DOI:
10.1039/D3CC02790B
(Highlight)
Chem. Commun., 2023,
59, 9424-9444
C–C bond formation via photocatalytic direct functionalization of simple alkanes
Received
9th June 2023
, Accepted 28th June 2023
First published on 29th June 2023
Abstract
The direct functionalization of alkanes represents a very important challenge in the goal to develop more atom-efficient and clean C–C bond forming reactions. These processes, however, are hampered by the low reactivity of the aliphatic C–H bonds. Photocatalytic processes based on hydrogen atom transfer C–H bond activation strategies have become a useful tool to activate and functionalize these inert compounds. In this article, we summarize the main achievements in this field applied to the development of C–C bond forming reactions, and we discuss the key mechanistic features that enable these transformations.

Álvaro Velasco-Rubio
| Álvaro Velasco-Rubio, born in Salamanca (Spain), received his BSc in chemistry from the University of Salamanca, Spain, in 2015. He completed his MSc in 2016 and his PhD thesis in 2021 under the supervision of Prof. Carlos Saá and Jesús A. Varela. He spent a predoctoral research stay at Caltech under the supervision of Prof. Brian M. Stoltz. In 2022, he joined the group of Prof. Martín Fañanás-Mastral as a postdoctoral researcher. In 2023, he joined the group of Prof. Rubén Martín as a Juan de la Cierva Researcher. |

Pol Martínez-Balart
| Pol Martinez-Balart completed his BSc in Chemistry in 2019 at the University of Barcelona where he conducted research in asymmetric hydrogenations under the supervision of Prof. Antoni Riera at the IRB. After a stay at the company Enantia, Pol continued his MSc studies in Organic Chemistry at the University of Santiago de Compostela under the supervision of Prof. José Luís Mascareñas working on Rh-catalysed C–H activation of heterocycles. Currently, he is PhD candidate in the Fañanás-Mastral group at CiQUS working on bimetallic functionalization of light alkanes employing photocatalysis. |

Andrés M. Álvarez-Constantino
| Andrés M. Álvarez-Constantino obtained his BSc in Chemistry (2019) and MSc in Chemistry at the Interface with Biology and Materials Science (2021) from the University of Santiago de Compostela, where he worked in Rh-catalysed C–H activation and DFT mechanistic studies under the supervision of Prof. Jesús A. Varela and Prof. Carlos Saá at CiQUS. After that, he joined the Fañanás-Mastral group, and he is currently a PhD candidate (Xunta de Galicia predoctoral fellowship) working on the functionalization of light hydrocarbons by means of organometallic photocatalysis as well as in computational mechanisms elucidation. |

Martín Fañanás-Mastral
| Martín Fañanás-Mastral obtained his PhD from the University of Oviedo (Spain) in 2007. During that time, he carried out a short stay at the group of Prof. Steven Ley at the University of Cambridge (UK). In 2009 he joined the group of Prof. Ben L. Feringa at the University of Groningen (The Netherlands) as a postdoctoral researcher. In 2014 he moved to CiQUS (University of Santiago de Compostela, Spain), where he leads the “Sustainable Catalysis and Asymmetric Synthesis” group. His current research interests focus on the development of synthetic methods towards the stereoselective carboboration of hydrocarbons and the direct functionalization of alkanes. |
Introduction
Carbon–carbon bond formation plays a central role in the synthesis of natural products, pharmaceuticals, agrochemicals, and organic materials. Thus, it lies at the heart of chemical sciences. Traditionally, C–C bond formation reactions have involved the coupling of organic electrophiles with pre-made organometallic reagents.1 Despite their great utility, most of these transformations involve multistep procedures associated with the preparation and purification of the organometallic reagent prior to cross-coupling. Moreover, they are limited to the inherent reactivity and basicity of the organometallic reagent which can impose limitations with respect to the functional-group tolerance. In this context, the direct functionalization of C–H bonds offers a more atom-efficient and straightforward way to incorporate a carbon framework into readily available substrates. While the selective C–H functionalization of hydrocarbons has seen major advances in recent decades,2 the direct functionalization of C(sp3)–H bonds in alkanes still represents one of the foremost challenges in synthetic chemistry. In contrast to unsaturated substrates, the lack of π-electrons and vacant π* molecular orbitals in alkanes makes them poor nucleophiles and hampers their coordination to metals, thus it is more difficult to activate and functionalize the alkane C–H bonds. Another important challenge in the functionalization of saturated hydrocarbons is selectivity. C–H bond dissociation energies increase in the order R3C–H < R2HC–H < RH2C–H < H3C–H in the range of approximately 95–104 kcal mol−1. This difference in C–H BDEs makes cleavage of tertiary C–H bonds more facile, although terminal C–H bonds are generally more accessible. The presence of a heteroatom in the hydrocarbon structure can facilitate C–H activation since the hetero-functionality can serve to coordinate the substrate to the catalyst, and the functional group can activate the C–H bond towards cleavage.3 However, activation and thus functionalization of simple alkanes lacking hetero-functionality represents a more substantial challenge. Nevertheless, different strategies have been reported for the selective C–H to C–C functionalization of alkanes.
The electrophilic activation of alkanes represents an important strategy for the conversion of saturated hydrocarbons into acids or esters through carbonylation reactions, albeit they typically require high temperature and/or harsh acidic media.4 Carbenes have also been applied in alkane functionalization with the formation of a C–C bond, mainly upon transition metal-mediated carbene insertion into C–H bonds.5 Besides alkane carbonylation and carbene insertion reactions, which have been thoroughly reviewed,4,5 photocatalysis processes in which saturated hydrocarbons are activated either by single electron transfer (SET) or hydrogen atom transfer (HAT) have emerged as a very powerful tool for the direct functionalization of alkanes.
The scope of this article is to cover the state-of-the-art techniques in the photocatalytic and photo-electrocatalytic functionalization of simple alkanes involving direct C–C bond formation, with the aim of highlighting the main achievements, their key mechanistic features, and the future challenges within the field. We have divided the review based on the nature of the species that is used to activate the aliphatic C–H bond.
Photocatalytic reactions involving a halogen radical as the HAT reagent
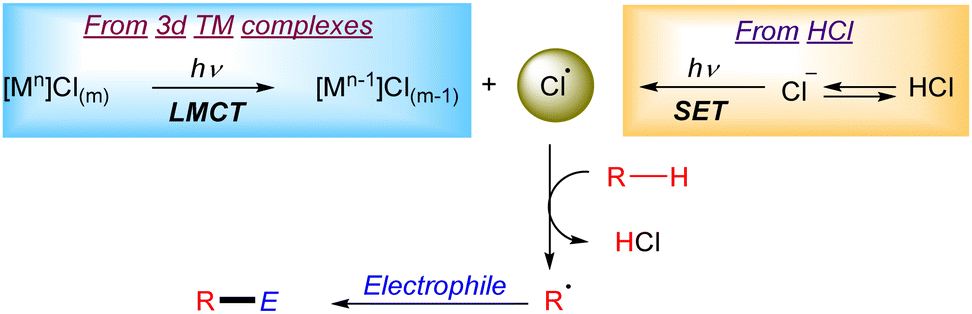
Chlorine radicals (Cl˙) have been broadly explored as HAT reagents in the formation of new C–C bonds (Scheme 1). The generation of Cl radicals is often based on a Ligand to Metal Charge Transfer (LMCT) process in a 3d transition metal complex (i.e. Ni, Cu, Fe, Ce or Ti).6 The general mechanism of this transformation often relies on the formation of a 3d transition metal–chloride complex which after irradiation generates an active Cl˙ in the reaction media through a photoinduced homolytic M–Cl cleavage via LMCT. The generated Cl radical can abstract a hydrogen atom from alkanes to form a carbon centred radical which then reacts with the corresponding electrophile to form the new C–C bonds. Additionally, the Cl˙ can be suitably generated upon reaction of a nickel–chloride complex with an excited photocatalyst (mainly iridium complexes) and subsequent LMCT on the oxidized nickel complex.7 Another useful strategy to generate Cl radicals is based on photoredox catalysis. In this case the photocatalyst oxidizes a chloride anion (Cl−) to Cl˙ via an outer-sphere electron transfer mechanism.
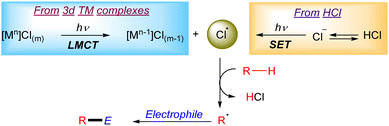 |
| | Scheme 1 General mechanisms for the generation of Cl radicals. | |
Photocatalytic generation of halogen radicals from HX via SET
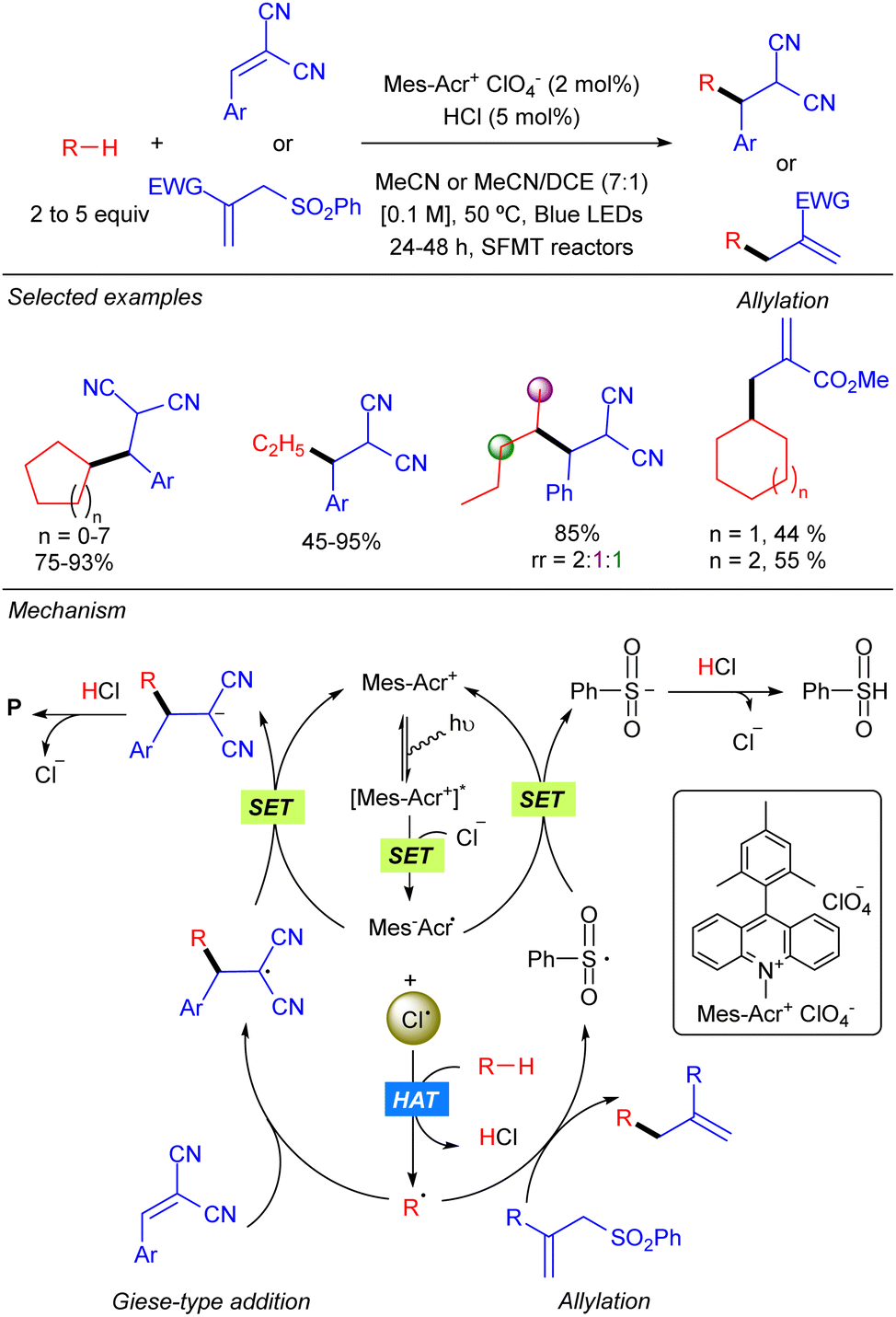
In 2018 the group of Wu reported the functionalization of alkanes using HCl as a HAT catalyst precursor.8 In this reaction the generation of new C–C bonds was demonstrated by a Giese-type reaction with Michael acceptors and by allylation with allylic sulfones using a stop-flow microtubing reactor (Scheme 2). Cl˙ is generated after the reaction of Cl− with the photoexcited Mes–Acr photocatalyst. Hydrogen atom abstraction produces a carbon centred radical which adds to a Michael acceptor generating a new carbon radical which undergoes SET with the reduced photocatalyst to regenerate the active species. Final protonation with HCl delivers the desired product and releases the chloride anion. For allylic sulfones, SH2′-addition of the carbon radical produces a benzene sulfinyl radical which reacts with the reduced photocatalyst in a similar manner. Both reactions showed a broad alkane scope, including ethane, which led to the corresponding products in very good yields. Regarding the electrophile, the scope was limited to activated Michael acceptors and activated allylic sulfones.
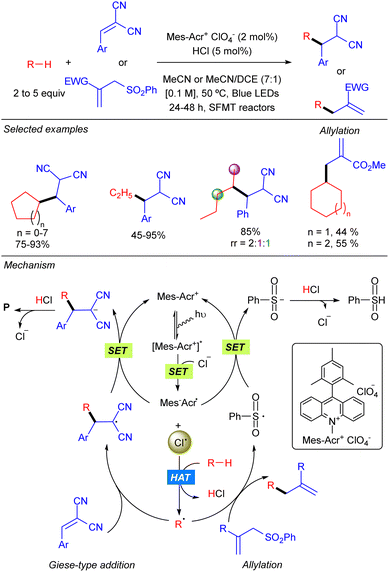 |
| | Scheme 2 Hydro-alkylation/allylation driven by HCl as the HAT catalyst. | |
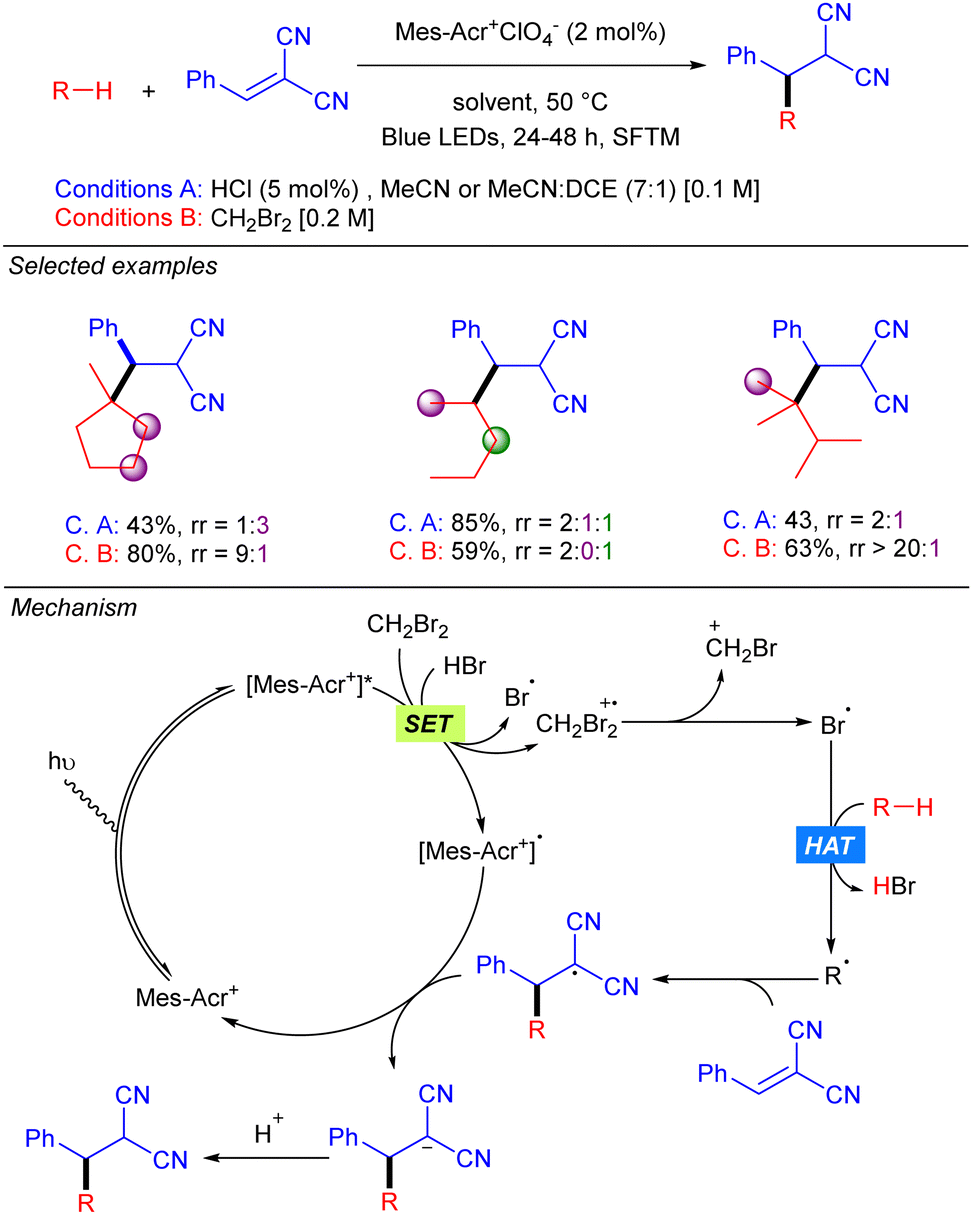
Later, the same group developed a similar protocol based on bromine radical photo-HAT processes, instead of chlorine (Scheme 3).9 It was shown that bromine radicals display excellent reactivity and selectivity for tertiary C–H bonds, whereas chlorine radicals showed almost no selectivity. Thus, alkanes such as methyl-cyclopentane or 2,3-dimethyl-butane could be alkylated in excellent yields and regioselectivities under these new bromine radical-based conditions.
 |
| | Scheme 3 Hydro-alkylation of Michael acceptors using Br˙ as the HAT reagent. | |
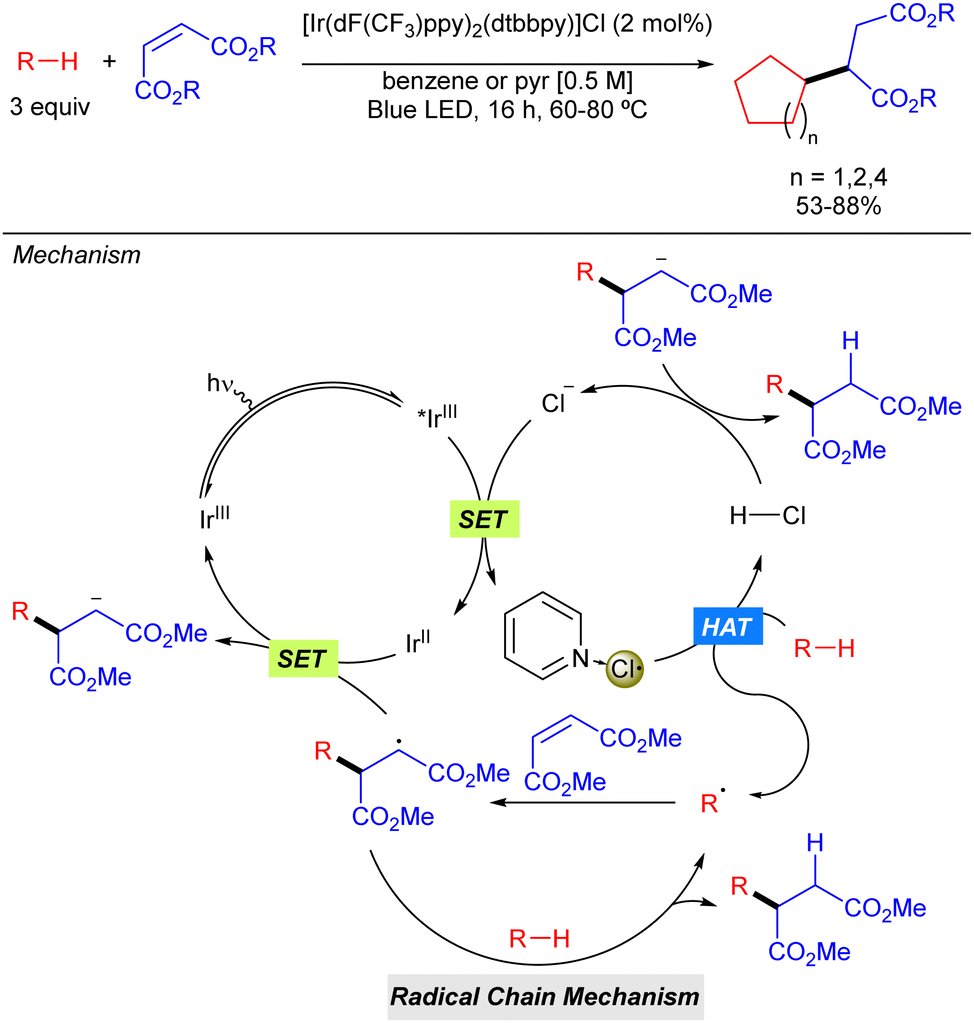
In the same year, the group of Barriault reported hydro-alkylation of Michael acceptors that exploits the use of Cl˙ as the HAT reagent in combination with Ir photocatalysis (Scheme 4).10 In this case, after excitation of the iridium catalyst with blue light, a SET process takes place between the excited iridium complex (*IrIII) with its counter anion (Cl−) to form the electrophilic Cl˙ which may be stabilized by coordination of the pyridine base, or benzene, attenuating its high reactivity in order to promote a more selective process. Generation of the carbon radical via HAT with the alkane, followed by addition to the corresponding Michael acceptor via Giese type reaction and subsequent SET process with the reduced form of the Ir catalysts, restarting the Ir catalytic cycle and producing the carbanion which gets protonated with HCl. The reaction was limited to activated diester Michael acceptors and to the use of cycloalkanes such as cyclopentane, cyclohexane and cyclooctane that provided the corresponding products in good to excellent yields.
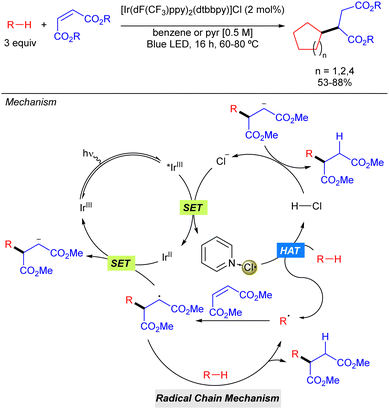 |
| | Scheme 4 Hydro-alkylation of Michael acceptors enabled by the generation of Cl˙ via photoredox catalysis. | |
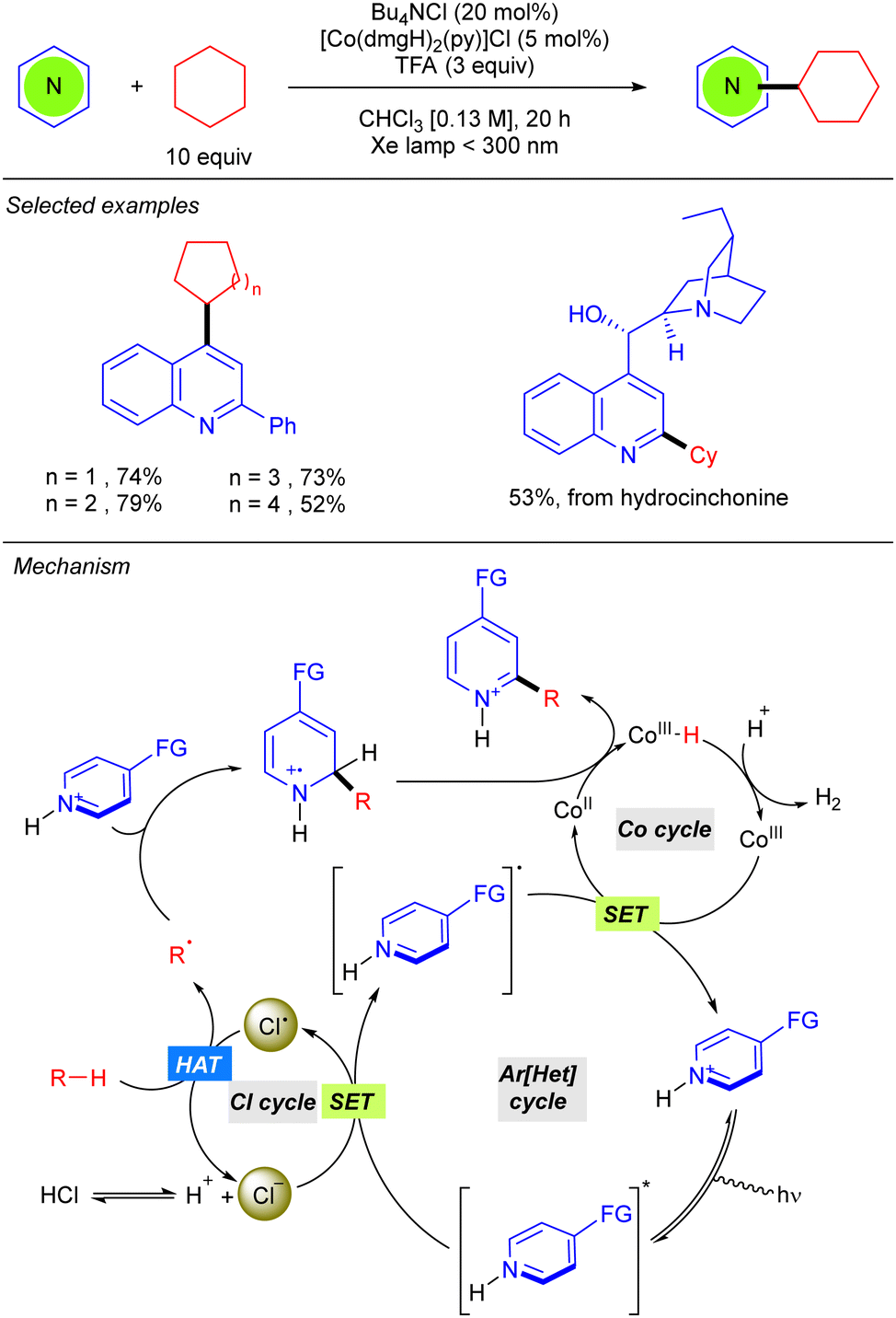
Besides the use of a Michael acceptor in Giese-type reactions, Li reported the first Minisci-type reaction with alkanes through a cooperative catalysis merging the photoredox generation of chlorine radical and a cobaloxime catalyst, which enables hydrogen evolution for catalytic turnover (Scheme 5).11 This Minisci-type alkylation is limited to cyclic alkanes and promotes a cross-dehydrogenative C(sp3)–H heteroarylation on position 4 of the 2-phenylquinoline under far UV-C irradiation, in chloroform as solvent in the presence of TFA. This strong acid was shown to be indispensable for the reaction since mechanistic studies revealed that protonation of the heteroarene and subsequent excitation is required for the oxidation of the Cl anion (that comes from the Bu4NCl) via the SET process. The chlorine radical promotes the HAT step regenerating the Cl− and the new alkyl radical. R˙ is added over the position 2, or 4, of the heteroarene leading to the N-centred radical-cation. This species will lastly undergo oxidation with the CoII complex, promoting the hydrogen evolution process, and leading to the final product.
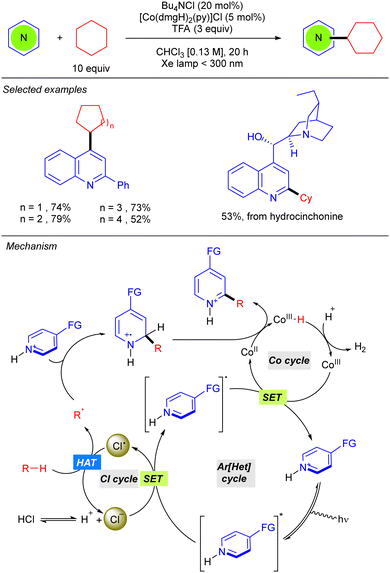 |
| | Scheme 5 Minisci-type alkylation through dual cobalt/Cl˙ catalysis. | |
LMCT: Ni/Ir catalysis
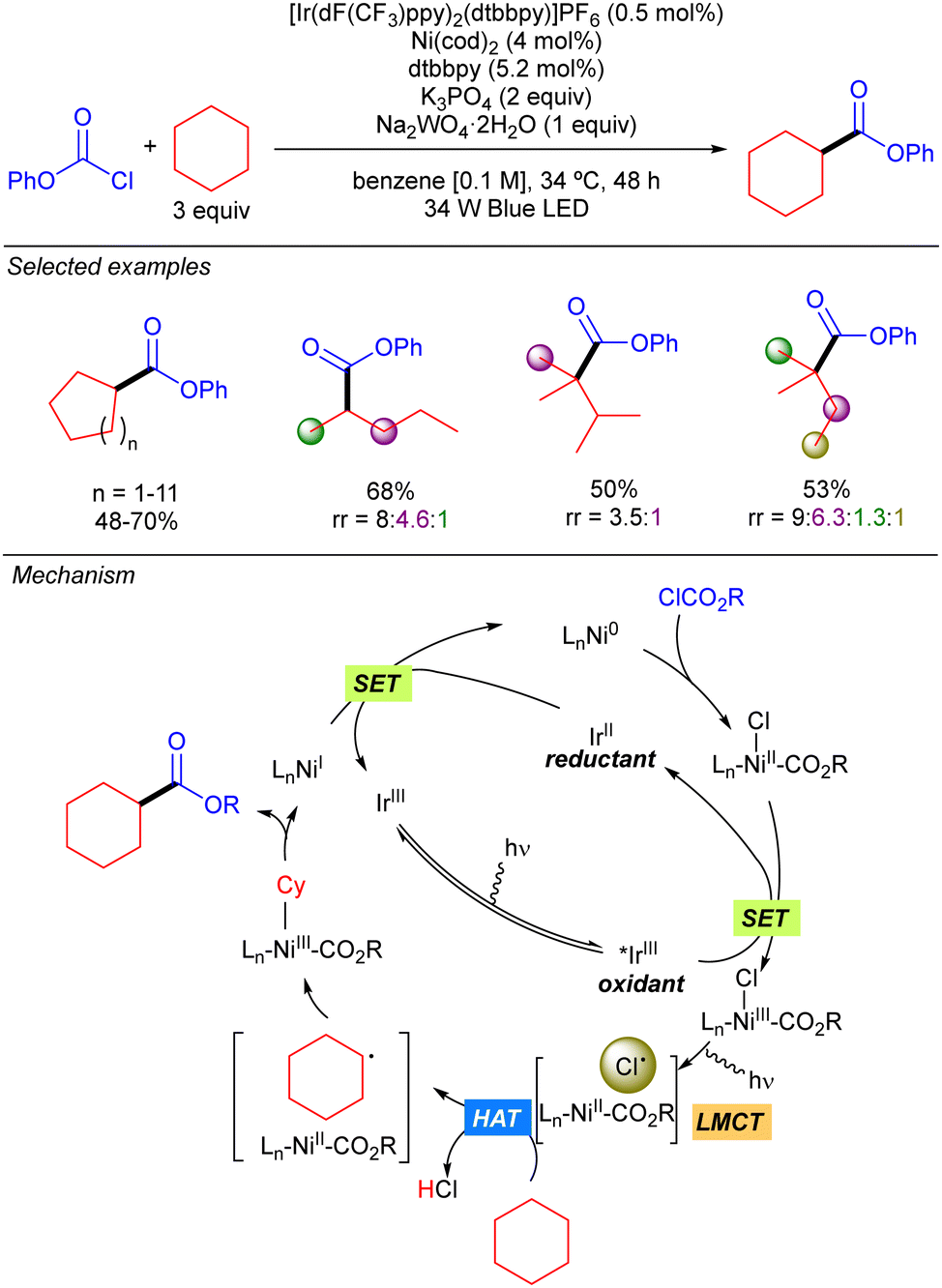
In 2016 the group of Doyle reported a new methodology for direct C(sp3)–H cross-coupling enabled by catalytic formation of Cl˙ from LnNi(III)(CO2R)Cl intermediates. This methodology was mainly applied to the direct arylation of ethers but one example on the use of cyclohexane was reported.12 Two years later, this group expanded the methodology to the direct esterification of simple alkanes with chloroformates (Scheme 6).13 The proposed mechanism for this transformation starts with the oxidative addition of Ni(0) to chloroformate to generate a Ni(II)(Cl)(CO2R) complex. Concomitantly, the Ir(III) complex is photoexcited to the active triplet Ir(III)* which undergoes a SET with the Ni(II) complex to produce a Ni(III)(Cl)(CO2R) species. The photolysis of this intermediate delivers Cl˙ and LnNi(II)(CO2R). The Cl˙ abstracts a hydrogen from the alkane to generate an alkyl radical which recombines with Ni(II)(CO2R) species to form a Ni(III) intermediate that releases the desired product after reductive elimination. Finally, a SET process takes places between the resulting Ni(I) species and Ir(II) to restart both catalytic cycles. Cyclic and acyclic alkanes worked in good to excellent yields. Regarding selectivity, it was compromised with linear and branched alkanes, showing a site preference according to the stability of the formed carbon radical (3° > 2° > 1°).
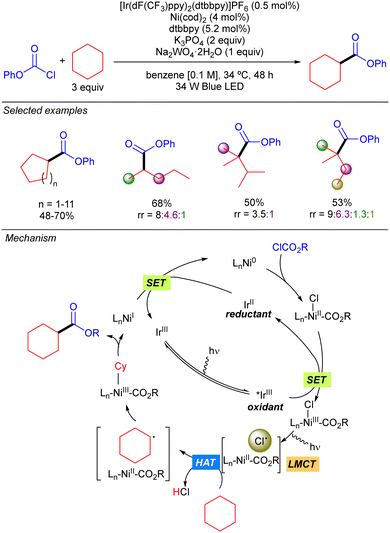 |
| | Scheme 6 Ni-catalysed esterification of alkanes. | |
LMCT: Fe catalysis
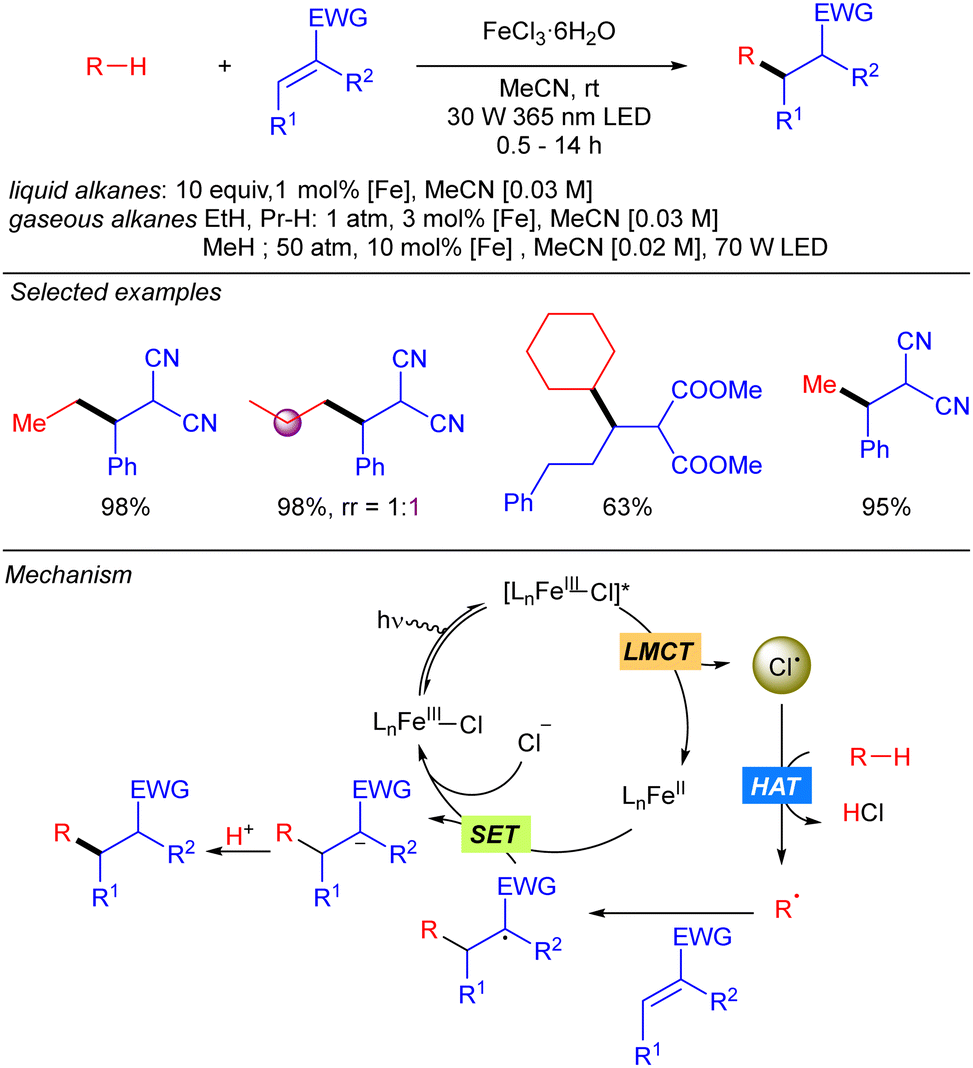
In 2021, the group of Jin and Duan reported the iron-catalysed functionalization of alkanes using Michael acceptors and/or diazocarboxylates as coupling partners via Cl˙ generation through LMCT from FeCl3 (Scheme 7).14 The reaction shows a broad scope of cyclic and linear alkanes including ethane and propane, with low regioselectivity for linear alkanes. In fact, regioselectivity on linear systems depends on the stability of the corresponding generated carbon radicals (3° > 2° > 1°) and the number of available hydrogen atoms with the same chemical environment. Moreover, branched alkanes were also tested in the reaction conditions, but no selectivity was achieved over primary/tertiary hydrogens. In the cases of ethane and propane, due to the low solubility in MeCN at ambient temperature and pressure, catalyst loading was required to be increased and the reaction was diluted to increase the relative concentration of the gaseous alkane. In 2022, the same group extended this methodology to the use of methane.15 The use of the simplest alkane requires higher catalyst loadings, as well as higher pressure and higher dilution to increase the relative concentration of the gas. Mechanistically, this transformation starts with the photoexcitation of the Fe(III)–Cl complex to [Fe(III)–Cl]* under UV-A irradiation. Then, homolysis of the Fe–Cl bond provides Cl˙ which reacts with the corresponding alkane to obtain the alkyl radical. This radical undergoes a Giese type reaction to obtain a new carbon centred radical that after a SET process provides the carbanion and regenerates the Fe–Cl active species. Finally, protonolysis gives rise to the desired product.
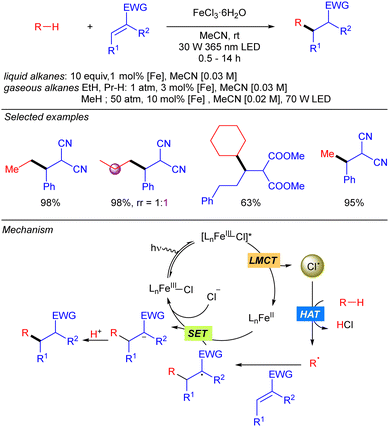 |
| | Scheme 7 Iron-catalysed functionalization of alkanes enabled by LCMT Cl˙ generation. | |
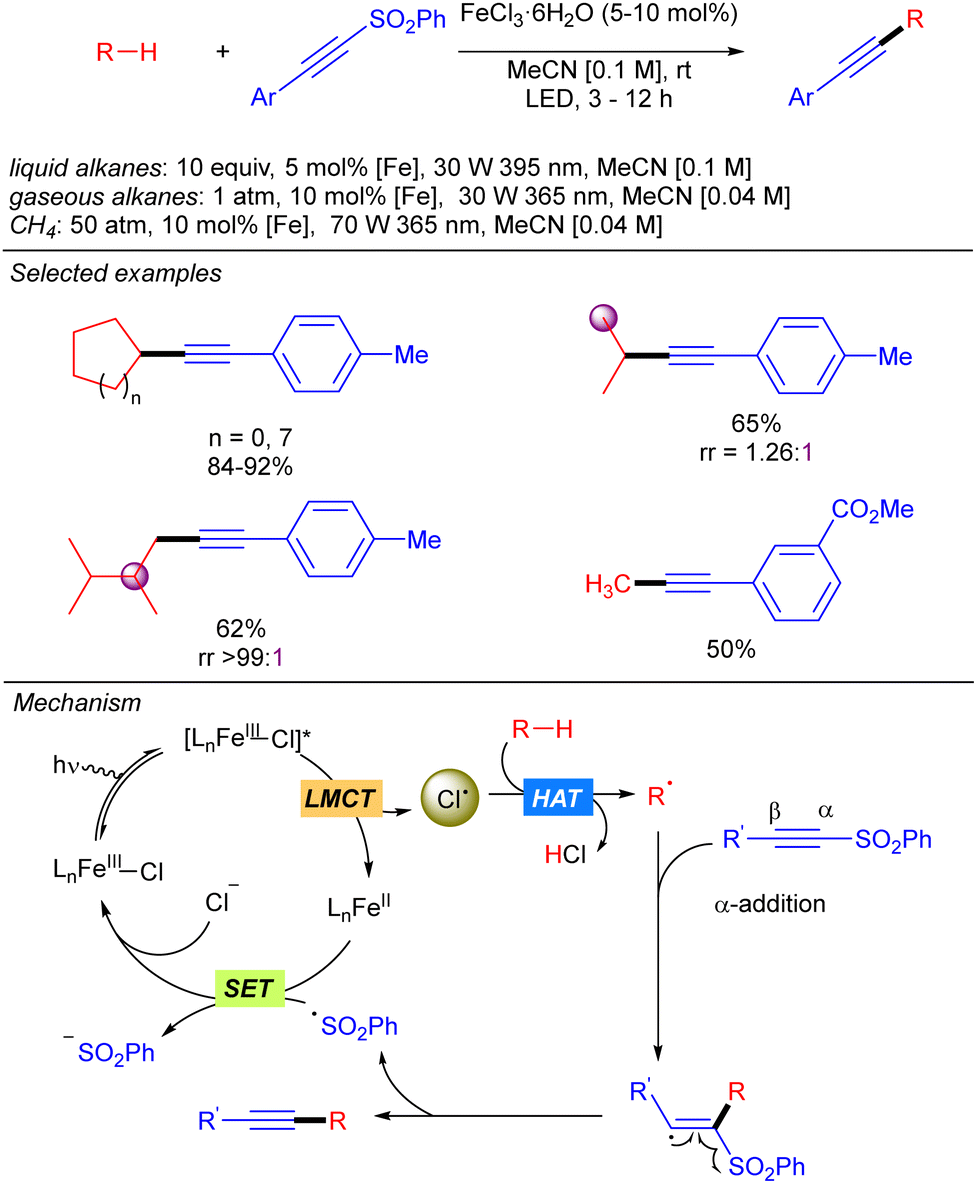
The group of Jin and Duan also reported in the same year iron-catalysed direct alkynylation of methane and other light alkanes using ethynyl phenyl sulfones (Scheme 8).16 This methodology can be applied to cyclic, linear and branched alkanes. In this last case, in sterically hindered systems, excellent selectivity towards the functionalization of primary hydrogens was observed. Mechanistically, the alkyl radical generated by Cl˙ promoted alkane hydrogen atom abstraction reaction with the alkyne via α-addition followed by a sulphonyl radical elimination to release the desired alkynylated product. The sulphonyl radical reacts via a SET process with Fe(II) species to restart the iron catalytic cycle. This mechanism was supported by experimental studies and DFT calculations.
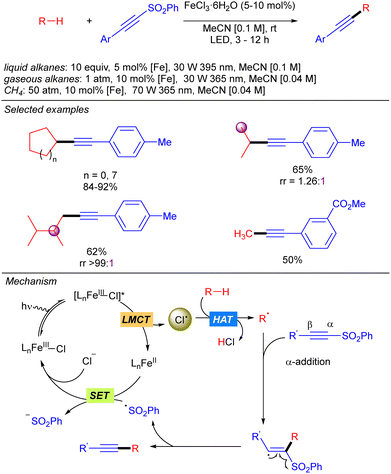 |
| | Scheme 8 Iron-catalysed alkynylation of alkanes enabled by LCMT Cl˙ generation. | |
LMCT: Cu catalysis
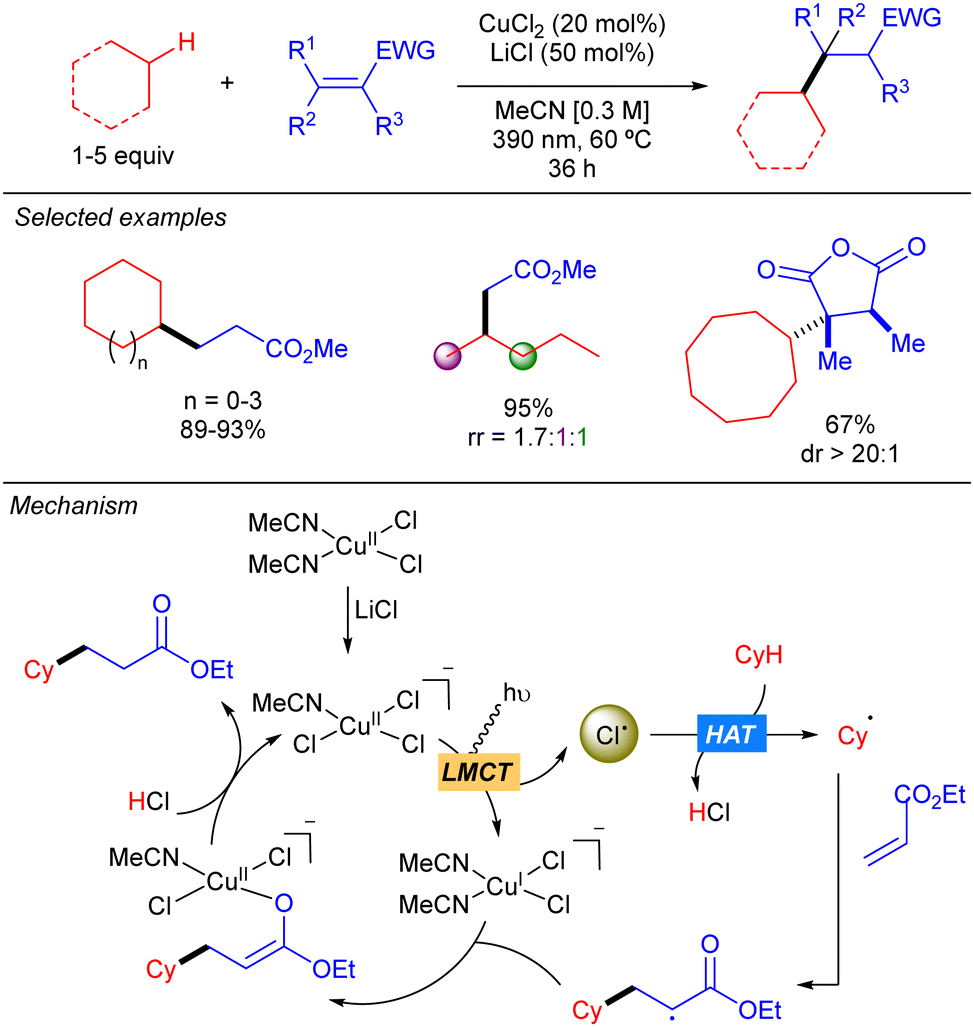
In 2021, the group of Rovis reported the copper(II)-catalysed coupling of alkanes with Michael acceptors (Scheme 9).17 The method is based on the use of a mixture of CuCl2 and LiCl that provides chlorocuprate species [CuIICl3]− which undergoes LMCT to generate the Cl˙ and [CuICl2]− upon irradiation with UV-A light. The Cl˙ abstracts the hydrogen to generate the carbon centred radical which undergoes a Giese-type reaction to obtain a more stable α-radical ester which recombines with [CuICl2]− to form a CuII-enolate that after proto-demetallation with HCl releases the desired product and restarts the catalytic cycle. The transformation features a broad scope of C–H donors. Among them, different simple alkanes were proven to be efficient partners for this reaction, albeit low regioselectivity was observed when a linear substrate such as pentane was used. Remarkably, activated endocyclic alkenes, such as acid anhydrides, furnish the corresponding products with high diastereoselectivity. This stereocontrol is proposed to be modulated by the formation of the CuII-enolate which undergoes stereoselective protonation to regenerate the oxidized copper catalyst.
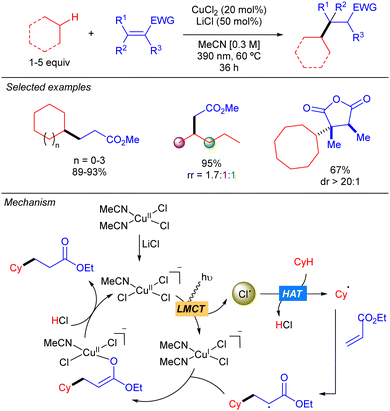 |
| | Scheme 9 Cu-catalysed functionalization of alkanes. | |
LMCT: Ti catalysis
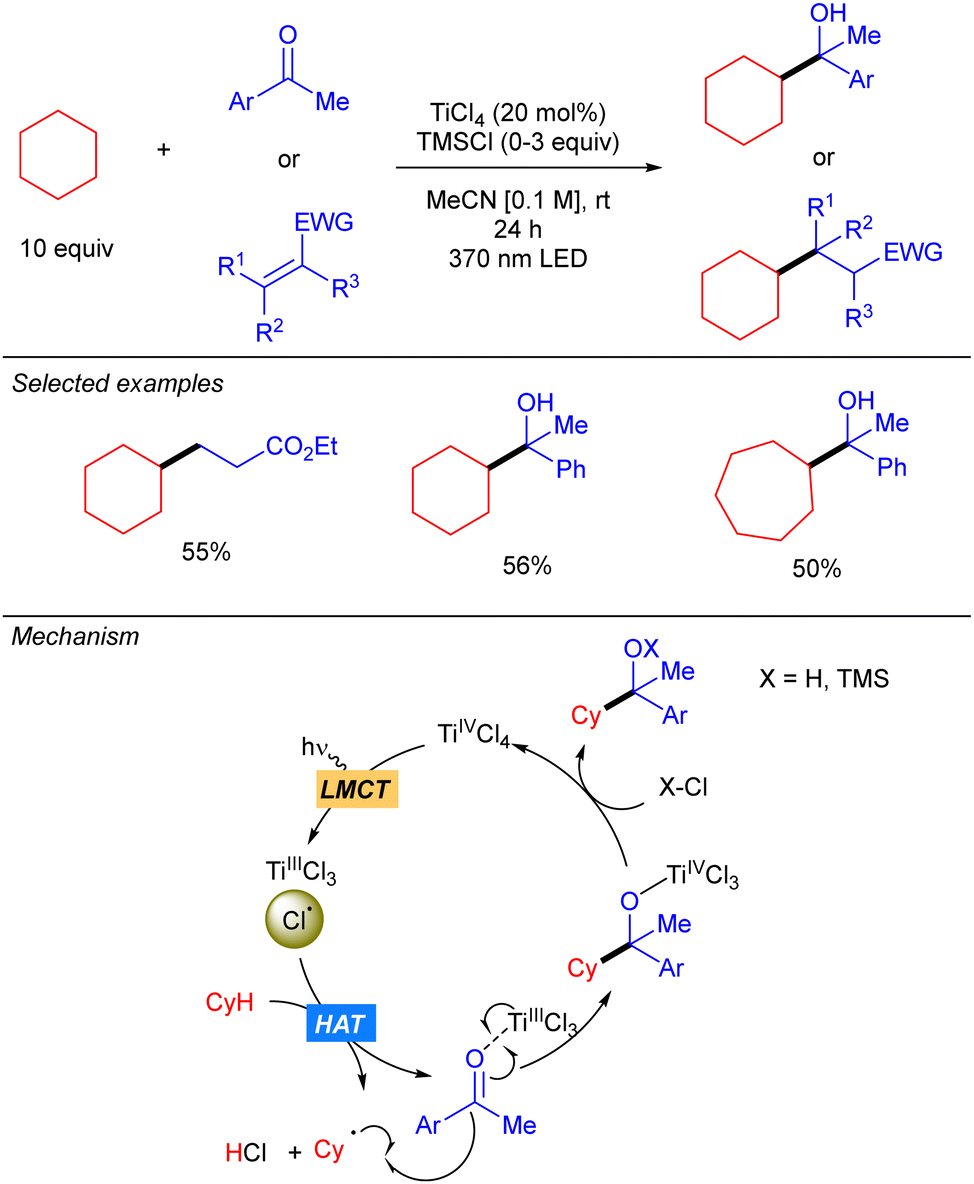
One year later, the group of Mitsunuma and Kanai reported the Ti-catalysed functionalization of cyclic alkanes using ketones or Michael acceptors as coupling partners (Scheme 10).18 The proposed mechanism starts with the excitation of TiCl4 which after a LMCT provides Cl˙ and TiCl3. When Michael acceptors are used as the electrophile, a typical Giese type reaction takes place. However, when ketones are used as the electrophile the generated TiCl3 acts as Lewis acid, enhancing the electrophilicity of the ketone. Therefore, after the addition of the alkyl radical a titanium alkoxide is obtained, which after proto-demetallation with HCl or TMSCl releases the desired product, and the catalytic cycle is restarted.
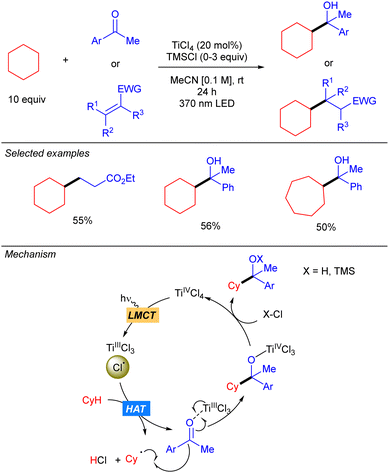 |
| | Scheme 10 TiCl4-catalysed functionalization of alkanes. | |
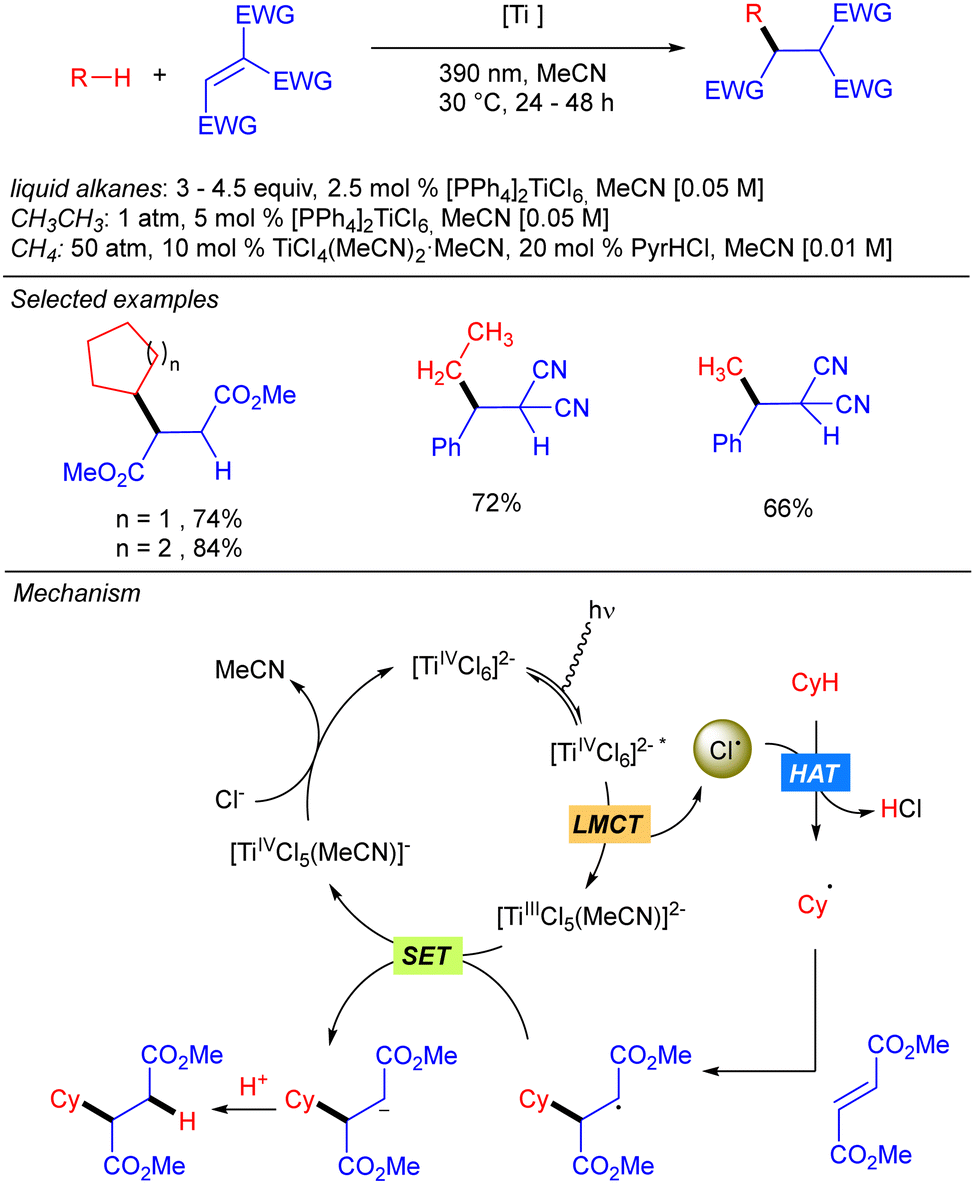
Simultaneously to the work of Mitsunuma and Kanai, Schelter and co-workers reported the reactivity of dianionic species Ti(IV)Cl62− in C(sp3)–H bond functionalization of light alkanes, including methane and ethane (Scheme 11).19 [PPh4]2TiCl6 was found to be significantly more air and moisture stable than TiCl4. The use of methane required an alternative catalytic system involving a combination of TiCl4(MeCN)2 (10 mol%) and pyridine hydrochloride (PyrHCl, 20 mol%) as the source of the Ti(IV)Cl62− anion. Mechanistic studies indicated that photoexcitation into the LMCT band of Ti(IV)Cl62− leads to the excited complex. Upon vibrational relaxation, the formation of Cl˙ together with Ti(III)Cl52− takes place. Cl˙ promotes the HAT and after alkyl radical addition to the electron-poor olefin, the resulting organoradical undergoes SET with Ti(III)Cl52−, leading to Ti(IV)Cl5− and the corresponding carbanion. This gets protonated with in situ formed HCl to yield the product and photoactive Ti(IV)Cl62− catalyst regeneration.
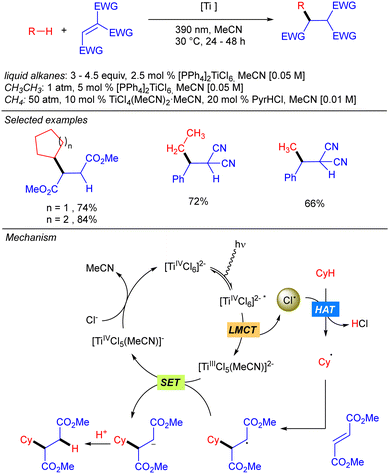 |
| | Scheme 11 [PPh4]2TiCl6-catalysed functionalization of alkanes. | |
LMCT: Ce catalysis
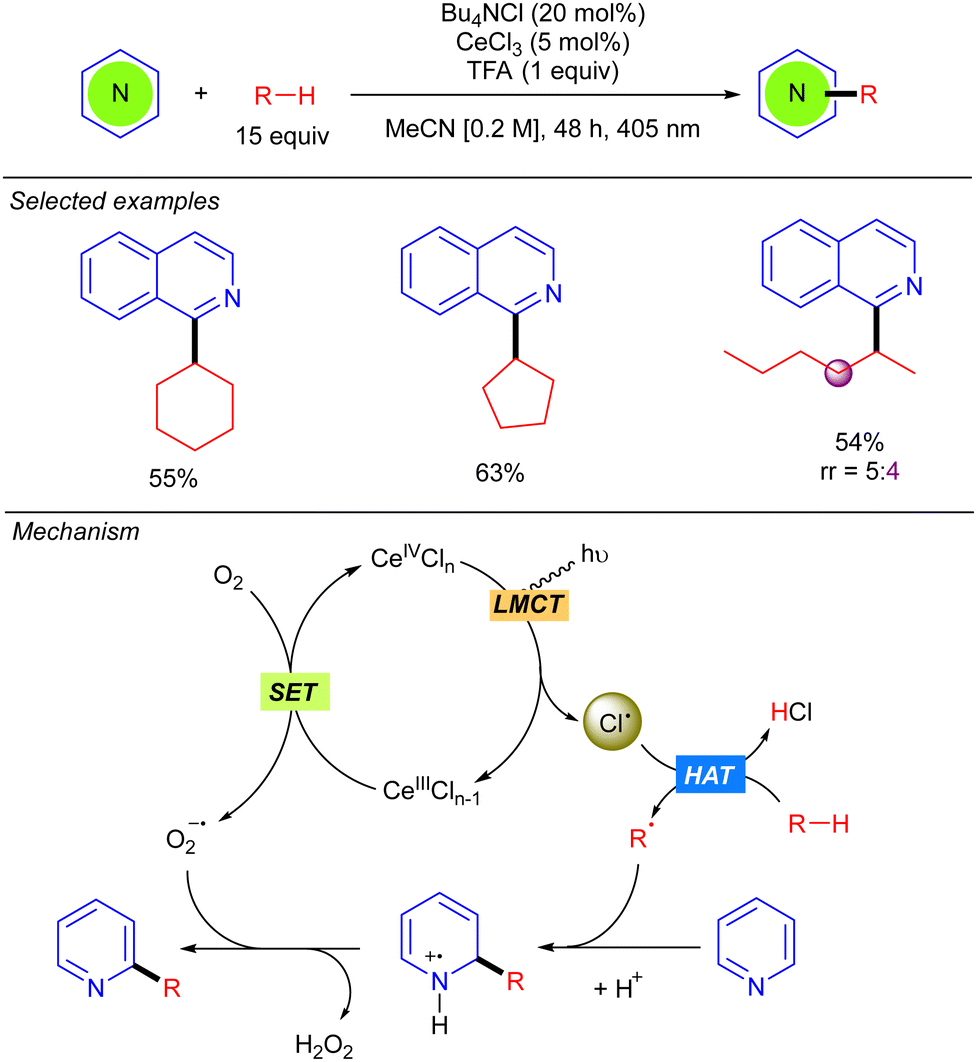
In 2021, Zhang and Liu developed a Minisci-type reaction with alkanes by using CeCl3 as the photocatalyst (Scheme 12).20 Compared to the methodology previously reported by Li based on cooperative photoredox/cobaloxime catalysis (see Scheme 5),11 this protocol allows the reaction to be carried out under milder conditions with visible light (405 nm) irradiation. Beyond cyclic alkanes, a linear alkane such as n-hexane also proved to be compatible, albeit with poor regioselectivity. The catalytic cycle begins with the oxidation of Ce(III)Cln−1 with O2 to generate Ce(IV)Cln and a superoxide radical anion (O2˙−). Ce(IV)Cln undergoes a photoinduced LMCT that generates the Cl˙ and the Ce(III)Cln−1 species. The alkyl radical R˙, formed by Cl˙-mediated hydrogen abstraction, adds to the protonated heteroarene at the 2-position leading to a N-centred radical-cation, which undergoes a subsequent rearomatization with the formed oxygen radical-anion yielding the desired product along H2O2. The role of O2 was analysed and discussed, leading to the conclusion that singlet oxygen (1O2) is not involved in the reaction mechanism.
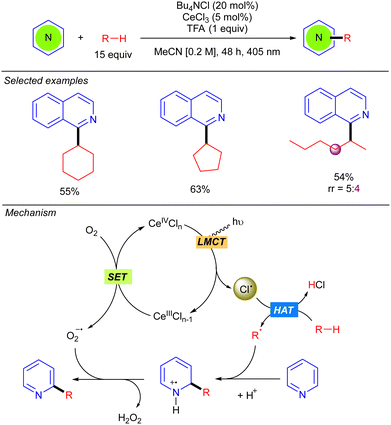 |
| | Scheme 12 Minisci-type alkylation through dual cerium/Cl˙ catalysis. | |
Other methods involving Cl˙ as the HAT reagent
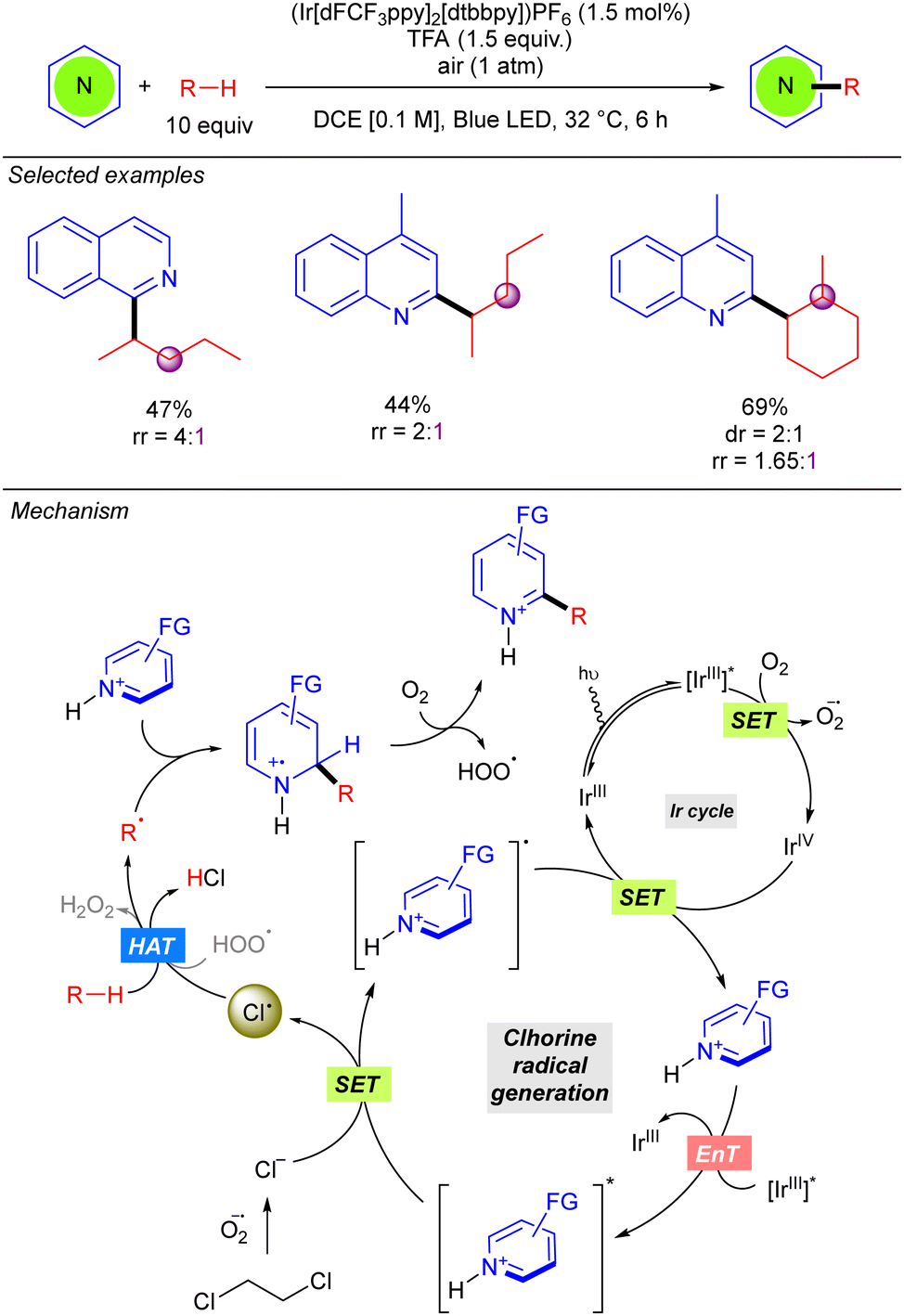
In 2022, the Guin group reported an alternative methodology to the direct coupling of alkanes and nitrogen heterocycles by iridium photocatalysis which involves a non-traditional generation of Cl˙ under aerobic conditions (Scheme 13).21 This transformation involves oxygen reduction to the superoxide radical anion (O2˙−) by the photoexcited Ir(III) complex. This superoxide species provides the chloride anion (Cl−) from DCE, via nucleophilic substitution or an electron-transfer pathway. Oxidation of the Cl− with the photoexcited heteroarene via SET provides the Cl˙ which abstracts a hydrogen atom from the alkane to generate the key alkyl radical. Addition to the protonated nitrogen heterocycle provides a radical cation species which upon O2-mediated aromatization, furnishes the alkylated heterocycle along with hydroperoxide radical (HOO˙). The in situ generated HOO˙ was proposed to propagate a radical chain pathway via HAT from the alkane. This methodology presented a relevant improvement in terms of regioselectivity, leading to better regioisomeric ratios in linear alkanes. Furthermore, branched alkanes are also tolerated although they furnish the corresponding product in poor regioisomeric ratios. Interestingly, the reaction with this type of alkanes exhibits certain diastereoselectivity.
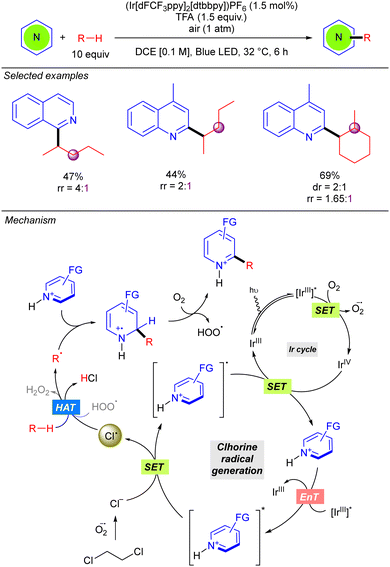 |
| | Scheme 13 Minisci-type alkylation through dual iridium/Cl˙ catalysis. | |
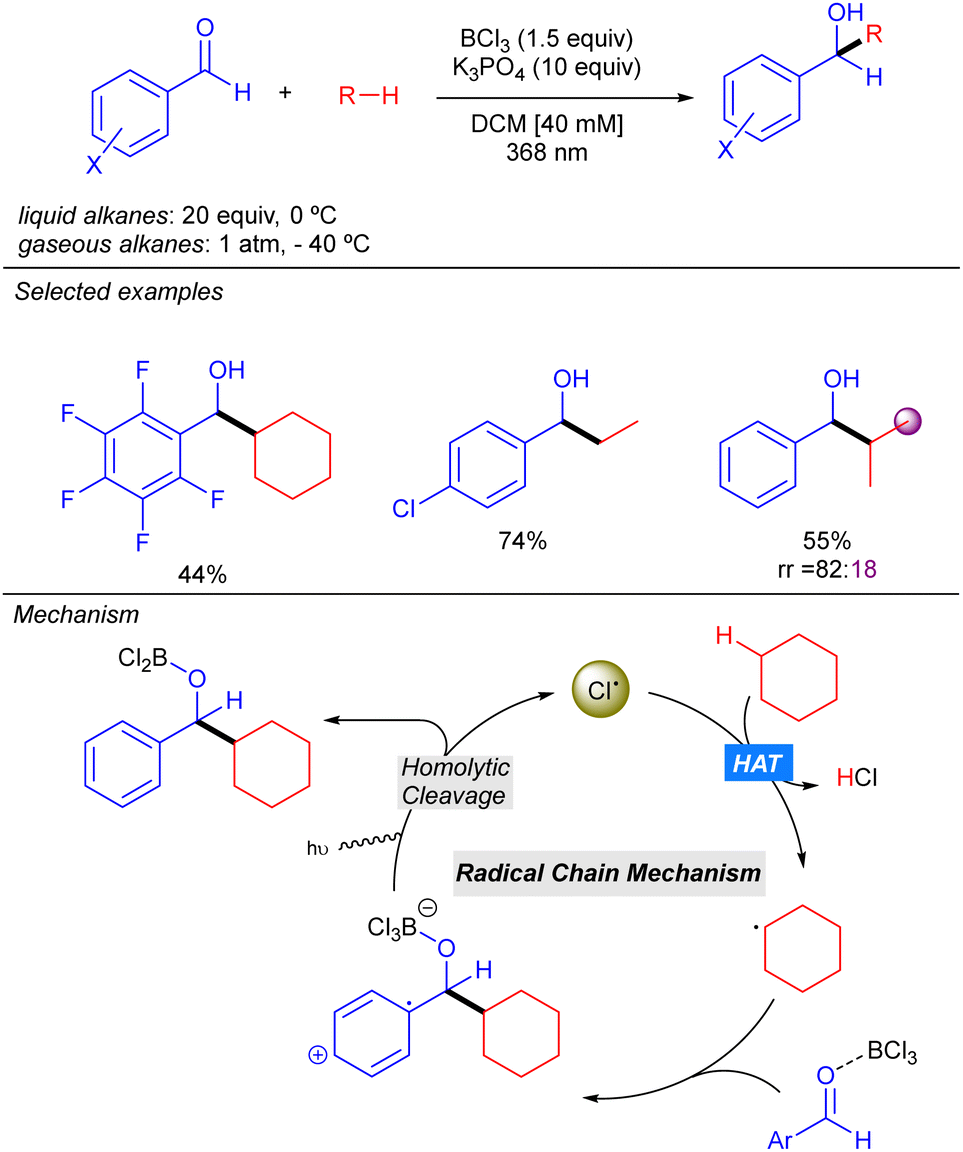
Very recently, the groups of Bach, Riedle and Hauer reported BCl3-promoted alkylation of aldehydes using alkanes as C–H donors (Scheme 14).22 Experimental and computational studies revealed that the process involves a radical chain mechanism. DFT calculations showed that upon irradiation, the boron–aldehyde complex generates Cl˙ which undergoes HAT with the alkane to afford the alkyl radical. Afterwards, a radical addition to the BCl3–aldehyde complex furnishes a stabilized radical carbocation where homolytic B–Cl dissociation leads to the boron alkoxide product and restarts the Cl˙ catalytic cycle. In contrast to other methodologies where the key step is the LMCT for generating the Cl˙, no redox–active transition metal is required here. Instead, an electron is transferred from the bounded chlorine to the aromatic π-system of the aldehyde, weakening the B–Cl bond. The reaction worked in good to excellent yields with a variety of alkanes (including ethane and propane with good regioselectivity) and several benzaldehyde derivatives.
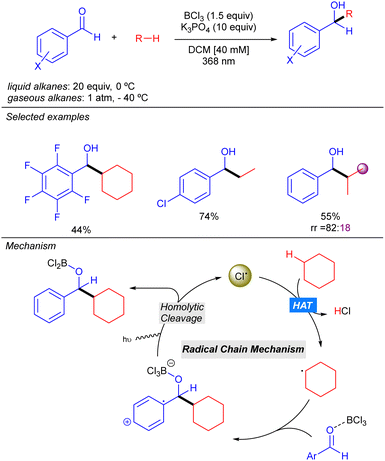 |
| | Scheme 14 BCl3-promoted alkylation of aldehydes. | |
Photocatalytic reactions mediated by an oxygen-centred radical
Oxygen-centred radicals have been thoroughly studied and applied in several classic radical chemical transformations by using stoichiometric amounts of peroxide oxidants, such as peroxides or molecular oxygen.23 Over the last decade, advances in photoredox chemistry have allowed the catalytic generation and use of this reactive species as HAT agents in selective C(sp3)–H functionalization. The generation of this O-centred radical relies on two different processes: LMCT or SET. LMCT processes involve a Ce–alkoxide complex that undergoes photoinduced homolytic Ce–OR cleavage releasing catalytic amounts of RO˙. On the other hand, SET methodology provides different O-centred radicals after single electron reduction, or oxidation, of selected HAT agents –such as hypervalent iodine compounds, H2O2 or N-oxides –in combination with excited organic or metal-based photocatalysts.
LMCT: Ce catalysis with free alcohols
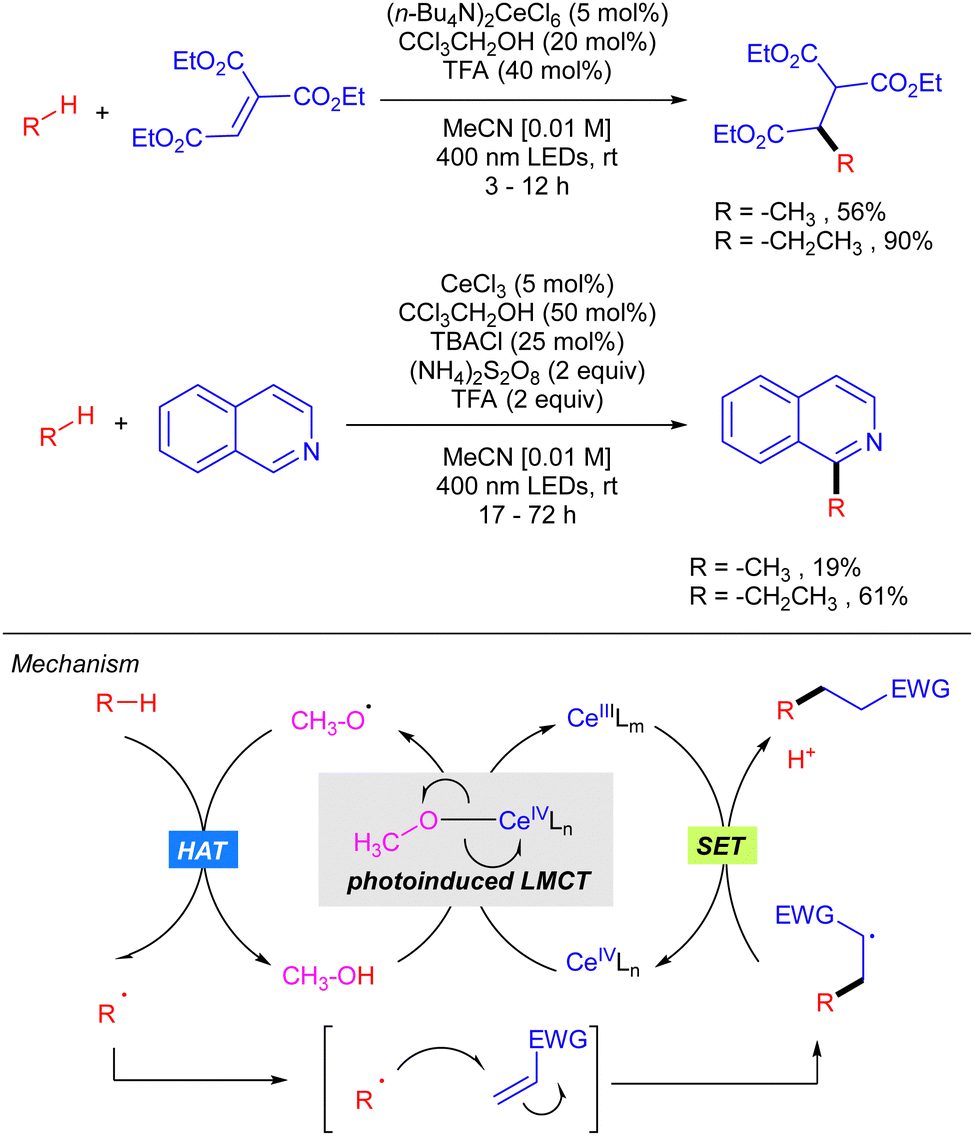
In 2018, Zuo demonstrated how the use of CeCl3 catalysis in combination with alcohols could be efficiently applied to the functionalization of light alkanes mediated by alkoxy radicals (Scheme 15).24 The reaction was shown to be effective for alkylation and heteroarylation of the gaseous alkanes using Michael acceptors and isoquinolines, respectively. The proposed mechanism, which has been recently supported by extensive mechanistic studies,25 involves the formation of a Ce(IV)–alkoxide complex ([Ce(OMe)Cl5]2−) that undergoes photoinduced LMCT leading to the selective formation of alkoxy radical that is responsible for the rate-limiting alkane hydrogen abstraction. Subsequent radical addition to the Michael acceptor forms a new carbon centred radical which reacts with the Ce(III) intermediate through a SET process furnishing the product with concomitant regeneration of the active Ce(IV) catalyst.
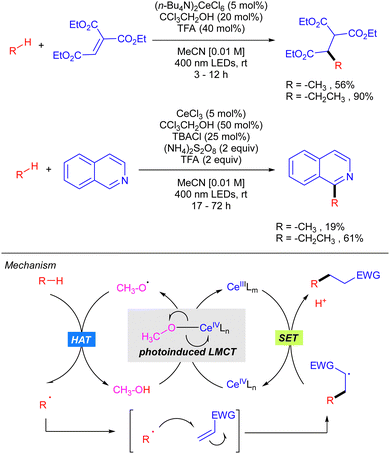 |
| | Scheme 15 Ce-catalysed functionalization of methane and ethane using alcohols as HAT agents. | |
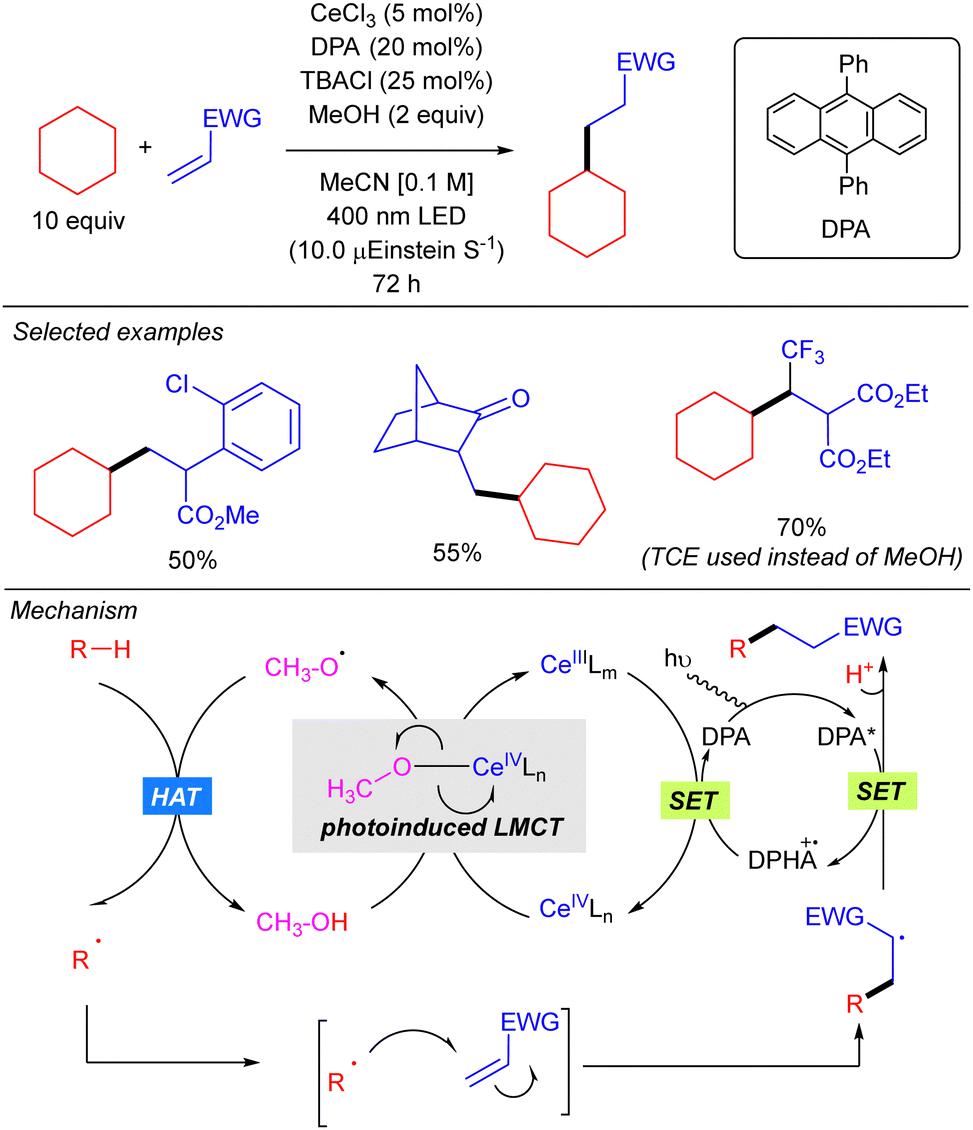
In 2020, the same group extended this concept to the functionalization of higher linear and cyclic alkanes by means of a Ce-catalysed C–H amination using DBAD. In the case of C–H alkylation, cyclohexane was the only alkane tested with various electron-poor olefins as electrophilic coupling partners (Scheme 16).26
 |
| | Scheme 16 Ce-catalysed functionalization of alkanes. | |
In this work, the role of the alcohol as the HAT reagent was evaluated. It was found that MeOH is selective towards the functionalization of 3° C–H bonds, while the use of bulkier alcohols such as tBuOH decreases the regioselectivity. Alcohols bearing electron-withdrawing substituents like 2,2,2-trichloroethanol (TCE) proved to be efficient although they led to almost equimolar 3°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2° ratios when lineal alkanes were used. The effect of additives in the regeneration of the catalytic species CeIVLn was also studied. It was observed that reaction requires catalytic amounts of TBACl for maintaining the high turnover efficiency, probably due to the necessity of an external chloride which would act as the supporting ligand in the formation of [Ce(OR)Cln] species, rather than the insoluble Ce(OR)4. Additionally, it was observed that the use of 9,10-diphenylanthracene (DPA) in the C–H alkylation reaction promoted an acceleration on the reaction likely because of a photoinduced electron transfer to close the cerium catalytic cycle.
:2° ratios when lineal alkanes were used. The effect of additives in the regeneration of the catalytic species CeIVLn was also studied. It was observed that reaction requires catalytic amounts of TBACl for maintaining the high turnover efficiency, probably due to the necessity of an external chloride which would act as the supporting ligand in the formation of [Ce(OR)Cln] species, rather than the insoluble Ce(OR)4. Additionally, it was observed that the use of 9,10-diphenylanthracene (DPA) in the C–H alkylation reaction promoted an acceleration on the reaction likely because of a photoinduced electron transfer to close the cerium catalytic cycle.
SET: Ru catalysis with hypervalent iodine(III) reagents
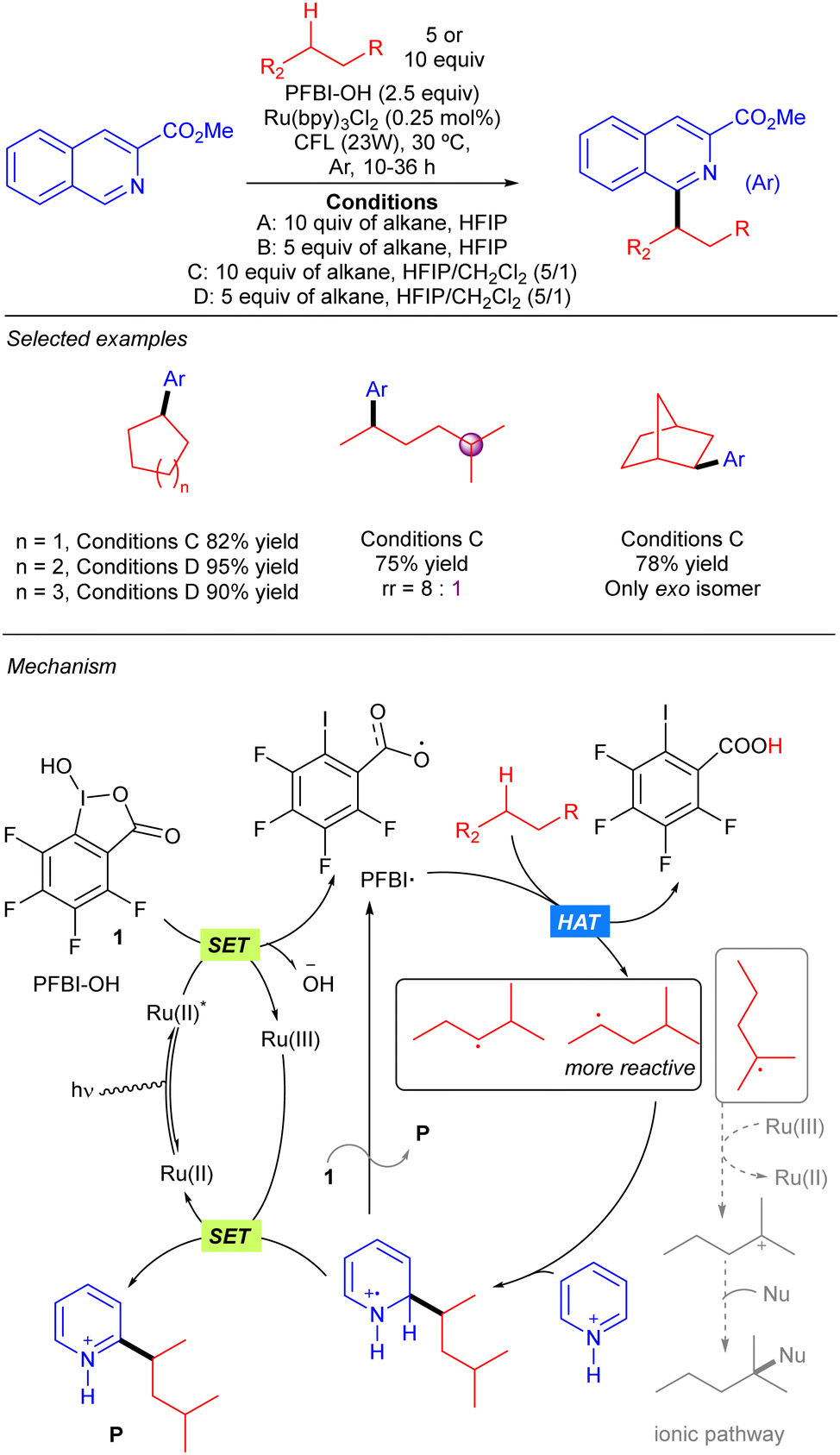
Chen and colleagues reported a strategy to produce benzoyloxy radicals from hydroxyl benziodoxole species PFBI-OH to achieve Minisci-type alkane heteroarylation processes (Scheme 17).27 PFBI-OH displayed a significant degree of steric sensitivity for alkane hydrogen abstraction, being more selective towards the more sterically accessible 2° C–H bonds over the weaker 3° ones. A large variety of cyclic and acyclic alkanes were shown to undergo this transformation with excellent yields. The proposed mechanism for the reaction starts with the oxidation of PFBI-OH upon the photoexcitation of the Ru(II) complex followed by a SET process, generating PFBI˙ radical and Ru(III) species; PFBI˙ promotes HAT over the alkane to generate the alkyl radical and generates a carboxylic acid as a side product. The alkyl radical adds to the N-protonated heteroaryl ring forming an intermediate that evolves to the final product by a SET mediated rearomatization process. An alternative ionic pathway involving re-oxidation of Ru(II) with the alkyl radical and generation of a tertiary carbocation followed by nucleophilic trapping was also considered but seems to be less feasible.
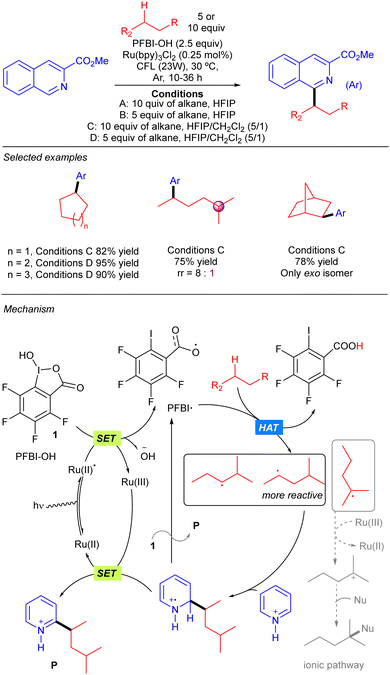 |
| | Scheme 17 Photoredox-mediated Minisci-type alkylation of N-heteroarenes with alkanes. | |
SET: 4CzIPN catalysis with H2O2 as the oxidant and HAT reagent
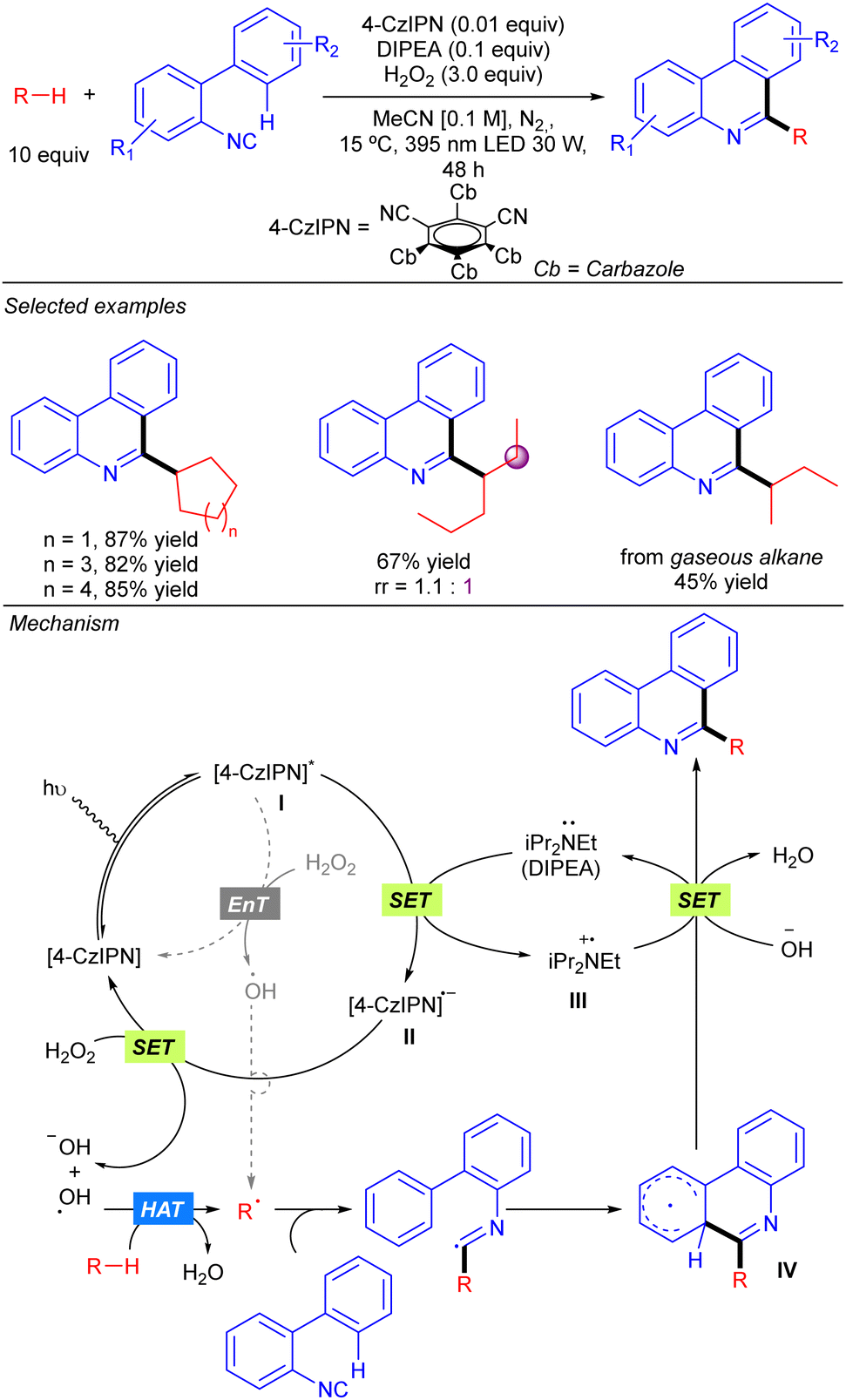
Duan and co-workers reported a visible light-induced metal-free synthesis of substituted phenanthridines by the reaction of simple alkanes with 2-isocyanobiaryls using aqueous H2O2 as the terminal oxidant (Scheme 18).28 This approach enabled the use of cyclic alkanes and several linear ones such as n-hexane or 3-methylpentane with good regioselectivity in some cases. The use of gaseous alkanes such as n-butane and iso-butane was also permitted. This transformation was proposed to proceed via the photoexcitation of the 4-CzIPN catalyst, followed by a SET from DIPEA that provides radical species II and III. Subsequent SET between II and H2O2 generates a hydroxyl anion and a HAT-active hydroxyl radical that provides the alkyl radical by alkane hydrogen abstraction. An alternative direct formation of the O-centred radical would be possible through an energy transfer process (EnT) from excited photocatalyst with H2O2. Addition of the alkyl radical to the 2-isocyanobiaryl affords an imidoyl radical that evolves to a delocalized radical intermediate IV through an intramolecular aromatic addition. A final SET between III and IV and proton abstraction by the hydroxyl anion provided the final product, regenerating DIPEA (Scheme 20).
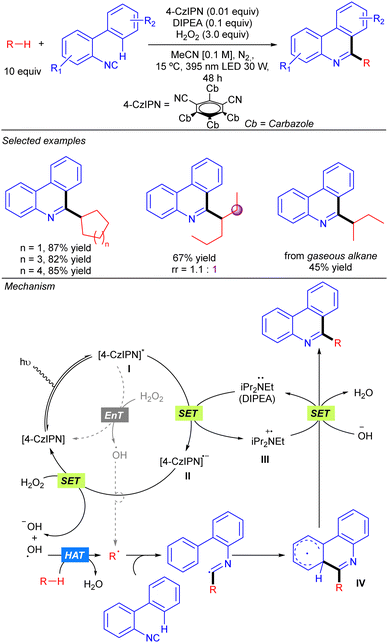 |
| | Scheme 18 Metal-free alkylation of phenanthridines with H2O2 as the terminal oxidant. | |
SET: Acridinium catalysis with N-oxide as the HAT catalyst
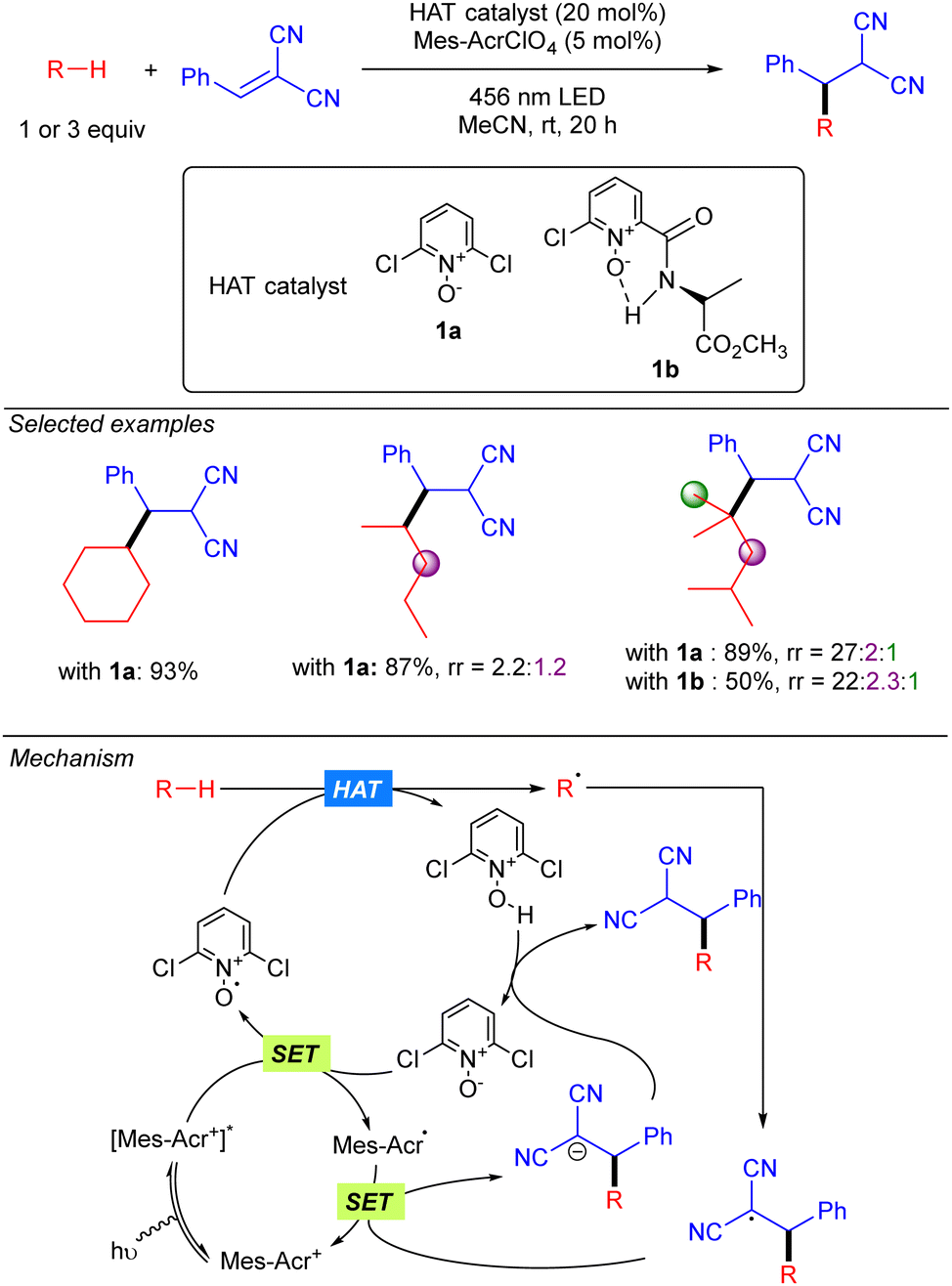
In 2022, the group of Deng published a Giese-type addition of unactivated alkanes to electron-poor olefins using N-oxides as HAT photocatalysts (Scheme 19).29 The transformation allows the use of cyclic and acyclic alkanes in excellent yields. Regarding regioselectivity, n-hexane was shown to yield a statistical mixture of 2° C–H bond functionalized products. Branched alkanes showed excellent to good selectivity was observed, displaying a preferential 3° > 2° > 1° functionalization. Mechanistically, the reaction relies on the SET from the N-oxide to the excited acridinium photocatalyst (Mes-Acr+*). The formed electrophilic N-oxide radical cation can promote the HAT over the alkane leading to an alkyl radical, which reacts with the radical acceptor, and a protonated N-oxide. The new C-centred radical formed promotes a second SET with the reduced form of the acridinium catalyst, resetting the photocatalytic cycle, and the formed anion is neutralized by the protonated N-oxide to regenerate the HAT catalyst.
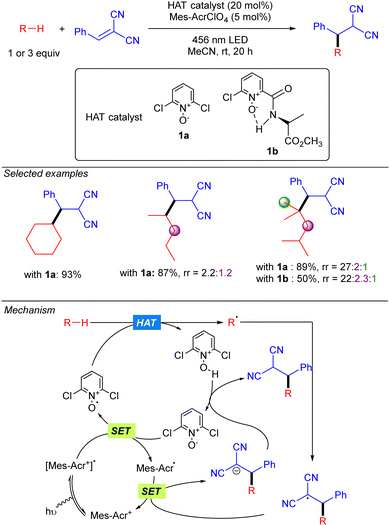 |
| | Scheme 19 Giese-type addition of unactivated alkanes to electron-poor substituted olefins using N-oxide photocatalyst (Deng and co-workers). | |
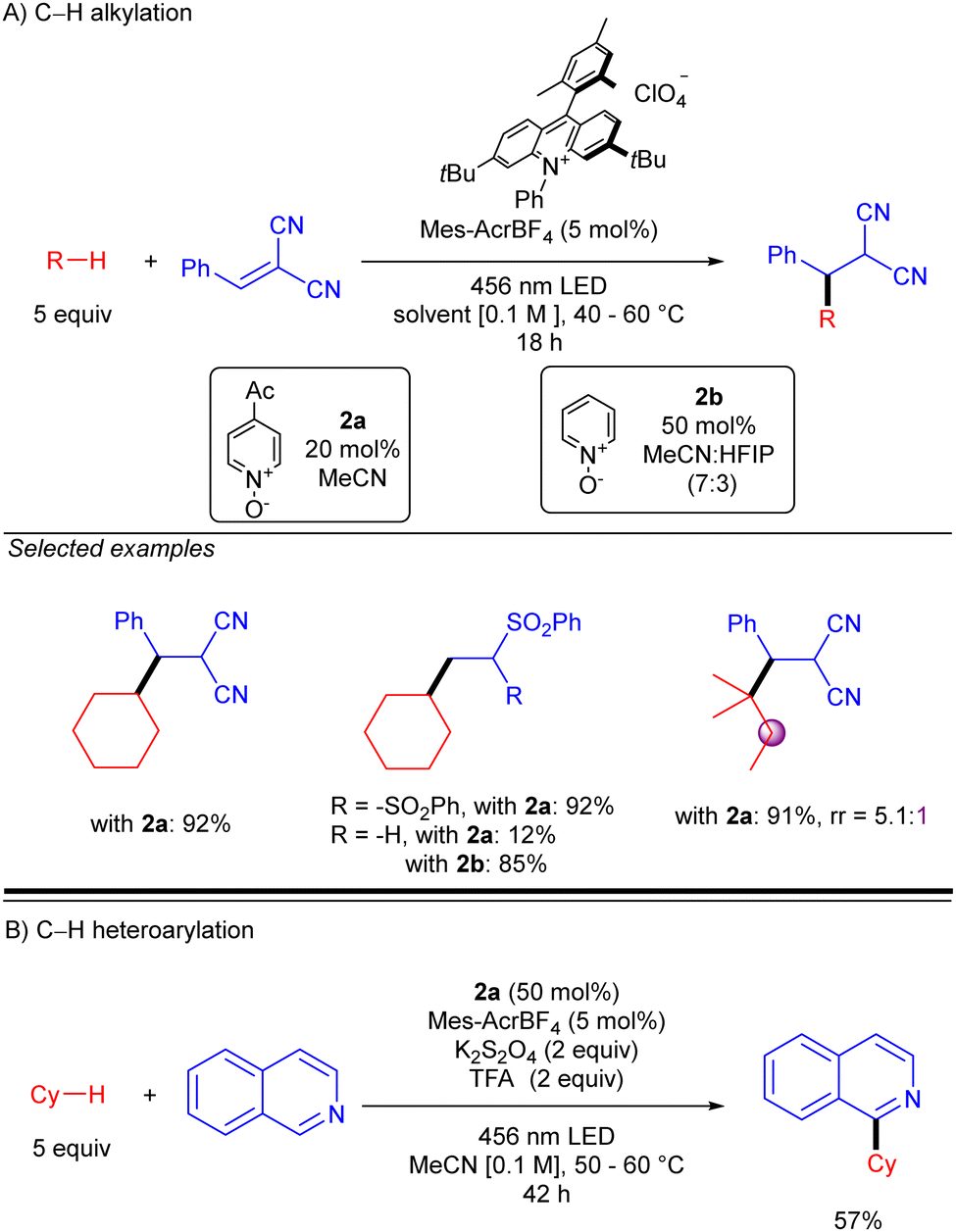
Simultaneously to the work of Deng, the group of Nicewicz reported a similar strategy but with a different HAT photocatalyst combination (Scheme 20).30 This procedure was not only demonstrated with Michael acceptors but also with Minisci-type reactions, leading to good to excellent yields in both cases. Regarding Giese-type additions, two different HAT reagents (2a or 2b) were used depending on the nature of the acceptor. The reaction was shown to be effective with cyclic and acyclic branched alkanes, showing moderate selectivity for tertiary C–H bonds. In the case of the C–H heteroarylation, reaction conditions were modified adding K2S2O8 as the terminal oxidant and TFA for heteroarene activation.
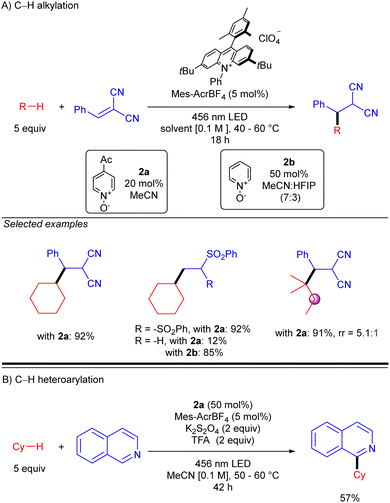 |
| | Scheme 20 Giese- and Minisci-type reaction of unactivated alkanes using N-oxide photocatalyst (Nicewicz and co-workers). | |
Alkane functionalization using TBADT as the HAT photocatalyst
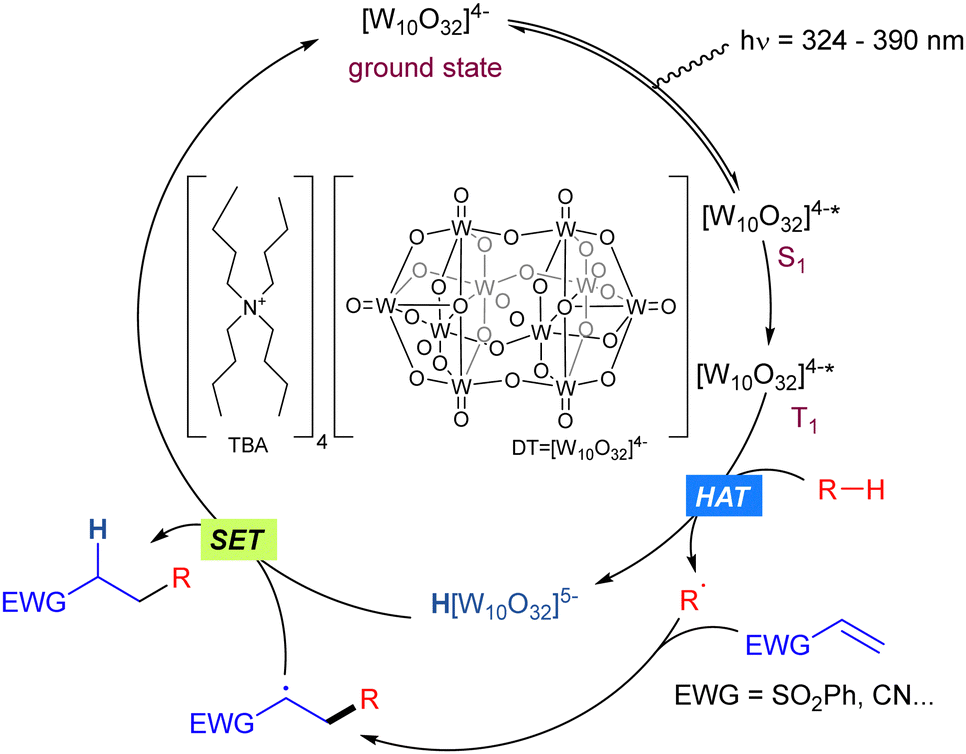
Tetrabutylammonium decatungstate (TBADT) is a polyoxometalate (tungsten-based oxygen-anion cluster) with high photocatalytic activity which has found widespread application in synthetic transformations involving the C–H bond activation in organic molecules such as alcohols, aldehydes or alkanes.31 The general mechanism for the reactions is given by the photoexcitation of the decatungstate anion (max absorption band 324 nm),32 due to an allowed HOMO–LUMO transition with a marked LMCT character, where electrons are moved from oxygen centres to tungsten atoms, to obtain the excited state of the anion. The singlet excited state (S1) initially formed rapidly decays to the reactive state.33 This relaxed excited state, is probably a triplet in multiplicity (T1). Once in the excited state, those species can perform a HAT (Hydrogen Atom Transfer) process34 or SET (Single Electron Transfer) process depending on the redox potential of the substrate. When it matches that of TBADT (E [W10O32]4−*/[W10O32]5− = +2.44 V vs. SCE), a SET event may take place, giving access to the radical cation of the starting substrate. On the other hand, when the redox potential of the substrate does not match the above-mentioned value, the generation of a R˙ radical via HAT is the most feasible pathway. Both processes would lead to the reduced form of the anion (H[W10O32]5−, blue colour).31c Finally, the starting active photocatalyst is recovered through SET process(es) (Scheme 21).
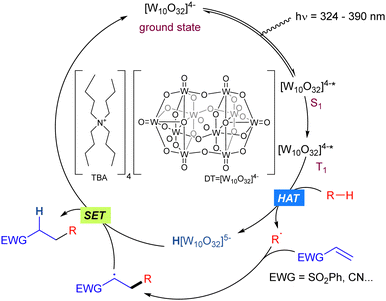 |
| | Scheme 21 General photocatalytic action mechanism of TBADT. | |
The rate of hydrogen abstraction kR–H in photocatalytic C–H bond functionalization processes promoted by TBADT highly depends on the C–H bond polarity of the transition state and the steric hindrance. Bond dissociation energies are not necessarily reflected in that constant. Given the size and the electrophilic nature of the active excited TBADT* species, there is a preferred selectivity in the hydrogen abstraction of more hydridic and sterically accessible C–H bonds.31a,b
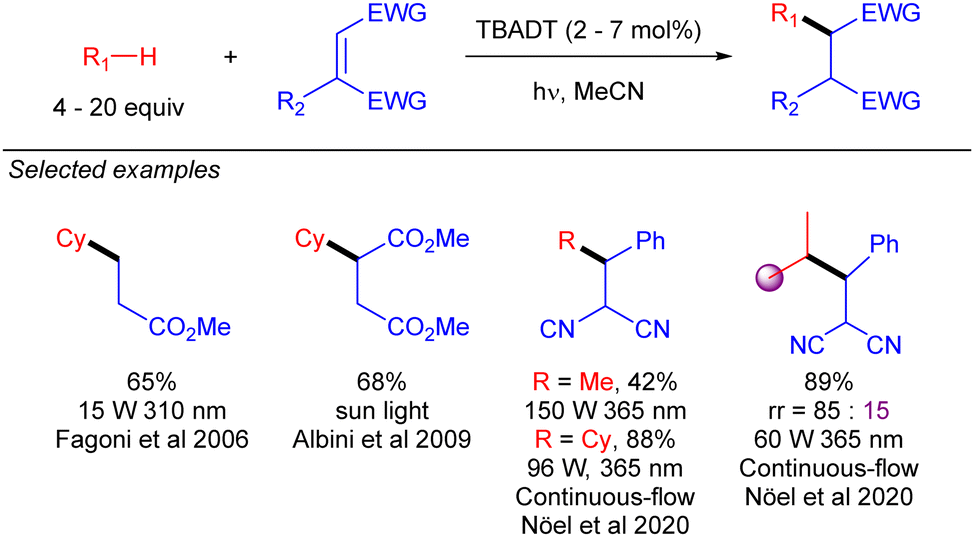
First reactions with TBADT-promoted photo-generation of alkyl radicals were conjugate additions to electron-deficient olefins. Early examples of this chemistry were reported by Fagnoni and co-workers.35 Some years later, Albini extended this transformation to cyclopentane and cyclohexane under sun light irradiation (Scheme 22).36 Several years later, Noël and co-workers used continuous-flow chemistry to make this transformation to cyclohexane feasible in the gram-scale,37 and even to the functionalization of gaseous light alkanes such as methane, ethane, or propane.38
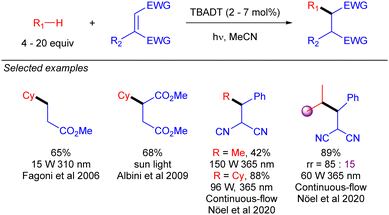 |
| | Scheme 22 TBADT-catalysed radical conjugate addition of alkanes. | |
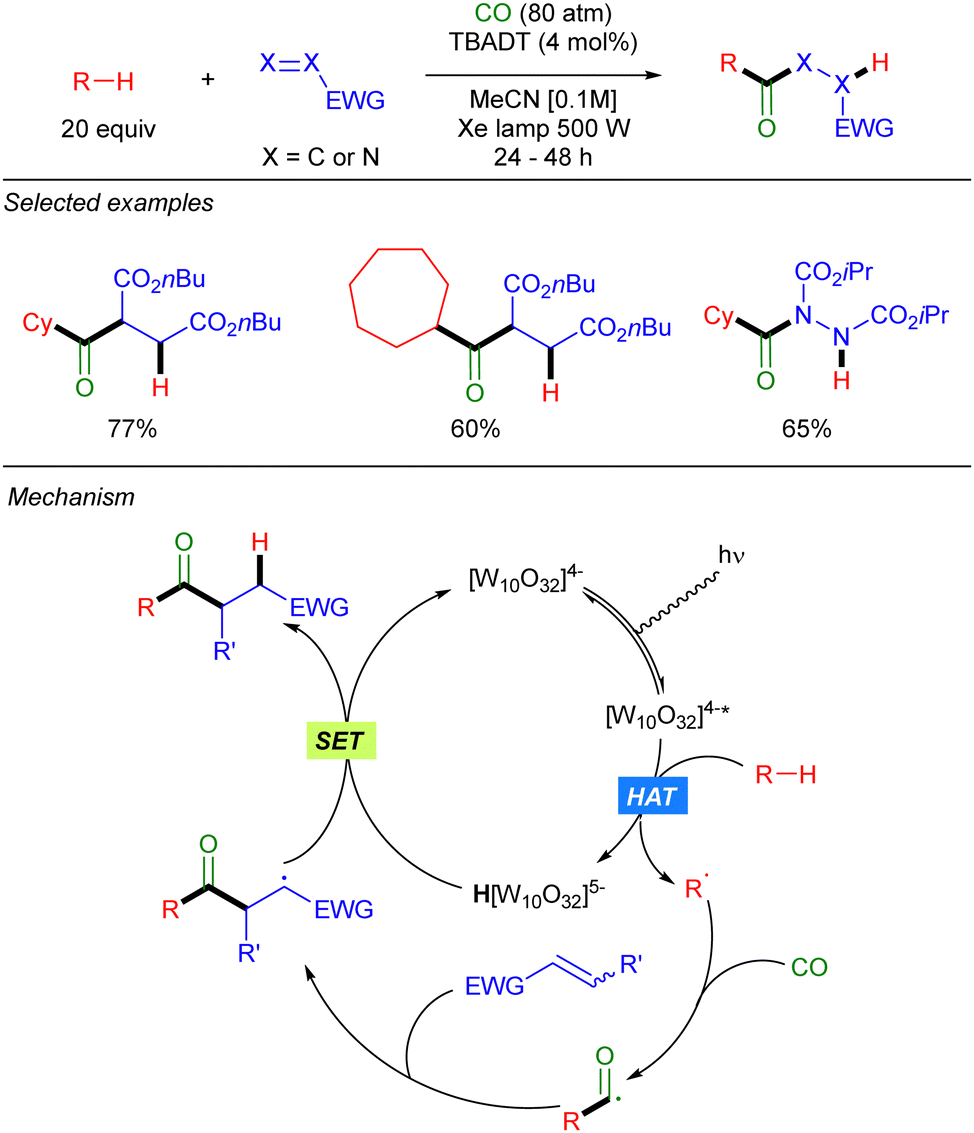
The groups of Fagnoni and Ryu39 and Ravelli and Protti,40 also showed that carbon monoxide can be used in this photocatalytic transformation to first trap the alkyl radical to generate a carbonyl radical, that undergoes a Giese-type reaction to result in a formal acylation of the alkane (Scheme 23).
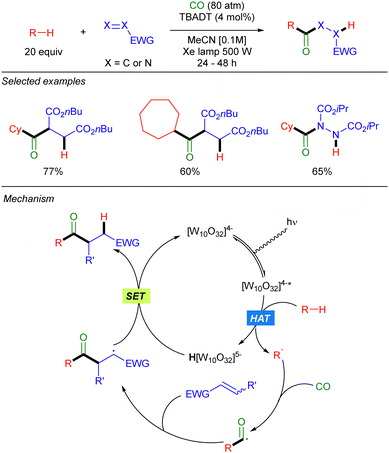 |
| | Scheme 23 TBADT-catalysed acylation of alkanes. | |
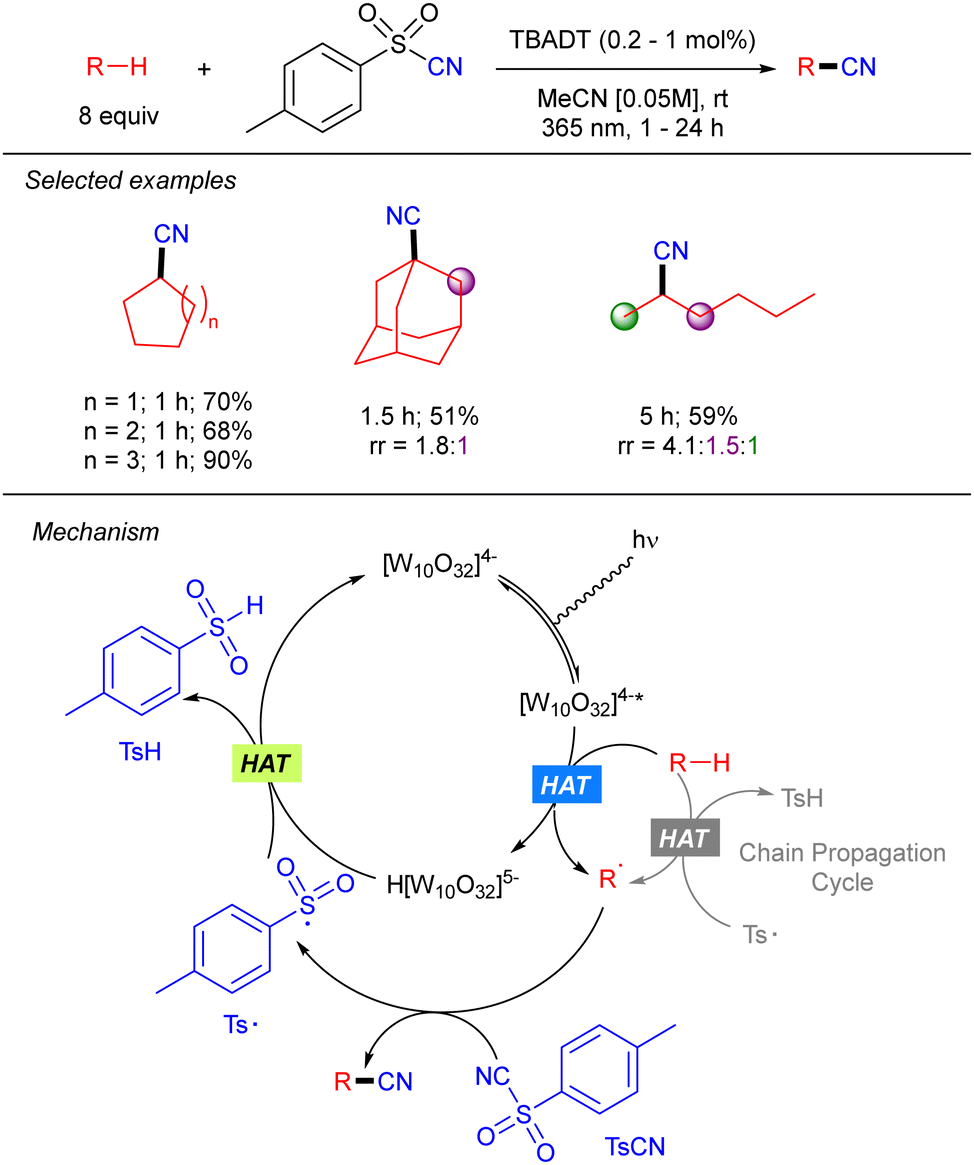
Beyond the application of TBADT catalysis in Giese-type reactions, Hong and co-workers reported a direct C(sp3)–H cyanation combining the TBADT-promoted alkyl radical generation with benzenesulfonyl-cyanides (Scheme 24).41 The reaction worked in good to excellent yields with a variety of alkanes including n-hexane with moderate regioselectivity. The transformation involves the electrophilic trapping of the alkyl radical by the tosyl cyanide that generates the product and the tosyl radical. The latter undergoes SET with the reduced TBADT to regenerate the photocatalyst. The authors performed experiments that showed that when catalyst concentration is significantly lowered the reaction between the reduced catalyst and tosyl radical is slow so that a radical chain process becomes dominant.
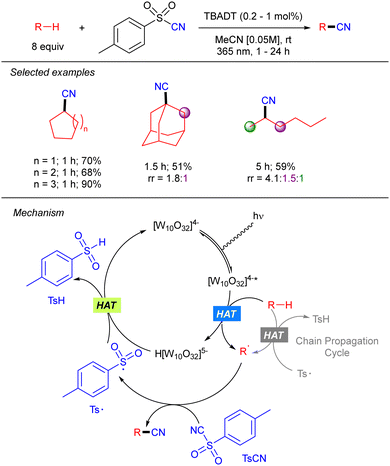 |
| | Scheme 24 TBADT-catalysed cyanation of alkanes. | |
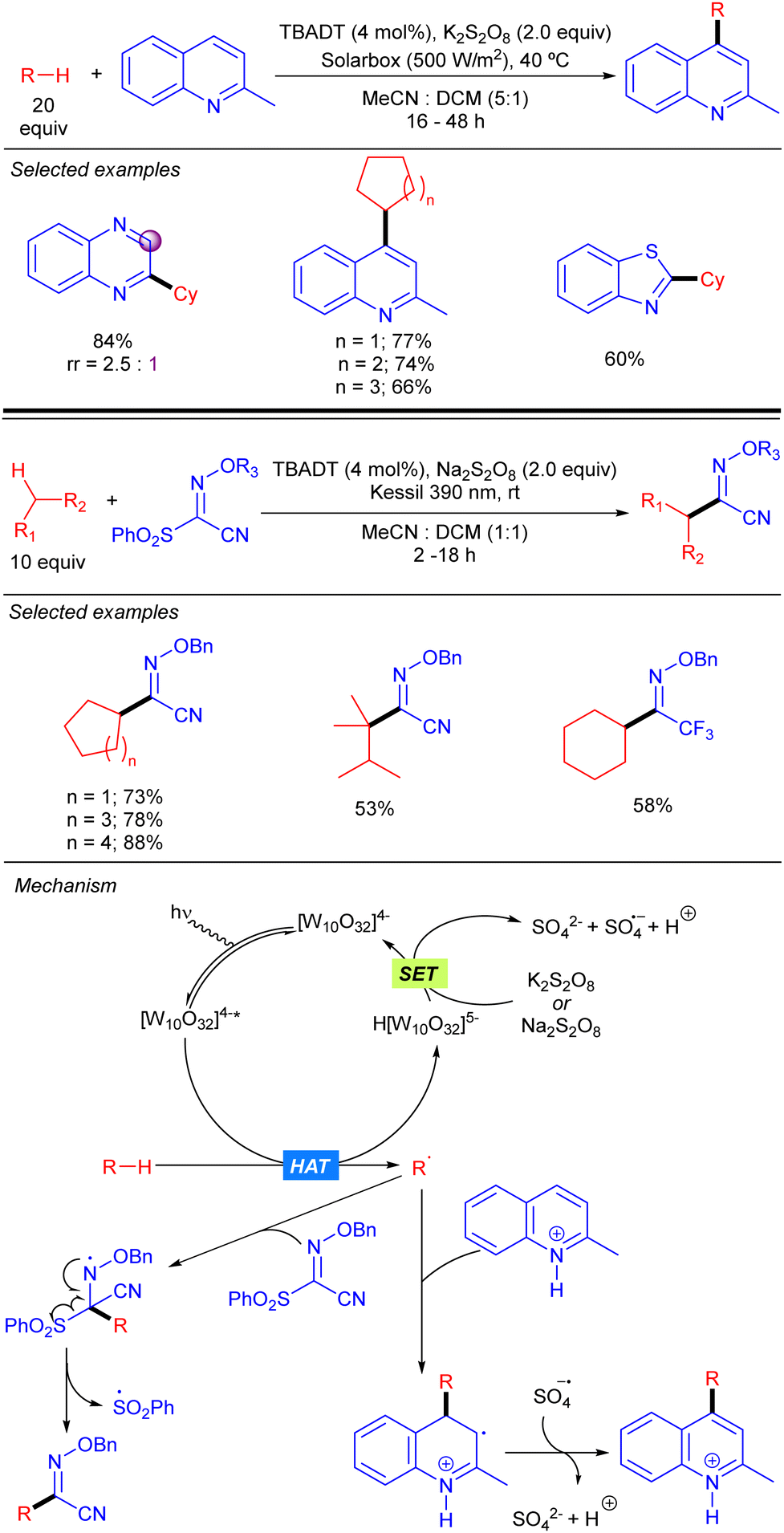
A protocol for heteroarylation of light alkanes with solar light and isoquinolines was described by Ryu and co-workers combining TBADT and K2S2O8 (Scheme 25).42 Some years later, in 2021, Wang and co-workers introduced the same alkyl moieties into oximes with a similar catalytic system but using 390 nm LED as the light source to obtain high valuable building blocks.43 Both reactions allowed different cyclic and acyclic alkanes to be functionalized in moderate to very good yields. The reaction was based in the formation of the alkyl radical via TBADT photocatalysis followed by the attack towards the C–C or C–N double bond. That process generated a N-centred radical that evolves differently according to the nature of the substrate and the reaction conditions. For isoquinolines, the reaction followed a typical Minisci-type reaction mechanism. The radical adds to the para position of the pyridine ring and the resulting radical undergoes SET with the sulphate radical anion promoting the re-aromatization of the final product. On the other hand, for the oxime substrate, after the addition of the radical, the resulting N-centred radical undergoes an electronic rearrangement that ends up in the C–S bond dissociation, forming the product and a sulphonyl radical.
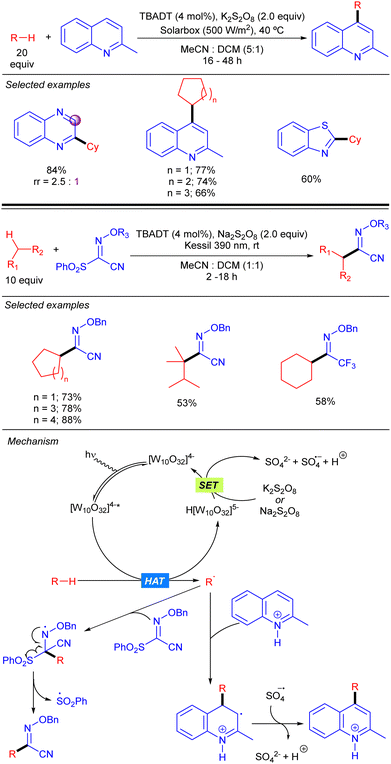 |
| | Scheme 25 Heteroarylation of alkanes based on TBADT/S2O82− catalysis. | |
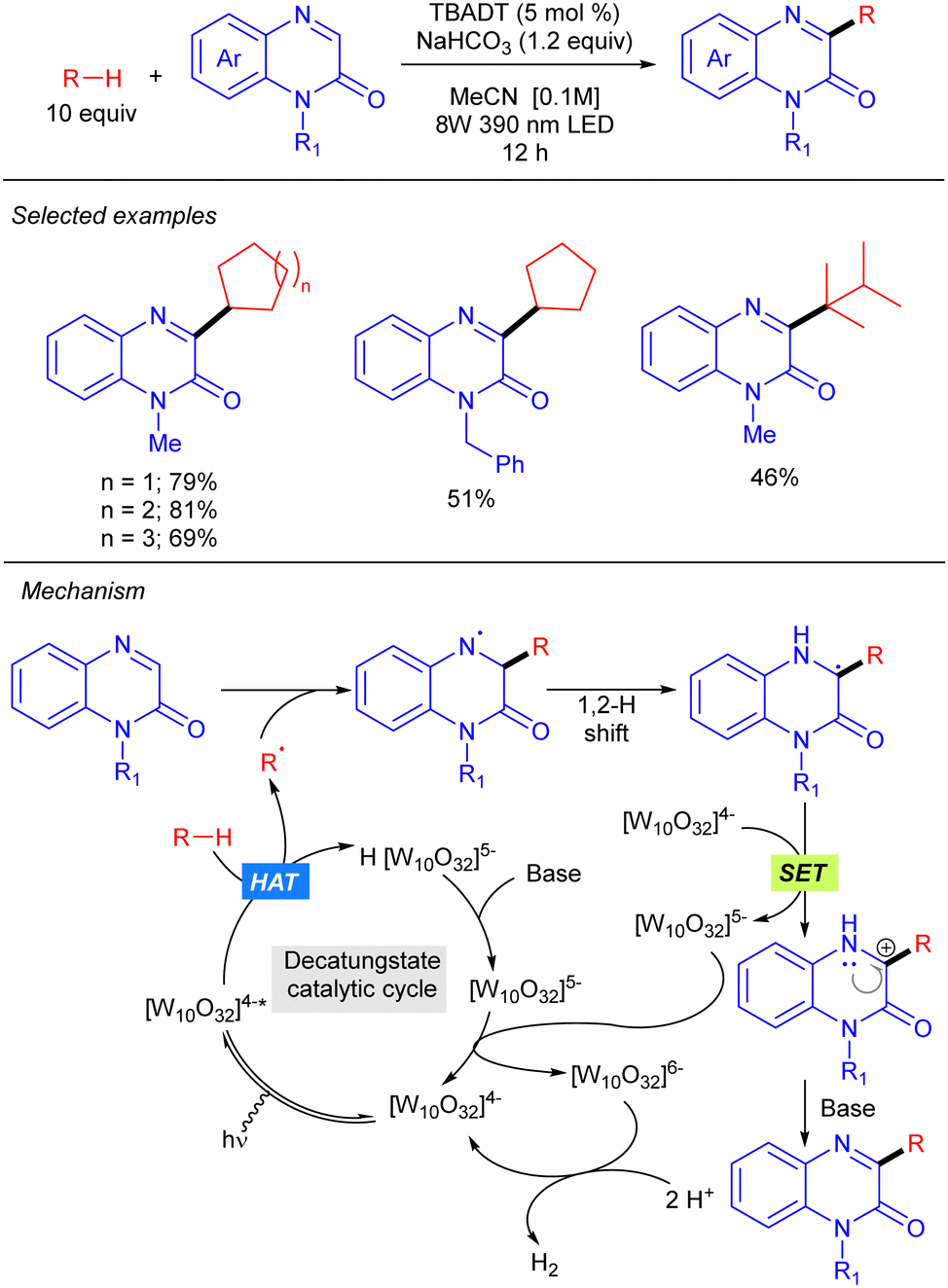
Quinoxalinones also proved to be efficient partners for the heteroarylation of alkanes under TBADT catalysis (Scheme 26).44 A mechanism involving alkyl radical generation via [W10O32]4−*-mediated HAT, addition to quinoxaline followed by 1,2-hydrogen shift, SET with [W10O32]4− and final deprotonation was proposed.
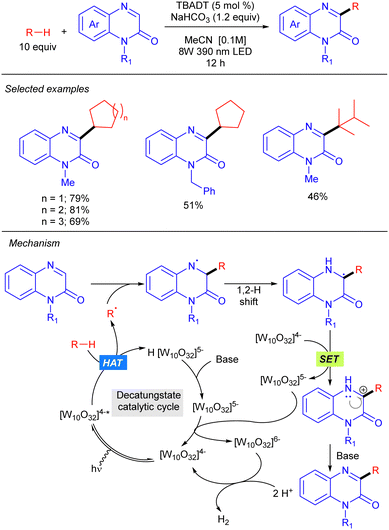 |
| | Scheme 26 TBADT-catalysed heteroarylation of alkanes with quinoxalines. | |
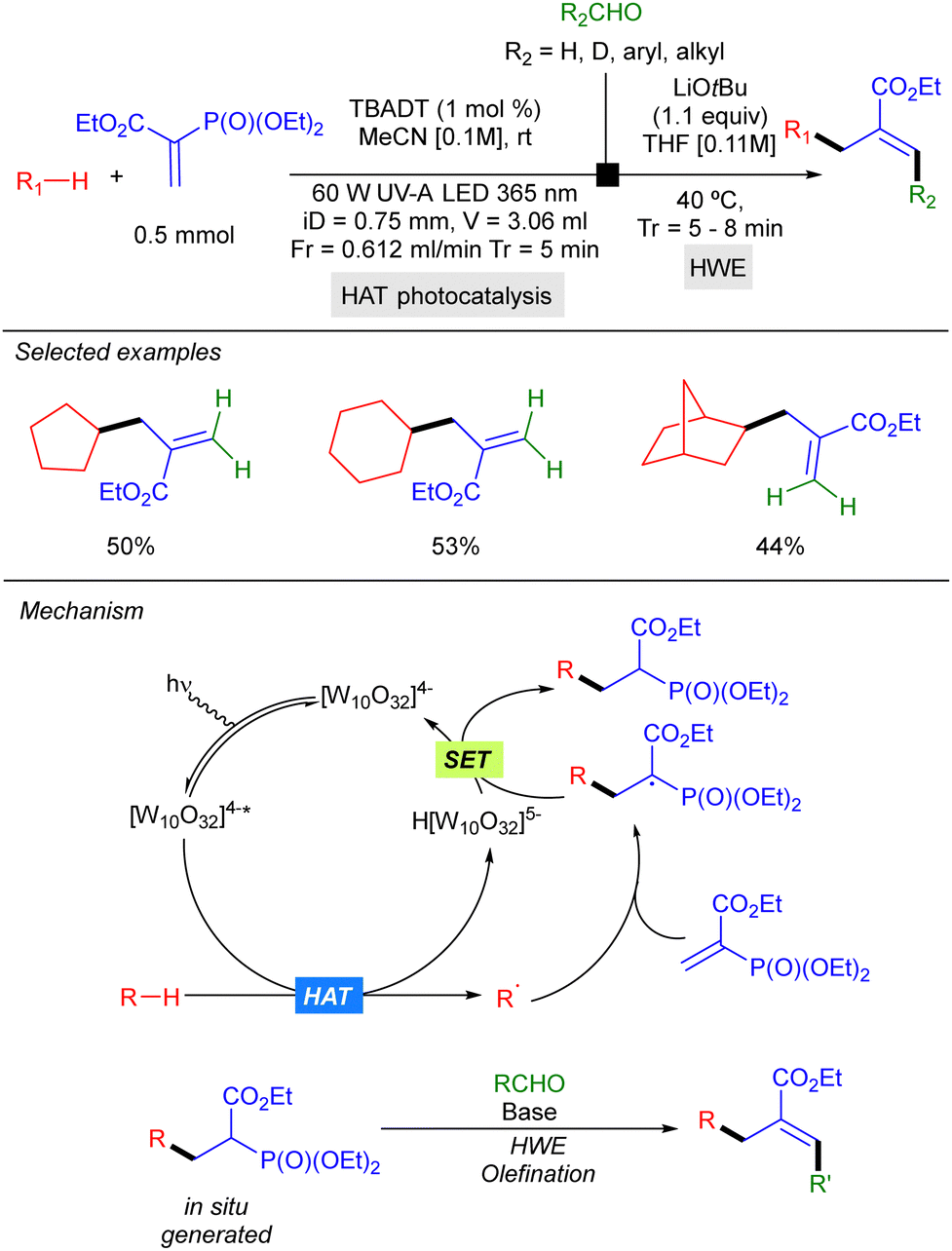
In 2022, Noël and co-workers reported a two-step strategy for aliphatic C–H bond allylation that involves trapping of the alkyl radical generated by HAT photocatalysis with a vinyl phosphonate, followed by a Horner–Wadsworth–Emmons olefination of the resulting radical addition product, under continuous flow conditions. This protocol could be applied to cyclic alkanes such as cyclopentane and cyclohexane (Scheme 27).45
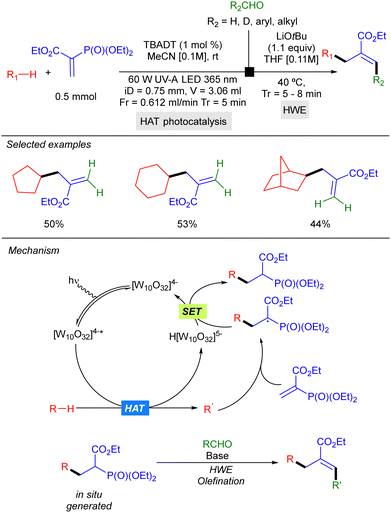 |
| | Scheme 27 Two-step photocatalytic Michel addition/HWE olefination in flow. | |
Merging of TBADT photocatalysis with transition metal catalysis
Although TBADT photocatalysis activity has provided a wide range of new C–C bond forming reactions, the use of synergistic cooperative bimetallic systems has emerged as a powerful tool to increase the chemical space of these transformations.
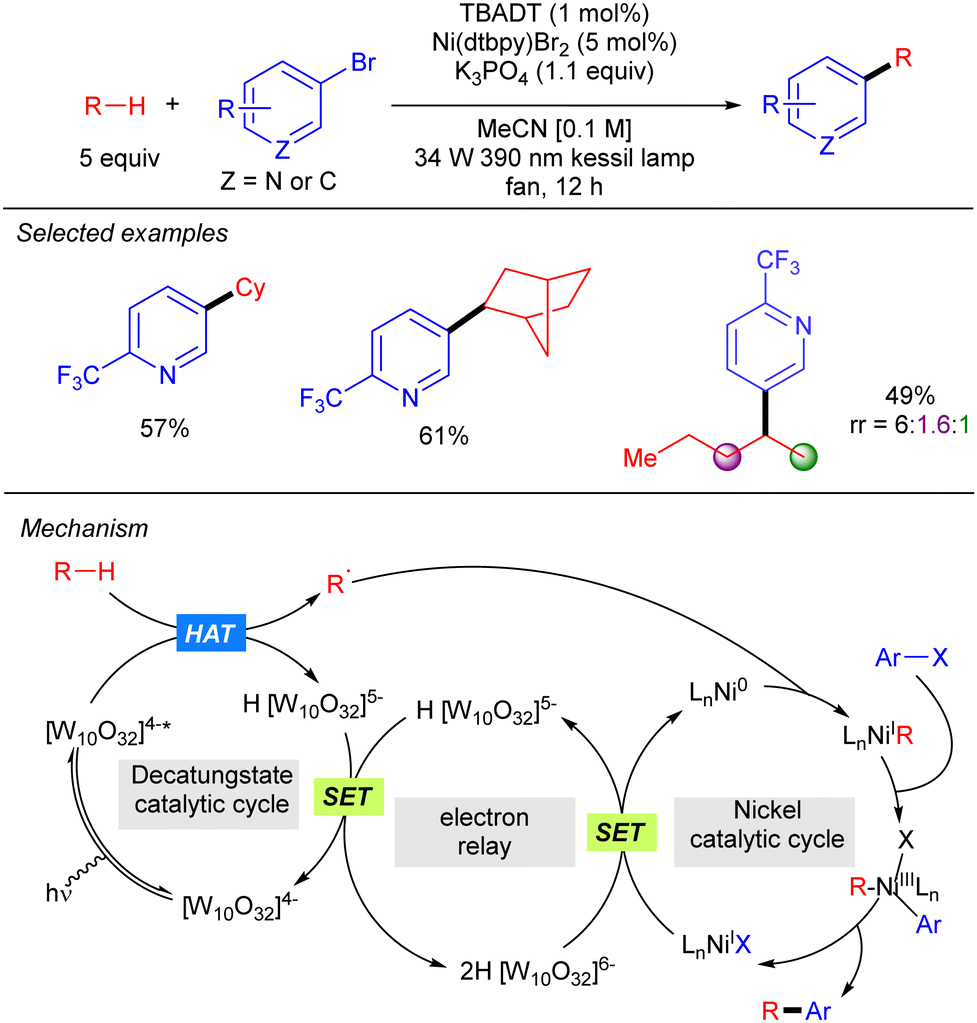
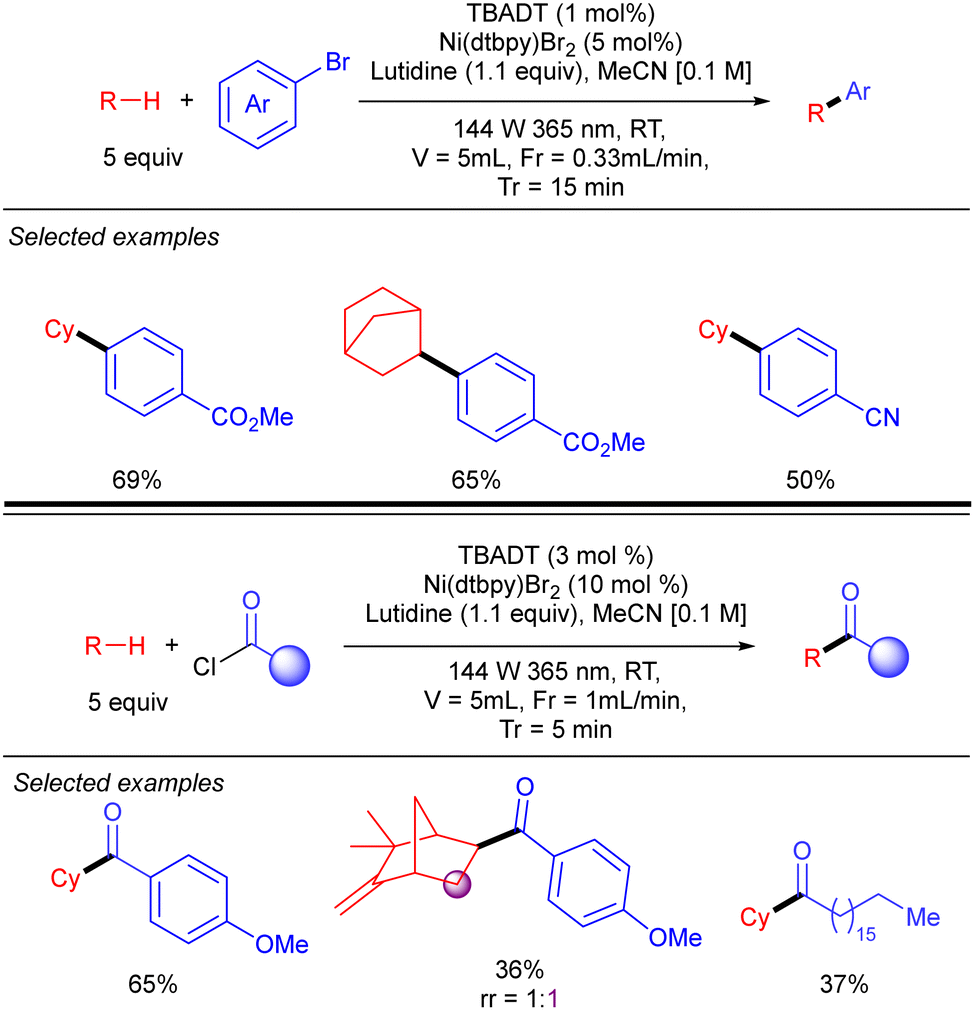
A new strategy to perform direct C(sp3)-arylation using nickel catalysis in conjunction with TBADT photocatalysis was described by MacMillan and co-workers, setting one of the first examples of photocatalytic synergistic strategies (Scheme 28).46 The method worked for a wide variety of aliphatic chemical frameworks containing C–H bonds, such as cyclic and acyclic alkanes with excellent selectivity. This arylation reaction was proposed to occur by initial hydrogen abstraction by excited state decatungstate that affords the alky radical and the singly reduced decatungstate H[W10O32]5−. Disproportionation of the latter regenerates the active ground-state photocatalyst [W10O32]4− and forms doubly reduced decatungstate 2H[W10O32]6−. Concomitantly, an in situ reduced Ni(0) species combines with the alkyl radical leading to a Ni(I)–alkyl intermediate that after oxidative addition of the aryl bromide affords a Ni(III) (aryl)(alkyl) species. Reductive elimination furnishes the product and a new Ni(I) species that undergoes a SET with the doubly reduced polyoxometalate to regenerate the active Ni(0) catalyst. An alternative mechanism involving the oxidative addition of the aryl bromide to the Ni(0) catalyst was also considered.
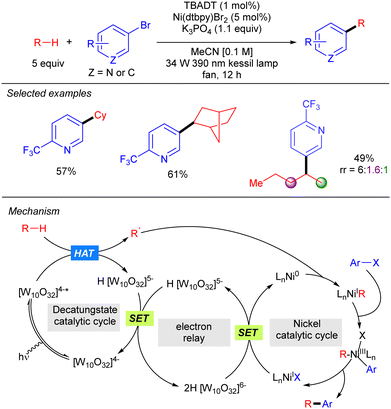 |
| | Scheme 28 Direct arylation of strong aliphatic C–H bonds by cooperative TBADT/Ni catalysis. | |
Noël and co-workers further demonstrated this transformation under continuous flow conditions. These new conditions allowed us to obtain the arylation products in good yield and selectivity in very short reaction times and could also be extended to the direct acylation of alkanes (Scheme 29).47
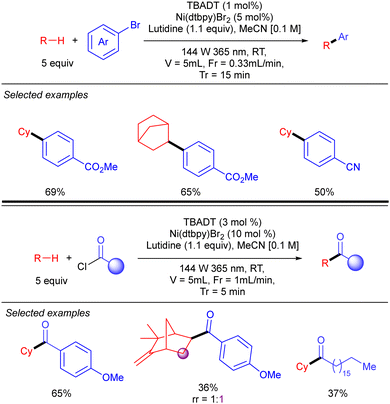 |
| | Scheme 29 Ni/TBADT-catalysed arylation and acylation of simple alkanes under flow chemistry conditions. | |
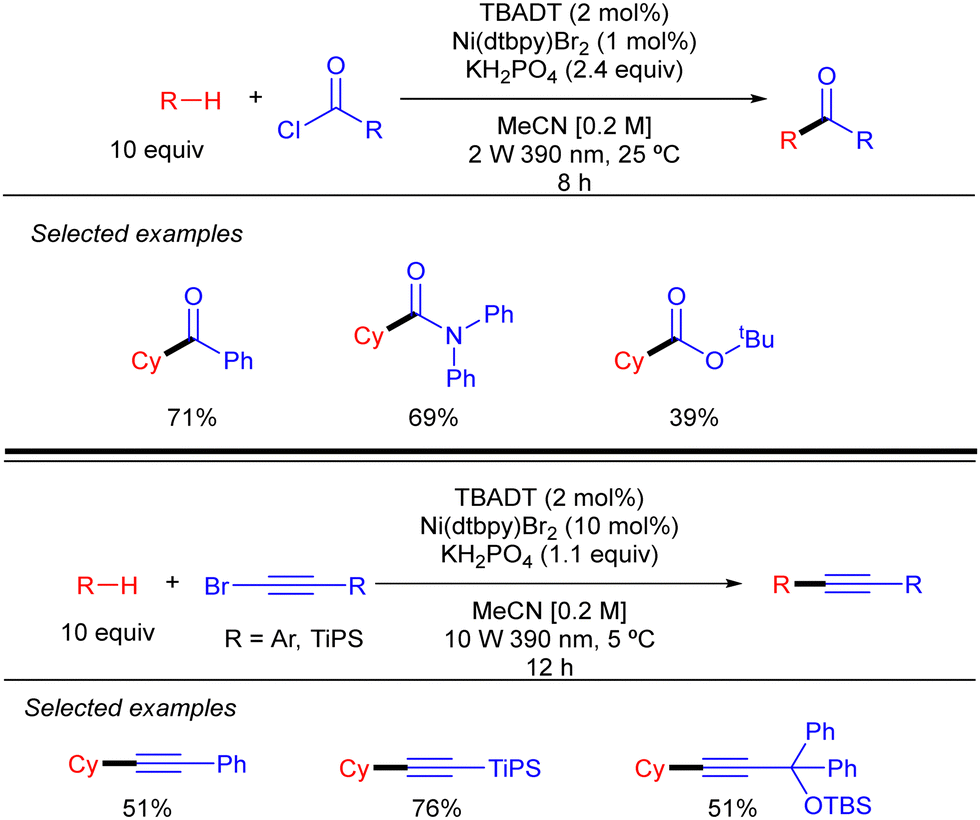
By using a related Ni/TBADT synergistic photocatalytic system, the group of Kong expanded the acylation of alkanes to the synthesis of amide and esters. Furthermore, this catalytic strategy was also applied to coupling of alkanes with bromoalkynes (Scheme 30).48 Both transformations were demonstrated for cyclic alkanes and a range of carbonyl and alkynyl electrophiles. A similar mechanism as the one depicted in Scheme 28 (in this case just involving the singly reduced decatungstate anion) was proposed to explain both reactions.
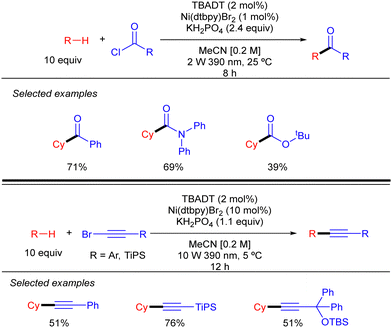 |
| | Scheme 30 Alkynylation and acylation of alkanes with Ni/TBADT synergistic photocatalysis. | |
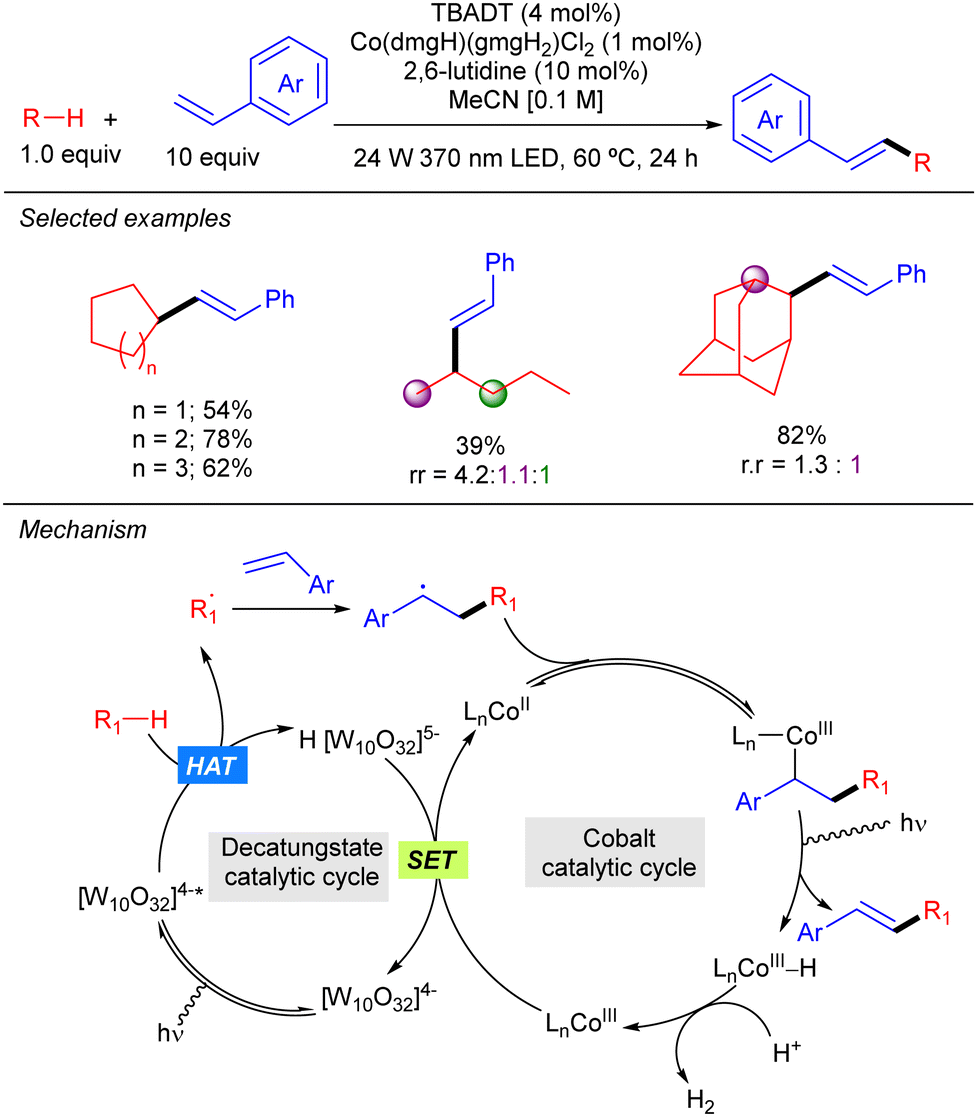
TBADT photocatalysis has also been combined with Co catalysis as illustrated by the work of Wu on the dehydrogenative cross-coupling of alkanes and styrene derivatives (Scheme 31).49 The reaction represents a C–H alkenylation that operates in the absence of any external oxidant and provides access to a wide variety of 1,2-disubstituted alkenes with excellent E selectivity. This method relies on the synergistic interaction of HAT photocatalysis and a cobaloxime-mediated hydrogen-evolution cross-coupling. As a mechanistic proposal, it was proposed that the secondary radical resulting from the addition of the alkyl radical to the styrene derivative combines with the Co(II) catalyst to afford an alkyl–Co(III) intermediate that undergoes β-hydride elimination to furnish the product and a Co(III)–H complex that gets protonated forming H2 and a new Co(III) species that reacts with the reduced H[W10O32]5− through a SET process to restart both catalytic cycles.
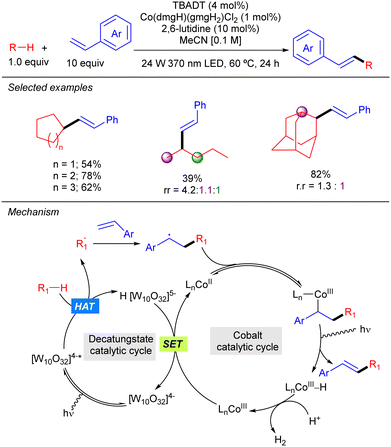 |
| | Scheme 31 Alkenylation of alkanes by Co/TBADT-catalysis. | |
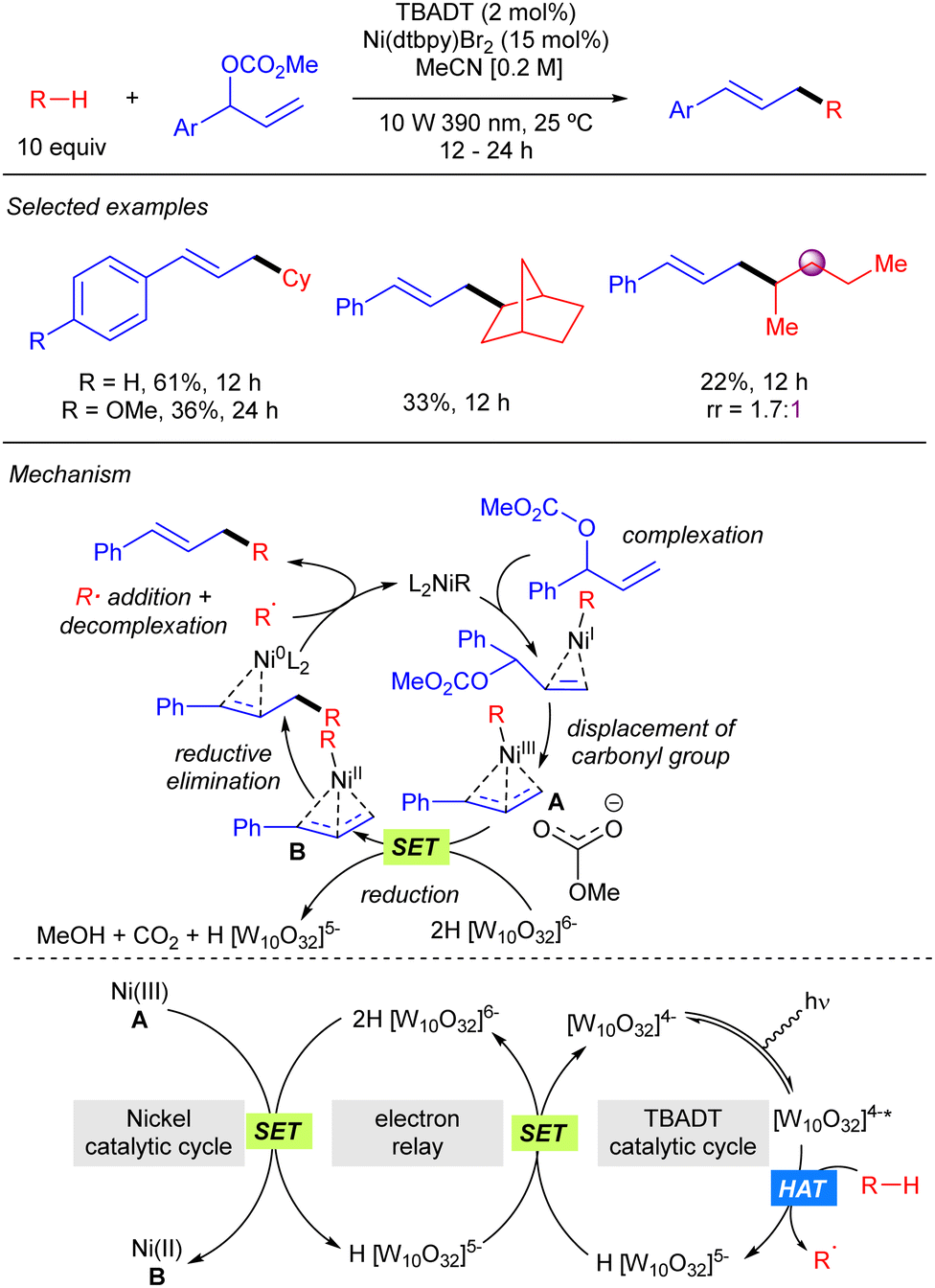
TBADT-mediated HAT photocatalysis has also found application in direct C–H allylation of alkanes. In 2022, Gong reported the cooperative use of TBADT and nickel catalysis for the allylation of alkanes with branched allylic carbonates (Scheme 32).50 The reaction provides linear products and works for cyclic alkanes and allylic carbonates bearing aromatic groups. The reaction was also applied to one linear alkane such as pentane, although it proceeded in low yield and regioselectivity. The mechanism of the reaction was studied by DFT calculations. These studies suggest that the reaction proceeds through the formation of an alkyl–Ni(I) by combination of the alkyl radical with the catalytically active Ni(0) complex. Subsequent oxidative addition of the allylic carbonate affords an allyl–Ni(III) intermediate that reacts with the doubly reduced decatungstate 2H[W10O32]6−via SET to generate an allyl–Ni(II) complex that provides after reductive elimination the product and the active Ni(0) species.
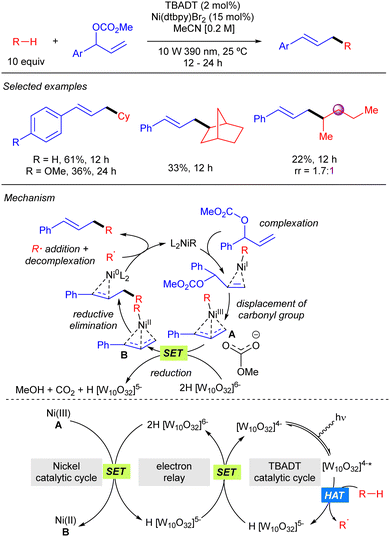 |
| | Scheme 32 Synergistic Ni/TBADT photocatalysis for the C–H allylation of alkanes. | |
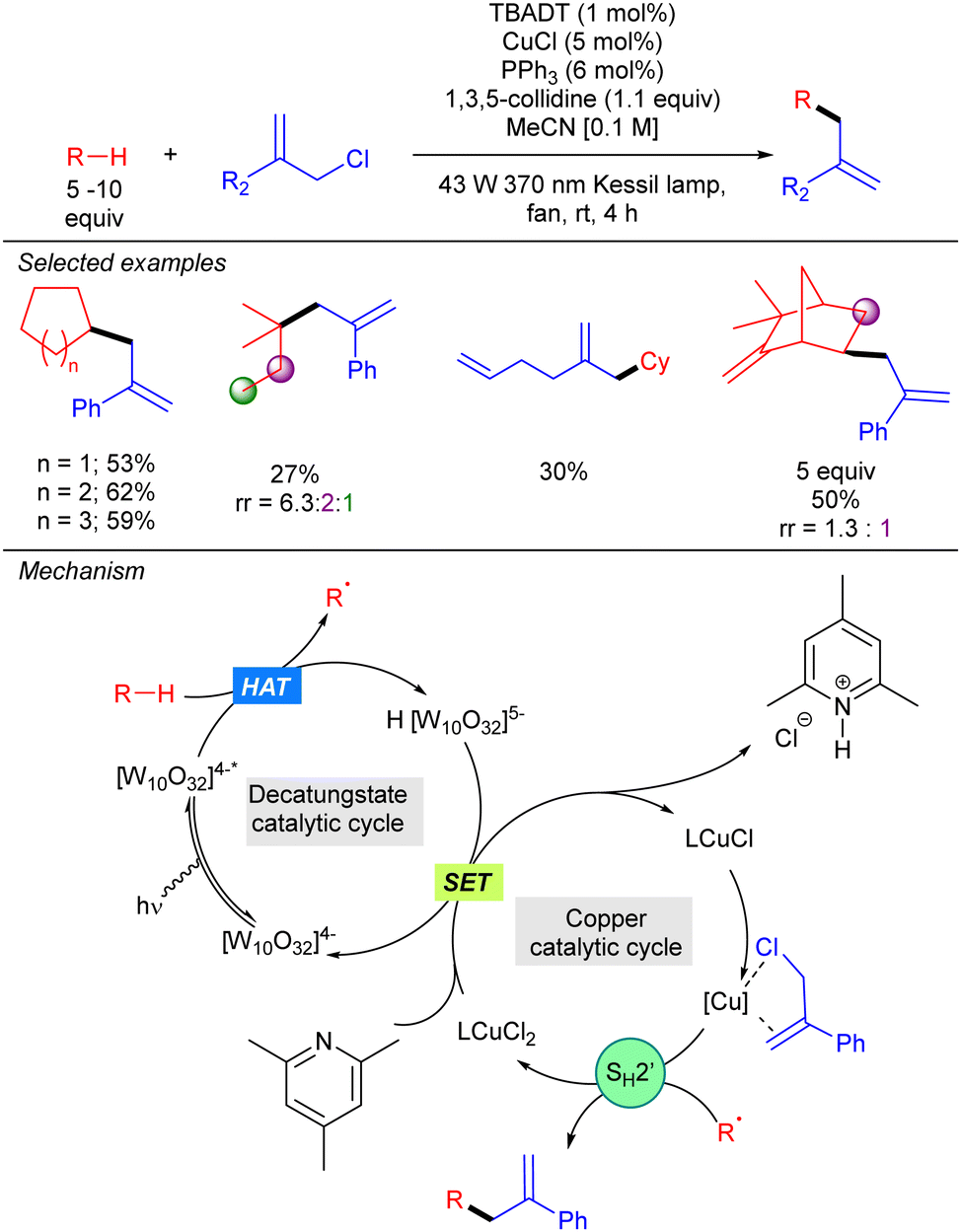
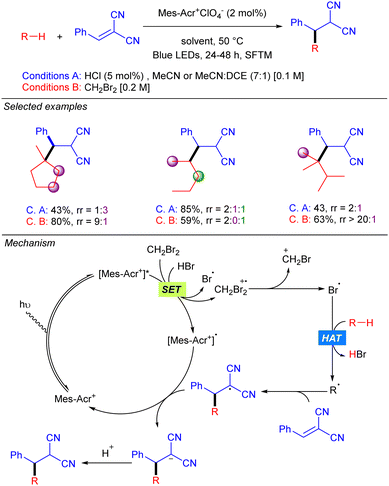
In the same year, our group described a photocatalytic strategy that allowed alkanes to be directly coupled with allylic chlorides (Scheme 33).51 The process was based on a synergistic interaction between TBADT and a Cu(I) co-catalyst. In this transformation, the alkyl radical generated by HAT photocatalysis engages in a SH2′ reaction with an activated allylic π-olefin-copper complex. This step generates a LCu(II)Cl2 complex that undergoes SET with the singly reduced decatungstate anion to regenerate both catalytic cycles. NMR and UV experiments supported this mechanism and the activation role of the copper catalyst. This dual catalysis allowed the functionalization of a wide variety of simple cyclic and acyclic alkanes, and also aliphatic natural products. Allylic chlorides bearing both aromatic and aliphatic substituents on the 2-position proved to be efficient for this transformation.
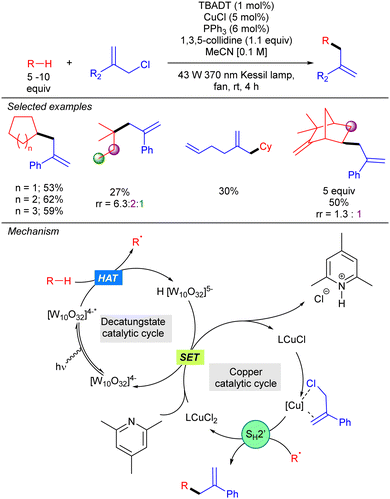 |
| | Scheme 33 TBADT/Cu-catalysed direct C–H allylation of unactivated alkanes. | |
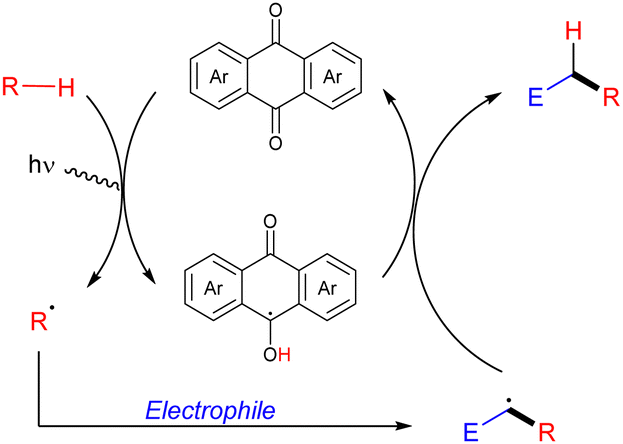
Organic molecules as HAT photocatalysts in alkane functionalization
Organic molecules also have the ability to get excited under certain light irradiation wavelengths.52 Thus, these simple molecules have also been successfully employed as HAT photocatalysts in alkane functionalization. Arylketones represent the main type of organophotocatalysts. Depending on their structure, these easily tuneable molecules can get selectively excited upon irradiation to promote a HAT process with an alkane to generate the carbon centred radical and a ketyl radical intermediate. In analogy with the processes described above, the alky radical addition to an electrophile furnishes a new carbon centred radical which oxidizes the ketyl radical to restart the catalytic cycle and deliver the coupling product (Scheme 34).
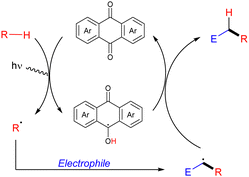 |
| | Scheme 34 General mechanism of aryl ketones as HAT catalysts. | |
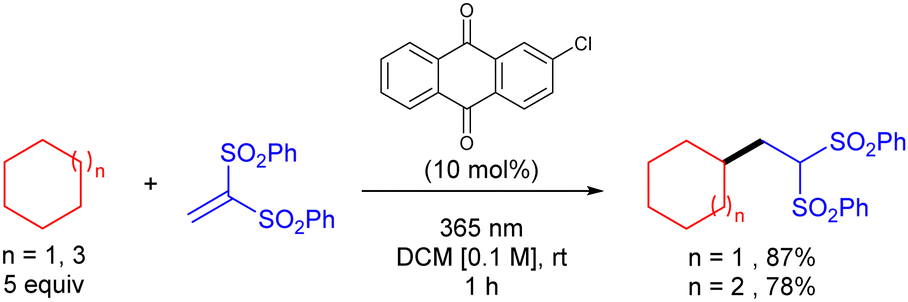
In 2016, Kamijo and co-workers reported the use of 2-chloroantraquinone (2-ClAQ) to promote the photocatalytic (365 nm light irradiation) addition of cyclohexane and cyclooctane to 1,1-bis(phenylsulfonyl)ethylene (Scheme 35).53
 |
| | Scheme 35 Arylketone-catalysed Michael addition of cycloalkanes to 1,1-bis(phenylsulfonyl)ethylene. | |
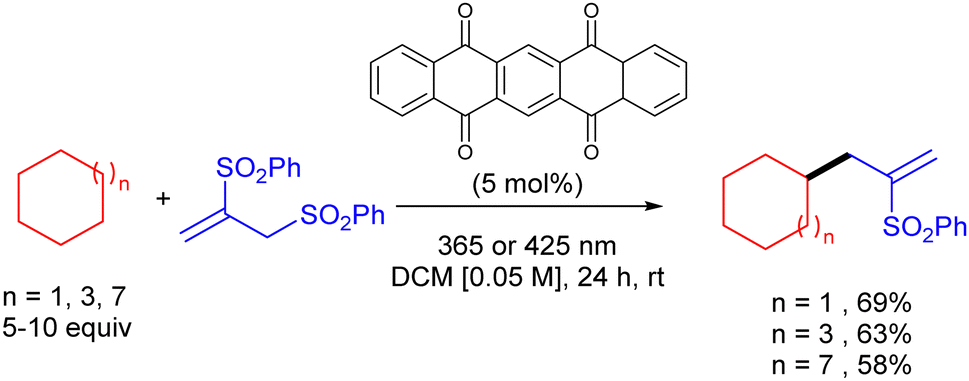
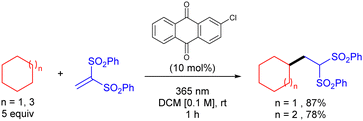
The same group reported the use of 5,7,12,14-pentacenetetrone (PT) as a HAT photocatalyst for the C–H allylation of cyclic alkanes with activated allyl sulfones as in (Scheme 36).54 The use of this arylketone as the HAT photocatalyst allowed the reaction to be efficiently carried out both under UV-A (365 nm) and even visible light (425 nm) irradiation.
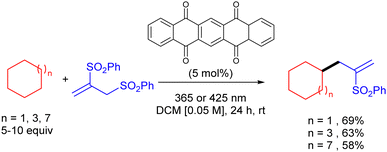 |
| | Scheme 36 Arylketone-catalysed radical C–H allylation of cyclic alkanes. | |
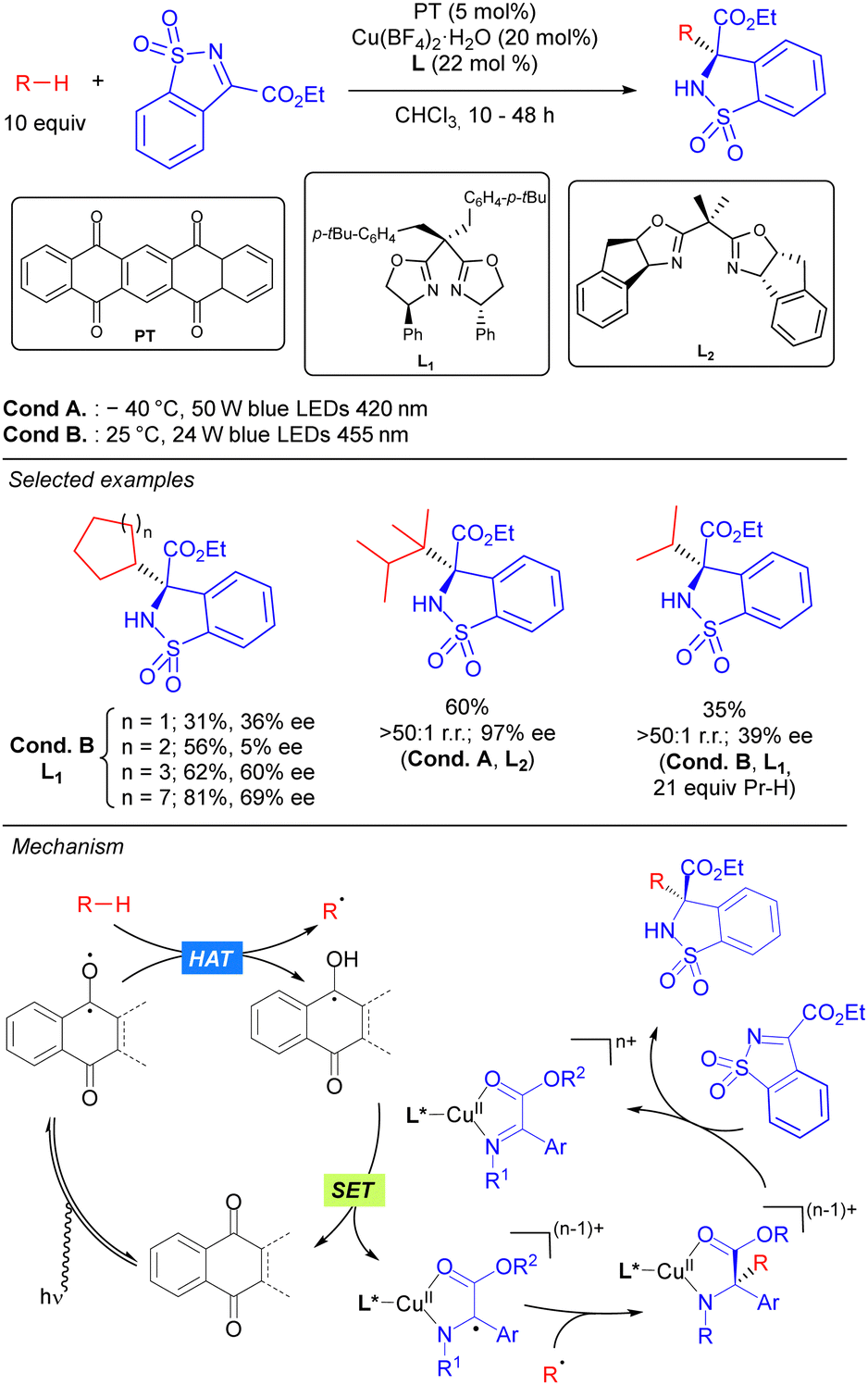
In 2019, the group of Gong reported the first asymmetric C–C bond formation in the field of photocatalytic direct alkane functionalization. This was achieved by merging HAT photocatalytic activation and chiral copper catalysis in the enantioselective alkylation of cyclic sulfonimines (Scheme 37).55 In this transformation, PT is used as the HAT photocatalyst for the generation of the alkyl radical under visible light irradiation, while a chiral bisoxazoline/Cu complex acting as a Lewis acid catalyst. It was proposed that the coordinated Cu-imine species undergoes SET with the ketyl radical intermediate restarting the photocatalytic cycle and generating a metal-stabilized carbon radical that evolves through intermolecular radical–radical coupling in which the regio- and stereoselectivity are sterically governed by the chiral ligand-transition metal moiety. Beyond functionalization of benzylic and allylic substrates, this methodology could also be efficiently applied to unactivated simple cyclic and acyclic alkanes with moderate to excellent enantioselectivity and very good selectivity towards the functionalization of the more substituted carbons. The high regioselectivity observed in compounds with tertiary C(sp3)–H bonds was attributed not only to the different formation rates of primary, secondary and tertiary carbon radicals, but also to the steric recognition by the HAT photocatalyst and transition-metal catalyst.
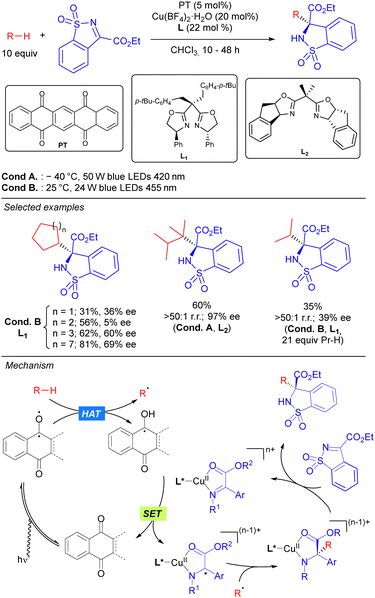 |
| | Scheme 37 Enantioselective Cu-(BOX)/PT-catalysed functionalization of alkanes. | |
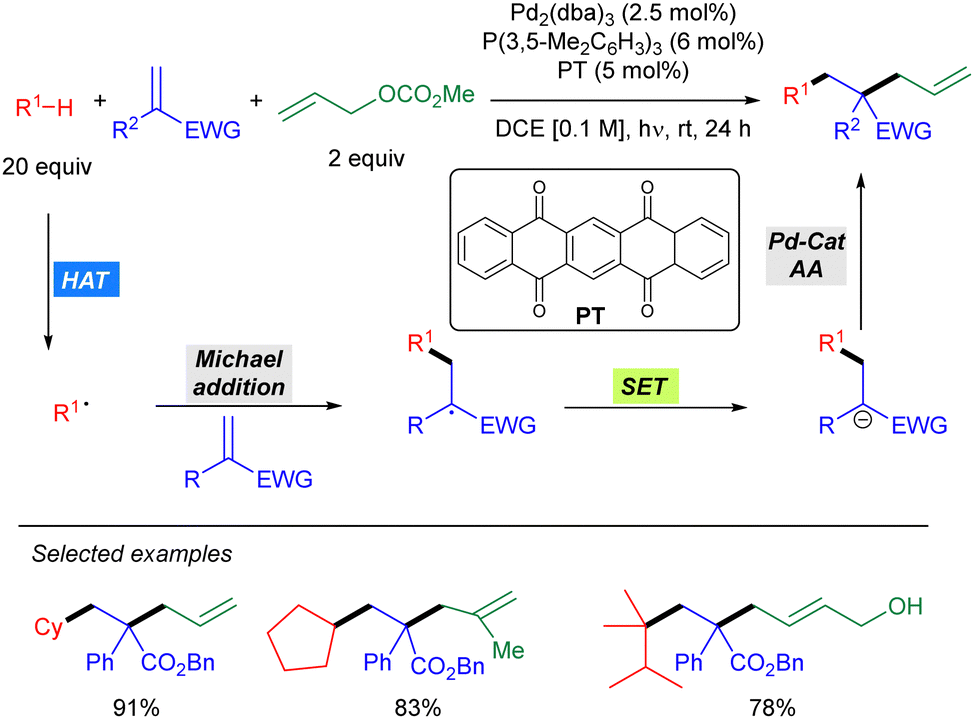
In 2021, Wang and co-workers reported a Pd-catalysed allylic alkylation involving a transient radical nucleophile (Scheme 38).56 The combination of 5,7,12,14-pentacenetetrone (PT) as a HAT photocatalyst with a Pd(0) catalyst promoted the three-component coupling between an alkane, a Michael acceptor and an allylic carbonate. Mechanistically, the alkyl radical generated via PT-mediated HAT adds to the Michael acceptor generating a new carbon-centred radical which undergoes SET with the ketyl radical leading to a carbanion which acts as the nucleophile in the Pd-catalysed allylic alkylation of the carbonate. This methodology was shown to be effective for cyclic alkanes and branched acyclic ones, displaying high levels of site selectivity for tertiary carbons. The utility of this protocol was highlighted by the concise synthesis of (±)-Mesembrine.
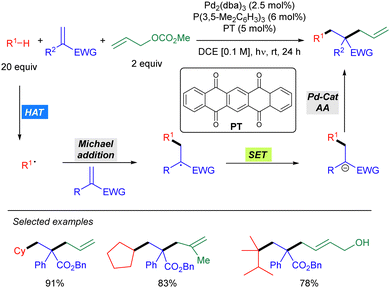 |
| | Scheme 38 Pd-catalysed allylic alkylation through photocatalytic generation of the nucleophile. | |
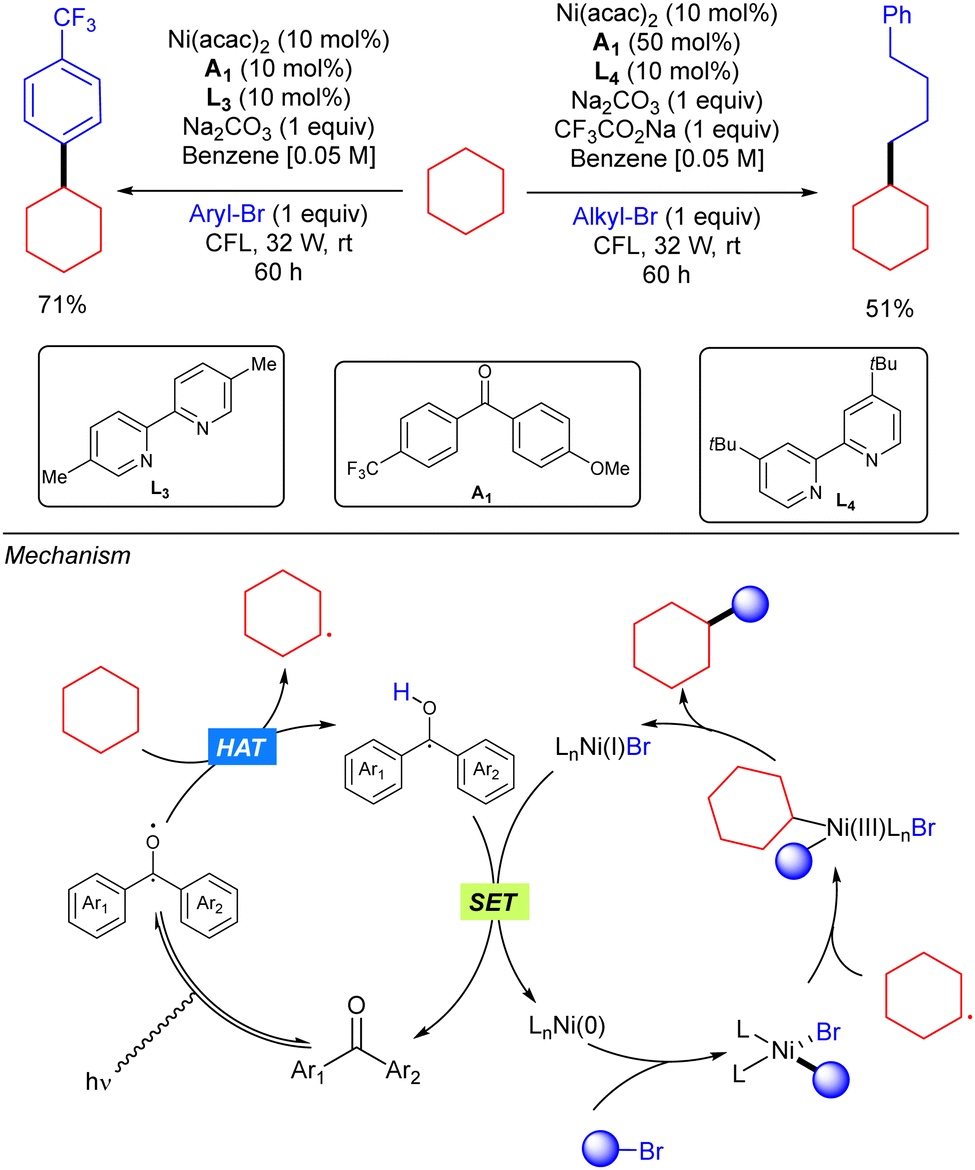
In 2018, Martin and co-workers demonstrated how the merging of a diarylketone photocalyst and a Ni catalyst resulted as an efficient system for alkane C(sp3)–H arylation and alkylation with organic bromides (Scheme 39).57 In this strategy, the triplet photoexcited diarylketone enables the homolytic cleavage of the C(sp3)–H bond to generate a carbon-centred radical that combines with a Ni(II)Ar intermediate, generated from oxidative addition of the organic bromide to the Ni(0) catalyst, to form a Ni(III) intermediate. This intermediate undergoes reductive elimination to give rise to the product. A final SET process releases both the catalytically active arylketone and the Ni(0) species.
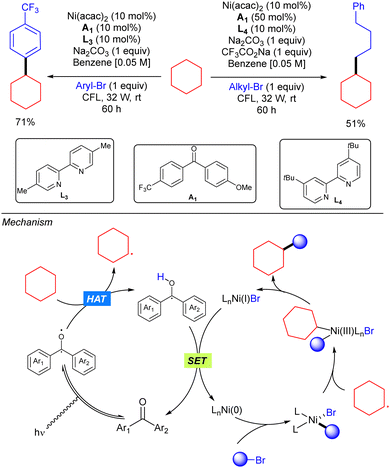 |
| | Scheme 39 Ni/ketone-catalysed C(sp3)–H arylation and alkylation of alkanes. | |
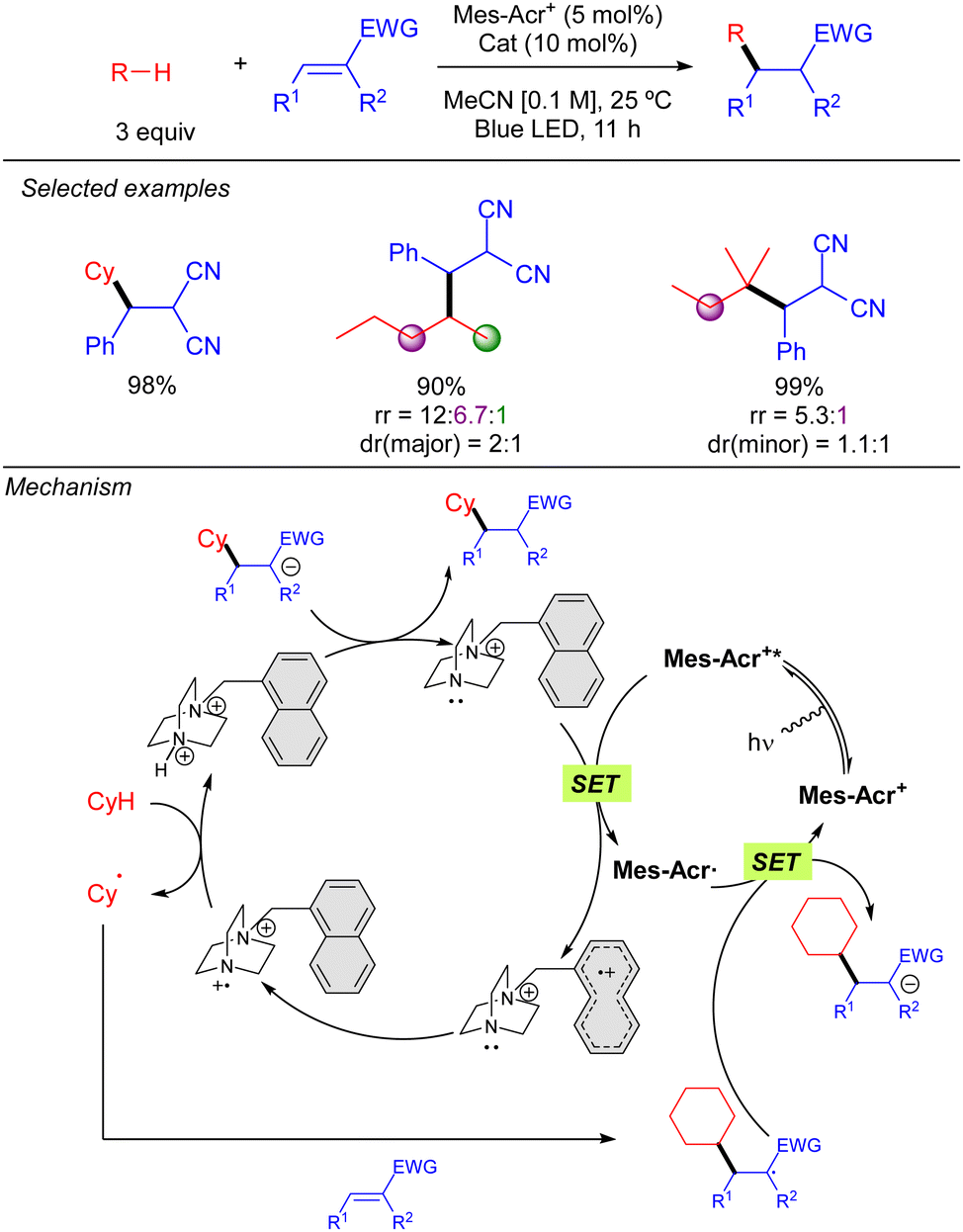
In 2022, Maruoka and co-workers described a new cationic DABCO derivative as a HAT catalyst for radical Michael addition of alkanes to trisubstituted electron deficient olefins (Scheme 40).58 The proposed mechanism, supported by experimental studies, starts with the oxidation of the cationic DABCO-based catalyst by [Mes–Acr+*]. The single electron oxidation occurs at the naphthyl pendant group. Then, the nitrogen transfers intramolecularly an electron to the naphthyl group to obtain the active radical species which undergoes HAT over the alkane. Subsequently, a Michael addition step takes place to generate a carbon-centred radical which undergoes a SET from the reduced [Mes–Acr˙] to release the product.
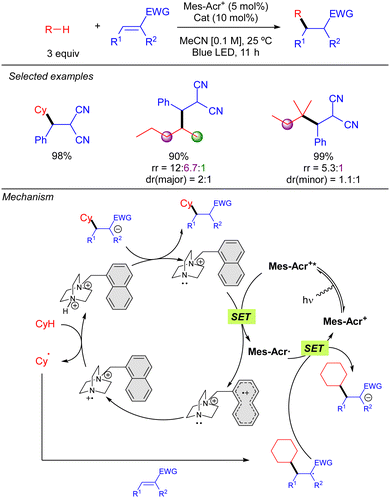 |
| | Scheme 40 Cationic DABCO-based HAT photocatalyst for the conjugate addition of alkanes. | |
Direct C–C bond formation in alkanes through (photo)electrocatalysis
Electrochemical reactions in which redox transformations are achieved with traceless electricity, instead of oxidants or reductants, have gained considerable attention in the past few years due to its inherent sustainability and tunability.59 In fact, electro- and photocatalysis are powerful methodologies in organic synthesis. Both share essential aspects since the same open–shell intermediates are generated upon one electron exchange between the organic molecule and the photocatalyst, or electrode. However, each field presents some drawbacks like the energy limitation for photoredox transformations (constrained by the used wavelength) or the overoxidation of the generated radical species under electrochemical conditions.60 Very recently, the combination of both has received attention as it was shown to be capable of generating new synthetic pathways, enabling milder conditions and better functional group tolerance and chemoselectivity in several examples.60
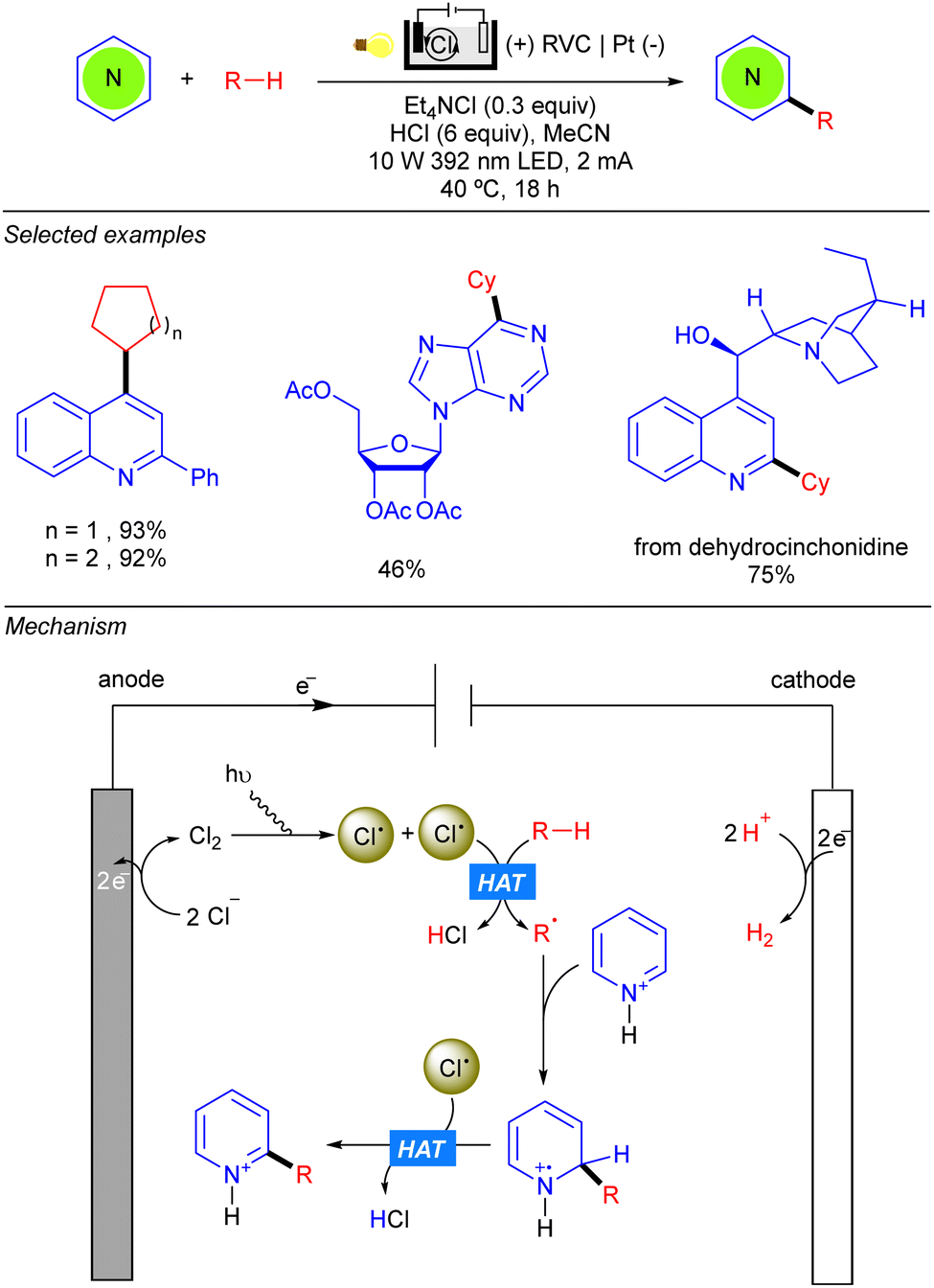
Within the context of simple alkane functionalization through C–C bond forming reactions, new Minisci-type transformations have been developed using (photo)electrocatalysis. The merging of both techniques not only allows the replacement of terminal oxidants with electrochemistry, but also allows us to obviate the required Ir- or Co-based co-catalyst for closing catalytic cycles, that can interfere with the reduction and elimination of alkyl radicals, limiting the scope of the photochemical methods. In 2020, Xu and co-workers were able to merge both photo- and electrocatalysis, reporting the first photoelectrochemical method that achieves dehydrogenative cross-coupling between a range of heteroarenes with cyclopentane and cyclohexane in good to excellent yields (Scheme 41).61 Regarding the reaction setup, they conducted the reaction in an undivided electrolytic cell using a reticulated vitreous carbon (RVC) anode and a Pt cathode. Mechanistically anodic oxidation of Cl− and subsequent homoleptic rupture under light irradiation, leads to a tow unit of Cl˙. The radical performs the HAT over the alkane leading to the alkyl radical (R˙) that adds to the heteroarene, yielding an N-centred radical–cation intermediate. Subsequent HAT between the second Cl˙ and the heteroarene leads to the final product.
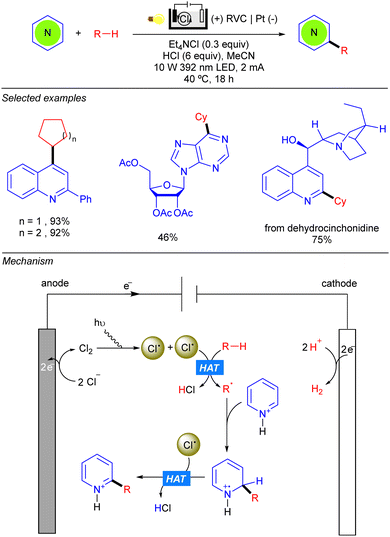 |
| | Scheme 41 Photoelectrochemical alkylation of heteroarenes promoted by the formation of Cl˙ radical. | |
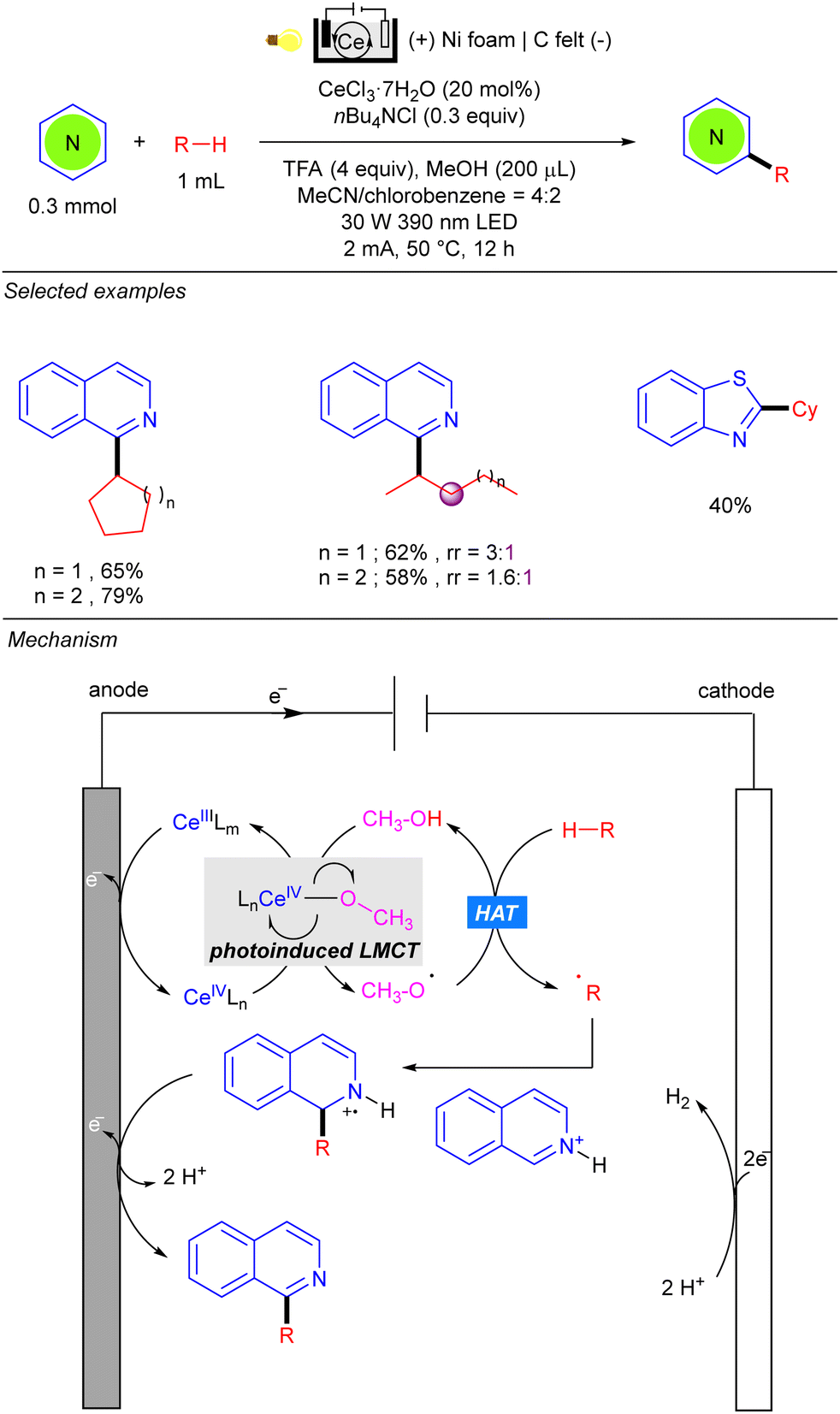
One year later, the group of Zeng reported the photo-electrocatalytic heteroarylation of alkanes through Ce catalysis (Scheme 42).62 Reaction setup was also based on an undivided electrolytic cell, albeit the choice of electrode differed from the work of Xu, and comprised a carbon felt (C felt) anode and a nickel foam (Ni foam) cathode. The reaction resembles some mechanistic features of the Ce-catalysed alkane functionalization reported by Zuo.24–26 Anodic oxidation of Ce(III) in the presence of nBu4NCl leads to the Ce(IV) photoactive species. The alkoxy radical generated by photoinduced LMCT promotes the HAT over the alkane, and the alkyl radical is added over the activated heteroarene. Anodic oxidation and deprotonation yield the desired product as well as the required H+ for cathodic hydrogen evolution. Regarding the alkane scope, the reaction tolerates a variety of cycloalkanes and linear ones such as n-pentane and n-hexane, the heteroarylation at the C-2 position being the preferred regioselectivity.
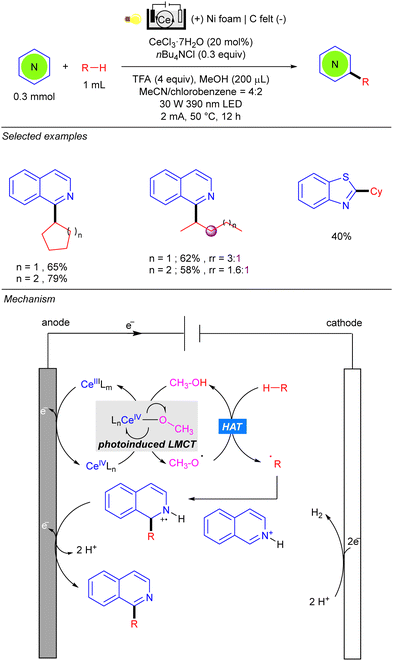 |
| | Scheme 42 Cerium-catalysed photoelectrochemical alkylation of nitrogen heterocycles. | |
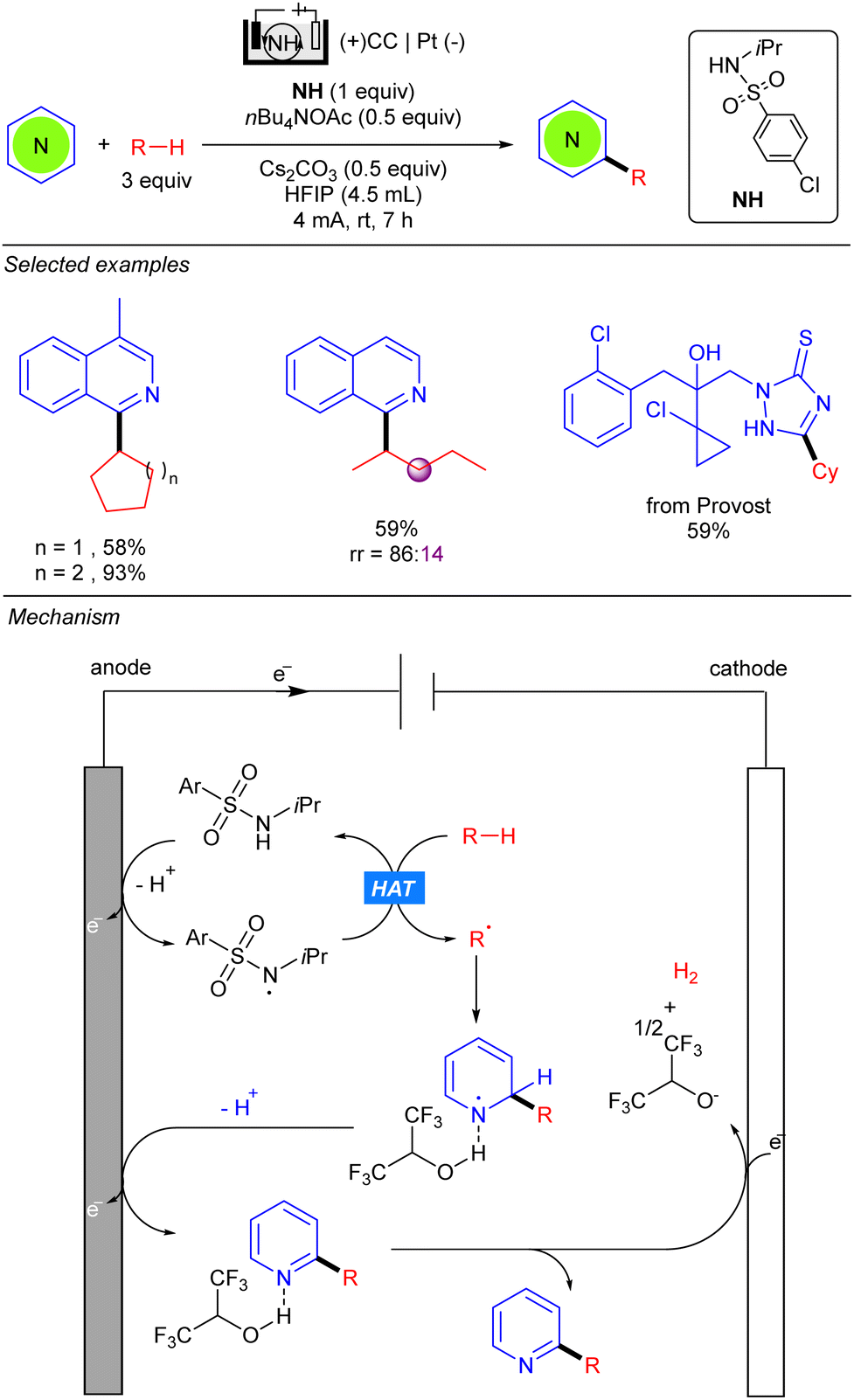
The group of Qi simultaneously reported the electrocatalytic radical alkylation of heteroarenes, using sulfonamides as HAT reagents (Scheme 43).63 Similar to previous examples, an undivided electrolytic cell comprising a carbon-based anode and a metal-based cathode was used. Opposite to the methodologies of Xu and Qui, light is not required for this reaction since anodic oxidation of sulfonamide, assisted by the base, leads to the corresponding N-centred radical responsible for the HAT process. The resulting alkyl radical adds to the 2-position of the activated heteroarene yielding a new N-centred radical. Second deprotonation and anodic oxidation releases the final product. Finally, HFIP undergoes a cathodic reduction thus promoting the hydrogen evolution process. Kinetics studies suggested that the generation of the N-centred radical was likely to be the rate-determining step, therefore a stoichiometric amount of the sulfonamide was used for increasing reaction rates. This methodology allows the use of both cyclic and linear alkanes, displaying good regioselectivity for the latter. However, sterically bulky branched alkanes, such as 2,3-dimethyl-butane, proved not to be efficient for this transformation.
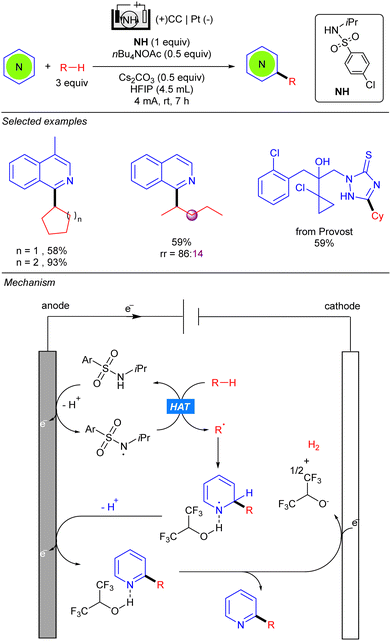 |
| | Scheme 43 Electrochemical alkylation of nitrogen heterocycles using an N-alkyl sulfonamide as the HAT reagent. | |
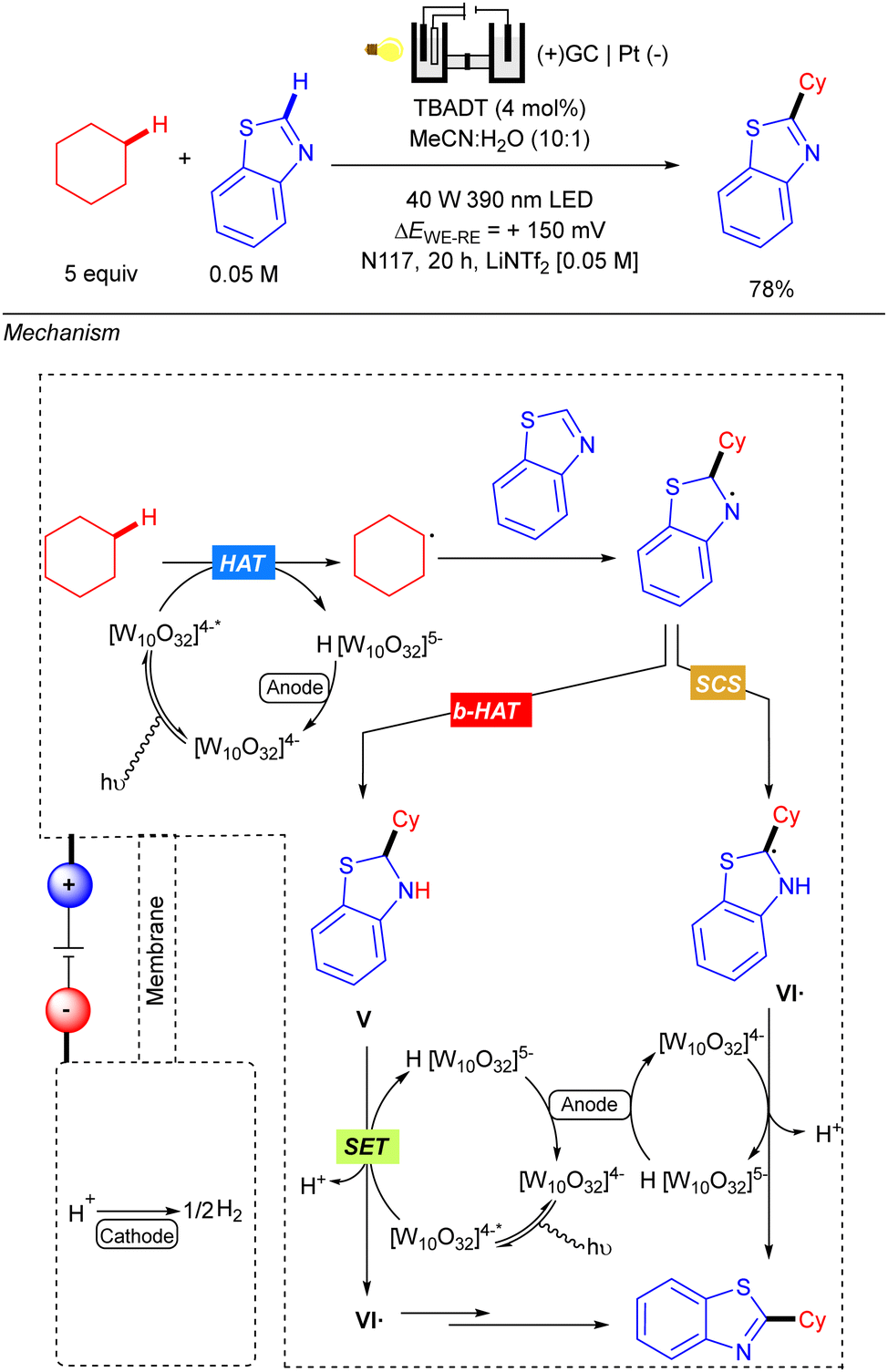
Also in 2021, Ravelli reported the C–H alkylation of benzothiazoles with cyclic alkanes via the combination of TBADT-photocatalysis under UV-A irradiation and electrochemistry (Scheme 44).64 Reaction setup differs from previous examples since an undivided cell did not work properly for this transformation. Therefore, a standard H-type electrochemical cell (Nafions N-117 polymeric membrane) equipped with a three-electrode system was used instead. Anolyte was composed of a LiNTf2 [0.05 M] MeCN/H2O (10:1) solution along with the starting materials and TBADT, separated into the catholyte by the polymeric membrane, which was composed of a LiNTf2 [0.05 M] water solution. The N-117 membrane preserves both different chemical environments around each electrode and only allows the proton exchange from anode to cathode for the cathodic H2 evolution process. The proposed reaction mechanism relies on the three-fold role of the decatungstate anion: HAT photocatalyst, photoredox catalyst and electrocatalyst. The reaction starts with a first HAT process catalysed by TBADT and subsequent alkyl radical addition over the benzothiazole. The resulting N-centred radical intermediate was proposed to evolve through two possible routes: a back-HAT (b-Hat) or a spin-centre shift (SCS). The first alternative involves a b-HAT from the reduced form of the decatungstate anion H [W10O32]5− to give neutral benzothiazoline V with regeneration of the [W10O32]4− species. Then, intermediate V would undergo a photoelectrochemical sequence, where TBADT acts as photoredox catalyst promoting the oxidative SET and later regeneration after cathodic reduction; to deliver radical species VI˙. The alternative SCS pathway would involve proton-mediated formation of intermediate VI˙. The last step from VI˙ relies on the role of decatungstate anion as an electrocatalyst to oxidize the radical and yield the final product.
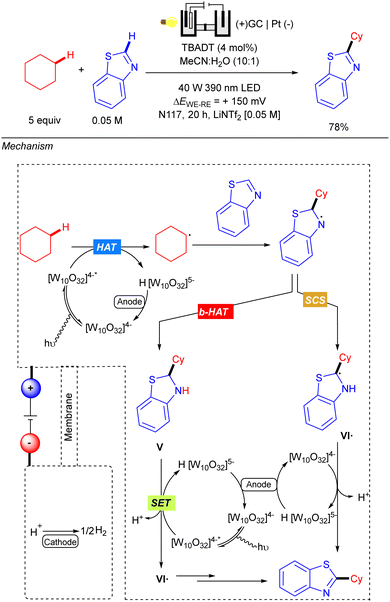 |
| | Scheme 44 TBADT-catalysed photoelectrochemical alkylation of benzothiazoles. | |
Conclusions
The area of photocatalytic alkane C–H functionalization has grown significantly in recent years. Several groups have reported different catalytic strategies, mainly based on a hydrogen atom transfer C(sp3)–H activation event, for the development of atom-efficient C–C bond forming reactions. In this article, we have outlined these transformations that are enabled either by halogen or oxygen-centred radicals, photoexcited species or electrocatalysis. Moreover, we have also emphasized their key mechanistic features.
Despite these great advances, several challenges still need to be met. This field is largely dominated by Giese- and Minisci-type reactions. Future efforts should be directed towards the development of catalytic strategies that allow the use of different radical trapping reagents that could expand the chemical space of these transformations. Another important issue to be solved is the regioselectivity of the HAT step, especially when linear alkanes are used. The design of new HAT agents that offer the possibility to tune site selectivity upon modification of steric or electronic properties offers exciting opportunities. Finally, the enantioselective direct C–H functionalization of alkanes represents an almost uncharted territory. New synthetic tools to achieve this goal across several transformations will have tremendous impact in the important field of asymmetric C–C bond forming reactions.
Author contributions
All the authors conceptualized and discussed the concept of this article. A. V.-R., P. M.-B. and A. M. A.-C. performed the literature research and wrote the article (equal contributions). The manuscript was co-written and corrected by M. F.-M.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Financial support from the European Research Council (ERC-CoG 863914-BECAME), AEI (PID2020-118237RB-I00), Xunta de Galicia (ED431C 2022/27; Centro singular de investigación de Galicia accreditation 2019–2022, ED431G 2019/03) and the European Regional Development Fund (ERDF) is gratefully acknowledged. A. M. A.-C. thanks Xunta de Galicia for a predoctoral fellowship.
Notes and references
-
(a) E. Negishi, Angew. Chem., Int. Ed., 2011, 50, 6738–6764 CrossRef CAS;
(b) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed;
(c) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587–9652 CrossRef CAS.
-
(a) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed;
(b) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC;
(c) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed;
(d) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 133, 15901–15924 CrossRef.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
-
(a) G. A. Olah, Acc. Chem. Res., 1987, 20, 422–428 CrossRef CAS;
(b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879–2932 CrossRef CAS PubMed;
(c) N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS;
(d)
A. J. L. Pombeiro, in Alkane Functionalization, ed. A. J. L. Pombeiro and M. F. C. Guedes da Silva, Wiley, Oxford, 2019, ch. 1, pp.1–15 CrossRef.
-
(a) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417–424 CrossRef CAS;
(b)
P. J. Pérez, Alkane C−H Activation by Single-Site Metal Catalysis, 2012 CrossRef;
(c) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704–724 CrossRef CAS.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef CAS.
- H.-P. Deng, Q. Zhou and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 12661–12665 CrossRef CAS PubMed.
- P. Jia, Q. Li, W. C. Poh, H. Jiang, H. Liu, H. Deng and J. Wu, Chem, 2020, 6, 1766–1776 CAS.
- S. Rohe, A. O. Morris, T. McCallum and L. Barriault, Angew. Chem., Int. Ed., 2018, 57, 15664–15669 CrossRef CAS.
- C.-Y. Huang, J. Li and C.-J. Li, Nat. Commun., 2021, 12, 4010 CrossRef CAS.
- B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef CAS.
- L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed.
- Y. Jin, Q. Zhang, L. Wang, X. Wang, C. Meng and C. Duan, Green Chem., 2021, 23, 6984–6989 RSC.
- Q. Zhang, S. Liu, J. Lei, Y. Zhang, C. Meng, C. Duan and Y. Jin, Org. Lett., 2022, 24, 1901–1906 CrossRef CAS PubMed.
- Y. Jin, L. Wang, Q. Zhang, Y. Zhang, Q. Liao and C. Duan, Green Chem., 2021, 23, 9406–9411 RSC.
- S. M. Treacy and T. Rovis, J. Am. Chem. Soc., 2021, 143, 2729–2735 CrossRef CAS PubMed.
- M. Yamane, Y. Kanzaki, H. Mitsunuma and M. Kanai, Org. Lett., 2022, 24, 1486–1490 CrossRef CAS PubMed.
- G. B. Panetti, Q. Yang, M. R. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. Catal., 2022, 2, 853–866 CrossRef CAS.
- J. Xu, H. Cai, J. Shen, C. Shen, J. Wu, P. Zhang and X. Liu, J. Org. Chem., 2021, 86, 17816–17832 CrossRef CAS PubMed.
- M. Bhakat, B. Khatua and J. Guin, Org. Lett., 2022, 24, 5276–5280 CrossRef CAS PubMed.
- D. P. Schwinger, M. T. Peschel, T. Rigotti, P. Kabaciński, T. Knoll, E. Thyrhaug, G. Cerullo, J. Hauer, R. de Vivie-Riedle and T. Bach, J. Am. Chem. Soc., 2022, 144, 18927–18937 CrossRef CAS PubMed.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2023, 145, 359–376 CrossRef CAS PubMed.
- Q. An, Z. Wang, Y. Chen, X. Wang, K. Zhang, H. Pan, W. Liu and Z. Zuo, J. Am. Chem. Soc., 2020, 142, 6216–6226 CrossRef CAS PubMed.
- G.-X. Li, X. Hu, G. He and G. Chen, ACS Catal., 2018, 8, 11847–11853 CrossRef CAS.
- Y. Zhang, Y. Jin, L. Wang, Q. Zhang, C. Meng and C. Duan, Green Chem., 2021, 23, 6926–6930 RSC.
- B. Wang, C. A. Pettenuzzo, J. Singh, G. E. Mccabe, L. Clark, R. Young, J. Pu and Y. Deng, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS.
- M. Schlegel, S. Qian and D. A. Nicewicz, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS.
-
(a) M. D. Tzirakis, I. N. Lykakis and M. Orfanopoulos, Chem. Soc. Rev., 2009, 38, 2609–2621 RSC;
(b) V. D. Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, ACS Catal., 2016, 6, 7174–7182 CrossRef CAS;
(c) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- D. C. Duncan, T. L. Netzel and C. L. Hill, Inorg. Chem., 1995, 34, 4640–4646 CrossRef CAS.
- C. Tanielian, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS.
- S. Bonciolini, T. Noël and L. Capaldo, Eur. J. Org. Chem., 2022, e202200417 CAS.
- D. Dondi, M. Fagnoni and A. Albini, Eur. J. Chem., 2006, 12, 4153–4163 CrossRef CAS PubMed.
- S. Protti, D. Ravelli, M. Fagnoni and A. Albini, Chem. Commun., 2009, 7351–7353 RSC.
- Z. Wen, A. Maheshwari, C. Sambiagio, Y. Deng, G. Laudadio, K. Van Aken, Y. Sun, H. P. L. Gemoets and T. Noël, Org. Process Res. Dev., 2020, 24, 2356–2361 CrossRef CAS PubMed.
- G. Laudadio, Y. Deng, K. van der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, Science, 2020, 369, 92–96 CrossRef CAS PubMed.
-
(a) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2011, 50, 1869–1872 CrossRef CAS PubMed;
(b) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed.
- M. Bergami, S. Protti, D. Ravelli and M. Fagnoni, Adv. Synth. Catal., 2015, 357, 3687–3695 CrossRef.
- K. Kim, S. Lee and S. H. Hong, Org. Lett., 2021, 23, 5501–5505 CrossRef CAS PubMed.
- M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnonia and I. Ryu, Chem. Commun., 2017, 53, 2335–2338 RSC.
- X. Wang, M. Yu, H. Song, Y. Liu and Q. Wang, Org. Lett., 2021, 23, 8353–8358 CrossRef CAS PubMed.
- X.-Y. Yuan, Y.-F. Si, X. Li, S.-J. Wu, F.-L. Zeng, Q.-Y. Lv and B. Yu, Org. Chem. Front., 2022, 9, 2728–2733 RSC.
- L. Capaldo, S. Bonciolini, A. Pulcinella, M. Nuño and T. Noël, Chem. Sci., 2022, 13, 7325–7331 RSC.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noël, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS PubMed.
- W. Liu, Y. Ke, C. Liu and W. Kong, Chem. Commun., 2022, 58, 11937–11940 RSC.
- H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed.
- Y. Jin, E. W. H. Ng, T. Fan, H. Hirao and L.-Z. Gong, ACS Catal., 2022, 12, 10039–10046 CrossRef CAS.
- P. Martínez-Balart, B. L. Tóth, Á. Velasco-Rubio and M. Fañanás-Mastral, Org. Lett., 2022, 24, 6874–6879 CrossRef PubMed.
-
(a) I. Loeff, A. Treinin and H. Linschitz, J. Phys. Chem., 1983, 87, 2536–2544 CrossRef CAS;
(b) S. A. Carlson and D. M. Hercules, Photochem. Photobiol., 1973, 17, 123–131 CrossRef CAS;
(c) Y. Hou, L. A. Hucka and P. Wan, Photochem. Photobiol. Sci., 2009, 8, 1408–1415 CrossRef CAS PubMed.
- S. Kamijo, G. Takao, K. Kamijo, T. Tsuno, K. Ishiguro and T. Murafuji, Org. Lett., 2016, 18, 4912–4915 CrossRef CAS PubMed.
- S. Kamijo, K. Kamijo, K. Maruoka and T. Murafuji, Org. Lett., 2016, 18, 6516–6519 CrossRef CAS PubMed.
- Y. Li, M. Lei and L. Gong, Nat. Catal., 2019, 2, 1016–1026 CrossRef CAS.
- Y. Shen, Z.-Y. Dai, C. Zhang and P.-S. Wang, ACS Catal., 2021, 11, 6757–6762 CrossRef CAS.
- Y. Shen, Y. Gu and R. Martin, J. Am. Chem. Soc., 2018, 140, 12200–12209 CrossRef CAS PubMed.
- A. Matsumoto, M. Yamamoto and K. Maruoka, ACS Catal., 2022, 12, 2045–2051 CrossRef CAS.
-
(a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed;
(b) J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed;
(c) T. H. Meyer, L. H. Finger, P. Gandeepan and L. Ackermann, Trends Chem., 2019, 1, 63–76 CrossRef CAS.
-
(a) L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS PubMed;
(b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed;
(c) Y. Yu, P. Guo, J.-S. Zhong, Y. Yuan and K.-Y. Ye, Org. Chem. Front., 2020, 7, 131–135 RSC.
- P. Xu, P.-Y. Chen and H.-C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- Z. Tan, X. He, K. Xu and C. Zeng, ChemSusChem, 2022, 15, e202102360 CAS.
- Y. Liu, B. Shi, Z. Liu, R. Gao, C. Huang, H. Alhumade, S. Wang, X. Qi and A. Lei, J. Am. Chem. Soc., 2021, 143, 20863–20872 CrossRef CAS PubMed.
- L. Capaldo, L. L. Quadri, D. Merli and D. Ravelli, Chem. Commun., 2021, 57, 4424–4427 RSC.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2023 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Pol
Martínez-Balart†
,
Pol
Martínez-Balart†