
Impact of dynamic covalent chemistry and precise linker length on crystallization kinetics and morphology in ethylene vitrimers†
Bhaskar
Soman
ab,
Yoo Kyung
Go
ab,
Chengtian
Shen
bc,
Cecilia
Leal
 ab and
Christopher M.
Evans
*ab
ab and
Christopher M.
Evans
*ab
aDepartment of Materials Science and Engineering, Urbana, Illinois 61801, USA. E-mail: cme365@illinois.edu
bFrederick Seitz Materials Research Laboratory, Urbana, Illinois 61801, USA
cDepartment of Chemistry University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 16th December 2021
Abstract
Vitrimers, dynamic polymer networks with topology conserving exchange reactions, have emerged as a promising platform for sustainable and reprocessable materials. While prior work has documented how dynamic bonds impact stress relaxation and viscosity, their role on crystallization has not been systematically explored. Precise ethylene vitrimers with 8, 10, or 12 methylene units between boronic ester junctions were investigated to understand the impact of bond exchange on crystallization kinetics and morphology. Compared to linear polyethylene which has been heavily investigated for decades, a long induction period for crystallization is seen in the vitrimers ultimately taking weeks in the densest networks. An increase in melting temperatures (Tm) of 25–30 K is observed with isothermal crystallization over 30 days. Both C10 and C12 networks initially form hexagonal crystals, while the C10 network transforms to orthorhombic over the 30 day window as observed with wide angle X-ray scattering (WAXS) and optical microscopy (OM). After 150 days of isothermal crystallization, the three linker lengths led to double diamond (C8), orthorhombic (C10), and hexagonal (C12) crystals indicating the importance of precision on final morphology. Control experiments on a precise, permanent network implicate dynamic bonds as the cause of long-time rearrangements of the crystals, which is critical to understand for applications of semi-crystalline vitrimers. The dynamic bonds also allow the networks to dissolve in water and alcohol-based solvents to monomers, followed by repolymerization while preserving the mechanical properties and melting temperatures.
Introduction
The development of sustainable and recyclable plastics is a major challenge to handle the massive volume of products that end up in landfills.1 Polymer networks, either elastomers or thermosets, are not processesable with traditional chemistries but can be made recyclable via the incorporation of dynamic covalent bonds into the polymer. Notably, Leibler and coworkers described dynamic polyester networks which exhibit glass-like processability due to conserved ester exchange reactions, now commonly called vitrimers.2–5 This concept has been widely applied to a range of dynamic bonds2,6–19 including boronic esters and boroxine.20–29 This ever growing toolbox of dynamic covalent bonds which conserve the network topology is described in recent review articles.30,31 A major focus thus far has been to understand how the exchange kinetics, crosslink density, and polymer backbone chemistry control the stress relaxation and reprocessability of vitrimers. The glass transition temperature (Tg) and a hypothetical topology freezing temperature (Tv) have been discussed to understand the interplay with bond exchange processes which governs the temperature dependent viscoelasticity. In contrast, an understanding of crystallization phenomena and melting temperatures (Tm) in vitrimers is currently lacking. Dynamic bonds provide a mechanism for chain rearrangements within a polymer network, as well as a new timescale which can potentially facilitate crystal perfection and growth. Knowledge of how dynamic bonds impact crystallization kinetics and morphology will be critical to the development of new polymers which are easier to recycle and reprocess while still retaining desirable physical and optical properties.The crystallinity of commodity polymers such as polyethylene (PE) and polypropylene is key to their electrical breakdown strength,32 mechanical properties,33–35 and thermal conductivity.36 Electron37,38 and ion39–41 conducting functional polymers also show performance which is critically related to the crystal structure and amorphous fraction. In all cases, processing conditions can lead to vastly different material properties depending on the timescales of material relaxation and rate of deformation during pressing or extrusion. With the introduction of dynamic bonds in a network, an additional timescale is generated which may facilitate crystal formation by allowing smaller strands to rearrange rather than an entire polymer chain. An understanding of how bond exchange affects kinetics and final morphology is currently lacking. While some prior work has focused on semi-crystalline vitrimers based on polyethylene,18,23–25,27,42,43 and poly(lactic acid),44 they did not investigate any temporal evolution of crystallinity or morphology due to dynamic bond rearrangements within the matrix. Little is known about the role of bond exchange on crystallization in dynamic networks.
Another key factor in determining crystal structure and melting is the presence of precise motifs. Here the term precise refers to the exact number of carbons in the linker following a definition used in prior work on “precise” polymers.45,46 For example, the melting temperatures of telechelic alkanes show a pronounced odd–even effect depending on the number of carbons between functional groups.47–49 In polymers, precise polyethylenes50 and polyacetals51 have been made as linear polymers, including materials with periodic ionic groups which crystallize and show enhanced conductivity.52,53 Precise permanent networks with alkane chain linkers have shown odd–even effects on the glass transition temperature and ionic conductivity.54 All of these studies point to the potentially critical role of precision on crystallization, which has not been investigated in vitrimers. In addition to being important from an application point of view, understanding the fundamental roles of precision and dynamic bond exchange on semi-crystalline polymers could provide new insights into the design and application of such materials.
Here, we report the synthesis and systematic investigation of ethylene vitrimers with precise carbon spacing between dynamic boronic ester crosslinks. Boronic esters are a popular dynamic bond because of the relative ease by which they can be incorporated into conventional polymers. In addition, boronic esters tend to be a fast exchanging bond, even at ambient temperatures and without catalyst.29 This allows one to observe the effects of dynamic bond exchange on crystallization at room temperature. This model vitrimer was chosen because linear PE has been widely studied and the crystal structure evolution is well known.55,56 The initial crystallization of vitrimers is inhibited due to the network architecture, and longtime crystallization studies indicate that Tm grows continually over 30 days. An unexpected crystal-crystal phase transition is also observed via X-ray scattering, and is further tracked via polarized optical microscopy. This morphological transition, as well as the long time evolution of Tm, is not present in linear PE and a precise, permanent ethylene network shows no crystallization even after one week, implicating dynamic bonds as the origin of this phenomena. This is an extension of our previous work on the rheological investigation of ethylene vitrimers in the amorphous state.29
Results and discussion
Precise ethylene vitrimers were synthesized by the step growth polymerization of telechelic alkane diols and boric acid following our prior work.29 Networks with exactly 8, 10 or 12 carbons (C8, C10, C12) between boronic ester crosslinking junctions were prepared by mixing the diol and acid, followed by heating to melt the diol and then application of vacuum to drive off water condensate (Fig. 1a). The term precise only refers to the carbon spacing, not the network topology, following prior work.29,57 Fourier transform infrared spectroscopy (FTIR) of the networks was performed at 80 °C to confirm the absence of the broad OH peak in the 3000 cm−1 to 3500 cm−1 range corresponding to unreacted species on the diols, boric acid, and water. Within the sensitivity of the instrument, no OH peak is detected indicating that the network formation reached high conversion for all samples. While the samples can take up 4–5 wt% of water under ambient conditions, they still form self-standing networks. Nevertheless, all crystallization was done in a glove box to avoid issues of moisture on kinetics. We do not claim 100% conversion of the network because the FTIR instrument would not be able to detect ∼0.1% impurities within the sample. Additionally, the purity of the monomers varies from 98% to 99% which is accounted for in our stoichiometric calculations. Thus free OH groups are thought to be present below the detection limit which enable exchange reactions in the network.29 It is worth noting that boronic esters can also exchange via methathesis reactions27 without free alcohol groups which can also lead to bond exchange. The emergence of a sharp peak at 1300 cm−1 corresponds to the asymmetric B–O stretch58 indicating the formation of the boronic ester crosslinks. Differential scanning calorimetry (DSC) was performed using a rapid quench from 150 to −80 °C, and only a glass transition is observed in C8 and C10 networks on the cooling curve while C12 shows a weak crystallization peak. In contrast, a linear PE standard crystallizes instantly (Fig. S1b and c, ESI†).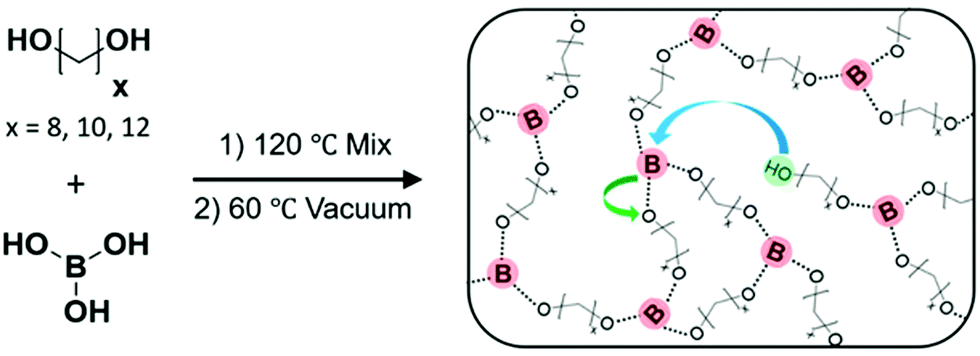 | ||
| Fig. 1 Step growth polymerization of telechelic alkane diols and boric acid is carried out to make ethylene vitrimers. By using diol with an exact number of carbon atoms, networks with precise spacing between crosslinks were synthesized. Schematic has been reproduced from ref. 29 with permission. | ||
The networks are termed vitrimers due to the well-known conserved exchange reaction of boronic esters in small molecules59 and polymers.20–28 In our prior work, oscillatory shear rheology was used to probe the storage (G′) and loss (G′′) modulus of the ethylene networks in their fully amorphous state to determine the characteristic timescale for flow. Frequency sweeps were performed in 10 °C intervals from 140–40 °C with networks showing rubbery behavior at higher frequencies and terminal relaxation at lower frequencies (Fig. S1d, ESI†) due to dynamic bond exchange.29 As expected, the modulus tracked with crosslink density/linker length. For a constant crosslink density, the modulus should increase with temperature due to entropic elasticity,60 which is observed in the ethylene networks supporting the presence of topology conserving exchange reactions. We have previously observed that denser networks have longer relaxation times and slightly higher activation energies which is a function of both bond exchange kinetics and also the ability of dynamic bonds to find each other. A detailed discussion on the trends of relaxation times can be found in our previous report on amorphous ethylene dynamic networks.29 The viscosity was calculated from the slope of G′′ vs. ω in the low frequency limit and plotted against inverse temperature, which showed the anticipated Arrhenius behavior. This method of zero-shear viscosity determination has been compared to the complex viscosity and gives the same values and temperature dependent trends.29 All of these measurements are consistent with a network held together by associative dynamic covalent bonds.
The glass transition temperatures (Tg) of the networks increases monotonically from −43 °C (C8) to −41 °C (C10) to −32 °C (C12), meaning there is mobility to allow the networks to crystallize at room temperature. Crystallization is observed in C12 networks on the cooling curve (Fig. S1b, ESI†), indicating that crystals will be present on first heating. Conversely, the C8 network shows no initial crystallinity while the C10 and C12 networks have a small melting transition at 25 and 35 °C. A separate batch of samples was prepared and quenched directly to room temperature from 150 °C. In the C8 network, no melting was observed on the initial heating ramp (Fig. S2, ESI†) while C10 and C12 gradually develop a melting peak at room temperature over 300 min. After 1 day of isothermal crystallization at room temperature, both C10 and C12 networks show a clear melting transition with larger enthalpy and shifted to higher temperature relative to the samples directly quenched to −80 °C, whereas the C8 vitrimer remains amorphous (Fig. 2). We attribute the latter result to the fact that this vitrimer has the highest crosslink density, which inhibits the ability of linkers to pack, as well as the highest viscosity which reduces translational mobility of network strands.
 | ||
| Fig. 2 After 1 day of isothermal crystallization at room temperature, C10 and C12 show clear melting peaks whereas C8 still exhibits no melting transition. | ||
The melting transitions of C10 and C12 vitrimers were monitored with DSC as a function of isothermal crystallization time at room temperature over 30 days, and a monotonic increase in the peak melting temperature (Tm) of the networks was observed (Fig. 3a and b). The C10 network shows a melting peak splitting around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 080 min, which eventually coalesces after 43200 min. In contrast, C12 vitrimers only show one peak for the entire series. Optical microscopy, to be discussed in detail subsequently, shows the growth of dendritic structures which appear only in the C10 networks and are assigned as the cause of peak splitting. A sigmoidal curve is typically seen for processes governed by a nucleation and growth mechanism,61,62 which is observed in the C12 networks when plotting the peak melting temperatures over 43200 min (Fig. 3c). The C10 network has not yet plateaued even after ∼104 min. In both cases, there is an initial induction period where Tm is flat, followed by an upturn at 100 and 1000 min for the C10 and C12 networks, respectively. The slow crystallization kinetics in C10 as compared to C12 can be rationalized because Tm is lower for C10 networks, thus they have a smaller thermodynamic driving force for crystallization. Additionally, a higher crosslink density in C10 vitrimers means that the system is more frustrated which would also impede crystallization and Tm evolution.
080 min, which eventually coalesces after 43200 min. In contrast, C12 vitrimers only show one peak for the entire series. Optical microscopy, to be discussed in detail subsequently, shows the growth of dendritic structures which appear only in the C10 networks and are assigned as the cause of peak splitting. A sigmoidal curve is typically seen for processes governed by a nucleation and growth mechanism,61,62 which is observed in the C12 networks when plotting the peak melting temperatures over 43200 min (Fig. 3c). The C10 network has not yet plateaued even after ∼104 min. In both cases, there is an initial induction period where Tm is flat, followed by an upturn at 100 and 1000 min for the C10 and C12 networks, respectively. The slow crystallization kinetics in C10 as compared to C12 can be rationalized because Tm is lower for C10 networks, thus they have a smaller thermodynamic driving force for crystallization. Additionally, a higher crosslink density in C10 vitrimers means that the system is more frustrated which would also impede crystallization and Tm evolution.
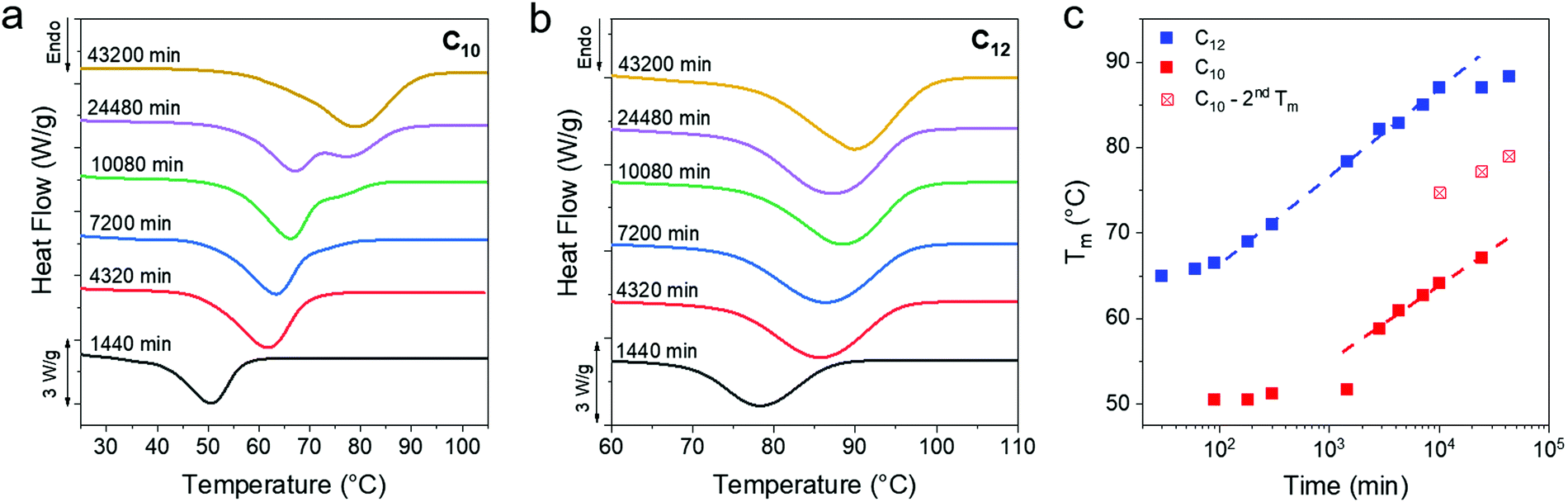 | ||
| Fig. 3 (a and b) DSC of C10 and C12 after long time crystallization at room temperature. A monotonic increase in the Tm is evident from the rightward shift of the melting curve. DSC data for less than 1 day of crystallization time can be found in Fig. S2 (ESI†). (c) A plot of Tmvs. time shows similar slopes for the two networks. The 7, 17 and 30 day samples of C10 show two melting peaks, the lower one shown as solid squares and the higher peak as crossed squares. | ||
Over the observed period of 43 200 min a ΔTm of 25 °C for C12 and 30 °C for C10 is observed. An increase in the enthalpy of melting (and thus crystallinity) accompanies the Tm evolution, and nearly doubles for the C12 network (Fig. S3a, ESI†). The C10 network shows an ∼70% increase in enthalpy over the same time period. Only the relative change in crystallinity is reported and not the absolute crystallinity, as there is no appropriate reference enthalpy. Polyethylene consists of only methylene repeat units, while the present networks include a periodic placement of boronic ester crosslinks which will affect the enthalpy of fusion. Ethylene vitrimer crystals undergo a polymorphic transition, discussed later, which makes an absolute degree of crystallization determination difficult. After 150 days, the Tm reaches 82 °C and 91 °C for the C10 and C12 networks, respectively (Fig. S3b, ESI†). There are no free OH groups within the resolution of the instrument after crystallization (Fig. S3d, ESI†). Thus, we conclude that the boronic esters are incorporated into the vitrimers crystals.
At intermediate times the Tm shows a power law dependence with crystallization time. Generally, a power law slope is attributed to the logarithmic growth of the lamellar thickness which in turn causes a logarithmic growth in Tm.63 The apparent power law dependence was fit to the equation Tm(tA) = T0Dlog(tA/t0) used in prior work where tA is annealing time and t0 is the initial time.64 The D values were 10.3 and 8.8 for C12 and C10 networks using only data beyond 180 and 1440 min, respectively. These values are similar and attributed to the fact that the same dynamic bond exchange is controlling the growth of the crystal. Future work will incorporate dynamic bonds with much faster exchange reactions to understand how D and the time evolution of Tm are affected.
More analysis is required for a detailed molecular explanation, but we presently assign the slow evolution and emergence of crystals to dynamic bond facilitated reorganization of strands in the amorphous region into the crystalline phase. Different dynamic bonds with variable exchange kinetics are expected to yield different slopes for Tm evolution. Two key control experiments were performed, the first on linear PE (SRM 1475, 52000 g mol−1) which shows instantaneous crystallization and no substantial long-term Tm evolution (Fig. S1c, ESI†). This is expected as PE exhibits fast crystallization kinetics, and Tm evolution has only been observed using flash differential scanning calorimetry with time resolution as fast as 10−4 s.65 The second control involved the synthesis of a precise, permanent ethylene network with C10 linkers as described in the ESI† (Fig. S4). In the absence of dynamic bonds, this network does not crystallize even after a week while the dynamic analogue shows crystallization within the first day (Fig. S5a, ESI†). A linear C10 polymer with the same amide linkages was also synthesized which crystallizes immediately upon cooling and indicates that the network topology, not the specific junction chemistry, are preventing crystallization (Fig. S5b, ESI†). Thus, the presence of dynamic bonds is critical to the melting temperature evolution in semi-crystalline vitrimers.
The long time evolution of Tm is atypical for polymer crystals. Increases in properties like Tm, density and lamellar thickness have been previously reported for polyethylene66,67 and polyethylene terephthalate64,68–71 but only with annealing or crystallization at elevated pressure. Slow reordering would require molecular motions of the chains in the amorphous regions which can reorganize to add to the crystalline interface. Such a reorganization in local structure would be possible only if mobility is available for strands to translate. Is it noteworthy that previous reports on increasing Tm emphasize the role of an external stimuli, while the present networks sit quiescently at room temperature. In addition, these reports are on linear polymers and little is known about the role of network architectures on crystallization, particularly at such high crosslink densities.72
As mentioned, along with the upward shift in Tm the melting curves for C10 show a transition from a single peak to a double peak and then revert to a single peak (Fig. 3a). The second peak is visible first as a small shoulder in the 5 day measurement and progressively increases in intensity in the 10080 min and 24480 min measurements at the expense of the first peak. This single to double peak transition is not seen in C12. To probe the nature of this phenomena, wide-angle X-ray scattering (WAXS) was performed on the networks. Patterns collected over 1440 min (1 day) and 150 days of isothermal crystallization time at room temperature in an Argon glove box are shown in Fig. 4. Scattering patterns were also collected more frequently on a separate benchtop XRD at room temperature for samples crystallized under ambient conditions (Fig. S6, ESI†) and show the same general phenomena to be discussed. The presence of atmospheric moisture slightly modifies the kinetics of crystal evolution, while the patterns in Fig. 4a–c correspond to samples crystallized in a glovebox. The final crystal structures are not affected by ambient water. Prior work on PE has indicated that the semi-crystalline state at room temperature has three types of unit cells: orthorhombic, monoclinic and hexagonal.73–75 The orthorhombic PE crystal phase is the densest and most commonly observed phase in PE, while the monoclinic PE crystal structure may coexist with orthorhombic.50,53,57 The hexagonal phase is typically seen under high pressure crystallization.76,77 The X-ray scattering patterns at 1 day show a single Bragg peak at 15 nm−1 for both C10 and C12 arising from the diffraction of planes (20), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1), and (11) of a PE hexagonal unit cell (lattice parameters: a = 4.7 Å, b = 8.2 Å; P6m space group). With increasing isothermal crystallization at room temperature, a transition to a two peak pattern is observed for C10 (Fig. 4a). The two Bragg peaks at 14.8 nm−1 and 16.4 nm−1 can be respectively assigned to the diffraction of planes (110) and (200) of a PE orthorhombic unit cell (lattice parameters: a = 7.1 Å, b = 4.9 Å, c = 2.5 Å; in a Pnam space group).78 The C12 network only shows one peak after 150 days in the glovebox but annealing at 65 °C for 2 days can drive a transition to the orthorhombic phase (Fig. 4b). In both cases, the networks evolve from a metastable phase given sufficient time and temperature to orthorhombic with a coexistence of monoclinic. In prior work on ionic telechelic ethylenes, a hexagonal to orthorhombic transition was observed on cooling and the monoclinic phase coexisted with orthorhombic phase, which may explain the shoulder observed in the scattering result of the 150 days and annealed samples. In contrast, a linear PE standard immediately forms an orthorhombic crystal structure (Fig. S7, ESI†). In the low q range the lamellar (100) and (200) reflections are observed for C10 and C12 indicating formation of lamellar stacks. The X-ray scattering patterns were also collected for C8, which shows a single dominant peak even after 150 days of isothermal crystallization (Fig. 4c). Analysis of the scattering data indicates a tetrahedral arrangement of short rods, or the C8 alkyl chain, interconnected in a cubic crystal lattice with a lattice parameter of a = 1.55 nm and symmetry of a Pn3m space group. A double diamond cubic crystal structure has not been previously reported in polyethylene or in polymer networks. This network showed only a faint melting peak by DSC after 30 days (Fig. S3c, ESI†) due to the extremely slow kinetics. The crystal structures and proposed packing of the linkers is illustrated in Fig. 4d. The peaks for all three networks shift to higher q with annealing indicating densification of the crystalline networks, and a key finding of this work is that the crystal structure depends strongly on the choice of linker length in precise, dynamic networks.
1), and (11) of a PE hexagonal unit cell (lattice parameters: a = 4.7 Å, b = 8.2 Å; P6m space group). With increasing isothermal crystallization at room temperature, a transition to a two peak pattern is observed for C10 (Fig. 4a). The two Bragg peaks at 14.8 nm−1 and 16.4 nm−1 can be respectively assigned to the diffraction of planes (110) and (200) of a PE orthorhombic unit cell (lattice parameters: a = 7.1 Å, b = 4.9 Å, c = 2.5 Å; in a Pnam space group).78 The C12 network only shows one peak after 150 days in the glovebox but annealing at 65 °C for 2 days can drive a transition to the orthorhombic phase (Fig. 4b). In both cases, the networks evolve from a metastable phase given sufficient time and temperature to orthorhombic with a coexistence of monoclinic. In prior work on ionic telechelic ethylenes, a hexagonal to orthorhombic transition was observed on cooling and the monoclinic phase coexisted with orthorhombic phase, which may explain the shoulder observed in the scattering result of the 150 days and annealed samples. In contrast, a linear PE standard immediately forms an orthorhombic crystal structure (Fig. S7, ESI†). In the low q range the lamellar (100) and (200) reflections are observed for C10 and C12 indicating formation of lamellar stacks. The X-ray scattering patterns were also collected for C8, which shows a single dominant peak even after 150 days of isothermal crystallization (Fig. 4c). Analysis of the scattering data indicates a tetrahedral arrangement of short rods, or the C8 alkyl chain, interconnected in a cubic crystal lattice with a lattice parameter of a = 1.55 nm and symmetry of a Pn3m space group. A double diamond cubic crystal structure has not been previously reported in polyethylene or in polymer networks. This network showed only a faint melting peak by DSC after 30 days (Fig. S3c, ESI†) due to the extremely slow kinetics. The crystal structures and proposed packing of the linkers is illustrated in Fig. 4d. The peaks for all three networks shift to higher q with annealing indicating densification of the crystalline networks, and a key finding of this work is that the crystal structure depends strongly on the choice of linker length in precise, dynamic networks.
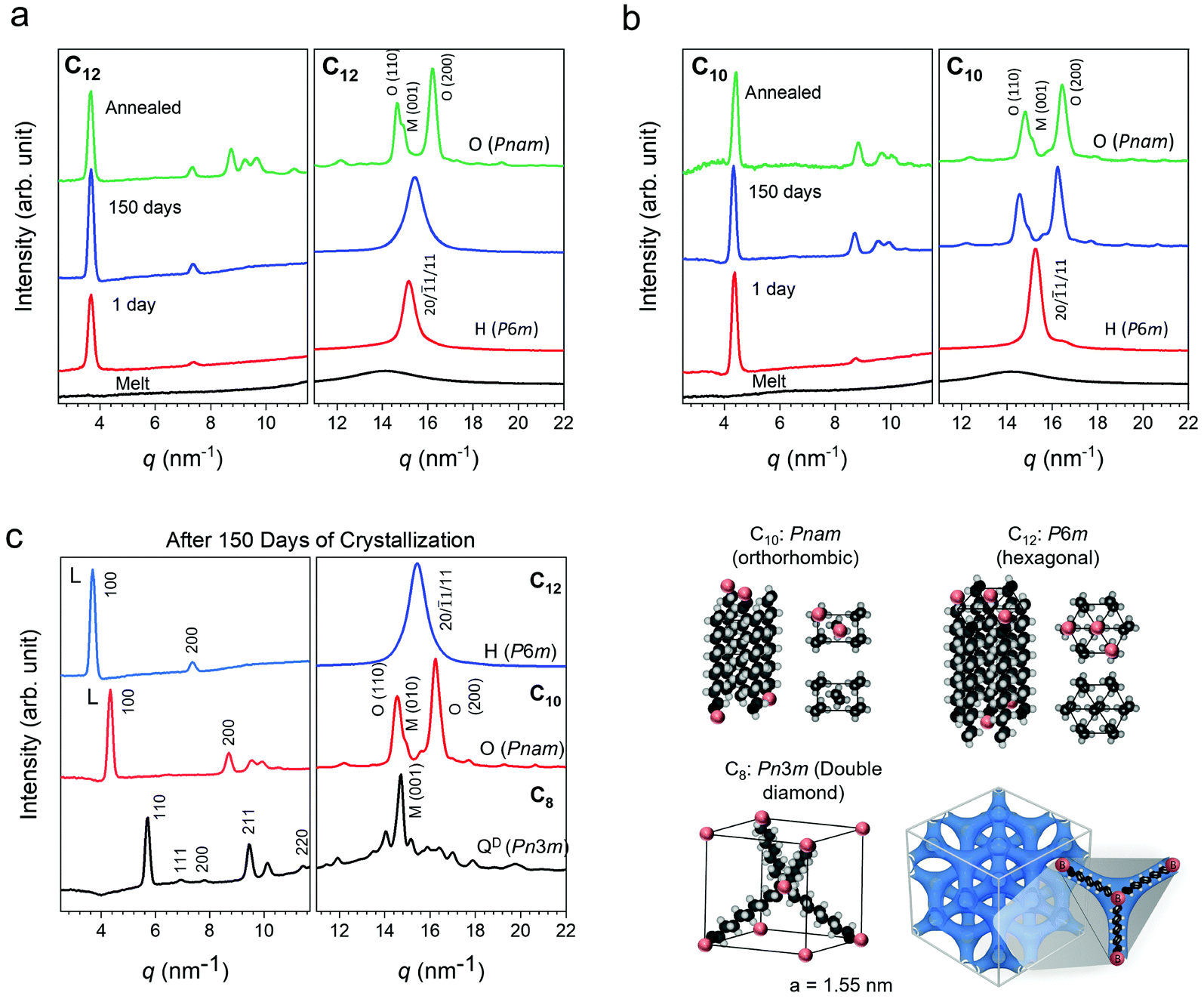 | ||
| Fig. 4 (a) and (b) Room temperature WAXS patterns of C10 and C12 networks crystallized in a glove box. Samples were sandwiched and sealed between Kapton to prevent exposure to moisture. After 150 days of crystallization, samples were annealed at 50 °C (C10) and 65 °C (C12) for 2 days for the “annealed” data set. (c) A comparison of the WAXS patterns after 150 days of crystallization. (d) Schematic representation of the hexagonal (P6m), orthorhombic (Pnam) and double diamond (Pn3m) crystal lattices adopted by the C12, C10, and C8 polymers, respectively. | ||
A comparison of the scattering patterns for 150 days-crystallized networks reveals a monotonic shift of the lower q peak with increasing linker length. This is attributed to the boron-boron correlation length along the backbone based on a comparison with the calculated d-spacing (d = 2π/q) which is slightly shorter than that of an all-trans configuration of the ethylene linkers (Table 1). The correlation length of the crystals was also calculated using the Scherer equation ε = 2π/Δq where Δq is the full width at half maximum of the peak corresponding to the spacer length. This length is ∼30 nm and decreases slightly as the linker length increases.
| Network | q (nm−1) | d (nm) | All-trans linker length (nm) | Correlation length, ε (nm) |
|---|---|---|---|---|
| C8 | 5.71 | 1.09 | 1.23 | 31.5 |
| C10 | 4.33 | 1.44 | 1.54 | 28.1 |
| C12 | 3.68 | 1.70 | 1.84 | 27.4 |
To confirm that the network is intact after months of crystallization, FTIR spectra were collected on samples crystallized and tested inside the glovebox (Fig. S3d, ESI†). No free OH groups are observed indicating an intact network. Additionally, the scattering patterns of the starting alkane diols are shown in Fig. S9b (ESI†), which are clearly distinct from the network patterns. Thus, the alkanes are not disengaging from the network and crystallizing separately over time.
An increase in Tm can occur for multiple reasons. First, the growth of crystallites raises the melting temperature due to the well-known Gibbs–Thomson effect and surface destabilization of a small crystal.79–82 Second, a decreasing number of defects improves the crystal perfection64 and decreases the free energy, leading to an increase in Tm. The long-term evolution of Tm in vitrimers is a consequence of the bond exchange reactions allowing for local rearrangements of network strands in the amorphous regions or at the crystal–amorphous interfaces which increase both the size and perfection of the crystals. To examine morphological evolution over time, cross polarization microscopy was used to observe changes in the crystal structure. The formation of uniformly distributed spherulites is observed in the C10 network during the initial 720 min of crystallization (Fig. 5a) which is typical of linear polymers. After 1440 min of crystallization under ambient conditions, a new crystal structure emerges (Fig. 5b). With time these crystals proliferate, while the growth of the spherulites is arrested (Fig. 5c and d). Eventually the new crystals dominate and the spherulites are no longer observable. We assign the initial crystals as a metastable hexagonal phase which transitions into the final orthorhombic crystal. The morphological development observed in optical microscopy is qualitatively consistent with the WAXS evolution further supporting a crystal–crystal transition in the absence of external stimuli and occurring over multiple days. Quantitatively relating these slow transformations to dynamic bond rearrangements is an important avenue for future work.
 | ||
| Fig. 5 Cross polarization microscopy of C10, crystallization at room temperature under ambient condition. We follow the evolution in crystal structure as a function of time (from left to right). (a) Initial formation of spherulites after 720 min of crystallization time. (b) A new crystal structure is seen alongside the spherulites at 1440 min. (c) and (d) At 2400 min and 3600 min, the new crystals grow and uniformly span the image while the size of the spherulites remains constant with time. | ||
In select regions of the initial micrographs for C10 networks, dendritic structures are observed alongside the spherulites (Fig. 6a and b). When annealed at 50 °C for 2880 min, the dendrites grow and eventually span the entire crystal domain (Fig. 6c and d). Comparing the morphological changes with the appearance of the second melting peak in C10, we assign the second melting peak in DSC to the percolated dendritic crystal once it grows large enough to contribute to the heat flow. Compared to C10, the crystal morphology of C12 networks is noticeably different by optical microscopy (Fig. S8, ESI†). Spherulites are not resolvable in C12 networks indicating their size is below the resolution of our microscope. Nevertheless, WAXS confirms the presence of the same initial hexagonal crystal. Dendrites are not observed in C12 at any time in agreement with the absence of a second melting peak in C12 network DSC curves.
 | ||
| Fig. 6 (a) and (b) Dendritic structures are seen in other locations of the same C10 sample along with the crystals shown in Fig. 5. (c) and (d) After annealing at 50 °C for 2 days the dendrites span the entire crystal domain and the spherulites are no longer visible. | ||
One open question for the present ethylene vitrimers is how high the Tm can be with annealing protocols. Higher temperature annealing of the C10 and C12 networks at 50 °C and 65 °C, respectively, was performed after the initial isothermal crystallization for 43200 min at room temperature. Annealing for 2 days resulted in an increase of Tm to 89 °C for C10 and 100 °C for C12 (Fig. 7a). Future work will investigate more complex annealing protocols for manipulating the final percent crystallinity and Tm. It is worth noting that the evolved Tm of the networks is above the Tm of their corresponding monomers and the WAXS patterns of the diols do not match those of the ethylene networks (Fig. S9, ESI†). This indicates that the alkane diols are not simply being expelled from the network and crystallizing as the small molecule.
 | ||
| Fig. 7 (a) DSC of the annealed samples. After annealing for 2 days an increase in Tm to 89 °C and 100 °C is observed for C10 and C12 respectively. (b) Networks were dissolved in ethanol and recovered by solvent evaporation. (c) DSC of as synthesized vs. recycled C10 and C12. The Tg and Tm of the recovered networks match the virgin networks. (d) Comparison of the rheology of recycled C12 and as synthesized C12 network indicates that there is minimal degradation in material properties. | ||
An attractive property of vitrimers is their recyclability and the ability to recover monomer in some cases.20,83–87 The present ethylene vitrimers are crosslinked by boronic esters, and water or alcohol based solvent can attack the boron and dissolve the network. As a proof of concept, ethanol was used to dissolve both a C10 and C12 network, followed by removal of solvent under heat and vacuum (Fig. 7b). The recovered networks were tested by DSC and oscillatory shear rheology to compare properties of freshly prepared networks (‘as synthesized’) to the recovered networks. As shown in Fig. 7c, the same Tg and Tm are reported for the recovered networks indicating that semi-crystalline vitrimers can be easily broken down to monomer and recovered. Fig. 7d shows that the rheological properties of the networks are mostly retained, although the modulus does drop from 4.9 to 3.2 MPa which may be due to the volatility of boric acid at elevated temperature. Future materials can incorporate bonds which are more or less sensitive to the desired solvent for recovery.
Conclusions
Ethylene vitrimers with boronic ester bonds are shown to evolve their crystal morphology and Tm over long time periods relative to linear PE or permanent networks. With increasing crosslink density, the initial crystallization is frustrated and there is a longer induction period before Tm increases logarithmically with isothermal annealing time. In the C10 networks, a crystal – crystal transformation is observed by WAXS from hexagonal to orthorhombic structure which does not occur in linear PE under ambient conditions. Depending on linker length, either double diamond (C8), orthorhombic (C10), or hexagonal (C12) crystals are formed after 150 days of isothermal room temperature crystallization in a glovebox indicating the importance of precision on the final structure. The long-time evolution of Tm, morphology transformation, and dendrite formation are all attributed to dynamic bond exchange as they are not present in linear PE or a precise, permanent ethylene network control sample. Annealing the vitrimers at higher temperatures following the initial crystallization leads to a further increase in Tm which reaches 100 °C for a C12 network. Vitrimer recyclability is demonstrated by dissolving the networks in common laboratory solvents and then recovered by solvent evaporation. These findings on the role of dynamic bond exchange on crystallization are critical to the design, processing, and use of vitrimers as both commodity polymers, and functional materials, which are more sustainable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding to support this work was provided by the National Science Foundation (CBET-2029928 and DMR-1554435). We acknowledge the use of facilities at the Materials Research Laboratory and the NMR lab of School of Chemical Science at University of Illinois at Urbana Champaign.References
- R. Geyer, J. R. Jambeck and K. L. Law, Production, use, and fate of all plastics ever made, Sci. Adv., 2017, 3(7), e1700782 CrossRef PubMed.
- Y.-X. Lu, F. Tournilhac, L. Leibler and Z. Guan, Making Insoluble Polymer Networks Malleable via Olefin Metathesis, J. Am. Chem. Soc., 2012, 134(20), 8424–8427 CrossRef CAS PubMed.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-Like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965 CrossRef CAS PubMed.
- S. Ciarella, R. A. Biezemans and L. M. C. Janssen, Understanding, predicting, and tuning the fragility of vitrimeric polymers, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(50), 25013 CrossRef CAS.
- Q.-L. Lei, X. Xia, J. Yang, M. Pica Ciamarra and R. Ni, Entropy-controlled cross-linking in linker-mediated vitrimers, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(44), 27111 CrossRef CAS PubMed.
- F. Lossada, D. Jiao, X. Yao and A. Walther, Waterborne Methacrylate-Based Vitrimers, ACS Macro Lett., 2020, 9(1), 70–76 CrossRef CAS.
- J. L. Self, C. S. Sample, A. E. Levi, K. Li, R. Xie, J. R. de Alaniz and C. M. Bates, Dynamic Bottlebrush Polymer Networks: Self-Healing in Super-Soft Materials, J. Am. Chem. Soc., 2020, 142(16), 7567–7573 CrossRef CAS PubMed.
- Y. Yang, Z. Pei, Z. Li, Y. Wei and Y. Ji, Making and Remaking Dynamic 3D Structures by Shining Light on Flat Liquid Crystalline Vitrimer Films without a Mold, J. Am. Chem. Soc., 2016, 138(7), 2118–2121 CrossRef CAS PubMed.
- M. Guerre, C. Taplan, R. Nicolaÿ, J. M. Winne and F. E. Du Prez, Fluorinated Vitrimer Elastomers with a Dual Temperature Response, J. Am. Chem. Soc., 2018, 140(41), 13272–13284 CrossRef CAS PubMed.
- J. J. Lessard, G. M. Scheutz, R. W. Hughes and B. S. Sumerlin, Polystyrene-Based Vitrimers: Inexpensive and Recyclable Thermosets, ACS Appl. Polym. Mater., 2020, 3044–3048 CrossRef CAS.
- J. J. Lessard, G. M. Scheutz, S. H. Sung, K. A. Lantz, T. H. Epps and B. S. Sumerlin, Block Copolymer Vitrimers, J. Am. Chem. Soc., 2020, 142(1), 283–289 CrossRef CAS PubMed.
- Y. Spiesschaert, M. Guerre, I. De Baere, W. Van Paepegem, J. M. Winne and F. E. Du Prez, Dynamic Curing Agents for Amine-Hardened Epoxy Vitrimers with Short (Re)processing Times, Macromolecules, 2020, 53(7), 2485–2495 CrossRef CAS.
- W.-X. Liu, C. Zhang, H. Zhang, N. Zhao, Z.-X. Yu and J. Xu, Oxime-Based and Catalyst-Free Dynamic Covalent Polyurethanes, J. Am. Chem. Soc., 2017, 139(25), 8678–8684 CrossRef CAS PubMed.
- C. He, S. Shi, D. Wang, B. A. Helms and T. P. Russell, Poly(oxime–ester) Vitrimers with Catalyst-Free Bond Exchange, J. Am. Chem. Soc., 2019, 141(35), 13753–13757 CrossRef CAS PubMed.
- H. Liu, A. Z. Nelson, Y. Ren, K. Yang, R. H. Ewoldt and J. S. Moore, Dynamic Remodeling of Covalent Networks via Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2018, 7(8), 933–937 CrossRef CAS.
- M. M. Obadia, B. P. Mudraboyina, A. Serghei, D. Montarnal and E. Drockenmuller, Reprocessing and Recycling of Highly Cross-Linked Ion-Conducting Networks through Transalkylation Exchanges of C–N Bonds, J. Am. Chem. Soc., 2015, 137(18), 6078–6083 CrossRef CAS PubMed.
- C. A. Tretbar, J. A. Neal and Z. Guan, Direct Silyl Ether Metathesis for Vitrimers with Exceptional Thermal Stability, J. Am. Chem. Soc., 2019, 141(42), 16595–16599 CrossRef CAS PubMed.
- A. Zych, R. Pinalli, M. Soliman, J. Vachon and E. Dalcanale, Polyethylene vitrimers via silyl ether exchange reaction, Polymer, 2020, 199, 122567 CrossRef CAS.
- B. M. El-Zaatari, J. S. A. Ishibashi and J. A. Kalow, Cross-linker control of vitrimer flow, Polym. Chem., 2020, 5339–5345 RSC.
- B. B. Jing and C. M. Evans, Catalyst-Free Dynamic Networks for Recyclable, Self-Healing Solid Polymer Electrolytes, J. Am. Chem. Soc., 2019, 141(48), 18932–18937 CrossRef CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Room-Temperature Self-Healing Polymers Based on Dynamic-Covalent Boronic Esters, Macromolecules, 2015, 48(7), 2098–2106 CrossRef CAS.
- O. R. Cromwell, J. Chung and Z. Guan, Malleable and Self-Healing Covalent Polymer Networks through Tunable Dynamic Boronic Ester Bonds, J. Am. Chem. Soc., 2015, 137(20), 6492–6495 CrossRef CAS PubMed.
- R. G. Ricarte, F. Tournilhac, M. Cloître and L. Leibler, Linear Viscoelasticity and Flow of Self-Assembled Vitrimers: The Case of a Polyethylene/Dioxaborolane System, Macromolecules, 2020, 53(5), 1852–1866 CrossRef CAS.
- R. G. Ricarte, F. Tournilhac and L. Leibler, Phase Separation and Self-Assembly in Vitrimers: Hierarchical Morphology of Molten and Semicrystalline Polyethylene/Dioxaborolane Maleimide Systems, Macromolecules, 2019, 52(2), 432–443 CrossRef CAS.
- F. Caffy and R. Nicolaÿ, Transformation of polyethylene into a vitrimer by nitroxide radical coupling of a bis-dioxaborolane, Polym. Chem., 2019, 10(23), 3107–3115 RSC.
- M. O. Saed, A. Gablier and E. M. Terentejv, Liquid Crystalline Vitrimers with Full or Partial Boronic-Ester Bond Exchange, Adv. Funct. Mater., 2020, 30(3), 1906458 CrossRef CAS.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, High-performance vitrimers from commodity thermoplastics through dioxaborolane metathesis, Science, 2017, 356(6333), 62–65 CrossRef PubMed.
- W. A. Ogden and Z. Guan, Recyclable, Strong, and Highly Malleable Thermosets Based on Boroxine Networks, J. Am. Chem. Soc., 2018, 140(20), 6217–6220 CrossRef CAS PubMed.
- B. Soman and C. M. Evans, Effect of precise linker length, bond density, and broad temperature window on the rheological properties of ethylene vitrimers, Soft Matter, 2020, 3569–3577 Search PubMed.
- W. Denissen, J. M. Winne and F. E. Du Prez, Vitrimers: Permanent organic networks with glass-like fluidity, Chem. Sci., 2016, 7(1), 30–38 RSC.
- N. J. Van Zee and R. Nicolaÿ, Vitrimers: Permanently crosslinked polymers with dynamic network topology, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- F. Ziaie, M. Borhani, G. Mirjalili and M. A. Bolourizadeh, Effect of crystallinity on electrical properties of electron beam irradiated LDPE and HDPE, Radiat. Phys. Chem., 2007, 76(11), 1684–1687 CrossRef CAS.
- R. K. Krishnaswamy, Q. Yang, L. Fernandez-Ballester and J. A. Kornfield, Effect of the Distribution of Short-Chain Branches on Crystallization Kinetics and Mechanical Properties of High-Density Polyethylene, Macromolecules, 2008, 41(5), 1693–1704 CrossRef CAS.
- F. M. Mirabella Jr, S. P. Westphal, P. L. Fernando, E. A. Ford and J. G. Williams, Morphological explanation of the extraordinary fracture toughness of linear low density polyethylenes, J. Polym. Sci., Part B: Polym. Phys., 1988, 26(9), 1995–2005 CrossRef.
- K. S. Simis, A. Bistolfi, A. Bellare and L. A. Pruitt, The combined effects of crosslinking and high crystallinity on the microstructural and mechanical properties of ultra high molecular weight polyethylene, Biomaterials, 2006, 27(9), 1688–1694 CrossRef CAS PubMed.
- M. Borhani Zarandi, H. Amrollahi Bioki, Z.-A. Mirbagheri, F. Tabbakh and G. Mirjalili, Effect of crystallinity and irradiation on thermal properties and specific heat capacity of LDPE & LDPE/EVA, Appl. Radiat. Isot., 2012, 70(1), 1–5 CrossRef PubMed.
- S. Y. Son, Y. Kim, J. Lee, G.-Y. Lee, W.-T. Park, Y.-Y. Noh, C. E. Park and T. Park, High-Field-Effect Mobility of Low-Crystallinity Conjugated Polymers with Localized Aggregates, J. Am. Chem. Soc., 2016, 138(26), 8096–8103 CrossRef CAS PubMed.
- Y. Wu, S. Schneider, C. Walter, A. H. Chowdhury, B. Bahrami, H.-C. Wu, Q. Qiao, M. F. Toney and Z. Bao, Fine-Tuning Semiconducting Polymer Self-Aggregation and Crystallinity Enables Optimal Morphology and High-Performance Printed All-Polymer Solar Cells, J. Am. Chem. Soc., 2020, 142(1), 392–406 CrossRef CAS PubMed.
- J. Sun, X. Liao, A. M. Minor, N. P. Balsara and R. N. Zuckermann, Morphology-Conductivity Relationship in Crystalline and Amorphous Sequence-Defined Peptoid Block Copolymer Electrolytes, J. Am. Chem. Soc., 2014, 136(42), 14990–14997 CrossRef CAS PubMed.
- L. Yan, C. Rank, S. Mecking and K. I. Winey, Gyroid and Other Ordered Morphologies in Single-Ion Conducting Polymers and Their Impact on Ion Conductivity, J. Am. Chem. Soc., 2020, 142(2), 857–866 CrossRef CAS PubMed.
- Z. Stoeva, I. Martin-Litas, E. Staunton, Y. G. Andreev and P. G. Bruce, Ionic Conductivity in the Crystalline Polymer Electrolytes PEO6:LiXF6, X = P, As, Sb, J. Am. Chem. Soc., 2003, 125(15), 4619–4626 CrossRef CAS PubMed.
- F. Ji, X. Liu, C. Lin, Y. Zhou, L. Dong, S. Xu, D. Sheng and Y. Yang, Reprocessable and Recyclable Crosslinked Polyethylene with Triple Shape Memory Effect, Macromol. Mater. Eng., 2019, 304(3), 1800528 CrossRef.
- M. Maaz, A. Riba-Bremerch, C. Guibert, N. J. Van Zee and R. Nicolaÿ, Synthesis of Polyethylene Vitrimers in a Single Step: Consequences of Graft Structure, Reactive Extrusion Conditions, and Processing Aids, Macromolecules, 2021, 54(5), 2213–2225 CrossRef CAS.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, Polylactide Vitrimers, ACS Macro Lett., 2014, 3(7), 607–610 CrossRef CAS.
- L. Yan, K. C. Bustillo, O. Panova, A. M. Minor and K. I. Winey, Solution-grown crystals of precise acid-and ion-containing polyethylenes, Polymer, 2018, 135, 111–119 CrossRef CAS.
- L. Yan, M. Häußler, J. Bauer, S. Mecking and K. I. Winey, Monodisperse and telechelic polyethylenes form extended chain crystals with ionic layers, Macromolecules, 2019, 52(13), 4949–4956 CrossRef CAS.
- K. Yang, Z. Cai, A. Jaiswal, M. Tyagi, J. S. Moore and Y. Zhang, Dynamic Odd–Even Effect in Liquid n-Alkanes near Their Melting Points, Angew. Chem., Int. Ed., 2016, 55(45), 14090–14095 CrossRef CAS PubMed.
- E. Badea, G. D. Gatta, D. D’Angelo, B. Brunetti and Z. Rečková, Odd–even effect in melting properties of 12 alkane-α,ω-diamides, J. Chem. Thermodyn., 2006, 38(12), 1546–1552 CrossRef CAS.
- V. R. Thalladi, M. Nüsse and R. Boese, The Melting Point Alternation in α,ω-Alkanedicarboxylic Acids, J. Am. Chem. Soc., 2000, 122(38), 9227–9236 CrossRef CAS.
- L. Yan, M. Häußler, J. Bauer, S. Mecking and K. I. Winey, Monodisperse and Telechelic Polyethylenes Form Extended Chain Crystals with Ionic Layers, Macromolecules, 2019, 52(13), 4949–4956 CrossRef CAS.
- X. Zhang, X. Zuo, P. Ortmann, S. Mecking and R. G. Alamo, Crystallization of Long-Spaced Precision Polyacetals I: Melting and Recrystallization of Rapidly Formed Crystallites, Macromolecules, 2019, 52(13), 4934–4948 CrossRef CAS.
- E. B. Trigg, T. W. Gaines, M. Maréchal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Self-assembled highly ordered acid layers in precisely sulfonated polyethylene produce efficient proton transport, Nat. Mater., 2018, 17(8), 725–731 CrossRef CAS PubMed.
- C. Rank, L. Yan, S. Mecking and K. I. Winey, Periodic Polyethylene Sulfonates from Polyesterification: Bulk and Nanoparticle Morphologies and Ionic Conductivities, Macromolecules, 2019, 52(21), 8466–8475 CrossRef CAS.
- C. Shen, Q. Zhao and C. M. Evans, Ion specific, odd–even glass transition temperatures and conductivities in precise network polymerized ionic liquids, Mol. Syst. Des. Eng., 2019, 4(2), 332–341 RSC.
- D. C. Bassett, Chain-extended polyethylene in context: a review, Polymer, 1976, 17(6), 460–470 CrossRef CAS.
- B. Wunderlich and T. Davidson, Extended-chain crystals. I. General crystallization conditions and review of pressure crystallization of polyethylene, J. Polym. Sci., Part A-2, 1969, 7(12), 2043–2050 CrossRef CAS.
- L. R. Middleton, E. B. Trigg, E. Schwartz, K. L. Opper, T. W. Baughman, K. B. Wagener and K. I. Winey, Role of periodicity and acid chemistry on the morphological evolution and strength in precise polyethylenes, Macromolecules, 2016, 49(21), 8209–8218 CrossRef CAS.
- M. K. Smith and B. H. Northrop, Vibrational Properties of Boroxine Anhydride and Boronate Ester Materials: Model Systems for the Diagnostic Characterization of Covalent Organic Frameworks, Chem. Mater., 2014, 26(12), 3781–3795 CrossRef CAS.
- G. Springsteen and B. Wang, A detailed examination of boronic acid–diol complexation, Tetrahedron, 2002, 58(26), 5291–5300 CrossRef CAS.
- P. J. Flory, Principles of polymer chemistry, Cornell University Press, 1953 Search PubMed.
- Y.-L. Loo, R. A. Register and A. J. Ryan, Polymer Crystallization in 25 nm Spheres, Phys. Rev. Lett., 2000, 84(18), 4120–4123 CrossRef CAS PubMed.
- L. Mandelkern, Crystallization kinetics of homopolymers: overall crystallization: a review, Biophys. Chem., 2004, 112(2), 109–116 CrossRef CAS PubMed.
- E. Woo and T. Ko, A differential scanning calorimetry study on poly (ethylene terephthalate) isothermally crystallized at stepwise temperatures: multiple melting behavior re-investigated, Colloid Polym. Sci., 1996, 274(4), 309–315 CrossRef CAS.
- G. C. Alfonso, E. Pedemonte and L. Ponzetti, Mechanism of densification and crystal perfection of poly(ethylene terephthalate), Polymer, 1979, 20(1), 104–112 CrossRef CAS.
- E. Zhuravlev, V. Madhavi, A. Lustiger, R. Androsch and C. Schick, Crystallization of Polyethylene at Large Undercooling, ACS Macro Lett., 2016, 5(3), 365–370 CrossRef CAS.
- M. Tian and J. Loos, Investigations of morphological changes during annealing of polyethylene single crystals, J. Polym. Sci., Part B: Polym. Phys., 2001, 39(7), 763–770 CrossRef CAS.
- S. C. Moyses and J. Zukermann-Schpector, Annealing in Low Density Polyethylene at Several Temperatures, Polym. J., 2004, 36(9), 679–683 CrossRef CAS.
- P. J. Holdsworth and A. Turner-Jones, The melting behaviour of heat crystallized poly(ethylene terephthalate), Polymer, 1971, 12(3), 195–208 CrossRef CAS.
- N. Hiramatsu and S. Hirakawa, Melting Behavior of Poly(ethylene terephthalate) Crystallized and Annealed under Elevated Pressure, Polym. J., 1980, 12(2), 105–111 CrossRef CAS.
- D. L. Nealy, T. G. Davis and C. J. Kibler, Thermal history and melting behavior of poly(ethylene terephthalate), J. Polym. Sci., Part A-2, 1970, 8(12), 2141–2151 CrossRef CAS.
- A. Mehta, U. Gaur and B. Wunderlich, Equilibrium melting parameters of poly(ethylene terephthalate), J. Polym. Sci., Polym. Phys. Ed., 1978, 16(2), 289–296 CrossRef CAS.
- Q. Li, J. Zhou, M. Vatankhah-Varnoosfaderani, D. Nykypanchuk, O. Gang and S. S. Sheiko, Advancing Reversible Shape Memory by Tuning the Polymer Network Architecture, Macromolecules, 2016, 49(4), 1383–1391 CrossRef CAS.
- P. Ingram and A. Schindler, Morphology of as-polymerized polythylene II. Electron microscopy, Makromol. Chem., 1968, 111(1), 267–270 CrossRef CAS.
- P. H. Geil, F. R. Anderson, B. Wunderlich and T. Arakawa, Morphology of polyethylene crystallized from the melt under pressure, J. Polym. Sci., Part A: Gen. Pap., 1964, 2(8), 3707–3720 CrossRef CAS.
- C. M. L. Atkinson and M. J. Richardson, Thermodynamic properties of ideally crystalline polyethylene, Trans. Faraday Soc., 1969, 65(0), 1764–1773 RSC.
- S. Tsubakihara, A. Nakamura and M. Yasuniwa, Hexagonal Phase of Polyethylene Fibers under High Pressure, Polym. J., 1991, 23(11), 1317–1324 CrossRef CAS.
- D. C. Bassett, S. Block and G. J. Piermarini, A high-pressure phase of polyethylene and chain-extended growth, J. Appl. Phys., 1974, 45(10), 4146–4150 CrossRef CAS.
- J. A. O. Bruno, N. L. Allan, T. H. K. Barron and A. D. Turner, Thermal expansion of polymers: Mechanisms in orthorhombic polyethylene, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(13), 8416–8427 CrossRef CAS.
- K. Yamada, M. Hikosaka, A. Toda, S. Yamazaki and K. Tagashira, Equilibrium Melting Temperature of Isotactic Polypropylene with High Tacticity: 1. Determination by Differential Scanning Calorimetry, Macromolecules, 2003, 36(13), 4790–4801 CrossRef CAS.
- J. D. Hoffman and J. I. Lauritzen, Jr., Crystallization of Bulk Polymers With Chain Folding: Theory of Growth of Lamellar Spherulites, J. Res. Natl. Bur. Stand. A Phys. Chem., 1961, 65A(4), 297–336 CrossRef CAS PubMed.
- N. Goldenfeld, Theory of spherulitic crystallization, J. Cryst. Growth, 1987, 84(4), 601–608 CrossRef CAS.
- C. T. Campbell, S. C. Parker and D. E. Starr, The Effect of Size-Dependent Nanoparticle Energetics on Catalyst Sintering, Science, 2002, 298(5594), 811 CrossRef CAS PubMed.
- S. Wang, S. Ma, Q. Li, W. Yuan, B. Wang and J. Zhu, Robust, Fire-Safe, Monomer-Recovery, Highly Malleable Thermosets from Renewable Bioresources, Macromolecules, 2018, 51(20), 8001–8012 CrossRef CAS.
- F. Lossada, D. Jiao, D. Hoenders and A. Walther, Recyclable and Light-Adaptive Vitrimer-Based Nacre-Mimetic Nanocomposites, ACS Nano, 2021, 15, 5043–5055 CrossRef CAS PubMed.
- L. Cheng, S. Liu and W. Yu, Recyclable ethylene-vinyl acetate copolymer vitrimer foams, Polymer, 2021, 123662 CrossRef CAS.
- G. Li, P. Zhang, S. Huo, Y. Fu, L. Chen, Y. Wu, Y. Zhang, M. Chen, X. Zhao and P. Song, Mechanically Strong, Thermally Healable, and Recyclable Epoxy Vitrimers Enabled by ZnAl-Layer Double Hydroxides, ACS Sustainable Chem. Eng., 2021, 9(6), 2580–2590 CrossRef CAS.
- M. Häußler, M. Eck, D. Rothauer and S. Mecking, Closed-loop recycling of polyethylene-like materials, Nature, 2021, 590(7846), 423–427 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures including synthesis, FTIR, rheology, DSC, and NMR. See DOI: 10.1039/d1sm01288f |
| This journal is © The Royal Society of Chemistry 2022 |