 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alkene reactions with superoxide radical anions in flow electrochemistry†
Rojan
Ali
 ,
Tuhin
Patra‡
and
Thomas
Wirth
*
,
Tuhin
Patra‡
and
Thomas
Wirth
*
School of Chemistry, Cardiff University, Park Place, Main Building, Cardiff, CF10 3AT, UK. E-mail: wirth@cf.ac.uk
First published on 6th March 2023
Abstract
Alkenes were cleaved to ketones by using dioxygen in an electrochemical flow set-up. The pressurised system allowed efficient gas–liquid mixing with a stabilised flow. This mild and straightforward approach avoids the use of transition metals and harsh oxidants.
Introduction
Significant progress has been made in the electrochemical reduction of oxygen to provide hydrogen peroxide for wastewater treatment or as a bleaching agent.1 The coupling of electrochemical oxygen reduction with preparative organic chemistry and the set-up of a flow system for such paired synthetic transformations is, however, a largely unexplored area. Flow electrochemistry has been a focus area of research recently.2 We have developed electrochemical flow reactors, which also have been commercialised.3 Biphasic reactions in microreactors have already attracted a lot of interest due to increased mixing abilities.4 Even in electrochemical microreactors such biphasic flow systems have been investigated and analysed.5 The gas/liquid biphasic reaction system we propose to use in this research will allow an intense mixing. As oxygen is used as a gas, the precise control of the potential in the electrochemical flow reactor will result in a convenient generation of the superoxide radical anion (O2˙−), which can be used for proton uptake and hydrogen atom abstraction.6 However, the use of a non-conductive gas as the second phase in such biphasic mixtures might require additional optimisation in the flow set-up.Results and discussion
Here we describe how a biphasic gas/liquid solution can be employed in electrochemical flow reactors to effect oxygen activation and perform a direct reaction with alkenes as exemplary substrates. Electrochemically generated superoxide radical anions (O2˙−) have been reported previously and used in reactions with esters.7 The electrochemical flow experiments were stimulated via the corresponding batch electrochemical procedure which has been described by Chiba and co-authors.8 Due to the small dimensions of the electrochemical microreactor, the use of oxygen does not pose any safety hazards as the total amount of hazardous material is low. A back-pressure regulator was employed to maintain a pressurised system resulting in an efficient gas–liquid mixing, while allowing a stable flow rate at the same time. The electrochemical flow experiments were performed at room temperature in an undivided, commercially available electrochemical flow reactor.3 The flow of the oxygen gas was controlled by use of a mass flow controller.9 The resulting biphasic reaction mixture was pumped into the electrochemical reactor with a peristaltic pump.10 This pump controlled the combined flow rate of the biphasic reaction mixture and can be used to monitor the back-pressure created via a tuneable back-pressure regulator (BPR) positioned after the electrochemical reactor, as shown in the set-up in Fig. 1.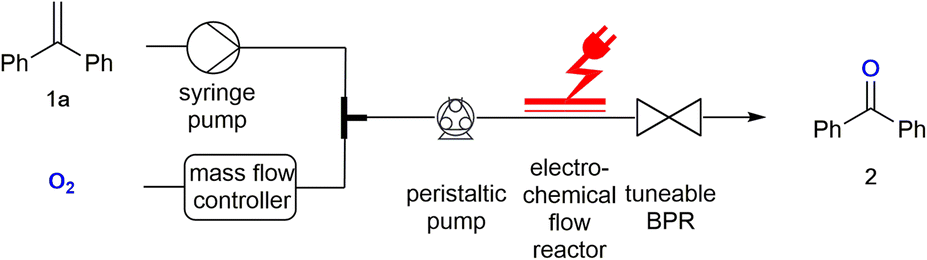 | ||
| Fig. 1 Experimental set-up for the electrochemical flow reactions with oxygen. | ||
For the flow experiments, platinum electrodes with an active surface area of 12 cm2 each were separated by a 0.5 mm fluorinated ethylene propylene (FEP) spacer, creating a 0.6 mL volume channel in the reactor. The reactions were performed under constant current electrolysis. 1,1-Diphenylethylene 1a was investigated to determine the optimal conditions required for its electrolysis to benzophenone 2 (Table 1). Initially, the reaction was performed in acetonitrile using platinum electrodes. The alkene solution and oxygen gas were pumped with a flow rate of 0.05 mL min−1 each resulting in a combined flow rate of 0.1 mL min−1. The molar ratio of oxygen![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :alkene was at least 4.5:2.5 (no back-pressure). With an applied charge of 3 F and a back-pressure of 2.8 bar, the desired product 2 was obtained in 25% yield (Table 1, entry 1). GC-MS analysis of the crude reaction mixture showed several undesired side products, which were generated due to the solvent participating in the reaction. The formation of these side products was suppressed by changing acetonitrile to isobutyronitrile (iPrCN) allowing 2 to be obtained in 36% yield (Table 1, entry 2) (see ESI†). On increasing the flow rate to 0.2 mL min−1, and decreasing the applied charge to 2 F, the yield of 2 was increased to 53% (Table 1, entry 3). The yield remained unaffected at lower back-pressures of 2.2 and 1.1 bar (Table 1, entries 4 and 5), while a lack of back-pressure (Table 1, entry 6) reduced the yield to 41%. Finally, on increasing the charge applied from 2 F to 2.5 F and 3 F, the yield of 2 decreased (Table 1, entries 7 and 8).
:alkene was at least 4.5:2.5 (no back-pressure). With an applied charge of 3 F and a back-pressure of 2.8 bar, the desired product 2 was obtained in 25% yield (Table 1, entry 1). GC-MS analysis of the crude reaction mixture showed several undesired side products, which were generated due to the solvent participating in the reaction. The formation of these side products was suppressed by changing acetonitrile to isobutyronitrile (iPrCN) allowing 2 to be obtained in 36% yield (Table 1, entry 2) (see ESI†). On increasing the flow rate to 0.2 mL min−1, and decreasing the applied charge to 2 F, the yield of 2 was increased to 53% (Table 1, entry 3). The yield remained unaffected at lower back-pressures of 2.2 and 1.1 bar (Table 1, entries 4 and 5), while a lack of back-pressure (Table 1, entry 6) reduced the yield to 41%. Finally, on increasing the charge applied from 2 F to 2.5 F and 3 F, the yield of 2 decreased (Table 1, entries 7 and 8).
| Entry | Solvent | Combined flow rate (mL min−1) | Charge (F) | Current (mA) | Back-pressure (bar) | Yield 2 (%) |
|---|---|---|---|---|---|---|
| a Standard reaction conditions: undivided flow cell, Pt anode and cathode (active surface area: 12 cm2 each), interelectrode distance: 0.5 mm, 1a (0.025 M) and nBu4NClO4 (0.05 M) in solvent. Yield determined by gas chromatography-flame ionization detection (GC-FID) using benzonitrile as an internal standard. | ||||||
| 1 | MeCN | 0.1 | 3 | 12 | 2.8 | 25 |
| 2 | iPrCN | 0.1 | 3 | 12 | 2.8 | 36 |
| 3 | iPrCN | 0.2 | 2 | 16 | 2.8 | 53 |
| 4 | iPrCN | 0.2 | 2 | 16 | 2.2 | 51 |
| 5 | iPrCN | 0.2 | 2 | 16 | 1.1 | 51 |
| 6 | iPrCN | 0.2 | 2 | 16 | 0 | 41 |
| 7 | iPrCN | 0.2 | 2.5 | 20 | 1.1 | 46 |
| 8 | iPrCN | 0.2 | 3 | 24 | 1.1 | 35 |
Using the optimised reaction conditions, several di- and tri-substituted alkenes were converted into their corresponding carbonyl derivatives (Fig. 2). Starting from the disubstituted alkene 1,1-diphenylethylene 1a, benzophenone 2 was obtained in 45% yield. Tri-substituted olefins 1b and 1c produced 2 in 33% and 27% yields, respectively. The reaction of α-methylstyrene 1d resulted in the formation of acetophenone 3 in 39% yield. Additionally, indene and 4-bromo- and 4-fluorostyrene were investigated in this reaction. Although these starting materials were fully consumed, the corresponding aldehydes were not obtained. This might not be completely surprising as it has been reported that superoxide radical anions can decompose aromatic aldehydes through a base-catalysed process.11
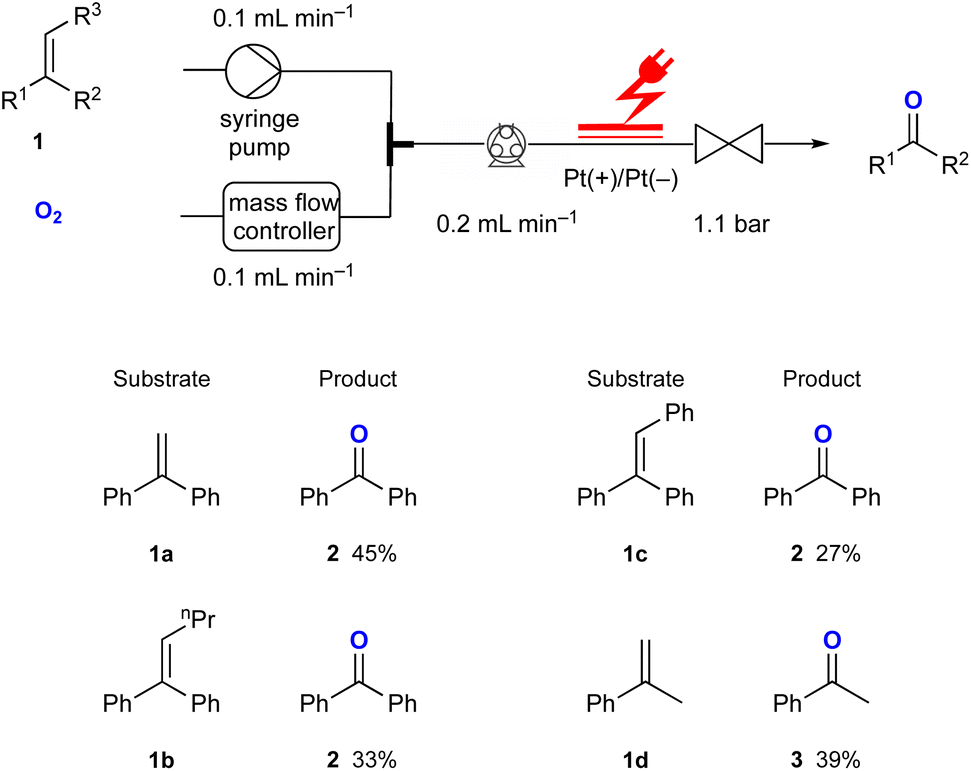 | ||
| Fig. 2 Substrate scope for the electrochemical synthesis of carbonyl compounds from di- and tri-substituted alkene derivatives in flow. | ||
The proposed reaction mechanism is shown in Fig. 3. Initially, oxygen undergoes a one-electron reduction to form the corresponding superoxide radical anion (O2˙−).12 An aprotic solution containing the superoxide radical anion has been reported to be stable at room temperature for several hours.13 Therefore, O2˙− is stable enough to capture the radical cation of the alkene (1a˙+) generated by the one-electron oxidation of the alkene. This could form the intermediate A followed by the generation of dioxetane intermediate B, which decomposes to the corresponding carbonyl compound.14
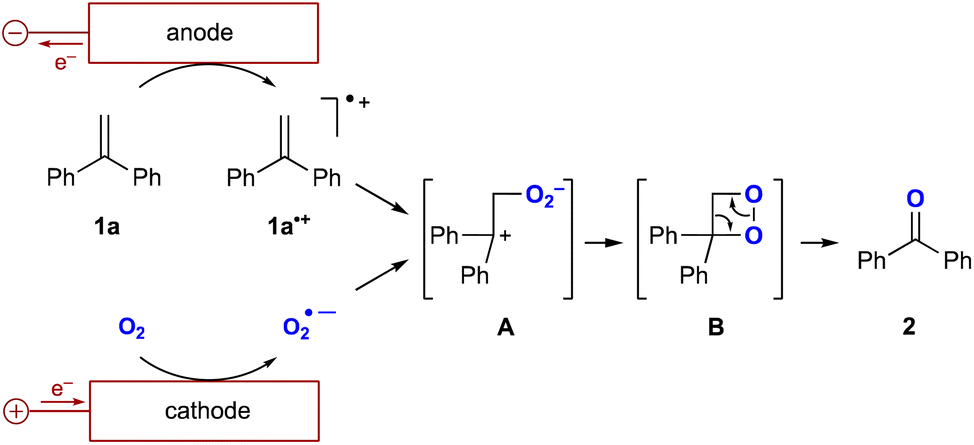 | ||
| Fig. 3 Proposed reaction mechanism. | ||
Conclusions
In conclusion, a continuous biphasic electrochemical flow method was developed to efficiently generate superoxide radical anions and oxidatively cleave di- and trisubstituted alkenes into their corresponding carbonyl compounds. This is a mild and straightforward approach that uses molecular oxygen as a green oxygen source in combination with flow electrochemistry as an alternative to transition metals and harsh oxidants. Moreover, an intrinsic advantage of flow chemistry is that the procedures can be easily scaled up. Finally, this method allowed the synthesis of several ketones in moderate yields.Author contributions
The authors confirm contributions to the paper as follows: study conception and design: T. Wirth; experiments and data collection: R. Ali and T. Patra; analysis and interpretation of results: all authors; draft manuscript preparation: T. Patra, R. Ali and T. Wirth. All authors reviewed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge the financial support provided by the Welsh Government (SMART Expertise grant).References
- (a) Y. Pang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, J. Mater. Chem. A, 2020, 8, 24996 RSC; (b) N. Wang, S. Ma, P. Zuo, J. Duan and B. Hou, Adv. Sci., 2021, 8, 2100076 CrossRef CAS PubMed.
- (a) N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed; (b) M. Elsherbini and T. Wirth, Acc. Chem. Res., 2019, 52, 3287 CrossRef CAS PubMed; (c) T. Noël, Y. Cao and G. Laudadio, Acc. Chem. Res., 2019, 52, 2858 CrossRef PubMed; (d) N. Kockmann, P. Thenée, C. Fleischer-Trebes, G. Laudadio and T. Noël, React. Chem. Eng., 2017, 2, 258 RSC; (e) M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811 CrossRef CAS PubMed; (f) D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573 CrossRef CAS PubMed.
- Ion electrochemical reactor, https://www.vapourtec.com/products/flow-reactors/ion-electrochemical-reactor-features/, accessed February 2023.
- M. J. Hutchings, B. Ahmed-Omer and T. Wirth, in Microreactors in Organic Chemistry and Catalysis, ed. T Wirth, Wiley-VCH, Weinheim 2013, p. 197 Search PubMed.
- Y. Cao, N. Padoin, C. Soares and T. Noël, Chem. Eng. J., 2022, 427, 131443 CrossRef CAS.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029 CrossRef CAS PubMed.
- (a) F. Magno and G. Bontempelli, J. Electroanal. Chem., 1976, 68, 337 CrossRef CAS; (b) D. Bauer and J. P. Beck, J. Electroanal. Chem., 1972, 40, 233 CrossRef CAS.
- Y. Imada, Y. Okada, K. Noguchi and K. Chiba, Angew. Chem., Int. Ed., 2019, 58, 125 CrossRef CAS PubMed.
- Gas Mass flowmeter and controllers, https://www.omega.co.uk/pptst/FMA5400A_5500A.html, accessed February 2023.
- SF-10 reagent pump, https://www.vapourtec.com/products/sf-10-pump-features/, accessed February 2023.
- M. J. Gibian, D. T. Sawyer, T. Ungermann, R. Tangpoonpholvivat and M. M. Morrison, J. Am. Chem. Soc., 1979, 101, 640 CrossRef CAS.
- (a) D. L. Maricle and W. G. Hodgson, Anal. Chem., 1965, 37, 1562 CrossRef CAS; (b) Q. Li, C. Batchelor-McAuley, N. S. Lawrence, R. S. Hartshorne and R. G. Compton, J. Electroanal. Chem., 2013, 688, 328 CrossRef CAS.
- J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049 CrossRef CAS PubMed.
- A. A. Frimer, Chem. Rev., 1979, 79, 359 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3fd00050h |
| ‡ Present address: School of Basic Sciences, Indian Institute of Technology Bhubaneswar, Argul, Odisha 752050, India. |
| This journal is © The Royal Society of Chemistry 2023 |