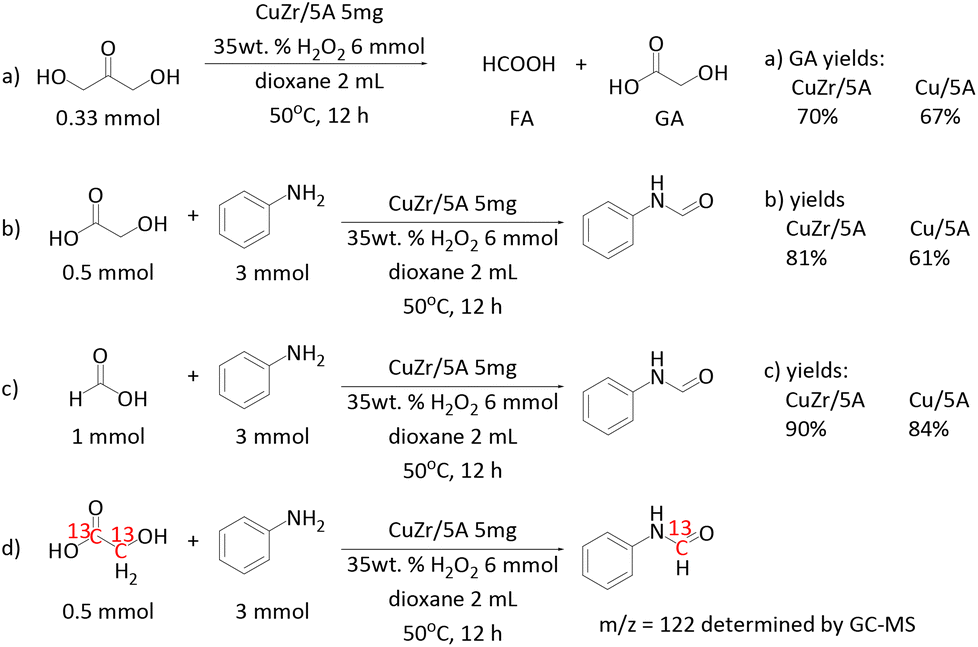 | ||
| Scheme 2 Control experiments. | ||
To learn more about the beneficial role of Zr in the catalytic reaction, the performances of Cu/5A and CuZr/5A were compared in the C–C bond cleavage of DHA and GA, and in C–N bond formation (Scheme 2a–c). The CuZr/5A catalyst gave a little higher GA yield than Cu/5A (Scheme 2a), suggesting a slightly better activity in the C–C bond cleavage of DHA. A similar positive effect was also observed in the reaction of aniline and FA (Scheme 2c), implying a certain promotion of C–N bond formation by Zr. An even more positive effect of Zr is evident in the reaction of GA and aniline (Scheme 2b). Here the yield of formanilide increased from 61% to 81% after introducing Zr. This suggests that Zr might especially promote the C–C bond cleavage of GA.
For elucidating the origin of carbon atoms in the carbonyl group of the formamide product, isotopic labelling experiments are the method of choice. Unfortunately, 13C labelled DHA was not available. However, as mentioned above, no formanilide was formed by the reaction of H2O2 with aniline in 1,4-dioxane without DHA. This suggests that it is DHA and not the solvent that acts as the carbonyl source. In our previous work we have shown that the reaction of aniline and DHA can enable the co-synthesis of GA and formanilide, indicating that one of the three carbon atoms in DHA can be converted and transferred into the formanilide product.47 Furthermore, the reaction of 13C labelled GA and aniline resulted in a formanilide product giving the m/z = 122 fragment in mass spectrometry (Scheme 2d). This confirms clearly that the carbonyl carbon atoms in the formanilide product stem from GA, which was also confirmed by 13C NMR (Fig. S10†). Based on these results, we conclude that all carbon atoms in DHA are converted and transferred into the formamide product.
Considering all the above findings, we propose a possible reaction mechanism (Scheme 3). First, H2O2 is activated by the catalyst to produce ˙OH radicals that selectively split a C–C bond in DHA resulting in two organic carbon-centered radical intermediates ˙C(O)CH2OH and ˙CH2OH. These react with ˙OH radicals to give intermediates GA and FA, respectively. The latter reacts further with the aniline substrate to afford the desired formamide product even without any catalyst.21,55,75 The formation of GA and FA intermediates has been confirmed by operando ATR-FTIR and 1H-NMR. Besides, ˙OOH radicals (according to eqn (1)) can cut the C–C bond in GA to form FA and new carbon-centered radicals, most probably ˙COOH. In parallel, ˙OOH can also react with aniline to form ˙NHPh radicals, which combine with ˙COOH to give EPR-silent formamide, as indicated by EPR spin trapping experiments with DMPO.
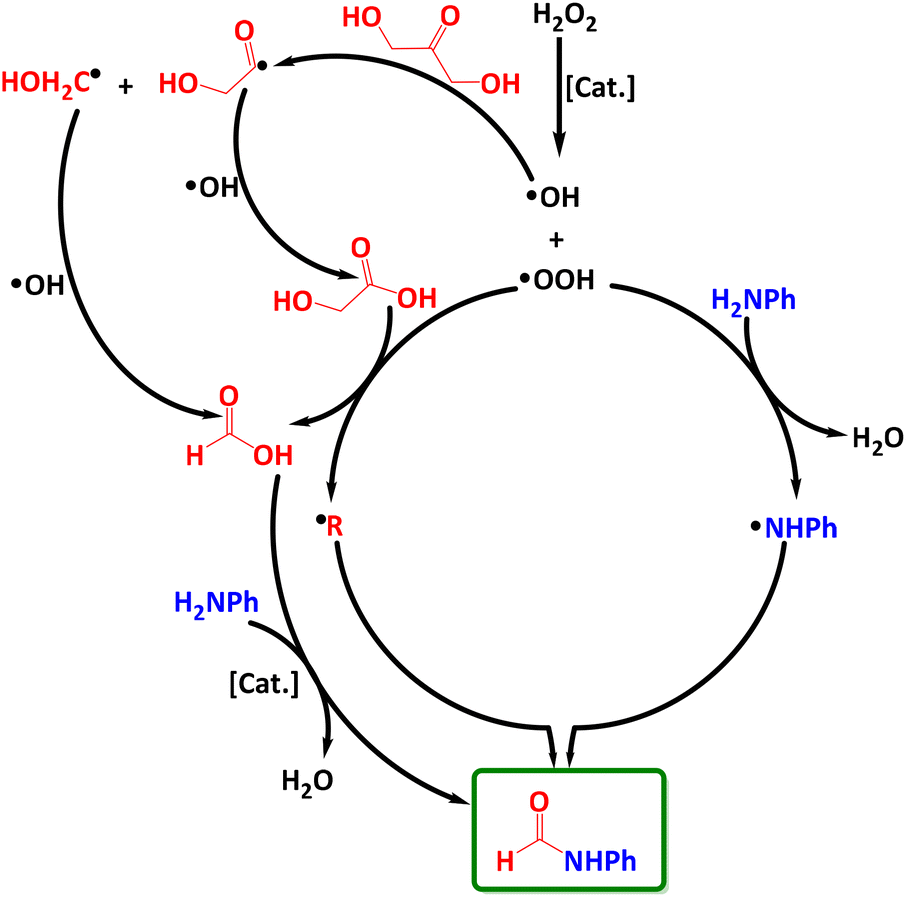 | ||
| Scheme 3 Proposed reaction mechanism. | ||
Conclusions
In this work, we developed active CuM/5A (M = Ni, Zr, Ag, Pd and Rh) catalysts for the sustainable synthesis of valuable formamide by the reaction of amines with glycerol derivatives as the carbonyl source. Compared to Cu/5A, the introduction of the second metals Zr and Ag increased the yield of the formamide target products while it was lowered by introducing other second metals. The most active catalyst CuZr/5A is recyclable and can be consecutively run 4 times without any obvious activity loss, but re-calcination is necessary to restore the activity by removing surface deposits. It has been shown that various amines with diverse structures can react with DHA to provide the corresponding formamide in good yields. Besides, other glycerol derivatives GLA and GA are also well tolerated. Isotopic labelling and control experiments confirmed that all carbon atoms of the formyl group in the formamide product stem from DHA in the reaction of amines and DHA. We found that the incorporation of Zr reduced the amount of surface acidic sites, in particular, the medium–strong acidic sites and led to the selective formation of more ˙OOH radicals, which might be responsible for the better catalytic performance of CuZr/5A than Cu/5A. EPR spin-trapping, operando ATR-IR and control experiments indicated that the reaction of aniline and DHA proceeds probably via a radical relay mechanism in which the C–C bond is split by ˙OH and ˙OOH radicals successively. The formation of formamide involved the intermediates GA, FA ˙CH2OH, ˙C(O)CH2OH and ˙NHPh radicals. The concept of using glycerol derivatives directly as the carbonyl source via oxidative C–C bond cleavage to synthesize valuable formamides can be extended to the valorization of other biomass-based platform molecules.
Author contributions
Xingchao Dai (investigation, methodology, supervision, and writing – original draft); Xinzhi Wang (investigation and methodology); Carsten Kreyenschulte (methodology); Hangkong Yuan (investigation); Angelika Brückner (conceptualization, funding acquisition, supervision, and writing – review & editing); Feng Shi (conceptualization, funding acquisition, supervision, and writing – review & editing); Jabor Rabeah (conceptualization, supervision, and writing – review & editing).Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The National Natural Science Foundation of China (21961132025 and 21925207), the Light of West China of CAS, Gansu Science and Technology Major Project (21ZD4WA021) and Deutsche Forschungsgemeinschaft (grant no. BR 1380/27-1) are gratefully acknowledged.References
- L. C. Meher, D. Vidya Sagar and S. N. Naik, Renewable Sustainable Energy Rev., 2006, 10, 248–268 CrossRef CAS.
- F. Karaosmanoǧlu, K. B. Cıǧızoǧlu, M. Tüter and S. Ertekin, Energy Fuels, 1996, 10, 890–895 CrossRef.
- J. Chowdhury and K. Fouhy, Chem. Eng., 1993, 100, 35–39 CAS.
- A. Talebian-Kiakalaieh, N. A. S. Amin, K. Rajaei and S. Tarighi, Appl. Energy, 2018, 230, 1347–1379 CrossRef CAS.
- P. M. Walgode, R. P. V. Faria and A. E. Rodrigues, Catal. Rev., 2021, 63, 422–511 CrossRef CAS.
- Z. He, X. Ning, G. Yang, H. Wang, Y. Cao, F. Peng and H. Yu, Catal. Today, 2021, 365, 162–171 CrossRef CAS.
- L. Yang, X. Li, P. Chen and Z. Hou, Chin. J. Catal., 2019, 40, 1020–1034 CrossRef CAS.
- G. Dodekatos, S. Schünemann and H. Tüysüz, ACS Catal., 2018, 8, 6301–6333 CrossRef CAS.
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, L. Prati and G. J. Hutchings, Acc. Chem. Res., 2015, 48, 1403–1412 CrossRef CAS PubMed.
- M. Pera-Titus and F. Shi, ChemSusChem, 2014, 7, 720–722 CrossRef CAS PubMed.
- M. Besson, P. Gallezot and C. Pinel, Chem. Rev., 2014, 114, 1827–1870 CrossRef CAS PubMed.
- B. Katryniok, H. Kimura, E. Skrzyńska, J.-S. Girardon, P. Fongarland, M. Capron, R. Ducoulombier, N. Mimura, S. Paul and F. Dumeignil, Green Chem., 2011, 13, 1960–1979 RSC.
- C.-H. Zhou, J. N. Beltramini, Y.-X. Fan and G. Q. Lu, Chem. Soc. Rev., 2008, 37, 527–549 RSC.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- E. Arunan, J. Phys. Chem. A, 1997, 101, 4838–4839 CrossRef CAS.
- J. Omprakash Rathi and G. Subray Shankarling, ChemistrySelect, 2020, 5, 6861–6893 CrossRef CAS.
- Y. Zhang, X. Dai, H. Wang and F. Shi, Acta Phys.-Chim. Sin., 2018, 34, 845–857 CAS.
- A. Tlili, E. Blondiaux, X. Frogneux and T. Cantat, Green Chem., 2015, 17, 157–168 RSC.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- N. Lukasik and E. Wagner-Wysiecka, Curr. Org. Synth., 2014, 11, 592–604 CrossRef CAS.
- C. J. Gerack and L. McElwee-White, Molecules, 2014, 19, 7689–7713 CrossRef PubMed.
- X. J. Yang and Y. S. Zhang, Res. Chem. Intermed., 2013, 39, 2843–2848 CrossRef CAS.
- R. M. Lanigan and T. D. Sheppard, Eur. J. Org. Chem., 2013, 7453–7465 CrossRef CAS.
- S. Ding and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 9226–9237 CrossRef CAS PubMed.
- C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- G. A. Olah, L. Ohannesian and M. Arvanaghi, Chem. Rev., 1987, 87, 671–686 CrossRef CAS.
- M. Nasrollahzadeh, N. Motahharifar, M. Sajjadi, A. M. Aghbolagh, M. Shokouhimehr and R. S. Varma, Green Chem., 2019, 21, 5144–5167 RSC.
- K. Beydoun and J. Klankermayer, Recent Advances on CO2 Utilization as C1 Building Block in C-N and C-O Bond Formation, Springer International Publishing, Cham, 2019.
- K. Dong, R. Razzaq, Y. Hu and K. Ding, Top. Curr. Chem., 2017, 375, 23 CrossRef PubMed.
- Z. Li, Z. Yu, X. Luo, C. Li, H. Wu, W. Zhao, H. Li and S. Yang, RSC Adv., 2020, 10, 33972–34005 RSC.
- M. Hulla and P. J. Dyson, Angew. Chem., Int. Ed., 2020, 59, 1002–1017 CrossRef CAS PubMed.
- X.-F. Liu, X.-Y. Li and L.-N. He, Eur. J. Org. Chem., 2019, 2437–2447 CrossRef CAS.
- G. Yuan, C. Qi, W. Wu and H. Jiang, Curr. Opin. Green Sustainable Chem., 2017, 3, 22–27 CrossRef.
- K. P. Dhake, P. J. Tambade, R. S. Singhal and B. M. Bhanage, Green Chem. Lett. Rev., 2011, 4, 151–157 CrossRef CAS.
- I. Metaxas, E. Vasilikogiannaki and M. Stratakis, Nanomaterials, 2017, 7, 440 CrossRef PubMed.
- Z. Ke, Y. Zhang, X. Cui and F. Shi, Green Chem., 2016, 18, 808–816 RSC.
- N. Shah, E. Gravel, D. V. Jawale, E. Doris and I. N. N. Namboothiri, ChemCatChem, 2014, 6, 2201–2205 CrossRef CAS.
- L. Zhou, C. G. Freyschlag, B. Xu, C. M. Friend and R. J. Madix, Chem. Commun., 2010, 46, 704–706 RSC.
- B. Xu, L. Zhou, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2010, 49, 394–398 CrossRef CAS PubMed.
- O. Saidi, M. J. Bamford, A. J. Blacker, J. Lynch, S. P. Marsden, P. Plucinski, R. J. Watson and J. M. J. Williams, Tetrahedron Lett., 2010, 51, 5804–5806 CrossRef CAS.
- P. Preedasuriyachai, H. Kitahara, W. Chavasiri and H. Sakurai, Chem. Lett., 2010, 39, 1174–1176 CrossRef CAS.
- X. Dai and F. Shi, Curr. Opin. Green Sustainable Chem., 2020, 22, 1–6 CrossRef.
- K. Natte, H. Neumann, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2017, 56, 6384–6394 CrossRef CAS PubMed.
- L. M. Kabadwal, S. Bera and D. Banerjee, Org. Chem. Front., 2021, 8, 7077–7096 RSC.
- S. Jalwal, V. Atreya, T. Singh and S. Chakraborty, Tetrahedron Lett., 2021, 82, 153362 CrossRef CAS.
- X. Dai, S. Adomeit, J. Rabeah, C. Kreyenschulte, A. Brückner, H. Wang and F. Shi, Angew. Chem., Int. Ed., 2019, 58, 5251–5255 CrossRef CAS PubMed.
- X. Dai, J. Rabeah, H. Yuan, A. Brückner, X. Cui and F. Shi, ChemSusChem, 2016, 9, 3133–3138 CrossRef CAS PubMed.
- X. Dai, X. Cui, Y. Deng and F. Shi, RSC Adv., 2015, 5, 43589–43593 RSC.
- X. Cui, Y. Deng and F. Shi, ACS Catal., 2013, 3, 808–811 CrossRef CAS.
- X. Cui, C. Zhang, F. Shi and Y. Deng, Chem. Commun., 2012, 48, 9391–9393 RSC.
- X. Dai, X. Wang, J. Rabeah, C. Kreyenschulte, A. Brückner and F. Shi, Chem. – Eur. J., 2021, 27, 16889–16895 CrossRef CAS PubMed.
- J. Wang, X. Dai, H. Wang, H. Liu, J. Rabeah, A. Brückner, F. Shi, M. Gong and X. Yang, Nat. Commun., 2021, 12, 6840 CrossRef CAS PubMed.
- P. Pullanikat, S. J. Jung, K. S. Yoo and K. W. Jung, Tetrahedron Lett., 2010, 51, 6192–6194 CrossRef CAS PubMed.
- B. Das, M. Krishnaiah, P. Balasubramanyam, B. Veeranjaneyulu and D. Nandan Kumar, Tetrahedron Lett., 2008, 49, 2225–2227 CrossRef CAS.
- G. Brahmachari and S. Laskar, Tetrahedron Lett., 2010, 51, 2319–2322 CrossRef CAS.
- B. Krishnakumar and M. Swaminathan, J. Mol. Catal. A: Chem., 2011, 334, 98–102 CrossRef CAS.
- M. Nasrollahzadeh, S. M. Sajadi and A. Hatamifard, J. Colloid Interface Sci., 2015, 460, 146–153 CrossRef CAS PubMed.
- S. Xu, H. Zhu, W. Cao, Z. Wen, J. Wang, C. P. François-Xavier and T. Wintgens, Appl. Catal., B, 2018, 234, 223–233 CrossRef CAS.
- K. Pierloot, A. Delabie, M. H. Groothaert and R. A. Schoonheydt, Phys. Chem. Chem. Phys., 2001, 3, 2174–2183 RSC.
- J. C. Klein, C. P. Li, D. M. Hercules and J. F. Black, Appl. Spectrosc., 1984, 38, 729–734 CrossRef CAS.
- J. C. Klein, A. Proctor, D. M. Hercules and J. F. Black, Anal. Chem., 1983, 55, 2055–2059 CrossRef CAS.
- G. Mattogno, R. Zanoni, D. Giusto, G. Russo and L. Sisti, Inorg. Chim. Acta, 1985, 104, 9–13 CrossRef CAS.
- A. P. Dementjev, O. P. Ivanova, L. A. Vasilyev, A. V. Naumkin, D. M. Nemirovsky and D. Y. Shalaev, J. Vac. Sci. Technol., A, 1994, 12, 423–427 CrossRef CAS.
- V. K. Kaushik, J. Electron Spectrosc. Relat. Phenom., 1991, 56, 273–277 CrossRef CAS.
- G. Kumar, J. Blackburn, R. Albridge, W. Moddeman and M. Jones, Inorg. Chem., 1972, 11, 296–300 CrossRef CAS.
- J. L. G. Fierro, J. M. Palacios and F. Tomas, Surf. Interface Anal., 1988, 13, 25–32 CrossRef CAS.
- A. M. Dennis, R. A. Howard, K. M. Kadish, J. L. Bear, J. Brace and N. Winograd, Inorg. Chim. Acta, 1980, 44, L139–L141 CrossRef CAS.
- A. Gervasini, M. Manzoli, G. Martra, A. Ponti, N. Ravasio, L. Sordelli and F. Zaccheria, J. Phys. Chem. B, 2006, 110, 7851–7861 CrossRef CAS PubMed.
- P. M. Schosseler, B. Wehrli and A. Schweiger, Geochim. Cosmochim. Acta, 1999, 63, 1955–1967 CrossRef CAS.
- M. Monte, D. Gamarra, A. López Cámara, S. B. Rasmussen, N. Gyorffy, Z. Schay, A. Martínez-Arias and J. C. Conesa, Catal. Today, 2014, 229, 104–113 CrossRef CAS.
- R. V. Lloyd, P. M. Hanna and R. P. Mason, Free Radical Biol. Med., 1997, 22, 885–888 CrossRef CAS PubMed.
- D. W. McKee, J. Catal., 1969, 14, 355–364 CrossRef CAS.
- K. Reszka, A. Naghipur and J. W. Lownz, Free Radical Res. Commun., 1990, 10, 47–56 CrossRef CAS PubMed.
- M. Rahman, D. Kundu, A. Hajra and A. Majee, Tetrahedron Lett., 2010, 51, 2896–2899 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03993a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |