
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a new 3D bioprinted antibiotic delivery system based on a cross-linked gelatin–alginate hydrogel
Adam
Mirek
ab,
Habib
Belaid
b,
Fanny
Barranger
b,
Marcin
Grzeczkowicz
a,
Yasmine
Bouden
c,
Vincent
Cavaillès
c,
Dorota
Lewińska
a and
Mikhael
Bechelany
 *b
*b
aNalecz Institute of Biocybernetics and Biomedical Engineering, Polish Academy of Sciences, 4 Ks. Trojdena St., 02-109 Warsaw, Poland
bInstitut Européen des Membranes, IEM, UMR 5635, Univ Montpellier, CNRS, ENSCM Place Eugène Bataillon, 34095 Montpellier cedex 5, France. E-mail: mikhael.bechelany@umontpellier.fr; Fax: +33467149119; Tel: +33467149167
cIRCM, Institut de Recherche en Cancérologie de Montpellier, INSERM U1194, Université Montpellier, Montpellier F-34298, France
First published on 5th August 2022
Abstract
3D bioprinting uses bioink deposited directly on a collector to create any previously designed 3D model. One of the most common and the easiest to operate bioinks is gelatin–alginate hydrogel. The present study aimed to combine 3D bioprinting with different cross-linking techniques to develop a new stable and biodegradable gelatin–alginate hydrogel matrix for drug delivery applications. The matrix-building biopolymers were crosslinked by ionotropic gelation with Ca2+ ions, chemical crosslinking with GTA or a combination of the two crosslinkers at various concentrations. The influence of the crosslinking method on the hydrogel properties, stability and structure was examined using scanning electron and optical microscopy, differential scanning calorimetry and thermogravimetric analysis. Analyses included tests of hydrogel equilibrium swelling ratio and release of marker substance. Subsequently, biological properties of the matrices loaded with the antibiotic chlorhexidine were studied, including cytotoxicity on HaCAT cells and antibacterial activity on Staphylococcus aureus and Escherichia coli bacteria. The conducted study confirmed that the 3D bioprinted cross-linked drug-loaded alginate–gelatin hydrogel is a good and satisfying material for potential use as a drug delivery system.
1. Introduction
3D bioprinting has developed intensively since the beginning of 2010s and has gained significant interest in both medicine and pharmaceutics.1 It is used for advanced tissue engineering,2–5 drug delivery6 and drug screening,7 wound dressings,8–10 and cancer research.11 The most common 3D bioprinters use material jetting (noncontact bioprinting, generating picolitre droplets and firing at a frequency of thousands times per second),12 vat polymerization (photohardening of bioresins)13 or extrusion.2,3,14 In this technique controlled volumes of liquid are delivered to predefined locations.15 The material known as a bioink is deposited directly on a collector creating any previously designed 3D model. A wide variety of bio-artificial tissues has been printed including bone, cardiac, lung, pancreas, skin, and vascular tissues.16 However, not only can bioink be loaded with living cells but also enriched with various bioactive substances. For instance, Jingjunjiao and co-workers bioprinted drug delivery systems based on chitosan-pectin hydrogel with lidocaine hydrochloride for wound dressings.17Hydrogels seem to be the best bioinks, derived from both synthetic or natural polymers,1 the latter being obtained from mammalian (collagen, gelatin, fibrin, elastin) or non-mammalian sources (agarose, alginate, chitosan).18 They meet lots of requirements including good cell adhesion, expected biodegradation rate, non-cytotoxicity, suitable rheological properties (sufficiently viscous to be dispensed as a filament), fast gelation time and adequate strength and stiffness. One of the most common and the easiest to operate bioinks is gelatin–alginate hydrogel.10,19–21 Its printing temperature (37 °C) makes it suitable for use with living cells and heat-sensitive substances such as drugs. It is solid in room temperature but the solidification process can be sped up by lowering the temperature of the collector.21 Once the gelatin is dissolved in warm water, it is transformed into a hydrogel when cooled to room temperature as a result of forming weak hydrogen bond network. Moreover, gelatin addition into the alginate solution improves the viscosity and stiffness for extrusion.19
Depending on the composition, a hydrogel bioink can undergo various gelation processes. The biopolymer molecules can be assembled by non-covalent or covalent bonds.1 A printable material can often be yielded thanks to weak binding forces between polymer chains (hydrogen-bonding, π-stacking and van der Waals forces). Another way to link polymer molecules is to use an ionic bond – e.g. for polysaccharides such as alginate, pectin and gellan gum that require bivalent cations (e.g. Ca2+) to form intermolecular bonds between polymer chains. Non-covalent bonds are fast to form but due to their weakness and reversibility they cannot be used for long-acting systems or scaffolds. However, they can be a good starting point to create a 3D hydrogel model, which could then be subjected to post-processing action aimed at creating covalent bonds and thus strengthening the structure through photo or chemical cross-linking. For gelatin–alginate hydrogels, this can be achieved in two ways – ionotropic gelation creating links between anionic alginate chains (COO− groups) by means of calcium cations (Ca2+) or chemical crosslinking of gelatin chains with the use of glutaraldehyde (GTA). GTA undergoes a Schiff's base reaction with the amino groups of the amino acids present in gelatin. As a result –CO–NH– bonds are formed creating a sustainable network of gelatin–gelatin and gelatin–alginate chains.22
Alginate–gelatin hydrogel has been used in biomedical engineering for a long time, it is also widely used for the 3D printing of drug- and cell-loaded scaffolds.10,19–21,23 Its printing temperature (37 °C) makes it suitable for use with living cells and mild to sensitive substances such as drugs. It is solid in room temperature but the solidification process can be sped up by lowering the temperature of a Petri dish where a printout is collected.21 Once the gelatin is dissolved in warm water, it can transform into a hydrogel when is cooled to room temperature as a result of forming weak hydrogen bond network. Also, the addition of gelatin into alginate solution improves the viscosity and stiffness for extrusion.19 Therefore, it is possible to print a 3D model quickly and easily and then strengthen it by crosslinking. In the context of novel drug delivery systems, it seems interesting that, by using different crosslinking techniques in different conditions, the degradation time of the printed hydrogel matrix could be controlled, and this can directly translate into the time of drug release. The highly cross-linked matrix-building biopolymer chains create a more compact structure, limiting swelling of hydrogel, and thus slowing down the substance release process. Moreover, the substance is released from such a material not only by diffusion, but also during the degradation or dissolution of the material – this phenomenon can be reduced by cross-linking, thus limiting the burst effect.24,25
Not only are hydrogels useful as drug delivery systems due to the cross-linking-based control over the substance release, but also because they can absorb wound exudates. This is a desirable feature of modern dressings due to the harmfulness of some enzymes present in such exudates which can delay the healing process by affecting cell proliferation and activity.26 In addition, the high water content of hydrogel matrices used as wound dressing could ensure adequate hydration of the wound area, which can prevent desiccation and cell death, promote tissue regeneration and enhance epidermal migration.27
Based on these considerations, the present study aimed to combine 3D bioprinting technology with controlled release of chlorhexidine acetate in order to develop a novel stable biodegradable and antibacterial 3D bioprinted gelatin–alginate hydrogel matrix for potential wound dressing applications. Two different methods of cross-linking were used based on ionotropic gelation with Ca2+ ions or chemical crosslinking with GTA. Their influence on the gelatin–alginate matrices (GAMs) properties including equilibrium swelling ratio, drug release, cytocompatibility and antibacterial activity was examined using multiple techniques. The material developed in the work can be used as a new drug delivery system.
2. Materials and methods
Materials
Gelatin type B (CAS Number: 9000-70-8) from bovine skin (gel strength 225 g Bloom), sodium alginate (CAS Number: 9005-38-3), calcium chloride (CaCl2, CAS Number: 10043-52-4, ≥97%), sodium chloride (NaCl, CAS Number: 7647-14-5, ≥99%), potassium chloride (KCl, CAS Number: 7447-40-7, ≥99%), potassium phosphate monobasic (KH2PO4, CAS Number: 7778-77-0, ≥99%), sodium phosphate dibasic (Na2HPO4, CAS Number: 7558-79-4, ≥99) and agar (CAS Number: 9002-18-0) were purchased from Sigma-Aldrich. Aqueous 25% glutaraldehyde (GTA) solution (CAS Number: 111-30-8) was purchased from Alfa Aesar. Glycine (CAS Number: 56-40-6) was purchased from Sigma-Aldrich and used as a neutralization agent for GTA. Rhodamine 640 perchlorate (CAS Number: 72102-91-1) was purchased from Exciton. Chlorhexidine acetate (CAS Number: 56-95-1) was obtained from Fluka Chemicals. The non-pathogenic Gram positive Staphylococcus aureus and Gram negative Pseudomonas aeruginosa (K12 DSM 423, from DSMZ, Germany) were chosen as a model microorganism. The culture medium was a Tryptone Salt Broth (TSB, Sigma Aldrich). The chemicals were used without further purification. Water at 18 MΩ was produced by MilliQ (Millipore).Bioink preparation
The bioink used during 3D bioprinting was prepared, using the procedure described by Pan et al.21 Sodium alginate (0.04 g mL−1) was dissolved in reverse-osmosis water at a room temperature. Gelatin powder (0.2 g mL−1) was added to the alginate solution and stirred at 40 °C for 2 hours. The matrices for in vitro release tests were prepared by adding rhodamine 640 (0.1 mg mL−1) to pure water before the sodium alginate and it was stirred at a room temperature until rhodamine crystals were completely dissolved. The hydrogel matrices with drug for bioactivity studies were prepared using bioink loaded with chlorhexidine acetate (0.1 and 1 mg mL−1) which was added to pure water before sodium alginate and completely dissolved (analogous to rhodamine). Each bioink was centrifuged for 2 minutes at 1500 rpm to remove air and carefully transferred to the bioprinting syringe. After preparation of the alginate–gelatin bioink and placing in the extrusion syringe, it can be stored in a fridge. Once it is set in the heating holder, bioink liquefies at 37 °C within 15–20 minutes, and then 3D model can be printed.3D bioprinting of gelatin–alginate hydrogel matrices (GAMs) with rhodamine and drug
A model of matrices was designed using DesignSpark Mechanical 4.0 software as a cubic mesh (20 mm × 20 mm × 12 mm) made of 6 layers, each of height 0.2 mm; the grid size was set as 1 mm and line width as 0.2 mm (Fig. 1). The model was exported as an STL file, sliced by Repetier-Host/Slic3r software and converted into G-code. The gelatin–alginate matrices were manufactured using Allevi 2 3D bioprinter (Philadelphia, USA). The scheme of the experimental setup is shown in Fig. 1. The scheme of 3D bioprinting setup with a digital 3D mesh matrix design. The following settings were used: G30 nozzle with inner diameter of 0.164 mm, layer height of 0.2 mm, printing speed of 10 mm s−1, temperature of the bioink in the syringe of 37 °C, temperature of the collector Petri dish of ∼15 °C, and printing pressure of 380 kPa (55 psi). After printing, the samples were stored in a refrigerator (5 °C).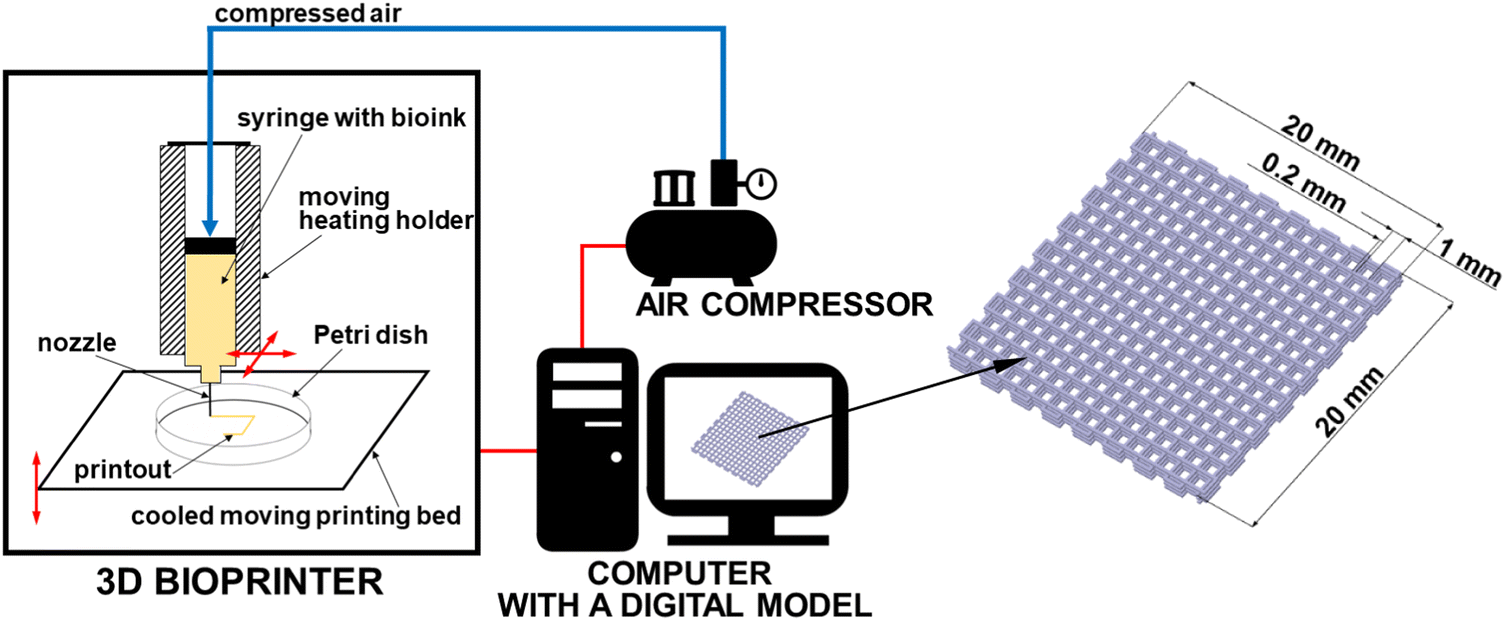 | ||
| Fig. 1 The scheme of 3D bioprinting setup with a digital 3D mesh matrix design. To simplify the picture, only one syringe holder is shown in the diagram (the Allevi 2 3D bioprinter model has two holders that can be used simultaneously during one print). | ||
Cross-linking of the matrices
The obtained GAMs were treated using Ca2+ ions due to the cross-linking of alginate present in the structure and glutaraldehyde (GTA) due to the gelatin. In order to find the best cross-linking method for the hydrogel matrices produced, a series of experiments was carried out using various cross-linking methods. To provide calcium ions, the samples placed in the glass Petri dishes were sprayed with 2% or 5% solution on each side and left to cross-link for 10 minutes (method 1). Finally, the samples were dried by gently placing them on a tissue and touching them with the other piece of paper from the other side. For GTA cross-linking, the GAMs were treated with the method similar to the one by Zhang et al.28 with use of the GTA aqueous solutions of concentrations 0.5%, 5% and 25%. The solution (20 mL) was placed in hermetic containers equipped with a plastic grate located about 5 mm above the surface of the solution. The GAMs were conditioned in GTA vapors for 24 h at 37 °C (method 2). After that, in order to remove and neutralize GTA residues, each sample was slightly rinsed with RO water, placed for 48 hours in PBS solution and then for next 24 hours in 20% glycine solution.In the series of 12 experiments, the samples of matrices were cross-linked in different ways – using the first method, using the second method or using a combination of both methods. The obtained GAMs are summarized in Table 1 with a proper denotation for each of them (the first number corresponds to the concentration of calcium ions, the second number corresponds to the concentration of GTA in the solutions used for cross-linking). The GAMs for studies requiring anhydrous samples were frozen (8 h at −20 °C) and lyophilized (24 h at 12 Pa, −70 °C).
| Sample | Cross-linking procedure |
|---|---|
| 0_0_GAM | No cross-linking |
| 0_0.5_GAM | 0.5% GTA vapors |
| 0_5_GAM | 5% GTA vapors |
| 0_25_GAM | 25% GTA vapors |
| 2_0_GAM | 2% Ca2+ solution spraying |
| 2_0.5_GAM | 2% Ca2+ solution spraying + 0.5% GTA vapors |
| 2_5_GAM | 2% Ca2+ solution spraying + 5% GTA vapors |
| 2_25_GAM | 2% Ca2+ solution spraying + 25% GTA vapors |
| 5_0_GAM | 5% Ca2+ solution spraying |
| 5_0.5_GAM | 5% Ca2+ solution spraying + 0.5% GTA vapors |
| 5_5_GAM | 5% Ca2+ solution spraying + 5% GTA vapors |
| 5_25_GAM | 5% Ca2+ solution spraying + 25% GTA vapors |
Characterization of the cross-linked GAMs
The morphology of the cross-linked and lyophilized GAMs was observed using scanning electron microscopy (SEM, HITACHI S4800), optical microscopy and fluorescent microscopy systems. The samples were covered with 10 nm layer of gold before observations with SEM.Thermal properties
Differential scanning calorimetry (DSC) was used to investigate the thermal properties of GAMs. The thermal transition points and enthalpies (calculated as an area under the peak) of the differently cross-linked GAMs were analyzed using differential scanning calorimeter (DSC, TA Instruments 2920) equipped with a RCS90 cooling system. The samples were weighed (∼2–3 mg) in an aluminum TA pan and sealed. An empty sealed pan was used as a reference. The samples were cooled to −80 °C and then heated up to 200 °C with a heating rate of 20 °C min−1. Moreover, the thermogravimetric analysis (TGA, TA Instruments TGA G500) was performed under oxygen flow of 60 mL min−1 to define the cross-linking influence on a thermal stability of matrices. The samples of ∼6–10 mg were heated up to 1000 °C at a heating rate of 10 °C min−1.Swelling of the matrices
The equilibrium swelling ratio (ESR) of cross-linked lyophilized GAMs in phosphate buffered saline (PBS) at 37 °C was determined using a gravimetric method.29 Samples were weighed (∼15 mg) and placed in a tightly closed bottle with 10 ml of swelling medium (PBS). At the preset time intervals, the samples were withdrawn, surface water was gently removed with a tissue and the GAMs were immediately weighed and placed again in PBS. The experiments were continued until a constant weight was observed. The ESR was calculated using eqn (1):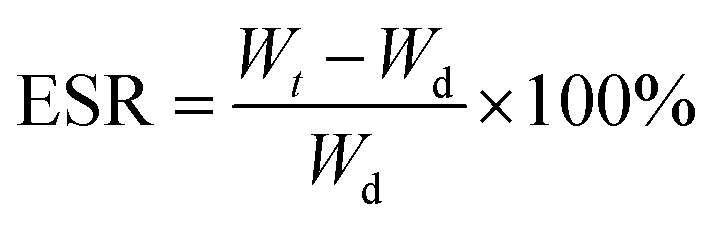 | (1) |
Three samples of the same hydrogel matrix were used to perform the experiment and count average ESR value.
Model drug molecule (rhodamine) release
The release profile of rhodamine 640 from GAMs was determined using a flow spectrophotometric method. Light wavelength: 574 nm. A sample of 5.0–7.5 mg was cut from each freeze-dried GAM and placed in a glass container with 2.0–3.5 mL of deionized water. The content of the container was stirred all the time. Measurements of absorbance of the solution were made every 2 minutes for the first 6 hours, every 30 minutes for the next 2 hours, and the last measurement was made after 9 hours. The volume of the tested liquid did not change during the process. Such a method was proposed by Grzeczkowicz et al.30 On the basis of the obtained data, the profiles of rhodamine release from GAMs were plotted on graphs, and then the curves were fitted to them using the OriginPro software, using the linear and exponential fit with the ExpDec1 function available in the program library. Selected samples were transferred with water to sealed tubes and their concentration was tested several times with a time interval of several weeks to check the long-term profile of rhodamine release from GAMs. All measeurements were repeated three times and the average value of absorbance for each sample was calculated.Cell culture and cytotoxicity assays
HaCaT cells (immortalized human keratinocytes) were cultured using DMEM (Dulbecco's Modified Eagle Medium α) (Gibco 31331-028) media supplemented with 10% (v/v) fetal bovine serum (FBS) (Eurobio CVFSVF00-01). Cells were cultured at 37 °C in 5% CO2 in a 10 cm diameter Petri dish and trypsinized using 0.05% Trypsin-EDTA (Gibco 25300-054). HaCat cells (104 cells per well) were seeded in 96 well plates and allowed to attach for 24 hours. After sterilization with 70% (w/v) ethanol for 30 min and UV irradiation for 1 h, the hydrogels were dried and added to the cell culture. The sterilized samples were added in the wells and incubated for 1, 4, 6 and 8 days. Cell viability was analyzed using MTT assay carried out by incubating 100 μL of 0.5 mg mL−1 of MTT solution on the cells for 3 h. Purple coloured formazan crystals were dissolved using 100 μL of dimethyl sulfoxide (DMSO, BDH Prolab 23486.297) and the absorbance was recorded at 560 nm using Multiskan plat reader (thermos, USA).Antibacterial activity of the samples loaded with chlorhexidine acetate
The antibacterial activity of drug-loaded GAMs was examined against Staphylococcus aureus (S. aureus, Gram positive) and Pseudomonas aeruginosa (P. aeruginosa, Gram negative). These bacteria were selected because they are among the primary causes of delayed healing and infection in both acute and chronic wounds.31 For the cultivation, bacteria aliquots frozen at −20 °C were used. Fresh Tryptone Salt Broth (TSB) medium was inoculated by bacteria and incubated overnight at 37 °C under constant rotation and in aerobic conditions. Once the stationary phase was reached, cells were harvested by centrifugation and resuspended in a TSB. The optical density at 620 nm (OD600) of the bacterial suspension was then adjusted to 0.75 ± 0.01 for S. aureus and 0.80 ± 0.01 for P. aeruginosa by dilution.For the antibacterial tests three samples were prepared – cross-linked with 2% Ca2+ and 0.5% GTA and neutralized with 20% glycine solution with the chlorhexidine acetate content: 0 mg mL−1, 0.1 mg mL−1 and 1 mg mL−1. The samples were sterilized by exposition to UVC light (10 min for each sample side). Mueller–Hinton agar (GMH) plates were prepared by adding microbiological agar (15 g·L−1) to PBS medium; two rectangular dishes were used, each for one type of bacteria. GMH agar plates were inoculated individually with 1 mL of S. aureus or P. aeruginosa suspension. Immediately after, the samples of GAMs were put onto the inoculated GMH plates to characterize the ability of the materials to prevent bacterial growth around the samples. The plates were then incubated overnight at 37 °C in aerobic conditions to allow the formation of a bacterial biofilm. Plates were pictured to check the presence of clear zones (inhibited bacterial growth) around the samples and within the biofilm formed.
3. Results and discussion
3D bioprinting feasibility
A total of 12 different GAM were produced with different cross-linking methods (Table 1). When the hydrogel is extruded onto a cooled Petri dish at a temperature of about 10–15 °C, it solidifies quickly, allowing the printing of desired shapes. For example, freshly printed hydrogel matrix is shown in Fig. 2(A) – the digital model of the matrix was presented in the previous section (Fig. 1). The gelatin–alginate bioink shows good printability with visible grid lines with its form so tight that it can be easily removed from the Petri dish, it has adequate mechanical integrity and maintains a shape. Fig. 2(B) presents GAM filled with rhodamine 640 – the addition of a marker does not affect bioink printability. The cross-linking process affects the macroscopic structure of hydrogel matrices. GAMs cross-linked with GTA vapors slightly shrunked and changed their color to yellow-orange (Fig. 2(C)) which is due to the establishment of aldimine bonds (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) between the free amine groups of gelatin proteins and glutaraldehyde.32–34 After processing, the GAMs become visibly harder and stronger but they do not lose their flexibility which improves the potential contact with wound and is beneficial for comfort of use.
N) between the free amine groups of gelatin proteins and glutaraldehyde.32–34 After processing, the GAMs become visibly harder and stronger but they do not lose their flexibility which improves the potential contact with wound and is beneficial for comfort of use.
 | ||
| Fig. 2 3D bioprinted gelatin–alginate hydrogel (gelatin 0.2 g mL−1, sodium alginate 0.04 g mL−1) matrices before lyophilization – (A)–(C) phone camera pictures: (A) immediately after printing, bioink without additives, no cross-linking, (B) bioink with rhodamine, no cross-linking, (C) bioink without additives, after cross-linking with 2% Ca2+ and 25% GTA solutions; (D) optical microscopy picture, magnification 200×, bioink without additives, after cross-linking with 2% Ca2+ and 25% GTA solutions; (E) and (F) fluorescent microscopy pictures, bioink without additives, after cross-linking with 2% Ca2+ and 25% GTA solutions: (E) magnification 200×, (F) magnification 20×, (G) fluorescent microscopy picture, magnification 20×, bioink without additives, after cross-linking with 25% GTA solution. | ||
During printing, each printed matrix layer solidified before the next layer was plotted on it, as illustrated by optical (Fig. 2(D)) and fluorescent (Fig. 2(E)) microscopy pictures. It is easy to see that the top layer (horizontal) covers the bottom layer (vertical). They do not fuse with each other. Unfortunately, not all matrices printed one after the other are identical. Despite the same model and printing parameters, the continuity of grid happens to be broken, which can be seen by comparing Fig. 2(F) and (G) (red circle). The reason for this may be over- or underliquefying (too little or too much hydrogel is supplied) or blocking the nozzle, which was also observed by Long et al.17 with chitosan-pectin hydrogel. The structure of the matrix has been retained after printing, but the sizes of model elements (pores, grids) are not consistent with those designed in the computer model (Fig. 1). Adjusting the printing speed or greater control over the viscosity of the bioink (e.g. by changing the process and surrounding temperature) could solve those problems. The speed of the moving printing nozzle and the viscosity of the bioink are of great importance for ensuring the continuity of the printout.1
Effect of chemical cross-linking and lyophilization on the structure of GAMs
The surface morphology of 3D printed GAMs was examined using scanning electron microscopy (SEM). The SEM technique requires the samples to be dehydrated, so all of the matrices were lyophilized as described in the Materials and methods section. Analysis of SEM images (Fig. 3) and comparison with images from optical and fluorescent microscopes (Fig. 2(D)–(G)) showed a clear effect of freeze-drying on GAMs structure. Indeed, SEM showed matrices that are not clearly multi-layered (subsequent layers have fused together). In addition, the hydrogel surface appeared not smooth and depended on the crosslinking method (see smaller pictures provided in Fig. 3). The only smooth surface was the underside, which was deposited directly on the slide during printing as a first layer (Fig. 3(E)).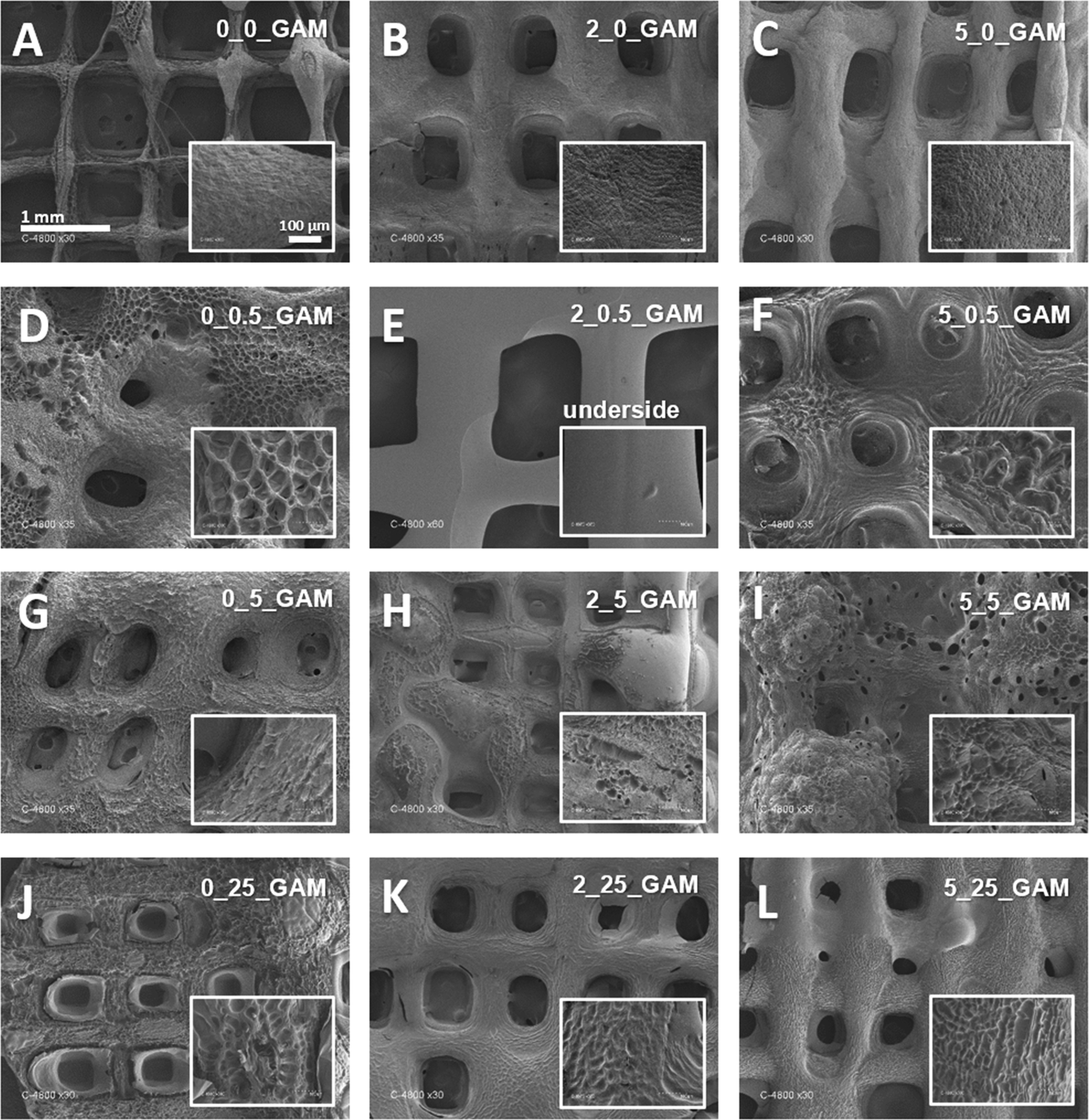 | ||
| Fig. 3 SEM pictures of gelatin–alginate matrices cross-linked with agents of different concentration: (A) no crosslinking, (B) 2% Ca2+, no GTA, (C) 5% Ca2+, no GTA, (D) no Ca2+, 0.5% GTA, (E) 2% Ca2+, 0.5% GTA, (F) 5% Ca2+, 0.5% GTA, (G) no Ca2+, 5% GTA, (H) 2% Ca2+, 5% GTA, (I) 5% Ca2+, 5% GTA, (J) no Ca2+, 25% GTA, (K) 2% Ca2+, 25% GTA, (L) 5% Ca2+, 25% GTA – magnification 30×; surface structures – magnification 300×. | ||
As shown in Fig. 3, GAMs that were not cross-linked retained a regular mesh structure, with slightly visible layers (Fig. 3(A)). The surface of the matrix cross-linked with calcium ions looked similar (Fig. 3(B) and (C)) i.e. rough, slightly porous, but uniform and compact. Cross-linking of GAMs with GTA caused significant changes in their structure. One modification is that subsequent layers of hydrogel fused with each other (Fig. 3(D) and (F)). Moreover, the holes in the matrix lose their rectangular shape and became rounded or disappeared, which is clearly visible especially in the case of matrices cross-linked with calcium ions and GTA vapors (Fig. 3(I) and (L)). Altogether, GTA vapor crosslinking impacted the uniformity and smoothness of GAMs surfaces, which become porous with pores relatively big as compared to those of non-crosslinked matrices (Fig. 3(A)).
Fig. 4 shows the differential scanning calorimetry and thermogravimetric analysis curves of GAMs samples. Additionally, the graphs were enriched with the curves for pure gel and sodium alginate as well as the temperatures of thermal transitions. Thermal transition enthalpies (ΔH1 – gel–sol transition, ΔH2 – glass transition, ΔH3 – melting) with the total enthalpy change ΔH for pure gelatine, pure sodium alginate and the GAMs obtained from the DSC analysis are presented in Table 2. For sake of clarity of the data, it was decided to present only the ones for samples 0_0_GAM, 0_5_GAM, 2_0_GAM and 2_5_GAM, as there were no significant differences to the other samples or additional information that could be derived from an extended analysis.
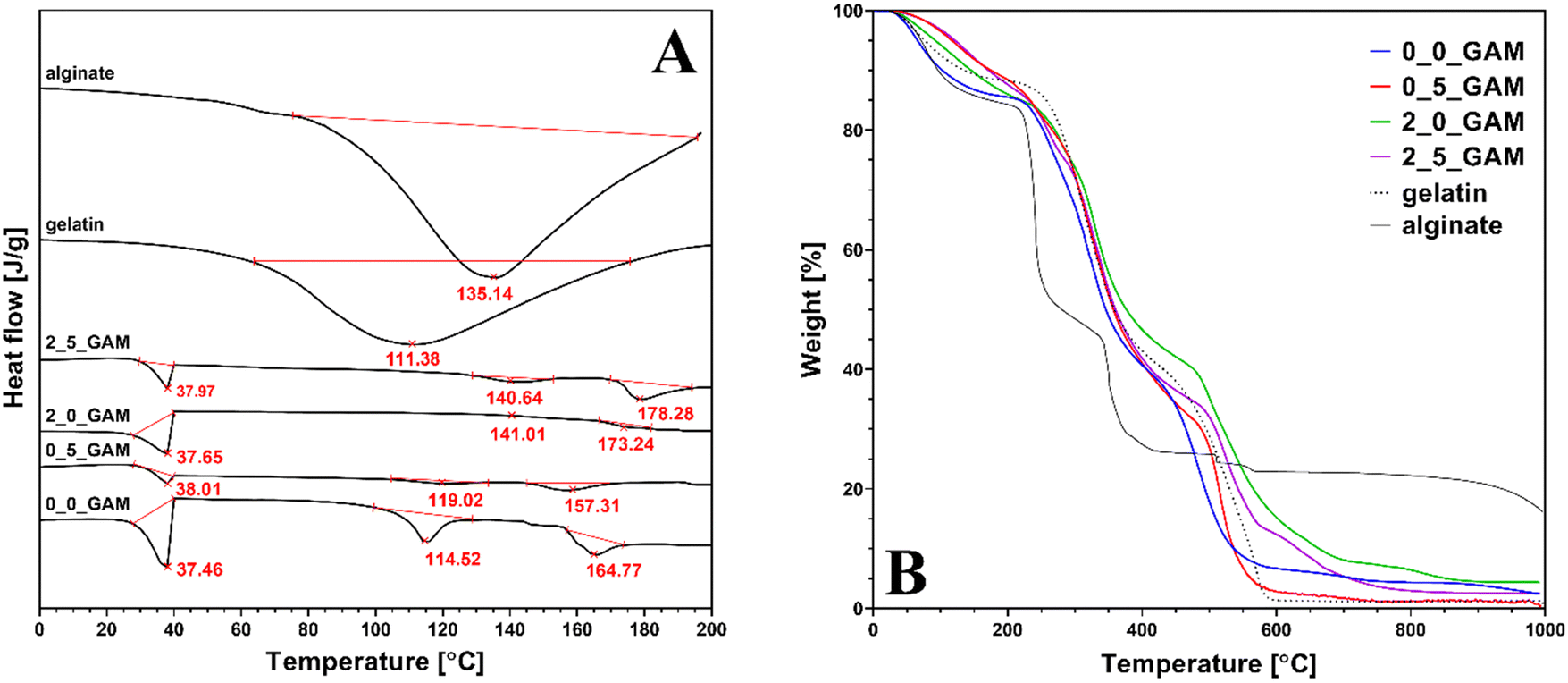 | ||
| Fig. 4 (A) Differential scanning calorimetry and (B) thermogravimetric analysis of pure gelatin, sodium alginate and GAMs results. | ||
| Sample | ΔH1 [J g−1] | ΔH2 [J g−1] | ΔH3 [J g−1] | ΔH [J g−1] |
|---|---|---|---|---|
| Gel powder | — | 294.3 ± 8.8 | — | 294.3 ± 8.8 |
| Alg powder | — | — | 289.5 ± 8.7 | 289.5 ± 8.7 |
| 0_0_GAM | 17.7 ± 0.5 | 14.6 ± 0.4 | 13.9 ± 0.4 | 46.2 ± 1.4 |
| 0_5_GAM | 3.0 ± 0.1 | 2.8 ± 0.1 | 8.1 ± 0.2 | 13.9 ± 0.4 |
| 2_0_GAM | 5.5 ± 0.2 | 0.5 ± 0.1 | 1.3 ± 0.1 | 7.3 ± 0.2 |
| 2_5_GAM | 3.6 ± 0.1 | 4.6 ± 0.1 | 8.4 ± 0.3 | 16.6 ± 0.5 |
For DSC research pure gelatin and sodium alginate showed wide endothermic peaks in the temperature range of 40–180 °C, with endothermic enthalpy change of 294.3 and 289.5 J g−1, respectively, representing the dehydration of the polymers and changes in the structure of their chains.35–37 The endothermic peaks observed in the GAMs hydrogel with reduced endothermic enthalpy of 7.31–46.11 J g−1 indicate that the formation of hydrogen bonds is correlated to a better organization of polymer chains and stronger bonding. It also suggests that the freeze-drying process successfully reduced the moisture content in the GAMs.
There are no low-temperature downward peaks situated around 0 °C due to the melting enthalpy of ice – all free and freezable water was removed as they are bound to the matrix less closely.38 Although nonfreezable water itself is so strongly related to the polymer network that it does not show phase transition in calorimetric analysis, in the case of GAMs, its presence in the hydrogel is indirectly confirmed by the peaks seen in the graphs at around 37 °C. This is the temperature at which the alginate–gelatin hydrogel undergoes thermal transition. In the case of lyophilized GAMs, non-crystallized water acted as a plasticizer, increasing the mobility of the polymer chains and reducing the glass transition temperature, leading to the transformation of the hydrogel into sol. A similar phenomenon was described by Yoshida et al. for various polysaccharide hydrogels.39 The differences that appear between differently crosslinked hydrogels result from the over-all hydrogen bonding ability (HBA) of the polymer. The only covalently cross-linked sample (0_5_GAM) shows the smallest peak due to the greatest reduction of said HBA. In the case of an ionotropically cross-linked sample (2_0_GAM), the cross-linking strength is lower, thus reducing HBA to a lesser extent. The under-curve area for the sample cross-linked with both methods (2_5_GAM) is between the peaks for the two samples described above, which may suggest that the two cross-linking methods do not always strengthen the polymer network simultaneously, but may be in opposition, e.g. by limited access to –COO– groups, needed in both Ca2+ ion cross-linking and GTA vapors covalent one, which also results from the work of Sun et al.40
The second peak appearing in the DSC plots can be attributed to the material's second glass transition temperature, Tg, caused by the helix-coil transition of gelatin and the release of bound water. With increasing temperature, alginate shows an increase in the overall hydrogen bonding ability due to the increased dissociation of the ions pairs, which was confirmed for other hydrophilic polymers.35 The largest endothermic peak can be observed for the sample that was not cross-linked (0_0_GAM), so it can be concluded that in this case hydrogen bonds are involved in maintaining the triple helix structure. In other cases, cross-linked by any method, the peaks are barely marked on the DSC curve, which means that the structure is stabilized by stronger bonds than hydrogen ones – Ca2+ bridges and covalent –CO–NH– bonds from Schiff's base reaction. The reduction in the degree of unfolding of the triple helix structure of GTA vapor cross-linked gelatin compared to non-cross-linked gelatin was also demonstrated by Nagarajan et al.41
The third peak represents the melting temperature Tm of the material. Last endothermic peak in thermogram may be related to water tightly bonded through polar interactions with carboxylate. The presence of such bound water in polysaccharides has been reported by El-Houssiny et al.42 and Sabater et al.43 The highest peak was again observed for the non-cross-linked sample, which had the most unbound –COOH groups that could react with water molecules. The sample cross-linked only with GTA vapors (0_5_GAM) showed a smaller peak than the non-cross-linked one, suggesting that some of the carboxyl groups from alginate were used to cross-link the hydrogel with bonds between GTA-treated gelatin and alginate. The samples for which Ca2+ ions were used (2_0_GAM and 2_5_GAM) did not show any peak, indicating strong links between the alginate and gelatin chains as well as Ca2+ ions, which resulted in a high crosslinked network with few free reactive groups able to bind to water molecules.
The results of the TGA analysis presented in Fig. 4(B) confirm that the composition of the hydrogel did not change throughout the entire treatment. The first weight loss (10–15%) is observed between 100 and 200 °C. This decrease is related to the release of structural water and it is much slighter in cross-linked samples and occurs later than in the case of pure hydrogel components (sodium alginate and gelatin) or non-cross-linked samples. This confirms that in the case of cross-linked GAMs the bounded water is immobilized stronger. The second major weight loss observed between 200 and 400 °C shows the degradation of gelatin molecules. The third major weight loss observed from ∼520 °C is due to the thermochemical decomposition of remaining organic content in the case of gelatin and GAMs. Although all GAMs samples degrade at much higher temperatures and in this matter the alginate content in the matrices does not affect their properties, it makes the GAMs leave more residue at the end of the analysis than pure gelatin. At the end of the TGA analysis, the difference between the amount of residual mass after burning the gelatin (dashed line) and after burning the GAMs (colored lines) is about 3–4 percentage points. The only exception is the weight of the sample cross-linked only with GTA vapor (0_5_GAM, red line) which is equal to the weight of pure gelatin. This may suggest that cross-linked gelatin has a much greater effect on the thermal properties of the hydrogel than non-cross-linked sodium alginate. Likewise, the mass of the sample cross-linked only with calcium ions (2_0_GAM, blue line) at the end of the process is much greater than the mass of pure gelatin or the rest of the samples with a final mass value somewhere in between. This confirms the previous observation that the effects of hydrogel cross-linking with calcium ions and GTA vapors do not reinforce each other when they are used simultaneously, but are rather competitive.
Swelling of the matrices
Considering that the method used to cross-link the gelatin–alginate hydrogel has a large impact on its structure, it can be presumed that it will also affect its other properties. It is therefore important to properly select the concentrations of the cross-linking agents in order to obtain a desired material. A hydrogel suitable for drug delivery system for wound dressing (the purpose of this study) should be characterized by a high equilibrium swelling ratio because it directly affecets the hydrogel ability for absorbing the wound exudates which can contain possibly harmful substances.26 The equilibrium swelling ratio (ESR) of GAMs is presented in Fig. 5.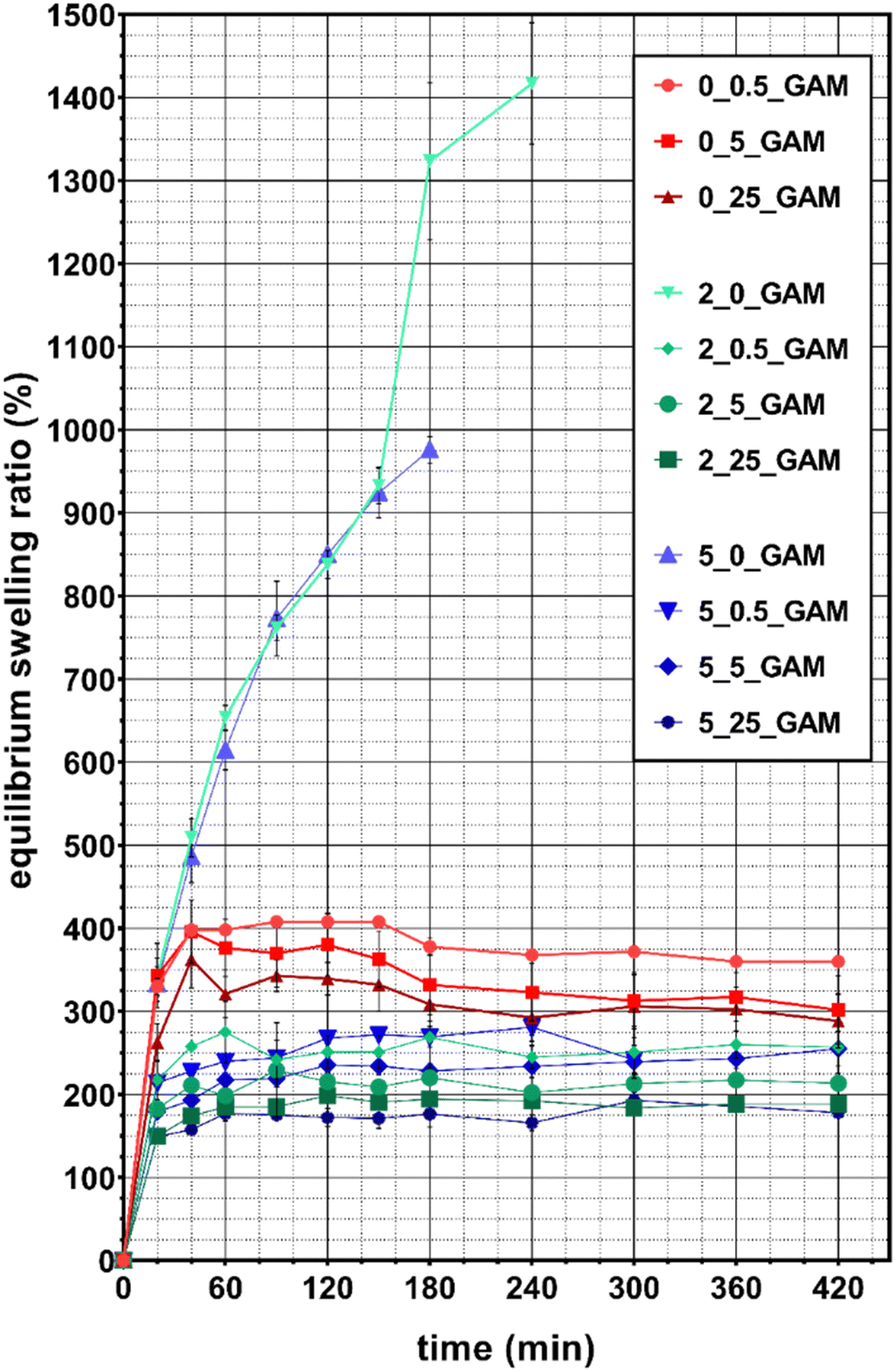 | ||
| Fig. 5 Equilibrium swelling ratio of lyophilized GAMs depending on the cross-linking method and the concentrations of cross-linking agents. A chart uses points connected by line segments from left to right to demonstrate changes in value and to facilitate tracking of them. | ||
Almost all lyophilized hydrogel matrices swelled rapidly and reached an equilibrium state after 40–60 min. The only exceptions were 2_0_GAM and 5_0_GAM samples cross-linked only with calcium ions. They did not reach equilibrium state before degradation, which occurred after 3 hours (for GAM cross-linked with the solution containing 2% Ca2+) and after 4 hours (for the one cross-linked with the 5% Ca2+ solution) – this is visible as a sudden break in the curves due to missing data (sample decay). During the last measurement, the samples reached 980% and 1420% hydration, respectively. The samples degradation was caused by the displacement of calcium ions from the polymer network by monocations (Na+) contained in the PBS solution. The greater stability was observed for the sample cross-linked with 5% Ca2+ and thus with more intermolecular bonds between alginate chains. Consequently, this sample absorbed more water being able to spend more time in the swelling medium.
For all other samples the equilibrium state was reached, although average equilibrium water content (EWC) values were slightly fluctuating. This value was the highest for samples 0_0.5_GAM, 0_5_GAM and 0_25_GAM cross-linked only with glutaraldehyde vapors – after 40 minutes, EWC for these samples reached approximately 400%, 380% and 325%, respectively. The matrices cross-linked by both methods (calcium ions and GTA vapors) showed reduced ability to absorb water. EWC for samples 2_0.5_GAM and 5_0.5_GAM were comparable and amounted to about 260–280%. Similarly, other samples can be combined into pairs – for the 2_5_GAM and 5_5_GAM samples, EWC was 200–240%, while for the 2_25_GAM and 5_25_GAM samples, it was the lowest and its value reached 180–200%. It can be easily noticed that a higher concentration of GTA vapors results in lower hydrogel swelling capability. In addition, if this crosslinking agent is used together with calcium ions, then Ca2+ crosslinking has a negligible effect on matrix swelling properties. During the tests, it was also observed that after about 3 hours, the weight of samples cross-linked only with GTA vapors decreased due to their slow degradation (data not shown). This phenomenon does not occur for samples cross-linked by both methods (Ca2+ and GTA).
Rhodamine release from GAMs
The substance controlled release is an important ability of the hydrogel from the point of view of its potential use as a wound dressing. It is important that the release proceeds without a burst effect at the beginning and continues as long as possible. It is also useful to be able to control the release properties, for example by selecting the hydrogel cross-linking method. The marker dye rhodamine 640 was used due to its highly sensitive ability to absorb light at a wavelength of 574 nm. Moreover, the size of its molecule is comparable to the size of the chlorhexidine molecule, which was used in the study of the antibacterial properties of GAMs (591 g mol−1 for Rod and 625 g mol−1 for CA), it may therefore mimic the release of this drug.Fig. 6 and 7 show the rhodamine release profiles from hydrogel matrices over the period of 9 hours. The curves are divided into three groups differing in colors, red for samples cross-linked only with GTA vapors (with various concentrations ranging from 0.5–25%), green and blue for cross-link with GTA vapors and a 2% or 5% solution of Ca2+ ions, respectively.
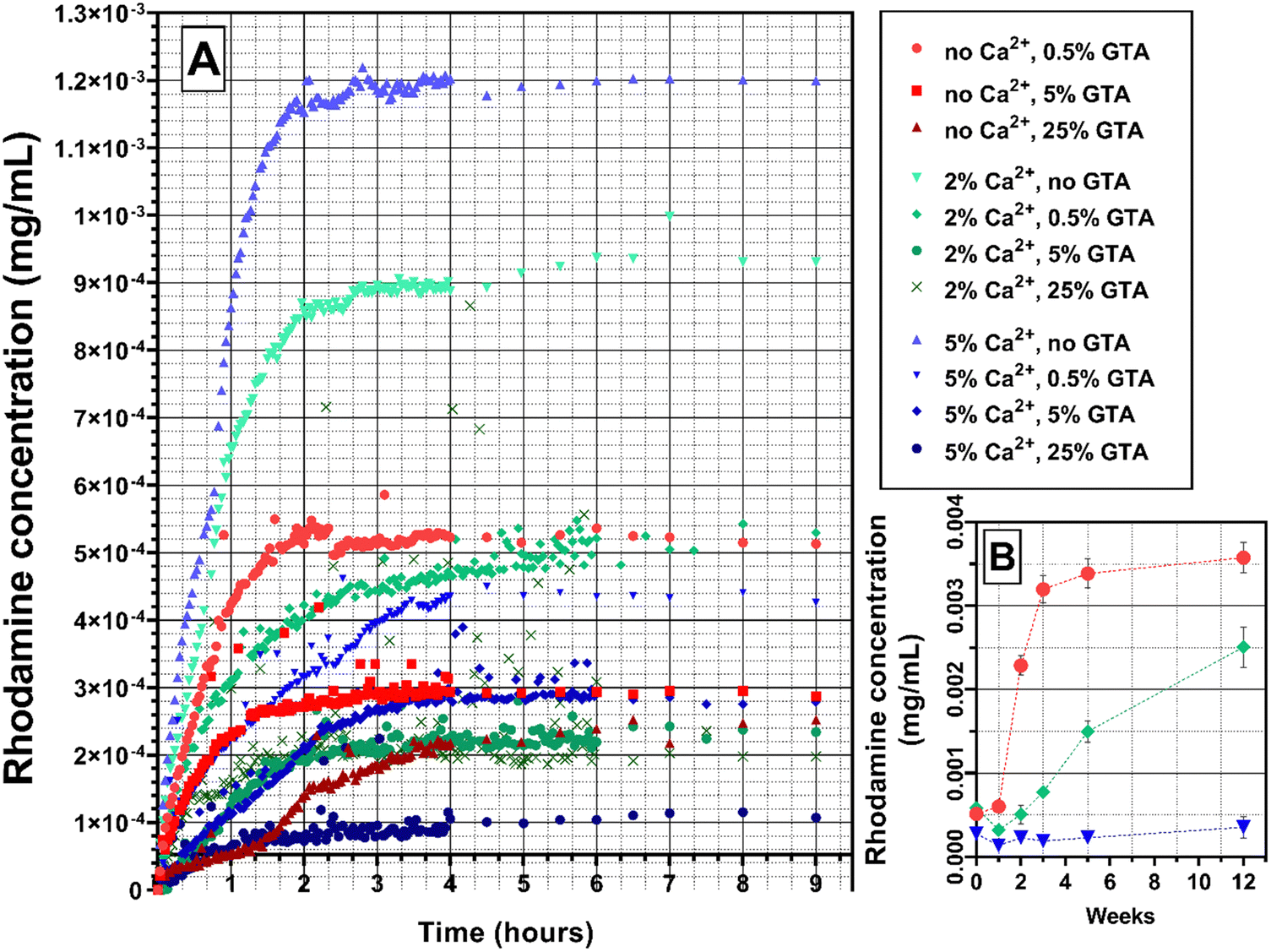 | ||
| Fig. 6 Rhodamine 640 release profile from GAMs cross-linked with different methods: (A) initial release, 9 hours; (B) long-term release, selected samples, 12 weeks. A chart uses points connected by line segments from left to right to demonstrate changes in value and to facilitate tracking of them. | ||
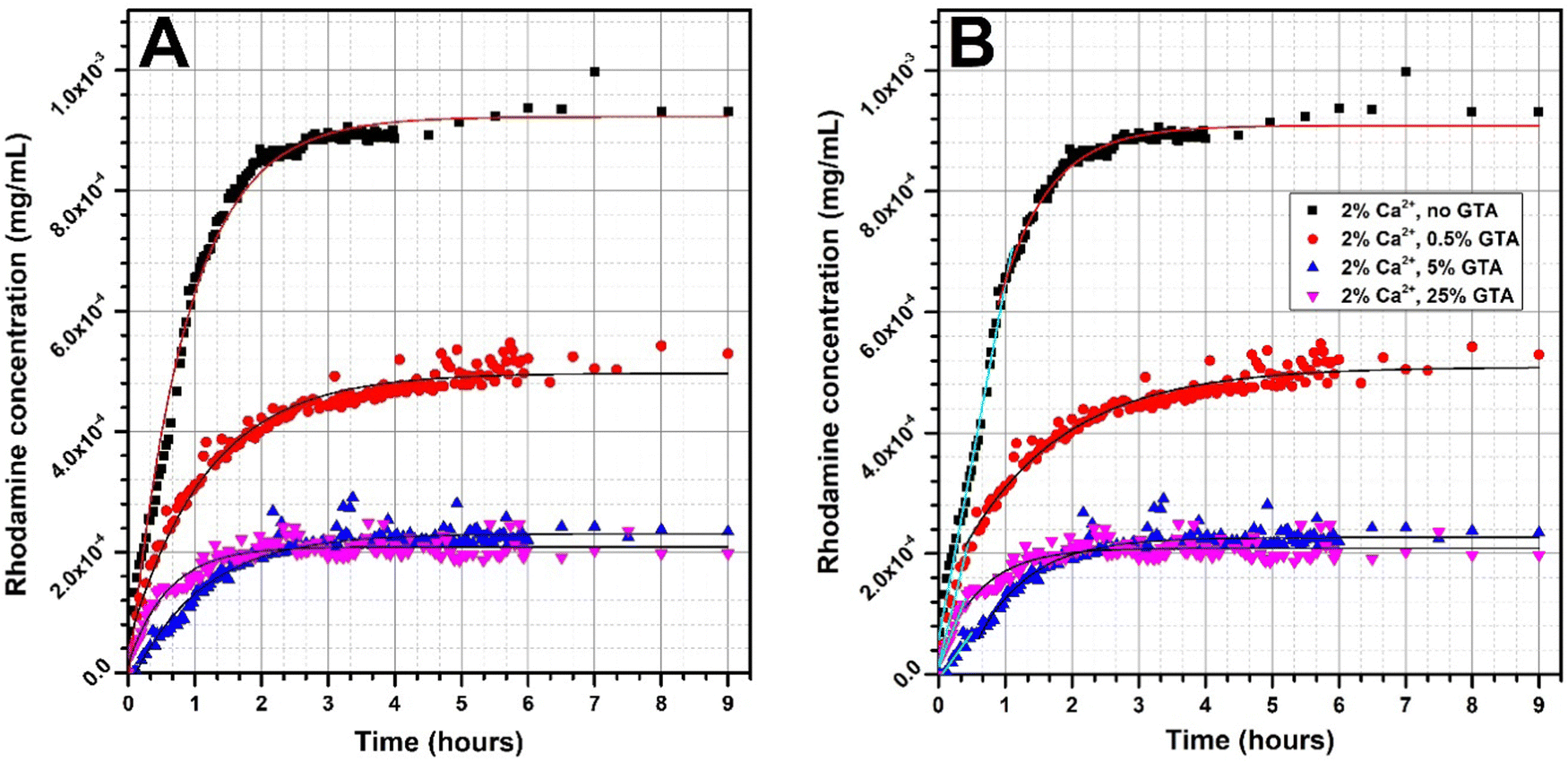 | ||
| Fig. 7 Rhodamine 640 release profile from GAMs cross-linked with different methods with model fitted curves: (A) exponential fit for all the data, (B) linear fit for the initial period of releasing and exponential fit for the following data. | ||
In all three groups of samples mentioned, increasing the concentration of GTA vapors or calcium ions used for cross-linking causes a slower release rate of rhodamine. Additionally, for samples cross-linked with higher GTA concentrations, the lower concentrations of rhodamine are reached meaning that less rhodamine is released. The theoretical content of rhodamine in the matrices is the same. As shown in Fig. 6(A), the release of rhodamine is 10−4 mg mL−1 for the 5_25_GAM and 1.2 × 10−3 mg mL−1 for the 5_0_GAM. The hydrogel matrix 2_0.5_GAM, which was cross-linked by both methods, but under the mildest conditions, did not reach the equilibrium state even after 12 weeks (Fig. 6(B), green squares), unlike the other two matrices presented in the graph (0_0.5_GAM and 5_0.5_GAM). For the sample cross-linked only with 0.5% GTA vapors without Ca2+ (red circles), the equilibrium state is reached around week 5, for the sample cross-linked with 5% Ca2+ and 0.5% GTA vapors (blue triangles), the equilibrium state reached at the beginning of rhodamine release (burst release) is maintained until the week 12.
The shape of the rhodamine release profile from GAMs is of particular interest. In most cases, the release of rhodamine appears to be two-step. In order to better illustrate this phenomenon, curves were fitted to the experimental data, and the results are presented in Fig. 7. Since the matrices cross-linked with 2% and 5% of calcium ions showed similar properties, it was decided to further analyze the data only for samples cross-linked 2% of Ca2+.
When a drug or model substance is released, it is possible to acquire a mathematical description of the relationship between release rate and time. Such a release profile can be usually described by a linear or an exponential function (zero- or first-order kinetics). The fit of an expotential curve to the experimental data of rhodamine releasing from GAMs is shown in Fig. 7(A). In general, this fit is good as indicated by the values of the coefficient of determination R2exp1 which ranged from 0.8994 to 0.9892 (Table 3). However, this fit did not cover exactly all the experimental points, especially in the initial period of rhodamine release.
| Sample | A | B | |||
|---|---|---|---|---|---|
| Expotential fit | Linear fit | Expotential fit | |||
| R 2exp1 [−] | Range [h] | R 2lin [−] | Range [h] | R 2exp2 [−] | |
| 2% Ca2+, no GTA | 0.9892 | 0–1.10 | 0.9908 | 1.10–9.00 | 0.9674 |
| 2% Ca2+, 0.5% GTA | 0.9725 | 0–0.47 | 0.9466 | 0.47–9.00 | 0.9605 |
| 2% Ca2+, 5% GTA | 0.9419 | 0–0.50 | 0.9701 | 0.50–9.00 | 0.8776 |
| 2% Ca2+, 25% GTA | 0.8994 | 0–0.40 | 0.9113 | 0.40–9.00 | 0.6746 |
Therefore, the data was devided into two ranges in order to adjust the curve independently to the experimental points in each of them, as shown in Fig. 7(B). In the initial period of rhodamine release, a linear fit was applied. The values of the coefficient of determination R2lin for such a fit ranged from 0.9113 to 0.9908 (Table 3) and are higher than for the exponential fit covering all points. An exponential curve was fitted to the data following the linear range, however, in only two cases, the R2exp2 was higher than 0.95 for this fit. In order to model the release of rhodamine from freeze-dried GAMs, a linear fit should be used in the initial release period and then an exponential fit for the resting data range.
It can therefore be concluded that the release of rhodamine from the freeze-dried GAMs initially follows zero-order kinetics. In this case, the release of the active substance is only a function of time, and the process proceeds at a constant rate regardless of the concentration of the agent. Comparing the time range in which the release takes place with zero-order kinetics (Table 3(B)) with the GAMs swelling over time graph (Fig. 5) leads to the observation that they correspond to the initial period of the fastest swelling of the matrices. When GAM is placed in the solution, the water is imbibed into the hydrogel structure dissolving the rhodamine. The active ingredient is first dissolved in the hydrating water and only then can be released into the solution. The constant release rate is due to the saturation of the hydrogel, because only the swollen matrix can be permeable to rhodamine, so its release rate depends on the swelling. Such behavior of active substance-releasing membranes according to zero-order kinetics after placing in a liquid has been previously described.24,25 Once the full hydration is achieved, release of rhodamine from the hydrogel begins to follow first-order kinetics.24 Systems that exhibit such a subtance release mechanism are suitable for controlled sustained drug delivery because due to the zero-order kinetics in the initial subtance release period, the burst release effect is limited.25
Cytotoxicity of GAMs
In order to validate the potential use of the 3D bioprinted gelatin–alginate hydrogel matrices for wound dressing application, we aimed to demonstrate its biocompatibility on human keratinocytes. The biocompatibility of the GAMs was analyzed after 1, 4, 6 and/or 8 days of culture using HaCat cells. As shown in Fig. 8(A), a clear cytotoxicity was observed upon increasing concentrations of GTA used for gelatin cross-link. This effect was strongly attenuated by glycine neutralization (Fig. 8(B)) and we measured a significant increase in cell proliferation between 1 and 8 days in the presence of neutralized GAMs thus demonstrating their cytocompatibility.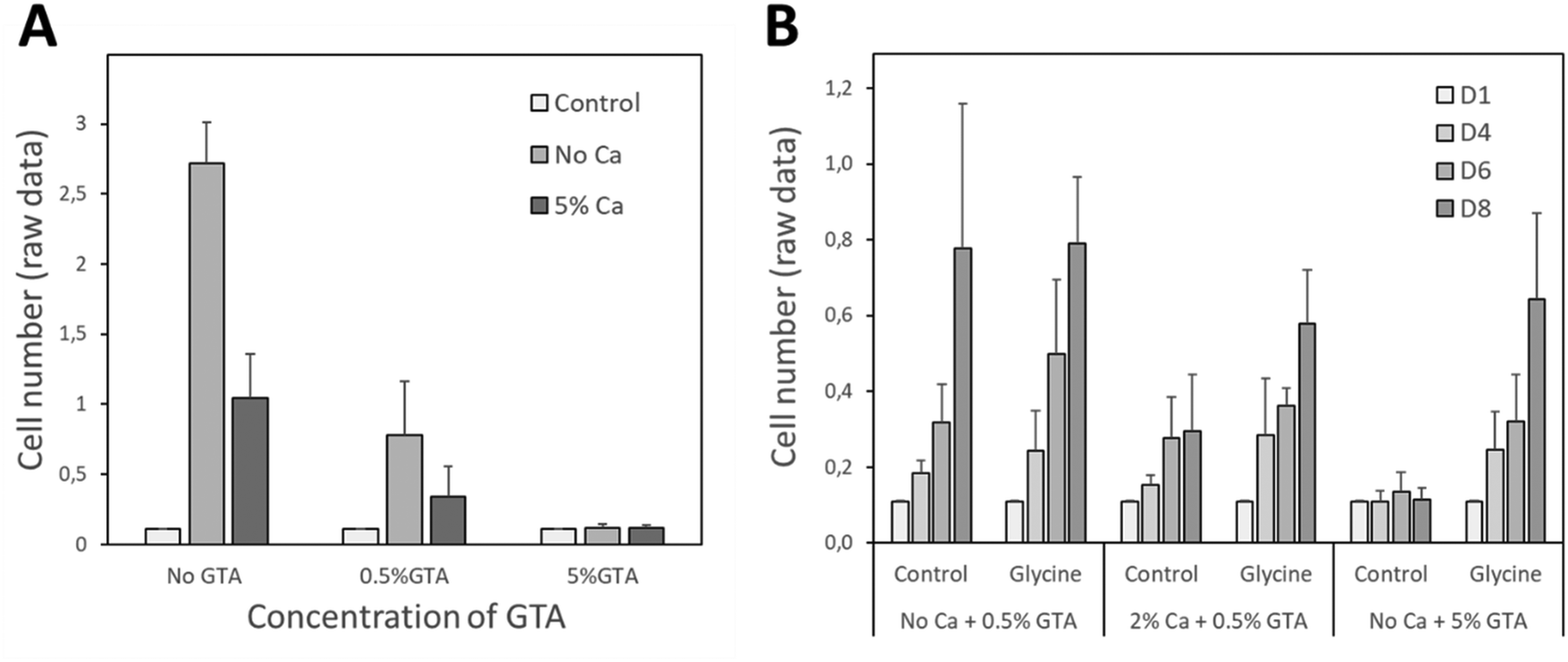 | ||
| Fig. 8 Analysis of GAMs cytocompatibility using hFOB1.19 cells. (A) The cytotoxicity of GAMs cross-linked or not with 5% Ca2+ and 0.5% or 5% GTA tested at day 8 after seeding. (B) Effect of glycine neutralization on the cytotoxicity of GAMs cross-linking with or without 2% Ca2+ and 0.5% or 5% GTA analyzed. The analysis was performed 4, 6 and 8 days after cell seeding. | ||
Antibacterial activity of chlorhexidine-loaded GAMs
Gelatin–alginate hydrogel matrices with antibiotic activity is useful for wound dressing applications regarding the need to maintain sterility at the wound site. In that frame, we loaded GAMs with different concentration of chlorhexidine acetate (CA) and characterized their antimicrobial activity using an agar diffusion inhibitory growth test. CA is a widely used well-known bisbiguanide compound with rapid bactericidal activity against both Gram-positive and Gram-negative organism.44 It was used in our work due to its application in disinfection of the skin, as an additive to creams, toothpaste, deodorants, and antiperspirants as well as in pharmaceutical products.45The antibacterial activity tests were carried out with the sample 2_0.5_GAM neutralized with a 20% glycine solution. Each sample was deposited on the surface of nutrient agar plate previously inoculated with S. aureus or E. coli at a concentration of 108 CFU mL−1 and incubated for 24 h. Fig. 9 illustrates the results of the anibacterial properties study.
 | ||
| Fig. 9 Bacteriostatic tests of CA-loaded GAMs on nutrient agar plates covered with S. aureus and E. coli biofilms after 24 h of material treatment. GAMs cross-linked with 2% Ca2+ and 0.5% GTA, neutralized with glycine with no drug, 0.1 mg mL−1 CA or 1 mg mL−1 CA. | ||
In all cases, the concentration of CA was sufficient to suppress bacterial growth. The area of growth inhibition zones (clear zones) were visible around the materials for S. aureus and E. coli. They were greater for material with a higher drug concentration. A tenfold increase in drug content (1 mg mL−1, Fig. 9) resulted in a significant increase in the clear zone size. This observation was made only for drug-loaded GAMs, and not for the drug-free one, which means that there are no antibacterial glutaraldehyde residues in the material. This suggests that the GTA vapor concentration had been properly selected and the matrix had been neutralised. The antibacterial action of the material loaded with chlorhexidine was rapid (with a significant effect visible from 3 h of treatment) and persistent over at least 72 h. The antibacterial activity of the GAMs against the bacteria evidenced that the integrity of the drug had not been affected by the temperature or pressure during bioink preparation and 3D bioprinting. Hence, these types of GAMs are suitable materials for wound dressing applications to treat infected sites or to prevent infection.
4. Conclusions
The gelatin–alginate bioink with the compunds concentrations of 0.2 g mL−1 and 0.04 g mL−1, respectively, is a good and satisfying material for use in 3D bioprinting. Easy to prepare from the readily available substances, the hydrogel can be conveniently bioprinted in mild conditions. It represents an excellent basis for creating a novel stable biodegradable 3D bioprinted material for drug delivery purposes, potentially wound dressing applications. The gelatin–alginate hydrogels can be successfully cross-linked by exposure to calcium ions or glutaraldehyde vapors. The chosen method and concentrations of crosslinkers have an impact on the properties of the obtained materials including the structure of freeze-dried hydrogels and the morphology of their surface. The high equilibrium swelling ratio (ESR) and equilibrium water content (EWC) values of GAMs indicated their potential high effectiveness in absorbing wound exudates, a desirable feature of dressings. The study of rhodamine release showed that it can be described with a zero-order kinetics in the initial release period followed by a first-order kinetics release after 30–60 minutes, the burst release effect being limited. The data showed that the cross-linking method has an influence on the release rate.In the present study, a compromise was made between physicochemical properties and cytotoxicity, leading to the selection of the combined method (ionotropic-covalent) with the lowest concentrations of cross-linking agents (2% Ca2+ and 0.5% GTA). The biocompatibility of the glycine-neutralized cross-linked GAMs and the antibacterial properties of chlorexidin-loaded hydrogels against both Gram positive and Gram negative bacteria were validated. The work presents coherently the 3D bioprinted cross-linked drug-loaded gelatin–alginate hydrogel matrices and a detailed description of various properties important for a potential use as wound dressings. Such a material can be potentially applied in biomedical engineering, especially as controlled drug delivery systems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was supported by European Social Fund [POWR.03.02.00-00-1028/17-00] as part of the POWER Och!Dok program and partialy supported by “Cancéropôle Grand Sud-Ouest” through a project “Emergence de projets 2021”. AM would like to thank Campus France for the funding through a French Government Scholarship. Authors acknowledge the financial support of project H2020-MSCA-RISE-2017, ‘Novel 1D photonic metal oxide nanostructures for early stage cancer detection’ (Project number: 778157).References
- L. Valot, J. Martinez, A. Mehdi and G. Subra, Chemical insights into bioinks for 3D printing, Chem. Soc. Rev., 2019, 48(15), 4049–4086 RSC.
- K. Jakab, C. Norotte, F. Marga, K. Murphy, G. Vunjak-Novakovic and G. Forgacs, Tissue engineering by self-assembly and bio-printing of living cells, Biofabrication, 2010, 2, 2001 CrossRef PubMed.
- L. Moroni, J. de Wijnab and C. van Blitterswijka, 3D fiber-deposited scaffolds for tissue engineering: Influence of pores geometry and architecture on dynamic mechanical properties, Biomaterials, 2006, 27(7), 974–985 CrossRef CAS PubMed.
- W. Long Ng, C. Kai Chua and Y.-F. Shen, Print me an organ! why we are not there yet, Prog. Polym. Sci., 2019, 97, 101145 CrossRef.
- I. Matai, G. Kaur, A. Seyedsalehi, A. McClinton and C. T. Laurencin, Progress in 3D bioprinting technology for tissue/organ regenerative engineering, Biomaterials, 2019, 226, 119536 CrossRef PubMed.
- P. Shende and S. Agrawal, Integration of 3D printing with dosage forms: A new perspective for modern healthcare, Biomed. Pharmacother., 2018, 107, 146–154 CrossRef PubMed.
- W. Peng, P. Datta, B. Ayan, V. Ozbolat, D. Sosnoski and I. T. Ozbolat, 3D bioprinting for drug discovery and development in pharmaceutics, Acta Biomater., 2017, 57, 26–46 CrossRef CAS PubMed.
- A. Rees, L. C. Powell, G. Chinga-Carrasco, D. T. Gethin, K. Syverud and K. E. Hill, 3D bioprinting of carboxymethylated-periodate oxidized nanocellulose constructs for wound dressing applications, BioMed Res. Int., 2015, 1–7 CAS.
- P. He, J. Zhao, J. Zhang, B. Li, Z. Gou, M. Gou and X. Li, Bioprinting of skin constructs for wound healing, Burns Trauma, 2018, 6, 5 Search PubMed.
- B. Stubbe, A. Mignon, H. Declercq, S. van Vlierberghe and P. Dubruel, Development of gelatin–alginate hydrogels for burn wound treatment, Macromol. Biosci., 2019, 19(8), e1900123 CrossRef PubMed.
- S. M. King, S. C. Presnell and D. G. Nguyen, Development of 3D bioprinted human breast cancer for in vitro drug screening, Cancer Res., 2014, 74(19), 2034 CrossRef.
- X. Li, B. Liu, B. Pei, J. Chen, D. Zhou, J. Peng and T. Xu, Inkjet bioprinting of biomaterials, Chem. Rev., 2020, 120(19), 10793–10833 CrossRef CAS PubMed.
- W. L. Ng, J. M. Lee, M. Zhou, Y.-W. Chen, K.-X. A. Lee, W. Yeong and Y.-F. Shen, Vat polymerization-based bioprinting – process, materials, applications and regulatory challenges, Biofabrication, 2019, 12(2), 022001 CrossRef PubMed.
- T. Jiang, J. G. Munguia-Lopez, S. Flores-Torres, J. Kort-Mascort and J. M. Kinsella, Extrusion bioprinting of soft materials: An emerging technique for biological model fabrication, Appl. Phys. Rev., 2019, 6(1), 011310 Search PubMed.
- S. Murphy and A. Atala, 3D printing of tissues and organs, Nat. Biotechnol., 2014, 32(8), 773–785 CrossRef CAS PubMed.
- I. Ozbolat, W. Peng and V. Ozbolat, Application areas of 3D bioprinting, Drug Discovery Today, 2016, 21(8), 1257–1271 CrossRef CAS PubMed.
- J. Long, A. E. Etxeberria, A. V. Nand, C. R. Bunt, S. Ray and A. Seyfoddin, A 3D printed chitosan-pectin hydrogel wound dressing for lidocaine hydrochloride delivery, Mater. Sci. Eng., C, 2019, 104, 109873 CrossRef CAS PubMed.
- G. Poologasundarampillai and A. Nommeots-Nomm, Materials for 3D printing in medicine, in 3D Printing in Medicine, 2017, pp. 43–71 Search PubMed.
- B. Duan, L. A. Hockaday, K. H. Kang and J. T. Butcher, 3D Bioprinting of heterogeneous aortic valve conduits with alginate/gelatin hydrogels, J. Biomed. Mater. Res., Part A, 2012, 101A(5), 1255–1264 CrossRef PubMed.
- T. Gao, G. J. Gillispie, J. S. Copus, A. K. Pr, Y.-J. Seol and A. Atala, Optimization of gelatin–alginate composite bioink printability using rheological parameters: A systematic approach, Biofabrication, 2018, 10(3), 034106 CrossRef PubMed.
- T. Pan, W. Song, X. Cao and Y. Wang, 3D bioplotting of gelatin/alginate scaffolds for tissue engineering: Influence of crosslinking degree and pore architecture on physicochemical properties, J. Mater. Sci. Technol., 2016, 32(9), 889 CrossRef CAS.
- I. Migneault, C. Dartiguenave, M. J. Bertrand and K. C. Waldron, Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking, Biotechniques, 2004, 37(5), 790–802 CrossRef CAS PubMed.
- J. H. Y. Chung, S. Naficy, Z. Yue, R. Kapsa, A. Quigley, S. E. Moulton and G. G. Wallace, Bio-ink properties and printability for extrusion printing living cells, Biomater. Sci., 2013, 1(7), 763 RSC.
- M.-L. Laracuente, M. H. Yu and K. J. McHugh, Zero-order drug delivery: State of the art and future prospects, J. Controlled Release, 2020, 327, 834–856 CrossRef CAS PubMed.
- M. L. Bruschi, Mathematical models of drug release, Strategies to Modify the Drug Release from Pharmaceutical Systems, Woodhead Publishing, Cambridge, 2015, pp. 63–86 Search PubMed.
- J. S. Boateng, K. H. Matthews, H. N. E. Stevens and G. M. Eccleston, Wound healing dressings and drug delivery systems: A Review, J. Pharm. Sci., 2008, 97(8), 2892–2923 CrossRef CAS PubMed.
- D. Okan, K. Woo, E. A. Ayello and G. Sibbald, The role of moisture balance in wound healing, Adv. Skin Wound Care, 2007, 20(1), 39–53 CrossRef PubMed.
- Y. Zhang, J. Venugopal, Z. Huang, C. Lim and S. Ramakrishna, Crosslinking of the electrospun gelatin nanofibers, Polymer, 2006, 47, 2911–2917 CrossRef CAS.
- J. R. Khurma, D. R. Rohindra and A. V. Nand, Synthesis and properties of hydrogels based on chitosan and poly(vinyl alcohol) crosslinked by genipin, J. Macromol. Sci., Part A: Pure Appl. Chem., 2006, 43, 749–758 CrossRef CAS.
- M. Grzeczkowicz and D. Lewińska, A method for investigating transport properties of partly biodegradable spherical membranes using vitamin B12 as the marker, Desalin. Water Treat., 2018, 128, 170–178 CrossRef CAS.
- P. Bowler, B. Buerden and D. Armstron, Wound microbiology and ssociated approaches to wound management, Clin. Microbiol. Rev., 2001, 14(2), 244–269 CrossRef CAS PubMed.
- O. Damnik, P. J. Dijkstra, M. Luyn, L. O. Damink, P. Dijkstra, M. J. A. Van Luyn, P. B. Van Wachem, P. Nieuwenhuis and J. Feijen, Glutaraldehyde as a crosslinking agent for collagen-based biomaterials, J. Mater. Sci.: Mater. Med., 1995, 6(8), 460–472 CrossRef.
- Y. Z. Zhang, J. Venugopal, Z.-M. Huang, C. T. Lim and S. Ramakrishna, Crosslinking of the electrospun gelatin nanofibers, Polymer, 2006, 47(8), 2911–2917 CrossRef CAS.
- T. Chaochai, Y. Imai, T. Furuike and H. Tamura, Preparation and properties of gelatin fibers fabricated by dry spinning, Fibers, 2016, 4(4), 2 CrossRef.
- Z. Ping, Q. Nguyen, S. Chen, J. Zhou and Y. Ding, States of water in different hydrophilic polymers—DSC and FTIR studies, Polymer, 2001, 42(20), 8461–8467 CrossRef CAS.
- D. Chi-An, C. Yi-Fan and L. Ming-Wei, Thermal properties measurements of renatured gelatin using conventional and temperature modulated differential scanning calorimetry, J. Appl. Polym. Sci., 2006, 99(4), 1795–1801 CrossRef.
- H. Hatakeyama and T. Hatakeyama, Interaction between water and hydrophilic polymers, Thermochim. Acta, 1998, 308(1-2), 3–22 CrossRef CAS.
- M. U. Joardder, M. Mourshed and M. Hasan, Bound water measurement techniques, State of bound water: Measurement and significance in food processing, Springer International Publishing, NYC, 2019, pp. 47–82 Search PubMed.
- H. Yoshida, T. Hatakeyama and H. Hatakeyama, Effect of water on the main chain motion of polysaccharide hydrogels, ACS Symp. Ser., 1992, 489, 217–230 CrossRef CAS.
- J. Sun and H. Tan, Alginate-based biomaterials for regenerative medicine applications, Materials, 2013, 6(4), 1285–1309 CrossRef CAS PubMed.
- S. Nagarajan, H. Belaid, C. Pochat-Bohatier, C. Teyssier, I. Iatsunskyi, E. Coy, S. Balme, D. Cornu, P. Miele, N. S. Kalkura, V. Cavailles and M. Bechelany, Besign of boron nitride/gelatin electrospun nanofibers for bone tissue engineering, ACS Appl. Mater. Interfaces, 2017, 9, 33695–33706 CrossRef CAS PubMed.
- A. El-Houssiny, A. Ward, D. Mostafa, S. Abd-El-Messieh, K. Abdel-Nour, M. Darwish and W. Khalil, Drug-polymer interaction between glucosamine sulfate and alginate nanoparticles: FTIR, DSC and dielectric spectroscopy studies, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2016, 7, 025014 Search PubMed.
- R. Sabater i Serra, J. Molina-Mateo, C. Torregrosa-Cabanilles, A. Andrio-Balado, J. M. Meseguer Duenas and A. Serrano-Aroca, Bio-nanocomposite hydrogel based on zinc alginate/graphene oxide: Morphology, structural confirmation, thermal behavior/degradation, and dielectric properties, Polymers, 2020, 12(3), 702 CrossRef PubMed.
- I. R. Sanchez, K. E. Nusbaum, S. F. Swaim, A. S. Hale, R. A. Henderson and J. A. Mcguire, Chlorhexidine diacetate and povidone-iodine cytotoxicity to canine embryonic fibroblasts and staphylococcus aureus, Vet. Surg., 1988, 17(4), 182–185 CrossRef CAS PubMed.
- T. Güthner, Guanidine and Derivatives, in Ullman's Encyclopedia of Industrial Chemistry, Wiley, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |