
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoelectrochemical alcohols oxidation over polymeric carbon nitride photoanodes with simultaneous H2 production†
Neeta
Karjule
 a,
Ravindra S.
Phatake
a,
Shmuel
Barzilai
b,
Biswajit
Mondal
a,
Adi
Azoulay
a,
Alexander I.
Shames
c,
Michael
Volokh
a,
Josep
Albero
d,
Hermenegildo
García
d and
Menny
Shalom
*a
a,
Ravindra S.
Phatake
a,
Shmuel
Barzilai
b,
Biswajit
Mondal
a,
Adi
Azoulay
a,
Alexander I.
Shames
c,
Michael
Volokh
a,
Josep
Albero
d,
Hermenegildo
García
d and
Menny
Shalom
*a
aDepartment of Chemistry and Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel. E-mail: mennysh@bgu.ac.il
bDepartment of Chemistry, Nuclear Research Centre-Negev, P.O. Box 9001, Beer-Sheva 84910, Israel
cDepartment of Physics, Ben-Gurion University of the Negev, Beer-Sheva 8410501, Israel
dInstituto Universitario de Tecnología Química (ITQ), Consejo Superior de Investigaciones, Científicas (CSIC), Universitat Politècnica de València (UPV), Avda. de Los Narajos s/n, Valencia 46022, Spain
First published on 25th July 2022
Abstract
The photoelectrochemical oxidation of organic molecules into valuable chemicals is a promising technology, but its development is hampered by the poor stability of photoanodic materials in aqueous solutions, low faradaic efficiency, low product selectivity, and a narrow working pH range. Here, we demonstrate the synthesis of value-added aldehydes and carboxylic acids with clean hydrogen (H2) production in water using a photoelectrochemical cell based solely on polymeric carbon nitride (CN) as the photoanode. Isotope labeling measurements and DFT calculations reveal a preferential adsorption of benzyl alcohol and molecular oxygen to the CN layer, enabling fast proton abstraction and oxygen reduction, which leads to the synthesis of an aldehyde at the first step. Further oxidation affords the corresponding acid. The CN photoanode exhibits excellent stability (>40 h) and activity for the oxidation of a wide range of substituted benzyl alcohols with high yield, selectivity (up to 99%), and faradaic efficiency (>90%).
1. Introduction
The photoelectrochemical (PEC) generation of solar fuel—typically hydrogen (H2)—is a promising technology for energy management. However, it is often coupled with oxygen (O2) production, a low-value product. Hence, some research efforts have been dedicated towards combining H2 production with the transformation of various chemicals into value-added products, which should occur on the surface of the photoanode.1–5 In particular, aldehydes and carboxylic acids are valuable industrial and medicinal compounds on their own and may also be used for further chemical syntheses.6,7 Their synthesis usually involves the selective oxidation of an alcohol using environmentally unfriendly oxidants, such as permanganates, persulfates, dichromats, and strong inorganic acids.8,9 Organic synthesis via PEC could be a greener route than traditional methods for the selective oxidation of alcohols.10,11 To date, only a few key reports have demonstrated the utility of photoanodic oxidation for the synthesis of valuable chemicals in parallel with H2 formation at the cathode.2,12Using a semiconductor film at a solid/liquid junction, where the active material both absorbs solar radiation and catalyzes the photoelectrochemical reaction, is a simple and scalable technique.13 Recently, BiVO4, WO3, and several nanocomposites-based photoelectrodes have been successfully used for oxidation, C–H functionalization, and cross-coupling reactions.14–16 Despite the progress in this field, the production of valuable chemicals using PEC is still impeded by low faradaic efficiency, low stability in aqueous solutions, narrow pH ranges (owing to the instability of the involved photoanode materials), low product selectivity, and moderate control over the oxidation process (in case of a sequential oxidation).17,18 To improve the activity and stability of the photoanode, two approaches are frequently used: (i) a miscible mixture of a non-aqueous solvent and water is used as the electrolyte solution,4,16 and (ii) a cocatalyst is deposited.19,20 However, the cocatalyst enhances the oxidative reaction rate at the photoanode, and it also increases O2 production, resulting in low product formation and low faradaic efficiency. In addition, in some cases, the density of reaction sites for the adsorption and activation of reactants on the cocatalysts is limited.21 Changing the solvent composition may augment chemical production and selectivity but decrease in the rate of the H2 evolution reaction (HER) is inevitable. To overcome these challenges, new catalytic materials for the photoanode should be introduced.
Polymeric carbon nitride (CN) is a promising photoanodic semiconductor material for water-splitting PEC because it is composed of abundant and benign elements and has appropriate properties such as suitable band edges for water splitting, an adjustable electronic structure, and UV-visible light absorption capability up to 460 nm.22 In addition, other characteristic material features of CN photoanodes—such as morphology, porosity, specific surface area, and crystallinity also significantly impact the PEC water-splitting performance.23 However, O2 evolution over CN materials is less common due to the challenges posed by the valence band (VB) position and the sluggish oxygen evolution reaction (OER) reaction kinetics. In our previous findings,24 cocatalysts were deposited into porous CN photoanodes to overcome the OER kinetic barrier and increase the consumption rate of photogenerated charges, particularly the holes. This, in turn, increased the photoanode's stability versus (vs.) photo-corrosion.
Despite very significant progress,25 low-cost CN-based photoanodes in water-splitting PEC are still an order of magnitude less performant than state-of-the-art photoanodes (based on heterojunctions that contain BiVO4, WO3, Fe2O3, TiO2, etc.) because of sluggish oxygen production.26,27 In a water-splitting process using CN, hole (h+) extraction and O2 production are considered more difficult steps because water oxidation is slow since it involves four electron–hole (e−–h+) pairs.28 This limitation of CN becomes a key advantage for PEC alcohol oxidation. In contrast to water oxidation, the oxidation of organic substrates is typically easier, therefore requiring lower photovoltage.29 Under illumination, CN photoanodes can directly oxidize an organic substrate in an aqueous electrolyte, while suppressing the competing water oxidation reaction. Additionally, CN is stable in a wide pH range with a high affinity for organic molecules on its surface and a suitable VB position for alcohol oxidation;24,30 hence, a CN-based photoanode is an ideal candidate for such PECs.
Herein, we report the use of a CN-based photoanode in PEC for the selective oxidation of benzyl alcohols (BnOH) to benzaldehydes (BZD) and benzoic acids (BZA). The optimized CN photoanode demonstrates good stability and high efficiency: it affords complete conversion of reactant (100%) with excellent faradaic yield (>99%) for the oxidation of BnOH to benzoic acid at the photoanode (in aqueous media at room temperature) together with good H2 production at the cathode. The PEC oxidation reaction mechanism was elucidated with the help of transient absorption spectroscopy (TAS), electron paramagnetic resonance (EPR) measurements, isotope-labeling experiments, and density functional theory (DFT) calculations. The scope of suitable substrate molecules was successfully enlarged by the oxidation of several other aryl alcohols to their corresponding carboxylic acids under the same conditions. Finally, we demonstrated the compatibility of the reported PEC system with heteroatom-containing substrates by preparing heterocyclic acids commonly used in pharmaceutical applications.
2. Results and discussion
2.1 PEC oxidation of benzyl alcohol on the CN photoanode
CN electrodes were prepared by a two-step method consisting of doctor-blading followed by thermal treatment (see the experimental section for the detailed procedure). The UV-vis absorption spectra, surface morphology, X-ray diffraction (XRD) patterns, and X-ray photoelectron spectroscopy (XPS) of the films are consistent with our previous reports (Fig. S1–S4†).24,28 Here, we used CN electrodes modified with reduced graphene oxide (rGO) for their porous structure, good electron (e−) mobility, and high electrochemically active surface area. In our previous work,24,28 without the addition of rGO, a CN photoanode showed very low photocurrent density and was unstable. Upon rGO integration, the photocurrent density and stability increased, mainly because of the conductivity and e− diffusion improvement due to the presence of rGO (Fig. S5†). Therefore, the metal-free CN electrodes modified with rGO were chosen for PEC BnOH oxidations. As illustrated in Fig. 1a, we propose to use CN photoanodes for the PEC oxidation of BnOH and its derivatives to aldehydes or carboxylic acids with molecular O2 as the oxidant. The BnOH oxidation reaction was performed under 1-sun illumination (AM 1.5 G, 100 mW cm−2) using a CN photoanode, 1 M NaOH aqueous solution (pH = 13.9) saturated with O2 as an electrolyte, a Pt-foil counter electrode (cathode), and an Ag/AgCl reference electrode.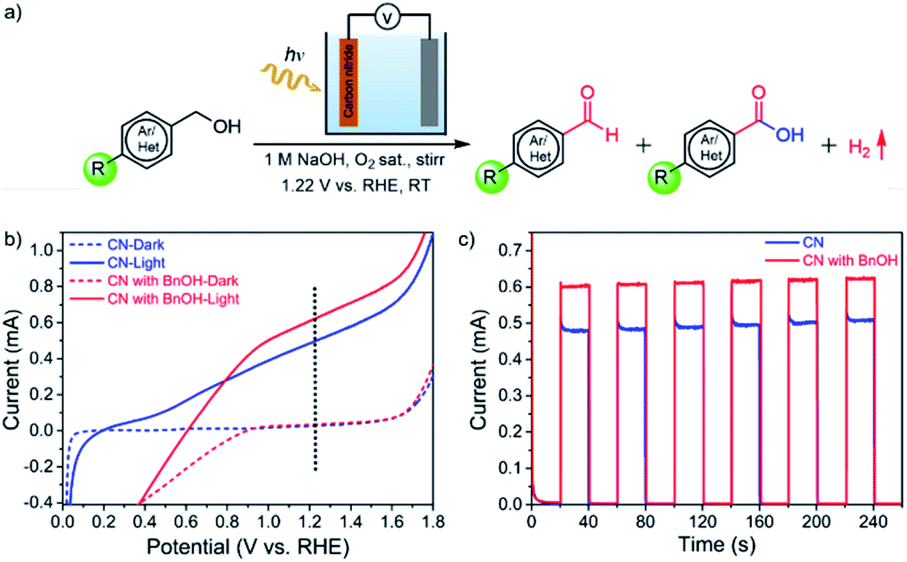 | ||
| Fig. 1 PEC oxidation: reaction conditions and characterization of photoanodes. (a) PEC oxidation scheme of primary alcohols using a CN film on a transparent conductive fluorine-doped tin oxide (FTO) electrode as the photoanode in the presence of O2 as the oxidant and a Pt cathode. (b) Linear sweep voltammetry (LSV) curves of CN without (blue) (under argon) and with BnOH (red) (in the presence of O2) under illumination (continuous lines) or in the dark (dashed lines) at a scan rate of 20 mV s−1. (c) Chronoamperometry (current vs. time) of CN electrode in 1 M NaOH aqueous solution (aqueous solution, pH = 13.9) without (blue) and with (red) BnOH (substrate concentration: 10 mM BnOH, 1 M NaOH aqueous solution, in the presence of O2). | ||
Fig. 1b shows the linear sweep voltammograms (LSV) under dark and illumination conditions. The CN photoanode has a low onset potential, with a measured photocurrent of 0.50 ± 0.03 mA at 1.22 V vs. a reversible hydrogen electrode (RHE). In the presence of BnOH and saturated with O2, the photocurrent increases to 0.60 ± 0.02 mA, suggesting that BnOH is more easily oxidized at the photoanode than water (Fig. 1b). We note that the current decay below 0.9 V is due to concurrent oxygen reduction reaction (ORR). Periodic on/off illumination of CN without and with BnOH-containing electrolytes (saturated O2 solution) at a fixed potential of 1.22 V vs. RHE affords a stable photocurrent: 0.62 ± 0.02 mA under illumination in the presence of BnOH (Fig. 1c). Under chopped light, the CN photoresponse of the electrode is consistent with the LSVs (Fig. S6a†). From this point onwards, all reported organic oxidation reactions were conducted under an applied voltage of 1.22 V (vs. RHE). Because of the lack of a cocatalyst, at this voltage bias, the production of molecular O2 from the oxygen evolution reaction (OER) is negligible.
2.2 Optimization studies
The oxidation of BnOH over the CN photoanode at different reaction times (or charge passed, Q) was carried out at 1.22 V vs. RHE under light irradiation and in the presence of O2 (Fig. S6b†). Fig. 2a shows the conversion of BnOH and charge passed vs. reaction time. As the reaction proceeds, the concentration of BnOH decreases while the concentration of generated BZD increases. The selectivity for BZD after 12 h and 24 h is 100% and ∼98%, respectively (Table S1†). After 36 h, the BnOH oxidation reaction conversion reaches 90%, and the selectivity towards BZD is >85% (Fig. 2b). Longer reaction times lead to further oxidation of BZD to BZA until full conversion of the starting BnOH. After 48 h, the major product is BZA (87.1% selectivity). A complete selectivity towards BZA is obtained after a reaction time of 58 h with an excellent faradaic yield of >99% (Fig. 2b). The PEC process enables the control of product selectivity with time because BnOH is initially oxidized to BZD (up to a selectivity of 85.0%) and then further oxidized to BZA (selectivity of 100%, indicating the complete PEC oxidation of BnOH to BZA).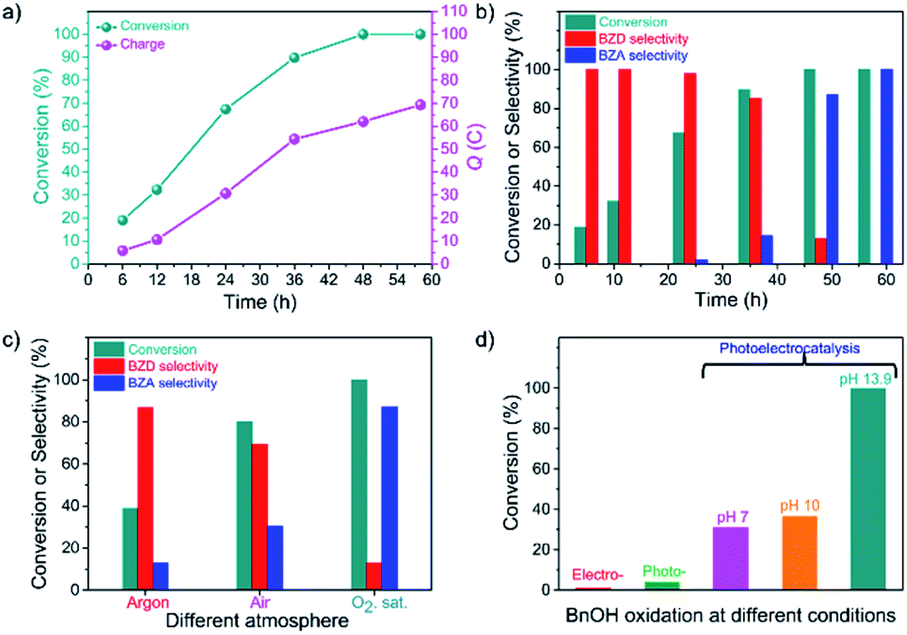 | ||
| Fig. 2 PEC oxidation of BnOH over CN photoanodes. (a) Conversion and charge passed (Q) vs. reaction time. (b) Conversion and corresponding selectivity of BZD or BZA vs. reaction time. (c) Conversion and corresponding selectivity of BZD or BZA under different reaction atmospheres (argon, air, and O2) at the same reaction time (substrate concentration: 10 mM BnOH, 1 M NaOH aqueous solution). (d) Comparison between the conversion rate of BnOH oxidation under different conditions: electrocatalysis (no illumination), photocatalysis (no applied voltage), and photoelectrocatalysis at different pH values (pH = 7, 10, and 13.9). | ||
To explore the reaction mechanism and optimize the reaction conditions, we conducted the BnOH oxidation under air and argon environments (identical reaction times, Table S2†).12 LSVs under O2, air, and Ar are provided in Fig. S7a:† the observed photocurrent varies depending on the atmosphere, leading to dissimilar conversion and selectivity between BZD and BZA formation (Fig. 2c). In the absence of O2—under Ar—the conversion rate of BnOH is low (39%), whereas it increases to 80% in air because of the presence of O2. However, in the absence of O2, the initial photocurrent is lower than in the presence of O2, and a slow decrease is observed after a few hours (Fig. S7b†). In the Ar atmosphere, conversion of BnOH to BZA over CN significantly decreases since electrons cannot activate O2 to produce ˙O2− radicals. A dramatic increase in photocurrent is achieved in a solution saturated with O2; a reaction conversion of almost 100% is achieved, with 13% and 87% selectivity towards BZD and BZA, respectively, revealing that O2 has a vital role in the oxidation reaction.31
Different experimental methods were used to distinguish the respective contributions of electrocatalysis and photocatalysis (Fig. 2d).32 The BnOH oxidation performance is summarized in Table S3.† In electrocatalytic mode (without illumination; with a voltage bias of 1.22 V vs. RHE), BnOH oxidation was negligible. Under photocatalytic conditions (under illumination; without a voltage bias), barely any BnOH was consumed (4%). These results indicate that in the electrocatalytic route, molecular O2 (i.e., without activation) cannot directly participate in the BnOH oxidation reaction and that in the photocatalytic route, BnOH undergoes sluggish oxidation with poor reaction conversion. In contrast, high conversion of BnOH (100%) is obtained via the PEC process, demonstrating that the combination of photoexcitation and voltage bias is necessary for the reaction to occur at an acceptable rate.
We investigated the effect of pH on the BnOH PEC oxidation, unveiling that the oxidation reaction slows down at lower pHs (Fig. 2d).33 The slow oxidation kinetics at low pH allows the obtention of BZD from BnOH with high selectivity (>99%). At high pH, in contrast, a higher photocurrent with good long-term stability allows further oxidation to BZA (Table S3†). Resembling water oxidation,24 in BnOH oxidation, higher photocurrent and higher rates are observed at higher pH. The conversion of alcohols to acids is achieved only at pH 13 mainly because the generation rate of reactive intermediates as ˙O2− is much higher (due to increased stability) in an alkaline medium, which ultimately increases the reaction rate.34,35
2.3 Isotope-labeling experiments
To uncover the source of the oxygen atoms involved in the oxidation process, systematic D2O and 18O2 isotope-labeling experiments were conducted (Fig. 3).31,36,37 D2O labeling experiments were carried out by replacing H2O with D2O in the 1 M NaOH electrolyte solution. The GC-MS results show that deuterium incorporates into BnOH and BZD by hydrogen–deuterium exchange (Fig. 3a–b and Fig. S8–S9†).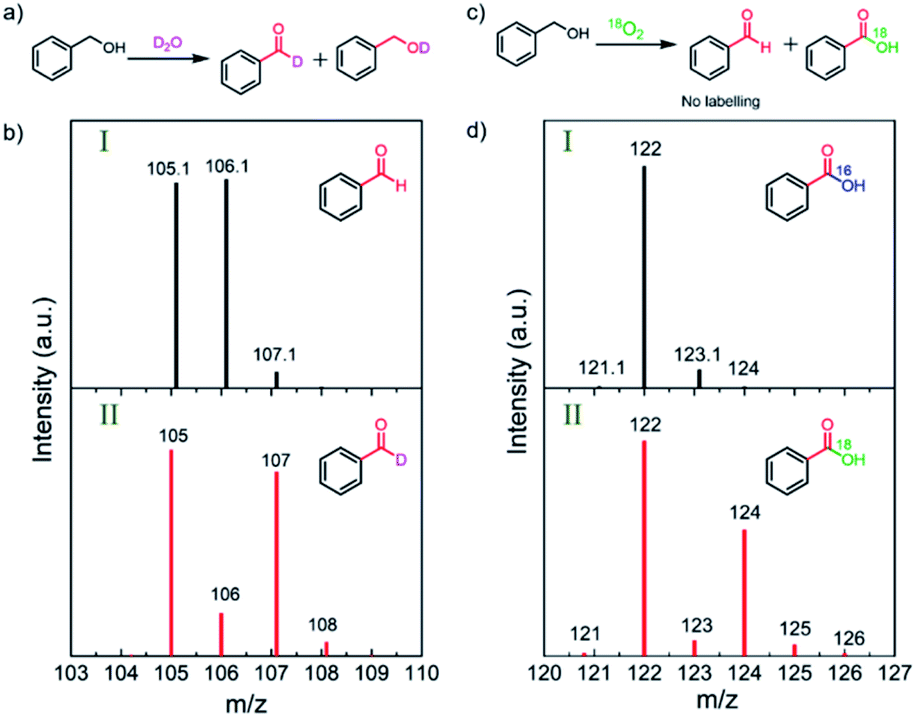 | ||
| Fig. 3 D2O and 18O2 labeling experiments for BnOH oxidation. (a) PEC oxidation of BnOH using a D2O solution containing 1 M NaOH. (b) Mass spectra of BZD produced in (I) H2O and (II) D2O showing labeling in BZD. (c) PEC oxidation reaction of BnOH in 18O-labeled oxygen gas (18O2). (d) Mass spectra of BZD produced in (I) 16O2 and (II) 18O2 showing labeling in benzoic acid (18OH group of the carboxylic acid). The analyzed reaction conditions are illuminated CN photoanode at 1.22 V vs. RHE for 48 h; the substrate concentration is 10 mM BnOH in 1 M NaOH aqueous solution. | ||
To further clarify the exact role of molecular O2 in the BnOH oxidation, the PEC oxidation of BnOH under an isotope-labeled 18O atmosphere was performed. In the first oxidation process, no 18O-labeled BZD was detected after photoelectrocatalysis, ruling out the inclusion of O atoms from O2 or ˙O2− radicals into BZD (Fig. S10†).38 However, 18O2 labeling experiments reveal that the second oxygen in BZA originates from O2 and not from water (Fig. 3c–d, marked in green in the chemical structure).
2.4 Mechanistic investigations
A series of control experiments were conducted by adding different scavengers to the system to further elucidate the reaction mechanism (Fig. 4a).39,40 In the presence of 1,4-benzoquinone (BQ), an ˙O2− radical scavenger, the conversion of BnOH remained similar. However, the yield of BZA was reduced significantly, indicating that the oxidation step of BZD to BZA involves ˙O2− radicals, in line with the 18O2 labeling measurements. The addition of tert-butyl alcohol (TBA), a hydroxyl-radical (˙OH) scavenger, had little influence, disclosing that ˙OH has a negligible effect on the reaction and, hence, on the BZA yield (Table S4†).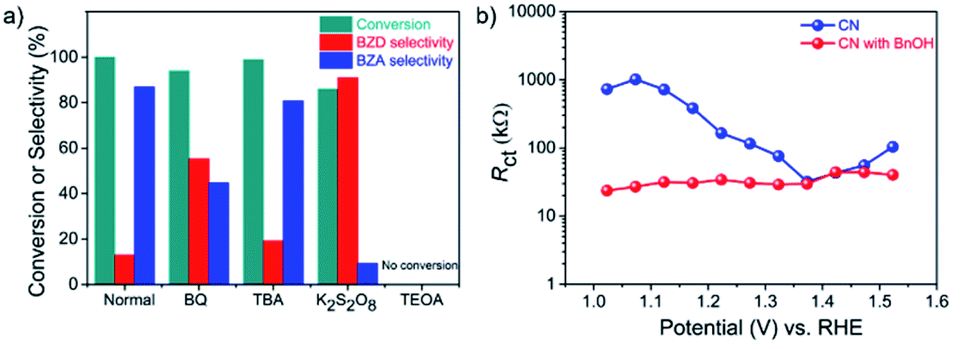 | ||
| Fig. 4 Mechanistic and electrochemical investigations. (a) BnOH oxidation reaction over a CN photoanode in the presence of various radical scavengers for 48 h (“Normal” was conducted without scavenger); BQ as a ˙O2− radical scavenger, 0.5 mM; TBA as an ˙OH radical scavenger, 1 mM; K2S2O8 as an e− scavenger, 1 mM; TEOA as a h+ scavenger, (10% v/v). (b) Charge transfer resistance (Rct) of CN photoanode at various applied potentials without and with BnOH (in the presence of O2). | ||
In contrast, the photocurrent density and the yield of BZA were significantly decreased by the addition of potassium persulfate (K2S2O8), an e− scavenger, confirming the importance of O2 activation to form ˙O2− radicals by photogenerated e− in BnOH oxidation. Finally, the addition of triethanolamine (TEOA), a h+ scavenger, completely suppressed the BnOH conversion, indicating that photogenerated h+ play a key role in the oxidation of BnOH. Summarizing this section, h+ and ˙O2− radicals are the main active species for the PEC BnOH oxidation reaction under visible light illumination. Electrochemical impedance spectroscopy (EIS) measurements reveal that the presence of BnOH lowers the charge transfer resistance (Rct) of CN (Fig. S11 and S12†).41 EIS studies at applied potentials of 1.02–1.52 V vs. RHE indicate better h+ extraction properties from the electrode to the solution in the presence of BnOH over a wide potential range (Fig. 4b).
In order to gain information of the BnOH oxidation mechanism, transient absorption (TA) measurements have been performed in a CN dispersion in acetonitrile (355 nm laser excitation) under N2 and O2 atmosphere (Fig. S13 and S14,† respectively).42 Under an O2 saturated atmosphere (Fig. S14†), the CN TA spectrum shows a continuous band along the measured spectrum, with a relative maximum intensity around 660 nm. In our results, the slow component in the CN TA under O2 atmosphere can be attributed to long lived photogenerated h+ in the CN valence band (VB).
In order to further investigate the PEC reaction mechanism, TA experiments have been also carried out using CN electrodes under 355 nm laser excitation in the presence of BnOH after O2 purging. The TA kinetics of the CN electrode were measured at 660 nm with either no applied bias voltage or 1.2 V vs. NHE (Fig. 5a).43 Under no bias, the TA decay follows the expected bimodal behavior. The calculated lifetimes after experimental data fitting to eqn (1) were 120.6 ns and 2.03 μs for the fast and slow components, respectively. However, under applied bias voltage 1.2 V vs. NHE, the TA signal was completely quenched. These results indicate that light-induced h+ in the CN VB could not have enough oxidation potential to oxidize BnOH, while addition of the applied bias voltage would promote the prompt h+ transfer to BnOH.44 These results are in good agreement with our control experiments, where photocatalytic and electrocatalytic BnOH oxidation showed negligible reaction conversion.
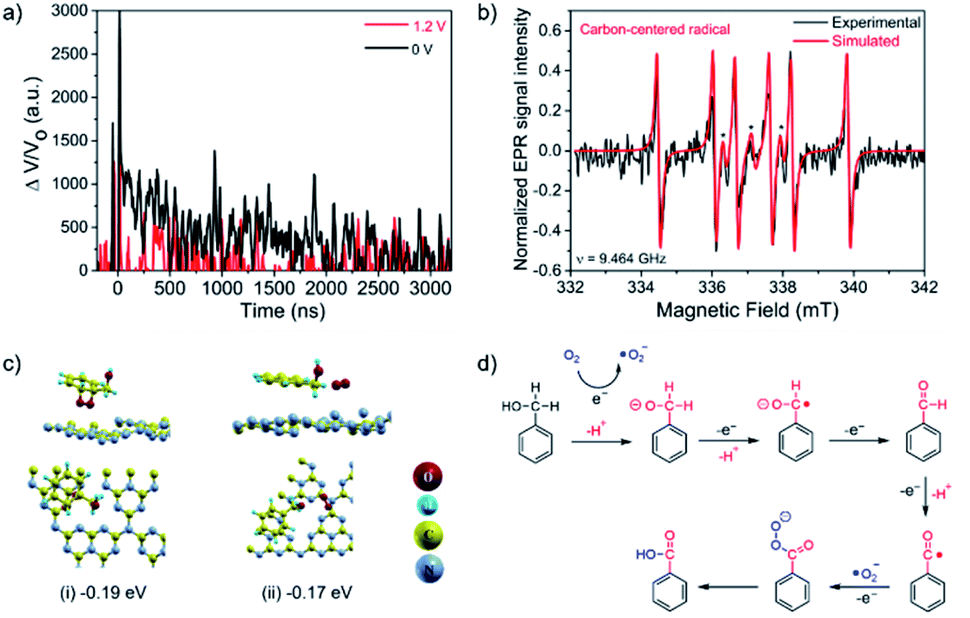 | ||
| Fig. 5 Mechanistic studies: experiments and calculations. (a) TA kinetics of CN photoelectrode in O2-saturated LiClO4 solution (0.1 M in acetonitrile) and 64 mM BnOH upon no applied bias voltage (black) and 1.2 V vs. NHE (red). Pt wire and Ag/AgCl electrode were used as counter and reference electrodes, respectively. Laser excitation 355 nm. Monitored wavelength of 660 nm. (b) EPR detection of carbon-centered radicals formed during BnOH oxidation using DMPO as the spin-trapping agent (black asterisks indicate characteristic DMPO peaks). (c) DFT calculations showing the favorable configuration of BnOH and O2 over CN; The adsorption energy beneath each illustration was computed relative to the energy sum of the relaxed CN and the isolated relaxed moiety. (d) Proposed mechanism of the PEC oxidation of BnOH over a CN photoanode. | ||
Overall, the combination of the photogenerated e− in the CN conduction band (CB) and the applied oxidation potential of 1.22 V vs. RHE are the driving forces of the outstanding performance of the CN photoanodes for the PEC BnOH oxidation under these conditions. Electron paramagnetic resonance (EPR) spectroscopy was performed under an O2 atmosphere by adding the radical spin trapping agent DMPO (5,5-dimethyl-1-pyrroline N-oxide) to the electrolyte, confirming the existence of a radical intermediate (Fig. 5b).
In the EPR results, we didn't detect ˙O2− radicals; it could be that reactive oxygen species, such as ˙O2− radicals, have short lifetimes in an aqueous solution with high pH.34 The six new peaks in the fitted results are attributed to carbon-centered radicals, implying that these intermediates are formed during the reaction.45 Typically, carbon-cantered radicals, which can be produced by reactive ˙O2− radicals or h+, are regarded as the crucial intermediates for PEC BnOH oxidation.31
The first essential step in most heterogeneous catalytic reactions is the adsorption of reactants on the catalyst surface.46,47 Therefore, DFT calculations were preformed to compute the adsorption energies of the initial moieties ((a) molecular O2, (b) BnOH, and (c) BnOH adjacent to O2) and intermediate reaction products ((d) BZD, (e) BZA) over CN (Fig. 5c and S15†). From an energetic viewpoint, the adsorption energy of O2 is about −0.22 eV with a preferred location above the meso pore (Fig. S15a†). It is worth mentioning that for a dissociated molecule, the adsorption energy was relatively high, exceeding −1.5 eV. The same site is also preferred by the BnOH, with an adsorption energy of −0.12 eV (Fig. S15b†). When both moieties are located above this site, each moiety changes its preferred location, and the obtained integral adsorption energy is −0.17 eV. Thus, the adsorbed O2 molecule (Oads2) can effectively bind to the CN photoanode surface, even in the vicinity of a BnOH molecule.
From an energetic viewpoint, Oads2 reacts with the BnOH on graphitic carbon nitride48 with a low adsorption energy of −0.17 eV (Fig. 5c). In this configuration, and under both illumination and applied potential, the CN photoanode, which generates e− and h+ charge carriers, can efficiently catalyze the reduction of Oads2 into ˙O2− radicals using the available e−. Hence, the effective reaction rate for the BnOH oxidation into BZA depends to a large extent on the rate of adsorption and activation of BnOH and O2 molecules on the CN surface (Fig. S16†).
We propose a mechanism for the PEC oxidation of BnOH over CN photoanodes (Fig. 5d). Under the combined illumination and applied potential, the CN photoanode generates e− and h+ charge carriers.49 The photogenerated e− (at the CB) reduces O2 molecules to ˙O2− radicals while the h+ (at the VB) oxidizes the BnOH molecules to the corresponding carbon-centered radicals (Fig. S17†).50 Simultaneously, ˙O2− radicals abstract protons from the BnOH molecules to form carbon-centered radicals, which are then oxidized to BZD. The ˙O2− radicals play the important role of promoting the rapid consumption of photogenerated e− in the reaction, thus accelerating the separation of photogenerated e− and h+, which is beneficial for the BnOH oxidation.51,52 Subsequently, the BZD molecules are oxidized by h+ at the electrode to form carbon-centered radicals, which themselves react with ˙O2− to form BZA. During the oxidation process, most e− transfer to the Pt counter electrode (the cathode), thanks to the applied bias, where they reduce the adsorbed hydrogen (Hads) formed by water dissociation into H2.53 It is important to note that the uptake of e− by O2 at the photoanode enhances the stability of the CN electrodes because it removes e− that are far from the conductive substrate. Thus, less recombination occurs and the photogenerated h+ lifetime is extended.
After finding the optimal reaction conditions and understanding the underlying oxidation mechanism, we extended the scope of demonstrated PEC oxidation substrates to BnOH substituted with –CH3, –OCH3, –Cl, –CF3, and –NO2 groups (Fig. 6 and Table S5†). The reaction conversion and amount of oxidation product (3) were determined by GC-MS and 1H NMR measurements. BnOH derivatives substituted with electron-donating groups (–CH3, –OCH3) and electron-withdrawing groups (–CF3, –Cl, and –NO2) are efficiently oxidized into the corresponding carboxylic acids (100% conversion)
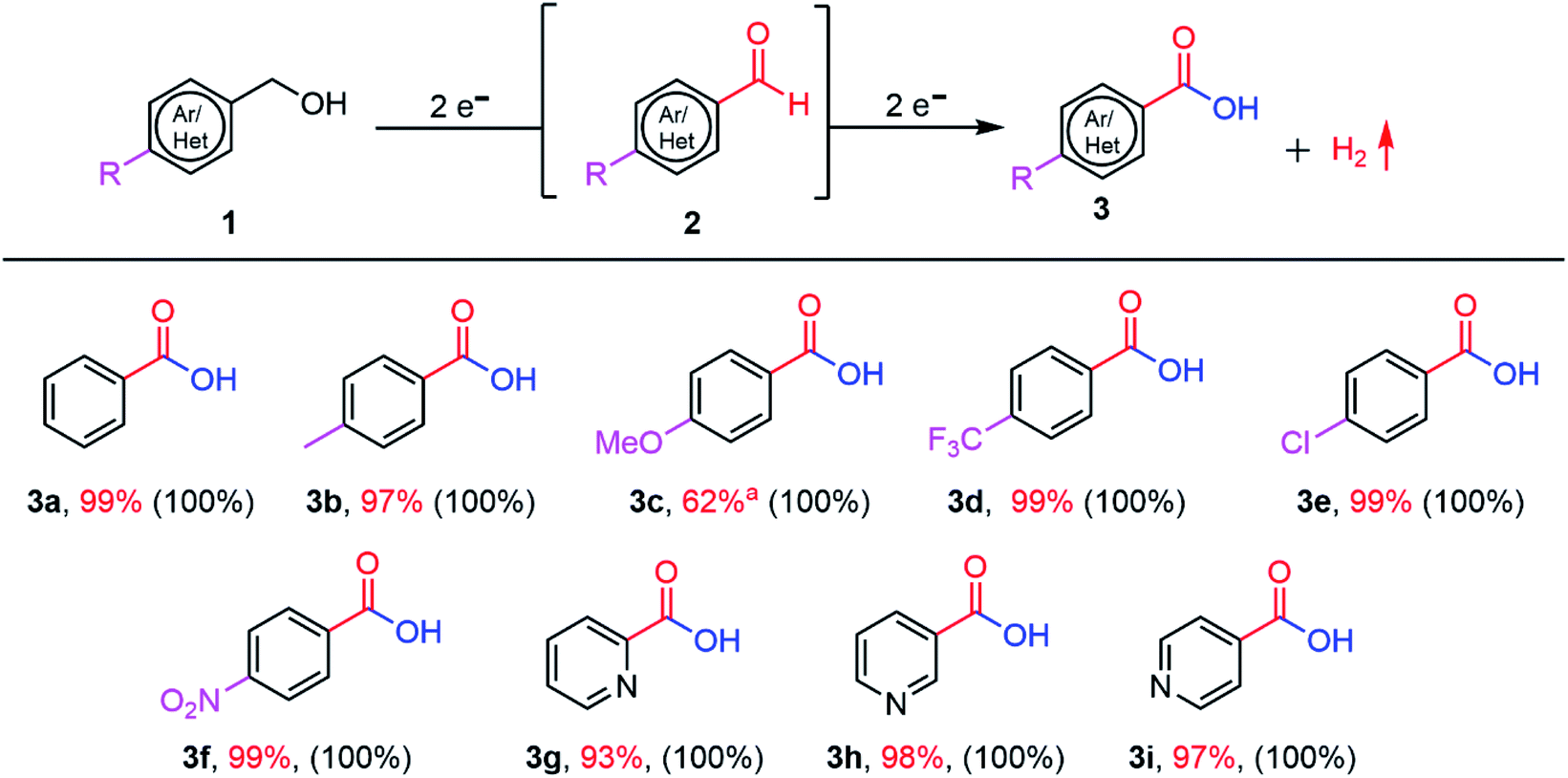 | ||
| Fig. 6 Scope of the substituted BnOH used as substrates. Reaction conditions: 10 mM of alcohol (0.18 mmol) in 1 M NaOH aqueous solution (pH 13.9) saturated with O2 as an electrolyte, CN photoanode as the working electrode. PEC was conducted in constant potential mode (1.22 V vs. RHE) under 1-sun illumination. The reaction conversion (in parentheses) was determined by GC-MS and 1H NMR. The amount of oxidation product (3) was determined by 1H NMR using dibromomethane as an internal standard. aThe oxidation of 4-methoxybenzyl alcohol yields 38% 4-methoxybenzaldehyde. | ||
The observed photocurrent varies depending on the substrate identity; hence, the PEC time to achieve complete conversion itself varies (a total charge of around 69–70 C passed is necessary to complete the 4e− oxidation reactions).
In the case of BnOH substituted with –OCH3 (4-methoxybenzyl alcohol), the reactant is fully consumed (100% conversion), but the corresponding carboxylic acid is obtained in low yield (62%). –OCH3 is a strong electron-donating group; therefore, the resonant structures of the 4-methoxybenzyl alcohol radicals are more stabilized in the para orientation, which does not promote effective aldehyde-to-carboxylic acids conversion.54 Heteroatom-containing substrates (pyridine derivatives 3g–3i) can also be successfully converted to the corresponding carboxylic acids in high yield (≥93%). Overall, all the substrates show 100% conversion and faradaic yields ≥92% (except 4-methoxybenzyl alcohol: faradaic yield of 81.3%). The cathodic reaction, that is, HER, was measured during BnOH oxidation in a two-compartment cell (Fig. S18†).12 The concentration of generated H2 increases steadily with time. After 10 h of PEC operation time, 35.6 μmol H2 is produced (faradaic yield (%) of 75 ± 8). The non-optimal faradaic yield stems from the reaction of e− with O2.
A detailed characterization of the electrodes after 48 h of PEC operation reveals changes in the morphology and the thickness of the CN film (stability measurement, Fig. S19–S20†). XRD and Fourier-transform infrared (FTIR) measurements of CN after a long reaction time indicate structural changes in the CN films (Fig. S21†). FTIR shows that the characteristic peaks of CN slowly disappear with increasing PEC operation time (the strong band at 880–1640 cm−1, attributed to the typical stretching vibration of C–N and C–NH heterocycles and the band near 808 cm−1, related to the s-triazine). The XRD measurements suggest that the CN film on FTO has a lower crystallinity the characteristic peaks of CN are much weaker than before PEC operation. The intensity of the diffraction reflection at ∼27.7° (002 plane of CN) decreases, which indicates that some CN detaches from the FTO surface during operation. Complementary XPS measurements suggest partial oxidation of the CN framework during the PEC oxidation reaction of BnOH (Fig. S22†). In the C 1 s spectrum, the peak corresponding to C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C decreases, revealing an increase in the C-to-N atomic ratio (from 0.87 to 2.13) (Table S6†). Hence, the main reason behind the slow decrease in photocurrent lies in the leaching of CN from the FTO surface during operation; we intend to direct future research efforts towards stabilizing the CN film under the working conditions.
C decreases, revealing an increase in the C-to-N atomic ratio (from 0.87 to 2.13) (Table S6†). Hence, the main reason behind the slow decrease in photocurrent lies in the leaching of CN from the FTO surface during operation; we intend to direct future research efforts towards stabilizing the CN film under the working conditions.
3. Conclusions
In conclusion, the utilization of CN-based photoanode in PEC has been successfully demonstrated with an organic synthesis producing value-added aldehydes and carboxylic acids while H2 is produced at the cathode. Under visible light, excited e− from the semiconductor CN surface activate O2 molecules into ˙O2− radicals, whereas photogenerated h+ oxidize BnOH to BZD or BZA. This setup allows the successful oxidation of multiple substituted aryl alcohols into their corresponding carboxylic acids with excellent yields, up to 99%. The faradaic yields of the oxidation reactions were very high for all substrates (>90%). In the proposed mechanism (backed by TAS, EPR, and isotope-labeling experiments), after a proton is abstracted from BnOH (and then BDZ), oxidation by ˙O2− radicals forms carbon-centered radicals as the reaction intermediates towards the BZA product. DFT calculations verified that CN has an excellent ability to adsorb BnOH and O2, whereas the rapid reduction of O2 by photoexcited e− is responsible for the excellent charge separation. This study shows the compelling potential of utilizing metal-free polymeric CN materials for photoelectrochemical organic transformations. These promising results may lead to safer, more selective and cost-effective chemical reactions.4. Experimental
4.1 Materials and reagents
All chemicals were purchased from commercial sources and used as received. Melamine (99%), potassium chloride (KCl, ReagentPlus®, ≥99%), 4-methoxybenzyl alcohol (98%), 4-(trifluoromethyl)benzyl alcohol (98%), 4-chlorobenzyl alcohol (99%), 4-methylbenzyl alcohol (98%), 4-nitrobenzyl alcohol (99%), 4-pyridinemethanol (99.5%), 3-pyridinemethanol (98%), 2-pyridinemethanol (98%), oxygen-18O2 (99 at% 18O, 99% (CP)), deuterium oxide (D2O, 99.9 at% D), 1,4-benzoquinone (BQ, ≥98%), tert-butyl alcohol (TBA, ≥99.5%), and 5,5-dimethyl-1-pyrroline N-oxide (DMPO, ≥97%) for electron paramagnetic resonance (EPR) spectroscopy were purchased from Sigma-Aldrich. Ethylene glycol (EMSURE® Reag. Ph. Eur, Reag. USP) and potassium persulfate (K2S2O8, ≥99%) were purchased from Merck. Dichloromethane (CH2Cl2, 99.5% amylene-stabilized), ethanol (≥99.9%), acetone (99.5%), and hydrochloric acid (32 wt%, AR grade) were purchased from Bio-Lab Ltd, Israel. Sodium hydroxide (NaOH, 99%), sodium sulphate anhydrous (Na2SO4, 99%), and potassium hydroxide pellets (KOH, 85%) were purchased from Loba Chemie, India. Benzyl alcohol (BnOH, ≥99.5%) was purchased from Acros Organics. Dibromomethane (CH2Br2, 99%) was purchased from TCI chemicals. Chloroform-D (CDCl3, >99.8%) was purchased from Fluorochem, UK. Dimethyl sulphoxide-d6 (DMSO-d6, 99.9%) was obtained from Cambridge Isotope Laboratories. Graphene oxide (GO, 0.4 wt%, >95%) aqueous suspension was purchased from University Wafer Inc., USA (C89/GOSD18004/D). TEOA (99%) was purchased from Glentham Life Sciences, UK. Fluorine-doped tin oxide (FTO) coated glass (TEC15, 12–14 Ω sq−1) was purchased from Xop Glass Company, Spain. De-ionized water (DI) was purified using a Millipore Direct-Q3 water purification system (18.2 MΩ cm resistivity).4.2 Materials characterization
UV-vis absorption spectra were measured on a Cary 100 spectrophotometer using the transmittance mode for films over a transparent substrate to measure T and a diffuse reflectance accessory (ATR) to estimate the reflectance, R. Fourier-transform infrared spectroscopy (FTIR) was performed on a Thermo Scientific Nicolet iS5 FTIR spectrometer (equipped with a Si attenuated total reflection (ATR) accessory). Scanning electron microscopy (SEM) images of the CN electrodes were obtained using an FEI Verios 460L high-resolution SEM equipped with a FEG source and operated at an accelerating voltage, U0 = 3.5 kV (after sputtering with Pt, ∼10 nm using a Quorum Q150T ES system). X-ray diffraction (XRD) patterns were obtained using a PANalytical's Empyrean diffractometer equipped with a position-sensitive detector X'Celerator. Data were collected with a scanning time of ∼7 min for a 5–60° 2θ range using Cu Kα radiation (λ = 1.54178 Å, 40 kV, 30 mA). X-ray photoelectron spectroscopy (XPS) data were obtained from an X-ray photoelectron spectrometer ESCALAB 250 ultrahigh vacuum (1 × 10−9 bar) device with an Al Kα X-ray source and a monochromator. The X-ray beam size was 500 μm, survey spectra were recorded with a pass energy (PE) of 150 eV, and high energy resolution spectra were recorded with a PE of 20 eV. To correct for charging effects, we calibrated all spectra relative to the carbon C 1 s peak positioned at 284.8 eV. The XPS results were processed using the Thermo Scientific™ Avantage software. Continuous-wave X-band (9.4 GHz) EPR measurements were carried out at room temperature (T ∼295 K) using a Bruker EMX-220 spectrometer equipped with an Agilent 53150A frequency counter. For the EPR measurements, 2 mL of the sample was collected from the reaction mixture and mixed with DMPO (0.04 mmol) as a radical spin trapping agent.4.3 Transient absorption spectroscopy (TAS)
Laser flash photolysis (LFP) measurements were performed using a Q-switched Nd:YAG laser (Quantel Brilliant, 355 nm, 20 mJ per pulse, 5 ns fwhm) coupled to a mLFP-111 Luzchem miniaturized equipment. This transient absorption spectrometer includes a ceramic xenon light source, a 125 mm monochromator, a Tektronix 9 bit digitizer TDS-3000 series with 300 MHz bandwidth, a compact photomultiplier and a power supply, a cell holder and fiber optic connectors, a fiber optic sensor for laser-sensing pretrigger signal, computer interfaces, and a software package developed in the LabVIEW environment from National Instruments. The LFP equipment supplies 5 V trigger pulses with a programmable frequency and delay. The TA experiments of CN dispersions in acetonitrile were recorded using 10 mm × 10 mm quartz cells with a capacity of 4 mL, while the TA experiments of CN photoelectrodes were recorded using a home-made quartz cell suitable for photoelectrochemistry experiments. A Pt wire (1 mm thick) and Ag/AgCl (saturated KCl) electrodes were used as counter and references electrodes respectively. LiClO4 (0.1 M in acetonitrile) was used as the electrolyte. In addition, BnOH (64 mM) was also added to the electrolyte solution and the whole system was O2 purged. The absorbance value of the CN samples in acetonitrile dispersions was around 0.6 at the laser excitation wavelength (355 nm). The lifetimes were calculated after experimental data fitting to eqn (1).| F(t) = a1 e−τ1t + a2 eτ2t | (1) |
4.4 DFT methodology
Based on previous experimental characterization, we considered a supercell with one layer of graphitic carbon nitride (CN) arranged in a tri-s-triazine geometry and containing 24 carbon atoms, 32 nitrogen atoms, and a top vacuum layer of ≈28.6 Å. To define the preferred location of the BnOH moiety over the CN surface and to compute its adsorption energies, all the atoms in this supercell were allowed to relax until the residual forces of all the atoms were less than 10−3 Ry/B3 and the change in energy was less than 5 × 10−5 Ry. For the final relaxed structures, a self-consistent convergence criterion of 10−6 Ry was imposed. The adsorption energy (Eads) of each configuration was computed according to eqn (2).| Eads = Esysrlx − (ECNrlx + EBnOHrlx) | (2) |
The preferred adsorbed configuration of BnOH molecules over the CN surface was used for further calculations to explore the preferred location and configuration of molecular oxygen (O2) over a CN surface covered with adsorbed BnOH. The calculations were carried out using the Quantum-Espresso package55 and performed on a periodically repeated supercell. The exchange–correlation potential was treated within the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA)56 and a k-mesh of 2 × 2 × 1 was constructed according to the Monkhorst and Pack scheme.57 The ion core was described by plane wave (PAW) pseudopotentials58 and the valence electrons (1s electron for the H atoms, 2s and 2p electrons for C and N atoms, and 2s and 2p electrons for O2) were treated explicitly with a 350 Ry cut-off for the charge density and kinetic 50 Ry for the wave function. To ensure that the results of the calculations are directly comparable, identical conditions and convergence criteria were employed for all systems.
4.5 Carbon nitride electrode preparation24,28
A melamine–graphene oxide supramolecular paste was formed by blending melamine powder (2.0 g) in ethylene glycol (1.4 mL) with a graphene-oxide aqueous suspension (1.0 mL of a 0.8 wt% GO suspension prepared by concentrating a commercial solution of GO at 0.4 wt%). The formed paste was doctor-bladed (with 2 scotch tape layers) onto FTO, subsequently dried at 90 °C on a hot plate, and finally transferred into a closed (by aluminum foil) 25 mm-diameter glass test tube. (Note: FTO substrates were cleaned by sonication in an aqueous 1% w/v Alconox solution, ethanol, and acetone, and finally dried before usage).4.6 Synthesis of CN
In a 25 mm-diameter glass test tube, 0.2 g of melamine powder were deposited at the bottom of the tube, after which two electrodes (electrode size (length × width): 1.7 cm × 2 cm) were placed in its middle. The test tube was then purged very carefully with N2 (g) for 5 min and then covered with aluminum foil. Subsequently, the sample was heated up to 550 °C at a ramp rate of 5 °C min−1 and kept at 550 °C for 4 h under a constant N2 flow before it was naturally cooled down to room temperature.4.7 Photoelectrochemical (PEC) measurements
All electrochemical measurements were performed using a three-electrode system and an Autolab potentiostat (Metrohm, PGSTAT302N). Pt foil (0.75 cm2) and Ag/AgCl (saturated KCl) were used as the counter and reference electrodes, respectively. The electrolyte was a 1 M NaOH aqueous solution (pH = 13.9). All the potentials were converted to reversible hydrogen electrode (RHE) values using eqn (3).| VRHE = VAg/AgCl + 0.059 × pH + 0.197 (V) | (3) |
Photocurrents were measured at 1.22 V vs. RHE under the illumination of a solar simulator (Newport 300 W Xe arc lamp, equipped with an AM 1.5G and water filters) at a power density of 100 mW cm−2, which was calibrated using a thermopile power meter (Model 919-P, Newport), that is, 1-sun conditions. To report photocurrents, a standard deviation (σ) from three independent experiments (electrode preparation in separate batches and different PEC measurements with new electrolytes) was calculated, and the values are presented as the average photocurrent ± σ. Nyquist plots of the samples were measured in the frequency range from 100 kHz to 10 mHz at an applied voltage of 1.22 V vs. RHE. Mott–Schottky measurements were performed in 1.0 M Na2SO4 aqueous solution (pH = 7) using the same potentiostat at 2000 Hz. Here, the voltage difference between the conduction band (CB) and the flat band potential is set to be 0.2 eV.
4.8 PEC oxidation of aryl alcohols
Bulk photoelectrolysis experiments were performed in a 30 mL undivided PEC cell. All the reactions were performed in aqueous solutions. A carbon nitride (CN) electrode (1.7 cm × 2 cm) was used as the working electrode and Pt foil (0.75 cm2) and Ag/AgCl (saturated KCl) were used as the counter and reference electrodes, respectively. Photoelectrolysis reactions were performed at a constant potential mode of 1.22 V vs. RHE. A PTFE-coated magnetic stir bar (1 cm) was used to stir the reaction mixture at 500 rpm during the reactions. All reactions were performed with a substrate concentration (alcohols) of 10 mM in 18 mL of 1 M NaOH aqueous solution (0.18 mmol). Before each experiment, O2 gas (or any other appropriate gas for test reactions) was bubbled into the solution for 10 min at a flow rate of 15 mL min−1, then filled for 3 min assuming the concentration of O2 reached saturation in the headspace. The area under the chronoamperogram corresponds to the charge passed during the PEC oxidation reaction. The characterization of the products and the reaction yields are given in the product isolation and yield determination section.4.9 GC-MS and NMR
Typically, the reaction mixture was dispersed in CH2Cl2. GC data were obtained using an Agilent 6850 GC equipped with an Agilent 5973 MSD, working under standard conditions and an Agilent HP5-MS (30 × 0.25 × 0.25) column. The column oven temperature was ramped to 80 °C and held for 2 min, then raised to 300 °C and held for 8 min (heating rate: 30 °C min−1). Ultrapure helium (≥99.99%) was adopted as the carrier gas at a flow rate of 1.0 mL min−1. The temperatures of the injection port, MS transfer line, and ion source were set at 230 °C, 250 °C, and 300 °C, respectively. A sample volume of 1 μL with a split ratio of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was used. Selected ion monitoring mode (SIM) was adopted for the analysis. GC-MS spectra of the products are provided below (GC-MS data section). Similarly, the samples were extracted, concentrated in a rotary evaporator, and then dispersed in CDCl3 or DMSO-d6 for further NMR detection. The 1H and 13C NMR spectra were recorded using a Bruker DPX 400 instrument; the chemical shifts, given in ppm, are referenced to the residual solvent peak. Alcohol conversion and product selectivity were calculated according to eqn (4) and (5).
:1 was used. Selected ion monitoring mode (SIM) was adopted for the analysis. GC-MS spectra of the products are provided below (GC-MS data section). Similarly, the samples were extracted, concentrated in a rotary evaporator, and then dispersed in CDCl3 or DMSO-d6 for further NMR detection. The 1H and 13C NMR spectra were recorded using a Bruker DPX 400 instrument; the chemical shifts, given in ppm, are referenced to the residual solvent peak. Alcohol conversion and product selectivity were calculated according to eqn (4) and (5).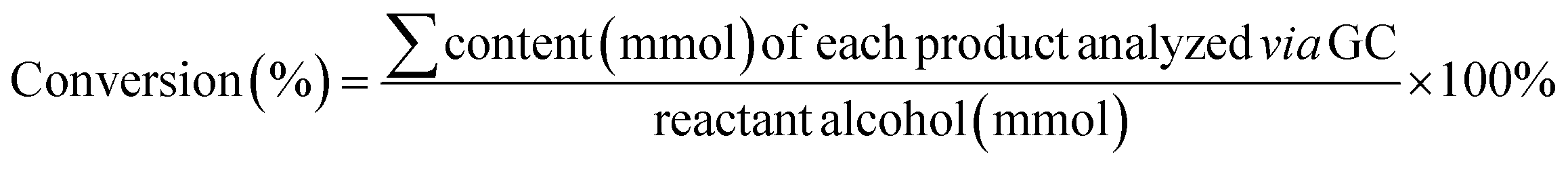 | (4) |
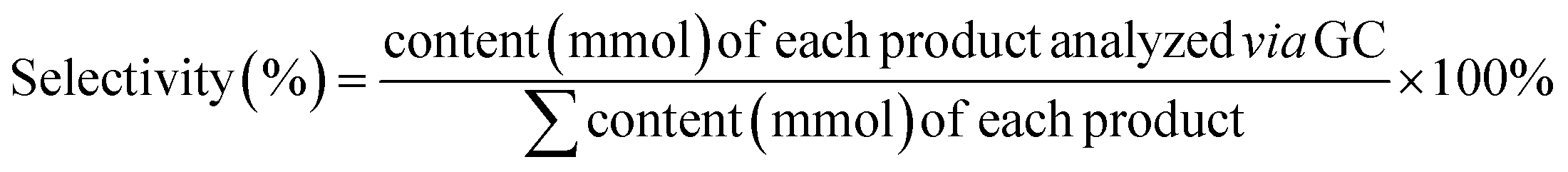 | (5) |
4.10 Hydrogen (H2) measurements
The amount of photogenerated H2 in the reactor headspace was analyzed using an Agilent 7820 GC system equipped with a thermal conductivity detector (TCD). Samples of gases were intermittently withdrawn every 2 h with an A-2 Luer lock gas syringe series purchased from VICI® precision sampling (Pressure-lok® precision analytical syringe). Two-compartment cells were thoroughly sealed with rubber septa and parafilm to prevent any gas leakage. Before the experiment, O2 gas was bubbled into the anode solution for 30 min; the solution was then filled with O2 for 3 min, assuming the O2 concentration reached saturation in the headspace. Meanwhile, Ar (g) was purged into the cathode solution for 30 min to remove any dissolved O2. The electrode was held at 1.22 V (vs. RHE) under illumination. Faradaic efficiency was calculated using eqn (6).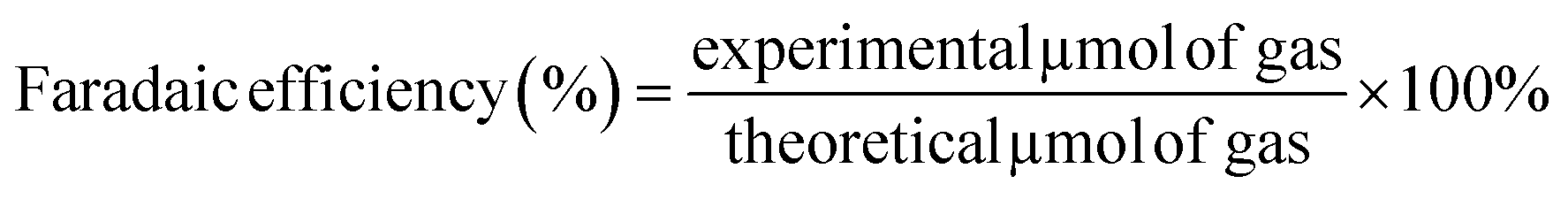 | (6) |
The theoretical amount of gas was calculated from Faraday's law, eqn (7).
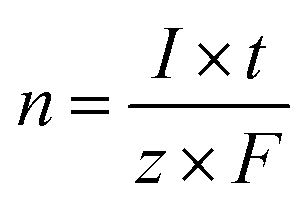 | (7) |
485 C mol−1).
Author contributions
N. K. performed most of the experiments, analyzed the data, and wrote the initial draft of the manuscript. R. P. measured and analyzed the GC-MS and NMR data. S. B. performed the DFT calculations. B. M. assisted with the PEC measurements. A. A. and M. V. performed material characterization. A. S. measured and analyzed EPR data. M. V. took part in analysis and writing. J. A. and H. G. performed TAS measurements. M. S. supervised the study and co-wrote the paper. All the authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No. [849068]). This work was also partially supported by the joint Israel Science Foundation–National Science Foundation of China (ISF–NSFC), grant No. 2969/19, the ISF grant No.601/21, and by the Pazy Foundation (grant No. 119-2020). J.A. and H.G. gratefully acknowledge financial support by the Spanish Ministry of Science and Innovation (RTI2018-98237-CO2-1) and Generalitat Valenciana (Prometeo 2021/038). The authors thank Liel Abisdris, Jonathan Tzadikov, Ayelet Tashakory, and Rotem Geva for help with material characterization. We thank Dr Chabanne for fruitful discussion.References
- J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed.
- L. Luo, Z. j. Wang, X. Xiang, D. Yan and J. Ye, ACS Catal., 2020, 10, 4906–4913 CrossRef CAS.
- P. Lianos, Appl. Catal., B, 2017, 210, 235–254 CrossRef CAS.
- T. Li, T. Kasahara, J. He, K. E. Dettelbach, G. M. Sammis and C. P. Berlinguette, Nat. Commun., 2017, 8, 390 CrossRef PubMed.
- T. Li, J. Y. Mo, D. M. Weekes, K. E. Dettelbach, R. P. Jansonius, G. M. Sammis and C. P. Berlinguette, ChemSusChem, 2020, 13, 3622–3626 CrossRef CAS PubMed.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- Z. Guo, B. Liu, Q. Zhang, W. Deng, Y. Wang and Y. Yang, Chem. Soc. Rev., 2014, 43, 3480–3524 RSC.
- M. Zhao, J. Li, Z. Song, R. Desmond, D. M. Tschaen, E. J. Grabowski and P. J. Reider, Tetrahedron Lett., 1998, 39, 5323–5326 CrossRef CAS.
- A. Mahmood, G. E. Robinson and L. Powell, Org. Process Res. Dev., 1999, 3, 363–364 CrossRef CAS.
- L. Capaldo, L. L. Quadri and D. Ravelli, Angew. Chem., Int. Ed., 2019, 58, 17508–17510 CrossRef CAS PubMed.
- N. E. S. Tay, D. Lehnherr and T. Rovis, Chem. Rev., 2021, 122, 2487–2649 CrossRef PubMed.
- Z. Zhou, Y. N. Xie, W. Zhu, H. Zhao, N. Yang and G. Zhao, Appl. Catal., B, 2021, 286, 119868 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- H. Tateno, Y. Miseki and K. Sayama, ChemElectroChem, 2017, 4, 3283–3287 CrossRef CAS.
- H. Tateno, S. Iguchi, Y. Miseki and K. Sayama, Angew. Chem., Int. Ed., 2018, 57, 11238–11241 CrossRef CAS PubMed.
- L. Zhang, L. Liardet, J. Luo, D. Ren, M. Grätzel and X. Hu, Nat. Catal., 2019, 2, 366–373 CrossRef CAS PubMed.
- T. Hardwick, A. Qurashi, B. Shirinfar and N. Ahmed, ChemSusChem, 2020, 13, 1967–1973 CrossRef CAS PubMed.
- Y. C. Wu, R. J. Song and J. H. Li, Org. Chem. Front., 2020, 7, 1895–1902 RSC.
- C. Ding, J. Shi, Z. Wang and C. Li, ACS Catal., 2017, 7, 675–688 CrossRef CAS.
- D. J. Chadderdon, L. P. Wu, Z. A. McGraw, M. Panthani and W. Li, ChemElectroChem, 2019, 6, 3387–3392 CrossRef CAS.
- S. Zhong, Y. Xi, S. Wu, Q. Liu, L. Zhao and S. Bai, J. Mater. Chem. A, 2020, 8, 14863–14894 RSC.
- M. Volokh, G. Peng, J. Barrio and M. Shalom, Angew. Chem., Int. Ed., 2019, 58, 6138–6151 CrossRef CAS PubMed.
- F. K. Kessler, Y. Zheng, D. Schwarz, C. Merschjann, W. Schnick, X. Wang and M. J. Bojdys, Nat. Rev. Mater., 2017, 2, 1703 Search PubMed.
- N. Karjule, C. Singh, J. Barrio, J. Tzadikov, I. Liberman, M. Volokh, E. Palomares, I. Hod and M. Shalom, Adv. Funct. Mater., 2021, 31, 2101724 CrossRef CAS.
- W. Xiong, F. Huang and R. Q. Zhang, Sustainable Energy Fuels, 2020, 4, 485–503 RSC.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- R. Tang, S. Zhou, Z. Zhang, R. Zheng and J. Huang, Adv. Mater., 2021, 33, 2005389 CrossRef CAS PubMed.
- N. Karjule, J. Barrio, L. Xing, M. Volokh and M. Shalom, Nano Lett., 2020, 20, 4618–4624 CrossRef CAS PubMed.
- C. R. Lhermitte, N. Plainpan, P. Canjura, F. Boudoire and K. Sivula, RSC Adv., 2021, 11, 198–202 RSC.
- J. Xia, N. Karjule, L. Abisdris, M. Volokh and M. Shalom, Chem. Mater., 2020, 32, 5845–5853 CrossRef CAS.
- H. Li, F. Qin, Z. Yang, X. Cui, J. Wang and L. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- R. Zhang, M. Shao, Z. Li, F. Ning, M. Wei, D. G. Evans and X. Duan, Chem. - Eur. J., 2017, 23, 8142–8147 CrossRef CAS PubMed.
- P. Wu, J. Wang, J. Zhao, L. Guo and F. E. Osterloh, Chem. Commun., 2014, 50, 15521–15524 RSC.
- M. Hayyan, M. A. Hashim and I. M. AlNashef, Chem. Rev., 2016, 116, 3029–3085 CrossRef CAS PubMed.
- D. R. Paul, R. Sharma, S. P. Nehra and A. Sharma, RSC Adv., 2019, 9, 15381–15391 RSC.
- C. Xu, Y. Pan, G. Wan, H. Liu, L. Wang, H. Zhou, S. H. Yu and H. L. Jiang, J. Am. Chem. Soc., 2019, 141, 19110–19117 CrossRef CAS PubMed.
- J. Dong, X. Wang, Z. Wang, H. Song, Y. Liu and Q. Wang, Chem. Sci., 2020, 11, 1026–1031 RSC.
- X. Xiao, J. Jiang and L. Zhang, Appl. Catal., B, 2013, 142–143, 487–493 CrossRef CAS.
- S. Yang, Q. Wang, Q. Wang, G. Li, T. Zhao, P. Chen, F. Liu and S. F. Yin, J. Mater. Chem. A, 2021, 9, 21732–21740 RSC.
- M. Zhou, P. Yang, S. Wang, Z. Luo, C. Huang and X. Wang, ChemSusChem, 2018, 11, 3949–3955 CrossRef CAS PubMed.
- C. Xiao, L. Zhang, H. Hao and W. Wang, ACS Sustainable Chem. Eng., 2019, 7, 7268–7276 CrossRef CAS.
- F. Ichihara, F. Sieland, H. Pang, D. Philo, A. T. Duong, K. Chang, T. Kako, D. W. Bahnemann and J. Ye, J. Phys. Chem. C, 2020, 124, 1292–1302 CrossRef CAS.
- C. Ye, J. X. Li, Z. J. Li, X. B. Li, X. B. Fan, L. P. Zhang, B. Chen, C. H. Tung and L. Z. Wu, ACS Catal., 2015, 5, 6973–6979 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- D. Wang, P. Wang, S. Wang, Y. H. Chen, H. Zhang and A. Lei, Nat. Commun., 2019, 10, 2796 CrossRef PubMed.
- C. Huang, Y. Wen, J. Ma, D. Dong, Y. Shen, S. Liu, H. Ma and Y. Zhang, Nat. Commun., 2021, 12, 320 CrossRef CAS PubMed.
- B. Zhang, T. J. Zhao and H. H. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 34922–34929 CrossRef CAS PubMed.
- V. W. h. Lau and B. V. Lotsch, Adv. Energy Mater., 2022, 12, 2101078 CrossRef CAS.
- X. Zou, Z. Sun and Y. H. Hu, J. Mater. Chem. A, 2020, 8, 21474–21502 RSC.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- M. Du, G. Zeng, J. Huang, D. Sun, Q. Li, G. Wang and X. Li, ACS Sustainable Chem. Eng., 2019, 7, 9717–9726 CrossRef CAS.
- A. Jena, C. J. Chen, H. Chang, S. F. Hu and R. S. Liu, J. Mater. Chem. A, 2021, 9, 3767–3785 RSC.
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188 CrossRef.
- J. P. Desclaux, Comput. Phys. Commun., 1970, 1, 216–222 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: supplementary methods, characterization and additional GC-MS and NMR spectra. See https://doi.org/10.1039/d2ta03660f |
| This journal is © The Royal Society of Chemistry 2022 |