
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Does HNO3 dissociate on gas-phase ice nanoparticles?†
Anastasiya
Khramchenkova
a,
Andriy
Pysanenko
 b,
Jozef
Ďurana
b,
Barbora
Kocábková
b,
Michal
Fárník
*b and
Jozef
Lengyel
*a
b,
Jozef
Ďurana
b,
Barbora
Kocábková
b,
Michal
Fárník
*b and
Jozef
Lengyel
*a
aLehrstuhl für Physikalische Chemie, TUM School of Natural Sciences, Technische Universität München, Lichtenbergstraße 4, 85748 Garching, Germany. E-mail: jozef.lengyel@tum.de
bJ. Heyrovský Institute of Physical Chemistry v.v.i., Czech Academy of Sciences, Dolejškova 3, 18223 Prague, Czech Republic. E-mail: michal.farnik@jh-inst.cas.cz
First published on 11th July 2023
Abstract
We investigated the dissociation of nitric acid on large water clusters (H2O)N, ![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif) ≈ 30–500, i.e., ice nanoparticles with diameters of 1–3 nm, in a molecular beam. The (H2O)N clusters were doped with single HNO3 molecules in a pickup cell and probed by mass spectrometry after a low-energy (1.5–15 eV) electron attachment. The negative ion mass spectra provided direct evidence for HNO3 dissociation with the formation of NO3−⋯H3O+ ion pairs, but over half of the observed cluster ions originated from non-dissociated HNO3 molecules. This behavior is in contrast with the complete dissociation of nitric acid on amorphous ice surfaces above 100 K. Thus, the proton transfer is significantly suppressed on nanometer-sized particles compared to macroscopic ice surfaces. This can have considerable implications for heterogeneous processes on atmospheric ice particles.
≈ 30–500, i.e., ice nanoparticles with diameters of 1–3 nm, in a molecular beam. The (H2O)N clusters were doped with single HNO3 molecules in a pickup cell and probed by mass spectrometry after a low-energy (1.5–15 eV) electron attachment. The negative ion mass spectra provided direct evidence for HNO3 dissociation with the formation of NO3−⋯H3O+ ion pairs, but over half of the observed cluster ions originated from non-dissociated HNO3 molecules. This behavior is in contrast with the complete dissociation of nitric acid on amorphous ice surfaces above 100 K. Thus, the proton transfer is significantly suppressed on nanometer-sized particles compared to macroscopic ice surfaces. This can have considerable implications for heterogeneous processes on atmospheric ice particles.
1 Introduction
Acid dissociation is a fundamental chemical process wherein an acid molecule releases a proton, which associates with water as an oxonium ion.1,2 Understanding the equilibrium involved in acid dissociation is essential for assessing the behavior of acids and their interactions with other molecules in aqueous solutions or on ice surfaces.1,3 Of particular significance is the study of acid dissociation on ice particles, which differs from its behavior in bulk water, as it holds substantial relevance to atmospheric phenomena.4 For example, ice particles containing nitric acid actively contribute to the formation of polar stratospheric clouds (PSCs) and facilitate heterogeneous reactions on their surfaces, which lead to ozone depletion in the polar regions.5–8 Hence, investigating factors that determine whether an acid exists in its molecular or dissociated form in/on these ices is crucial to predict the behavior of these species in the atmosphere.Nitric acid (HNO3) is known to be a strong acid in an aqueous solution that is essentially fully dissociated, yielding hydrated NO3− and H3O+ ions.9 Also, it acts as a strong oxidizing agent.9 Apart from being an essential component of PSCs, nitric acid is also involved in various other atmospheric processes. These include the aging of naturally emitted aerosols, such as sea salt particles,10,11 and the formation of new particles in the upper troposphere.12–14 In particular, HNO3-mediated particle formation is driven by an acid–base proton transfer, thereby enhancing particle stability and formation rate.15 Though laboratory experiments have shown particle formation based on binary HNO3–NH3 to be less efficient compared to H2SO4–NH3,16 the slower particle formation rates with nitric acid are compensated by its significantly greater atmospheric abundance, which is several orders of magnitude higher than the concentration of sulfuric acid.17 Recent computational study has shown that nitric acid could initiate new particle formation just as well as sulfuric acid under certain conditions.18 Furthermore, the injection of HNO3 into the H2SO4–NH3 nucleation has been found to result in synergistic effects that significantly increase particle formation rates.19 Likewise, a substantial enhancement in nucleation rates by nitric acid has also been reported for the sulfuric acid–dimethylamine nucleation in the polluted boundary layer.20
The dissociation mechanism of nitric acid in an aqueous solution has been the focus of many experimental and computational studies. X-ray photoelectron spectroscopy experiments have demonstrated the complete dissociation of HNO3 in bulk water solutions, with a 20% decrease in dissociation at the liquid/vapor interface.21 The presence of non-dissociated nitric acid at the surface is caused by incomplete solvation.22–24 Further experimental evidence for this weak acid behavior at the liquid/vapour boundary has been reported using sum-frequency generation (SFG) and infrared spectroscopies.25–28 In general, acid dissociation equilibrium is strongly dependent on temperature, and for most acids, such as HNO3, strong entropic contributions can suppress the exothermic nature of the reaction.29 Therefore, HNO3 is likely to be more acidic at lower temperatures, i.e., at the liquid/vapor interface it behaves as a weak acid at room temperature,21 whereas on the surface of amorphous ice it dissociates completely down to the temperatures of 100–120 K.30,31 At even lower temperatures, however, this thermodynamically favored channel becomes inhibited kinetically. For example, the IR spectroscopy of HNO3 deposited onto the amorphous ice revealed substantial acid dissociation even at 45 K, but some molecular HNO3 patterns were identified in the spectrum at these low temperatures as well. Nevertheless, they disappeared with annealing the ice substrate above 120 K confirming the complete dissociation at these temperatures.30
Herein, our aim is to address the question of how the HNO3 dissociation proceeds on/in the finite-size clusters. It is well established that HNO3 dissociation in small HNO3(H2O)N clusters is strongly dependent on the degree of hydration with an onset at N = 5–6, determined experimentally32,33 and by computations.2,34,35 However, more relevant to atmospheric chemistry is the question of the HNO3 dissociation on nanometer-sized ice particles, as they can mimic the ultra-fine ice particles in the atmosphere.
The experimental tool employed in the present study to reveal the state of HNO3 molecule on the ice nanoparticle is the slow electron attachment. Our previous studies showed that the electron-induced processes in HNO3 are strongly influenced by the presence of water molecules. The dissociative electron attachment to an isolated gaseous HNO3 molecule led to the dominant formation of NO2− ions with an overall yield of more than 96%.36,37 As the electron was hydrated to form a (H2O)n− cluster anion that reacts with gaseous HNO3 molecules in an ion trap mass spectrometer, the OH− formation was the only primary reaction channel.38 This drastic change in the reaction was explained by the greater hydration energy of OH− with respect to NO2−. There are two channels driven by thermochemistry, namely NO2− and OH−, which depend on the degree of hydration. In principle, the NO3− formation channel can also be considered, but it is much higher in energy than the other two channels, and was observed to yield a negligible 0.03% of NO3− from the gas phase HNO3 molecules.36 However, as soon as the acid is sufficiently hydrated, the ion pair NO3−⋯H3O+ is formed, as demonstrated by the electron attachment experiments with the mixed (HNO3)M(H2O)N clusters.36,37 The incoming free electron recombines efficiently with the H3O+ moiety generating water and releasing the H atom resulting in (H2O)nNO3− cluster ions. In other words, regardless of how much a particular channel was preferred by the thermochemistry, the NO3− was formed. Thus, the electron attachment to the mixed (HNO3)M(H2O)N clusters was very different from the other two cases and resulted in the prominent formation of NO3− containing cluster ions prevailing the yield of NO2− and OH− containing species.36 Further, adding bases like ammonia39 or dimethylamine40 into the hydrated HNO3 clusters resulted in even higher relative yield of the NO3− containing cluster ions due to their high proton affinity.
In the present experiment, gas-phase HNO3 molecules are picked up by pure (H2O)N clusters, and we let them interact with the free electrons with well-defined kinetic energies. We compare the present results to our previous experiments, where the mixed (HNO3)M(H2O)N clusters were produced by co-expansion of HNO3/H2O vapor in He buffer gas. In contrast to the co-expansion experiments, the pickup technique enables us to control the number of HNO3 molecules taken up by the ice nanoparticles.41 Our previous understanding of the hydration effects on electron interactions with HNO3 serves as a powerful probe to determine the molecular or dissociated form of the acid on the ice nanoparticle: The hydrated HNO3 produces OH− containing fragments in the mass spectrum, whereas NO3− containing clusters indicate the presence of an ion pair in the cluster.
2 Experimental
The cluster experiments were carried out on the CLUster Beam (CLUB) apparatus in the J. Heyrovsky Institute of Physical Chemistry in Prague. The experimental setup and procedures were described in our previous reviews39,41,42 and references cited therein. Details of the present experiments are given in ESI.† The water clusters were produced by a supersonic expansion of water vapor through a divergent conical nozzle into a high vacuum. The water reservoir with the attached nozzle were placed in a vacuum chamber and heated to controlled temperatures TR and TN, respectively, which determine the neutral water cluster (H2O)N mean size.43 After passing through a skimmer, the water clusters entered a differentially pumped pickup chamber filled with HNO3/H2O vapor. The pickup conditions are analyzed in ESI† and correspond to the probability of less than one for the pickup of an HNO3 molecule by an average (H2O)N cluster. The expansion conditions to produce the mixed HNO3/H2O clusters in the comparative experiments were essentially the same as in our previous studies.36,37 The clusters were generated in the co-expansion of vapor from HNO3 solution with He buffer gas (see ESI† for details).
The cluster beam passed through three differentially pumped vacuum chambers (≈1.5 m flight path in total corresponding to a flight time of about 1 ms) until it reached the ionization region of the perpendicularly mounted reflectron time-of-flight mass spectrometer (TOF). The cluster beam was crossed by an electron beam from a pulsed electron gun. The TOF can operate either in a positive or negative ion mode. After the extraction and acceleration, the ions passed through an ≈95 cm long TOF flight path, and the spectra were recorded. The negative ion mass spectra were recorded in electron energy scanning mode from 0 eV to 15 eV with a 0.20 eV step. From these spectra, the electron energy dependent ion yield curves were obtained for different ions. For a higher signal-to-noise ratio, the mass spectra presented here were recorded for a longer time at a constant electron energy of 1.5 eV.
3 Experimental results
3.1 Mass spectra
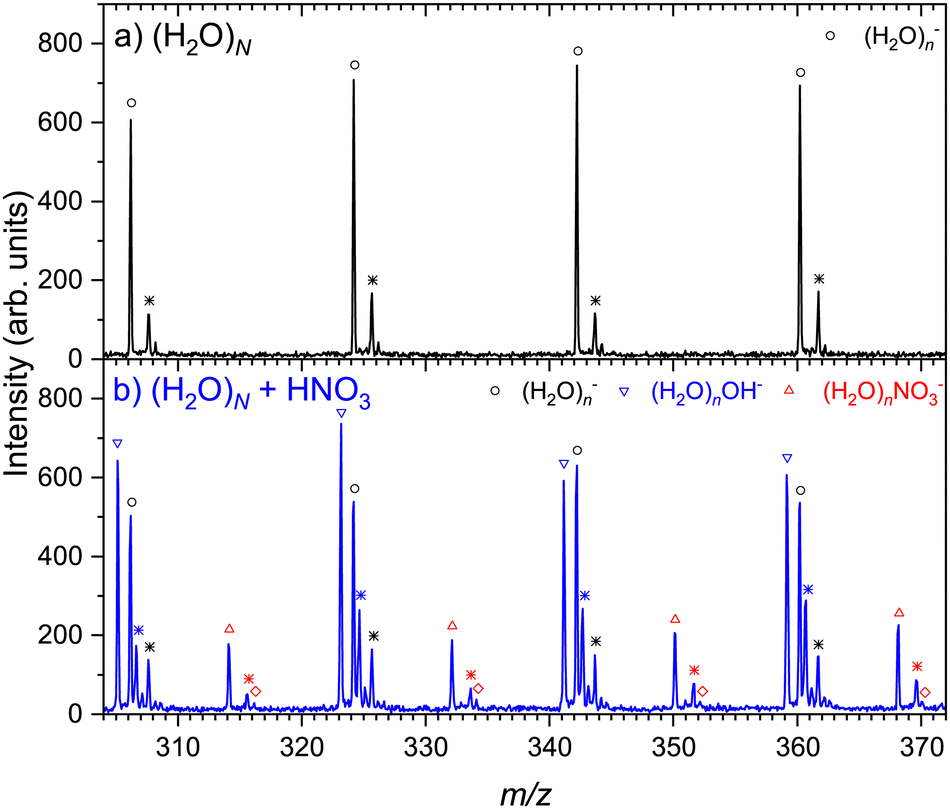
Fig. 1 shows an example of the negative ion mass spectra recorded at 1.5 eV electron energy. The expansion conditions corresponded to the mean neutral cluster size of ≈ 180. The top spectrum (a) shows the pure water clusters without any pickup. The spectrum exhibits only the (H2O)n− series, attributed to the well-known low energy electron attachment to pure water clusters.44 The (H2O)nOH− ions are only produced by the dissociative electron attachment (DEA) at the electron energies above 6 eV. The spectrum exhibits only small fragments n ≪ due to the limited mass range dictated by the perpendicular TOF arrangement (see ESI† for explanation). The bottom spectrum (b) was recorded under the same experimental conditions with the nitric acid vapor introduced into the pickup cell. In this experiment, the pickup pressure was set so that the pickup probability for a (H2O)N cluster of an average size ≈ 180 cluster was less than one to assure that we probe HNO3(H2O)N clusters with a single HNO3 molecule (see ESI† for the estimate of the number of adsorbed molecules). This step is crucial to avoid the contribution to NO3− moiety that could be generated by the reaction between OH− and a second HNO3.38,45 Two new pronounced series occur after the pickup: the (H2O)n− series (black open circles) is accompanied by (H2O)nOH− (blue downward triangles), and (H2O)nNO3− ions (red upward triangles) appear in between the water peaks (note that the possible mass coincidences are discussed in ESI†).
 | ||
| Fig. 1 Negative mass spectra at 1.5 eV for (a) pure water and (b) water with HNO3 pickup. | ||
The details of the mass spectra presented in Fig. 1 are depicted in Fig. 2. Upon closer investigation, the spectrum of water clusters shown in Fig. 2(a) exhibits a second series of peaks labeled by stars, which corresponds to metastable water evaporation. Observation of metastable cluster ion decay in reflectron TOF mass spectrometers for various clusters was described elsewhere,46,47 and the metastable fragmentation of positively charged water clusters was investigated in previous studies.48 Recently, we have investigated this effect in detail for the negatively charged pure water clusters as well, confirming the labeled peaks to be due to the metastable clusters (this point is also discussed in ESI†). In addition, there are much smaller peaks in the mass spectra corresponding to the contribution of naturally occurring isotopes.
 | ||
| Fig. 2 Negative mass spectra at 1.5 eV for pure water (top) and with HNO3 pickup (bottom) closeup. | ||
All features present in the pure water cluster spectrum in Fig. 2(a) are reproduced in the bottom spectrum (b) after the HNO3 pickup. Additionally, there are clearly separated (H2O)nOH− and (H2O)n− series, and the (H2O)nNO3− series. Aside these previously mentioned series, there are further peaks labeled by stars. Analogous to the pure water spectrum exhibiting metastable cluster ion fragments (black stars), the peaks labeled by blue and orange stars correspond to the metastable cluster ions in (H2O)nOH− and (H2O)nNO3− series, respectively. It should be noted that even though the displacement of the metastable peak next to the (H2O)nNO3− ion is close to Δm/z ≈ 1, it is not exactly 1 and changes slightly but regularly with m/z in accordance with the behavior of the metastable ion peaks. Thus, these peaks correspond to the metastable (H2O)nNO3− ions rather than (H2O)nHNO3− (although a small contribution of the latter ions cannot be excluded completely due to the overlap with the metastable peak). In addition, there is a very small series labeled by open red diamonds, which can be attributed to the (H2O)nNO2− ions. Nevertheless, their assignment is uncertain due to their low intensities and overlap with the isotope contributions. In summary, the unambiguously assigned ions resulting from the electron attachment to (H2O)NHNO3 clusters are (H2O)nOH− and (H2O)nNO3−.
To investigate the dependence of the observed processes on cluster size, we have measured the mass spectra for different expansion conditions corresponding to the (H2O)N cluster mean sizes from ≈ 30 to 470. Qualitatively, the mass spectra are essentially the same, i.e., they exhibit the same ion series (see ESI†). It ought to be mentioned that the clusters generated under varying expansion conditions might have different temperatures between approximately 90 K and 180 K. However, these cluster temperatures are only approximate, based on a semiempirical model,49 and cannot be determined experimentally. Thus, we refrain from drawing any conclusions regarding the temperature dependence of the pickup mass spectra.
3.2 Energy dependence
The above discussed spectra were recorded at the electron energy of 1.5 eV corresponding to the maximum negative ion yield. In addition, the electron energy-dependent mass spectra were measured between 0–15 eV in steps of 0.20 eV. Fig. 3 shows the electron energy dependent ion yield for selected (H2O)n−, (H2O)nOH−, and (H2O)nNO3− ions. The ion yield was qualitatively independent of the cluster ion size n apart from the intensity and was therefore integrated for n = 16–26 to achieve a better signal-to-noise ratio.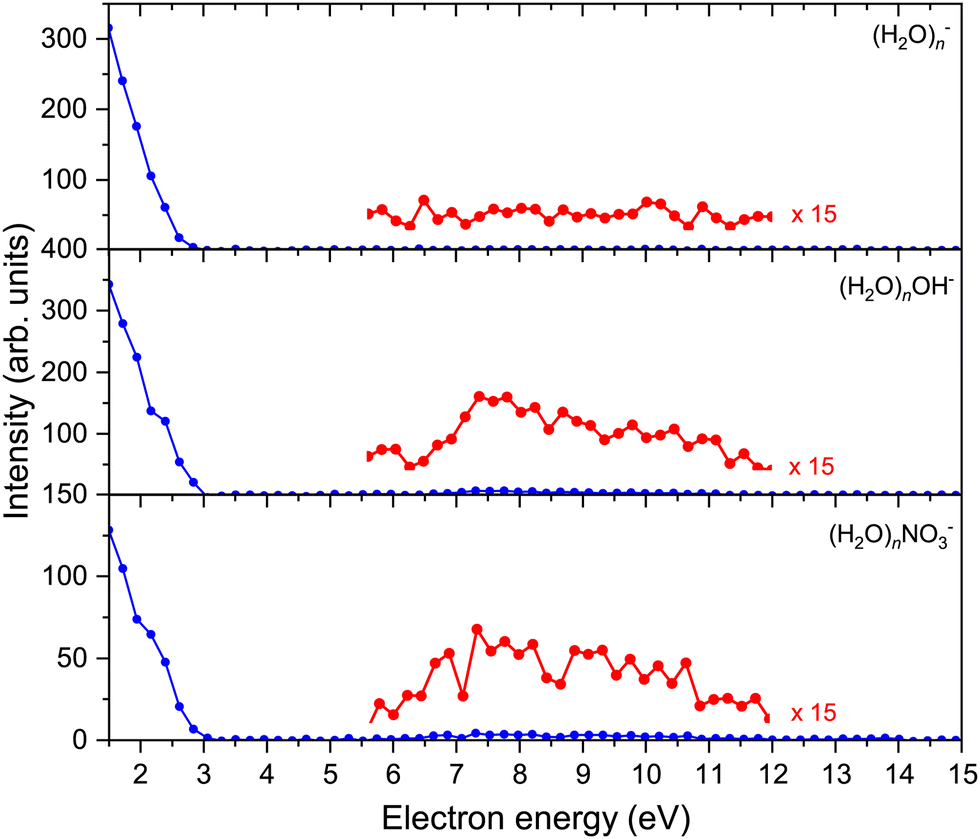 | ||
| Fig. 3 Intensities of all major series in dependence of electron energy. The depicted graphs are an average of n = 16–26 spectra for a better signal-to-noise ratio. | ||
The spectra start at 1.5 eV since our electron gun provides reliable data above this value (see ESI†). Therefore, all the observed cluster ion fragments have a maximum at electron energies of 1.5 eV or lower. Upon closer look, the (H2O)nOH− and (H2O)nNO3− ions exhibit a slightly increasing intensity above 6 eV. At these higher energies, OH− can be generated by the DEA to (H2O)N44 and NO3− generation was observed in our previous investigation of the (HNO3)M(H2O)N clusters generated in co-expansion,37 where the energy dependencies of individual ion yields were discussed in detail.
3.3 Co-expansion vs. pickup
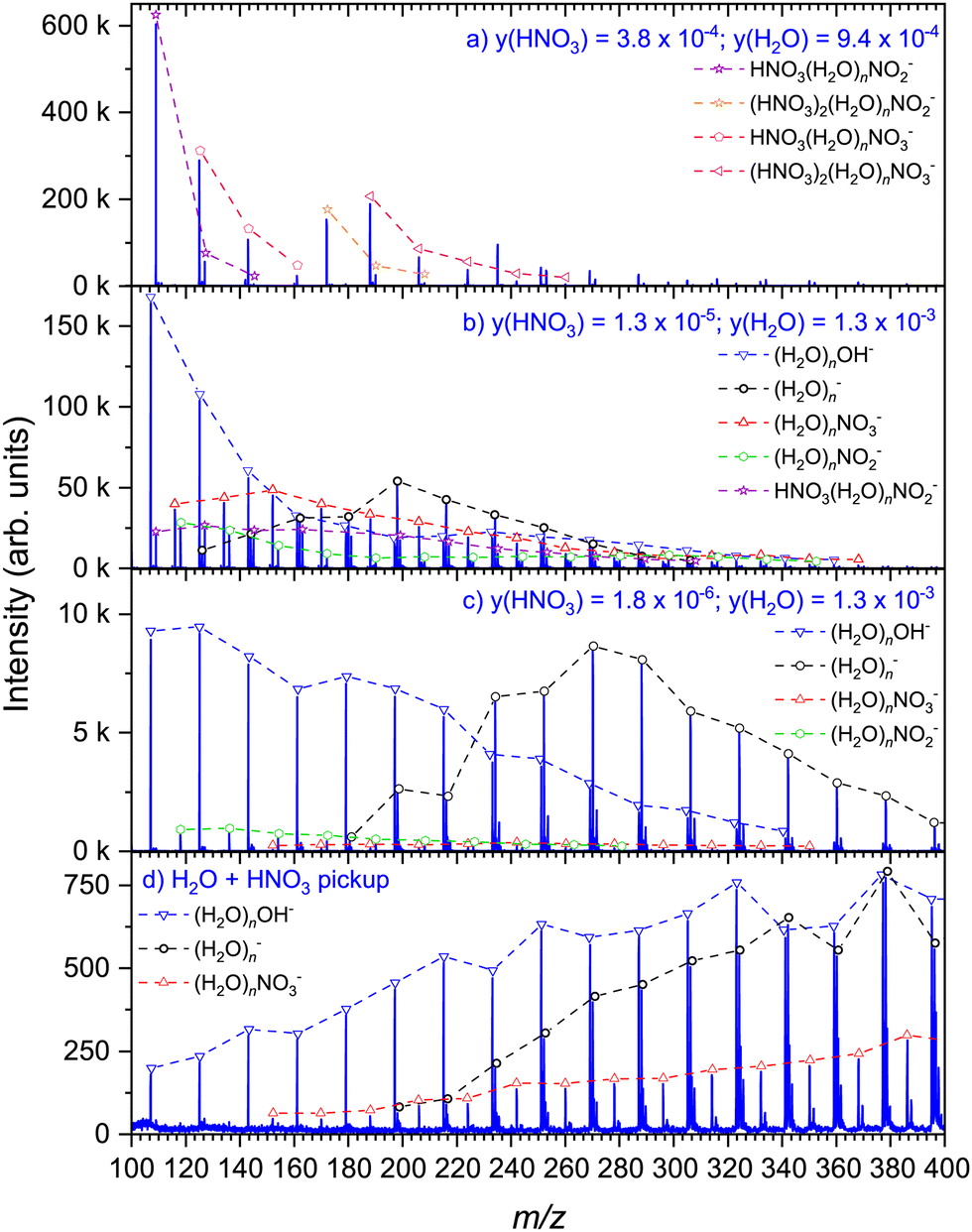
We compare the present experiment to the mass spectra of the hydrated nitric acid clusters generated in co-expansion previously.36,37 Here, we extend the previous investigations by changing the concentration of nitric acid in a similar manner as it was done for positive ion mass spectrometry earlier.33 To this end, the nitric acid solution of known concentration is filled in the source reservoir at a constant temperature TR = 70 °C, and the vapor is carried with He buffer gas at a stagnation pressure of 1 bar through the nozzle, where the hydrated HNO3 clusters form (see ESI† for details). The mole fraction of HNO3 in the vapor can be determined from acid concentration in solution, reservoir temperature, and stagnation pressure.50Fig. 4 shows the dependence of the mass spectra on the HNO3 concentration in co-expansion (a)–(c) in comparison with the present pickup spectrum (d). At the highest concentration, Fig. 4(a), the spectrum is dominated by the (HNO3)m(H2O)nNO2− and (HNO3)m(H2O)nNO3− series, corresponding with observations in our previous investigation under similar conditions.36 Each pronounced series contains at least one HNO3 molecule, i.e., m ≥ 1. As the nitric acid is diluted to y(HNO3) = 1.3 × 10−5, Fig. 4(b), series with m = 0 occur, i.e., (H2O)nNO2− and (H2O)nNO3−, with the latter one having a higher intensity. In contrast to the relatively abundant HNO3(H2O)nNO2− ion yield in Fig. 4(a), the prevailing ions in Fig. 4(b) are (H2O)nOH− and (H2O)n− series. We suggest the former one originating most likely from the clusters containing a single HNO3 molecule, and the latter one attributed to the clusters of pure water without HNO3. This is even more pronounced upon further dilution of the nitric acid, Fig. 4(c), where these two series are absolutely dominating the spectrum. Aside, there are minor ion series (H2O)nNO2− and (H2O)nNO3− present. The pickup spectrum, Fig. 4(d), as previously discussed, consists of the (H2O)n−, (H2O)nOH−, and (H2O)nNO3− ions.
 | ||
| Fig. 4 Mass spectra of HNO3 co-expansion with water (top) and of HNO3 pickup (bottom). | ||
4. Discussion
We can start the discussion with the spectrum of the pickup experiment displayed in Fig. 1(b). The water ion series (H2O)n− originates from the attachment of slow electrons to the pure water clusters. Since the probability of picking up an HNO3 molecule for an average cluster is less than one, there are also clusters with no adsorbed HNO3 molecules in the beam. The electron attachment to these bare clusters yields the (H2O)n− ions.44 The clusters with HNO3 can in principle contribute to this signal, if HNO3 evaporates after the electron attachment. The (H2O)nOH− ions, on the other hand, cannot be produced from pure water clusters at the given electron energy of 1.5 eV.44 Thus, the (H2O)nOH− signal originates from the clusters containing HNO3 prior to the electron attachment. Since the clusters with HNO3 consist of approximately 180 water molecules and only a single HNO3, a previously shown38 possible pathway involves the generation of a hydrated electron eaq− in the water cluster and a subsequent reaction of eaq− with HNO3 leading to the (H2O)nOH− ion formation. We also observed (H2O)nOH− ion production as a result of the electron attachment to the mixed (HNO3)M (H2O)N clusters generated by co-expansion in our previous study.36 The energetically favorable pathway to OH− was calculated to lead through the non-dissociated HNO3 solvated with H2O molecules. Thus, in any case, the OH− series originates from the non-dissociated HNO3 in the cluster. On the other hand, when the electron attachment occurs to the cluster containing the ion pair, the most probable reaction is the neutralization of the oxonium ion H3O+, resulting in the observed (H2O)nNO3− or (HNO3)m(H2O)nNO3− ions after evaporation of the H radical and eventually some water from the cluster, as was also concluded in our previous work.36,37The energy spectra in Fig. 3 show that all the observed ions are formed mainly at very low electron energies below 3 eV. This is consistent with the generation of (H2O)n− ions in pure water clusters,44 as well as with the production of (H2O)nOH− ions in the reaction of the hydrated electron with an HNO3 molecule.38 Also the (H2O)nNO3− ions were formed mainly by the low kinetic energy attachment to the mixed (HNO3)M(H2O)N clusters.36,37
Next, we can compare the pickup and co-expansion spectra with regard to the resulting products and possible underlying chemical processes. Working backwards through the co-expansion spectra, the most diluted one, Fig. 4(c), is dominated by the same ion series as the pickup spectrum in Fig. 4(d), namely (H2O)n− and (H2O)nOH−. This can be expected, as there are many pure water clusters in the beam at very low nitric acid concentration, as demonstrated also in our previous investigation of the positive (HNO3)M(H2O)N cluster ionization.33 Thus, the (H2O)n− ions originate primarily from the pure water clusters in the beam, while the generation of the (H2O)nOH− ions at 1.5 eV electron energy requires the presence of an HNO3 molecule in the clusters. The spectrum suggests that the clusters containing HNO3 molecule(s) produced by co-expansion are significantly smaller than those in the pickup experiment. We assume that the difference in the mean cluster size between the two experiments is the primary reason for the presence of (H2O)nNO2− in the co-expansion experiment, while this series could not be unambiguously confirmed in the pickup spectrum. Given the thermodynamic preference for NO2− as the product for less hydrated clusters, its absence in the ion spectrum resulting from clusters containing on average 180 water molecules can be expected.36,51
With increasing HNO3 concentration in the co-expansion, Fig. 4(b), the contribution of (H2O)n− ions decreases, pointing to the lower abundance of pure water clusters in the beam. In addition, the now most prominent OH− series indicates alongside (H2O)nNO2− that the majority of clusters contains at least one single non-dissociated HNO3. Moreover, it is important to note, that while the presence of (H2O)nNO3− in the co-expansion spectra might be the result of acidic dissociation, as is the case in the previously discussed pickup experiment, NO3− can also be the result of an intracluster reaction of NO2− or OH− with a second HNO3 molecule.36 This can occur in the experiments with higher nitric acid concentration, since clusters containing more than one HNO3 molecule are confirmed by the HNO3(H2O)nNO2− series in this spectrum. However, it cannot be excluded with certainty for the previously discussed spectrum of the lowest HNO3 concentration, where the neutral clusters with more than one HNO3 molecules can be also present although we do not see them in the mass spectra due to the fragmentation after the DEA.
Lastly, at the highest concentration, Fig. 4(a), there is no evidence for the presence of pure water clusters in the beam, and there are no OH− containing ions in the cluster mass range above m/z 100. This observation is clear evidence for the majority of clusters containing more than one HNO3 molecule. Previous investigations32,33 and our calculations36 have shown that acidic dissociation is energetically favourable in clusters with more HNO3 molecules. Thus, the majority of the (HNO3)M(H2O)N clusters generated in co-expansion of concentrated nitric acid most likely contains acidically dissociated HNO3.
Since we have established that in the pickup experiments the (H2O)nOH− ions originate from the clusters containing a non-dissociated HNO3 molecule, while the (H2O)nNO3− ions are generated in the clusters with an acidically dissociated HNO3 molecule, we may speculate to which extent the HNO3 molecules dissociate after landing on the ice nanoparticles. The ratio of the integrated intensities of the NO3− series to the OH− series varied between 0.3 and 0.5 for different expansion conditions, i.e., different (H2O)N cluster sizes between ≈ 30 and 470 (and possibly different cluster temperatures between 90 K and 180 K). However, one has to be cautious with quantitative analysis, as the ion intensities reflect not only the abundance of the clusters with dissociated vs. molecular HNO3, but also their ionization and detection probabilities. In particular, the probability of the electron attachment to clusters with ion pairs can be significantly higher due to the ion pair dipole than to the clusters with the covalently bound molecules. Thus, the observed ratios can be interpreted qualitatively, that the acid dissociation occurs on ice nanoparticles less frequently than the non-dissociated events.
Our observation that molecular HNO3 accounts for 50–70% of the amount present in ice nanoparticles, as determined by mass spectra (but can be even higher when the electron attachment cross section is accounted for), is notable. It is particularly interesting when compared to experiments on amorphous ice, in which substantial quantities of dissociated HNO3 have been identified at temperatures as low as 45 K, with traces of molecular HNO3 completely disappearing at higher temperatures of about 120 K.30 In contrast to these results, our mass spectrometry analysis revealed that we primarily observe HNO3 in its molecular form on the ice nanoparticles, even though we are far above the dissociation onset (recall the estimated cluster temperature of 90–180 K49). In cluster experiments, the immediate environment of an HNO3 molecule can be considered very similar to that investigated in the bulk ice studies. The structure of the ice nanoparticles is expected to be amorphous, although some crystalline core may emerge for our largest measured particle size ( ≈ 470).52,53 Unlike in macroscopic ice, the HNO3 molecule appears to exhibit weak acid behavior when adsorbed onto ice nanoparticles. This suggests that significant size effects may occur as particles shrink to the nanometer scales, leading most likely to a kinetic inhibition of the acid dissociation.
Further, we discuss our present results in the light of the quantum chemical computations on small HNO3(H2O)N clusters, which predict the onset for the HNO3 dissociation at N ≥ 5.35 The present clusters are much larger, yet over half of the HNO3 remains in the molecular form even when adsorbed on the clusters with an average size of about 500 molecules. The first principles molecular simulations21 demonstrated that complete solvation led to the dissociation, while the HNO3 molecules at the water–air interface did not fully dissociate. They also showed that at higher concentrations, nitric acid generated hydrogen bonds without dissociating. This somewhat contradicted the structural calculations of small (HNO3)M(H2O)N clusters,36 showing that even less than 5 water molecules were required for the dissociation in the clusters with M ≥ 2. In any case, the present experiments are performed upon single collision conditions, i.e., the pickup of more than one HNO3 molecule by the cluster is unlikely. Previous molecular dynamics simulations of pickup experiments demonstrated that most dopant molecules interacted with polar water molecules upon uptake, which prevented their mobility and migration within the particle.54,55 The dopants remained isolated at the surface and only adjusted their orientation with respect to the neighboring water molecules. Consequently, the acid dissociation can be suppressed by the incomplete solvation in the present case. Upon collision, however, some molecules may submerge into the cluster55 and dissociate.
Furthermore, the temperature is also a well-known factor influencing proton transfer reactions. For example, ab initio simulations of small hydrogen chloride–water clusters HCl(H2O)N with sizes at the onset of dissociation, i.e. N = 4, demonstrated that the acid dissociated and formed ion pairs at low temperatures, whereas it recombined back to the molecular form, which was more stable as the temperature increased.56 However, for HCl adsorbed on a bulk ice the temperature trend is reversed,57 and similar to the behavior of HNO3 on ice discussed in the introduction. Our present case of ice nanoparticles is probably somewhere between the small clusters and bulk ice. Considering that the estimated temperatures of the larger clusters are considerably lower compared to the smaller ones (see Table S1 in ESI†),49 it becomes apparent that the temperature may play a role in addition to the size effects. This opens up opportunities for further theoretical exploration of the interplay between cluster size and temperature on the dynamics of the acid dissociation in finite size clusters.
5 Conclusions
We have examined dissociative electron attachment of water clusters containing a single HNO3 molecule. The aim was twofold: (i) to investigate the fundamental process of acid dissociation on an ice nanoparticle surface at a molecular level; and (ii) to mimic gas phase processes that may occur on ice particles in the atmosphere. The products were analyzed using negative ion mass spectrometry and compared to experimental results obtained with mixed nitric acid–water clusters (HNO3)M(H2O)N produced in co-expansion.The electron attachment at 1.5 eV electron energy yielded three ion series: (1) (H2O)n− ions resulting from pure water clusters in the beam; (2) (H2O)nOH− ions originating from clusters containing a molecular HNO3; and (3) (H2O)nNO3− ions generated from the clusters with the NO3−⋯H3O+ ion pairs. We have demonstrated that the HNO3 molecules landing on the ice nanoparticles can dissociate to a limited extent and the majority of HNO3 molecules remains non-dissociated on the ice nanoparticles. The ice nanoparticle temperature cannot be determined exactly, however, according to various models and previous experiments,49 it can be safely assumed between 90 and 180 K for the investigated mean cluster sizes of ≈ 30 to 470. Our observations are in interesting contrast to the bulk ice experiments, where the acid dissociation occurs already at 45 K and is complete at 100–120 K. Thus, the fact that we still see non-dissociated molecules on the ice nanoparticles after about 1 millisecond of the flight time in the molecular beam speaks for a kinetic inhibition of the acid dissociation on the nanometer size ice particles.
Author contributions
AK: performed experiment and data evaluation, writing; AP: performed experiment and data evaluation; JĎ: helped data acquisition, review; BK: helped data acquisition, review; MF: supervised experiment, interpretation, writing; JL: proposed idea for the experiment, interpretation, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; Project LE 4583/1-1) and Czech Science Foundation (Project 21-07062S).References
- M. Eigen, Angew. Chem., Int. Ed. Engl., 1964, 3, 1–72 CrossRef.
- K. R. Leopold, Annu. Rev. Phys. Chem., 2011, 62, 327 CrossRef CAS PubMed.
- D. Marx, ChemPhysChem, 2006, 7, 1848–1870 CrossRef CAS PubMed.
- T. Huthwelker, M. Ammann and T. Peter, Chem. Rev., 2006, 106, 1375–1444 CrossRef CAS PubMed.
- T. Peter, Annu. Rev. Phys. Chem., 1997, 48, 785–822 CrossRef CAS PubMed.
- S. Solomon, Rev. Geophys., 1999, 37, 275–316 CrossRef CAS.
- A. J. Prenni and M. A. Tolbert, Acc. Chem. Res., 2001, 34, 545–553 CrossRef CAS PubMed.
- A. R. Douglass, P. A. Newman and S. Solomon, Phys. Today, 2014, 67, 42–48 CrossRef CAS.
- C. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson, Harlow, England, 4th edn, 2012 Search PubMed.
- B. J. Finlayson-Pitts, Chem. Rev., 2003, 103, 4801–4822 CrossRef CAS PubMed.
- Y. Liu, J. P. Cain, H. Wang and A. Laskin, J. Phys. Chem. A, 2007, 111, 10026–10043 CrossRef CAS PubMed.
- L. Liu, H. Li, H. Zhang, J. Zhong, Y. Bai, M. Ge, Z. Li, Y. Chen and X. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 17406–17414 RSC.
- M. Kumar, H. Li, X. Zhang, X. C. Zeng and J. S. Francisco, J. Am. Chem. Soc., 2018, 140, 6456–6466 CrossRef CAS PubMed.
- Y. Knattrup and J. Elm, ACS Omega, 2022, 7, 31551–31560 CrossRef CAS PubMed.
- C. N. Jen, P. H. McMurry and D. R. Hanson, J. Geophys. Res.: Atmos., 2014, 119, 7502–7514 CrossRef CAS.
- M. Wang, W. Kong, R. Marten, X.-C. He, D. Chen, J. Pfeifer, A. Heitto, J. Kontkanen, L. Dada and A. Kürten, et al. , Nature, 2020, 581, 184–189 CrossRef CAS PubMed.
- K. Acker, D. Möller, R. Auel, W. Wieprecht and D. Kalaß, Atmos. Res., 2005, 74, 507–524 CrossRef CAS.
- C. J. Bready, V. R. Fowler, L. A. Juechter, L. A. Kurfman, G. E. Mazaleski and G. C. Shields, Environ. Sci.: Atmos, 2022, 2, 1469–1486 CAS.
- M. Wang, M. Xiao, B. Bertozzi, G. Marie, B. Rörup, B. Schulze, R. Bardakov, X.-C. He, J. Shen and W. Scholz, et al. , Nature, 2022, 605, 483–489 CrossRef CAS PubMed.
- L. Liu, F. Yu, L. Du, Z. Yang, J. S. Francisco and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108384118 CrossRef CAS PubMed.
- T. Lewis, B. Winter, A. C. Stern, M. D. Baer, C. J. Mundy, D. J. Tobias and J. C. Hemminger, J. Phys. Chem. C, 2011, 115, 21183–21190 CrossRef CAS.
- E. S. Shamay, V. Buch, M. Parrinello and G. L. Richmond, J. Am. Chem. Soc., 2007, 129, 12910–12911 CrossRef CAS PubMed.
- R. Bianco, S. Wang and J. T. Hynes, J. Phys. Chem. A, 2008, 112, 9467–9476 CrossRef CAS PubMed.
- S. Wang, R. Bianco and J. T. Hynes, Phys. Chem. Chem. Phys., 2010, 12, 8241–8249 RSC.
- C. Schnitzer, S. Baldelli, D. J. Campbell and M. J. Shultz, J. Phys. Chem. A, 1999, 103, 6383–6386 CrossRef CAS.
- H. Yang and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2001, 105, 1890–1896 CrossRef CAS.
- K. A. Ramazan, L. M. Wingen, Y. Miller, G. M. Chaban, R. B. Gerber, S. S. Xantheas and B. J. Finlayson-Pitts, J. Phys. Chem. A, 2006, 110, 6886–6897 CrossRef CAS PubMed.
- M. C. K. Soule, P. G. Blower and G. L. Richmond, J. Phys. Chem. A, 2007, 111, 3349–3357 CrossRef PubMed.
- J. J. Christensen, L. D. Hansen and R. M. Izatt, Handbook of Proton Ionizaion Heats and Related Thermodynamis Quantities, John Wiley and Sons, New York, 1976 Search PubMed.
- P. Marchand, G. Marcotte and P. Ayotte, J. Phys. Chem. A, 2012, 116, 12112 CrossRef CAS PubMed.
- G. Marcotte, P. Ayotte, A. Bendounan, F. Sirotti, C. Laffon and P. Parent, J. Phys. Chem. Lett., 2013, 4, 2643–2648 CrossRef CAS.
- B. D. Kay, V. Hermann and A. W. Castleman Jr., Chem. Phys. Lett., 1981, 80, 469 CrossRef CAS.
- J. Lengyel, A. Pysanenko, J. Kočišek, V. Poterya, C. C. Pradzynski, T. Zeuch, P. Slavíček and M. Fárník, J. Phys. Chem. Lett., 2012, 3, 3096 CrossRef CAS PubMed.
- P. R. McCurdy, W. P. Hess and S. S. Xantheas, J. Phys. Chem. A, 2002, 106, 7628–7635 CrossRef CAS.
- J. R. Scott and J. B. Wright, J. Phys. Chem. A, 2004, 108, 10578 CrossRef CAS.
- J. Lengyel, M. Ončák, J. Fedor, J. Kočišek, A. Pysanenko, M. K. Beyer and M. Fárník, Phys. Chem. Chem. Phys., 2017, 19, 11753–11758 RSC.
- J. Lengyel, J. Fedor and M. Fárník, Phys. Chem. Chem. Phys., 2019, 21, 8691–8697 RSC.
- J. Lengyel, J. Med, P. Slavíček and M. K. Beyer, J. Chem. Phys., 2017, 147, 101101 CrossRef PubMed.
- M. Fárník, J. Phys. Chem. Lett., 2023, 14, 287–294 CrossRef PubMed.
- A. Pysanenko, K. Fárníková, J. Lengyel, E. Pluhařová and M. Fárník, Environ. Sci.: Atmos, 2022, 2, 1292–1302 CAS.
- M. Fárník, J. Fedor, J. Kočišek, J. Lengyel, E. Pluharová, V. Poterya and A. Pysanenko, Phys. Chem. Chem. Phys., 2021, 23, 3195–3213 RSC.
- M. Fárník and J. Lengyel, Mass Spectrom. Rev., 2018, 37, 630–651 CrossRef PubMed.
- C. Bobbert, S. Schütte, C. Steinbach and U. Buck, Eur. Phys. J. D, 2002, 19, 183–192 CAS.
- M. Knapp, O. Echt, D. Kreisle and E. Recknagel, J. Phys. Chem., 1987, 91, 2601–2607 CrossRef CAS.
- J. Lengyel, M. Ončák and M. K. Beyer, Chem. – Eur. J., 2020, 26, 7861–7868 CrossRef CAS PubMed.
- O. Echt, P. D. Dao, S. Morgan and A. W. Castleman, J. Chem. Phys., 1985, 82, 4076–4085 CrossRef CAS.
- S. Q. Wei and A. W. Castleman, Int. J. Mass Spectrom. Ion Processes, 1994, 131, 233–264 CrossRef CAS.
- L. Belau, K. R. Wilson, S. R. Leone and M. Ahmed, J. Phys. Chem. A, 2007, 111, 10075–10083 CrossRef CAS PubMed.
- D. Becker, C. W. Dierking, J. Suchan, F. Zurheide, J. Lengyel, M. Fárník, P. Slavíček, U. Buck and T. Zeuch, Phys. Chem. Chem. Phys., 2021, 23, 7682–7695 RSC.
- G. B. Taylor, Ind. Eng. Chem., 1925, 17, 633–635 CrossRef CAS.
- F. C. Fehsenfeld, C. J. Howard and A. L. Schmeltekopf, J. Chem. Phys., 1975, 63, 2835–2841 CrossRef CAS.
- V. Buch, B. Sigurd, J. P. Devlin, U. Buck and J. K. Kazimirski, Int. Rev. Phys. Chem., 2004, 23, 375–433 Search PubMed.
- C. C. Pradzynski, R. M. Forck, T. Zeuch, P. Slavícek and U. Buck, Science, 2012, 337, 1529–1532 CrossRef CAS PubMed.
- A. Pysanenko, A. Habartová, P. Svrčková, J. Lengyel, V. Poterya, M. Roeselová, J. Fedor and M. Fárník, J. Phys. Chem. A, 2015, 119, 8991–8999 CrossRef CAS PubMed.
- J. Poštulka, P. Slavícek, A. Pysanenko, V. Poterya and M. Fárník, J. Chem. Phys., 2022, 156, 054306 CrossRef PubMed.
- R. P. de Tudela and D. Marx, Phys. Rev. Lett., 2017, 119, 223001 CrossRef PubMed.
- S.-C. Park and H. Kang, J. Phys. Chem. B, 2005, 109, 5124–5132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details; analysis of metastable clusters and isobaric ions; experiments with different cluster sizes and temperatures. See DOI: https://doi.org/10.1039/d3cp02757k |
| This journal is © the Owner Societies 2023 |