
N-Haloimide-enabled halogenation via halogen-bond-assisted C–C activation of alkanols†
Yan
Geng
a,
Yue
Ma
a,
Rui
Huang
a,
Xingwei
Li
b and
Songjie
Yu
 *ab
*ab
aZhang Dayu School of Chemistry, Dalian University of Technology, Dalian, China. E-mail: yusongjie08@mails.ucas.ac.cn
bInstitute of Molecular Science and Engineering, Institute of Frontier and Interdisciplinary Sciences, Shandong University, Qingdao, China
First published on 25th November 2022
Abstract
The selective halogenation of the inert C–C bonds of alcohol-based feedstock is of tremendous significance in synthetic chemistry. Previous halogenation usually requires a metal catalyst and/or an oxidant and is limited to the cleavage of the C–C bonds of tertiary cycloalkanols. Herein, we report a broadly applicable strategy for the synthesis of iodoalkanes, bromoalkanes and chloroalkanes via the halogen-bond-assisted C–C activation of cyclic and acyclic alkanols in the absence of catalysts and oxidants, where the inexpensive N-haloimides act as bifunctional reagents to activate and halogenate alcohols. This redox-neutral protocol is a general method for the halogenation of the C–C bonds of primary, secondary, and tertiary alkanols, thus installing three types of halogen atoms and boronic esters through one-pot deiodination–borylation in a wide range of feedstock chemicals in a practical and sustainable fashion.
Introduction
Alkyl halides are highly valuable transformation handles in chemical synthesis given that carbon–halogen bonds are easily exploited in metal-catalysed C–C coupling reactions and the preparation of a variety of useful functional groups through versatile reaction modes including nucleophilic substitution reactions, elimination reactions, and reactions with metals.1 Developing mild methods for the halogenation of feedstocks to avoid the use of traditional halogen sources such as air-sensitive halides, toxic metal halides, corrosive halogens and halogenic acids2 is of great significance for streamlining the synthesis and derivatization of a broad class of functionalized molecules. Alcohol, an abundant native functional group and among the most prevalent organic molecules found in nature,3 has been shown to be an attractive feedstock in recent functional group transformations.4 For example, Suárez et al.5 reported an intramolecular alkoxy radical-assisted hydrogen abstraction reaction of alkanols in the presence of (diacetoxyiodo)benzene and iodine, affording functionalized tetrahydrofuran skeletons. Knowles et al.6 and Zuo et al.7 applied proton-coupled electron transfer and ligand-to-metal charge transfer to the C–C cleavage of alkanols, respectively, which paved the way for building carbon-centered radicals to construct ketone and amine derivatives. In this regard, the inert C–C bond β-scission of alcohol-based feedstocks in diverse halogen moieties through sustainable chemical manipulation also represents a highly desirable strategy for the synthetic community.8 Since the early work on the oxidative halogenation of strained cycloalkanols with stoichiometric Pb(IV) and Ce(IV),9 advances in this area have provided well-developed methods to access alkyl halides via the oxidation-triggered conversion of tertiary cycloalkanols (Scheme 1a, left).10 Recently, Zhu et al. reported the visible-light-catalysed C–C bromination of tertiary cycloalkanols via a combination of N-bromosuccinimide (NBS) and hypervalent iodide.11 Conversely, the C–C halogenation dominated by non-oxidation process was a rare event until Barluenga et al. developed the bis(pyridine)iodonium tetrafluoroborate-promoted iodination of cycloalkanols through successive photocatalysis and hydrolytic depyridination.12 However, this iodinating strategy requires super-stoichiometric Cs2CO3 additive (up to 10 equiv.) and extra hydrolysis procedure with strong acid and cannot allow access to bromination and chlorination due to the limitations of halogenating reagents. Consequently, to date, general access to all three types of halogenation (iodination, bromination and chlorination) of C–C bonds in different types of alkanols, especially acyclic alcohols, has been limited by the low compatibility of functional groups and strong oxidants/metal catalysts, the competing dehydration and dehydrogenation, and the lack of inexpensive bench-stable halogen sources. Based on the increasing demand for sustainable synthesis, together with the great synthetic utility of alkyl halides, we envisioned that it is worthwhile to explore the mild redox-neutral halogenation of inert C–C bonds of a wide range of primary, secondary, and tertiary alkanols in a catalyst- and oxidant-free manner (Scheme 1a, right).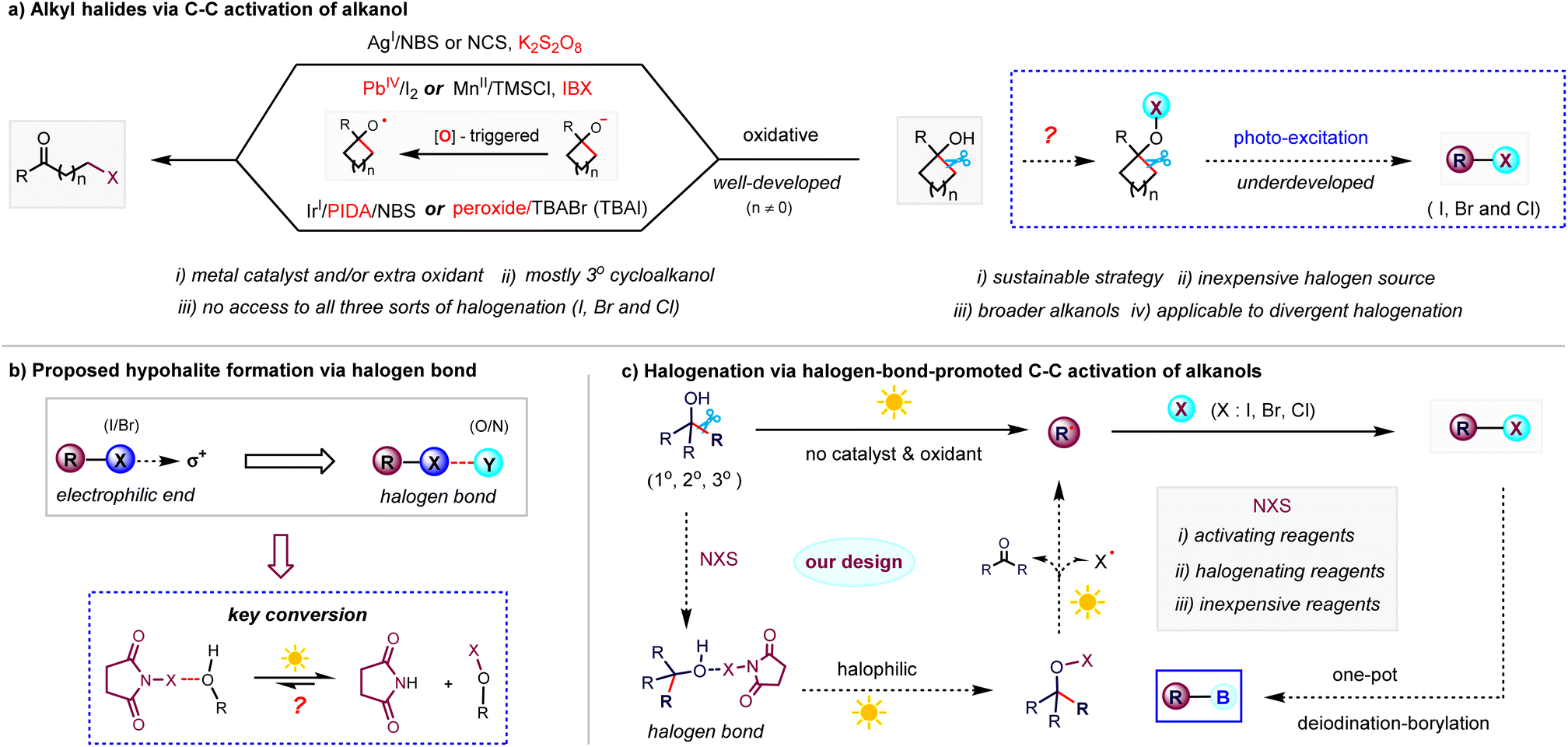 | ||
| Scheme 1 Alkanol halogenation via radical C–C cleavage. | ||
In the case of N-haloimides, a type of bench-stable and commercially available halogen sources in electrophilic halogenation reactions,13 it is commonly recognized that the anisotropic electrostatic potential from the electron density redistribution of the halogen atom can form a σ-hole, which undergoes linear interaction with N-,O-nucleophiles to form a halogen bond (Scheme 1b).14 Inspired by the formation of reactive hypobromites driven by halogen bonding interaction of phenols and NBS,15 we wondered whether analogous hypohalites could be generated in situ through a photo-excited halogen bonding complex generated from alkanol and N-haloimide.16 In this case, different from the previous oxidation-triggered alkyloxy radical formation, the resulting hypohalites will undergo halogen–oxygen homolysis under photo excitation to form an alkyloxy radical via a redox-neutral pathway (Scheme 1c). Then, the O-centered radicals undergo β-scission to form diverse alkyl radicals, which can be explored to prepare synthetically useful alkyl halides. The employment of a non-oxidation strategy instead of the previously used strong oxidants and transition metal catalysts makes this protocol very mild and practical with a broader scope for halogenation. Based on our continued interest in alcohol transformations,17 herein, we report the practical visible-light-induced halogenation via the N-haloimide-assisted C–C activation of alkanols to deliver an array of diverse alkyl halides and alkyl boronic esters through the one-pot halogenation–dehalogenation of free alcohols.
Results and discussion
With the above-mentioned hypothesis in mind, we initiated our investigations by identifying the optimal reaction conditions using bicyclo[1.1.1]pentanyl alcohol (BCP-OH) and N-iodosuccinimide (NIS) as model substrates (more details in the ESI†) because iodinated cyclobutanone scaffolds are important building blocks for the synthesis of biologically active molecules.18 Treatment of BCP-OH with NIS (1.2 equiv.) in MeCN solvent at ambient temperature under blue LED irradiation for 8 h gave the desired iodinated cyclobutanone (2) in 72% yield (entry 1, Table 1). Adding 10 mol% FeCl3 as an additive or diminishing the reaction time to 2 h both had a marginal effect on the reaction (entries 2 and 3). Further investigation of different light sources revealed that white light gave the best yield of iodinating product (entries 4–6), which is attributed to the low stability of alkyl iodide under high-energy light. Screening different solvents such as acetone, DCE, PhCF3 and HFIP showed that the optimal MeCN solvent afforded the corresponding product in 97% isolated yield (entries 7–11).|
|
|||
|---|---|---|---|
| Entry | Solvent | λ/nm | Yieldb/% |
| a Conditions: BCP-OH (0.1 mmol), NIS (0.12 mmol), solvent (3 mL), room temperature, 8 h, and N2 atmosphere. b GC yield determined with naphthalene as the internal standard. c 10 mol% FeCl3. d 2 h. e 0.3 mmol scale, isolated yield. | |||
| 1 | MeCN | 390 | 72 |
| 2c | MeCN | 390 | 73 |
| 3d | MeCN | 390 | 70 |
| 4d | MeCN | 450 | 82 |
| 5d | MeCN | 370 | 74 |
| 6d | MeCN | White light | 98 |
| 7d | Acetone | White light | 78 |
| 8d | DCE | White light | 57 |
| 9d | PhCF3 | White light | 71 |
| 10d | HFIP | White light | 73 |
| 11d,e | MeCN | White light | 97 |
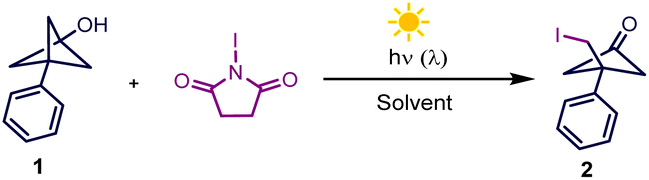
Having established the optimal conditions, we investigated the BCP-OH scope of the iodination reaction, as summarized in Scheme 2. Tethering both electron-rich and electron-deficient aromatic rings to BCP-OH was successful in this NIS-mediated iodination reaction, and the desired products were afforded in high yields (2–8). It should be noted that several BCP-OH bearing olefin fragments such as alkenyl (9) and allyl groups (10), which are reactive functional groups in NIS-assisted nucleophilic addition, were also tolerated under the reaction conditions, yielding the products in high efficiency. Installing different alkyl groups including benzyl (11), iso-butyl (12), n-butyl (13) and cycloalkyl groups (14–16) to BCP-OH showed that they were also compatible and gave the desired products in excellent yields. These products displayed particularly high functional group density including halogen, carbonyl, and strained four-membered ring, providing versatile handles for further downstream modification.
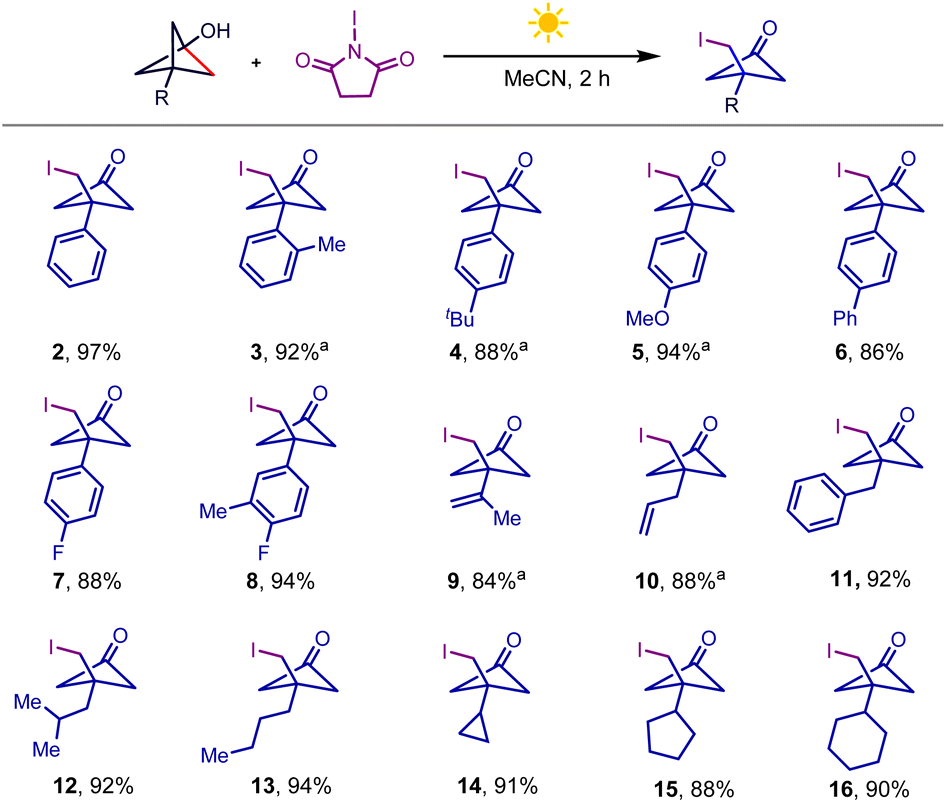 | ||
Scheme 2 BCP-OH scope of iodination conditions. BCP-OH (0.3 mmol), NIS (0.36 mmol), MeCN (3 mL), white light (8 W), room temperature, 2 h, isolated yield; a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) and 0.3 mmol NIS. and 0.3 mmol NIS. | ||
To examine the generality of this transformation, next we investigated the iodination efficiency of more readily available alkanols, as summarized in Scheme 3. The strained tertiary cyclic alcohols such as cyclobutanol and cyclopentanol derivatives underwent the iodination reaction smoothly to afford the corresponding remote ketone-functionalized alkyl iodides (17–20) in high yields (60–80%). In some cases, we found that the addition of a catalytic amount of base significantly inhibited the dehydration of alkanols. The unstrained cycloalkanols including substituted cyclohexanol (21 and 22),19 tetrahydro-2H-pyran-4-ol (23), cycloheptanol derivatives (24), adamantanol (25), and L-menthol-derived tertiary alcohol (26) were also successful substrates to afford a variety of distally iodinated alkyl ketones that are hard to synthesize otherwise. The C–C bond of the more complex polycyclic cedrol (28) could also be regioselectively cleaved to be iodinated in 81% yield. However, halogenation of the strained polycyclic corodane derivative (27) gave the corresponding alkyl halide with a regioselectivity ratio (rr) of 1.7:1, which is likely due to the kinetic control driven by the strain release. It should be noted that this iodinating protocol is applicable to acyclic tertiary alcohols bearing formyl (29), ether (30), and acetate (31) groups, giving the products in good yields. In addition to tertiary alkanols, the generality of substituted secondary alkanols has also been investigated extensively. Various secondary cycloalkanol skeletons and polycyclic and natural alcohols were compatible with this iodination reaction, incorporating an iodine atom into the alkyl chains in good yields (32–37). The more challenging acyclic secondary alkanols also exhibited a high level of reactivity in this iodination system, furnishing the alkyl iodides in good yields (38–41). However, in the case of a secondary alkanol containing an electron-rich aromatic ring as the substrate, a second iodine atom was embedded at the para-position of the phenyl ring via a follow-up electrophilic iodination (39). The primary alkanols could also undergo this iodination by releasing formaldehyde, albeit in a relatively low yield (42).
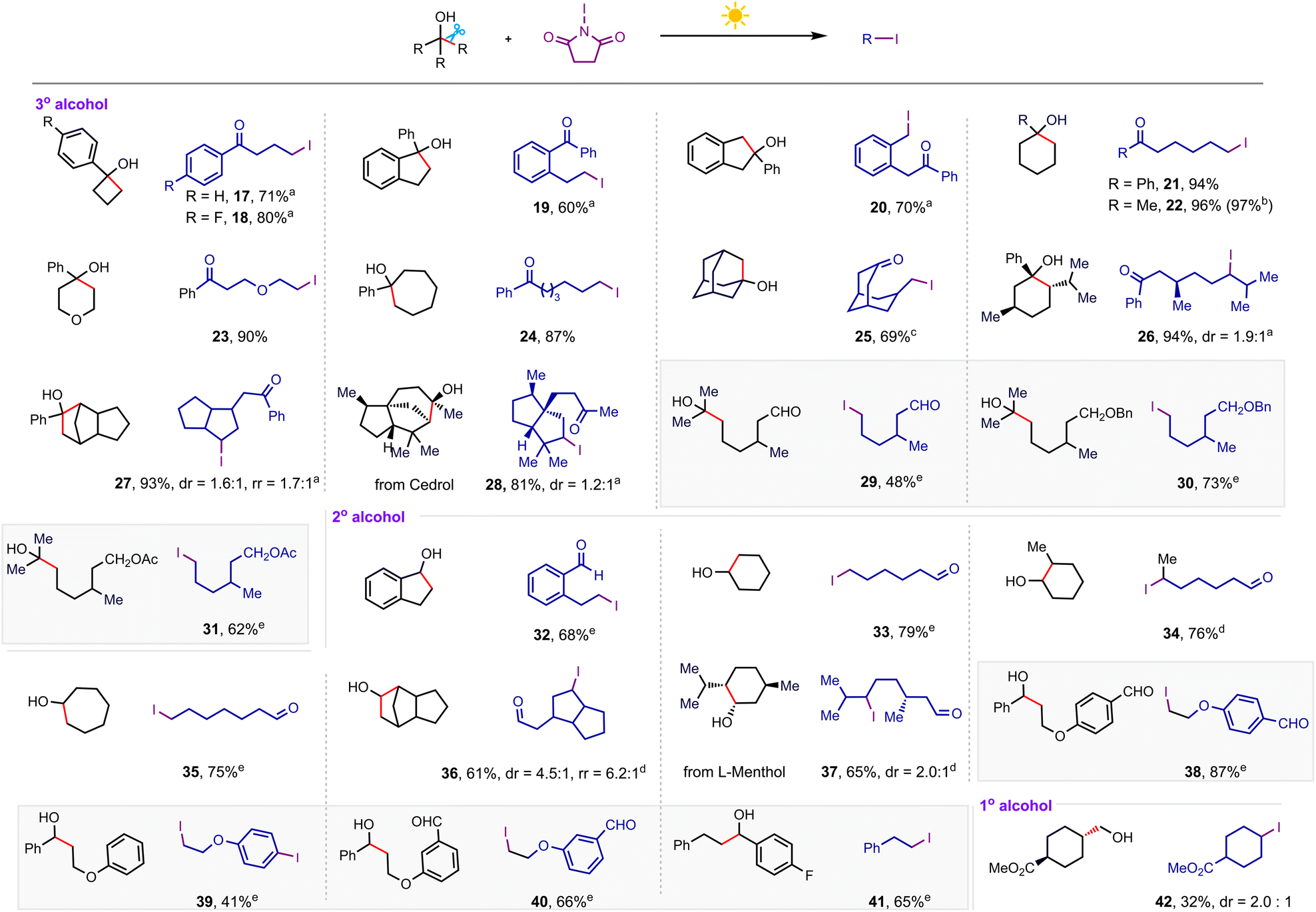 | ||
| Scheme 3 Alkanol scope of iodination reaction. Conditions: alkanol (0.3 mmol), NIS (0.45 mmol), MeCN (3 mL), white light (8 W), room temperature, 2 h, isolated yield. a30 mol% K3PO4. bRecrystallized NIS used as an iodinating reagent. c12 h. d30% K2CO3, HFIP (3 mL), 2 h. e60% K2CO3, HFIP (3 mL), 4 h. | ||
Next, we turned our attention to the bromination of alkanols by using NBS as the brominating reagent (Scheme 4), where the optimal solvent was switched to acetone, and NBS was added in two batches (detailed procedure presented in the ESI†). When examining a series of strained monocyclic and polycyclic tertiary alcohols, we found that the bromination process proceeded smoothly (43–47). Furthermore, the bromine atom could also be incorporated into longer alkyl chains even when the cycloalkanols were varied to unstrained six-membered and seven-membered rings (48–52) as well as polycyclic alcohol (53). Bromination of trans-methyl-4-(hydroxymethyl)cyclohexane-1-carboxylate gave the corresponding product in 31% yield (54). At the current stage, this bromination showed low efficiency for secondary alkanols due to the dehydrogenation-dominated side reaction.
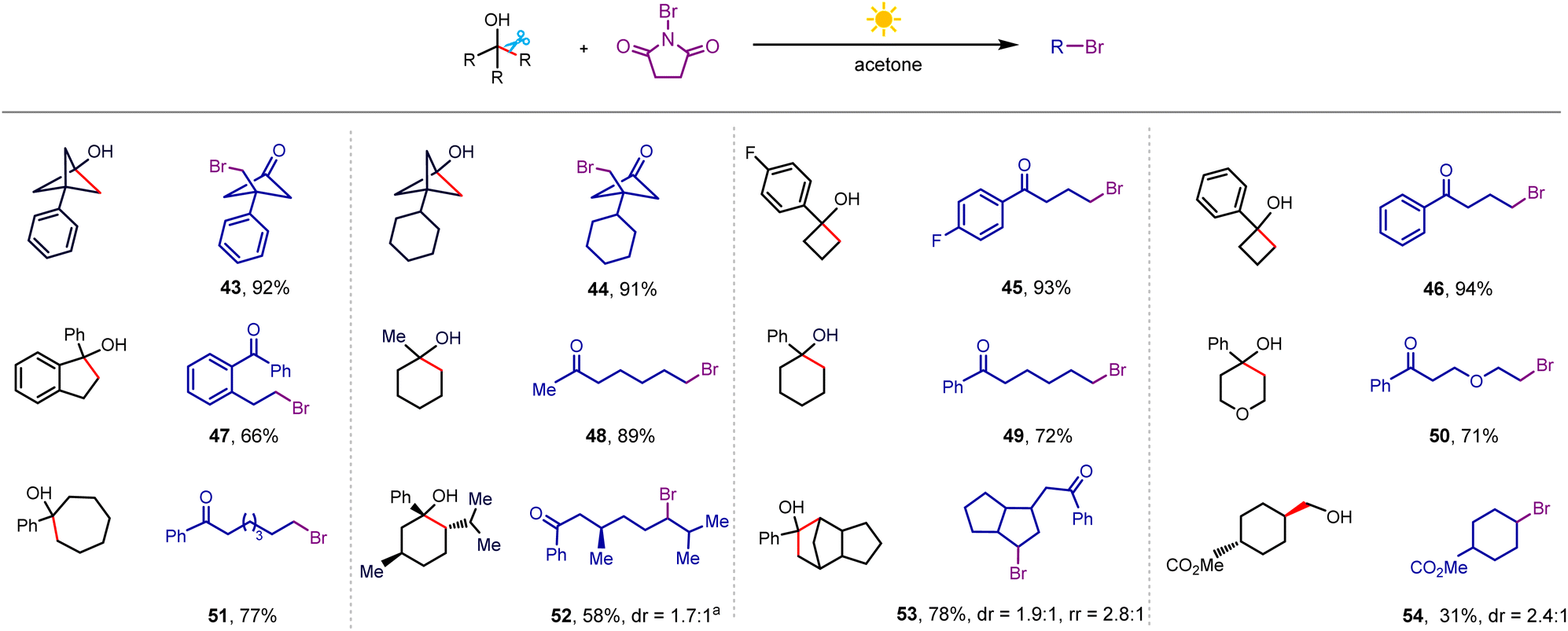 | ||
| Scheme 4 Bromination scope. Conditions: alkanol (0.3 mmol), NBS (0.72 mmol), 50 mol% K3PO4, acetone (3 mL), white light (8 W), room temperature, 4 h, isolated yield. a7 h. | ||
Exploring N-chlorosuccinimide (NCS) as a chlorinating reagent under the optimal conditions afforded a trace amount of desired product even with the addition of 0.5 equiv. of NIS as a radical initiator (Scheme 5), which is likely ascribed to the strong N–Cl bond and relatively weak electrophilicity of the Cl atom in NCS.20 However, switching the chlorinating reagent to trichloroisocyanuric acid (TCCA) afforded the desired alkyl chloride in 94% yield (55). This system well-tolerated strained cyclobutanol (57), cyclopentanol (58), and polycyclic alkanol (59), and unstrained cyclohexanol (55 and 56) and cycloheptanol (60), delivering alkyl chlorides in satisfactory yields.
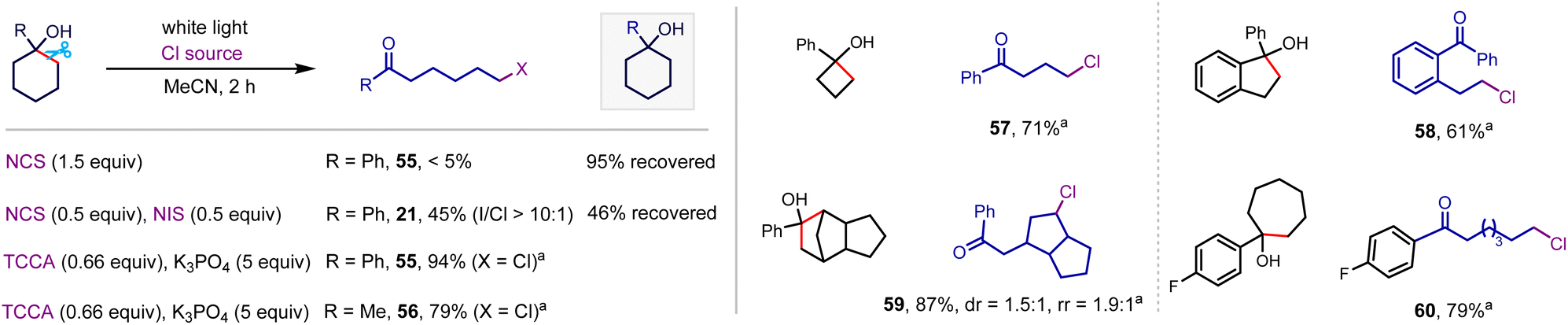 | ||
| Scheme 5 Alkanol chlorination. Conditions: alkanol (0.3 mmol), TCCA (0.2 mmol), K3PO4 (5 equiv.), MeCN (3 mL), white light (8 W), room temperature, 10 h, isolated yield. | ||
To demonstrate the synthetic utility of this protocol, we conducted the synthesis of the antipsychotic drug haloperidol from the brominated product 45 (Scheme 6a), giving the desired drug in 78% yield over two steps.21 In addition, we performed the elimination and reductive deiodination of product 28, generating two chiral polycyclic skeletons, which are prevalent in natural products (Scheme 6b).22 As shown in Scheme 6c, the reactions of easily accessible NXS (X = I and Br) and TCCA with bulky 1-methylcyclohexan-1-ol in the presence of white light irradiation furnished 7-iodoheptan-2-one, 7-bromoheptan-2-one, and 7-chloroheptan-2-one in good yields on a gram scale. Unlike alkyl halides, alkyl boronic esters are highly valuable nucleophilic precursors, which can also be transformed into a wide range of useful functional groups.23 Inspired by the radical borylation of alkyl iodides disclosed by Studer and coworkers,24 we investigated the sequential C–C and C–I cleavage to prepare alkyl boronic esters from free alcohols. Our initial studies focused on the one-pot borylation of 1-methylcyclohexanol through the procedure of our NIS-mediated iodination, followed by visible-light-induced deiodination–borylation (Scheme 6d). The addition of B2Cat2 and DMF to the reaction and changing white light to blue light upon the completion of the iodination step afforded the borylative product in 55% yield. Pleasingly, simply switching the solvent from MeCN to DMF in the borylation step resulted in an improved yield of the desired product of 66 of 76%, wherein this new transformation avoided the limitations including pre-activating alcohol and use of metal catalyst and tertiary cycloalkanol in the previous borylation.25 With the good yielding conditions in hand, subsequently we explored the scope of this practical borylation of free alcohols. Following the success of the model substrates, we were pleased to disclose that various tertiary and secondary cycloalkanols with different ring sizes were successful substrates, yielding linear borylative products bearing a distal carbonyl group in moderate to good yields (64–70 and 72–75). In addition to the borylation of cyclic alcohols, the protocol could cleave carbon-substituents from acyclic tertiary and secondary alcohols to generate alkyl boronic esters in good efficiency (71 and 76), proving the generality of this new protocol.
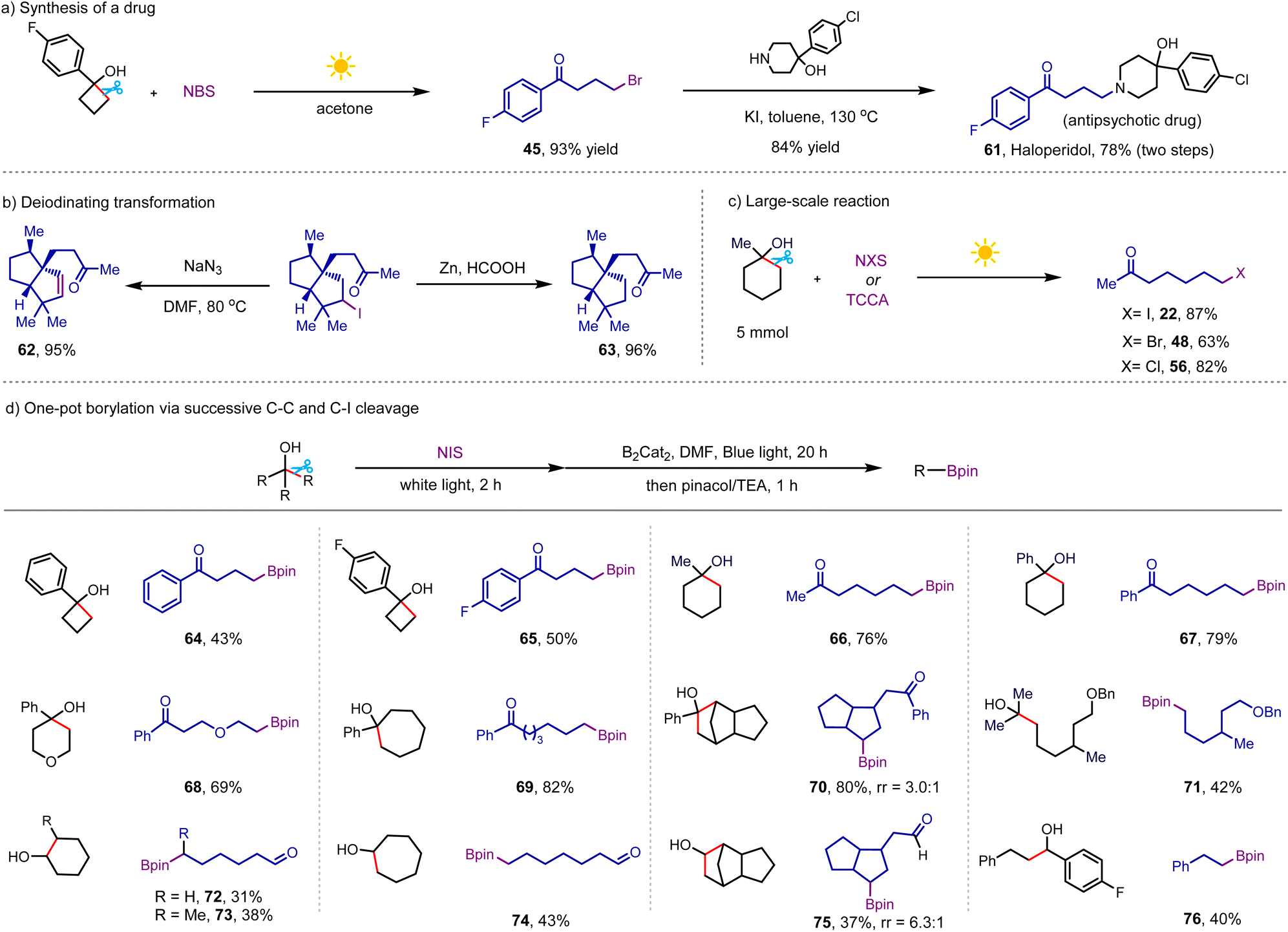 | ||
| Scheme 6 Applications of this protocol. Conditions: alcohol (0.3 mmol), NIS (0.45 mmol), MeCN (3 mL), white light (8 W), 2 h; then B2Cat2 (0.9 mmol), DMF (3 mL), blue light (8 W), 20 h (see detailed procedure in ESI†). | ||
To gain preliminary insight into the C–C bond halogenation, we focused on studying the mechanism of the halogenation. In the first step, we conducted a control reaction by exploring iodine as the iodinating reagent instead of NIS, resulting in the decomposition of the starting material and absence of the desired product (Scheme 7a), which could exclude the iodine-induced iodinating pathway. Conducting the reaction of 1-methylcyclohexan-1-ol and NIS under dark conditions failed to afford the corresponding iodinated product (Scheme 7b), even after heating the reaction at 50 °C for 2 h (Scheme 7b). Then, we performed an on/off experiment, where the reaction stopped when the light was off (more details in the ESI†). All these results demonstrate that irradiation with white light is required for the reaction to proceed. The reaction was carried out by adding TEMPO (1.5 equiv.) as a radical scavenger, and no product was detected with the recovery of the starting material in 91% yield (Scheme 7c). Complete inhibition of the reaction indicated that the reaction involved a radical pathway. The competing reaction between cyclobutanol and cyclohexanol showed a higher iodinating rate for more strained cycloalkanol, while the competing reaction of 1-phenylcyclohexan-1-ol and 1-methylcyclohexan-1-ol afforded 21 as the major product in a ratio of 15/1 (Scheme 7d and e, m respectively). Both results revealed that the extra driving force originating from the strain release and conjugated effect could facilitate the β-scission of the alkyloxy radical, which is consistent with previous computational studies of the oxidation-triggered ring opening of various cycloalkanols.10 Hypochlorite was prepared in a DCM solution, and subsequently irradiated under white light, giving the corresponding product in 46% yield over two steps (Scheme 7f). This result indicates that the hypochlorite is likely a potential intermediate. To gain further insight into the process of the iodination reaction, kinetic analyses were performed. The initial rates of the iodination reaction were investigated at various concentrations of the reactants under irradiation with white light to accelerate the kinetics (Scheme 7g). The rates showed a first-order dependence on the concentration of both the alkanol substrate and iodinating reagent (NIS), which indicated that the key intermediate in the rate-limiting step is interrelated with both alkanol and NIS. To elucidate the extent of the chain process, the measured quantum yield for iodinating 1-methylcyclohexan-1-ol under blue light is 131 (see details in the ESI†), indicating the predominant propagation pathway to product 22.
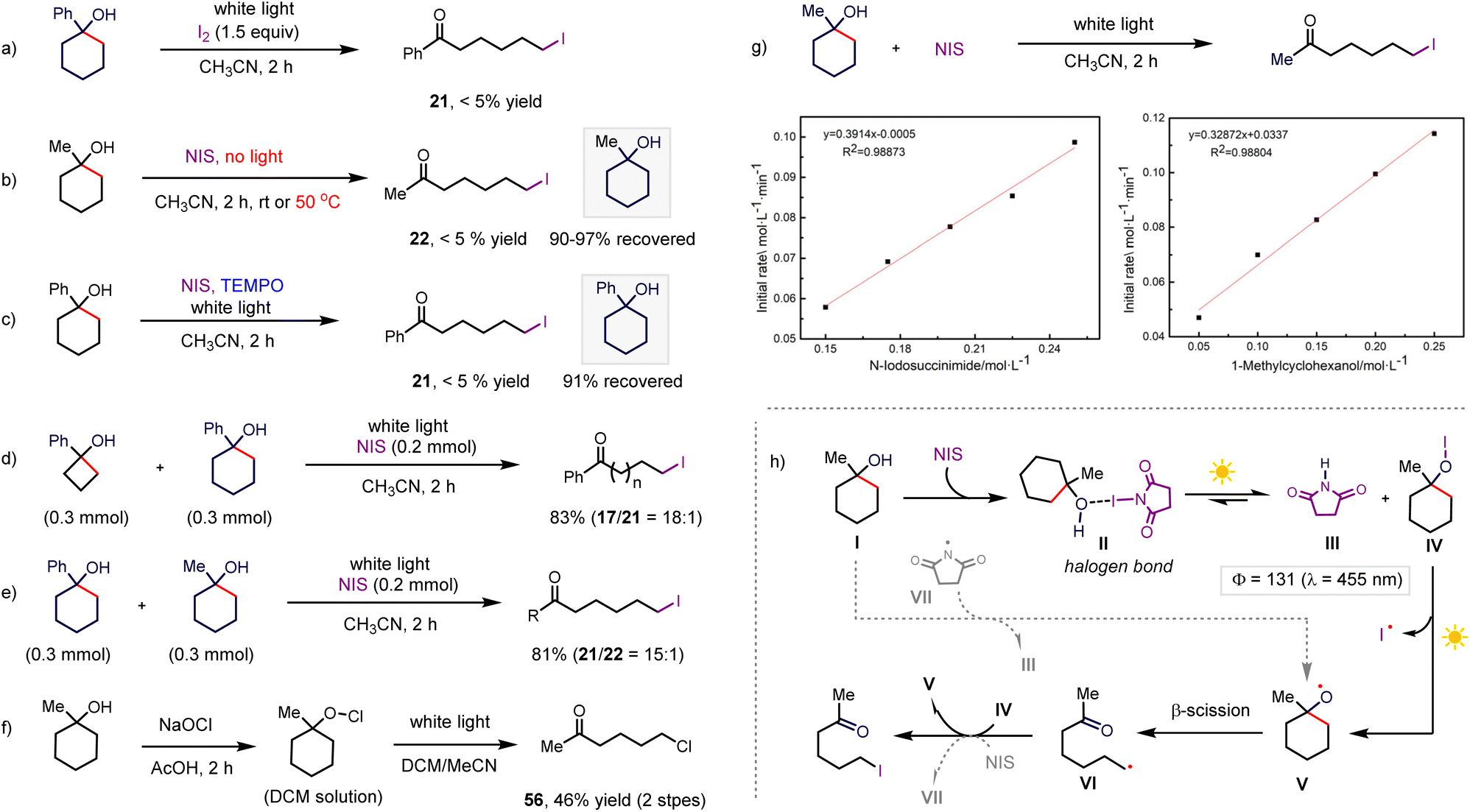 | ||
| Scheme 7 Mechanistic studies. | ||
Based on these experimental findings and previous work,26 a reaction pathway was proposed to form the alkoxy radical, which was assumed to be a crucial intermediate, as outlined in Scheme 7h. The halogen bonding interaction between alkanol and NIS could generate the reactive hypohalite intermediate VI under photo-irradiation. Then, the resulting hypohalite transformed to an alkyloxy radical through photo-excitation-promoted homolysis,16 followed by the β-scission of the high-energy alkyloxy radical to afford a distal ketone-substituted alkyl radical. Halogen atom transfer from hypohalite released the final product and regenerated the alkyloxy radical. In this system, we did not detect halogenation at the tertiary C(sp3)–H position of the alkanols via imidyl radical-mediated hydrogen atom transfer, which demonstrated that the formation of the alkyloxy radical by imidyl radical-induced hydrogen abstraction from O–H is less likely due to the much lower bond dissociation energy (BDE) of C(sp3)–H than O–H (BDE: imide N–H 107 kcal mol−1versus tertiary C–H 93 kcal mol−1 and t-alkanol O–H 105 kcal mol−1).27
Conclusions
In summary, we developed a catalyst- and oxidant-free halogenation system, wherein inexpensive bifunctional N-haloimide could be successfully applied to achieve the direct C–C activation of alkanols to yield all three types of synthetically useful alkyl halides by mild photocatalysis. This halogen-bond-assisted halogenation and one-pot deiodination–borylation exhibited a high tolerance and efficiency towards various alkanols including cyclic and acyclic alkanols. We envision that this robust alcohol-activation strategy can potentially serve as a practical and versatile platform for the functionalization of alkanols.Author contributions
Y. G. and Y. M. carried out the experiments. Y. G., R. H., X. L. and S. Y. analysed the data. Y. G. and S. Y. wrote the paper. S. Y. conceived and directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Dalian University of Technology, the Fundamental Research Funds for the Central Universities, and the Shandong Non-metallic Materials Institute for financial support. S. Y. thanks the Taishan Scholar of Shandong Province and the Qilu Young Scholar of Shandong University for funding. We thank Dr Hui Wang (AHNU) for helpful discussion.References
- (a) G. W. Gribble, Acc. Chem. Res., 1998, 31, 141–152 CrossRef CAS; (b) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (c) A. S. Dudnik and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 10693–10697 CrossRef CAS; (d) T. C. Atack and S. P. Cook, J. Am. Chem. Soc., 2016, 138, 6139–6142 CrossRef CAS PubMed; (e) T. Constantin, M. Zanini, A. Regni, N. S. Shikh, F. Julia and D. Leonori, Science, 2020, 367, 1021–1026 CrossRef CAS; (f) B. Gorski, A.-L. Barthelemy, J. J. Douglas, F. Julia and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef CAS.
- (a) A. Varenikov, E. Shapiro and M. Gandelman, Chem. Rev., 2021, 121, 412–484 CrossRef CAS; (b) W. Chung and C. D. Vanderwal, Angew. Chem., Int. Ed., 2016, 55, 4396–4434 CrossRef CAS PubMed; (c) J. R. Reyes and V. H. Rawal, Angew. Chem., Int. Ed., 2016, 55, 3077–3080 CrossRef CAS PubMed; (d) D. J. Wilger, J.-M. M. Grandjean, T. R. Lammert and D. A. Nicewicz, Nat. Chem., 2014, 6, 720–726 CrossRef CAS PubMed; (e) E. S. Lewis and C. E. Boozer, J. Am. Chem. Soc., 1952, 74, 308–311 CrossRef CAS; (f) B. Gaspar and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 5758–5760 CrossRef CAS; (g) S. M. King, X. Ma and S. B. Herzon, J. Am. Chem. Soc., 2014, 136, 6884–6887 CrossRef CAS; (h) M. A. Short, J. M. Blackburn and J. L. Roizen, Angew. Chem., Int. Ed., 2018, 57, 296–299 CrossRef CAS PubMed; (i) G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef CAS.
- T. Henkel, R. M. Brunne, H. Müller and F. Reichel, Angew. Chem., Int. Ed., 1999, 38, 643–647 CrossRef CAS.
- (a) X. Wu and C. Zhu, Chin. J. Chem., 2019, 37, 171–182 CAS; (b) X. Wu and C. Zhu, Chem. Commun., 2019, 55, 9747–9756 RSC; (c) E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC; (d) X. Wang, Y. Li and X. Wu, ACS Catal., 2022, 12, 3710–3718 CrossRef CAS; (e) S. Wang, L.-N. Guo, H. Wang and X.-H. Duan, Org. Lett., 2015, 17, 4798–4801 CrossRef CAS PubMed; (f) T. Xue, Z. Zhang and R. Zeng, Org. Lett., 2022, 24, 977–982 CrossRef CAS; (g) N. Xiong, Y. Li and R. Zeng, Org. Lett., 2021, 23, 8968–8972 CrossRef CAS PubMed; (h) W. Liu, Q. Wu, M. Wang, Y. Huang and P. Hu, Org. Lett., 2021, 23, 8413–8418 CrossRef CAS; (i) F. Cong, X.-Y. Lv, C. S. Day and R. Martin, J. Am. Chem. Soc., 2020, 142, 20594–20599 CrossRef CAS; (j) M. Lübbesmeyer, E. G. Mackay, M. A. R. Raycroft, J. Elfert, D. A. Pratt and A. Studer, J. Am. Chem. Soc., 2020, 142, 2609–2616 CrossRef PubMed; (k) J. Wu, R. M. Bär, L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2019, 58, 18830–18834 CrossRef CAS; (l) A. Cai, W. Yan and W. Liu, J. Am. Chem. Soc., 2021, 143, 9952–9960 CrossRef CAS PubMed; (m) H.-M. Guo and X. Wu, Nat. Commun., 2021, 12, 5365 CrossRef CAS; (n) F. W. Friese and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 9561–9564 CrossRef CAS; (o) H. A. Sakai and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6185–6192 CrossRef CAS; (p) J. Y. Su, D. C. Grunenfelder, K. Takeuchi and S. E. Reisman, Org. Lett., 2018, 20, 4912–4916 CrossRef CAS PubMed; (q) Y. Wei, B. Ben-zvi and T. Diao, Angew. Chem., Int. Ed., 2021, 60, 9433–9438 CrossRef CAS; (r) C. Chen, Z.-J. Wang, H. Lu, Y. Zhao and Z. Shi, Nat. Commun., 2021, 12, 4526 CrossRef CAS; (s) H. Zhao, X. Fan, J. Yu and C. Zhu, J. Am. Chem. Soc., 2015, 137, 3490–3493 CrossRef CAS PubMed; (t) R. Ren, H. Zhao, L. Huan and C. Zhu, Angew. Chem., Int. Ed., 2015, 54, 12692–12696 CrossRef CAS PubMed; (u) R. Ren, Z. Wu, Y. Xu and C. Zhu, Angew. Chem., Int. Ed., 2016, 55, 2866–2869 CrossRef CAS.
- (a) J. I. Concepción, C. G. Francisco, R. Hernández, J. A. Salazar and E. Suárez, Tetrahedron Lett., 1984, 25, 1953–1956 CrossRef; (b) A. Martín, J. A. Salazar and E. Suárez, J. Org. Chem., 1996, 61, 3999–4006 CrossRef.
- (a) S. T. Nguyen, E. A. McLoughlin, J. H. Cox, B. P. Fors and R. R. Knowles, J. Am. Chem. Soc., 2021, 143, 12268–12277 CrossRef CAS PubMed; (b) K. Zhao, G. Seidler and R. R. Knowles, Angew. Chem., Int. Ed., 2021, 60, 20190–20195 CrossRef CAS; (c) K. Zhao, K. Yamashita, J. E. Carpenter, T. C. Sherwood, W. R. Ewing, P. T. W. Cheng and R. R. Knowles, J. Am. Chem. Soc., 2019, 141, 8752–8757 CrossRef CAS; (d) H. G. Yayla, H. Wang, K. T. Tarantino, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2016, 138, 10794–10797 CrossRef CAS.
- (a) J.-J. Guo, A. Hu, Y. Chen, J. Sun, H. Tang and Z. Zuo, Angew. Chem., Int. Ed., 2016, 55, 15319–15322 CrossRef CAS; (b) A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef CAS PubMed; (c) K. Zhang, L. Chang, Q. An, X. Wang and Z. Zuo, J. Am. Chem. Soc., 2019, 141, 10556–10564 CrossRef CAS PubMed.
- (a) Y. Abderrazak, A. Bhattacharyy and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS PubMed; (b) L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- J. Jiao, L. X. Nguyen, D. R. Patterson and R. A. Flowers, Org. Lett., 2007, 9, 1323–1326 CrossRef CAS PubMed.
- (a) X. Fan, H. Zhao, J. Yu, X. Bao and C. Zhu, Org. Chem. Front., 2016, 3, 227–232 RSC; (b) L. Huan and C. Zhu, Org. Chem. Front., 2016, 3, 1467–1471 RSC; (c) K. Wang and R. Zeng, Org. Chem. Front., 2022, 9, 3692–3696 RSC; (d) R. Zhao, Y. Yao, D. Zhu, D. Chang, Y. Liu and L. Shi, Org. Lett., 2018, 20, 1228–1231 CrossRef CAS; (e) P. Wu and S. Ma, Org. Lett., 2021, 23, 2533–2537 CrossRef PubMed.
- D. Wang, J. Mao and C. Zhu, Chem. Sci., 2018, 9, 5805–5809 RSC.
- J. Barluenga, F. GonzaÂlez-Bobes, S. R. Ananthoju, M. A. García-Martín and J. M. GonzaÂlez, Angew. Chem., Int. Ed., 2001, 40, 3389–3392 CrossRef CAS.
- (a) H. Veisi and R. Ghorbani-Vaghei, Tetrahedron, 2010, 66, 7445–7463 CrossRef CAS; (b) W. Wang, X. Yang, R. Dai, Z. Yan, J. Wei, X. Dou, X. Qiu, H. Zhang, C. Wang, Y. Liu, S. Song and N. Jiao, J. Am. Chem. Soc., 2022, 144, 13415–13425 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- Y. L. Chow, D.-C. Zhao and C. I. Johansso, Can. J. Chem., 1988, 66, 2556–2564 CrossRef CAS.
- H. Wang, R.-W. Toh, X. Shi, T. Wang, X. Cong and J. Wu, Nat. Commun., 2020, 11, 4462 CrossRef CAS.
- S. Yu, Y. Ai, L. Hu, G. Lu, C. Duan and Y. Ma, Angew. Chem., Int. Ed., 2022, e202200052 CAS.
- (a) J. Li, K. Gao, M. Bian and H. Ding, Org. Chem. Front., 2020, 7, 136–154 RSC; (b) K. Tanino, M. Takahashi, Y. Tomata, H. Tokura, T. Uehara, T. Narabu and M. Miyashita, Nat. Chem., 2011, 3, 484–488 CrossRef CAS PubMed; (c) L. Deng, Y. Fu, S. Y. Lee, C. Wang, P. Liu and G. Dong, J. Am. Chem. Soc., 2019, 141, 16260–16265 CrossRef CAS.
- To reduce the effect of the potential impurities on the reaction, the iodination by using recrystallized NIS as an iodinating reagent was performed, giving a similar reactivity.
- P. Bovonsombat, S. Stone, M. Rossi and F. Caruso, Struct. Chem., 2019, 30, 2205–2215 CrossRef CAS.
- M. Lyles-Eggleston, R. Altundas, J. Xia, D. M. N. Sikazwe, P. Fan, Q. Yang, S. Li, W. Zhang, X. Zhu, A. W. Schmidt, M. Vanase-Frawley, A. Shrihkande, A. Villalobos, R. F. Borne and S. Y. Ablordeppey, J. Med. Chem., 2004, 47, 497–508 CrossRef CAS PubMed.
- E. J. Corey and R. D. Balanson, Tetrahedron Lett., 1973, 14, 3153–3156 CrossRef.
- (a) A. Suzuki, Acc. Chem. Res., 1982, 15, 178–184 CrossRef CAS; (b) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (c) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef CAS PubMed; (d) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (e) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed.
- Y. Cheng, C. Mück-Lichtenfeld and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 16832–16836 CrossRef CAS PubMed.
- (a) C. Shu, R. Madhavachary, A. Noble and V. K. Aggarwal, Org. Lett., 2020, 22, 7213–7218 CrossRef CAS PubMed; (b) J.-C. Yang, L. Chen, F. Yang, P. Li and L.-N. Guo, Org. Chem. Front., 2019, 6, 2792–2795 RSC.
- (a) H. Kurouchi, I. L. Andujar-De Sanctis and D. A. Singleton, J. Am. Chem. Soc., 2016, 138, 14534–14537 CrossRef CAS; (b) Y. Nakai, K. Moriyama and Hm Togo, Eur. J. Org. Chem., 2016, 768–772 CrossRef CAS; (c) Y. Tu, P. Shi and C. Bolm, Org. Lett., 2022, 24, 907–911 CrossRef CAS PubMed.
- (a) F. G. Bordwell and W. Z. Liu, J. Am. Chem. Soc., 1996, 118, 10819–10823 CrossRef CAS; (b) M. J. Bausch, B. David, V. Prasad, L. H. Wang, A. Vaughn and J. Phys, Org. Chem., 1992, 5, 1–6 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc03768h |
| This journal is © The Royal Society of Chemistry 2023 |