
Lanthanide-doped inorganic halide perovskites (CsPbX3): novel properties and emerging applications
Santosh
Kachhap
a,
Sachin
Singh
a,
Akhilesh Kumar
Singh
*b and
Sunil Kumar
Singh
 *a
*a
aDepartment of Physics, Indian Institute of Technology (Banaras Hindu University), Varanasi – 221005, India. E-mail: sunilks.app@iitbhu.ac.in
bDepartment of Physical Sciences, Banasthali Vidyapith, Banasthali, Rajasthan – 304022, India. E-mail: akhilesh_singh343@yahoo.com
First published on 2nd February 2022
Abstract
Inorganic halide perovskites (IHPs) have provoked intense research efforts because of their superior stability, excellent optoelectronic properties, cost-effectiveness, and striking optoelectronic applications. Recently, the doping of lanthanide ions in IHPs has opened new avenues, particularly for emerging applications like NIR and white light-emitting diodes, NIR emitters, NIR cameras, optical temperature sensing, optical data encoding, etc. Besides, lanthanide doping has also significantly improved the stability (thermal, photo, and phase), structure and optical properties of IHPs, which has resulted in improved device performance. However, a comprehensive review of this development for IHPs is rare. This review article is an attempt to fill this gap and is designed to provide important fundamental aspects as well as recent developments in the field. It comprises all the basics, synthesis strategies, crystal structure (including phase transition and phase stability), and optical properties (absorption, emission, lifetime, quantum yield, exciton binding energy, and anisotropy) for pristine IHPs with special reference to CsPbX3 (X = Cl, Br and I). The effects of lanthanide doping on the above-listed properties for IHPs are explored, and the doping of non-lanthanide metal ions such as alkali metals, alkaline earth metals, transition metals, and post-transition metals in all-IHPs are also covered for comparison. Furthermore, the review specifically outlines a few novel applications, which are due to the inherent merits of lanthanide doping in CsPbX3. Potential challenges and future perspectives are also discussed.
 Santosh Kachhap | Santosh Kachhap received the BSc degree in Physics in 2015 and the MSc degree in Physics (Electronics) in 2017 from Institute of Science, Banaras Hindu University, Varanasi. Uttar Pradesh, India. He was project fellow between January 2018–March 2019 at Indian Institute of Technology, Bhubaneswar, India. Currently he is working as a research scholar since 2019 in Luminescent Materials and Devices Development Laboratory, Department of Physics, Indian Institute of Technology, Banaras Hindu University, Varanasi, India. |
 Sachin Singh | Sachin Singh received the BSc degree in Physics in 2016 from University of Delhi, Delhi, India, and the MSc degree in Physics in 2018 from University of Allahabad, Uttar Pradesh, India. Currently he is working as a research scholar since 2019 in Luminescent Materials and Devices Development Laboratory, Department of Physics, Indian Institute of Technology, Banaras Hindu University, Varanasi, India. |
 Akhilesh Kumar Singh | Dr Akhilesh Kumar Singh is working as an Assistant Professor in the Department of Physical Sciences, Banasthali Vidyapith, Rajasthan, India. He holds MSc in Physics (2004) from V.B.S. Purvanchal University, Jaunpur, India and MTech (2007) and PhD (2012) in Materials Science and Technology from Indian Institute of Technology (Banaras Hindu University) Varanasi, India. He worked as a Postdoctoral Fellow in the UNAM, México, and University of Strathclyde, Glasgow. His research interests include hybrid halide perovskites, luminescent materials for energy harvesting, luminescent security inks, etc. He has published 43 peer-reviewed international journal papers and is a co-investigator of an Indian Patent. |
 Sunil Kumar Singh | Dr Sunil Kumar Singh is currently working as an Assistant Professor in the Department of Physics, Indian Institute of Technology (BHU), Varanasi, India (since 2018). His research is focused on spectroscopy of nanostructured materials and their technological applications in the area of photon upconversion, optical imaging, photovoltaics, optical and bio-sensors, etc. After completing PhD from Banaras Hindu University, India (2011), he carried-out post-doctoral studies from Changwon National University, South Korea (2011–12). He also served as DST-INSPIRE Faculty at the Department of Physics, IIT (BHU) during 2013–2017. Dr Singh has contributed more than 50 articles having citations >2100 and h index-27. |
1. Introduction
Oxide perovskites are well known for their versatile applications in many fields, for example, in piezoelectricity, magnetism, superconductivity,1–4etc. In the last two decades halide perovskites have shown a paradigm shift in the application of perovskites.5,6 Because of the direct bandgap, high absorption coefficient, high mobility, long carrier lifetime, cost-effectiveness, etc., the all-inorganic cesium-lead halide perovskites (CsPbX3, where X = Cl, Br, and I) have been considered as rising stars for photovoltaic applications.7–11 They are also used in making colored and white LEDs,12–15 fluorescence sensors,16 oxygen detection sensors,17 gas sensors,18 humidity sensors,19 field-effect transistors,20,21 filter-free color image sensors,22 display applications,23 photodetectors,24–26 spectro-chemical probes, explosive detectors,27 laser fabrication,28 X-ray detection,29,30etc. In this review, we mainly discuss the basics of halide perovskites, including the major advantages and challenges of halide perovskites. Detailed structural and optoelectronic properties of inorganic halide perovskites (IHPs) are also discussed. The study of the doping effect of lanthanide ions in the IHPs and their novel and emerging applications are the focus of this review. To study the various optoelectronic properties of all inorganic halide perovskites (IHPs), CsPbX3 (X = Cl, Br, and I) has been chosen as a model system. Overall, the review article provides important fundamental aspects, as well as recent developments, in the field of IHPs.Perovskites can be traced back to 1839 when a German scientist, Gustav Rose,31 encountered a new calcium titanate-based mineral in the Ural Mountain, on a journey to Russia. The mineral was then named “perovskite” in honor of the renowned Russian mineralogist Lev von Perovski. The materials with a similar structure to CaTiO3 are known as perovskites. The general formula of perovskites is ABX3.32 In the ABX3 structure, A occupies the corner position, B is at the body-centered position, and the face-centered positions are taken by the X atoms (see Fig. 1). The coordination number of B is 6, which is surrounded by the octahedral geometry formed by the X anions. The A atom has a coordination number 12 with cuboctahedral orientation. In general, the perovskites are characterized by the Goldschmidt tolerance factor, denoted as ‘t’, as follows:
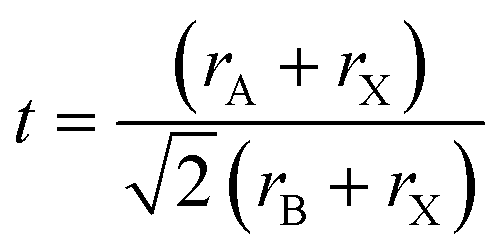
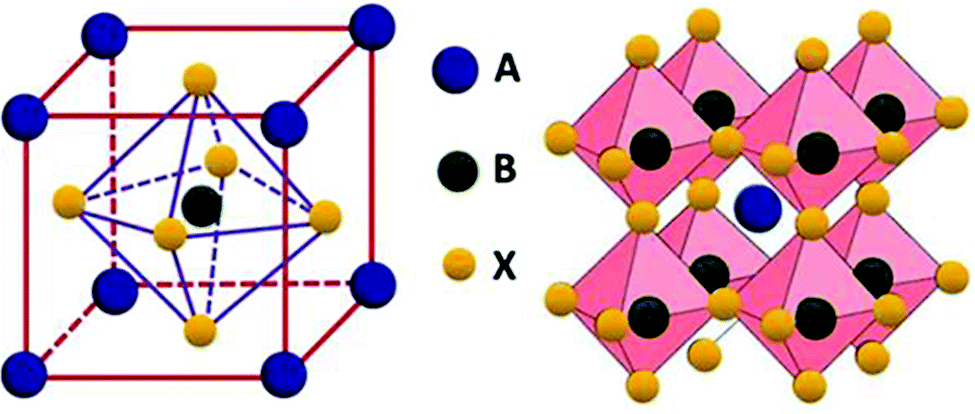 | ||
| Fig. 1 Structure of perovskite with formula ABX3. Reprinted from ref. 32. Open access (2019). | ||
In recent years, a class of perovskites with the general formula ABX3, where A and B are cations and X is the halide, has attracted a great deal of interest. In halide perovskites, A site atoms are either organic [MA+: methylammonium (CH3NH3+), FA+: formamidinium (HC(NH2)2+)] or inorganic cations (Cs+), whereas B is the metal cation (Pb2+, Sn2+, Ge2+, etc.), and the X-site can have halides (Cl−, Br−, I−).34–36 When an organic cation (MA+, FA+, etc.) remains present at the A-site, it is called an organic–inorganic hybrid halide perovskite (e.g., MAPbX3, FAPbX3).36–38 On the other hand, if an inorganic cation is present at the A-site, it is known as all-IHPs (for example, CsPbX3).39 The basic classes of perovskite structures, based on different types of atoms situated on A, B, and X sites, are listed in Table 1.
| Oxide perovskite | |||
| A = inorganic cation | |||
| B = inorganic cation | |||
| X = oxygen | |||
| SrTiO3,40 GdScO3,41 BaTiO342 | |||
| Organic–inorganic mixed halide perovskite (MA/FAPbCl3−x−yBrxIy) | |||
| A = organic cation | |||
| B = inorganic cation | |||
| X = two or more halide anions | |||
| CH3NH3Pb(BrxI1−x)3,43 CH3NH3PbCl0.5Br2.544 | |||
| Perovskite ABX3 | Hybrid perovskite | ||
| A = organic cation (MA+, FA+) | |||
| B = inorganic cation | |||
| X = halide anions | |||
| Halide perovskite | Organic–inorganic halide perovskite (MA/FAPbX3) | ||
| A = organic/inorganic cation (Cs+, MA+, FA+) | A = organic cation | ||
| B = inorganic cation (Pb2+) | B = inorganic cation | ||
| X = halide anions (Cl−, Br−, I−) | X = one halide anion | ||
| HC(NH2)2PbI3,45 CH3NH3PbCl346 | |||
| All-inorganic double perovskite (Cs2B1+B3+X6) | |||
| A = inorganic cation | |||
| B = two inorganic cations | |||
| X = one halide anion | |||
| Cs2AgBiBr6,47 Cs2InAgCl648 | |||
| All-inorganic perovskite | All-inorganic halide perovskite (CsPbX3) | ||
| A = inorganic cation (Cs+) | A = inorganic cation | ||
| B = inorganic cation | B = inorganic cation | ||
| X = halide anions | X = one halide anion | ||
| CsPbCl3,49 CsPbBr3,50 CsPbI351 | |||
| All-inorganic mixed halide perovskite (CsPbCl3−x−yBrxIy) | |||
| A = inorganic cation | |||
| B = inorganic cations | |||
| X = two or more halide anions | |||
| CsPbBr1I2,52 CsPbClxBr3−x53 |
It is obvious from Table 1 that the halide perovskites are broadly classified into two categories, i.e., hybrid (organic–inorganic) halide perovskites and all-IHPs. The first hybrid halide perovskite reported for visible-light harvesting was methylammonium lead iodide (CH3NH3PbI3) in 2009.11 It shows high efficiency in photovoltaic applications, however, it suffers from poor phase stability (particularly in a humid atmosphere) and poor photo-stability.54,55 It was found that the all-IHPs show better stability under different environments, e.g. humid atmospheres, light illumination, electric field, high temperature, and electron beam, etc.56 Li et al.10 carried out a comparative study of the stability of the hybrid halide perovskite and the all-IHPs in different atmospheres as listed in Table 2. The organic cations used in organic–inorganic halide perovskites, such as methylammonium and formamidinium, are among the major factors that result in phase instability.
| Environment | Hybrid halide perovskite | Inorganic halide perovskite |
|---|---|---|
| Humid | (1) Degrades into HI, PbI2, and volatile organics | (1) Induces a phase transition and reversibly transforms into its initial phase by heating to 350 °C under humid conditions |
| Illumination | (1) Decomposes into PbI2, metal lead, molecular iodine, and other volatile products | (1) Superior thermal and photochemical stability |
| (2) Superoxide reacts with the protonated MA+ and further decomposes its structure | (2) Acid protons are absent in Cs+ ions and make their structure more stable | |
| Electric field | (1) Migration barrier is lower under illumination | (1) Migration barrier is higher under illumination |
| (2) Rapidly and irreversibly decomposes to PbI2 under humid conditions | ||
| High temperature | (1) The loss of organic components; perovskite degrades into PbI2 | (1) Phase transition in four different phases |
| Electron-beam | (1) Defect formation due to irradiation damage, and phase transformation induced by electron-beam heating | (1) Remain stable and does not degrade under the electron beam |
| (2) Decomposes under the electron beam | (2) The intensity of the excitonic peak remains stable without the appearance of any high-energy peaks |
Both the hybrid and the all-IHPs show bandgap tunability with changing the halide composition and through the variation of particle size, which eventually covers the entire visible spectral region (410–700 nm).57–59 However, the all-IHPs show strong excitonic and spin–orbital coupling effects, resulting in spin-selective excitonic transitions, providing a new field for spintronics and quantum-optics applications.59,60 Thus, researchers are intensively looking at all-inorganic metal halide perovskites, in which the organic cations of the hybrid perovskites are substituted with cesium ions, such as CsPbX3 (X = Cl, Br, or I). The following sections in this review focus on the progress of CsPbX3 perovskites and highlight the emerging applications.
2. All-inorganic halide perovskite CsPbX3 (X = Cl, Br, or I)
2.1 Synthesis methods of CsPbX3
There are various methods for synthesizing bulk and nanostructured all-IHPs. Nasstrom et al.61 prepared CsPb(BrxI1−x)3 film, taking x between x = 0 and x = 2/3, using an inkjet printing technique, and x = 0.85 and x = 1 by spin coating. In another study, the efficient CsPbBr3 perovskite film was developed for solar cell applications by using a single-crucible vacuum thermal evaporation method.62 Recently, Borri et al.63 reported the synthesis of CsPbBr3 film of about 70 nm thickness by using RF-magnetron sputtering techniques on the glass substrate. In this method, the CsPbBr3 powder, prepared by mechano-chemistry, is pressed for 24 hours by a pneumatic press (working pressure 11.5 MPa) and sputtering deposition was carried out at room temperature. The RF power of 20 W was maintained in an argon environment with a flow rate of 20 atm cm3 min−1. This is one of the powerful techniques for thin-film deposition, both for conductive and non-conductive materials, and may attract researchers in the future. Zhang et al.64 synthesized CsPbBr1.2I1.8 nanocrystals (NCs) using the hot injection method. The colloidal solution of CsPbBr1.2I1.8 NCs was spin-coated at room temperature on a fused silica substrate. This formed a solid film and was used to study the light-induced phase segregations and optical studies. Liu et al.65 used modified hot injection techniques to synthesize all-IHPs quantum dots (QDs). In this technique, the organolead compound trioctylphosphine–PbI2 was swiftly injected into a 1-octadecene (ODE) solution, heated to 100–170 °C, which contained oleic acid (OA), oleylamine (OAm), and Cs-oleate.All these methods have certain advantages and disadvantages. One common challenge observed in all the methods discussed above is poor long-term stability against moisture. However, the film deposited using the thermal evaporation method showed comparatively longer film stability.66 The hot injection method, because of the requirements of high temperature and inert conditions, is neither cost-effective nor industrially viable.15 However, it shows high thermal stability and distinct optical features.57 The single-crucible vacuum thermal evaporation techniques used by Li et al.62 have advantages over multi-crucible vacuum thermal evaporation techniques. In multi-source (crucible) thermal techniques, the evaporation rate of the source material is highly dependent on parameters like the pressure of the vacuum chamber, the quality of evaporation materials, and the heating rate of the crucible. During the whole synthesis process, the adjustment of all these parameters for controlled evaporation is a difficult task so the single-crucible method is the best alternative to a multi-crucible method.
Thin-film deposition using the RF sputtering technique is suitable for both conductive and nonconductive materials.63 It also provides uniform thin-film limited contamination and ease of doping. The major challenge of this technique is the controlled monitoring of parameters like pressure, deposition rate, and RF power to get a uniform thin film. In the modified hot injection method used by Liu et al.,65 the particle size can be easily tuned by the injection temperature. In the modified hot-injection method, the organolead compound trioctylphosphine-PbI2 (TOP-PbI2) is taken as the reacting precursor. Fig. 2 shows a comparative study of conventional hot injection and modified hot injection methods based on Stokes shifts and PLQYs. The all-IHPs prepared by conventional methods show noticeable Stokes shifts that also vary with the particle size. However, the modified hot injection method shows almost constant Stokes shifts for the different sizes of QDs indicated in Fig. 2(a). The absorption coefficient of TOP-CsPbI3 QDs is higher than conventional OA/m-based CsPbI3 QDs as shown in Fig. 2(b). The photoluminescence quantum yields (PLQYs) are stable for different sizes of QDs in the modified method.
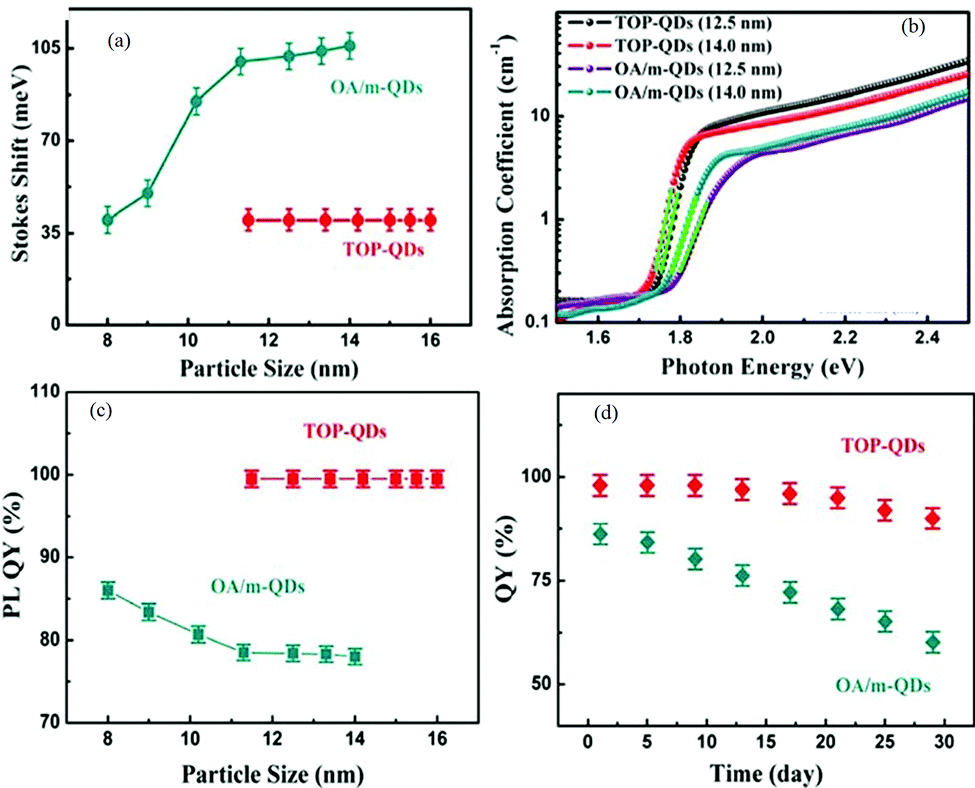 | ||
| Fig. 2 (a) Stokes shift of the TOP-QDs and OA/m-CsPbI3 QDs as a function of their particle sizes. (b) Optical absorption spectra of different sized TOP-QD and OA/m-CsPbI3. (c) PLQYs of OA/m-QDs and TOP-QDs as a function of their particle sizes. (d) Change in the PLQY of the OA/m-QDs, TOP-QDs versus storage duration. QD solutions synthesized at 140 °C were stored in a sealed bottle under ambient conditions. Before the QY measurement, all QD samples were washed twice using MeOAc. Reproduced with permission from ref. 65. Copyright©2017, American Chemical Society. | ||
The phosphine ligand-modified hot injection method was also reported by Li et al.67 to improve the emission intensity and stability of CsPbBr3 QDs under different atmospheres. The trioctylphosphine (TOP), tributylphosphine (TBP) and diphenylphosphine (DPP)-modified CsPbBr3 QDs show high PL intensity and improved stability as compared to CsPbBr3 QDs in ethanol, water and UV light irradiation. This may be attributed to the steric effect of the multi-branched phosphine ligand.
2.2 Phase and phase transitions of CsPbX3
Structure–property correlation makes it important to know about various phase transitions in halide perovskites. It affects the phase stability and the performance of devices like photovoltaic cells, light-emitting diodes, etc. The perovskite structure shows a stable cubic structure at high temperatures that switches to distorted metastable tetragonal and orthorhombic phases at low temperatures. The phase transition temperature depends on the perovskite composition and also to a lesser extent on sample preparation techniques. The formation enthalpy of different phases in the halide perovskite can be computed using the DFT method (even for the same composition) as follows:68| ΔHABX3 = EABX3 − μA − μB − 3μX | (1) |
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) were observed at 88 °C, and 130 °C, respectively. The neutron diffraction study carried out by Hirotsu et al.75 also indicated the transition temperature from the cubic to tetragonal phase and tetragonal to orthorhombic phase at 130 K and 88 K, respectively. Thus, the synthesis technique and synthesis temperature both affect the phase transition in CsPbBr3.
m space group) were observed at 88 °C, and 130 °C, respectively. The neutron diffraction study carried out by Hirotsu et al.75 also indicated the transition temperature from the cubic to tetragonal phase and tetragonal to orthorhombic phase at 130 K and 88 K, respectively. Thus, the synthesis technique and synthesis temperature both affect the phase transition in CsPbBr3.
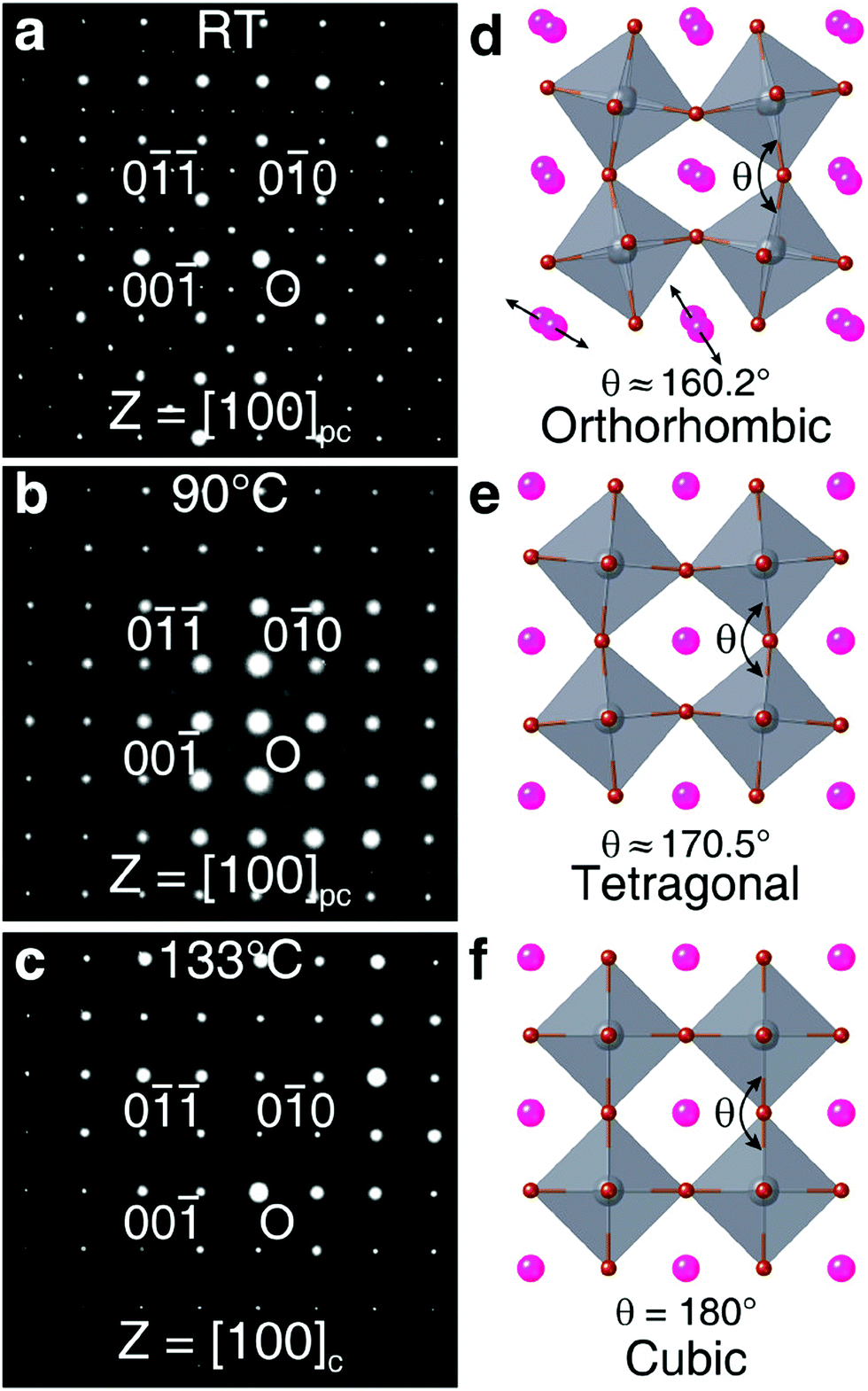 | ||
| Fig. 3 The SAED patterns obtained for the CsPbBr3 single-crystal at (a) room temperature, (b) 90 °C, and (c) 133 °C. (d–f) The orthorhombic, tetragonal, and cubic structures, respectively, for CsPbBr3. Reproduced with permission from ref. 74. Copyright©2020, American Chemical Society. | ||
m (cubic), β-P4/mbm (tetragonal), and γ-Pbnm (orthorhombic) and the fourth one δ-Pnma is a non-perovskite structure. The three perovskite phases of CsPbI3, namely, the α, β, and γ-phases, are also called “black phases” and the non-perovskite δ-phase is called the “yellow phase”.80,81
Fig. 4 shows the crystal structures of all these phases. It was observed that on increasing the temperature, a direct phase transition from the non-perovskite δ-Pnma to the perovskite cubic Pmm phase occurred at 595 K.79 This is a stable phase at high temperature. While cooling down, all the four crystal structures, cubic, tetragonal, orthorhombic, and non-perovskite, were observed. In the cooling process, the phase transition from the cubic perovskite structure α-Pmm phase to the tetragonal structure β-P4/mbm occurred at 539 K, the β-P4/mbm to orthorhombic structure γ-Pbnm was observed at 425 K and finally, the transition to the non-perovskite structure δ-Pnma phase occurred at room temperature. The bandgap of the non-perovskite yellow δ-Pnma phase was 3.01 eV and was stable at room temperature. The γ-phase orthorhombic structure was more stable under ambient conditions.82,83
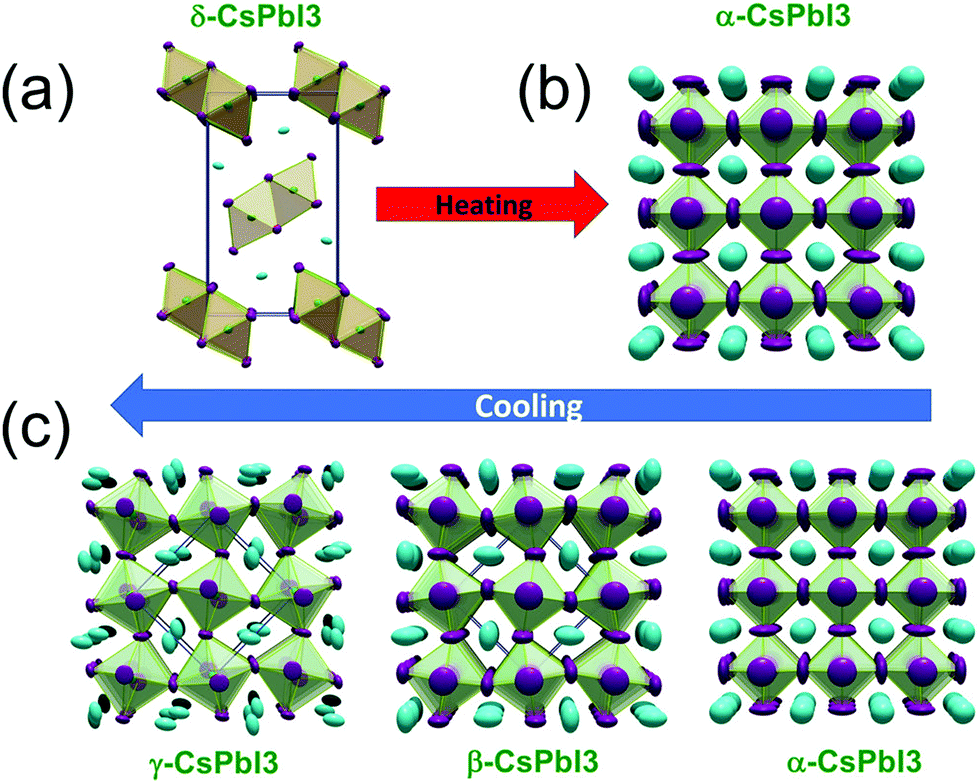 | ||
| Fig. 4 The phase transitions in CsPbI3: (a) the initial yellow non-perovskite δ-phase directly converts to (b) the black perovskite α-phase as the temperature exceeds the transition temperature on heating. (c) Upon cooling, the black perovskite α-phase undergoes various phase changes, namely, the tetragonal structure β-phase, orthorhombic structure γ-phase. These phases are metastable and finally transform into the thermodynamically stable δ-phase upon standing. Reproduced with permission from ref. 79. Copyright©2018, American Chemical Society. | ||
A comparative study of the phase transition in CsPbX3 indicates that the transition from the cubic structure to the tetragonal in CsPbCl3, CsPbBr3, and CsPbI3 takes place at 320 K, 406 K, and 595 K, respectively.68 The greater the size of the halide ion, the higher is the transition temperature. These transitions produce structural distortions in the crystal. The phase transition-induced structural distortions can be divided into three categories: (i) polar displacement of the A or B cations away from their high symmetry positions, (ii) rigid tilting of the corner-sharing BX6 octahedra, and (iii) the collective Jahn–Teller distortion of the BX6 octahedra.68 The corner-sharing BX6 octahedra tilting is associated with the size of the A-site cation and the ratio of the cavity volume to the A-site cation size. The smaller size A cation affects the Goldsmith's tolerance factor (t) and leads to a distortion in the network. Similarly, the larger the volume of the octahedral cavity to the size of the A-site cation ratio, the greater the tilting of the octahedra.
2.3 Stability of CsPbX3
The stability of halides is one of the major concerns that can be changed by modifying its internal structure; i.e., A and B-site cations, halide compositions, size reduction, and surface modification. As discussed, ions at A, B, and X-sites in the ABX3 halide perovskite affects the value of the Goldsmith tolerance factor “t”, and the octahedral factor (μ = rB/rX), which determines the structure formed. The value of t between 0.8 to 0.9 forms the distorted perovskite tetragonal or orthorhombic structure, and between 0.9 to 1.0 most likely forms a cubic structure with the most stable α-phase cubic structure at t = 1. 21,84,85 According to the definition of t, the larger A-site cations favor higher t-values that make the halide perovskite structure more stable.21,84 Cs+ is almost the largest inorganic cation in the periodic table. Therefore, we cannot find a better choice for its replacement in the periodic table for the synthesis of all-IHPs. Consequently, researchers have incorporated some organic cations into the CsPbX3 but it produces a negative effect on the stability of the structure. The t-value greater than 1.0 implies that the size of the B atom is too small or the X atom is too large. Similarly, for t-value less than 0.8, the size of the B atom is too large or the X atom is too small to form a perovskite structure. These conditions lead to the formation of a non-perovskite structure.86 In the ABX3 perovskite structure, the B atom remains situated at the center of the octahedra formed by B and X atoms. The octahedral factor “μ” has an important role in octahedral stability. For the perovskite structure, the μ value should be 0.414 < μ < 0.732.85 It plays a significant role in determining the size of the [BX6]4− octahedral and the cuboctahedral voids. Thus, the factor t and the factor μ are interconnected for the structural determination in all-IHPs. Hence, the substitution of smaller sized cations at the B-site decreases the size of the octahedra and thus increases the t value, which results in an increased stability.86 It has been reported that at room temperature, the stability of the halide perovskite increases from X = I to Br to Cl; i.e., for smaller X-site ions.87 All these halide perovskites are stable at the higher temperatures. The low stability of CsPbI3 at low temperature is because of the tilting of the [PbI6]4− octahedra, where the size of the cuboctahedral void available for the A-site atom is much larger than the Cs+ atom.The stability of all-IHPs is improved to some extent by the use of various organic surface-capping agents. Therefore, different organic compounds have been widely used to stabilize the perovskite structure.88 In the synthesis of CsPbX3 using the hot injection method, the Cs-oleate is injected in situ at elevated temperature into octadecene (ODE) solution containing PbI2, oleic acid (OA), and oleylamine (OAm). The particles are stabilized with both long-chain carboxylic and oleylammonium surface ligands.65 In CsPbI3 QDs, the ammonium ligands are easily lost because of the weak acid–base interactions arising between the I− and oleylammonium during the essential purification procedures, resulting in a fast agglomeration and consequently a phase transformation from the α-cubic phase to a non-perovskite δ-phase.
The stability of the halide perovskite under a humid atmosphere and at elevated temperatures is of great concern. This can be increased by the surface modification by complex layers such as moisture-resistant layer(s), encapsulation, passivation, or by the use of additives.89 Phenylethylammonium chlorine (PEACl), p-phenylenediammonium iodide (PPDI), and phenylethylammonium iodide (PEAI) have been widely used to address these concerns. They also improve the hydrophobic properties of all-IHPs and thereby stabilize the black phase perovskite structure. The hydrophobic alkyl chain or the conjugated heterocycle contained in the organic cations are believed to increase the humidity resistance.90 The XRD patterns of CsPbBrI2 and CsPbBrI2-PEI thin films under different environmental conditions are shown in Fig. 5(a–c). The XRD pattern of the CsPbBrI2-PEI thin film remains unaffected under 80 ± 5% relative humidity (RH) for 0.5 h and under heating up to 85 °C in an N2 glove box for 72 h. Whereas, the CsPbBrI2 treated under the same environmental conditions reflects significant changes in the intensity of XRD peaks, as well as the evolution of random peaks in the XRD pattern, which indicate poor stability under these environments. The significant improvements in the PL intensity for CsPbBrI2-PEI was also observed as indicated in Fig. 5(d). The CsPbBrI2-PEI demonstrated hydrophobic behavior due to the long alkyl chain present at the grain boundaries, as shown in Fig. 5(f). The extent of hydrophobic character was measured by the contact angle and was 81.3° for CsPbBrI2-PEI, and 43.2° for CsPbBrI2. The XRD pattern of the CsPbBrI2 film was completely transformed into the yellow phase under the relative humidity of 85% for 30 min but the CsPbBrI2-PEI pattern remained unchanged, which confirmed the hydrophobicity characteristics as indicated in Fig. 5(e).90 The CsPbBrI2 film suffered phase transition from the α-phase (black color) to the δ-phase (reddish-brown color) due to thermal degradation in the N2 environment. PEI effectively stabilized the CsPbBrI2-PEI film during the annealing process and the α-phase was retained at this temperature as shown in Fig. 5(a–c).90 The van der Waals forces among the PEI alkyl chains increased the crystal formation energy which resulted in enhanced phase stability. Polyethyleneimine (PEI) was used to reduce the defects in the all-IHPs, and it increased the phase as well as the structural stability of the CsPbBrI2 perovskite against humidity and temperature.
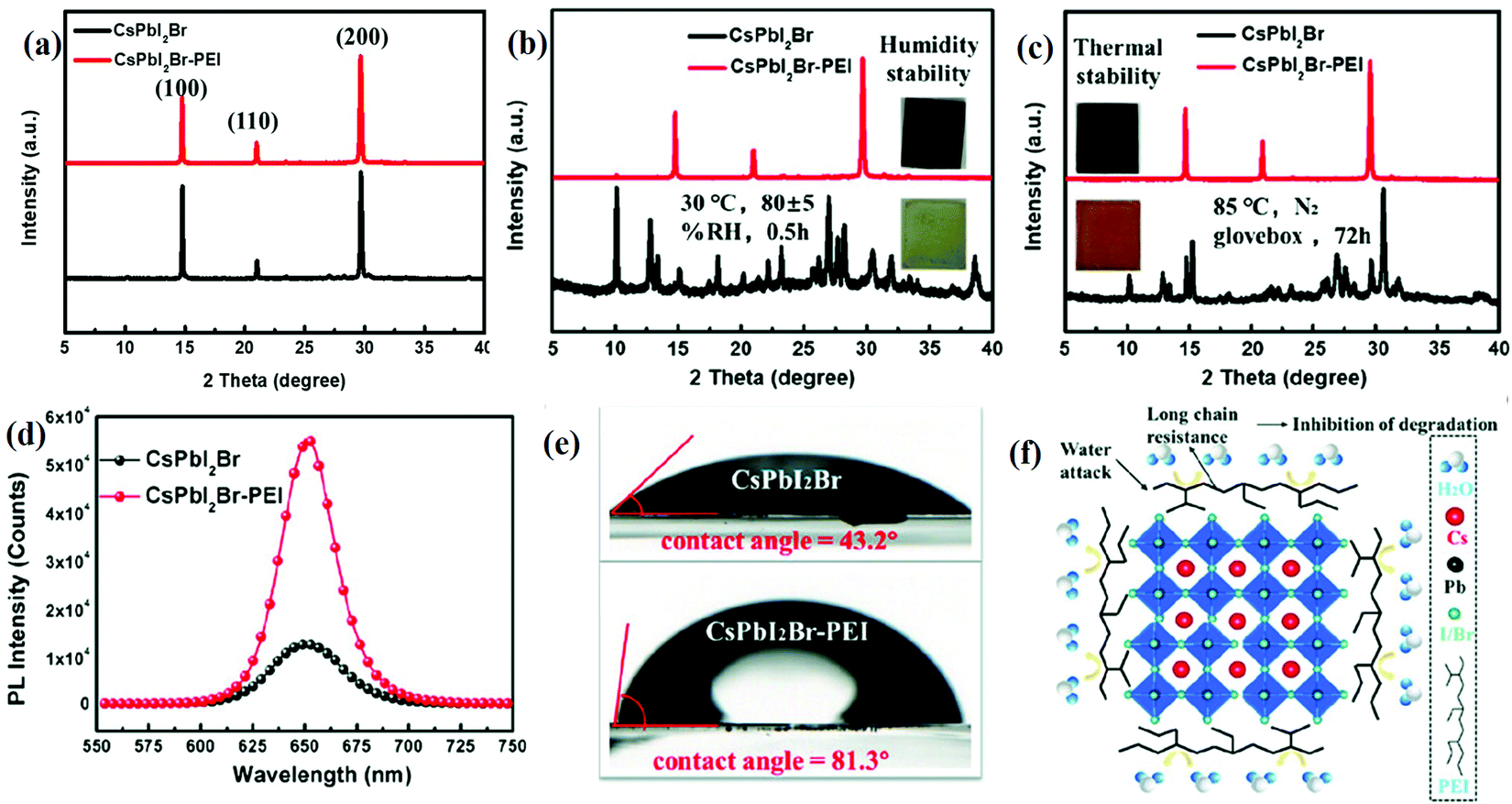 | ||
| Fig. 5 The XRD spectra of the (a) CsPbI2Br and CsPbI2Br-PEI thin-films. (b) CsPbI2Br and CsPbI2Br-PEI films after being exposed to 80 ± 5% RH for 0.5 h (the inset shows images of samples after being exposed to 80 ± 5% RH for 0.5 h). (c) CsPbI2Br and CsPbI2Br–PEI films heated at 85 °C in an N2 glovebox for 72 h (the inset shows images of samples after heating at 85 °C in an N2 glovebox for 72 h). (d) PL spectra of the CsPbI2Br and CsPbI2Br-PEI thin films. (e) The water contact angles of CsPbI2Br film and CsPbI2Br-PEI film. (f) A schematic diagram of the long-chain wrapping of CsPbI2Br grains. Reproduced with permission from ref. 90. Copyright©2020, Wiley Online Library. | ||
Zhang et al.91 used n-butylammonium iodide (BAI) in CsPbBr2I to improve factors like the crystallization and morphology of the structure. The BAI incorporation decreases the trap state density and nonradiative recombination and thereby facilitates preparation at the reduced annealing temperature. An aromatic bi-functional ligand L-phenylalanine (L-PHE) in the in situ management of the present surface ligand environment stabilizes the phase of CsPbI3 by coordinating with the cations and anions on the QD surface.92 The L-PHE incorporated CsPbI3 QDs also show improved PLQYs, and a longer lifetime, which provide longer diffusion length, high stability, and fewer trap states. This is advantageous in many applications like solar cells and LEDs, etc. Liu et al.65 improved the stability of CsPbI3 by using the organolead compound trioctylphosphine-PbI2 (TOP-PbI2) as a precursor. The room temperature quantum yield was up to 100%. The trapping defect centers, which contribute unwanted nonradiative recombination and result in luminescence quenching, are still a major problem in common quantum dots such as CdSe, and PbS, etc. Whereas, the TOP-PbI3 precursor used for the synthesis of the CsPbI3 QDs shows negligible electron or hole-trapping effects. This is evidenced by ultrafast kinetic analysis with time-resolved transient absorption spectroscopy.65,93 In summary, the stability of the perovskite is improved to some extent by the use of various organic surface capping agents. At the same time, dimensionality also affects the stability of the structures.13,87,94 The three black phases of CsPbI3, α, β, and γ are metastable and likely to transform into the non-perovskite black δ phase. Steele et al.94 reported that a thin film of the CsPbI3 black phase is stable as compared to the bulk powder sample. Lower-dimensional perovskites are more stable than bulk 3D perovskite towards humidity.95 This reflects that the stability can be improved by reducing the dimension of the structure.
2.4 Optical properties of CsPbX3
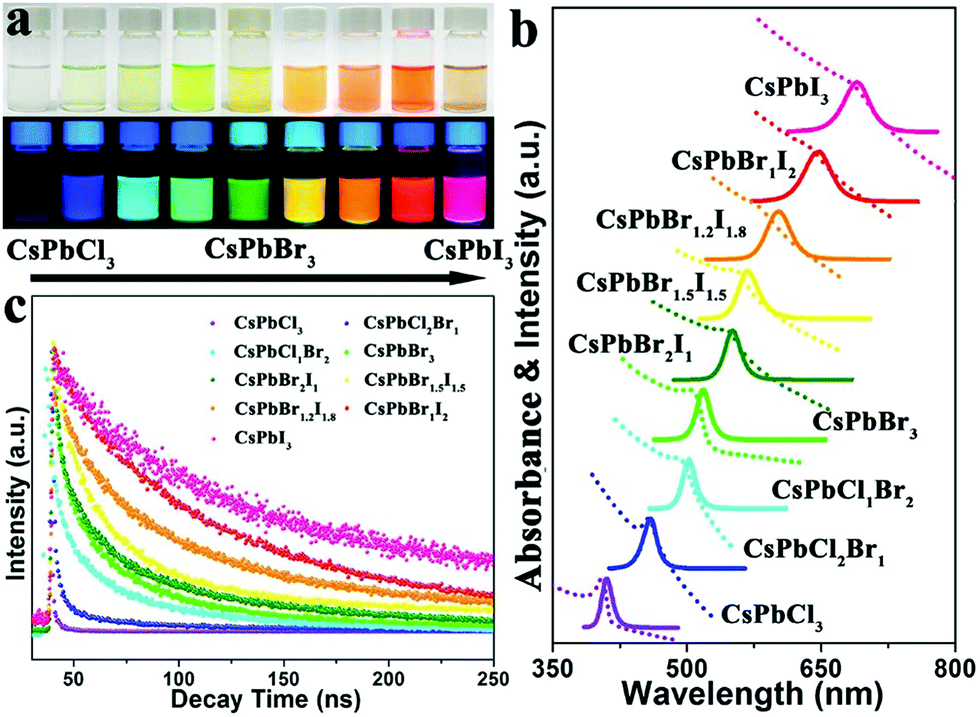 | ||
| Fig. 6 Colloidal CsPbX3 (X = Cl, Br, and I) NCs showing a change in the absorption and emission properties as a function of halide concentration. (a) Photographs of CsPbX3 in hexane solution illuminated under room light (top), and UV lamp (bottom). (b) UV-visible absorption (dotted line), and PL spectra of CsPbX3, and (c) PL lifetime of CsPbX3 NCs. Reproduced with permission from ref. 96. Copyright©2017, American Chemical Society. | ||
| Sample | Emission peak (nm) | FWHM (nm) | PLQY (%) |
|---|---|---|---|
| CsPbCl3 | 410 | 11.7 | 26.0 ± 0.3 |
| CsPbCl2Br | 444 | 15.5 | 35.6 ± 0.4 |
| CsPbCl1.5Br1.5 | 464 | 16.3 | 61.8 ± 0.4 |
| CsPbCl1Br2 | 481 | 17.9 | 61.4 ± 0.8 |
| CsPbBr3 | 520 | 20.3 | 79.8 ± 0.9 |
| CsPbBr2I1 | 587 | 28.2 | 71.2 ± 0.5 |
| CsPbBr1.5I1.5 | 631 | 29.4 | 62.7 ± 0.2 |
| CsPbBr1I2 | 660 | 29.9 | 58.5 ± 0.7 |
| CsPbI3 | 700 | 37.0 | 57.5 ± 0.4 |
Apart from the halide concentration, the size of the NCs also affects the optical properties of all-IHPs.57,101,102 With larger NCs, the emission peak and absorption band-edge shift towards longer wavelength. The size of the NCs also depends upon the cationic and anionic radii, larger ions increase the crystal size and vice versa for the same synthesis conditions. These effects were also observed in all-IHPs QDs. Fig. 7(a–c) show the variation in absorption and emission peaks of CsPbI3 as a function of QDs size, and the corresponding morphologies are indicated in Fig. 7(d–f).103 Similar variations in absorption and emission spectra were also observed in CsPbBr3 QDs and are indicated in Fig. 7(g).57
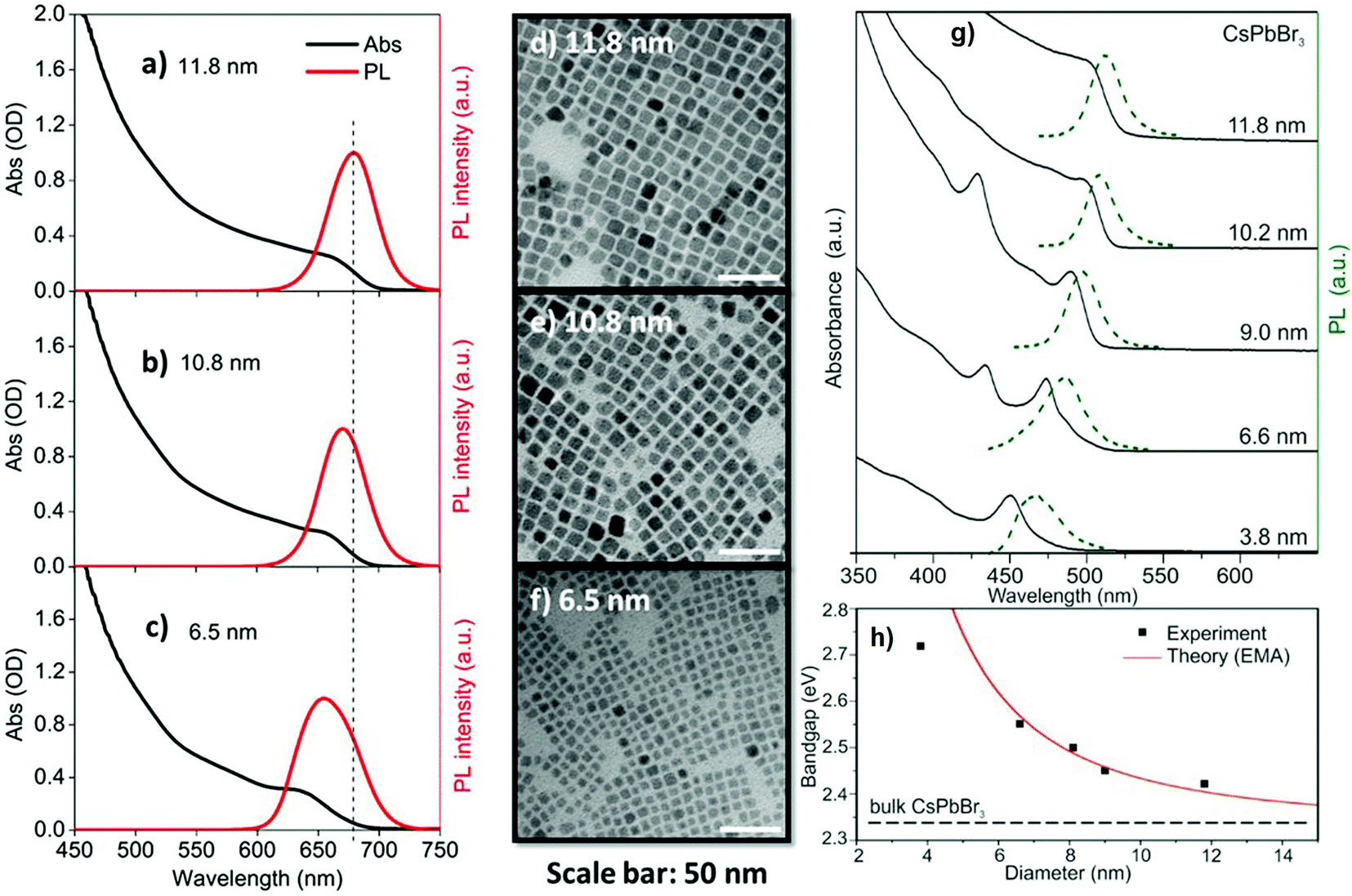 | ||
| Fig. 7 Optical properties and morphology of CsPbI3 perovskite QDs. Absorption (black) and emission (red) spectra [(a–c)] of CsPbI3 perovskite QDs with average sizes of 11.8 ± 1.6 nm, 10.8 ± 1.5 nm, and 6.5 ± 1.1 nm, respectively. (d–f) The corresponding TEM images of three QDs; scale bars in (d–f): 50 nm. Reproduced with permission from ref. 103. Copyright©2019, AIP Publishing. (g) Absorption and emission spectra of CsPbBr3 plotted in the same image as a function of quantum dot sizes. (h) Variation of the bandgap of CsPbBr3 as a function of quantum dot sizes, fitted with the EMA model. Reproduced with permission from ref. 57. Copyright©2015, American Chemical Society. | ||
The smaller the particle size, the higher the PLQYs, which is beneficial for light-emitting diodes.65,104 Whereas, for the perovskite QDs with size less than the exciton Bohr diameter (DB), the PLQYs decrease as observed by Kim et al.105 PLQYs for 5 nm perovskite QDs was about 68%, and 62% for 3 nm. For the small perovskite QDs, the surface-to-volume ratio increased, which promoted the creation of surface traps for the exciton. This led to trap-assisted nonradiative recombination at the surface and resulted in a decrease in PLQYs. The variation in the bandgap with quantum dot size can be modeled using effective mass approximation (EMA), where the confinement energy is given by the following:
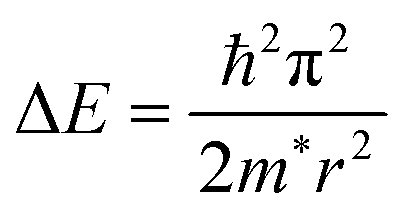 | (2) |
| Perovskite structure | Optical bandgap (eV) | Synthesis techniques |
|---|---|---|
| CsPbCl3 | 3.0 | Chemical vapor deposition110 |
| CsPbBr3 | 2.33–2.34 | Vacuum thermal evaporation62 |
| CsPbBr2I | 2.05 | Dual source thermal evaporation111 |
| CsPbI1.8Br1.2 | 1.88 | Spin coating112 |
| CsPbI2Br | 1.9 | One-step spray-coating113 |
| CsPbI2Br | 1.86 | Spin coating112 |
| CsPbI2.2Br0.8 | 1.83 | Spin coating112 |
| CsPbI2.4Br0.6 | 1.80 | Spin coating112 |
| CsPbI2.7Br0.3 | 1.76 | Spin coating112 |
| CsPbI3 | 1.7 | Spin coating114 |
The variation in the photoluminescence quantum yields (PLQYs) in CsPbX3 with halide composition has been studied by many groups. A literature survey indicated the clear trends of PLQYs from low to high with the partial or complete replacement of the Cl composition by Br, whereas, with the partial or complete replacement of Br by I, the PLQYs decreased. Based on the reported data, we predicted that the PLQY for the Cl-based all-inorganic halide perovskite (CsPbCl3) was low and increased towards the Br-based all-IHPs maximum for CsPbBr3; this again decreased towards I-based all-IHPs. As can be seen from Table 3, the PLQY value of the CsPbCl3 is a minimum of about 26%.58 It increased after the partial replacement of Cl− ions by Br− ions and attained the maximum value of about 80% for CsPbBr3. Further, the substitution of I-ions at Br-sites resulted in a decrease in PLQYs as CsPbI3 shows PLQYs of about 58%. However, the synthesis method and the type of precursor used for the synthesis of the particular CsPbX3 perovskite affected the PLQYs as well as the stability.115–118 Zhang et al.115 used the phenyl-phosphonic dichloride (PhPOCl2) as a “three precursor” source to synthesize the CsPbCl3 NCs using the hot injection (usually two precursors are used) method. The PhPOCl2 precursor-based CsPbCl3 NCs showed 71% PLQYs with increased stability. Similarly, the use of surface capping agents and the dimensionality of the perovskite structure also affects the PLQYs and stability even for the same CsPbX3 compositions.116,119–122 Dutta et al.116 reported the near-unity PLQYs in CsPbX3 (X = Cl, Br, I) NCs. To achieve near-unity PLQYs, the oleylammonium halide was used as a precursor. The oleylammonium halide acts as both a halide source and surface capping ligand and thus helps to increase the PLQYs of CsPbX3 (X = Cl, Br, I) NCs by controlling the reaction temperature. The near-unity PLQYs was also achieved by doping metal ions into the all-IHPs discussed in the lanthanide and other metal ion-doped CsPbX3 section.123,124
The photoexcited exciton lifetime is the time in which the number of electrons in the particular excited state is reduced to 1/e times the maximum population in that state. The radiative lifetime of the CsPbCl3 is low and increases towards a high atomic number halide ion with maximum radiative lifetime for CsPbI3.57,96,125,126 The PL lifetime variation trends in all-IHPs with different halide compositions are shown in Fig. 6(c). The PL decay lifetime for CsPbX3 in the band-edge exciton varies from 1.8–81.1 ns, measured by the pico-second pulsed laser of excitation wavelength 397 nm.58 The PL decay lifetimes for CsPbCl3−x−yBrxIy are 1.8 ns, 3.5 ns, 8.9 ns, 8.2 ns, 21.1 ns, 41.1 ns, 66.2 ns, 75.0 ns, 81.1 ns for (x, y) = (0.0, 0.0), (1.0, 0.0), (1.5, 0.0), (2.0, 0.0), (3.0, 0.0), (2.0, 1.0), (1.5, 1.5), (1.0, 2.0), and (0.0, 3.0), respectively. It was observed that the all-IHPs with a large bandgap (CsPbCl3) decays faster as compared to a low bandgap (CsPbI3).57 The radiative lifetime can be increased by reducing the thermal and concentration quenching effect. Thermal quenching is related to the temperature under analysis. Time-resolved PL study of CsPbX3 QDs, prepared by using the organolead compound precursor trioctylphosphine-PbI2 (TOP-PbI2), showed that the photoexcited exciton lifetime was much longer, which is a clear indicator of the reduction of the quenching defects.65
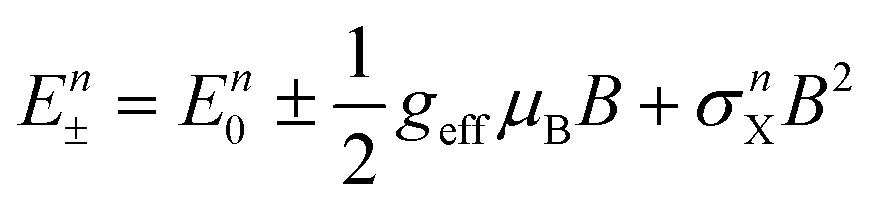 | (3) |
Another important aspect of CsPbX3 is their anisotropic behavior that indicates a change in the different properties in different crystallographic directions. Zhao et al.128 studied the Young's moduli of CsPbI3 and CsPbBr3 along different directions, which were found to be the same. On the other hand, CsPbBrI2 and CsPbBr2I showed the isotropic nature of Young's moduli along the [001] and [010] directions, and along the [100] and [010] directions, respectively (see Fig. 8). In CsPbBr2I and CsPbBrI2, the transport properties (electron and hole mobilities) are superior along [001] in both structures. In this direction, fewer carriers can be captured by the traps during transport. The superior properties of the crystal with [001] orientation may be more suitable for the applications of transistors, light-emitting diodes, photodetectors, solar cells, etc. Optical properties such as the extinction coefficient and refractive index of CsPbBr2I and CsPbBrI2 also show anisotropic behavior. The extinction coefficients of CsPbBr2I and CsPbBrI2 along the [001] direction are greater than that along the [100] direction. Therefore, photon absorption along the [001] direction is greater, resulting in the larger current density and better spectral responsibility that are required in optoelectronic applications.
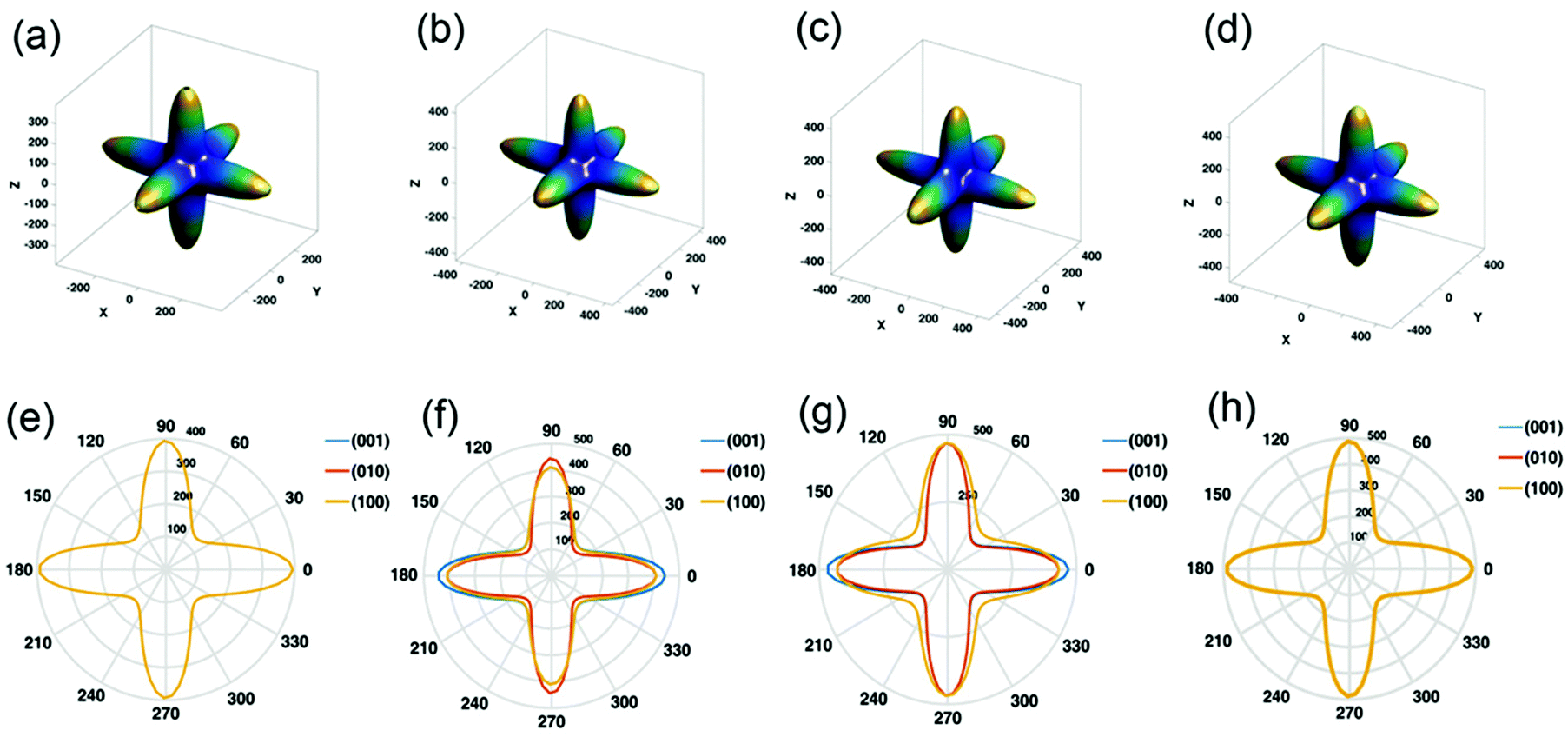 | ||
| Fig. 8 3D Young's modulus: (a) CsPbI3, (b) CsPbI2Br, (c) CsPbIBr2, and (d) CsPbBr3. 2D Young's modulus: (e) CsPbI3, (f) CsPbI2Br, (g) CsPbIBr2, and (h) CsPbBr3. Reproduced with permission from ref. 128. Copyright©2020, Wiley Online Library. | ||
The anisotropy-dependent photo-response properties in CsPbBr3 single-crystal thin-film have been studied by Wang et al.89 The CsPbBr3 film showed anisotropic behavior towards absorption and emission spectra, also verified by using first-principles studies. The absorption and emission intensity of CsPbBr3 film on ZnSe were recorded using circularly polarized light in different polarization directions. The absorption and emission reached their maximum values for the polarization angle 0° while the minimum was observed for 90°. On moving the polarization angle from 0–180°, these again reached a maximum value at 180°. The polarization-dependent photosensitivity of the sample reported was 200 A W−1, showing a fast photo-response time of 20 ms, and the current ratio for the on/off mode was about 104. The anisotropy-dependent absorption, emission and photoresponsivity of the film are indicated in Fig. 9(a), (b) and (c), respectively.
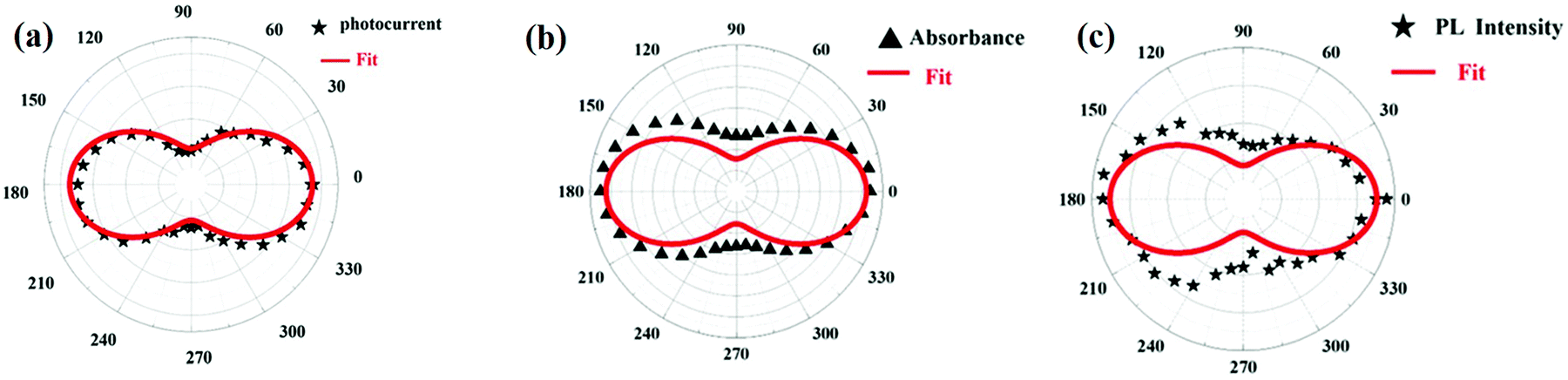 | ||
| Fig. 9 The polarization dependence polar plot of (a) absorption and (b) PL spectra for CsPbBr3 film. Black triangles and stars represent the experimental data and solid red lines are fitted curves. (c) The polarization effect on current intensity (bias voltage is 5 V and light intensity is 60 mW cm−2). Reproduced with permission from ref. 129. Copyright©2019, Wiley Online Library. | ||
The anisotropy-dependent optical parameter variations were also observed in the nanowires (NWs).130 The PL of CsPbBr3 NWs was studied by applying a polarized excitation field E0 perpendicular to the long axis of CsPbBr3 NWs. The PL spectra of 15 nm thick and 250 nm thick NWs excited at 405 nm for polarization angles of 0° and 90° are shown in Fig. 10(a). The polar plots of the polarization-dependent PL intensity of 15 nm thick and 250 nm thick NWs excited at 405 nm are shown in Fig. 10(b) and (c), respectively. Fig. 10(d and e) are the respective optical PL images of two NWs of thickness 15 nm (scale bar 250 nm), and 250 nm at different polarization directions. It was observed that the PL intensity in the parallel polarization is five times higher than the perpendicular polarization for 15 nm thick NWs, which confirmed the anisotropy effect. The thickness-dependence study of NWs showed that higher anisotropy was attained for 15 nm thick NWs as compared to 250 nm thick NWs.
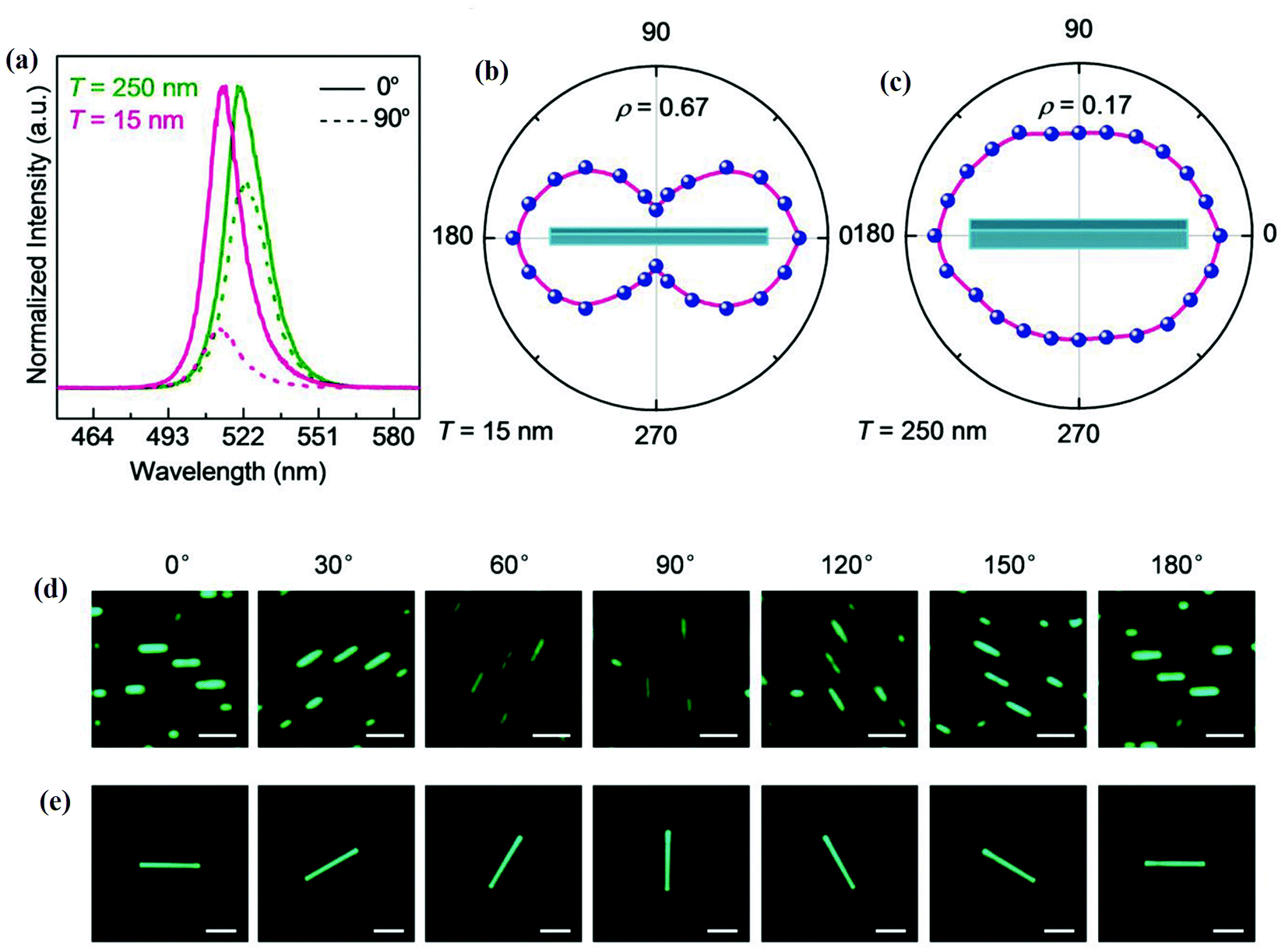 | ||
| Fig. 10 (a) PL spectra; (b) and (c) polarization dependence PL intensity polar plots (polarization angle 0° represents the polarization field parallel to the NWs) of 15 nm and 250 nm NWs excited at 405 nm. The optical PL images of NWs at different polarization directions of NWs thickness (d) 15 nm (scale bar 250 nm), and (e) 250 nm (scale bar 3 μm). Reproduced with permission from ref. 130. Copyright©2018, Wiley Online Library. | ||
3. Metal ion-doped CsPbX3
All IHPs have been doped with different types of metal ions such as alkali metals, alkaline earth metals, transition and post-transition metals, and lanthanides. Among these metal cations, lanthanide doping is unique in the sense that they exhibit discrete emission in a wide spectral region ranging from the UV-visible to the near-infrared (NIR) region. These additional optical features bring multifunctionality to IHPs, particularly for emerging applications like NIR and white light-emitting diodes, NIR emitters, NIR cameras, optical temperature sensing, optical data encoding, etc. On the contrary, non-lanthanide metal doping only has been used to improve the existing applications, particularly for LEDs/WLEDs and solar cells. In this review, we have covered metal doping in IHPs in two different sections, i.e., non-lanthanide metal-ion-doped IHPs (CsPbX3) and lanthanide-ion-doped IHPs. However, the main focus is to highlight the effect of lanthanide doping in more detail.3.1 Non-lanthanide metal ion-doped CsPbX3
Metal ions (M-ion), other than lanthanides, such as alkali metal cations Li+,131 Na+,132 K+,133,134 and Rb+,135 alkaline earth metal cations Mg2+,136 and Ca2+,137 transition metals Zn2+,138,139 Mn2+,140–143 Ni2+,144,145 Cu2+,146,147 Ti3+,148,149 Y3+,150 and Cd2+,124 post-transition metal Sn2+,151etc., have also been incorporated with the all-IHPs. These metal ions also improve the optoelectronic properties and the stability of IHPs against various environments. The alkali metal cations doped into the all-IHPs take the A-sites, i.e., the Cs+ position, whereas the alkaline earth metal cations, transition metals, and post transition metals take the B-site i.e., the Pb2+ position. B-site doping largely affects the optoelectronic properties as compared to A-site doping.152 The Mn2+ doping in place of Pb2+ into the CsPbX3 reduces the toxicity and increases the PLQYs and stability. As explained, the α-CsPbI3 perovskite, which shows good optoelectronic behavior, is unstable and is most likely to transform into the non-perovskite phase δ-CsPbI3 having poor optoelectronic properties. Mn2+ doped into CsPbI3 NCs stabilizes the α-CsPbI3 phase.153 It also improves the stability against UV irradiation as compared to the undoped CsPbI3 NCs. It exhibits band edge excitonic d-d transitions and results in orange luminescence bands. However, Mn2+-ion incorporation gives new emission bands, which result in impure light emission that limits practical applications. Sn2+ also takes the Pb2+ site and is less toxic than Pb2+; however, Sn2+ is highly unstable and is oxidized to Sn4+.3.1.1.1 Alkali metal ion-doped CsPbX3. The alkali metal ions such as Li+,131 Na+,132 K+,133,134 Rb+,135etc., have been doped in CsPbX3 to improve the PLQYs. The alkali metal ion takes the Cs+ position instead of Pb2+. The doping concentration of alkali metal ion in CsPbX3 improves the PLQYs up to a certain concentration limit, after which the PLQYs decreases. The average lifetime also varies in the same fashion as PLQYs with the doping of alkali metal ions in different concentrations. The PLQYs and the average lifetime of 1%Na:CsPbCl3 QDs are 56.8% and 26.5 ns, respectively.132 Both the PLQYs and the lifetime values gradually increase up to a doping of 4% Na ions and the maximum values are attained at 4% Na doping, i.e., 94.2% and 32.2 ns, respectively. Beyond 4% doping concentration of Na, the PLQYs and average lifetime both decrease and the values dropped to 50.4% and 13.1 ns, respectively for 7%Na:CsPbCl3 QDs. Similarly, the optimized doping concentration for Rb+ ions was 9% in CsPbCl3 for the maximum PLQYs and average lifetime.
3.1.1.2 Alkaline earth metal ion-doped CsPbX3. The alkaline earth metal ions are among the dopants for all-IHPs, which are used to improve the optoelectronic properties of the pristine samples. Alkaline earth metal cations take the Pb2+ position instead of Cs+, and have a large influence on the optoelectronic properties as compared to alkali metal ions that take the Cs+ position.152 Chen et al.154 reported that there was no shift in the diffraction peaks for Mg2+, Ca2+ and Sr2+-doped CsPbCl3 NCs as compared to pristine CsPbCl3 NCs, which confirmed that there is no structural change after doping. However, a slight shift in the diffraction peaks towards low angles for Ba2+-doped CsPbCl3 NCs was observed. Since the ionic radius of Ba2+ (1.35 Å) is larger than Pb2+ (1.19 Å), it is less probable that Ba2+ can take the Pb2+ position. However, Chen et al.154 confirmed experimentally and theoretically that Ba2+ can enter the core region of the lattice of NCs. Because of the large ionic radius, it produces more stress and lattice distortion and consequently, the diffraction peak shifts to lower angle as observed in the XRD pattern of Ba2+–CsPbCl3 NCs. The PLQYs of the Ca2+-doped CsPbBr3 NCs was higher as compared to the undoped sample.137,154 The X-ray diffraction (XRD) pattern of pristine CsPbX3 NCs and doped Ca2+:CsPbX3 NCs at different doping concentrations are shown in Fig. 11(a) and (b), respectively for X = Br, and X = Br1I2. Fig. 11(c) shows the PL spectra of pristine CsPbBr3 NCs and doped Ca2+:CsPbBr3 NCs, whereas Fig. 11(d) represents PL spectra of both pristine CsPbBr1I2 NCs and doped Ca2+:CsPbBr1I2 NCs. For the doping concentration of Ca2+-ion up to 40%, the PLQYs of the CsPbBr3 NCs as well as CsPbBr1I2 NCs increase with the concentration.137 For example, the values were 37.9% and 72.3% for Ca2+-ion doping concentrations of 10% and 40%, respectively in CsPbBr3 NCs, whereas the values were 44.0% and 78.7%, respectively in CsPbBr1I2 NCs. Further, for the doping concentrations of Ca2+-ion above 40%, a decrease in PLQYs was observed in both cases and values were 64.5% and 67.7%, respectively in CsPbBr3 NCs and CsPbBr1I2 NCs for Ca2+-ion concentration of 50%.
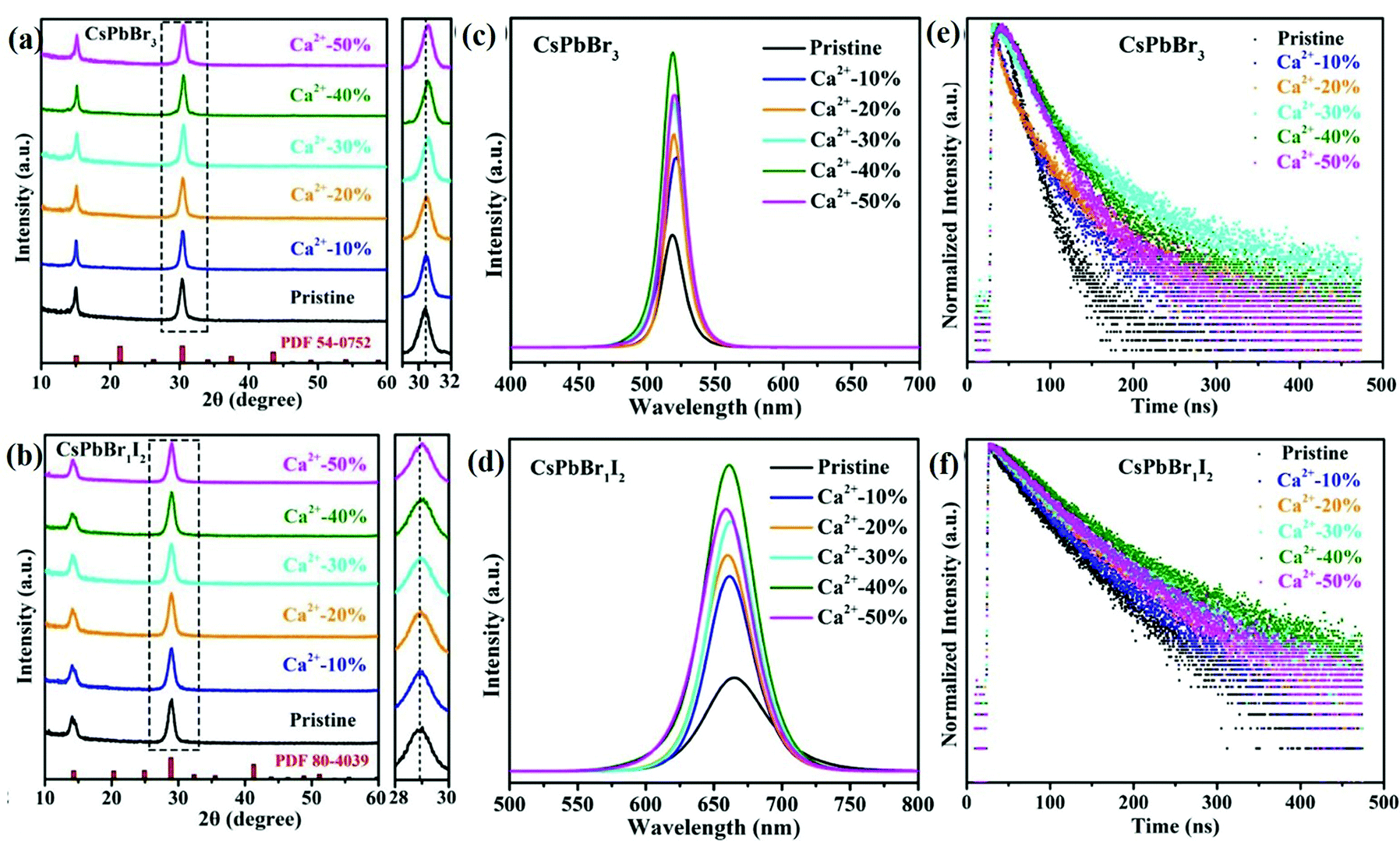 | ||
| Fig. 11 XRD patterns of (a) pristine CsPbBr3 NCs and Ca2+–CsPbBr3, (b) pristine CsPbBr1I2 NCs and Ca2+–CsPbBr1I2 NCs with different Ca2+ concentrations (enlarged views of the peaks marked by the dotted outline are shown in the right panels). PL spectra of (c) pristine CsPbBr3 NCs and Ca2+–CsPbBr3, (d) pristine CsPbBr1I2 NCs and Ca2+–CsPbBr1I2 NCs. PL lifetimes of (e) pristine CsPbBr3 NCs and Ca2+–CsPbBr3, (f) pristine CsPbBr1I2 NCs and Ca2+–CsPbBr1I2 NCs. Reproduced with permission from ref. 137. Copyright©2021, Elsevier. | ||
The same variation trend was also observed for the average lifetimes indicated in Fig. 11(e and f). The average lifetimes were 49.45 ns and 108.15 ns for Ca2+-ion doping concentrations of 10% and 40%, respectively, in CsPbBr3, whereas it was 80.07 ns and 146.25 ns, respectively in CsPbBr1I2. The doping concentration of Ca2+ ions above 40% showed a decrease in the average lifetime in both cases. The average lifetimes were 98.96 ns and 106.99 ns, respectively in CsPbBr3 and CsPbBr1I2 for Ca2+ ion concentration of 50%. Thus, the PLQYs and average lifetimes are higher in CsPbBr1I2 as compared to CsPbBr3 for the same doping concentration of Ca2+ ions. This result indicates that the PLQYs and average lifetime can be increased by the partial or complete substitution of Br or I in place of Cl in CsPbCl3 NCs. The optimized Ca2+ doping concentration for the high PLQYs and average lifetime in the host CsPbCl3 was 10.5%.154 For the higher doping concentration, the PLQYs and average lifetime decreased in a similar way as discussed above. Similarly, the optimized doping percentage of Mg2+ in CsPbCl3 was 37.9% at which the maximum PLQY value and average lifetime were observed and were 75.8% and 23.931 ns, respectively. For Mg2+ doping concentration higher than 75.8%, both the PLQYs and average lifetime decrease.136
3.1.1.3 Transition metal ion-doped CsPbX3. Transition metals such as Zn2+,138,139 Mn2+,140–143 Ni2+,144,145 Cu2+,146,147 Cd2+, Ti3+,148,149 Y3+,150 Cd2+,124etc., have been used as dopant elements for all-IHPs. The Mn2+ ion has been widely used for improving the PLQYs and stability and to reduce the toxicity of Pb-based all-IHPs. The schematic representation of Cs(Pb/Mn)Cl3 is shown in Fig. 12(a). The XRD patterns of Mn2+–CsPbCl3 QDs with different doping concentrations are shown in Fig. 12(b). Mn2+ doped into the CsPbCl3 exhibited two emission peaks; a narrow emission peak at around 400 nm (due to exciton recombination) and a broad band emission at 600 nm by the Mn2+ intrinsic band edge transition as shown in Fig. 12(c).140 These two peaks appeared at 460 nm and 610 nm for the Mn2+–CsPbBr3 nanoparticles.155 The orange luminescence was observed at 600 nm in the PL spectra due to the spin-forbidden d–d transition in Mn2+.140,143 The intensities of the emission peaks at 406 nm and 600 nm can be tailored by the Mn2+ doping concentration.140,156 For doping up to 20.4% Mn2+ (Mn
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Pb = 2:5) in CsPbCl3, the intensity corresponding to 406 nm was much higher as compared to the 600 nm emission peak as indicated in Fig. 12(c).140 For 37.6% Mn2+ (Mn:Pb = 1:2) doping, both emission peaks showed comparable PL intensities. At higher doping concentrations, the intensity of the emission peak at 406 nm was significantly reduced, whereas, the intensity of the emission peak at 600 nm increased.140,156 The increase in the intensity of the 600 nm emission peak at higher doping concentration was due to the effective energy transfer from CsPbCl3 to Mn2+.140 However, in the Mn2+-doped CsPbI3 NCs only one emission peak at 679 nm was reported and the peak position varied with the doping concentration.153
:Pb = 2:5) in CsPbCl3, the intensity corresponding to 406 nm was much higher as compared to the 600 nm emission peak as indicated in Fig. 12(c).140 For 37.6% Mn2+ (Mn:Pb = 1:2) doping, both emission peaks showed comparable PL intensities. At higher doping concentrations, the intensity of the emission peak at 406 nm was significantly reduced, whereas, the intensity of the emission peak at 600 nm increased.140,156 The increase in the intensity of the 600 nm emission peak at higher doping concentration was due to the effective energy transfer from CsPbCl3 to Mn2+.140 However, in the Mn2+-doped CsPbI3 NCs only one emission peak at 679 nm was reported and the peak position varied with the doping concentration.153
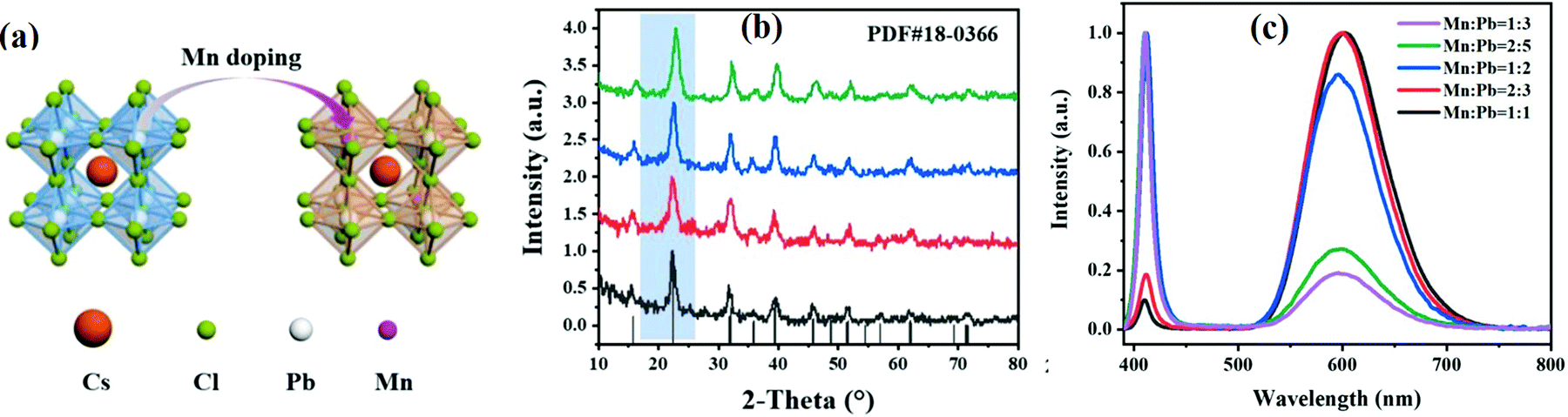 | ||
| Fig. 12 (a) Schematic representation of Cs(Pb/Mn)Cl3. (b) XRD pattern of the Mn2+-doped CsPbCl3 QDS for Mn to Pb ratios of 1:3 (black), 2:5 (red), 1:2 (blue), 1:1 (green). (c) PL spectra of the Mn2+:CsPbCl3 QDS prepared at different Mn:Pb ratios. Reproduced with permission from ref. 140. Copyright©2021, Elsevier. | ||
The PLQYs of the 5.0% Mn2+-doped CsPbI3 increased to 98%. To reduce the toxicity effect of Pb2+, a high doping percentage of non-toxic metal is required. Taking toxicity into consideration, Guo et al.139 reported the optimized doping of (Mn + Zn) into CsPbBr3. On the basis of “t” and “μ” values within the structural stability limit, the maximum values of y and (x + y) in CsPb1−x−yMnxZnyBr3 were 25% and 83%, respectively. However, (Mn + Zn) co-doping (x + y) beyond 50% reduced the visible optical absorption, defect tolerance, and carrier lifetime. Therefore, the maximum doping percentage of (Mn + Zn) in CsPb1−x−yMnxZnyBr3 was 50% without reducing the optoelectronic properties of the pristine CsPbCl3 film. The Mn2+ co-doping with Cu2+ in CsPbCl3 was also reported.49 The Mn2+–Cu2+ co-doped CsPbCl3 NCs boosted the emission efficiency and 8% PLQYs was achieved.
3.1.1.4 Post transition metal ion-doped CsPbX3. Post transition metal Sn2+ is also used as a dopant for all-IHPs.151 A major issue with the Sn2+-doped all-IHPs is related to its instability since it is readily oxidized to Sn4+, which limits the practical application of Sn2+-doped all-IHPs. However, researchers have adopted different synthesis approaches with different initial precursors to enhance the stability of Sn2+-doped all-IHPs. The variation trend for the PLQYs and average lifetime with the Sn2+ doping concentration in all-IHPs is the same as for other metal doped all-IHPs. As reported by Yan et al.,151 initially, the PLQYs and the average lifetime of the Sn2+-doped CsPbBr3 increased up to the doping concentration of 20% with maximum PLQYs and average lifetime values of 82.77% and 25.26 ns, respectively. For higher Sn2+-doping concentrations, both values decreased. Table 5 summarizes the optoelectronic properties of non-lanthanide metal ion-doped all-IHPs at optimized doping concentrations. However, the listed optoelectronic values of doped CsPbX3 are predominantly affected by the synthesis methods, precursors used for the synthesis, reaction temperature, reaction time, use of capping ligand, and the doping of different metal ions, etc.
| Doping ion | M:CsPbX3 | Band gap (eV) | Emission peak (nm) (for λexi (nm)) | Average lifetimes (ns) | Overall PLQYs (%) | Year |
|---|---|---|---|---|---|---|
| Li+ (NA) | 30.0%Li:CsPbBr3 NCs | — | 520 | 229.80 | 40.0 | 2021131 |
| Na+ (NA) | 4.0%Ca:CsPbBr3 QDs | — | 514 (372) | 32.20 | 94.2 | 2021132 |
| K+ (NA) | 4.0%K:CsPbCl3 QDs | 3.05 | 410 (365) | 12.98 | 10.3 | 2018133 |
| Rb+ (NA) | 9.0%Rb:CsPbCl3 QDs | 3.05 | 407 (365) | 10.20 | 13.0 | 2018135 |
| Mg2+ (0.72 Å) | 37.9%Mg:CsPbCl3 NCs | 3.084 | 401 | 23.931 | 75.8 | 2021136 |
| Ca2+ (1.0 Å) | 10.5%Ca:CsPbCl3 NCs | — | 407 | 17.86 | 77.1 | 2019154 |
| 40.0%Ca:CsPbBr3 NCs | — | 520.9 | 108.15 | 72.3 | 2021137 | |
| 40.0%Ca:CsPbBr1I2 NCs | — | 662.3 | 146.25 | 78.7 | 2021137 | |
| Cu2+ (0.73 Å) | 2.32%Cu:CsPbCl3 QDs | — | 412 (365) | — | 51.0 | 2021147 |
| Mn2+ (0.97 Å) | 37.6%Mn:CsPbCl3 QDs | 406, 600 | 10.702 | — | 2021140 | |
| x%Mn:CsPbBr3 NPs | — | 460, 610 (365) | 21.66 | 64.4 | 2021155 | |
| x%Mn:CsPb(Br/Cl)3 QDs | 463, 602 (365) | 2.66 | 72.5 | 2021141 | ||
| Ti3+ (0.67 Å) | 9.67%Ti:CsPbCl3 NCs | — | 410 | 22.75 | 48.4 | 2021149 |
| Ni2+ (0.69 Å) | 11.9%Ni:CsPbCl3 NCs | — | 407 | 18.39 | 96.5 | 2018144 |
| Cd2+ (0.93 Å) | x%Cd:CsPbCl3 NCs | — | 406 (375) | — | 98.0 | 2018124 |
| Sn2+ (NA) | 30.0%Sn:CsPbBr3 QDs | 514 | 25.26 | 82.77 | 2021151 |
Researchers have used various metal ion-doped all-IHPs for the fabrication of LEDs using various structural layers. The metal ion-doped LEDs were summarized in detail by Gao et al.157 It was reported that Zn2+-doped CsPbI3 with the LED structure ITO/ZnO/PEI/Zn:CsPbI3/TCTA/MoO3/Au showed a external quantum efficiency of 15.1%.158 The maximum luminescence described was 40000 cd m−2 for the Na+-doped CsPbBr3 with ITO/PeDOT:PSS/perovskite/TPBi/LiF/Al LED structure.159 Whereas, in the Mn2+-doped CsPbBr3, the maximum current efficiency of 6.41 cd A−1 was observed with layered structure ITO/PeDOT:PSS/poly-TPD/Mn:CsPbBr3/TPBi/LiF/Al.160
WLEDs have been constructed mainly by combining non-lanthanide metal ion-doped CsPbX3 with phosphor materials and LED chips. The CIE coordinates, luminescence efficiency (LE), color rendering index (CRI), and color temperature (CCT) of the non-lanthanide metal ion-doped CsPbX3 are listed in Table 6.
| WLED structure | CIE coordinate | LE (lm W−1) | CRI | CCT (K) | Year |
|---|---|---|---|---|---|
| Li:CsPbBr3 + Eu:BaMgAl10O17 + Eu:CaAlSiN3 | (0.3318, 0.3383) | — | — | 5523 | 2021131 |
| Y:CsPbBr1I2 + Ce:Y3Al5O12 + InGaN LED chip | (0.34, 0.35) | 61 | 83 | — | 2022150 |
| Ca:CsPbBr1I2 + Ca:CsPbBr3 + InGaN LED chip | (0.3366, 0.3376) | 92.4 | 92.2 | — | 2022137 |
| Mn:CsPb(Br/Cl)3 + InGaN LED chip | (0.39, 0.38) | — | — | — | 2021141 |
| Ni:CsPbBrxI3−x + Ce:YAG InGaN LED chip | (0.3621, 0.3458) | 113.20 | 94.9 | 4336 | 2021161 |
| Zn:CsPbBr3 + Mn:K2SiF6 + blue LED chip | (0.327, 0.336) | 36.0 | — | 5760 | 2021162 |
| Sn:CsPbBr3:AgInZnS:InGaN LED chip | (0.41, 0.48) | 43.2 | 89 | — | 2021151 |
Recently, Gao et al.157 summarized metal ion-doped all-IHPs solar cells also consisting of different combinations of structural layers. Comparative analysis indicated that Ca2+-doped CsPbBr1I2 had high solar cell parameter values with the solar cell structure FTO/TiO2/Ca:CsPbBr1I2/spiro-OMeTAD/Au. The photo-conversion efficiency (PCE), short circuit current (Jsc), open-circuit voltage (Voc) and fill factor (FF) of the FTO/TiO2/Ca:CsPbBr1I2/spiro-OMeTAD/Au structure were 16.79%, 15.32 mA cm−2, 1.32 V, and 83.29%, respectively.52 Other metal ion-doped all-IHPs also showed significant results for solar cell applications, and can be improved by selecting the different structural layers.
3.2 Lanthanide-doped CsPbX3: novel properties and emerging applications
Charge neutrality consideration: at first the basic conditions that should be fulfilled while doping with another ion are charge neutrality and the size of the dopant. The charge neutrality condition is that the cationic and anionic charges should be the same for the charge balance. However, in some cases, the presence of excess charge is also beneficial for particular applications like solar cells for the charge transport layer. Concerning charge neutrality, lanthanides are remarkable candidates for substitution into the ABX3 perovskite structure because of their comparable ionic radii with the Pb2+ ion (1.19 Å). Hence, in the doping process, the lanthanide replaces the Pb2+ ion because of its comparable ionic radius.164 This could be confirmed experimentally by high-resolution XPS spectra. For example, the high-resolution XPS spectra indicated a decrease in the binding energy of Pb2+ 4f5/2, and Pb2+ 4f7/2 when Eu3+ ions were doped.165 There was very little variation in the binding energies of Cs3+ 3d3/2, and Cs3+ 3d5/2. In Cl− 2p1/2 and Cl− 2p3/2, variation in binding energy was also very small. This indicates that Eu3+ ions occupy the Pb2+ lattice sites in the host lattice of CsPbCl3 instead of Cs+ and Cl− sites.
Realization of FRET pairs: lanthanide-doped nanoparticles and IHPs QDs can make a FRET (fluorescence resonance energy transfer) pair. In this process, the lanthanide-doped NPs convert low-energy photons (e.g., IR-photons) into photons of high energy through the UC process.166,167 These high energy photons are re-absorbed by the perovskite QDs through FRET and thereby, the electron and hole pairs are generated in the perovskite QDs. Finally, the emission of photons takes place from perovskite QDs through exciton recombination. Thus, the emission from perovskite QDs in principle can be achieved through IR excitation, which opens up a new area of applications.58,167
The FRET process in lanthanide-doped nanoparticles to perovskite QDs requires effective distance control between them, which is a difficult task. Further, the quenching of the upconversion (UC) luminescence intensity by QDs also depends on the absorbance coefficient of the perovskite QDs. Thus, the energy transfer efficiency (ηETE) depends on both the position of the energy level and the effective distance between the emitter and the absorber.58
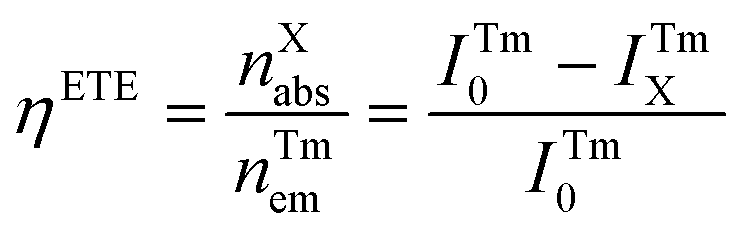 | (4) |
| Doping ion | N/A | Ce3+ | Sm3+ | Eu3+ | Tb3+ | Dy3+ | Er3+ | Yb3+ |
|---|---|---|---|---|---|---|---|---|
| a Emission decay was monitored at 410 nm. | ||||||||
| Ionic radius of dopant (Å) | N/A | 1.034 | 0.964 | 0.950 | 0.923 | 0.881 | 0.908 | 0.858 |
| Lattice constant (Å) | 3.96 | 3.94 | 3.93 | 3.92 | 3.90 | 3.89 | 3.88 | 3.87 |
| Absorption peak (nm) | 400 | 395 | 390 | 392 | 390 | 382 | 381 | 375 |
| Band gap (eV) | 2.87 | 2.91 | 2.94 | 2.98 | 3.03 | 3.05 | 3.08 | 3.10 |
| Excitation peak (nm) (for λem (nm)) | 368 (410) | 385 (430) | 470 (603) | 360 (620) | 400 (555) | 372 (575) | 400 (548) | 390 (982) |
| Average lifetimesa (ns) | 4.1 | 3.7 | 6.2 | 6.7 | 7.1 | 7.6 | 8.2 | 9.1 |
| Overall PLQYs (%) | 3.8 | 24.3 | 14.1 | 27.2 | 31.2 | 27.6 | 15.1 | 142.7 |
Multifunctionality and emerging applications: the halide perovskites were initially synthesized for perovskite-based solar cells because of their exceptional optoelectronic properties leading to the high efficiency of solar cells.168 The presence of volatile organic cations in the structure, however, reduces its stability. Therefore, efforts have been made to increase their stability and also to explore these optoelectronic properties for some other novel applications. One of the approaches for making them multifunctional involves the incorporation of lanthanide ions into the perovskite structure.169,170 This not only makes halide perovskite suitable for extended applications, but also increases their stability and improves the efficiency of solar cells.123,171,172 Huang et al.173 fabricated NIR light-emitting diodes by Yb3+-doped CsPbCl3 perovskite nanocrystals with excellent long-term stability. Zhang et al.174 reported Yb3+-doped CsPbCl3 nanocrystals that emit strong NIR light at 986 nm and the Yb3+/Er3+ co-doped CsPbCl3 nanocrystal has emission at 1533 nm. The PL quantum yield also increased from 5% to 127.8% on doping 2% Yb3+-into CsPbCl3. This shows a high NIR output irradiance of 112 mW cm−2@400 mA, which may have great potential in the future as a light source in night-vision cameras. The Eu3+ ion doped CsPbI2Br solar cell was reported by Chen et al.99 Solar cell efficiency of 12% was achieved with good thermal stability. It was also reported that the Eu3+ ion doping into CsPbI2Br stabilized the γ-CsPbI2Br phase, which also increased the device performance. The CsPbBr3 perovskite showed up-conversion phenomena by using BaYF5 as a sensitizer.167 The nano-composite of CsPbI3 and CaF2:Yb3+/Ho3+ showed both up-conversion and down-conversion behavior.166 This can be used in dual-mode bio-imaging applications.175 The newly emerging applications of lanthanide-doped all-IHPs are in improved solar cell applications (Section 3.2.4.1), the generation of WLEDs (Section 3.2.4.2), NIR-light emitters, NIR LEDs, and NIR cameras (Section 3.2.4.3), optical temperature sensing (Section 3.2.4.4), and as optical encoding material (Section 3.2.4.5).
The I−-based halide perovskite showed phase transformation from the perovskite structure α to β then to γ and finally to the non-perovskite structure δ phase. Jena et al.179 showed that the incorporation of Eu3+ ions stabilized the black α-CsPbI3 phase with improved photovoltaic performance. Zhang et al.174 reported that the CsPbCl3:Yb3+ NCs solution showed longer photostability than undoped CsPbCl3 NCs. To demonstrate this, both samples were placed under the continuous irradiation of a 6 W UV (365 nm) lamp and the PL was recorded as a function of time. The undoped CsPbCl3 NCs showed 80% decrease in PL intensity in 27 h, whereas, the same amount of decay in intensity was observed in 85 h for Yb3+-doped CsPbCl3 NCs. The lanthanide ions occupied the Pb2+ ion sites and also reduced the Cs+ ion vacancies.180 This in turn reduced the defect and trap states. Since the stability of halide perovskites and improving the efficiency of optoelectronic devices are still among the focus areas of research, further investigations on the integration of lanthanide ions with halide perovskites are expected to bring exciting outcomes.
3.2.3.1 The effects on UV-visible absorption and photoluminescence. The variation in the optical bandgap affects the band-to-band PL of semiconducting materials. The substitution of smaller ions (all lanthanides after Gd3+) for the Pb2+ ion leads to lattice shrinkage. This causes an increased ligand field within the [PbBr6]4− octahedra, leading to an increase in the bandgap. This effect can be monitored by recording the UV-visible absorption spectrum, which shows a shift in the absorption peak towards higher energy, i.e. a blue shift.181 Lanthanide ion doping also increases the stability of the perovskite structure. The effects of lanthanide ion doping on the structural and optical properties in CsPbCl3 were determined by Pan et al.165 The XRD patterns of the pristine CsPbCl3 NCs and Ln3+-doped CsPbCl3 NCs are shown in Fig. 13(a). These revealed that lanthanide ion doping preserves the structure of the host but the average crystallite size changes.165 The average crystallite size decreases linearly towards the higher atomic number of lanthanide ions.171 This also induces a blue shift in the PL spectra, i.e., an increase in the binding energy of the structure due to quantum confinements.165,171,182Fig. 13(b) shows that doping smaller-sized lanthanide ions into CsPbCl3 resulted in the blue shift of the absorption edge.165 This blue shift was also observed in the emission spectrum, indicating the increased bandgap of Ln3+-doped CsPbCl3 NCs. Table 7 summarizes the optoelectronic properties of the lanthanide-doped CsPbCl3 NCs.
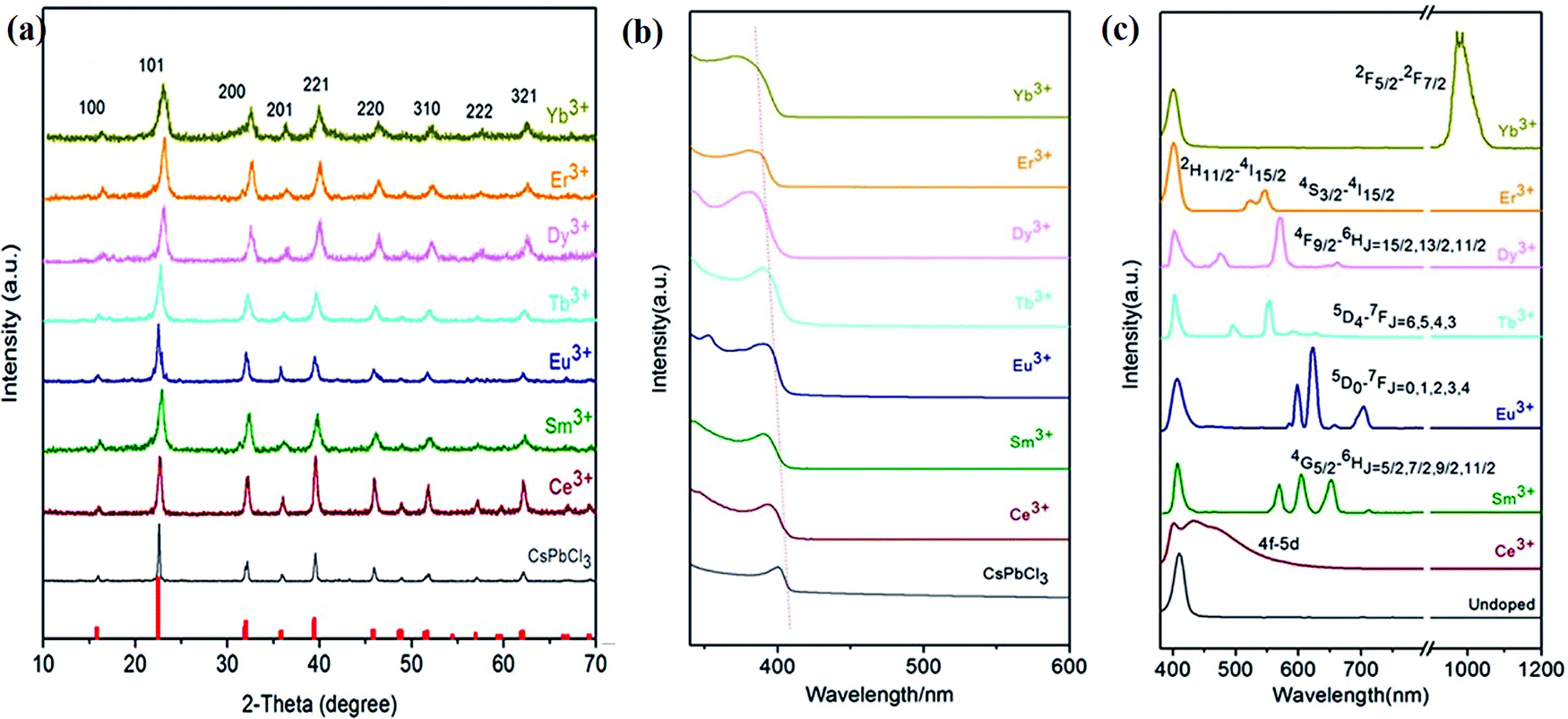 | ||
| Fig. 13 (a) XRD patterns, (b) absorption spectrum, (c) emission spectra of pristine CsPbCl3 NCs and lanthanide-doped CsPbCl3 NCs. Reproduced with permission from ref. 165. Copyright©2017, American Chemical Society. | ||
3.2.3.2 The effects on PL quantum yields and lifetimes. Lanthanide ion doping increases the PLQYs of CsPbCl3.165 In the lanthanide-doped CsPbCl3 structure, defect modification takes place and results in increased PLQYs. It should be noted that the PLQYs of the Yb3+-doped CsPbCl3 crystal was much higher as compared to the other lanthanide dopants.165,169 Zhou et al.123 considered the different host compositions for lanthanide ion doping. Table 8 shows various combinations of host and lanthanide with their quantum yields, which might be useful in selecting a particular combination of the host and lanthanide. Among the different host and dopant combinations, high PLQY was observed for the Yb3+–Pr3+–Ce3+-doped CsPbCl3−x−yBrxIy, and a relatively low PLQY for Yb3+–Dy3+-doped CsPbCl3−x−yBrxIy. The maximum and minimum PLQY were observed for Yb3+–Pr3+–Ce3+-doped CsPbClBr2 and Yb3+–Dy3+-doped CsPbI3.
| CsPbCl3 | CsPbCl1.5Br1.5 | CsPbClBr2 | CsPbBr3 | CsPbBr2I | CsPbBrI3 | CsPbI3 | |
|---|---|---|---|---|---|---|---|
| Yb3+ | 85 | 117 | 137 | 95 | 75 | 73 | 59 |
| Yb3+–Tb3+ | 63 | 102 | 114 | 89 | 64 | 59 | 51 |
| Yb3+–Dy3+ | 47 | 78 | 97 | 94 | 58 | 52 | 43 |
| Yb3+–Nd3+ | 80 | 115 | 132 | 93 | 71 | 67 | 56 |
| Yb3+–Ce3+ | 100 | 146 | 154 | 95 | 72 | 67 | 55 |
| Yb3+–Pr3+ | 110 | 148 | 163 | 97 | 73 | 65 | 63 |
| Yb3+–Pr3+–Ce3+ | 118 | 153 | 173 | 95 | 79 | 72 | 65 |
Time-resolved spectroscopy revealed that the lanthanide-doped CsPbCl3 has a longer average lifetime than 1.8 ns for undoped CsPbCl3.165 For Yb-doped CsPbCl3 NCs the average lifetime is 22 ns, which is larger than undoped CsPbCl3 NCs.183 Pan et al.182 reported that the average lifetime for 2.7%Ce:CsPbCl3 was 5.3 ns. For 0.1% Dy-doped CsPbBr3, the average lifetime was 27.88 ns, whereas for CsPbBr3 it was 21.22 ns.184 The average lifetime increased with Dy concentration and attained a maximum value 54.34 ns for 0.7% Dy-doped CsPbBr3 and then decreased with further increase in the Dy concentrations to 42.26 ns for 0.9% Dy:CsPbBr3. An initial increase in average lifetime indicates that the electronic defect-mediated non-radiative transition was effectively reduced due to Dy concentration. The decreased average lifetime at higher concentrations, i.e. at 0.9% Dy, was due to the concentration quenching effect of lanthanides. The average lifetime further increased with the higher atomic number of lanthanide ions for a particular CsPbX3 host. Longer average lifetimes, as well as increased PLQYs, are the consequence of the partial elimination of non-radiative channels in the lanthanide-doped CsPbCl3.
3.2.3.3 The effect of lanthanide doping concentration on the PL peak intensity of CsPbX3. The PL peak intensity is a measure of the number of emitted photons. The intensity of PL spectra depends on various parameters such as the type of material, the local surrounding temperature and doping concentration, etc. Doping concentration-dependent PL studies of Eu3+-doped CsPbCl3 have been carried out by Pan et al.92 The absorption edge and emission peak showed blue shifts with an increase in Eu3+ concentration as shown in Fig. 14. This blue shift is due to the lattice contraction with doping ion concentration.165,169,185 Zhou et al.169 confirmed that there was an increase in the bandgap with increasing the doping concentration. The intrinsic transition intensity corresponding to the Eu3+ ion increased with the doping concentration. This behavior is because the high doping concentration facilitates efficient energy transfer from the host CsPbCl3 to Eu3+ ions. The PLQYs of the doped CsPbCl3 initially increased and then decreased with the concentration of Eu3+ ions.165 The maximum PL of Eu3+ appeared for the doping concentration of 7.8 mole% (see Fig. 14). The effect of a blue shift in the absorption and emission spectra was observed with the Lu3+-doping concentration in Lu3+-doped CsPbBr3 NCs and in Yb3+-doped CsPbI3.177,185 Hence, this trend may also be predicted for Ln3+-doped CsPbX3.
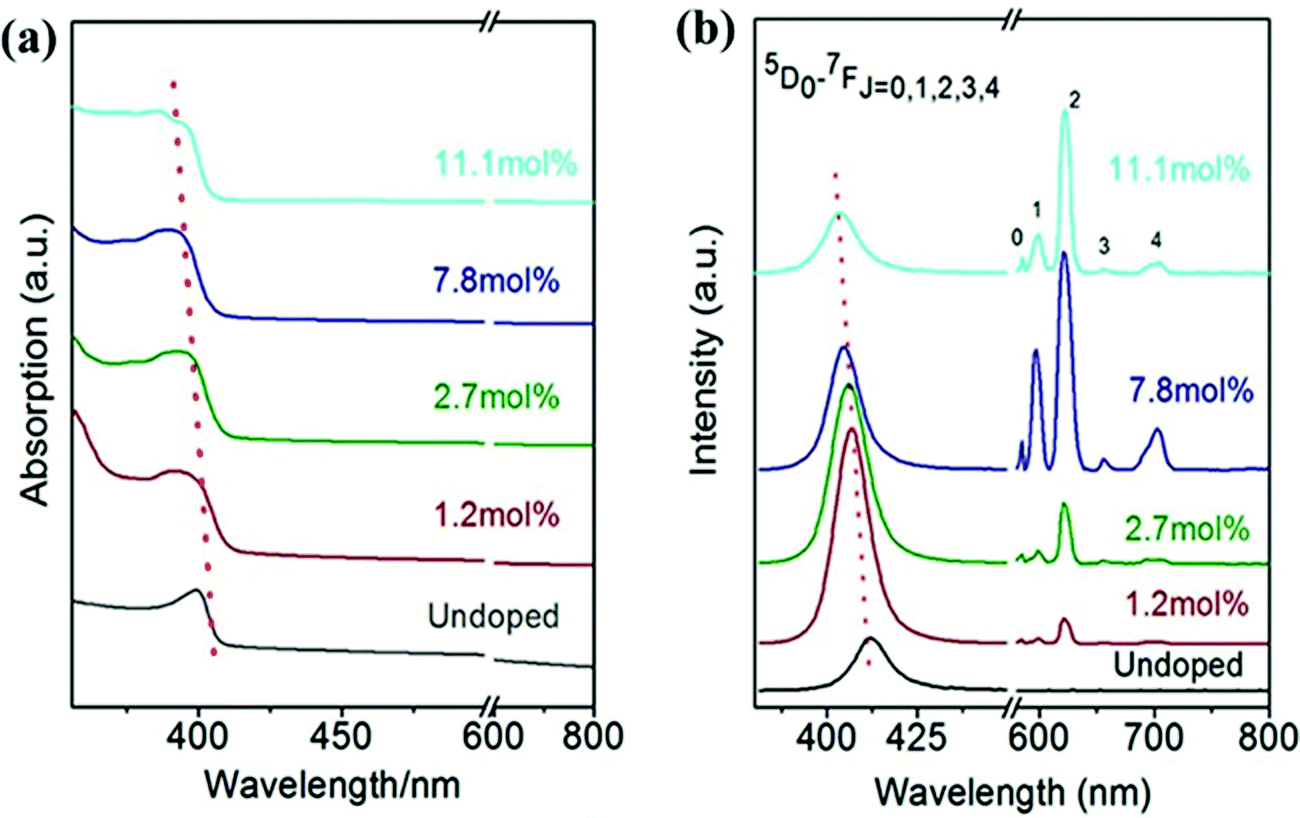 | ||
| Fig. 14 (a) Absorption and (b) emission spectra under 365 nm excitation of CsPbCl3 and different doping concentrations of Eu3+ in the CsPbCl3 structure under 365 nm excitation. Reproduced with permission from ref. 165. Copyright©2017, American Chemical Society. | ||
The optimized lanthanide doping concentration varies based on the CsPbX3 host. Also, in the lanthanide-doped CsPbX3, the PLQY and average lifetime initially increased with Ln3+-doping concentration and attained a maximum value. The PLQYs and the average lifetime subsequently decreased for higher doping concentrations. The variation trend for optoelectronic properties like PLQYs, average lifetime, etc., was the same as observed in the case of non-lanthanide metal ions-doped CsPbX3.
3.2.4.1 Advancements in solar cell application. To analyze the effects of lanthanide ion doping, Duan et al.171 used lanthanide ions (Sm3+, Tb3+, Ho3+, Er3+, and Yb3+) in CsPbBr3 in the fabrication of solar cells and calculated different solar cell parameters. Table 9 summarizes the solar cell parameters for different devices that use lanthanide-doped all-IHPs. The data indicate that lanthanide doping significantly improves cell performance. They found an ultrahigh open-circuit voltage of ∼1.594 V under the one sun illumination for lanthanide ion-doped all-IHPs. The lanthanide doping also improved thermal stability. On exposure to 80% relative humidity in an air atmosphere, the efficiency of the solar cell remained almost unchanged over 110 days. The Ln-doped ABX3 has been used as one of the layers of the solar cell structure, FTO/c-TiO2/m-TiO2/Ln:CsPbX3/carbon. The results from Table 9 indicate the potential of lanthanide-doped all-IHPs materials for solar cell applications.
| Halide perovskite | Dopant | Cell structure | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | Year |
|---|---|---|---|---|---|---|---|
| CsPbBr3 | Eu2+ | FTO/c-TiO2/m-TiO2/CsPb1−xEuxBr3/carbon | 6.33 | 1.4523 | 79.19 | 7.28 | 2021186 |
| CsPbBr3 | Yb3+ | FTO/c-TiO2/m-TiO2/CsPb0.97Yb0.03Br3/carbon | 7.45 | 1.536 | 80.20 | 9.20 | 2018171 |
| CsPbBr3 | Er3+ | FTO/c-TiO2/m-TiO2/CsPb0.97Er0.03Br3/carbon | 7.46 | 1.563 | 82.80 | 9.66 | 2018171 |
| CsPbBr3 | Ho3+ | FTO/c-TiO2/m-TiO2/CsPb0.97Ho0.03Br3/carbon | 7.45 | 1.572 | 83.20 | 9.75 | 2018171 |
| CsPbBr3 | Tb3+ | FTO/c-TiO2/m-TiO2/CsPb0.97Tb0.03Br3/carbon | 7.47 | 1.588 | 84.80 | 10.06 | 2018171 |
| CsPbBr3 | Sm3+ | FTO/c-TiO2/m-TiO2/CsPb0.97Sm0.03Br3/carbon | 7.48 | 1.594 | 85.10 | 10.14 | 2018171 |
| CsPbI2Br | Eu3+ | ITO/NiO/CsPb0.995Eu0.005I2Br/PC61BM/Ag | 15.30 | 1.130 | 70.50 | 12.10 | 2021172 |
| CsPbI2Br | Eu3+ | FTO/c-TiO2/m-TiO2/CsPb0.95Eu0.05I2Br/spiro-OMeTAD/Au | 14.63 | 1.223 | 76.60 | 13.71 | 2019187 |
| CsPbI3 | Eu3+ | FTO/TiO2/CsPb0.95Eu0.05I3/spiro-OMeTAD/Au | 11.10 | 0.890 | 68.00 | 6.80 | 2018179 |
| CsPbI3 | Yb3+ | FTO/TiO2/CsPb0.95Yb0.05I3/PTB7/MoO3/Ag | 13.25 | 1.250 | 75.00 | 12.42 | 2019180 |
| CsPbI3 | Yb3+ | FTO/TiO2/CsPbI3:5%YbCl3/spiro-OMeTAD/Ag | 18.40 | 1.130 | 60.00 | 12.40 | 2020188 |
3.2.4.2 Lanthanide-doped all-inorganic halide perovskites for WLEDs. The halide perovskite QDs show a very high PL quantum yield, which allows various novel optoelectronic applications. One of their fascinating applications is in the fabrication of tunable colored LEDs. In recent years, lanthanide-doped all-IHPs have also been actively explored for this purpose. For example, Nd3+ doping into CsPbBr3 NCs shifts the emission peak from 515 nm to 459 nm.189 After the encapsulation of green light-emitting CsPbBr3 NCs and blue-light emitting CsPbBr3:xNd3+ (x = 7.2%) NCs in the PMMA host and after their deposition on a UV-LED having a red-emitting CsPbBr1.2I1.8/PMMA, WLEDs can be realized. The fabricated WLEDs have excellent color coordinates (0.34, 0.33) and NTSC value of 122% with a cool-white correlated color temperature of 5310 K. Yuan et al.185 fabricated WLEDs by integrating Yb3+-doped CsPbI3 with the GaN LED blue-chip having the Y3Al5O12:Ce3+ phosphor. The PL spectrum of Yb3+-doped CsPbI3 coated over GaN is shown in Fig. 15(a). The schematic of the WLEDs and color coordinates for WLEDs are shown in Fig. 15(b and c), respectively.
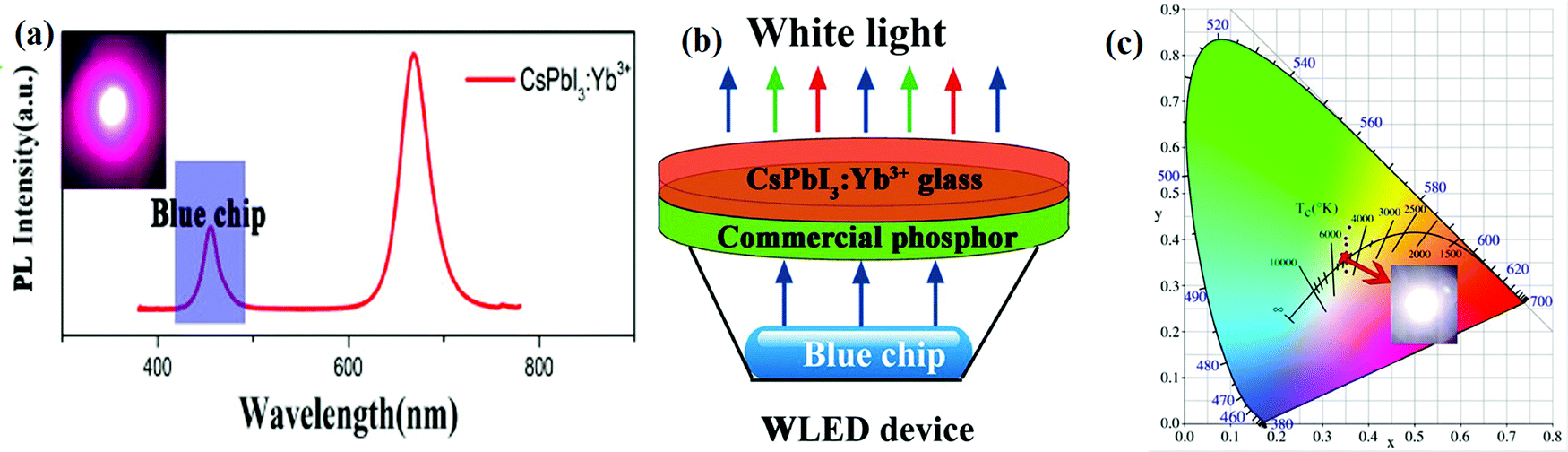 | ||
| Fig. 15 (a) PL spectrum of Yb3+-doped CsPbI3 NCs integrated with the GaN blue chip. (b) Schematic of the device structure for WLEDs. (c) The color coordinates of the WLEDs (0.3499, 0.3542). Reproduced with permission from ref. 185. Copyright©2020, Elsevier. | ||
Pan et al.182 compared the performance of WLEDs prepared using various lanthanide ion pairs in halide perovskites. They considered CsPbCl3:2.7% Ce3+:7.8% Eu3+, CsPbCl1.8Br1.2:2.7% Ce3+:7.8% Eu3+, CsPbCl3:2.7% Ce3+:7.8% Sm3+ and CsPbCl1.8Br1.2:2.7% Ce3+,7.8% Sm3+ for their studies. Among these four pairs, the maximum luminous efficiency (LE) and color rendering index (CRI) were 36 lm W−1 and 92, respectively for CsPbCl1.8B1r.2:2.7% Ce3+:7.8% Eu3+. Zhu et al.184 reported Dy3+-doped CsPbBr3 quantum dots to demonstrate WLEDs. For the generation of WLEDs, the green-emitting Dy3+-doped CsPbBr3 was incorporated with the red-emitting Mn2+-doped K2SiF6 phosphor and blue-emitting InGaN chips. The CIE coordinates of the fabricated WLEDs reported were (0.2881, 0.3001) and the NTSC value was 126.3% on application of a 40 mA drive current. The Ce3+ ion incorporation into the CsPbCl3 NCs enhanced the blue and green components.182 It also behaved as a sensitizer for ions such as Eu3+ and Sm3+ and promoted red emission. This opens up future applications for WLED based on phosphor converted all-IHPs. Table 10 summarizes the lanthanide ion-doped CsPbX3-based WLED parameters for the optimized lanthanide doping concentration.
| Ln-Doped WLED structure | CIE coordinates | LE (lm W−1) | CRI | CCT (K) | NTSC (%) | Year |
|---|---|---|---|---|---|---|
| Nd:CsPbBr3 + CsPbBr3 + CsPbBr1.2I1.8/PMMA film | (0.34, 0.33) | — | — | 122.0 | 2020189 | |
| Dy:CsPbBr3 + Mn:K2SiF6 + InGaN LED chip | (0.2881, 0.3001) | — | — | 126.3 | 2022184 | |
| Yb:CsPbI3 + Ce:Y3Al5O12 + GaN LED chip | (0.3499, 0.3542) | 57.3 | 85.4 | 3524 | — | 2020185 |
| Ce:Eu:CsPbCl3 + polystyrene + GaN LED chip | — | 23 | 73 | — | — | 2018182 |
| Ce:Eu:CsPbCl1.8Br1.2 + polystyrene + GaN LED chip | — | 36 | 92 | — | — | 2018182 |
| Ce:Sm:CsPbCl3 + polystyrene + GaN LED chip | — | 15 | 70 | — | — | 2018182 |
| Ce:Sm:CsPbCl1.8Br1.2 + polystyrene + GaN LED chip | — | 27 | 84 | — | — | 2018182 |
3.2.4.3 Lanthanide-doped halide perovskites for NIR emission and NIR LEDs. All-IHPs are known for their characteristic emission in the visible region. Lanthanide doping adds some UV and infrared components of the electromagnetic spectrum due to 4f–4f transitions of different lanthanide ions. For example, the Yb3+-doped CsPbBr3 NCs showed NIR emission;190 the NCs were encapsulated with SiO2. The strong NIR (985) emission occurred from Yb3+ ions. This is the intrinsic luminescence of Yb3+ and is pumped by the perovskite host. The excitation and emission spectra of CsPbBr3, Yb3+-doped CsPbBr3, and SiO2-encapsulated Yb3+-doped CsPbBr3 NCs are shown in Fig. 16(a). The energy transfer process from the CsPbBr3 to the Yb3+ ion is shown in Fig. 16(b).
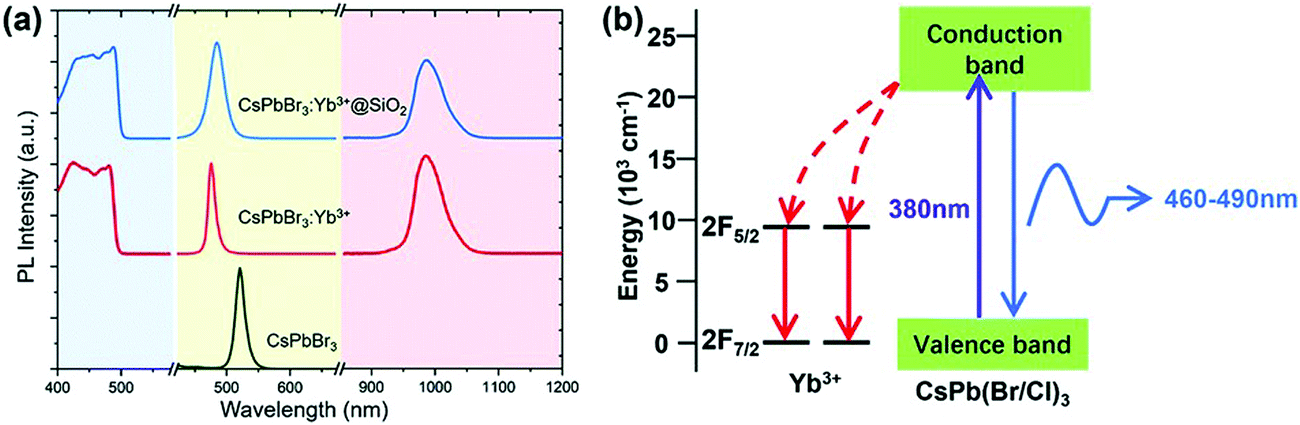 | ||
| Fig. 16 (a) Excitation spectra (emission 985 nm, left), visible PL spectra (middle), and infrared PL spectra (right) of the synthesized CsPbBr3, Yb3+-doped CsPbBr3, and SiO2-encapsulated Yb3+-doped CsPbBr3 NCs. (b) Schematic diagram of energy transfer in the Yb3+-doped CsPbBr3 NCs. Reproduced with permission from ref. 190. Copyright©2020, Royal Society of Chemistry. | ||
The synthesized Yb3+-doped CsPbBr3 NCs emits strong NIR emission. The emission peak is centered at 985 nm. This originated from the 2F5/2 → 2F7/2 transition in Yb3+ ions. The intrinsic transition in Yb3+ ions is excited by efficient energy transfer from the perovskite host. Since there is a charge difference between Yb3+ and Pb2+, the charge compensation defects Vpb were generated.73,190 The Vpb exists between the two Yb3+ ions in the form of Yb3+–Vpb–Yb3+. The Vpb can excite two Yb3+ ions nearby through the quantum-cutting process and thus a high PL quantum yield is obtained.191,192 The Yb3+-doped CsPbCl3 NCs emits 986 nm NIR light from the 2F5/2 → 2F7/2 transition, whereas Yb3+/Er3+ co-doped CsPbCl3 NCs emits NIR emission centered at 1533 nm for the transition from 4I13/2 → 4I15/2 of Er3+ ions.174 Zeng et al.193 reported various strategies to enhance the NIR emission by doping lanthanide ions into the CsPbCl3 NCs. It was observed that Er3+, Ho3+, and Nd3+ doping into CsPbCl3 NCs can emit intense NIR light through energy transfer mediated by Mn2+ ions. The emission wavelength centered at 1542 nm was observed for Er3+via a transition from 4I13/2 → 4I15/2. In Ho3+, the transition takes place from 5F5 → 5I7 and 5F5 → 5I6 and corresponding emission peaks are observed at 986 nm and 1484 nm, respectively. In the case of Nd3+ ions, three emission peaks were observed which were centered at 888 nm, 1064 nm, and 1339 nm corresponding to 4F3/2 → 4I9/2, 4F3/2 → 4I11/2, and 4F3/2 → 4I13/2 transitions, respectively. Lanthanide-doped all-IHPs NIR emission transitions and their peak wavelengths are listed in Table 11.
| Lanthanide-doped CsPbX3 | Transition | NIR emission peaks (nm) |
|---|---|---|
| Yb3+–CsPbBr3 NCs | 2F5/2 → 2F7/2 | 976,173 985,190 986174 |
| Yb3+/Er3+–CsPbBr3 NCs | 4I13/2 → 4I15/2 (Er3+) | 1533174 |
| Er3+–CsPbCl3 NCs | 4I13/2 → 4I15/2 | 1542193 |
| Ho3+–CsPbCl3 NCs | 5F5 → 5I7, 5F5 → 5I6 | 986, 1484193 |
| Nd3+–CsPbCl3 NCs | 4F3/2 → 4I9/2, 4F3/2 → 4I11/2, 4F3/2 → 4I13/2 | 888, 1064, 1339193 |
| Yb3+–CsPbI3 QDs | 2F5/2 → 2F7/2 | 988185 |
NIR-LEDs are useful in night vision cameras, bio-imaging, communications and food processing industries. Huang et al.173 proposed NIR-LEDs by doping ytterbium into CsPbCl3. The emission and absorption spectra of CsPbCl3 NCs and Yb3+ ion-doped CsPbCl3 NCs are shown in Fig. 17. The emission of Yb3+ ions at ∼1000 nm is suitable for NIR-LEDs. Fig. 18 demonstrates the application of Yb3+-doped CsPbCl3 as a NIR camera. The device shows an output irradiance of 112 mW cm−2@400 mA with excellent stability. For the analysis of Yb3+-doped CsPbCl3 as a NIR camera, photographs of fruits and a fist were taken in natural light and phosphor-converted NIR-LED light. When the NIR-LED is off, the fruits and fist are invisible to the eye, whereas when the NIR-LED is on, the black and white images of the fruits and fist are observed. The images recorded under natural light phosphor-converted Yb3+-doped CsPbCl3 NIR-LED light are shown in Fig. 18(a) and (b), respectively.
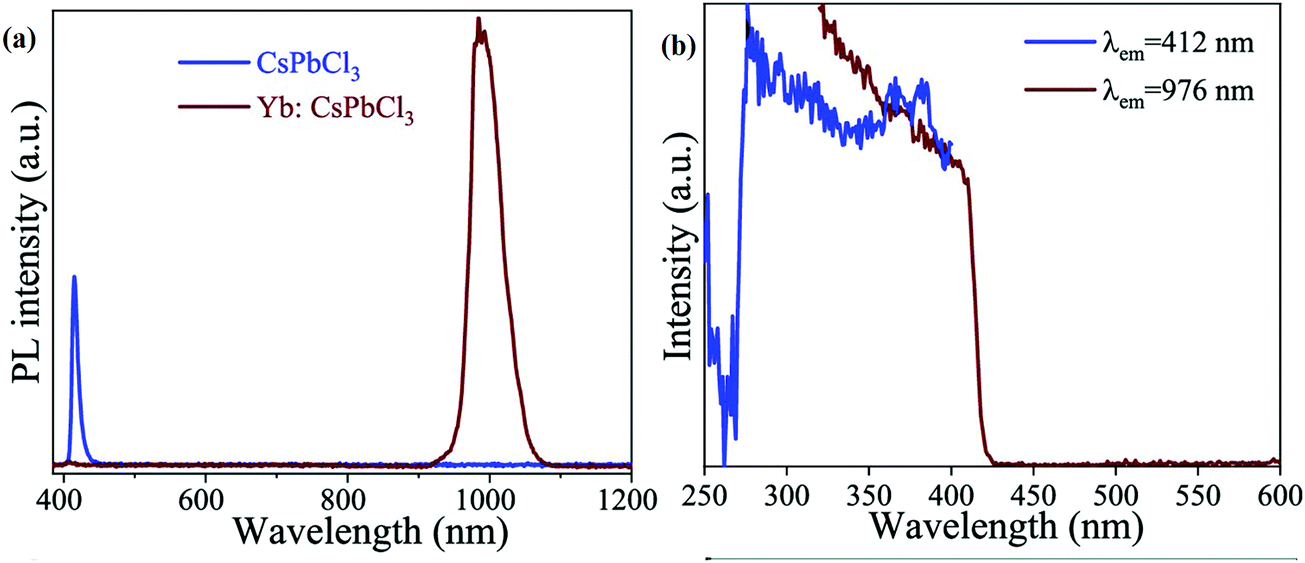 | ||
| Fig. 17 (a) Emission and absorption spectra of CsPbCl3 NCs and Yb3+-doped CsPbCl3 NCs. (b) Excitation spectra of CsPbCl3 NCs and Yb3+-doped CsPbCl3 NCs. Reproduced with permission from ref. 173. Copyright©2021, American Chemical Society. | ||
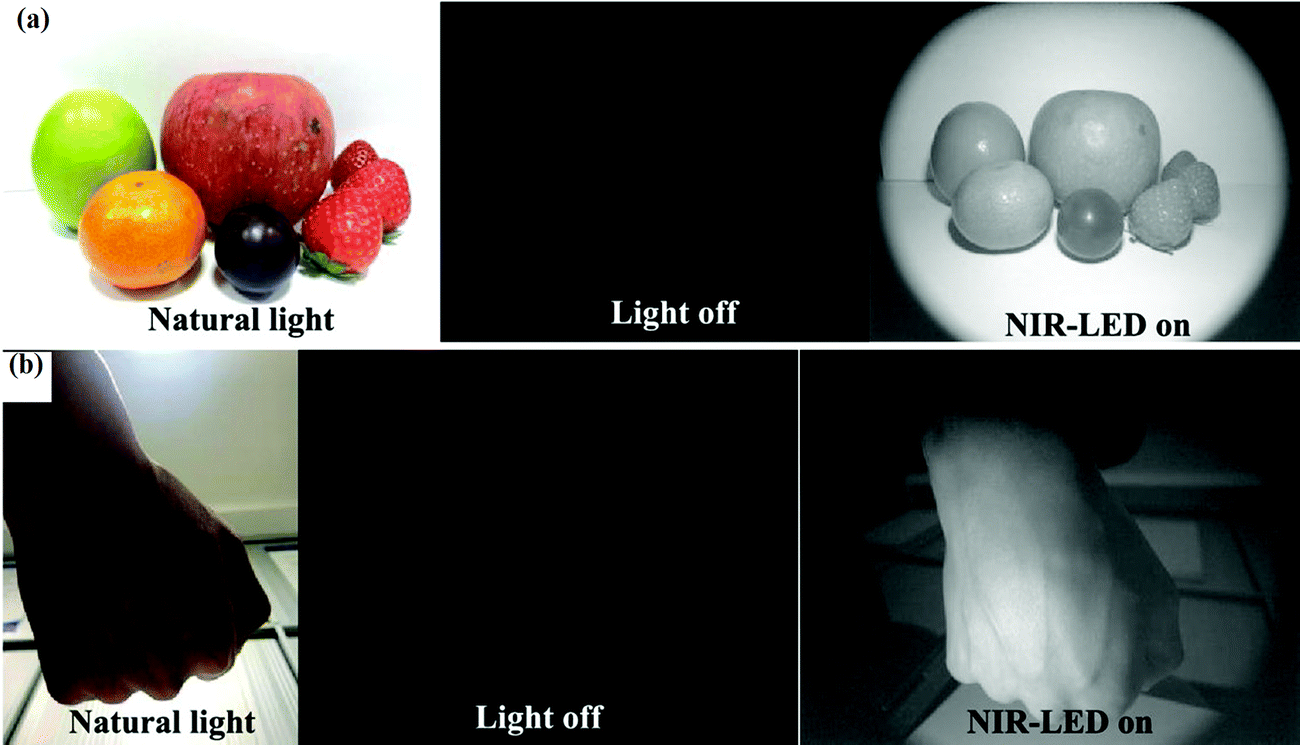 | ||
| Fig. 18 Images of fruits and a fist under natural light and NIR-LEDs light were recorded using a visible and NIR camera, respectively. Reproduced with permission from ref. 173. Copyright©2021, American Chemical Society. | ||
3.2.4.4 Emerging applications for optical temperature sensing. In addition to the optoelectronic device applications, lanthanide-doped all-IHPs have recently been explored for various types of sensing applications. In recent years, temperature-dependent PL spectra have been studied widely in lanthanide-doped materials. Lanthanide-doped all-IHPs showed a linear variation in PL peak intensity with a temperature variation. This characteristic of Ln3+-doped all-IHPs has attracted the attention of researchers for non-contact-based temperature sensing applications. In lanthanides and transition metals, the PL intensity variation occurs due to the electron population in the thermally coupled states.194 However, a very small energy gap between these thermally coupled states leads to the overlapping of the emission. This affects the temperature sensing accuracy. With an increase in temperature, the electron–phonon coupling becomes more and more prominent and leads to the thermal quenching of the PL intensity.
Zhang et al.195 demonstrated the application of Tb3+-doped CsPbI3 NCs for optical temperature sensing. The temperature-dependent emission spectra of Tb3+-doped CsPbI3 NCs are shown in Fig. 19(a). As the temperature increases, the emission intensities of both the Tb3+ ions and CsPbI3 NCs decrease systematically. Fig. 19(b) shows the integrated emission intensities corresponding to Tb3+ and CsPbI3 emission in Tb3+-doped CsPbI3 NCs at various temperatures. The fluorescence intensity ratio of Tb3+ at 544 nm and CsPbI3 at 677 nm with excitation at 480 nm is given by the following:
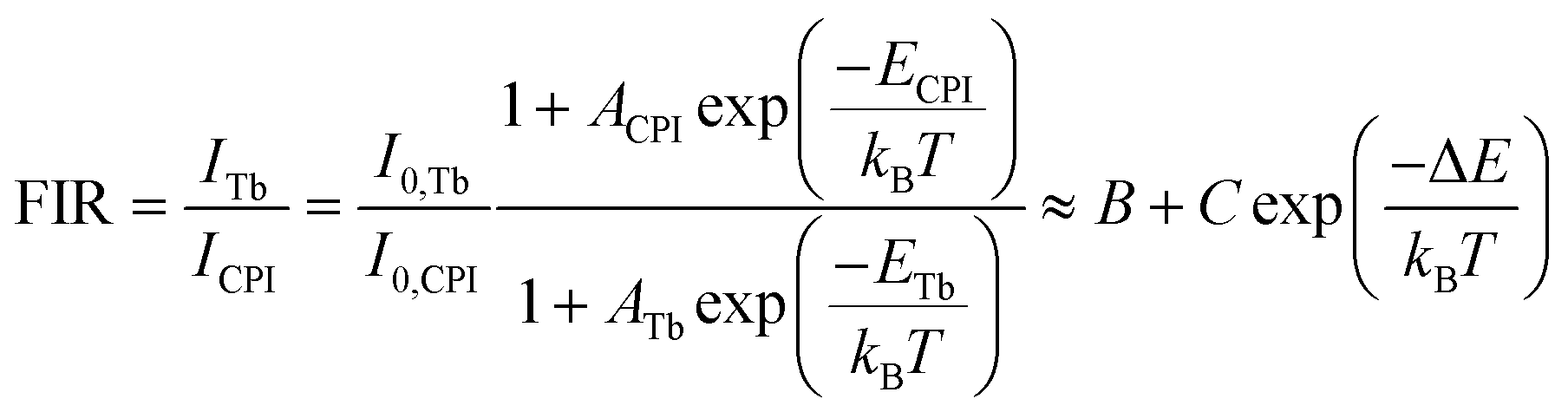 | (5) |
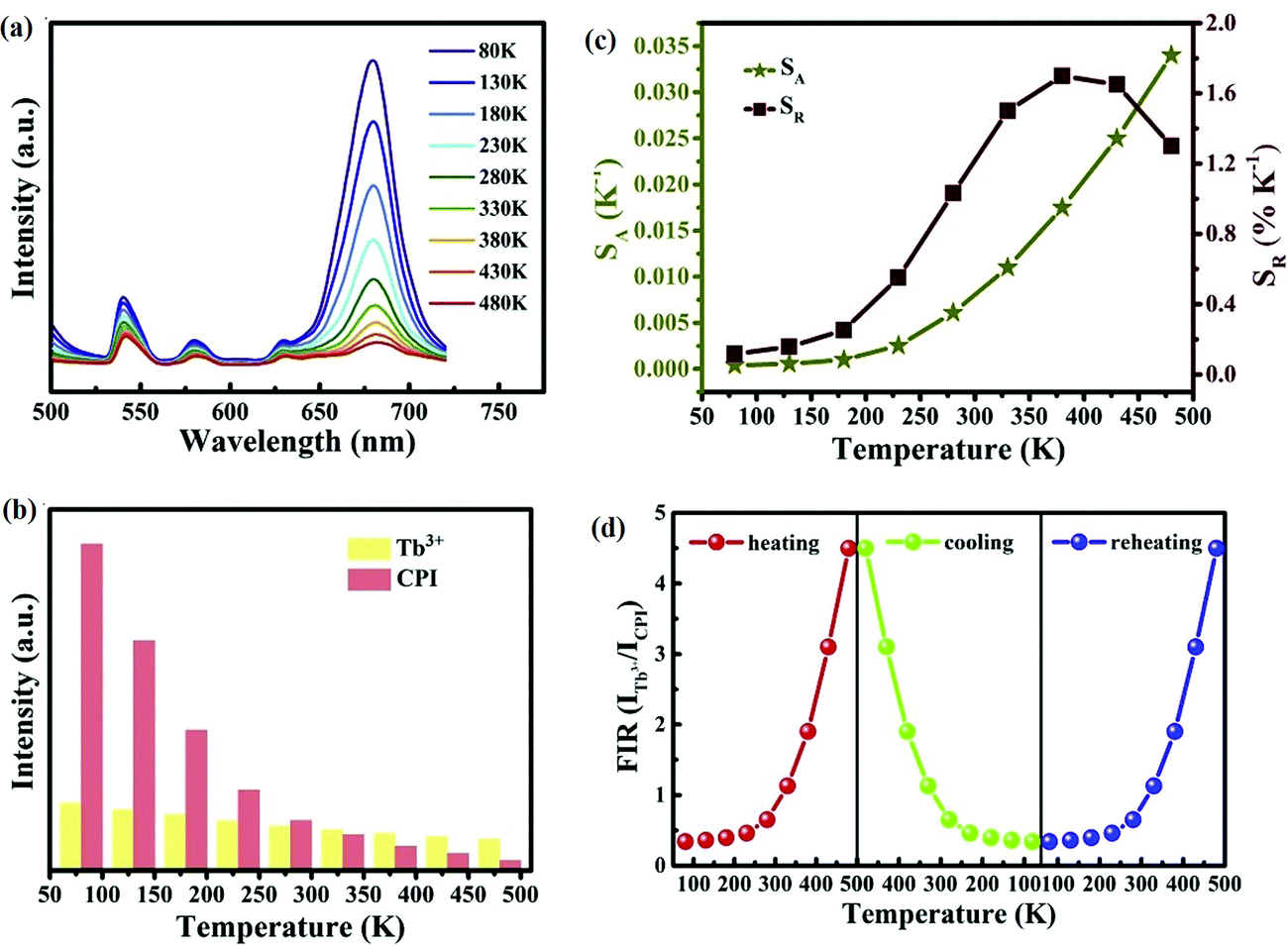 | ||
| Fig. 19 (a) Temperature-dependent emission spectra of Tb3+-doped CsPbI3 NCs excited at 480 nm. (b) Integrated emission intensities corresponding to Tb3+ and CsPbI3 emission in Tb3+-doped CsPbI3 NCs at various temperatures. (c) Absolute sensitivity SA and relative sensitivity SRversus T. (d) FIR in cyclic heating cooling and reheating. Reproduced with permission from ref. 195. Copyright©2020, Journal of European Ceramic Society. | ||
The absolute sensitivity is defined as 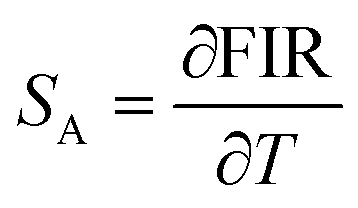 , and the relative sensitivity is
, and the relative sensitivity is 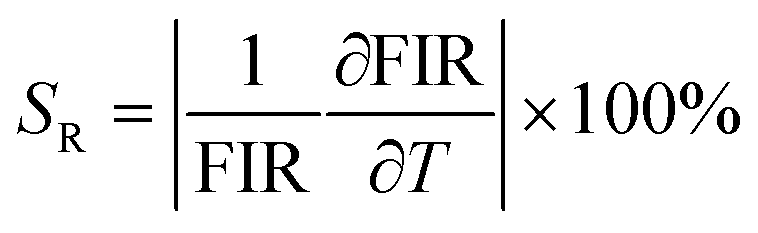 . The FIR value was calculated for the temperature range 80–480 K. The maximum reported values of SA and SR are 0.034 K−1 and 1.78% K−1 as shown in Fig. 19(c). The reversible effect on the FIR with temperature was also determined. The FIR remained almost the same in the cyclic temperature process as shown in Fig. 19(d). Yao et al.196 also reported Dy:CsPbBr3 QDs as a temperature sensor in the temperature range 80–298 K. For optimized Dy3+ doping, the Dy:CsPbBr3 QDs showed the maximum relative sensitivity 2.39% K−1 at 298 K.
. The FIR value was calculated for the temperature range 80–480 K. The maximum reported values of SA and SR are 0.034 K−1 and 1.78% K−1 as shown in Fig. 19(c). The reversible effect on the FIR with temperature was also determined. The FIR remained almost the same in the cyclic temperature process as shown in Fig. 19(d). Yao et al.196 also reported Dy:CsPbBr3 QDs as a temperature sensor in the temperature range 80–298 K. For optimized Dy3+ doping, the Dy:CsPbBr3 QDs showed the maximum relative sensitivity 2.39% K−1 at 298 K.
Table 12 summarizes the LED parameters of lanthanide-doped all-IHPs. Yb3+-doped CsPbCl3 showed a maximum SR of 3.31 K−1, whereas the wide temperature range response of 80–480 K was observed in Tb3+-doped CsPbCl3. Therefore, one may select the particular combination of host and dopant to achieve the requirements under study. These open up future applications for fluorescence intensity ratio-based non-contact efficient temperature sensing. Hence, Ln3+ ion-doped CsPbX3 shows prominent temperature sensitivity, which makes it a good candidate for temperature sensing applications.
3.2.4.5 Emerging application in optical encoding. In recent years, optical encoding research has become one of the most important fields of research in the scientific and technical industry for detecting counterfeiting. The poor materials, as well as their encoding in anti-counterfeiting technology, are easily depicted by the falsifier. It has been observed that stimuli-responsive fluorescence materials, such as lanthanide-doped all-IHPs, are very effective for optical data security with high recording density. Also, the fluorescence transitions switch between different states based on external factors such as temperature, dopant ions and their concentration, etc. Feng et al.198 reported a novel approach to modeling optical encoding materials using europium complex-decorated CsPbBr3 quantum dots with highly stable dual-stimuli-response. The schematic illustration of the formation of the europium complex Eu(PBA)3AA (HPBA = N-(2pyridinyl)benzoylacetamide, AA = 8-aminooctanoic acid)-decorated CsPbBr3 quantum dots is shown in Fig. 20(a and b). The temperature and pH responses by the CsPbBr3 (QDs) and Eu(PBA)3AA, respectively, were observed in the luminescence spectra. This is useful for this application also. The encryption and decryption mechanisms are shown in Fig. 20(c) and (d), which show the dual responsive optical recording and encryption of the designed material.
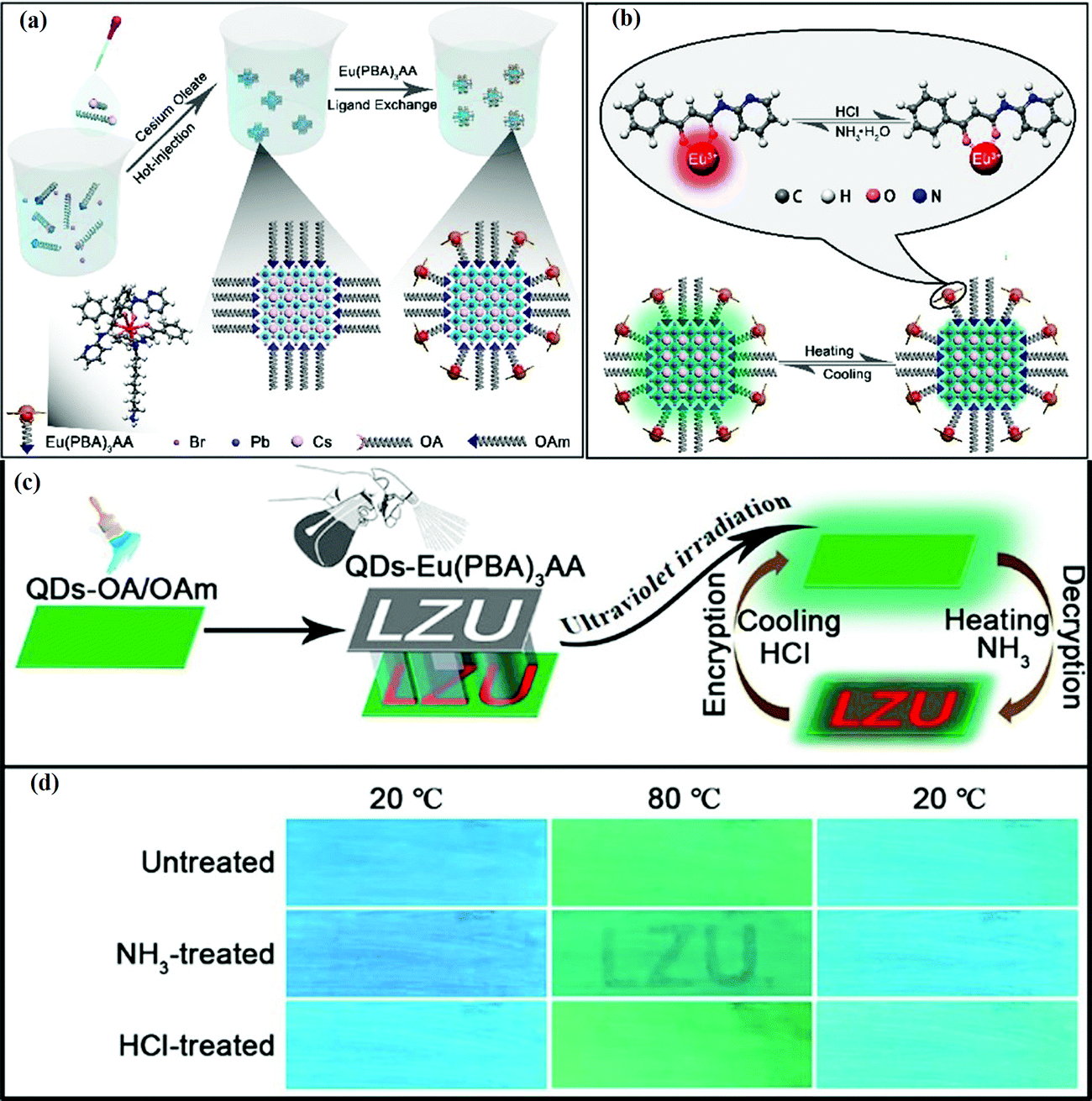 | ||
| Fig. 20 (a) Schematic illustration of the formation of QDs-Eu(PBA)3AA. (b) PL images of the dual-responsive optical recording and encryption. (c) The pattern design process and encryption and decryption mechanism. (d) PL images of patterns at different temperatures and pH. Reproduced with permission from ref. 198. Copyright©2021, American Chemical Society. | ||
OA/OAm-based QDs were painted on transfer paper and an encrypted word “LZU” was printed with Eu(PBA)3AA-QDs, which was decoded only when the fluorescence of Eu(PBA)3AA was enhanced by NH3 vapor stimulation and the fluorescence of CsPbBr3 was decreased by increasing the temperature to 80 °C as indicated in Fig. 20(d). The information “LZU” was doubly encrypted by temperature and pH and was readable only when these two values fulfilled the decryption conditions. Optical encryption and decryption were also reported by using Lu3+-doped CsPbBr3 but decryption occurred in UV light instead of NIR light.177 The information “ZJU” was encrypted using the blue-emitting ink of Lu3+:CsPbBr3 on white paper. It is not possible to decode the information “ZJU” under daylight; however, when it encounters UV light of wavelength 360 nm, decryption of the information takes place. Hence, europium complex-decorated CsPbBr3 quantum dots can be used as an optical encoding material in the future.
4. Summary and perspectives
The all-IHPs dominate organic–inorganic halide perovskites due to their better environmental stability, thus making them good candidates for optoelectronic applications. Conventionally, solid-state and liquid reaction synthesis methods have been used for the synthesis of bulk and nano-structured halide perovskites. Recently, researchers have reported several new synthesis techniques such as inject printing, spin coating, RF magnetron sputtering, single/multi crucible vacuum thermal evaporation, hot injection, etc. All these techniques have their advantages and disadvantages. Among these, the hot injection method is the most widely used method for the synthesis of all IHPs nanostructures with better stability. These reported synthesis methods are aimed toward the improved and stable structure of IHPs but none claim a completely stable phase synthesis. Therefore, the synthesis of cost-effective IHPs with long-term stability is still among the most challenging areas of research for IHPs.In general, three phases (α, β, and γ) are observed in all-IHPs except in the perovskites formed by the halide I-atom, where an additional non-perovskite δ phase occurs. The cubic phase with a Goldschmidt tolerance factor value of 1 is the most stable structure. The stability can be increased by tailoring the Goldschmidt tolerance factor value by using surfactants and dopant ions. One of the widely used dopants for IHPs is the lanthanide ion. The doping with lanthanide ions increases the stability (thermal stability, photostability, and phase stability) by reducing vacancies (defects and trap-states) in the structure and also improves optical properties. However, there is still a lot of scope for further improving the stability aspect of IHPs. A few frontline perspectives related to phase and stability issues can be outlined as follows. (i) Four phases are observed in all I-ion based IHPs. On heating, a direct transition from the α to δ phase occurs. However, in the cooling process, all fourα, β, γ, and δ phases are observed. Herein, the reason for the direct transition from α to δ phase is not well addressed and needs further attention. (ii) Phase kinetics depends on both temperature and pressure. However, the study of applied pressure-based phase transition is not well explored in pristine and lanthanide-doped all-IHPs. (iii) In CsPbX3, the toxicity of the Pb is unavoidable. Lead-free all-IHPs like CsGeX3, CsGnX3, etc., are reported but their phase stability is quite low as compared to CsPbX3. Replacing Pb2+ with any non-toxic cation considerably improves the stability and can open up an eco-friendly IHPs field. (iv) The development of single-crystal IHPs has not been tried so far. The single-crystal form of IHPs may also add new properties and should be explored in the future.
The all-IHPs provide a wide color gamut of emission spectra ranging from 400 nm to 700 nm by the compositional management of halide ions. The FWHM of the emission peaks at 400 nm for CsPbCl3 and 700 nm for CsPbI3 ranges between 12 nm to 42 nm, respectively. The bandgap of the structure can be tailored from ∼1.7 eV for CsPbI3 to ∼3.0 eV for CsPbCl3. The doping of lanthanide ions in IHPs bestows them with excellent optical properties. It not only adds unique 4f–4f luminescence of lanthanide ions but affects the characteristic optical features of IHPs. The substitution of smaller-sized lanthanide ions (all lanthanide after Gd3+) in place of Pb2+ ion leads to lattice shrinkage. This causes an increased ligand field within the [PbBr6]4− octahedra, leading to an increase in the bandgap, and a blue shift in the PL is observed. Further, the emission intensity of the band-to-band transition of the host initially increases on increasing the lanthanide concentration because of the improved defect state, but after a certain limit, it starts decreasing due to FRET from the host to the doped lanthanide ion. Time-resolved spectroscopy reveals that the lanthanide-doped CsPbX3 has a longer average lifetime and improved quantum yield as compared to the pristine CsPbX3. In general, the average lifetime increases towards the higher atomic number of lanthanide ions. Longer average lifetimes, as well as increased PL quantum yields, are the consequences of the partial elimination of nonradiative channels in the lanthanide-doped CsPbX3.
Anisotropic studies of Young's moduli of CsPbI3, CsPbBrI2, CsPbBr2I and CsPbBr3 structures have been reported. Young's moduli of CsPbI3 and CsPbBr3 along different directions are the same, whereas CsPbBrI2 showed isotropic nature along the [001] and [010] directions, and CsPbBr2I along the [100] and [010] directions. The anisotropic behaviors of CsPbBrI2 and CsPbBr2I are reflected in almost all transport properties, except for bandgap energy. Although the anisotropy exists along the [001] and [100] directions for CsPbBr2I and CsPbBrI2, respectively, the transport properties (electron and hole mobilities) are superior along [001] in both structures. In this direction, fewer carriers can be captured by the traps during transport. The superior properties of the crystal with the [001] orientation would be helpful in small devices for efficient output, such as transistors, light-emitting diodes, photodetectors, solar cells, etc. The extinction coefficients of CsPbBr2I and CsPbBrI2 along the [001] direction are also greater as compared to [100] direction. Therefore, photon absorption along the [001] direction is greater and results in large current density and better spectral responsibility, which are required in optoelectronic applications. Thus, the anisotropic response for different parameters under applications is the extended field for making the device compact and with high efficiency. The effect of lanthanides on this property has not been explored so far.
Although optical properties have been widely explored, a few challenges remain. A theoretical understanding of various phenomena, such as phonon energy and its effect on optoelectronic properties and phase transition, the effect of temperature on PL spectra, in-depth study on doping ion behavior, etc., is still lacking and needs to be addressed. Similarly, the tuning of the emission peak width with narrow FWHM is also one of the important ignored aspects. Narrow-band emitting IHPs can be used for switching and lasing applications. In addition to this, lanthanide-doped IHPs can give multi-modal emission: downconversion, UC, and quantum-cutting luminescence. Such multimodal emission has not been explored well in IHPs. This multi-modal emission can be achieved by incorporating lanthanide-based phosphors/organic complexes in combination with the IHPs. This facilitates FRET pair formation between the duo, which opens up applications for biomedical imaging and sensing. However, such biological applications require lead-free and stable lanthanide-doped inorganic perovskites.
Due to the outstanding optoelectronic properties of all IHPs, they have attracted attention in several application areas like solar cells, colored LEDs, display devices, spectro-chemical probes, explosive detectors, laser fabrication, X-ray detection, etc. However, lanthanide doping has further extended its application fields and is additionally being used for WLEDs, NIR light emitters, NIR-LEDs, NIR cameras, temperature sensors, optical encoding, etc. For example, Er3+/Ho3+/Nd3+-doping into CsPbCl3 NCs gives intense NIR light through mediated energy transfer by Mn2+ ions. The IR emission centered at 1542 nm was observed with Er3+ doping. The doping of Ho3+ ions gave emission peaks at 986 nm and 1484 nm. Due to the doping of Nd3+ ions, three emission peaks were observed at 888 nm, 1064 nm, and 1339 nm. Similarly, the NIR emission at 980 nm from Yb3+-doped CsPbCl3 has been used for the development of NIR cameras, which could be useful in night vision surveillance, bio-imaging, communication and food processing industries, etc. In another application, Tb3+ ion-doped CsPbI3 showed high-temperature sensitivity in the temperature range 80–480 K with the maximum values of absolute sensitivity (SA) and relative sensitivity (SR) 0.034 K−1 and 1.78% K−1, respectively. Similarly, Eu complex-decorated CsPbBr3 quantum dots have been used as an optical encoding material that has the potential for optical data storage and readout and anti-counterfeiting applications.
In conclusion, lanthanide doping not only improves the stability and optical properties of IHPs but also brings multi-functionality to them, beyond the well-established solar cell applications. Lanthanide-doped inorganic perovskites have great potential, and better theoretical and experimental studies may open up plenty of new application areas for them.
Conflicts of interest
There are no conflicts to declare.References
- H. Xiao, W. Dong, Y. Guo, Y. Wang, H. Zhong, Q. Li and M. Yang, Adv. Mater., 2019, 31, 1805802 CrossRef PubMed
.
- J.-S. Zhou, L. G. Marshall, Z.-Y. Li, X. Li and J.-M. He, Phys. Rev. B, 2020, 102, 104420 CrossRef CAS
- X. Zheng, L. Zhang, X. Wang, Y. Qing, J. Chen, Y. Wu, S. Deng, L. He, F. Liao and Y. Wang, Inorg. Chem. Front., 2020, 7, 3561–3570 RSC
- R. Russell, N. Ratcliff, K. Ahadi, L. Dong, S. Stemmer and J. W. Harter, Phys. Rev. Mater., 2019, 3, 91401 CrossRef CAS
- B.-B. Zhang, X. Liu, B. Xiao, A. Ben Hafsia, K. Gao, Y. Xu, J. Zhou and Y. Chen, J. Phys. Chem. Lett., 2019, 11, 432–437 CrossRef PubMed
- F. Zhou, Z. Li, W. Lan, Q. Wang, L. Ding and Z. Jin, Small Methods, 2020, 4, 2000506 CrossRef CAS
- A. K. Singh, Methods Appl. Fluoresc., 2020, 8, 45008 CrossRef CAS PubMed
- J. Wang, J. Zhang, Y. Zhou, H. Liu, Q. Xue, X. Li, C.-C. Chueh, H.-L. Yip, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2020, 11, 1–9 Search PubMed
- X. Gao, Q. Wang, Y. Zhang, C. Cui, N. Sui, X. Chi, H. Zhang, Q. Zhou, Y. Bao and Y. Wang, J. Phys. Chem. C, 2020, 124, 11239–11247 CrossRef CAS
- Z. Li, F. Zhou, Q. Wang, L. Ding and Z. Jin, Nano Energy, 2020, 71, 104634 CrossRef CAS
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed
- J. Ma, H. Wu, J. Qiu, J. Wang, Q. Wang, Y. Yang, D. Zhou and J. Han, J. Mater. Chem. C, 2019, 7, 3751–3755 RSC
- J. Dong, D. Song, J. Meng, Y. Lu, Y. Li, B. Qiao, S. Zhao and Z. Xu, J. Mater. Chem. C, 2020, 8, 6743–6748 RSC
- D. Yang and D. Huo, J. Mater. Chem. C, 2020, 8, 6640–6653 RSC
- Y. Xu, S. Lou, C. Xia, T. Xuan and H. Li, J. Lumin., 2020, 222, 117132 CrossRef CAS
- T. Wang, X. Wei, Y. Zong, S. Zhang and W. Guan, J. Mater. Chem. C, 2020, 8, 12196–12203 RSC
- W. Xing, Q. Yao, W. Zhu, H. Jiang, X. Zhang, Y. Ji, J. Shao, W. Xiong, B. Wang and B. Zhang, Small, 2021, 17, 2102733 CrossRef CAS PubMed
- E. Aznar, I. Sanchez-Alarcon, P. J. R. Cantó, F. Perez-Pla, J. P. Martínez-Pastor and R. Abargues, J. Mater. Chem. C, 2022, 10, 1754–1766 RSC
- Z. Wu, J. Yang, X. Sun, Y. Wu, L. Wang, G. Meng, D. Kuang, X. Guo, W. Qu and B. Du, Sens. Actuators, B, 2021, 337, 129772 CrossRef CAS
- J. Zhou, L. Xie, X. Song, Z. Wang, C. Huo, Y. Xiong, Z. Cheng, Y. Wang, S. Zhang, X. Chen and H. Zeng, J. Mater. Chem. C, 2020, 8, 12632–12637 RSC
- Y. Liu, P.-A. Chen and Y. Hu, J. Mater. Chem. C, 2020, 8, 16691–16715 RSC
- P. Zhang, Y. Hua, X. Li, L. Zhang, L. Liu, R. Li, G. Zhang and X. Tao, J. Mater. Chem. C, 2021, 9, 2840–2847 RSC
- Y. Lin, X. Zheng, Z. Shangguan, G. Chen, W. Huang, W. Guo, X. Fan, X. Yang, Z. Zhao and T. Wu, J. Mater. Chem. C, 2021, 9, 12303–12313 RSC
- Z. Yang, M. Jiang, L. Guo, G. Hu, Y. Gu, J. Xi, Z. Huo, F. Li, S. Wang and C. Pan, Nano Energy, 2021, 85, 105951 CrossRef CAS
- V. O. Eze, G. R. Adams, L. Braga Carani, R. J. Simpson and O. I. Okoli, J. Phys. Chem. C, 2020, 124, 20643–20653 CrossRef CAS
- M. I. Saleem, S. Yang, R. Zhi, M. Sulaman, P. V. Chandrasekar, Y. Jiang, Y. Tang, A. Batool and B. Zou, Adv. Mater. Interfaces, 2020, 7, 1–7 Search PubMed
- X. Chen, C. Sun, Y. Liu, L. Yu, K. Zhang, A. M. Asiri, H. M. Marwani, H. Tan, Y. Ai, X. Wang and S. Wang, Chem. – Eng. J., 2020, 379, 122360 CrossRef CAS
- T. Masuda, Y. Zhang, C. Ding, F. Liu, K. Sasaki, Q. Shen and M. Endo, J. Appl. Phys., 2020, 127, 243104 CrossRef CAS
- X. Wang, Y. Li, Y. Xu, Y. Pan, C. Zhu, D. Zhu, Y. Wu, G. Li, Q. Zhang, Q. Li, X. Zhang, J. Wu, J. Chen and W. Lei, Chem. Mater., 2020, 32, 4973–4983 CrossRef CAS
- B. Bin Zhang, X. Liu, B. Xiao, A. Ben Hafsia, K. Gao, Y. Xu, J. Zhou and Y. Chen, J. Phys. Chem. Lett., 2020, 11, 432–437 CrossRef PubMed
- A. R. Chakhmouradian and P. M. Woodward, Phys. Chem. Miner., 2014, 41, 387–391 CrossRef CAS
- Z. Yi, N. H. Ladi, X. Shai, H. Li, Y. Shen and M. Wang, Nanoscale Adv., 2019, 1, 1276–1289 RSC
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS
- A. K. Singh, S. Singh, V. N. Singh, G. Gupta and B. K. Gupta, J. Colloid Interface Sci., 2019, 554, 668–673 CrossRef CAS PubMed
- H. Wang, J. Lin, Y. Zhu, X. Zeng, H. Wei, P. Cheng, H. Lu, Y. Liu and R. Xiong, Adv. Electron. Mater., 2020, 6, 1–7 Search PubMed
- S. M. Shakouri, E. Bagherzadeh-Khajehmarjan and S. Ahmadi-Kandjani, Phys. Scr., 2019, 94, 55503 CrossRef CAS
- E. Bagherzadeh-Khajehmarjan, S. Mirershadi and S. Ahmadi-Kandjani, Appl. Phys. B, 2017, 123, 1–6 CrossRef CAS
- L. Guo, G. Tang and J. Hong, Chinese Phys. Lett., 2019, 36, 56201 CrossRef CAS
- S. Yang, L. Wang, L. Gao, J. Cao, Q. Han, F. Yu, Y. Kamata, C. Zhang, M. Fan, G. Wei and T. Ma, ACS Appl. Mater. Interfaces, 2020, 12, 13931–13940 CrossRef CAS PubMed
- M. Tyunina, L. L. Rusevich, E. A. Kotomin, O. Pacherova, T. Kocourek and A. Dejneka, J. Mater. Chem. C, 2021, 9, 1693–1700 RSC
- D. Hu, J. Dong, J. Tian, W. Wang, Q. Wang, Y. Xue, X. Xu and J. Xu, J. Lumin., 2021, 118243 CrossRef CAS
- Z. He, P. Yu, H. Wang, J. Gao, Y. Zhao, H. Zhang, Y. Zhao and Z. Miao, J. Am. Ceram. Soc., 2021, 104, 4389–4397 CrossRef CAS
- A. Mahapatra, D. Prochowicz, J. Kruszyńska, S. Satapathi, S. Akin, H. Kumari, P. Kumar, Z. Fazel, M. M. Tavakoli and P. Yadav, J. Mater. Chem. C, 2021, 9, 15189–15200 RSC
- Y. Chen, S. Tao, Y. Liu, X. Fu, M. Pei, X. Hou, H. Zhou, J. Yin and X. Zhang, J. Nanoelectron. Optoelectron., 2021, 16, 707–714 CrossRef CAS
- Z. Irshad, M. Adnan and J. K. Lee, J. Phys. Chem. Solids, 2022, 160, 110374 CrossRef CAS
- M. W. A. Sektiono, F. A. Permatasari, A. H. Aimon and F. Iskandar, RSC Adv., 2021, 11, 1360–1366 RSC
- X. Zhang, B.-B. Zhang, X. Ma, M. Fu, G. Zha and W. Jie, J. Phys. Chem. C, 2021, 125, 13033–13040 CrossRef CAS
- V.-A. Ha, G. Volonakis, H. Lee, M. Zacharias and F. Giustino, J. Phys. Chem. C, 2021, 125, 21689–21700 CrossRef CAS
- R. Chakraborty, A. Maiti, U. K. Ghorai and A. J. Pal, ACS Appl. Nano Mater., 2021, 4, 10155–10163 CrossRef CAS
- K. Enomoto, R. Oizumi, N. Aizawa, T. Chiba and Y.-J. Pu, J. Phys. Chem. C, 2021, 125, 19368–19373 CrossRef
- R. Han, Q. Zhao, J. Su, X. Zhou, X. Ye, X. Liang, J. Li, H. Cai, J. Ni and J. Zhang, J. Phys. Chem. C, 2021, 125, 8469–8478 CrossRef CAS
- Y. Han, H. Zhao, C. Duan, S. Yang, Z. Yang, Z. Liu and S. Liu, Adv. Funct. Mater., 2020, 30, 1909972 CrossRef CAS
- C. Quan, X. Xing, S. Huang, M. Jin, T. Shi, Z. Zhang, W. Xiang, Z. Wang and Y. Leng, Photonics Res., 2021, 9, 1767–1774 CrossRef
- E. J. Juarez-Perez, Z. Hawash, S. R. Raga, L. K. Ono and Y. Qi, Energy Environ. Sci., 2016, 9, 3406–3410 RSC
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2018, 119, 3418–3451 CrossRef PubMed
- T. Ma, S. Wang, Y. Zhang, K. Zhang and L. Yi, J. Mater. Sci., 2020, 55, 464–479 CrossRef CAS
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed
- W. Zheng, P. Huang, Z. Gong, D. Tu, J. Xu, Q. Zou, R. Li, W. You, J. C. G. Bünzli and X. Chen, Nat. Commun., 2018, 9, 1–9 CrossRef PubMed
- L. Chen, Y. Chuang, W.-D. Yang, K.-C. Tsai, C.-W. Chen and C.-D. Dong, J. Alloys Compd., 2021, 856, 157426 CrossRef CAS
- W. Zhao, R. Su, Y. Huang, J. Wu, C. F. Fong, J. Feng and Q. Xiong, Nat. Commun., 2020, 11, 1–7 Search PubMed
- H. Näsström, P. Becker, J. A. Márquez, O. Shargaieva, R. Mainz, E. Unger and T. Unold, J. Mater. Chem. A, 2020, 8, 22626–22631 RSC
- J. Li, R. Gao, F. Gao, J. Lei, H. Wang, X. Wu, J. Li, H. Liu, X. Hua and S. F. Liu, J. Alloys Compd., 2020, 818, 152903 CrossRef CAS
- C. Borri, N. Calisi, E. Galvanetto, N. Falsini, F. Biccari, A. Vinattieri, G. Cucinotta and S. Caporali, Nanomaterials, 2020, 10, 1–11 Search PubMed
- H. Zhang, X. Fu, Y. Tang, H. Wang, C. Zhang, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, Nat. Commun., 2019, 10, 1–8 CrossRef
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed
- L. Bai, S. Wang, Y. Zhang, K. Zhang, H. Li, K. Ou and L. Yi, J. Lumin., 2020, 226, 117422 CrossRef CAS
- Y. Li, X. Wang, W. Xue, W. Wang, W. Zhu and L. Zhao, Nano Res., 2019, 12, 785–789 CrossRef CAS
- R. X. Yang, J. M. Skelton, E. L. Da Silva, J. M. Frost and A. Walsh, J. Chem. Phys., 2020, 152, 24703 CrossRef CAS PubMed
- R. L. Armstrong, J. Magn. Reson., 1975, 20, 214–231 CAS
- S. Plesko, R. Kind and J. Roos, J. Phys. Soc. Jpn., 1978, 45, 553–557 CrossRef CAS
- M. I. Cohen, K. F. Young, T. Chang and W. S. Brower Jr, J. Appl. Phys., 1971, 42, 5267–5272 CrossRef CAS
- S. Hirotsu, J. Phys. Soc. Jpn., 1971, 31, 552–560 CrossRef CAS
- M. Stefanski, M. Ptak, A. Sieradzki and W. Strek, Chem. Eng. J., 2021, 408, 127347 CrossRef CAS
- X. Zhang, F. Wang, B. Bin Zhang, G. Zha and W. Jie, Cryst. Growth Des., 2020, 20, 4585–4592 CrossRef CAS
- S. Hirotsu, J. Harada, M. Iizumi and K. Gesi, J. Phys. Soc. Jpn., 1974, 37, 1393–1398 CrossRef CAS
- D. Malyshkin, V. Sereda, I. Ivanov, M. Mazurin, A. Sednev-Lugovets, D. Tsvetkov and A. Zuev, Mater. Lett., 2020, 278, 128458 CrossRef CAS
- M. Rodová, J. Brožek, K. Knížek and K. Nitsch, J. Therm. Anal. Calorim., 2003, 71, 667–673 CrossRef
- Š. Svirskas, S. Balčiūnas, M. Šimėnas, G. Usevičius, M. Kinka, M. Velička, D. Kubicki, M. E. Castillo, A. Karabanov and V. V. Shvartsman, J. Mater. Chem. A, 2020, 8, 14015–14022 RSC
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J. M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS PubMed
- M. Lai, Q. Kong, C. G. Bischak, Y. Yu, L. Dou, S. W. Eaton, N. S. Ginsberg and P. Yang, Nano Res., 2017, 10, 1107–1114 CrossRef CAS
- J.-K. Sun, S. Huang, X.-Z. Liu, Q. Xu, Q.-H. Zhang, W.-J. Jiang, D.-J. Xue, J.-C. Xu, J.-Y. Ma and J. Ding, J. Am. Chem. Soc., 2018, 140, 11705–11715 CrossRef CAS PubMed
- B. Zhao, S. F. Jin, S. Huang, N. Liu, J. Y. Ma, D. J. Xue, Q. Han, J. Ding, Q. Q. Ge, Y. Feng and J. S. Hu, J. Am. Chem. Soc., 2018, 140, 11716–11725 CrossRef CAS PubMed
- Z. Yang, A. Surrente, K. Galkowski, A. Miyata, O. Portugall, R. J. Sutton, A. A. Haghighirad, H. J. Snaith, D. K. Maude, P. Plochocka and R. J. Nicholas, ACS Energy Lett., 2017, 2, 1621–1627 CrossRef CAS
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed
- G. Wang, M. Lei, J. Liu, Q. He and W. Zhang, Sol. RRL, 2020, 4, 1–20 Search PubMed
- Y. Tang, A. Lesage and P. Schall, J. Mater. Chem. C, 2020, 8, 17139–17156 RSC
- J. T. Matondo, D. M. Malouangou, L. Bai, Y. Yang, J. N. Ondze, T. Bimenyimana and M. Guli, J. Mater. Chem. C, 2021, 9, 9377–9399 RSC
- F. Li and M. Liu, J. Mater. Chem. A, 2017, 5, 15447–15459 RSC
- H. Peng, M. Cai, J. Zhou, Y. Yang, X. Ding, Y. Tao, G. Wu, X. Liu, J. H. Pan and S. Dai, Sol. RRL, 2020, 4, 2000216 CrossRef CAS
- C. Zhang, K. Wang, Y. Wang, W. S. Subhani, X. Jiang, S. Wang, H. Bao, L. Liu, L. Wan and S. Liu, Sol. RRL, 2020, 4, 2000254 CrossRef CAS
- J. Shi, F. Li, Y. Jin, C. Liu, B. Cohen-Kleinstein, S. Yuan, Y. Li, Z. Wang, J. Yuan and W. Ma, Angew. Chem., 2020, 132, 22414–22421 CrossRef
- N. Mondal and A. Samanta, Nanoscale, 2017, 9, 1878–1885 RSC
- J. A. Steele, H. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. Yuan, C. Martin, E. Solano, K. Lejaeghere, S. M. J. Rogge, C. Notebaert, W. Vandezande, K. P. F. Janssen, B. Goderis, E. Debroye, Y. K. Wang, Y. Dong, D. Ma, M. Saidaminov, H. Tan, Z. Lu, V. Dyadkin, D. Chernyshov, V. Van Speybroeck, E. H. Sargent, J. Hofkens and M. B. J. Roeffaers, Science, 2019, 365, 679–684 CrossRef CAS PubMed
- H. Lin, C. Zhou, Y. Tian, T. Siegrist and B. Ma, ACS Energy Lett., 2017, 3, 54–62 CrossRef
- H. Liu, Z. Wu, H. Gao, J. Shao, H. Zou, D. Yao, Y. Liu, H. Zhang and B. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 42919–42927 CrossRef CAS PubMed
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed
- L. Yuan, Y. Zhou, Z. Wang, E. Mei, X. Liang and W. Xiang, Appl. Clay Sci., 2021, 211, 106158 CrossRef CAS
- C. Gai, D. He, Y. Wang, J. Wang and J. Li, Coatings, 2021, 11, 330 CrossRef CAS
- J. Deng, J. Xun and R. He, Opt. Mater., 2020, 99, 109528 CrossRef CAS
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS PubMed
- Q. Shang, A. L. Kaledin, Q. Li and T. Lian, J. Chem. Phys., 2019, 151, 74705 CrossRef PubMed
- H. Zhao, Z. Hu, L. Wei, P. Zeng, C. Kuang, X. Liu, S. Bai, F. Gao and M. Liu, Small, 2020, 16, 2003939 CrossRef CAS PubMed
- Y.-H. Kim, C. Wolf, Y.-T. Kim, H. Cho, W. Kwon, S. Do, A. Sadhanala, C. G. Park, S.-W. Rhee and S. H. Im, ACS Nano, 2017, 11, 6586–6593 CrossRef CAS PubMed
- R. E. Brandt, V. Stevanović, D. S. Ginley and T. Buonassisi, MRS Commun., 2015, 5, 265–275 CrossRef CAS
- Y. Song, X. Zhang, L. Li, Z. Mo, J. Xu, S. Yu, X. Liu and J. Zhang, Mater. Res. Express, 2019, 6, 115064 CrossRef CAS
- S. Xu, A. Libanori, G. Luo and J. Chen, Iscience, 2021, 24, 102235 CrossRef CAS PubMed
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed
- M. Branowski, P. Plochocka, R. Su, L. Legrand, T. Barisien, F. Bernardot, Q. Xiong, C. Testelin and M. Chamarro, Photonics Res., 2020, 8, A50–A55 CrossRef
- Q. Ma, S. Huang, X. Wen, M. A. Green and A. W. Y. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1502202 CrossRef
- Q. Wang, J. A. Smith, D. Skroblin, J. A. Steele, C. M. Wolff, P. Caprioglio, M. Stolterfoht, H. Köbler, M. Li, S. H. Turren-Cruz, C. Gollwitzer, D. Neher and A. Abate, Sol. RRL, 2020, 4, 1–9 Search PubMed
- Y.-T. Yu, S.-H. Yang, L.-H. Chou, I. Osaka, X.-F. Wang and C.-L. Liu, ACS Appl. Energy Mater., 2021, 4, 5466–5474 CrossRef CAS
- E. Breniaux, P. Dufour, S. Guillemet-Fritsch and C. Tenailleau, Eur. J. Inorg. Chem., 2021, 3059–3073 CrossRef CAS
- C. Zhang, Q. Wan, L. K. Ono, Y. Liu, W. Zheng, Q. Zhang, M. Liu, L. Kong, L. Li and Y. Qi, ACS Energy Lett., 2021, 6, 3545–3554 CrossRef CAS
- A. Dutta, R. K. Behera, P. Pal, S. Baitalik and N. Pradhan, Angew. Chem., Int. Ed., 2019, 58, 5552–5556 CrossRef CAS PubMed
- M. Imran, V. Caligiuri, M. Wang, L. Goldoni, M. Prato, R. Krahne, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 2656–2664 CrossRef CAS PubMed
- Y. Zhang, G. Li, C. She, S. Liu, F. Yue, C. Jing, Y. Cheng and J. Chu, Nano Res., 2021, 14, 2770–2775 CrossRef CAS
- H. Huang, W. Zhao, H. Yang, X. Zhang, J. Su, K. Hu, Z. Nie, Y. Li and J. Zhong, J. Mater. Chem. C, 2021, 9, 5535–5543 RSC
- L. Zhang, W. Liang, L. Xu, M. Zhu, X. Wang, J. Su, L. Li, N. Liu, Z. Zhang and Y. Gao, Chem. Eng. J., 2021, 417, 129349 CrossRef CAS
- S. Zhou, C. Zhou, J. Zhu, H. Huang, F. Hu, Q. Ye, J. Zhong, X. Yang and H. Mao, Appl. Surf. Sci., 2022, 578, 152116 CrossRef CAS
- D. Huang, J. Bo, R. Zheng, C. Luo, X. Sun, Q. Li, D. Shen, Z. Zheng, M. Chen and Y. Yang, Adv. Opt. Mater., 2021, 2100474 CrossRef CAS
- D. Zhou, R. Sun, W. Xu, N. Ding, D. Li, X. Chen, G. Pan, X. Bai and H. Song, Nano Lett., 2019, 19, 6904–6913 CrossRef CAS PubMed
- N. Mondal, A. De and A. Samanta, ACS Energy Lett., 2018, 4, 32–39 CrossRef
- J. Wei, W. Zheng, P. Huang, Z. Gong, Y. Liu, S. Lu, Z. Li and X. Chen, Nano Today, 2021, 39, 101179 CrossRef CAS
- S. Ye, M. Yu, M. Zhao, J. Song and J. Qu, J. Alloys Compd., 2018, 730, 62–70 CrossRef CAS
- S. Walck and T. Reinecke, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 9088–9096 CrossRef CAS
- P. Zhao, J. Su, Z. Lin, J. Wang, J. Zhang, Y. Hao, X. Ouyang and J. Chang, Adv. Theory Simul., 2020, 3, 1–9 Search PubMed
- Y. Wang, F. Yang, X. Li, F. Ru, P. Liu, L. Wang, W. Ji, J. Xia and X. Meng, Adv. Funct. Mater., 2019, 29, 1–9 Search PubMed
- Y. Gao, L. Zhao, Q. Shang, Y. Zhong, Z. Liu, J. Chen, Z. Zhang, J. Shi, W. Du and Y. Zhang, Adv. Mater., 2018, 30, 1801805 CrossRef PubMed
- D. Wang, J. Qiu, D. Zhou, S. Hu, Y. Wen, K. Zhang, Q. Wang, Y. Yang, H. Wu and Z. Long, Chem. Eng. J., 2021, 421, 127777 CrossRef CAS
- W. Shi, Y. Zhao, C. Xie and P. Yang, J. Lumin., 2021, 233, 117886 CrossRef CAS
- Y. Liu, G. Pan, R. Wang, H. Shao, H. Wang, W. Xu, H. Cui and H. Song, Nanoscale, 2018, 10, 14067–14072 RSC
- H. Lin, Q. Wei, K. W. Ng, J. Dong, J. Li, W. Liu, S. Yan, S. Chen, G. Xing and X. Tang, Small, 2021, 2101359 CrossRef CAS PubMed
- Z. Zhao, W. Xu, G. Pan, Y. Liu, M. Yang, S. Hua, X. Chen, H. Peng and H. Song, Mater. Res. Bull., 2019, 112, 142–146 CrossRef CAS
- Q. Hu, J. Guo, M. Lu, P. Lu, Y. Zhang, W. W. Yu and X. Bai, J. Phys. Chem. Lett., 2021, 12, 8203–8211 CrossRef CAS PubMed
- Z. He, X. Liang and W. Xiang, Chem. Eng. J., 2022, 427, 130964 CrossRef CAS
- X. Zhou, Y. Zhao, W. Huang, Y. Wu, Z. Wu and G. He, Org. Electron., 2021, 106253 CrossRef CAS
- Y. Guo, J. Su, L. Wang, Z. Lin, Y. Hao and J. Chang, J. Phys. Chem. Lett., 2021, 12, 3393–3400 CrossRef CAS PubMed
- K. Ma, Y. Sheng, G. Wang, X. Zhang, Y. Di, C. Liu, L. Yu, L. Dong and Z. Gan, J. Lumin., 2021, 118622 Search PubMed
- Z. Chen, H. He, Z. Wen, Z. Cui, S. Mei, D. Yang, X. Wei, W. Zhang, F. Xie and B. Yang, Mater. Sci. Eng. B, 2021, 273, 115426 CrossRef CAS
- H. Zhang, J. Yao, Y. Yang and H. Fu, Chem. Mater., 2021, 33, 2847–2854 CrossRef CAS
- A. Forde, S. A. Thomas, R. J. Petersen, S. L. Brown, D. S. Kilin and E. K. Hobbie, J. Phys. Chem. C, 2021, 125, 18849–18856 CrossRef CAS
- Z.-J. Yong, S.-Q. Guo, J.-P. Ma, J.-Y. Zhang, Z.-Y. Li, Y.-M. Chen, B.-B. Zhang, Y. Zhou, J. Shu and J.-L. Gu, J. Am. Chem. Soc., 2018, 140, 9942–9951 CrossRef CAS PubMed
- T. Chen, R. Liu, S. Tang, X. Li, S. Ye, X. Duan, R. Wang, Y. Shi, J. Yang and F. Qiu, AIP Adv., 2021, 11, 115008 CrossRef
- Y. Gao, C. Luo, C. Yan, W. Li, C. Liu and W. Yang, J. Colloid Interface Sci., 2022, 607, 1796–1804 CrossRef CAS PubMed
- R. Wu, Z. Bai, J. Jiang, H. Yao and S. Qin, RSC Adv., 2021, 11, 8430–8436 RSC
- Y. Tong, Q. Wang, X. Liu, E. Mei, X. Liang and W. Xiang, Chem. Eng. J., 2022, 429, 132391 CrossRef CAS
- Y. Li, Q. Liu, X. Liu, J. Feng, L. He, H. Li, C. Li and H. Zhang, J. Phys. Chem. Lett., 2021, 12, 10746–10752 CrossRef CAS PubMed
- X. Li, Y. Wei, P. Dang, X. Xiao, H. Xiao, G. Zhang, G. Li and J. Lin, Mater. Res. Bull., 2022, 146, 111592 CrossRef CAS
- D. Yan, Q. Mo, S. Zhao, W. Cai and Z. Zang, Nanoscale., 2021, 13, 9740–9746 RSC
- L. Xu, S. Yuan, H. Zeng and J. Song, Mater. Today Nano, 2019, 6, 100036 CrossRef
- Y. Ji, J.-B. Zhang, H.-R. Shen, Z. Su, H. Cui, T. Lan, J.-Q. Wang, Y.-H. Chen, L. Liu and K. Cao, ACS Omega, 2021, 6, 13831–13838 CrossRef CAS PubMed
- J.-K. Chen, J.-P. Ma, S.-Q. Guo, Y.-M. Chen, Q. Zhao, B.-B. Zhang, Z.-Y. Li, Y. Zhou, J. Hou and Y. Kuroiwa, Chem. Mater., 2019, 31, 3974–3983 CrossRef CAS
- Y. Zhao, C. Xie, X. Zhang, K. Matras-Postolek and P. Yang, ACS Appl. Nano Mater., 2021, 4, 6223–6230 CrossRef CAS
- Z. Chen, L. Dong, C. Zhou, B. Zhou, Z. Zheng, R. Chen and J. Zang, CrystEngComm, 2021, 23, 793–803 RSC
- Y. Gao, C. Yan, X. Peng, W. Li, J. Cao, Q. Wang, X. Zeng, X. Fu and W. Yang, Nanoscale, 2021, 13, 18010–18031 RSC
- X. Shen, Y. Zhang, S. V. Kershaw, T. Li, C. Wang, X. Zhang, W. Wang, D. Li, Y. Wang and M. Lu, Nano Lett., 2019, 19, 1552–1559 CrossRef CAS PubMed
- F. Qin, H. Tian, M. Yan, Y. Fang and D. Yang, J. Mater. Chem. C, 2021, 9, 2016–2023 RSC
- J. Chen, Z. Shen, P. Liu, Z. Sun, J. G. Liu, C. Shen, D. Song, S. Zhao and Z. Xu, Nanotechnology, 2021, 32, 325202 CrossRef CAS PubMed
- S. Yang, H. Zhu, E. Xu, J. Li, H. Yang, Y. Zhang, Z. Zhu and Y. Jiang, Nanotechnology, 2021, 32, 335601 CrossRef CAS PubMed
- R. Chen, Y. Xu, S. Wang, C. Xia, Y. Liu, B. Yu, T. Xuan and H. Li, J. Alloys Compd., 2021, 866, 158969 CrossRef CAS
- M. Rai, S. K. Singh, K. Mishra, R. Shankar, R. K. Srivastava and S. B. Rai, J. Mater. Chem. C, 2014, 2, 7918–7926 RSC
- Q. Hu, Z. Li, Z. Tan, H. Song, C. Ge, G. Niu, J. Han and J. Tang, Adv. Opt. Mater., 2018, 6, 1–5 RSC
- G. Pan, X. Bai, D. Yang, X. Chen, P. Jing, S. Qu, L. Zhang, D. Zhou, J. Zhu, W. Xu, B. Dong and H. Song, Nano Lett., 2017, 17, 8005–8011 CrossRef CAS PubMed
- L. Shao, D. Liu, J. Lyu, D. Zhou, N. Ding, R. Sun, W. Xu, N. Wang, S. Xu and B. Dong, Mater. Today Phys., 2021, 100495 CrossRef CAS
- M. Zeng, S. Singh, Z. Hens, J. Liu, F. Artizzu and R. Van Deun, J. Mater. Chem. C, 2019, 7, 2014–2021 RSC
- X. Xu, Y. Sun, D. He, Z. Liang, G. Liu, S. Xu, Z. Li, L. Zhu and X. Pan, J. Mater. Chem. C, 2021, 9, 208–213 RSC
- L. Zhou, T. Liu, J. Zheng, K. Yu, F. Yang, N. Wang, Y. Zuo, Z. Liu, C. Xue, C. Li, B. Cheng and Q. Wang, J. Phys. Chem. C, 2018, 122, 26825–26834 CrossRef CAS
- J. P. Ma, Y. M. Chen, L. M. Zhang, S. Q. Guo, J. D. Liu, H. Li, B. J. Ye, Z. Y. Li, Y. Zhou, B. Bin Zhang, O. M. Bakr, J. Y. Zhang and H. T. Sun, J. Mater. Chem. C, 2019, 7, 3037–3048 RSC
- J. Duan, Y. Zhao, X. Yang, Y. Wang, B. He and Q. Tang, Adv. Energy Mater., 2018, 8, 1–9 Search PubMed
- L. Chen, W. Wu, J. Wang, Z. Qian, R. Liu, Y. Niu, Y. Chen, X. Xie and H. Zhang, ACS Appl. Energy Mater., 2021, 4, 3937–3944 CrossRef CAS
- H. Huang, R. Li, S. Jin, Z. Li, P. Huang, J. Hong, S. Du, W. Zheng, X. Chen and D. Chen, ACS Appl. Mater. Interfaces, 2021, 13, 34561–34571 CrossRef CAS PubMed
- X. Zhang, Y. Zhang, X. Zhang, W. Yin, Y. Wang, H. Wang, M. Lu, Z. Li, Z. Gu and W. W. Yu, J. Mater. Chem. C, 2018, 6, 10101–10105 RSC
- M. Rai, S. K. Singh, A. K. Singh, R. Prasad, B. Koch, K. Mishra and S. B. Rai, ACS Appl. Mater. Interfaces, 2015, 7, 15339–15350 CrossRef CAS PubMed
- A. K. Singh, Coord. Chem. Rev., 2022, 455, 214365 CrossRef CAS
- Q. Cao, A. Ilyas, S. Zhang, Z. Ju, F. Sun, T. Liu, Y. M. Yang, Y. H. Lu and X. Liu, Nanoscale, 2021, 13, 11552–11560 RSC
- L. Wang, H. Zhou, J. Hu, B. Huang, M. Sun, B. Dong, G. Zheng, Y. Huang, Y. Chen and L. Li, Science, 2019, 363, 265–270 CrossRef CAS PubMed
- A. K. Jena, A. Kulkarni, Y. Sanehira, M. Ikegami and T. Miyasaka, Chem. Mater., 2018, 30, 6668–6674 CrossRef CAS
- J. Shi, F. Li, J. Yuan, X. Ling, S. Zhou, Y. Qian and W. Ma, J. Mater. Chem. A, 2019, 7, 20936–20944 RSC
- B. Wang, L. Liu, B. Liu, J. Li, B. Cao, Z. Zhao and Z. Liu, Phys. B, 2020, 599, 412488 CrossRef CAS
- G. Pan, X. Bai, W. Xu, X. Chen, D. Zhou, J. Zhu, H. Shao, Y. Zhai, B. Dong, L. Xu and H. Song, ACS Appl. Mater. Interfaces, 2018, 10, 39040–39048 CrossRef CAS PubMed
- J.-P. Ma, Y.-M. Chen, L.-M. Zhang, S.-Q. Guo, J.-D. Liu, H. Li, B.-J. Ye, Z.-Y. Li, Y. Zhou and B.-B. Zhang, J. Mater. Chem. C, 2019, 7, 3037–3048 RSC
- Y. Zhu, B. Yang, Q. Lu, L. Zhang, Y. Zhao, B. Xia, S. Mei, M. Shi, Y. Li and R. Hu, J. Non Cryst. Solids, 2022, 575, 121224 CrossRef CAS
- L. Yuan, L. Zhou, W. Xiang and X. Liang, J. Non Cryst. Solids, 2020, 545, 120232 CrossRef CAS
- S. K. Karunakaran, G. M. Arumugam, W. Yang, S. Ge, S. N. Khan, Y. Mai, X. Lin and G. Yang, Sol. RRL, 2020, 2000390 CrossRef CAS
- W. Xiang, Z. Wang, D. J. Kubicki, W. Tress, J. Luo, D. Prochowicz, S. Akin, L. Emsley, J. Zhou, G. Dietler, M. Grätzel and A. Hagfeldt, Joule, 2019, 3, 205–214 CrossRef CAS
- M. Wang, K. Deng, L. Meng and L. Li, Small Methods, 2020, 4, 1–8 Search PubMed
- Y. Xie, B. Peng, I. Bravić, Y. Yu, Y. Dong, R. Liang, Q. Ou, B. Monserrat and S. Zhang, Adv. Sci., 2020, 7, 28–30 Search PubMed
- C. Zhang, A. Zhang, T. Liu, L. Zhou, J. Zheng, Y. Zuo, Y. He and J. Li, RSC Adv., 2020, 10, 17635–17641 RSC
- W. J. Mir, T. Sheikh, H. Arfin, Z. Xia and A. Nag, NPG Asia Mater., 2020, 12, 1–9 CrossRef
- T. J. Milstein, D. M. Kroupa and D. R. Gamelin, Nano Lett., 2018, 18, 3792–3799 CrossRef CAS PubMed
- M. Zeng, F. Locardi, D. Mara, Z. Hens, R. Van Deun and F. Artizzu, Nanoscale, 2021, 13, 8118–8125 RSC
- X. Li, Y. Yu, J. Hong, Z. Feng, X. Guan, D. Chen and Z. Zheng, J. Lumin., 2020, 219, 116897 CrossRef CAS
- Y. Zhang, J. Liu, H. Zhang, Q. He, X. Liang and W. Xiang, J. Eur. Ceram. Soc., 2020, 40, 6023–6030 CrossRef CAS
- G. Yao, S. Li, D. Valiev, Q. Chen, Y. Hu, L. Jia, S. Stepanov, Y. Zhou, C. Li and Z. Su, Opt. Mater., 2021, 122, 111711 CrossRef
- Y. Yu, G. Shao, L. Ding, H. Zhang, X. Liang, J. Liu and W. Xiang, J. Rare Earths, 2021, 39, 1497–1505 CrossRef CAS
- P. Feng, X. Yang, X. Feng, G. Zhao, X. Li, J. Cao, Y. Tang and C.-H. Yan, ACS Nano, 2021, 15, 6266–6275 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2022 |