
Modulation of acetylcholinesterase activity using molecularly imprinted polymer nanoparticles†
Sergey A.
Piletsky
a,
Thomas S.
Bedwell
ab,
Rachele
Paoletti
a,
Kal
Karim
a,
Francesco
Canfarotta
b,
Rachel
Norman
c,
Donald J. L.
Jones
cd,
Nicholas W.
Turner
 e and
Elena V.
Piletska
*a
e and
Elena V.
Piletska
*a
aDepartment of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: ep219@le.ac.uk
bMIP Diagnostics Ltd, Colworth Science Park, Sharnbrook, MK44 1LQ, UK
cDepartment of Cardiovascular Sciences and NIHR Leicester Cardiovascular Biomedical Research Unit, Glenfield Hospital, Leicester, LE3 9QP, UK
dDepartment of Cancer Studies, University of Leicester, Leicester, LE2 7LX, UK
eSchool of Pharmacy, De Montfort University, Leicester, LE1 9BH, UK
First published on 25th March 2022
Abstract
Modulation of enzyme activity allows for control over many biological pathways and while strategies for the pharmaceutical design of inhibitors are well established; methods for promoting activation, that is an increase in enzymatic activity, are not. Here we demonstrate an innovative epitope mapping technique using molecular imprinting to identify four surface epitopes of acetylcholinesterase (AChE). These identified epitopes were then used as targets for the synthesis of molecularly imprinted nanoparticles (nanoMIPs). The enzymatic activity of AChE was increased upon exposure to these nanoMIPs, with one particular identified epitope nanoMIP leading to an increase in activity of 47× compared to enzyme only. The impact of nanoMIPs on the inhibited enzyme is also explored, with AChE activity recovering from 11% (following exposure to an organophosphate) to 73% (following the addition of nanoMIPs). By stabilizing the conformation of the protein rather than targeting the active site, the allosteric nature of MIP-induced reactivation suggests a new way to promote enzyme activity, even under the presence of an inhibitor. This method of enzyme activation shows promise to treat enzyme deficiency diseases or in medical emergencies where an external agent affects protein function.
Introduction
The capability to modulate enzymatic activity is a highly sought-after property and the goal of a considerable amount of pharmaceutical studies, as being able to control the activity and function of enzymes is key to the treatment of disease and other conditions.Acetylcholinesterase (AChE) is a serine hydrolase that causes the termination of neuronal transmission at the cholinergic synapse by hydrolyzing its natural substrate acetylcholine into choline and acetate ions.1,2 The active site of AChE contains a Glu/His/Ser catalytic triad, located at the center of a deep and narrow gorge.3 Inhibitors directed to the active site prevent the binding of a substrate molecule or its hydrolysis, either by occupying the site with a high affinity or by modifying the catalytic serine.
Whilst mild inhibition of AChE is desirable in the treatment of Alzheimer's disease,4–6 potent inhibition of AChE can have detrimental effects on the neuromuscular system. This is exploited in organophosphate (OP) nerve agents (e.g. Sarin, Soman, etc.) where irreversible inhibition of AChE leads to excessive cholinergic neurotransmission, resulting in cardiovascular and respiratory compromise, and ultimately death.7 In this case, an agent capable of increasing the activity of AChE during toxin exposure, or reactivating it afterwards would be of great interest in the clinical community.
Computational modeling suggests that allosteric AChE activators could provide a novel therapeutic route for treating OP intoxication;8 however, development of new therapeutics is currently focused on classic small molecule pharmacology with oxime-based reactivators the current field leaders. Unfortunately, these compounds suffer from a lack of broad-spectrum efficacy,9 and their inability to cross the blood–brain barrier limits their usefulness as emergency treatments.10
Nanoparticles (NPs) are considered one of the most promising drug delivery systems for targeting inaccessible regions of brain.11 They are able to pass the blood–brain barrier through tight junctions between endothelial cells12 using transcytosis13 or endocytosis.14 These features indicate strong potential for developing NPs with the ability to reach the target neural tissue after oral or percutaneous administration. NPs can be used not only as vehicles to deliver drugs but also as therapeutic agents capable of mimicking enzyme modulatory functions of antibodies.15,16 One of the most promising classes of therapeutic NPs are molecularly imprinted polymer nanoparticles (nanoMIPs).17 NanoMIPs are synthetic receptors, which are produced by self-assembly of complementary functional monomers around a target molecule that acts as a template. The ability of the nanoMIPs to recognize and re-bind the corresponding template is based on the spatial orientation of the functional groups present in the imprinted cavity, as well as its size and shape. It is known that nanoMIPs can function in vivo without triggering an immune response,18 and specific modulation of enzymes such as trypsin, thrombin and catalase by nanoMIPs has recently been documented.19–23 Given their ability to interact with protein templates in a targeted, selective manner, and that they exhibit low nM dissociation constants,24,25 these materials offer significant potential as active modulators, as both inhibitors or as activators.
In this study, we report the development of nanoMIPs that demonstrate site-specific binding to different regions of an enzyme's surface and provide modulation on its enzymatic activity, via a novel epitope target selection process. The Electrophorus electricus AChE (EeAChE) enzyme was used as a model to demonstrate this approach, with the intention of establishing nanoMIPs as novel therapeutics for both Alzheimer's disease and OP intoxication through allosteric inhibition and activation, respectively. The experimental method presented here can potentially be applied to any protein of interest, even in the absence of structural information.
Results and discussion
Identification of EeAChE epitopes
Due to their roles as natural receptors and enzymes, proteins have always been considered among the most important templates for the preparation of MIPs. At the same time, their intrinsic properties, large size and relatively low stability made them challenging targets for molecular imprinting.29 The aim when designing MIPs is to mimic the specificity of natural antibodies. One of the most advanced approaches for the production of MIPs specific for proteins is the ‘epitope approach’.30 In this method, instead of a whole protein, a peptide sequence is selected and used as a template for the imprinting. It was demonstrated that such peptide-specific MIPs are able to recognize a whole protein. This approach is analogous to protein recognition by antibodies, where an epitope of the immunogenic protein is the site of antibody binding. The binding of antigens to antibodies is well understood, and the nature of that interaction is as relevant to MIPs as it is to the natural molecules.The simplest way to decide if a protein fragment will make a good epitope for MIP design is to look at its position within the tertiary structure of the protein in the Protein Databank.31 There are many algorithms which have been written to predict antigenic regions and epitopes of proteins,32 although, unfortunately, their prediction power is far from optimal, with the rate of hits for predicted epitopes of human AChE by such software only reaching around 30%.33 It is also possible to consider imprinting known epitopes for AChE. There are approximately 65 known human AChE epitopes recorded in the Immune Epitope Database and Analysis Resource (https://www.iedb.org/). However, very few of their corresponding antibodies are able to modulate AChE.34,35
We have recently patented an experimental approach for using molecular imprinting to identify peptide sequences on the surface of proteins with potential antigenic properties.36 This method involves the synthesis of MIP NPs in the presence of a whole protein, partial proteolysis of the protein bound to the polymer, and subsequent sequencing of released peptides that were bound to the polymer (Fig. 1).
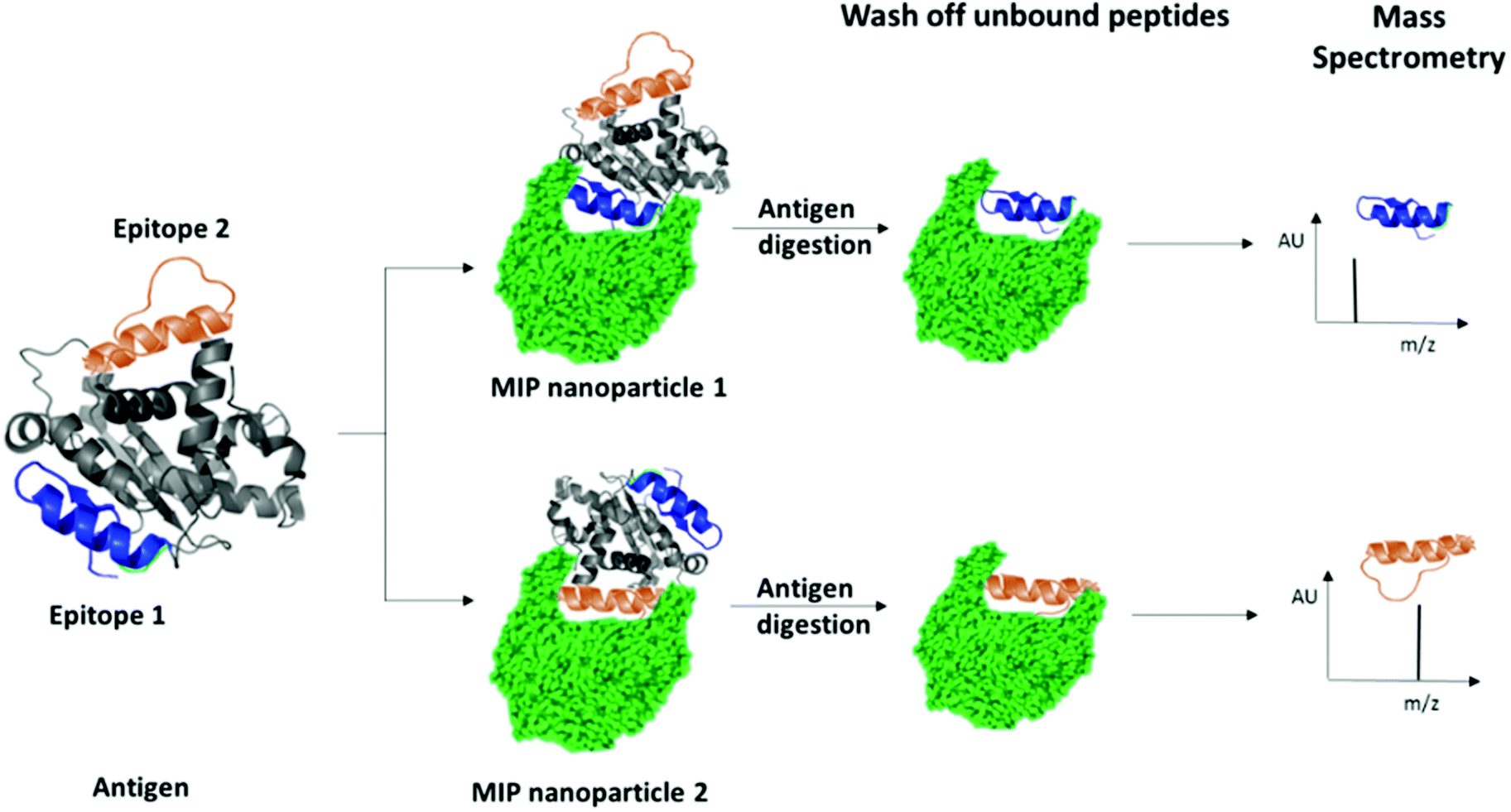 | ||
| Fig. 1 Schematic highlighting the principle of identification of peptide sequences exposed on protein surfaces using molecular imprinting and mass spectrometry. | ||
The central concept behind this principle relies on the assumption that MIPs that are synthesized in the presence of protein, protect the peptide sequences involved in MIP formation that are retained within binding sites from proteolysis. This approach allows for the identification of regions of the protein surface that have not been demonstrated to be antigenic in vivo, but which may offer improved affinity or alternative modulatory mechanisms for therapeutic applications. For the purposes of our research, we have selected EeAChE, which is available in sufficient quantity and molecular homogeneity to make it suitable for structural and functional studies. Cross-immunoreactions of antigens and antibodies have shown that anti-human brain AChE antiserum exhibited strong cross-immunoreactivity between AChEs from different species.33,37,38
In the first instance, MIPs were prepared to map the topography of the surface of EeAChE with the aim to identify peptide sequences for nanoMIP preparation using an epitope approach. MIP synthesis, enzyme proteolysis and peptide sequencing were performed as previously described.36
Fig. 2 shows the peptide sequences which were identified in the epitope selection stage (using molecular imprinting and mass spectrometry) that are the most prevalent (≥40% peak intensity) in the I-TASSER model.27 Four of the sequences identified using this method match those found in literature;33,39–41 however, three peptide sequences have not been previously identified as epitopes for acetylcholine esterase (Table 1). Out of the seven epitopes identified, four of the EeAChE sequences (200–217 LALQWVQDNIHFFGGNPK; 218–243 QVTIFGESAGAASVGMHLLSPDSRPK; 313–320 FRFSFVPV; 526–532 YWANFAR) have matches with human acetylcholine esterase (hAChE) and for this reason represent potential therapeutic interest. The positions of all four epitopes on the molecule of EeAChE in relation to an enzyme-binding site are shown on Fig. 2. All of these sequences are potentially allosteric in relation to the active site.
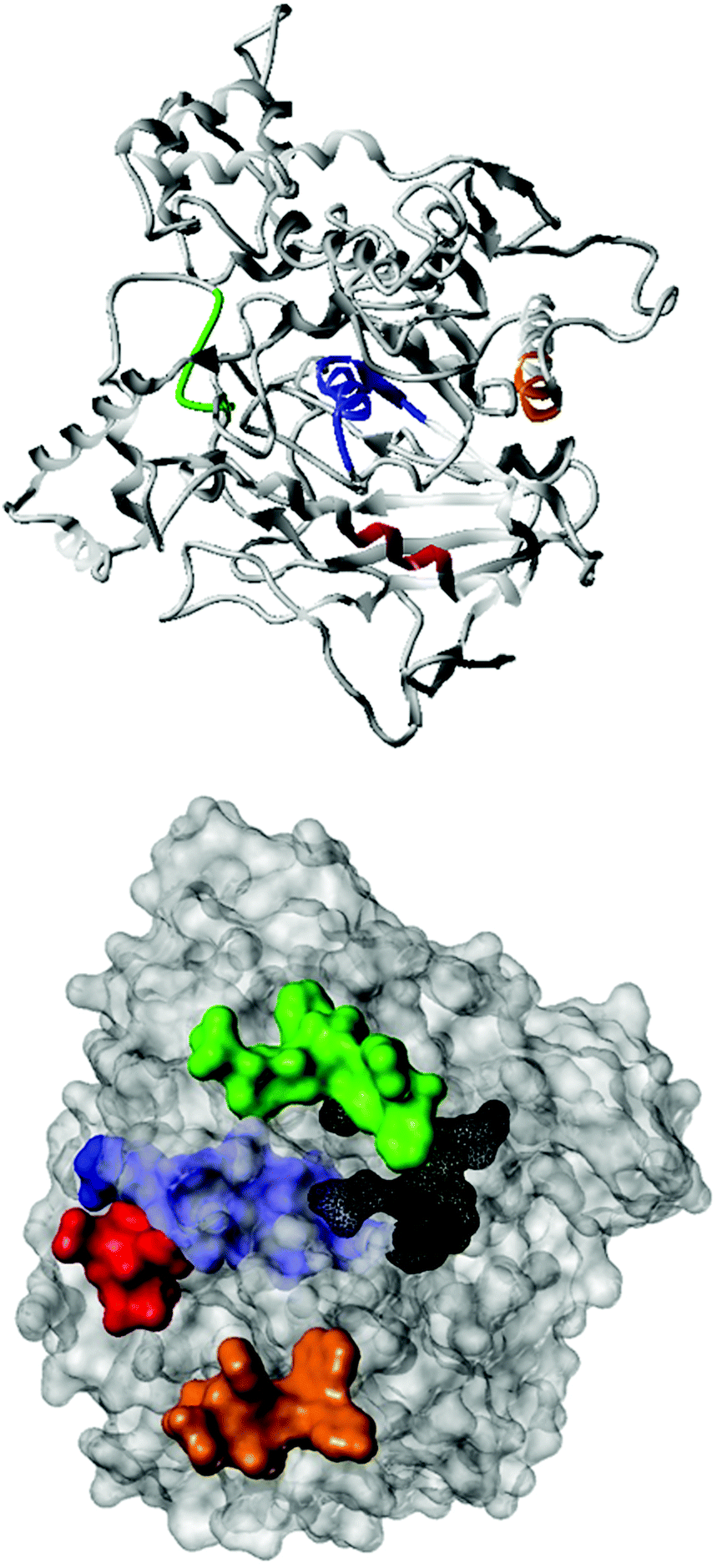 | ||
| Fig. 2 The relative positions of the four identified epitope sequences. LALQWVQDNIHFFGGNPK (red), FRFSFVPV (green), QVTIFGESAGAASVGMHLLSPDSRPK (blue), and YWANFAR (orange) are shown on the surface of EeAChE, as well as the location of the active site (black). | ||
| Position26,42,43 | Sequence identified using MIPs | Sequence of known epitopes |
|---|---|---|
| 200–217 |
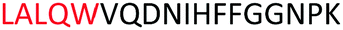
|

|
| 218–243 |

|
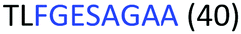
|
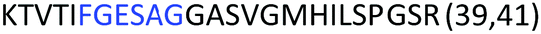
|
||
| 313–320 |

|

|
| 375–382 |
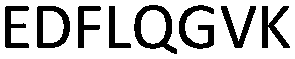
|
|
| 526–532 |

|

|
| 533–547 |

|
|
| 549–559 |

|
Synthesis and characterization of nanoMIPs
The corresponding peptides, with the addition of a Gly–Gly–Cys linker at the COOH terminus for covalent immobilization to glass and gold surfaces, were synthesized for use as templates by Zhejiang Ontores Biotechnologies Co., Ltd (China). A solid-phase approach described by Canfarotta et al.28 was adapted for MIP nanoparticle synthesis (Fig. S1, ESI†), where the terminal cysteine was used for immobilization onto the surface of amine-derivatized glass beads through succinimidyl iodoacetate (SIA) coupling (Fig. S1, ESI†).The polymer composition used was unaltered from the Canfarotta publication. This selection of the functional monomers was made specifically for the imprinting of peptides and proteins, and was successfully used in a number of studies since, with recognition being attributed to a combination of multiple weak electrostatic and hydrophobic interactions.44
Four batches of nanoparticles were synthesized for each identified epitope resulting in ‘LAL-MIP’ for LALQWVQDNIHFFGGNPK epitope, ‘FGE-MIP’ for QVTIFGESAGAASVGMHLLSPDSRPK epitope, ‘FRF-MIP’ for FRFSFVPV epitope and ‘YWA-MIP’ for YWANFAR epitope.
The concentration of nanoparticles was determined by weighing a lyophilized aliquot of a stock solution, with a typical synthesis yielding approximately 5 ± 2 mg of MIP. The size of nanoparticles was measured using dynamic light scattering (DLS) and transmission electron microscopy (Fig. S2–S6 and Table S1, ESI†) with the resultant materials comparable in size to nanoMIPs in prior literature.45,46
The affinity of MIPs imprinted against each identified epitope of AChE was assessed using a Surface Plasmon Resonance (SPR)-based instrument MP-SPR Navi 220A NAALI (BioNavis). For these experiments, MIPs were covalently immobilized on the sensor surface and a kinetic titration was performed, increasing the concentration of protein with each injection (Fig. S7–S10 and Table S2, ESI†).
All MIPs exhibited excellent affinity for EeAChE, with calculated KD values in the nanomolar range between 0.4 nM (for FRF-MIP) and 78.6 nM (for FGE-MIP) (Table S2, ESI†). These results are comparable to those observed in the literature for nanoMIPs targeting proteins through an epitope method.47,48
Modulation of enzyme activity
After establishing that the epitope-imprinted MIP NPs demonstrated strong affinities for AChE, the effect of this interaction on enzyme activity was then studied using the Ellman method. This utilizes 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB) to quantify the thiocholine produced from the hydrolysis of acetylthiocholine (ATCh) by AChE.49NanoMIPs specific for four epitopes were pre-incubated with AChE for 15 minutes, before simultaneous addition of DTNB and ATCh to initiate the reaction. Two further samples were used as controls. The first group of samples was spiked with tacrine, a known AChE inhibitor to act as a positive control, while the second group of samples contained the enzyme and substrate without the nanoMIPs, to act as a baseline. The results of this experiment clearly show the strong modulating activation effect of MIP binding (Fig. 3). The largest activation effect was demonstrated by YWA-MIPs, where the rate of hydrolysis increased 47-fold (Table 2). Interestingly, there is no clear correlation between affinity of the nanoMIP to the protein, and the observed activity.
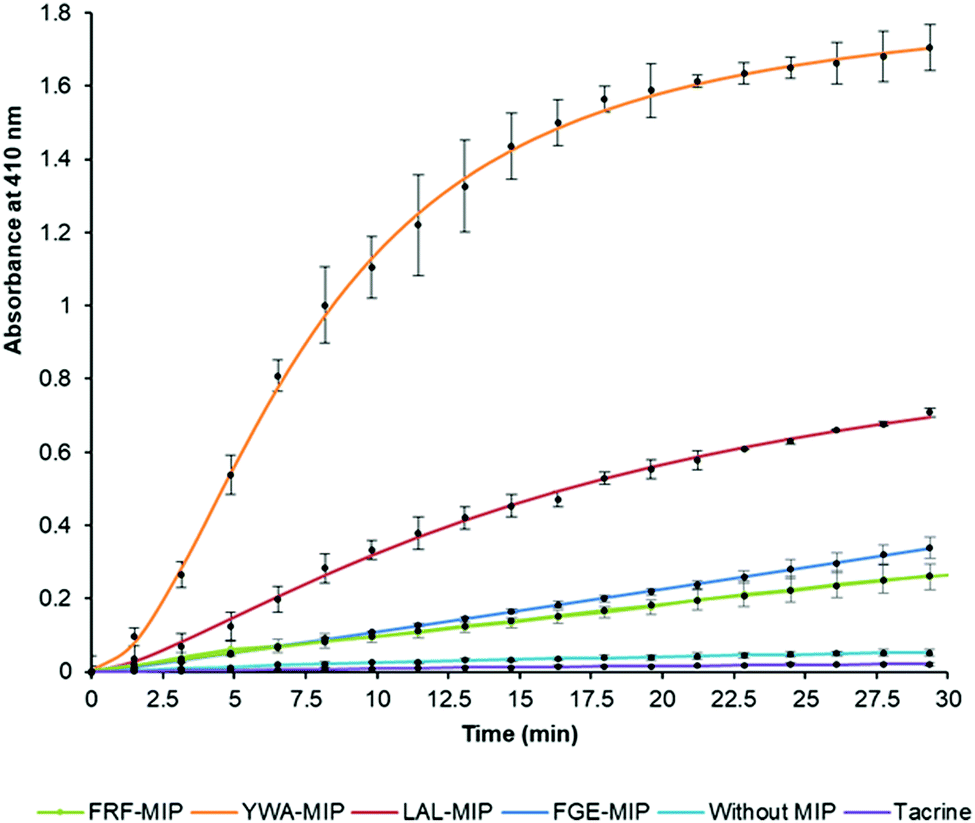 | ||
| Fig. 3 Hydrolysis of ATCh by EeAChE in the presence of select epitope-imprinted MIPs. Generation of thiocholine by AChE (25.0 mU mL−1) in the presence of epitope-imprinted MIPs (0.7 mg mL−1) or tacrine (50 nM); error bars indicate SD values. N = 3. | ||
| Sample | Concentrations of AChE, mU mL−1 | |||||
|---|---|---|---|---|---|---|
| 12.5 | 25 | 62.5 | ||||
| Rate | Relative rate (%) | Rate | Relative rate (%) | Rate | Relative rate (%) | |
| YWA-MIP | 7.96 × 104 | 3970 | 15.57 × 104 | 4773 | 6.51 × 104 | 235 |
| LAL-MIP | 4.16 × 104 | 2077 | 5.43 × 104 | 1664 | 7.54 × 104 | 272 |
| FGE-MIP | 1.55 × 104 | 772 | 1.71 × 104 | 524 | 4.38 × 104 | 158 |
| FRF-MIP | 5.90 × 105 | 294 | 1.19 × 104 | 365 | 2.84 × 104 | 103 |
| No MIP | 2.00 × 10 5 | 100 | 3.26 × 10 5 | 100 | 2.77 × 10 4 | 100 |
| Tacrine | 1.37 × 105 | 68 | 1.19 × 105 | 36 | 1.63 × 104 | 59 |
Likewise, there is no correlation between the size of the nanoMIP particle and the activity ruling out a simple steric effect. The importance of epitope imprinting is also supported by the control experiment with polymer nanoparticles (NIPs) that had the same composition as the nanoMIPs but in absence of targeted imprinting (in that they were created for non-AChE epitopes) did not show any activation effect on AChE. This strongly suggests that the activating effect is reliant on the specific position of the interaction of the epitope binding site introduced through imprinting – through an allosteric mechanism. It is also important to highlight that all AChE targeted nanoMIPs tested in this work were capable of activating AChE, irrespectively of their proximity to known inhibition sites.50
The increase in rate due to YWA-MIP is 40× (12.5 mU mL−1) and 47× (25 mU mL−1) that observed by the control of enzyme only, indicating that this is the maximum degree of activation possible by these MIPs. Whilst this increase in rate may seem unlikely for such an efficient enzyme, the specificity constant for hAChE (1.32 × 108 M−1 s−1) is still lower than that of a kinetically perfect enzyme (108 to 1010 M−1 s−1), suggesting it may be possible to increase AChE's rate between 10- and 100-fold higher than the native rate,51 in line with our observations.
Three trends emerge by comparing experiments with differing concentrations of enzyme. Firstly, the order of activation is consistent, as YWA-MIP clearly has the greatest effect, followed by LAL-MIP, FGE-MIP, and FRF-MIP. Secondly, as expected, the rate of reaction is proportional to enzyme concentration.
Finally, the degree of activation increases as enzyme concentration is reduced, which, again, is to be expected, given that the proportion of activated enzyme will be greater at lower AChE concentrations. Whilst Ellman's assay is a fast and cheap method of measuring cholinesterase activity, there are limitations52 that had to be mitigated in our study. Due to the uncommon nature of allosteric activation, the improved enzyme activity was further verified by directly measuring the substrate conversion in a kinetic mass spectrometry (MS) assay (Fig. S11, ESI†).
Originally, due to their size, it was expected that MIPs would inhibit AChE through steric occlusion of substrate access to the active center, similar to fasciculin,53 but no inhibition is observed, instead the opposite. It is possible that MIPs generated against other epitopes may indeed have this effect, especially those imprinted against sequences within the peripheral anionic site. However, in this work our efforts were focused on enzyme activation only and it is noticeable that selected sites are sterically removed from the active site (Fig. 2). Due to the highly optimized structure of the active site, it seems unlikely that any rearrangement of residues as a result of allosteric binding would be beneficial to the enzyme activity. Therefore, it was hypothesized that the activation effects observed were the result of alteration to the deep gorge leading to the enzyme active site, thus making it more easily accessible to substrate or for product removal. Exposure to a non-AChE specific MIP (imprinted using the same composition for a non-related epitope) exhibit no binding by SPR, and no activation effect (data not shown). This highlights the importance of selectivity of the target imprint and that this is not simply due to the presence of a nanoparticle.
In an attempt to observe such a conformational change, we employed circular dichroism to see if any changes could be observed. It is known that AChE is susceptible to conformational changes upon binding with ligands and inhibitors.54 Given the YWA-MIP was observed to have the greatest activity we used this nanoMIP to ensure maximal observation.
In a simple titration of YWA-MIP against a standard of AChE, changes to the protein's secondary structure observed (Fig. S12, ESI†). A noticeable shallowing of the overall trough from 200–240 nm was evident and showed a correlation with increase in MIP concentration. This suggests that the presence of MIPs is potentially loosening the structure of the protein into a more relaxed conformation, but there is no observed change in the shape of the curve suggesting that alpha-helice/beta-sheet balance is maintained. In the native enzyme, the entrance of a substrate molecule into the active site and the exit of products creates a traffic limitation to the catalytic turnover rate. Being a metastable, flexible protein,51,53,55 it has been suggested that substrate displacement is promoted via ‘breathing’ motions, leading to an increase in diameter of the gorge to allow better shuttling of molecules to the active site.55 Based on molecular dynamics, it has also been proposed that products could leave the active site through a ‘back door’, transitorily opened by concerted movements within the protein.56 These motions would involve contributions from a large fraction of the protein, so the binding of a relatively large MIP particle which stimulates a “relaxation” within the protein would be expected to significantly influence the kinetics of such processes.
Prevention and regeneration following inhibition
It was expected that MIP binding would affect the inhibition of AChE by way of irreversible inhibitors such as organophosphorus compounds. The possible mechanisms of such action involve improving substrate accessibility to the enzyme active site through MIP binding, as discussed above, and an increase in the rate of hydrolysis of the phosphoester-serine bond.40,57 In this scenario, the application of activating MIPs could protect the enzyme from these inhibitors.Malathion, a widely used OP pesticide, was employed as an irreversible inhibitor of AChE in order to investigate this hypothesis (Fig. 4). In order to test the ability of MIPs (0.35 mg mL−1) to prevent malathion from acting upon AChE (100 mU mL−1), a pre-incubation for 15 minutes was performed prior to addition of malathion (300 μM) and substrate (1 mM). The regenerative ability of MIPs was assessed in a similar manner.
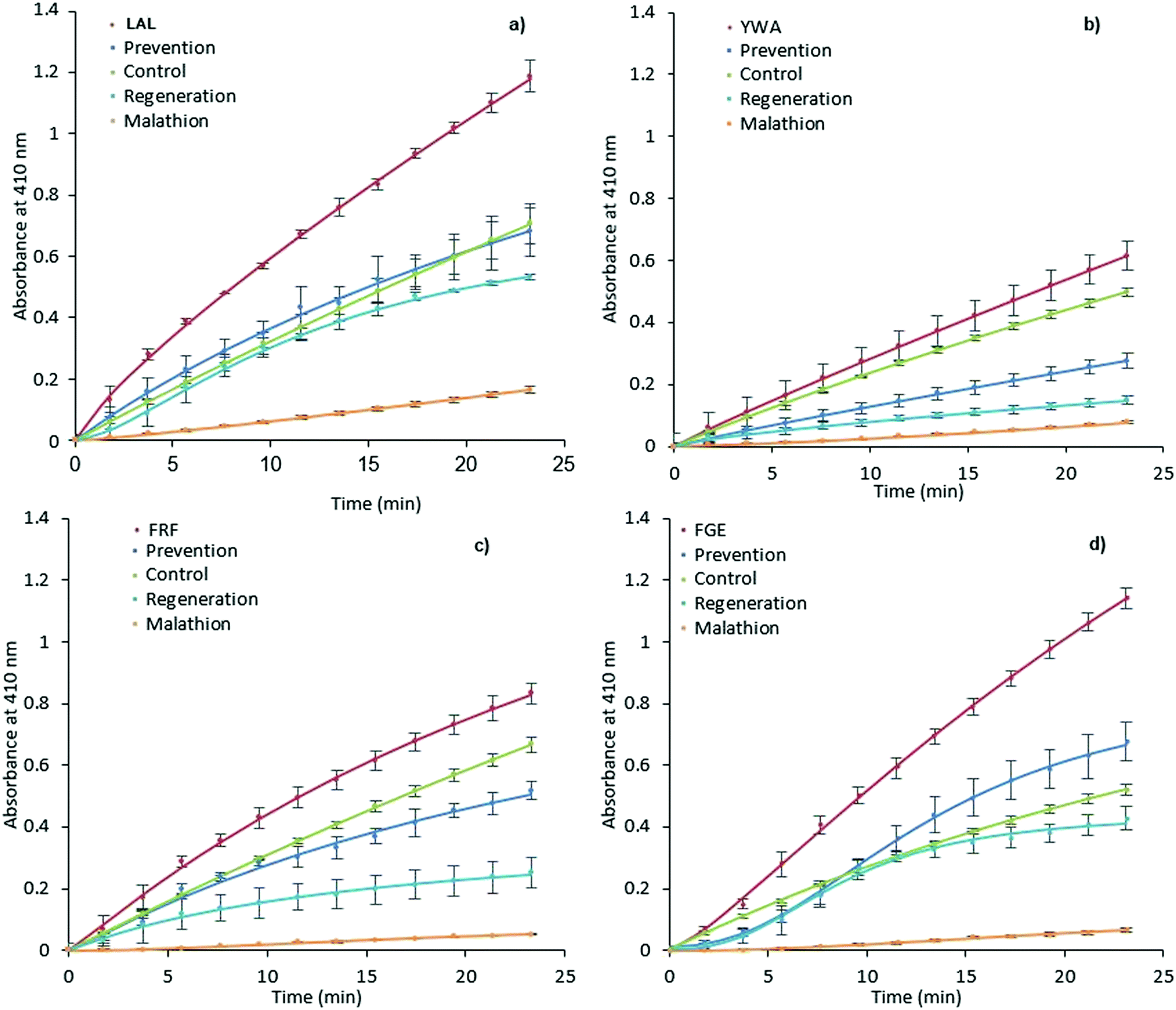 | ||
| Fig. 4 Prevention and regeneration of AChE by MIPs. LAL-MIP (a), YWA-MIP (b), FRF-MIP (c) and FGE-MIP (d)) following inhibition from malathion. Error bars indicate SD. | ||
However, AChE was incubated with malathion prior to the addition of MIP and substrate. Encouragingly, all MIPs appear to have a beneficial effect on retaining and restoring the enzyme activity; in the cases of LAL-MIP and FGE-MIP, the influence of malathion, which reduced the activity to as low as 11% of the native enzyme, was almost entirely negated.
Interestingly, the order of inhibition prevention for these epitopes (LAL/FGE > FRF > YWA – Fig. 4) is not the same as the activity improvement order (YWA > LAL > FGE > FRF – Fig. 3). The mechanism of action required for inhibition prevention will be different to that of activation can it has to consider the mechanism of the inhibition caused by the malathion so a difference while not predicted, is not unexpected.
Enzyme inactivation by OPs is a two-step process, with an intermediate Ser–OP conjugate following the initial reaction. This conjugate can then undergo two different hydrolysis reactions, with the leaving group determining the fate of the enzyme.58 If the Ser–OP bond is hydrolyzed, the enzyme is regenerated.
However, cleavage of the alternative phosphoester bond leads to strengthening of the Ser–OP bond that can no longer be hydrolyzed, a process known as ‘aging’, and is considered to be irreversible inhibition of the enzyme. Any change to the conformation of enzyme active site residues, triggered by MIP-AChE interactions, which could potentially weaken the Ser–OP bond of this intermediate conjugate, could make the cleavage of this bond preferential to that of phosphoester, and thus lead to a greater proportion of regeneration, as opposed to aging. We selected one of the MIPs and evaluated it for Michaelis Menten properties (FRF-MIP as it lies central to our set). The Michaelis constant (KM) of the enzyme in the presence of this MIP reveals an increase in KM, consistent with weaker multi-point substrate binding (Fig. S13 and Table S3, ESI†).
Looking specifically at the results in Fig. 4, we can see that the effect of prevention is always stronger than that of regeneration. This indicates that MIP binding affects the “non-aged” form of the phosphoester in addition to activating the remaining enzyme that has not been inactivated by malathion. This suggests that the MIP is actively countering the effects of the OP, rather than merely acting on the free enzyme.
Materials and methods
Materials
Acetylcholineesterase, from electric eel Electrophorus electricus (EeAChE), bovine pancrease trypsin, tacrine, malathion, N-isopropylacrylamide (NIPAm), N,N′-methylenebisacrylamide (BIS), N-tert-butylacrylamide (TBAm), acrylic acid (AAc), N-(3-aminopropyl)methacrylamide (APMA), phosphate buffered saline (PBS), ammonium persulfate (APS), N,N,N′,N′-tetramethylethylenediamine (TEMED), N-hydroxysuccinimide (NHS), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride, ethanolamine, sodium hydroxide, sulphuric acid, acetone, methanol, acetonitrile, toluene, (3-aminopropyl)triethoxysilane, 1,2-bis(triethoxysilyl)ethane, succinimidyl iodoacetate (SIA), ethylenediaminetetraacetic acid (EDTA), formic acid were purchased from Sigma-Aldrich, UK.The peptides (CLALQWVQDNIHFFGGNPK, CQVTIFGESAGAASVGMHLLSPDSRPK, CFRFSFVPV and CYWANFAR) were custom-made by Zhejiang Ontores Biotechnologies Co., Ltd (China). The cysteine residue was added to the carboxyl end of each peptide for immobilization using thiol coupling.
Epitope mapping of AChE
EeAChE (0.7 mL, 2.2 mg mL−1 in 10 mM phosphate buffered saline, pH 7.2 (PBS)) was mixed with a monomeric mixture (10 mL), consisting of NIPAm (19.5 mg), BIS (3 mg), TBAm (15 mg), AAc (50 μL of a 22 μL mL−1 solution in water), and APMA (3 mg) dissolved in 50 mL of PBS and deoxygenated (sparged) with nitrogen for 20 minutes. Polymerization was initiated by addition of APS (100 μL, 30 mg mL−1) and TEMED (6 μL), and allowed to react for 1 hour at room temperature (20 °C). To remove unreacted functional monomers and low-affinity particles, 3 × 15 mL of 10 mM PBS was added to the polymerized samples, prior to filtration through a 50 kDa centrifuge filter for 20 minutes at 3500 rpm. NanoMIPs bound to protein were reconstituted in 5 mL of 10 mM PBS containing trypsin (0.5 mg, bovine pancrease and incubated at room temperature for 36 hours. Free fragments of digested AChE and trypsin were removed by centrifugation of the samples using a 50 kDa centrifuge cartridge for 15 minutes at 3500 rpm followed by washing with 10 mM PBS (4 × 15 mL). The peptides bound to MIPs were separated from polymers using hot water (3 × 1 mL), lyophilized and reconstituted in 40 μL of 0.1% formic acid/3% acetonitrile.The resultant peptides were initially loaded onto a Waters 2G-V/M Symmetry C18 trap column (180 μm × 20 mm, 5 μm) to desalt and chromatographically focus the peptides prior to elution onto a Waters Acquity HSS T3 analytical UPLC column (75 μm × 250 mm, 1.8 μm). Single pump trapping was used with 99.9% solvent A and 0.1% solvent B at a flow rate of 5 μL min−1 for 3 min. Solvent A was LC-MS grade water containing 0.1% formic acid and solvent B was acetonitrile containing 0.1% formic acid. For the analytical column, the flow rate was set at 0.3 μL min−1 and the temperature maintained at 40 °C. The 50-minute run time gradient elution was initiated as the peptides were eluted from the trap column. The following gradient was used: 0 minute – 3% B, 30 minute – 40% B, 32 minute – 85% B, 40 minute – 85% B and 41 minute – 3% B. The NanoAcquity UPLC was coupled to a Waters Synapt G2 high definition mass spectrometer (HDMS), operating in positive electrospray ionization mode, with the capillary voltage set at 2.4 kV and cone voltage at 30 V. PicoTip emitters (New Objective, US) of 10 μm internal diameter) were used for the nanostage probe. A helium gas flow of 180 mL min−1 and ion mobility separator nitrogen gas flow of 90 mL min−1 with a pressure of 2.5 mbar were applied.
The ion mobility spectrometry (IMS) wave velocity was set at 650 m s−1 and the IMS wave height at 40 V. During the HDMS acquisition a low collision, induced dissociation energy of 4 V was applied across the transfer ion guide. For the high collision induced dissociation energy acquisition a ramp of 20 to 40 V was applied. Argon was used as the collision induced dissociation gas. Lockspray provided mass accuracy throughout the chromatographic run using [Glu1]-fibrinopeptide with m/z 785.8427. The data was acquired using MassLynx 4.1. All raw data were processed using ProteinLynx Global SERVER (Waters Corporation, Milford, Massachusetts, USA). ProteinLynx Global SERVER was used to assemble the data for alignment, peak picking, peptide and protein identification and limited upstream statistics. Data was searched against Uniprot Electrophorus electricus database (downloaded December 2016).
Structural modelling of AChE
The peptide sequence for AChE of Electrophorus electricus was obtained from the UniProt26 website (http://www.uniprot.org/) using the UniProt Knowledgebase under the code O42275 and saved in a FASTA format. The 3D structure of the peptide sequence was created using the structure prediction program I-TASSER.27 The peptide sequence was added in FASTA format and sent to the I-TASSER On-line Server.Five PDB structures were generated and downloaded from the Online Server and the C-score values were obtained. Only one structure had a positive C-score of +0.28 (the other four ranged from −1.41 to −2.62) and this structure was used as the 3D structure on the peptide sequence as an I-TASSER model and compared with the PDB ID: 4EY4.31 This was chosen as this was the most accurate structure available for EeAChE from X-ray diffraction (XRD) with the highest resolution of 2.16 Å for comparison with the I-TASSER model.
Synthesis of MIP nanoparticles (nanoMIPs) imprinted with AChE epitopes
A solid-phase approach described by Canfarotta et al.28 was adapted for nanoMIP synthesis. The surface of glass beads (200 g) was activated by boiling in sodium hydroxide (4 M, 160 mL) for 15 minutes prior to washing with water (3 × 200 mL). The beads were subsequently placed in a solution of sulfuric acid (50%, 160 mL) for 30 minutes before again washing with water (3 × 200 mL) and buffer (PBS, 3 × 200 mL), ensuring the final pH was between 6 and 8. Further washing with acetone (3 × 200 mL) was performed before drying under vacuum and placing the beads in an oven (150 °C) for 30 minutes. Activated beads were incubated in a solution of toluene (80 mL, anhydrous) with (3-aminopropyl)triethoxysilane (1.6 mL) and 1,2-bis(triethoxysilyl)ethane (0.27 mL) overnight at 70 °C.Beads were subsequently washed with methanol (3 × 200 mL) and acetone (5 × 200 mL) to remove any residual silane, before drying under vacuum and further oven drying for 30 minutes at 150 °C.
All epitopes were immobilized through their terminal cysteine modifications. SIA (10 mg) was added to silanized glass beads (60 g) in acetonitrile (25 mL) and incubated for 2 hours under exclusion of light, before washing with acetonitrile (5 × 25 mL). Thiolation buffer (pH 8.2) consisting of PBS (25 mL) and ethylenediaminetetraacetic acid (37 mg), was degassed and purged with nitrogen for 15 minutes prior to addition of peptide (2.5 mg). Incubation with SIA-linked glass beads (60 g) was allowed overnight with exclusion of light, followed by washing with water (10 × 60 mL) and drying under vacuum.
A monomer mixture consisting of NIPAM (39 mg), BIS (2 mg), TBAm (33 mg dissolved in 1 mL of ethanol), AAc (100 μL of a 22 μL mL−1 solution in water) and APMA (5.80 mg) was prepared in water (100 mL) and purged with nitrogen for 30 minutes. Following this, the monomeric mixture was added to the derivatized beads (60 g) and polymerization was initiated using a solution of APS (15 mg in 500 μL of water) and TEMED (8 μL). The polymerization was allowed to proceed for 1 hour, before quenching of the reaction by allowing oxygen into the system. The beads were subsequently washed with distilled water (9 × 30 mL) at room temperature to remove unreacted monomer and low affinity polymer before eluting high-affinity nanoparticles with hot HPLC grade water (100 mL, 60 °C).
Analysis of the size of nanoMIPs
Nanoparticle size was determined by DLS using a Zetasizer Nano (Nano-S) from Malvern Instruments Ltd (Malvern, UK) and images obtained using a JEM-2100 LaB6 TEM (JEOL, UK). Prior to DLS measurements samples were sonicated for 2 minutes, and measurements performed at 25 °C.Samples for TEM were prepared by placing 10 μL of the MIP NPs dispersion, previously sonicated for 2 minutes and filtered through a 1.2 μm glass fiber syringe filter, onto a carbon coated copper grid. The sample was left to dry overnight under a hood before imaging.
NanoMIP affinity measurements by SPR
SPR experiments were performed using a MP-SPR Navi 220A NAALI (BioNavis, UK). Bare gold sensor chips were incubated overnight with 11-mercaptoundecanoic acid (22 mg in 10 mL of ethanol) to obtain a carboxyl-functionalized surface, and then were rinsed with ethanol and dried under nitrogen immediately before use. All nanoMIPs were immobilized using amine-coupling chemistry. The surfaces of flow channels one and two were activated for 7 minutes with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of N-hydroxysuccinimide (NHS) (0.1 M) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (0.4 M) at a flow rate of 30 μL min−1. MIPs (10–200 μg mL−1 in 10 mM sodium acetate buffer, pH 5.0) and were then immobilized onto the second flow channel surface, with a control polymer of exactly the same monomer composition, imprinted for an unrelated peptide of similar size and isoelectric point immobilized on a first flow channel surface to serve as a reference surface.
:1 mixture of N-hydroxysuccinimide (NHS) (0.1 M) and N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) (0.4 M) at a flow rate of 30 μL min−1. MIPs (10–200 μg mL−1 in 10 mM sodium acetate buffer, pH 5.0) and were then immobilized onto the second flow channel surface, with a control polymer of exactly the same monomer composition, imprinted for an unrelated peptide of similar size and isoelectric point immobilized on a first flow channel surface to serve as a reference surface.
Both surfaces were subsequently blocked with a seven-minute injection of ethanolamine (1 M, pH 8.0). To collect kinetic binding data, analyte was injected over both flow cells at a rate of 15 μL min−1 at 25 °C, using ultrapure water as running buffer and for all analyte dilutions. A kinetic titration injection strategy was employed, with analyte allowed to associate and dissociate for 14 and 5 minutes respectively, before a final dissociation for 120 minutes. All data were reference subtracted and fit to a 1:2 interaction model using Tracedrawer 1.8 software.
Circular dichroism
Circular dichroism (CD) spectra were acquired using a Chirascan spectrometer from Applied Photophysics, UK. Free AChE (0.6 μM) and YWA-MIP (10 nM) in deionized water were added to a 1 mm path length cuvette and the signal allowed to stabilize. Six scans were then performed and averaged from 200–270 nm using 0.5 nm steps.Enzyme activity assay
Activity assays were adapted from the protocol booklet provided by Abcam (ab138871). Stock solutions were prepared as described in the assay kit. AChE (100 μL, 50–250 mU mL−1 in 50 mM PBS) was incubated with each nanoMIP (100 μL, 0.7 mg mL−1 in water) for 15 minutes before addition of 50 μL to wells in triplicate. 5,5′-dithio-bis-(2-nitrobenzoic acid) (2 mM in 50 mM PBS) and acetylthiocholine (2 mM in 50 mM PBS) were combined 1:1 before simultaneous addition to each of the test wells (50 μL) to initiate the reaction.
Measurements were run continuously using a Hidex Sense microplate reader (LabLogic, UK) at OD = 410 ± 5 nm for 2000 s. Regeneration and prevention experiments were performed in the same manner, however reagent concentrations were determined experimentally with the intention of reaching completion of substrate hydrolysis after 30 minutes under all scenarios tested. The final well concentrations were therefore as follows: hAChE (1–1000 mU), MIP (50 μg mL−1), DTNB (1 mM), ATCh (1 mM), malathion (4 mM), PBS (50 mM, pH 7.4). All samples were run in duplicate, and results subtracted against a reference.
Conclusions
Through a combination of whole protein imprinting and mass spectrometry, utilizing the protection offered by MIPs against proteolysis of the bound protein fragments, seven epitopes were identified for EeAChE. Of these, four were selected due to their presence in hAChE and used as templates for solid-phase imprinting. The resulting epitope-imprinted MIPs retained affinity for the native enzyme, demonstrating nanomolar binding constants. When assessed on their impact on enzyme activity, nanoMIPs were found to enhance the enzyme's catalytic rate up to 47-fold and this varied depending on the location of the targeted epitope. The ability of these compounds to protect and reverse the effects of a common OP pesticide was tested, with the AChE–MIP complexes able to withstand inhibition. The strength of this protection again varied depending on the location of the target epitope.We hypothesize that our MIPs are acting by a potential allosteric mechanism for two reasons. Firstly, they are not located in the proximity of binding site and for this reason cannot be acting directly promoting or restricting its access to substrate (a simple bulk effect). Secondly, we see pronounced change in CD spectrum in response to MIP addition, which is good indication of conformational changes typically characteristic for allosteric effects. Unfortunately, there are no known allosteric activation sites on AChE surface that we can compare with the structures of our epitopes. We are currently further exploring this hypothesis. We are also exploring the reason why a different pattern to activation and protection from inhibition is observed, through molecular dynamics.
Further experiments are necessary in order to assess the potential of MIPs as a therapeutic against a range of OPs at clinically relevant concentrations. However, the initial findings are encouraging. Alleviation of OP toxicity using this strategy is particularly advantageous. Since positive allosteric modulators do not interact directly with the active site, but instead alter the shape or dynamics of that site, using allosteric therapeutics may provide universal efficacy against nerve agents. In emergency treatment following self-poisoning, a biochemical terror attack, or exposure to pesticides, the value of a single antidote cannot be overstated. With the promising properties of MIPs for biological application and the need for new treatment options against OP intoxication, in vivo studies are being actively pursued.
The allosteric activation of enzymes by MIP NPs can potentially be exploited further for the treatment of diseases linked to enzyme deficiency. The translational nature of the strategy utilized for binding site identification and MIP generation in this work provides a blueprint for exploring the modulation of enzymes related to diseases using molecularly imprinted polymer nanoparticles.
This work leaves several questions that we are working towards answering, alongside several new and exciting avenues of study, highlighted above. We are currently exploring the nature of the observed inhibition/activation through further structural studies, and studying the observed activities with other enzymatic models, both in buffer and biological fluids. It is our intention to explore both in vitro and in vivo effects of these types of materials as our understanding of mode of action develops. We will seek to publish these studies in due course.
Author contributions
S. A. P.: conceptualization, methodology, funding acquisition and writing (first draft); T. S. B.: investigation and writing (first draft); F. C., R. P.: investigation and formal analysis; K. K.: investigation and visualisation; R. N., D. J. L. J.: investigation, methodology and formal analysis; E. P.: investigation, methodology, validation and writing (review and editing); N. T.: investigation, formal analysis and writing (review and editing).Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Massoulié, L. Pezzementi, S. Bon, E. Krejci and F.-M. Vallette, Prog. Neurobiol., 1993, 41, 31–91 Search PubMed
.
- I. Silman and J. L. Sussman, Chem. – Biol. Interact., 2008, 175, 3–10 Search PubMed
- J. L. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker and I. Sillman, Science, 1991, 253, 872–879 Search PubMed
- V. N. Talesa, Mech. Ageing Dev., 2001, 122, 1961–1969 Search PubMed
- K. Bergmann, B. E. Tomlinson, G. Blessed, P. H. Gibson and R. H. Perry, Br. Med. J., 1978, 2, 1457–1459 Search PubMed
- E. K. Perry, R. H. Perry, G. Blessed and B. E. Tomlinson, Neuropathol. Appl. Neurobiol., 1978, 4, 273–277 Search PubMed
- J. R. Roberts and J. R. Reigart, 2013. http://www2.epa.gov/pesticide-worker-safety. Accessed 25.12.2021.
- R. R. Chapleau, P. J. Robinson, J. J. Schlager and J. M. Gearhart, Theor. Biol. Med. Modell., 2014, 11, 42 Search PubMed
- F. Worek, T. Wille, M. Koller and H. Thiermann, Chem. – Biol. Interact., 2013, 203, 125–128 Search PubMed
- P. Masson, Toxicol. Lett., 2011, 206, 5–13 Search PubMed
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 Search PubMed
- X. Gao, J. Qian, S. Zheng, Y. Changyi, J. Zhang, S. Ju, J. Zhu and C. Li, ACS Nano, 2014, 8, 3678–3689 Search PubMed
- D. T. Wiley, P. Webster, A. Gale and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8662–8667 Search PubMed
- S. D. Kong, J. Lee, S. Ramachandran, B. P. Eliceiri, V. I. Shubayev, R. Lal and S. Jin, J. Controlled Release, 2012, 164, 49–57 Search PubMed
- S. H. Cha, J. Hong, M. McGuffie, B. Yeom, J. S. Vanepps and N. A. Kotov, ACS Nano, 2015, 9, 9097–9105 Search PubMed
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5018–5023 Search PubMed
- B. Sellergren, Nat. Chem., 2010, 2, 7–8 Search PubMed
- Y. Hoshino, H. Koide, T. Urakami, H. Kanazawa, T. Kodama, N. Oku and K. Shea, J. Am. Chem. Soc., 2010, 132, 6644–6645 Search PubMed
- A. Guerreiro, A. Poma, K. Karim, E. Moczko, J. Takarada, I. P. de Vargas-Sansalvador, N. W. Turner, E. Piletska, C. Schidmet de Magalhaes, N. Glazova, A. Serkova, A. Omelianova and S. Piletsky, Adv. Healthcare Mater., 2014, 3, 1426–1429 Search PubMed
- A. Cutivet, C. Schembri, J. Kovensky and K. Haupt, J. Am. Chem. Soc., 2009, 131, 14699–14702 Search PubMed
- J. Chen, K. Zeng, S. Lei, M. Wang, A. Asif and X. Ge, Part. Part. Syst. Charact., 2017, 34, 1600260 Search PubMed
- Y. J. Liao, Y. C. Shiang, C. C. Huang and H. T. Chang, Langmuir, 2012, 28, 8944–8951 Search PubMed
- H. Zhang, J. Jiang, H. Zhang, Y. Zhang and P. Sun, ACS Macro Lett., 2013, 2, 566–570 Search PubMed
- K. Smolinska-Kempisty, A. Guerreiro, F. Canfarotta, C. Cáceres, M. J. Whitcombe and S. Piletsky, Sci. Rep., 2016, 6, 37638 Search PubMed
- C. Esen, J. Czulak, T. Cowen, E. Piletska and S. A. Piletsky, Anal. Chem., 2019, 91, 958–964 Search PubMed
- UniProt Consortium, Nucleic Acids Res., 2015, 43, D204–D212 Search PubMed
- J. Yang, R. Yan, A. Roy, D. Xu, J. Poisson and Y. Zhang, Nat. Methods, 2014, 12, 7–8 Search PubMed
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 Search PubMed
- N. W. Turner, C. W. Jeans, K. R. Brain, C. J. Allender, V. Hlady and D. W. Britt, Biotechnol. Prog., 2006, 22, 1474–1489 Search PubMed
- A. Rachkov and N. Minoura, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1544, 255–266 Search PubMed
- H. M. Berman, P. E. Bourne and J. Westbrook, Curr. Proteomics, 2004, 1, 49–57 Search PubMed
- J. Ponomarenko, H.-H. Bui, W. Li, N. Fusseder, P. E. Bourne, A. Sette and B. Peters, BMC Bioinf., 2008, 9, 514 Search PubMed
- X. M. Zhang, G. Liu and M. J. Sun, Brain Res., 2000, 868, 157–164 Search PubMed
- C. E. Olson, V. Chhajlani, J. T. August and E. D. Schmell, Arch. Biochem. Biophys., 1990, 277, 361–367 Search PubMed
- Y. Bourne, L. Renault, S. Essono, G. Mondielli, P. Lamourette, D. Boquet, D. J. Grassi and P. Marchot, PLoS One, 2013, 8, e77226 Search PubMed
- S. Piletsky, E. Piletska, F. Canfarotta and D. Jones, 2018. https://patents.google.com/patent/WO2018178629A1/en. Accessed 25.12.2021.
- S. Bon, M. Vigny and J. Massoulie, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 2546–2550 Search PubMed
- X. M. Zhang, G. Liu and M. J. Sun, Brain Res., 2001, 895, 277–282 Search PubMed
- S. N. Abramson, M. H. Ellisman, T. J. Deerinck, Y. Maulet, M. K. Gentry, B. P. Doctor and P. Taylor, J. Cell Biol., 1989, 108, 2301–2311 Search PubMed
- K. M. George, T. Schule, L. E. Sandoval, L. L. Jennings, P. Taylor and C. M. Thompson, J. Biol. Chem., 2003, 278, 45512–45518 Search PubMed
- R. A. Ogert, M. K. Gentry, E. C. Richardson, C. D. De, Al, S.N. Abramson, C. R. Alving, P. Taylor and B. P. Doctor, J. Neurochem., 1990, 55, 756–763 Search PubMed
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 Search PubMed
- E. Boutet, D. Leiberherr, M. Tognolli, M. Schneider, P. Bansal, A. J. Bridge, S. Poux, L. Bougueleret and I. Xenarios, Methods Mol. Biol., 2016, 1374, 23–54 Search PubMed
- Y. Hoshino, T. Kodama, Y. Okahata and K. J. Shea, J. Am. Chem. Soc., 2008, 130, 15242–15243 Search PubMed
- C. Cáceres, F. Canfarotta, I. Chianella, E. Pereira, E. Moczko, C. Esen, C. A. Guerreiro, E. Piletska, M. J. Whitcombe and S. A. Piletsky, Analyst, 2016, 141, 1405–1412 Search PubMed
- T. S. Bedwell, N. Anjum, Y. Ma, J. Czulak, A. Poma, E. Piletska, M. J. Whitcombe and S. A. Piletsky, RSC Adv., 2019, 9, 27849–27855 Search PubMed
- E. Moczko, A. Guerreiro, C. Cáceres, E. Piletska, B. Sellergren and S. A. Piletsky, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1124, 1–6 Search PubMed
- S. S. Piletsky, E. Piletska, M. Poblocka, S. Macip, D. L. Jones, M. Braga, T. H. Cao, R. Singh, A. C. Spivey, E. O. Aboagye and S. A. Piletsky, Nano Today, 2021, 41, 101304 Search PubMed
- G. L. Ellman, Arch. Biochem. Biophys., 1958, 74, 443–450 Search PubMed
- C. Roca, C. Requena, V. Sebastián-Pérez, S. Malhotra, C. Radoux, C. Pérez, A. Martinez, J. A. Paez, T. L. Blundell and N. E. Campillo, J. Enzyme Inhib. Med. Chem., 2018, 33, 1034–1047 Search PubMed
- J. M. Bui and J. A. McCammon, Chem.-Biol. Interact., 2008, 175, 303–304 Search PubMed
- D. Dingova, J. Leroy, A. Check, V. Garaj, E. Krejci and A. Hrabovska, Anal. Biochem., 2014, 462, 67–75 Search PubMed
- M. Harel, G. J. Kleywegt, R. B. Ravelli, I. Silman and J. L. Sussman, Structure, 1995, 3, 1355–1366 Search PubMed
- D. N. Patil, S. A. Patil, S. Sistla and J. P. Jadhav, PLoS One, 2019, 14, e0215291 Search PubMed
- T. Shen, K. Tai, R. H. Henchman and J. A. McCammon, Acc. Chem. Res., 2002, 35, 332–340 Search PubMed
- Y. Xu, J. P. Colletier, M. Weik, G. Qin, H. Jiang, I. Silman and J. L. Sussman, Biophys. J., 2010, 99, 4003–4011 Search PubMed
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315–335 Search PubMed
-
C. Timperley, Best Synthetic Methods: Organophosphorus (V) Chemistry, Academic Press (Elsevier), London, 2015 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2tb00278g |
| This journal is © The Royal Society of Chemistry 2022 |