
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA highly contorted push–pull naphthalenediimide dimer and evidence of intramolecular singlet exciton fission†
Deepak
Bansal‡
 a,
Arup
Kundu‡
b,
Vijay Pal
Singh
a,
Arun K.
Pal
c,
Ayan
Datta
*c,
Jyotishman
Dasgupta
*b and
Pritam
Mukhopadhyay
*a
a,
Arup
Kundu‡
b,
Vijay Pal
Singh
a,
Arun K.
Pal
c,
Ayan
Datta
*c,
Jyotishman
Dasgupta
*b and
Pritam
Mukhopadhyay
*a
aSchool of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: m_pritam@mail.jnu.ac.in
bDepartment of Chemical Sciences, Tata Institute of Fundamental Research, Mumbai 400005, India. E-mail: dasgupta@tifr.res.in
cSchool of Chemical Sciences, Indian Association for the Cultivation of Science, Kolkata 700032, West Bengal, India. E-mail: spad@iacs.res.in
First published on 21st September 2022
Abstract
Singlet fission is a process by which two molecular triplet excitons are generated subsequent to the absorption of one photon. Molecules that enable singlet fission have triplet state energy at least half of the bright singlet state energy. This stringent energy criteria have challenged chemists to device new molecular and supramolecular design principles to modulate the singlet–triplet energy gap and build singlet fission systems from a wide range of organic chromophores. Herein, we report for the first time intramolecular singlet fission in the seminal naphthalenediimide (NDI) scaffold constrained in a push–pull cyclophane architecture, while individually the NDI chromophore does not satisfy the energy criterion. The challenging synthesis of this highly contorted push–pull cyclophane is possible from the preorganized pincer-like precursor. The special architecture establishes the shortest co-facial NDI⋯NDI contacts (3.084 Å) realized to date. Using broadband femtosecond transient absorption, we find that the correlated T–T pair forms rapidly within 380 fs of photoexcitation. Electronic structure calculations at the level of state-averaged CASSCF (ne,mo)/XMCQDPT2 support the existence of the multi-excitonic T–T pair state, thereby confirming the first example of singlet exciton fission in a NDI scaffold.
Introduction
Multiexciton generation in photostable molecular systems shows promise as a creative route to increase the efficiency of light-energy conversion devices. In the past decade, chemists have successfully demonstrated the generation of two triplet excitons upon photoexcitation in certain classes of pi-conjugated organic chromophores through a process called singlet exciton fission.1–4 In singlet fission (SF), the energy of the photogenerated singlet excited state gets redistributed with a neighbouring electronically coupled chromophore, via an intermediate multi-excitonic state called the correlated triplet–triplet (T–T) pair state,4 to generate two excited molecules in their independent triplet state. SF as a concept has been successfully exploited by the photovoltaic community to generate solar cells with efficiency higher than the theoretical maxima as calculated by Shockley and Queisser.5–8 However, expanding the limited number of successful chromophores for SF to a generic library is a major challenge,9 principally due to two reasons. Firstly, the molecule has to satisfy an energy criterion,1 which says that the first excited state singlet energy in the molecule should be greater/equal to twice of the triplet energy, i.e., E(S1) ≥ 2E(T1). Apart from the singlet-triplet energy requirement, it is necessary to tune the electronic coupling for these chromophores such that SF becomes the fastest mode of excited singlet state decay, thereby maximizing the quantum yield of triplets to be 200%.Intermolecular SF has been the predominant mechanism for multiexciton generation within molecular crystals and self-assembled molecular aggregates, and therefore prominently exploited in functional devices.10–15 As such, strong electronic coupling in the excited state within adjacent molecules is the key, which is dependent on noncovalent interactions such as Π–Π stacking, dipole or van der Waals forces that can proliferate and sustain long-range order.16 Recently seminal work by Campos and co-workers demonstrated that intramolecular SF in acene dimers can serve to benchmark the spatial dependence of electronic coupling and the interactions that favour ultrafast SF.13 Subsequently, different acene chromophores were covalently connected through rigid17–21 or flexible15 linkers to suitably control the distance and interchromophore orientation for successful intramolecular SF. It is observed that in all these molecular dimers that show SF the correlated T–T pair never dissociates to form free triplets due to fast recombination arising from the close proximity of the two chromophores. Apart from the covalent interactions in a dimer, it has been realized that molecular strain or backbone distortion in π-conjugated systems can be used to tune the S1 and T1 state energies to satisfy the SF energy criterion.22–25 This idea potentially can allow chromophores which do not satisfy the requisite SF energy criterion to be used as a SF layer in functional photovoltaic devices.
Napthalenediimide (NDI) and its derivatives are versatile photostable chromophores.26–35 The NDI scaffold has been diversely functionalized to modulate its excited state dynamics, establish artificial light-harvesting, and explore as conduits for electron and energy transfer as well as towards intriguing emissive systems. Recently, phosphorescence as well as delayed fluorescence was observed for the first time from the NDI scaffold.33,34 In addition, thermally activated delayed fluorescence (TADF) has also been discerned with the NDI scaffold.35 Importantly, NDI has an unfavourable singlet–triplet (S–T) gap for SF.36–38 The singlet state energies range from 2.5 eV to 3.5 eV, while triplets can be as high as 2 eV. As a result of this unfavourable S–T gap, SF has not been demonstrated from the NDI scaffold to date.
In our design, we envisaged that maximum electronic communication between the NDI units can be enforced in a cyclophane framework having very close co-facial contacts, which can allow for favourable evolution to the T–T pair state. The key to establishing such close contacts is stitching the NDI units from their core-/short-axis, compared to the synthetically less challenging cyclization from the axial-/long-axis.39 We therefore predicted significant inward arching of the naphthalene rings in the NDI units when rigid linkers like 1,2-diaminocyclohexane are integrated in the cyclophane framework (Scheme 1). Also, the integration of electron rich donor groups at the core positions of the electron deficient NDI will endow the cyclophane with a push–pull character. Herein, we report the synthesis of a highly contorted cyclophane constructed by fusing diamino linkers at the core positions of two NDI units, and having the shortest co-facial NDI⋯NDI contacts realized to date. We demonstrate ultrafast intramolecular SF in 380 fs in this novel NDI dimer, thereby providing the first report of a NDI scaffold hosting a T–T pair state.
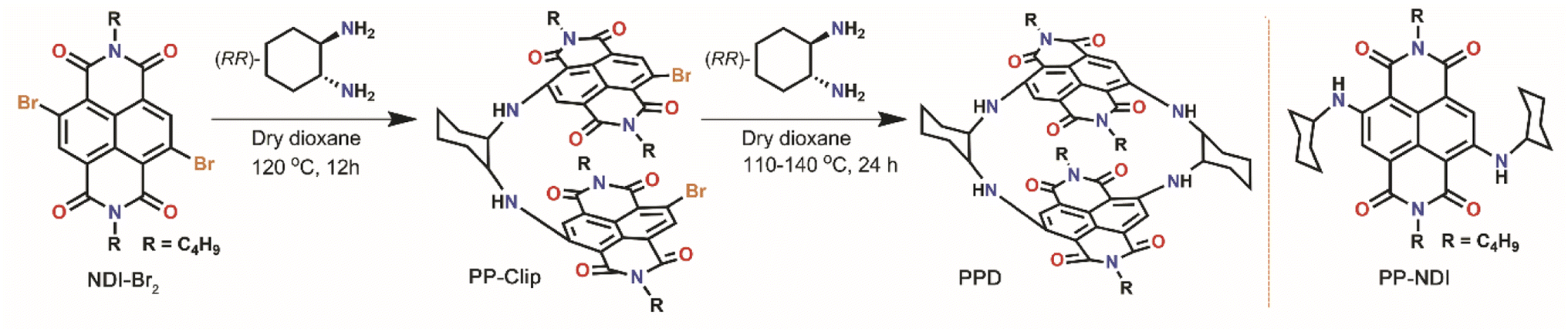 | ||
| Scheme 1 Schematic representation for the synthesis of the NDI core-extended push–pull clip (PP-Clip) and push–pull dimer (PPD), and structure of the reference molecule PP-NDI. | ||
Results and discussion
Synthesis and characterization of PPD
The NDI-dimer PPD was synthesized by reacting N,N-dibutyl-2,3-dibromo-naphthalene-1,4,5,8-tetracarboxylicacid-bis-imide (NDI-Br2) with (RR)-trans-1,2-cyclohexanediamine in a stepwise manner (Scheme 1). The challenging aspect of bringing the two electron-rich naphthalene units has been possible from the preorganized PP-Clip with a pincer-like architecture. Notably, this is the first example of a NDI cyclophane with donors substituted at the NDI-core and having a push–pull construct. The reference push–pull compound PP-NDI was synthesized from NDI-Br2.40 The synthesis of these compounds was confirmed by different spectroscopic techniques as well as by single crystal X-ray crystallography (Fig. S1–S7, ESI†). Interestingly, 1H NMR of PPD exhibits a peak corresponding to the NDI-core H's at 7.90 ppm, which is shielded by 0.25 ppm in comparison to PP-NDI (Fig. S3 and S6†). This clearly indicates that the naphthalene rings in PPD have developed a significant electron-rich character as a result of the co-facial stacking interactions (vide infra) compared to PP-NDI.In addition, from electrochemical studies we observed four distinct electron reduction and four oxidation waves at distinct potentials suggesting strong electronic communication between the NDI units (Fig. S8†). The oxidation and reduction potentials of PPD shift towards less positive and more negative values, respectively, compared to PP-NDI. This indicates the electron-rich nature of PPD and hence easier electron release from the cyclophane.
To have an insight into the structural aspects of PPD, we obtained single crystals by slow evaporation of its solution in chloroform–hexane. The crystal structure of PPD (see Table S5†) revealed highly contorted, co-facially stacked NDI units with maximal inward arching at the C atoms of the naphthalene rings bonded to the amino groups of cyclohexyldiamine linkers (Fig. 1a). The linkers connect the two NDI units through the alternate C atoms, i.e., C2 of one to C3 of the other NDI unit, while C6 is connected to C7 (Fig. 1b).
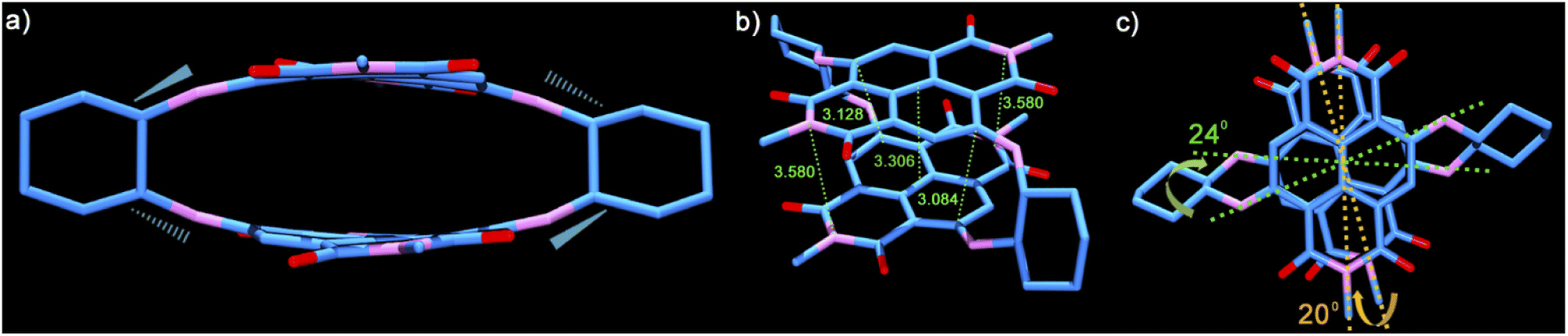 | ||
| Fig. 1 Single crystal structure of PPD. (a) Side view of the push–pull dimer depicting the diagonally positioned above- and below-the-plane cyclohexane (C–N) bonds. (b) Top-frontal view showing the contorted NDI units, the chair conformation of the cyclohexane rings and the various close contacts in Å (calculated using ORTEP-3). (c) Top view showing the slipped NDI Π-surfaces and the twisting along the long- and short-axes of PPD. The axial n-butyl groups and the H atoms have been removed for clarity. | ||
These C atoms make the shortest contacts of 3.084 Å and 3.128 Å (Fig. 1b), respectively (calculated using ORTEP-3), which are significantly lower than the sum of the van der Waals radii of C atoms (3.4 Å). The central region of PPD is arched in an outward fashion with centroid–centroid contacts of 3.306 Å formed between the mid-points of the annular bonds connecting the naphthalene rings. The openings at both the ends of the cyclophane are comparatively wider with the imide N atoms spaced at 3.580 Å. The conformational constraint enforced due to linking the NDI units along its core/short-axis is also evident from the non-eclipsed stacking arrangement of the two π-surfaces (Fig. 1c). The NDI units are twisted by 20° and 24° along the long- and short-axes, respectively (Fig. S9†). Therefore, linking NDI units from their core-/short-axis provides a definite route to realize the shortest contacts between the NDI units in any dimeric construct reported to date and establish a greater contortion of the π-surfaces.39
The steady-state absorption spectrum of PPD exhibits bands in the regions of 340–440 nm and 450–750 nm with λmax at 600 nm as shown in Fig. 2a. In contrast, PP-NDI exhibits bands at 325−420 nm and 450–660 nm due to the π–π* and ICT transitions, respectively (Fig. 2a). Thus, PPD exhibits significantly broadened absorption characteristics encompassing the UV and the whole visible region and endows it with a panchromatic character. Fluorescence quenching is observed for PPD as compared to its monomer counterpart PP-NDI. Notably, the ICT band in PPD exhibits a significantly decreased molar extinction coefficient (ε600 = 5530 M−1 cm−1) compared to that in PP-NDI (ε618 = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 440 M−1 cm−1) suggesting strong electronic coupling between the two co-facially π-stacked NDI units (Fig. S10†). Fig. 2b shows the comparative emission spectrum of PPD and PP-NDI. PPD shows significant quenching in the emission following excitation at 540 nm, suggesting that in the dimer there may be a different non-radiative decay pathway.
440 M−1 cm−1) suggesting strong electronic coupling between the two co-facially π-stacked NDI units (Fig. S10†). Fig. 2b shows the comparative emission spectrum of PPD and PP-NDI. PPD shows significant quenching in the emission following excitation at 540 nm, suggesting that in the dimer there may be a different non-radiative decay pathway.
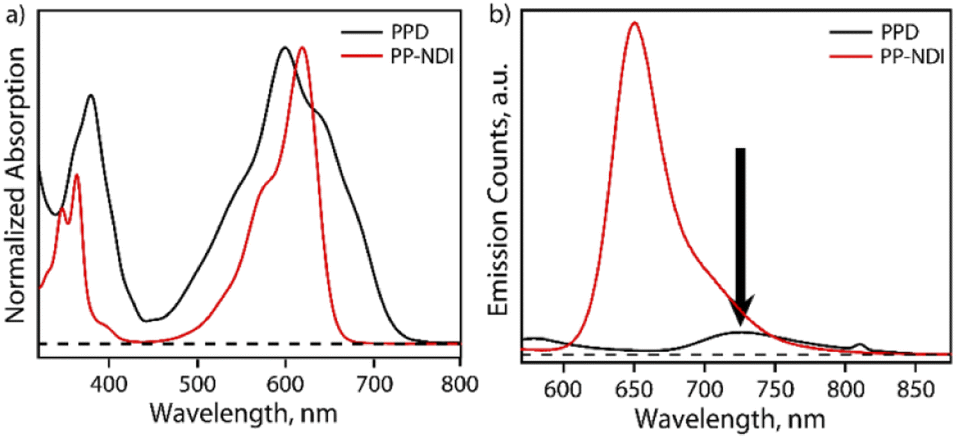 | ||
| Fig. 2 (a) Steady state absorption spectra of PPD (in black) and PP-NDI (in red) in DCM at 200 μM concentration, respectively. The PPD spectrum shows clear evidence of strong electronic coupling between the NDI scaffolds. (b) Steady state emission spectra of PPD (black) and PP-NDI (red) in DCM at 200 μM concentration, subsequent to excitation at 540 nm. | ||
Transient absorption spectroscopy
In order to test the feasibility of intramolecular SF in PPD, we probed the excited state dynamics of PPD with the help of transient absorption spectroscopy and compared it with PP-NDI. The excited state dynamics of PPD was probed using broadband white light continuum (∼430–1300 nm) subsequent to 400 nm femtosecond pump excitation. Fig. 3a shows the transient absorption spectra of PPD at different pump–probe delay time from negative 450 fs to 2 ns in the spectral range of 430 nm to 800 nm. After photoexcitation, we observed two positive excited state absorption (ESA) features centred at ∼500 nm and 740 nm and a negative ground state bleach (GSB) feature in the region of 540 nm to 650 nm. The ESA feature at 500 nm shows an initial rise of 300 fs followed by a mono-exponential decay of 68 ps (Fig. 3b). The initial feature at 500 nm is attributed to the absorption of the S1 state. Then it does an internal conversion to another state within the sub-picosecond timescale, which lives for ∼68 ps, as reflected in the single point kinetics at 493 nm, shown in Fig. 3b. On the other hand, the GSB feature at 593 nm shows an initial rise of 2.9 ps, followed by a decay of 66 ps (Fig. 3b and S11†). This concomitant rise in the GSB with the decay of S1 and appearance of another intermediate state is a direct proof of SF in PPD. The mismatch in the timescale of the decay of S1 and GSB rise from the single point kinetics analysis is due to the convolution of ESA and GSB features.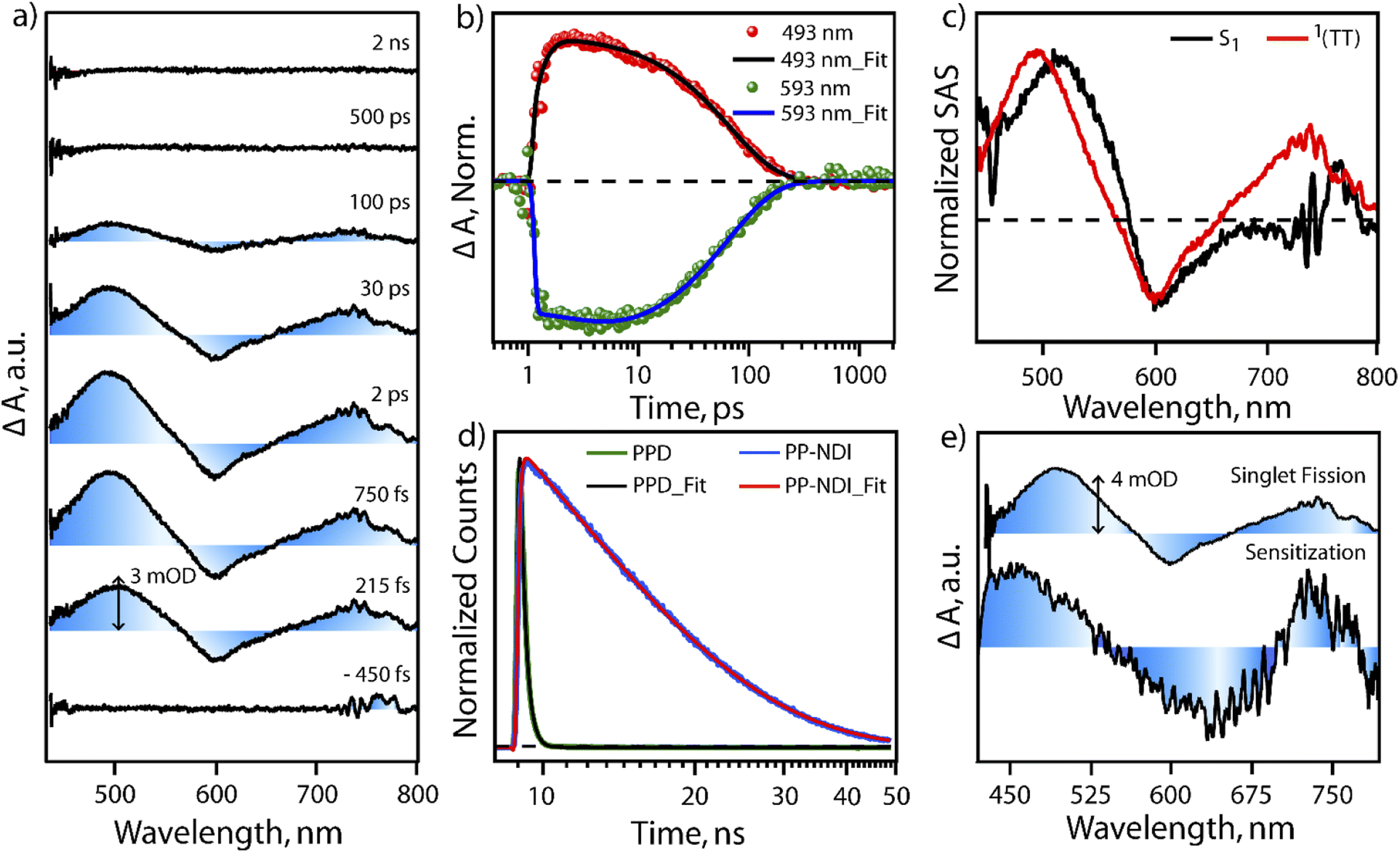 | ||
| Fig. 3 (a) Transient absorption of PPD (100 μM in ACN:DCM (1:1)) subsequent to 400 nm photoexcitation. (b) Single point kinetics at 593 nm (GSB) and 493 nm (T–T pair). (c) Normalized species associated spectra followed by two state sequential singular value decomposition of the TA spectra of PPD. Black is the S1 state and red is the T–T pair state. (d) TCSPC kinetics data for PPD and PP-NDI, emission collected at 720 nm, subsequent to excitation at 540 nm. (e) Triplet sensitization measurements of PPD. Top: T–T pair spectrum obtained from SF. Bottom: sensitized triplet spectrum of PPD. | ||
To understand the spectral features and the associated lifetimes of different states clearly, we performed singular value decomposition (SVD) analysis of the entire TA spectra of the 430 nm to 800 nm region with a two component sequential model. The normalized Species Associated Spectra (SAS) obtained from the SVD analysis are shown in Fig. 3c. The black trace represents the initially formed S1 state. Within 380 fs, S1 evolves to another state, represented by the red spectrum. The newly formed state could be assigned to a T–T pair state formed through SF. This undergoes a fast recombination with a lifetime of 70 ps. In the literature,41,42 it is well known that triplets formed through intramolecular SF in a dimeric system are prone to rapid recombination due to close proximity. On the other hand, the monomeric NDI derivative (PP-NDI) shows a pronounced emissive state that was confirmed using both TCSPC and transient absorption measurements. Fig. 3d shows the fluorescence decay dynamics of the dimer as compared to the monomer with an IRF of ∼100 ps by exciting at 540 nm. The data clearly show a lifetime of 10.6 ns for the monomer NDI, while the dimer shows a quenched emission with ∼240 ps average lifetime indicative of ultrafast SF. Femtosecond to nanosecond transient absorption spectroscopy on the monomer PP-NDI (Fig. S12(a) in ESI†) indicated that the excited state absorption and the stimulated emission spectral features at 470 nm and 650 nm, respectively, lived beyond nanoseconds (Fig. S13†). This clearly indicated that the excited state has a long lifetime, which was quantified precisely using nanosecond to microsecond transient absorption spectra (Fig. S12(b)†). The feature at 750 nm was used to ascertain the lifetime to be 10 ns (Fig. S14†). We therefore conclude that the monomer PP-NDI is emissive, while the dimer PPD shows a non-radiative SF process within 380 fs, dictating the formation of the T–T pair state.
To confirm the formation of the T–T pair state, we performed triplet-sensitization measurements with a Ruthenium sensitizer mixed in a 2:1 concentration ratio with PPD (details in the ESI, Fig. S15–S17†). Upon photoexcitation with 400 nm in the mixture, we observe the excited state spectral features of the [Ru-tris(bipy)3]2+-complex, which subsequently transfer excitation energy to generate triplets in PPD. However, it should be mentioned that we do also see a small fraction of NDI dimer dynamics, which can get triggered due to the overlapping absorption feature at 400 nm by direct photoexcitation (details explained in the ESI†). The NDI dimer dynamics decays within 100 ps, while the Ru-complex displays long-lived excited evolution. On the later timescale, microsecond time region, the Ru-complex does triplet energy transfer to the unexcited PPD molecules and a sensitized triplet is formed on the PPD molecule. Fig. 3e shows the spectral comparison of the SF induced triplet in PPD and the sensitized triplet on the NDI unit in one half of the dimer (PPD). It is clearly seen that the ESA features between 450 nm to 525 nm and 700 nm to 750 nm in the sensitized triplet spectra match well with the T–T pair spectra for the PPD photoexcitation. It is to be noted that we do not find any evidence for any other non-radiative pathways like symmetry breaking charge separation.43,44 The radical cation/anion signatures of NDI were not observed in our case. We therefore conclude that PPD does produce T–T species, which are weakly coupled to show almost similar spectra as the free triplet that can be hosted on the dimer. Also, our pump excitation wavelength dependent study following excitation at 620 nm in the ICT transition band shows similar spectral features (with better signal/noise) and timescales for SF in PPD. We do note that the lifetime of the T–T pair state is longer for the 620 nm excitation indicating that the detection of SF is more sensitive when exciting at 620 nm due to the larger extent of photoexcitation. We therefore infer that excitation at π–π* and the ICT band lead to the same T–T pair formation in PPD through the SF mechanism.
Electronic structure calculations
The multiexciton (ME) state in a SF process i.e. the correlated T–T pair state is a doubly excited state and hence, a single reference method such as TDDFT cannot properly describe this state. For that reason, we have employed static correlated SA3-CASSCF along with the dynamic correlated XMCQDPT2 method. The excitation energies for the ME state formally designated in computations as the lowest singlet excited state S1a (T–T state) while the first optically allowed singlet excited state (ICT) is designated as S2a are listed in Table 1.| (ne,mo) | S1a | χ | S2a | χ | T1 |
|---|---|---|---|---|---|
| (4,4) | 1.97 | ME | 2.63 | CT + LE | 1.84 |
| (6,6) | 2.13 | ME | 2.77 | CT + LE | 2.05 |
| (8,8) | 2.09 | ME | 2.71 | CT + LE | — |
| (10,10) | 2.24 | ME | 2.90 | CT + LE | — |
An equal weightage of the ground state and excited states was used to configure the wavefunctions for SA3-CASSCF calculations, where we have used C1 point group symmetry (computational details are provided in the ESI†). The S2a state is optically active as it carries the local excitation (LE) and charge transfer (CT) characters. The dark state which is 0.6 eV lower in energy with respect to the S2a state has an ME character and therefore leads to an exoergic singlet fission. The excitation energies for the S1a and S2a states increase with the increase in CAS active space and active orbitals. For instance, 2.63 eV is the energy for the S2a state with SA3-CAS(4,4), whereas it is 2.90 eV for the SA3-CAS(10,10) calculations.
From this observation, it is obvious that low-lying singlet excited states are well defined by HOMO-to-LUMO and HOMO−1-to-LUMO+1 transitions and the frontier molecular orbitals further lower in energies were treated as a rigid core.45,46 Therefore, in such cases, the active space requirement in SA-CAS beyond 4e–4o is not effective. However, we also performed CAS(12,10)/NEVPT2/def2-SVP calculations that predict the energy of the optically active excited singlet state as 2.13 eV, which is close to the experimental value (1.80 eV). The configuration state functions corresponding to the S1a and S2a states are shown in Table S1 of the ESI,† where the spin arrangements in ME, LE and CT states are also captured. The molecular orbital diagrams for the ME, LE and CT states are shown in Fig. 4. These calculations confirm the existence of a ME state close in energy to the LE with a highly mixed CT character.
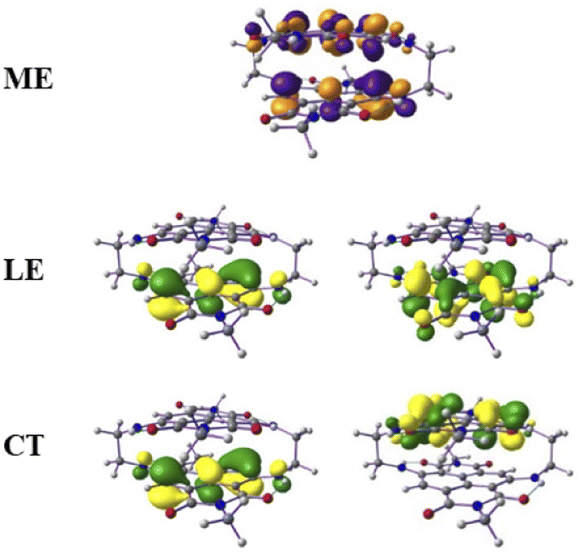 | ||
| Fig. 4 The molecular orbital (isovalues of ±0.04 au) corresponding to the ME configuration in the S1a (dark) state and LE and CT configurations in the S2a (bright) state. | ||
Furthermore, the S1 and T1 energies for the NDI monomer, i.e.PP-NDI, show that SF on the monomer backbone is endoergic as E(2T1) > E(S1) (Table S2†). Although the NDI monomer backbone does not thermodynamically support SF, we observe that our novel contorted NDI dimer, PPD, does allow for exoergic SF. Our observations, the first for the NDI class of molecules, are actually similar to covalently linked perylene diimide (PDI) dimers.25 However, for PDI dimers, the energy transfer is suppressed by the formation of a low-energy excimer.47,48 Consequently, Dover et al. have systematically explained that the challenging task is to design an endothermic SF material that avoids excimer formation.49 Therefore, for the present case, it is important to verify whether the formation of the ME state is trapped by an excimer formation or not. The excimer structure is obtained by calculating the energy of the S2a state where the centre-to-centre distance (d, in Å) of the PPD dimer is varied from 3.30 Å to 3.80 Å with an interval of 0.02 Å. These calculations are performed by the TDDFT method. The potential energy diagram is shown in Fig. 5, which suggests that the π-stacked structure with d = 3.50 Å belongs to the minima. Therefore, we have taken the structure as an excimer and the same procedure has been followed by Gao et al.50 The SA-CAS(4,4)/XMCQDPT2 calculated low-lying singlet excited states for the excimer structure are S1a = 1.98 eV (ME) and S2a = 2.64 eV (CT + LE), comparable with the values in Table 1. Hence, the S2a minima contain the characteristics of an excimer but do not act as a proper excimer, named the excimer-like structure. Such an intermediate before the formation of the ME state generally does not act as a trap and hence increases the triplet yield.51 The same calculations have been performed for planar PPD, where we have removed the bridges in PPD and reoptimized it. The dimer makes the two NDI units planar with a stacking distance of 3.49 Å (Fig. S20†). Interestingly, for this case, no such observation is found (see Fig. 5 and Table S3†).
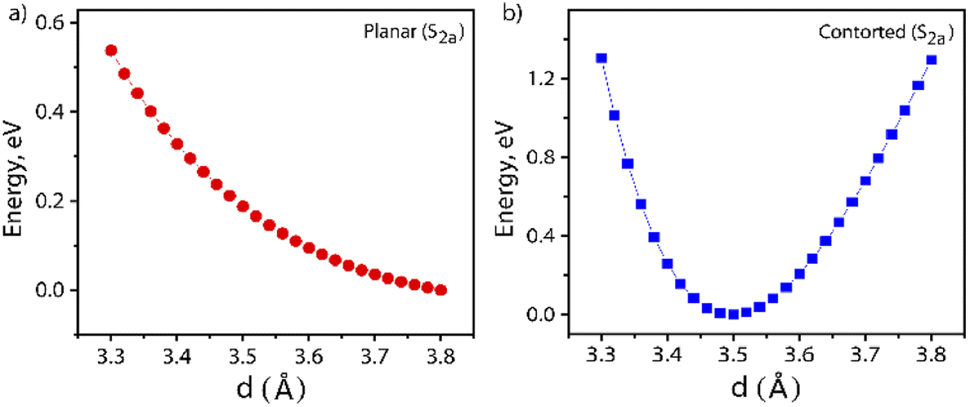 | ||
| Fig. 5 Potential energy surface plot for the geometry scan of the first excited singlet state (S2a) of: (a) planar and (b) contorted PPD. The centre-to-centre distance (d) of 3.50 Å shows the minima for the contorted PPD. | ||
To summarize, the NDI monomers used here have triplet energies that would not satisfy the SF energy criteria. However, by covalent stitching of these monomers in a dimer (PPD), we find singlet fission operative on an ultrafast timescale. The multiexcitonic T–T pair state is optimally stabilized in the dimer and avoided an excimer trap as analysed by electronic structure theory. We find that the photoexcitation of the π–π* (400 nm excitation to S3a) or ICT (620 nm excitation to S2a) features provides an ultrafast access to the multi-excitonic T–T pair state as shown in Fig. S21.† It is known that such excimer traps are also seen in other arylenediimides like PDI where endoergic SF has also been observed (Table S4†).52–55 This is in contrast to that of terylenediimides, where triplets follow the SF criteria to drive an exoergic SF (Table S4†).56–58 Our work, therefore, provides a framework for designing new SF systems even when triplet state energies are not ideal for hosting SF.
Conclusion
In conclusion, we have synthesized a new co-facially stacked NDI dimer having the shortest NDI⋯NDI contacts realized to date, enabling large electronic coupling of the monomer fragments. The short contacts and the contortion in the backbone visualized by the crystal structure of the NDI system allowed for stabilizing a multi-excitonic state, which has a substantial CT character, thereby allowing the excited molecular dimer to channel its excitation energy via SF. Therefore, our design enabled the first example of a NDI dimer that shows SF (see the kinetic model in Fig. S21†) within 380 fs due to the inherent backbone contortion. We believe that synthesizing such dimers with varying separation will allow for the optimal understanding and control of SF in NDI dimers. We envision that our exploration will provide a new way of tailoring a new generation of chromophores59,60 and make them amenable to host SF.Data availability
Crystallographic data for PP-NDI and PPD have been deposited at the CCDC under [2171334 and 2171335] and can be obtained from https://www.ccdc.cam.ac.uk. The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
P. M. and D. B. conceived the molecular design. J. D. provided with the experimental methods for measuring the excited state properties. A. D. provided the computational framework for understanding the results. D. B. carried out the synthesis, single crystal X-ray crystallography and electrochemistry experiments. A. K. performed the steady state and transient emission and absorption measurements along with triplet sensitization measurements. A. K. and J. D. analyzed the spectroscopic data. A. K. P. carried out all the theoretical calculations. V. P. S. carried out scaling-up of the product and related spectroscopic characterization. The manuscript was written by P. M., A. K., J. D., A. K. P., A. D. and D. B. The overall discussion and manuscript content was directed by A. D., J. D. and P. M. All the authors agreed to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. M. acknowledges SERB grant no. CRG/2021/008494 for partial funding. We thank AIRF, JNU for the instrumentation facilities and DST-FIST for the single crystal X-ray facility at SPS, JNU. D. B. thanks SERB-NPDF, India for his Postdoctoral Funding. A. K. and J. D. acknowledge support from the Department of Atomic Energy (DAE), Government of India, under Project no. 12 R&D-TFR-5.10-0100. A. D. acknowledges TRC-DST and SERB grant no. CRG/2020/000301 for partial funding. We thank Madhuri (TIFR Mumbai) for TCSPC measurements and the National NMR facility (TIFR Mumbai) for characterization.Notes and references
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891 CrossRef CAS PubMed.
- S. Singh, W. Jones, W. Siebrand, B. Stoicheff and W. Schneider, J. Chem. Phys., 1965, 42, 330 CrossRef CAS.
- N. Periasamy and K. Santhanam, Chem. Phys. Lett., 1977, 51, 442 CrossRef CAS.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Chem. Rev., 2019, 119, 4261 CrossRef CAS PubMed.
- D. N. Congreve, J. Lee, N. J. Thompson, E. Hontz, S. R. Yost, P. D. Reusswig, M. E. Bahlke, S. Reineke, T. Van Voorhis and M. A. Baldo, Science, 2013, 340, 334 CrossRef CAS PubMed.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90 CrossRef CAS PubMed.
- M. J. Tayebjee, A. A. Gray-Weale and T. W. Schmidt, J. Phys. Chem. Lett., 2012, 3, 2749 CrossRef CAS.
- I. Paci, J. C. Johnson, C. Chen, G. Rana, D. Popović, D. E. David, A. J. Nozik, M. A. Ratner and J. Michl, J. Am. Chem. Soc., 2006, 128, 16546 CrossRef CAS PubMed.
- K. M. Felter and F. C. Grozema, J. Phys. Chem. Lett., 2019, 10, 7208 CrossRef CAS PubMed.
- A. Kundu and J. Dasgupta, J. Phys. Chem. Lett., 2021, 12, 1468 CrossRef CAS PubMed.
- A. J. Musser, M. Maiuri, D. Brida, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2015, 137, 5130 CrossRef CAS PubMed.
- E. Busby, J. Xia, Q. Wu, J. Z. Low, R. Song, J. R. Miller, X. Zhu, L. M. Campos and M. Y. Sfeir, Nat. Mater., 2015, 14, 426 CrossRef CAS PubMed.
- A. J. Musser, M. Al-Hashimi, M. Maiuri, D. Brida, M. Heeney, G. Cerullo, R. H. Friend and J. Clark, J. Am. Chem. Soc., 2013, 135, 12747 CrossRef CAS PubMed.
- A. Aster, F. Zinna, C. Rumble, J. Lacour and E. Vauthey, J. Am. Chem. Soc., 2021, 143, 2361 CrossRef CAS PubMed.
- J. C. Johnson, A. J. Nozik and J. Michl, Acc. Chem. Res., 2013, 46, 1290 CrossRef CAS PubMed.
- S. N. Sanders, E. Kumarasamy, A. B. Pun, M. T. Trinh, B. Choi, J. Xia, E. J. Taffet, J. Z. Low, J. R. Miller and X. Roy, J. Am. Chem. Soc., 2015, 137, 8965 CrossRef CAS PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617 CrossRef CAS PubMed.
- K. Krishnapriya, P. Roy, B. Puttaraju, U. Salzner, A. J. Musser, M. Jain, J. Dasgupta and S. Patil, Nat. Commun., 2019, 10, 1 CrossRef PubMed.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2018, 140, 10179 CrossRef CAS PubMed.
- E. Kumarasamy, S. N Sanders, M. J. Tayebjee, A. Asadpoordarvish, T. J. Hele, E. G. Fuemmeler, A. B. Pun, L. M. Yablon, J. Z. Low and D. W. Paley, J. Am. Chem. Soc., 2017, 139, 12488 CrossRef CAS PubMed.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2016, 45, 1542 RSC.
- K. Nagarajan, A. R. Mallia, V. S. Reddy and M. Hariharan, J. Phys. Chem. C, 2016, 120, 8443 CrossRef CAS.
- Y. Hong, J. Kim, W. Kim, C. Kaufmann, H. Kim, F. Würthner and D. Kim, J. Am. Chem. Soc., 2020, 142, 7845 CrossRef CAS PubMed.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. H. Sánchez, B. Zhang, Q. Xu, M. L. Steigerwald, S. Xiao, H. Li, C. P. Nuckolls and X. Zhu, J. Am. Chem. Soc., 2019, 141, 13143 CrossRef CAS.
- S. V. Bhosale, M. Al. Kobaisi, R. W. Jadhav, P. P. Morajkar, L. A. Jones and S. George, Chem. Soc. Rev., 2021, 50, 9845 RSC.
- N. Sakai, R. Bhosale, D. Emery, J. Mareda and S. Matile, J. Am. Chem. Soc., 2010, 132, 6923 CrossRef CAS PubMed.
- S. Wu, F. Zhong, J. Zhao, S. Guo, W. Yang and T. Fyles, J. Phys. Chem. A, 2015, 119, 4787 CrossRef CAS.
- A. Aster, C. Rumble, A.-B. Bornhof, H.-H. Huang, N. Sakai, T. Solomek, S. Matile and E. Vauthey, Chem. Sci., 2021, 12, 4908 RSC.
- H. Kar, D. W. Gehrig, N. K. Allampally, G. Fernández, F. Laquai and S. Ghosh, Chem. Sci., 2016, 8, 1115 RSC.
- V. L. Gunderson, A. L. Smeigh, C. H. Kim, D. T. Co and M. R. Wasielewski, J. Am. Chem. Soc., 2012, 134, 4363 CrossRef CAS.
- S. Kumar, J. Shukla, K. Mandal, Y. Kumar, R. Prakash, P. Ram and P. Mukhopadhyay, Chem. Sci., 2019, 10, 6482 RSC.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 130, 17361 CrossRef.
- S. Kuila, A. Ghorai, P. K. Samanta, R. B. K. Siram, S. K. Pati, K. S. Narayan and S. J. George, Chem.–Eur. J., 2019, 25, 16007 CrossRef CAS PubMed.
- H. F. Higginbotham, P. Pander, R. Rybakiewicz, M. K. Etherington, S. Maniam, M. Zagorska, A. Pron, A. P. Monkman and P. Data, J. Mater. Chem. C, 2018, 6, 8219 RSC.
- M. Hussain, J. Zhao, W. Yang, F. Zhong, A. Karatay, H. G. Yaglioglu, E. A. Yildiz and M. Hayvali, J. Lumin., 2017, 192, 211 CrossRef CAS.
- S. Guo, W. Wu, H. Guo and J. Zhao, J. Org. Chem., 2012, 77, 3933 CrossRef CAS PubMed.
- S. Wu, F. Zhong, J. Zhao, S. Guo, W. Yang and T. Fyles, J. Phys. Chem. A, 2015, 119, 4787 CrossRef CAS PubMed.
- Y. Wu, M. Frasconi, D. M. Gardner, P. R. McGonigal, S. T. Schneebeli, M. R. Wasielewski and J. F. Stoddart, Angew. Chem., Int. Ed., 2014, 53, 9476 CrossRef CAS PubMed.
- M. Ajayakumar, D. Asthana and P. Mukhopadhyay, Org. Lett., 2012, 14, 4822 CrossRef CAS.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. H. N. Sánchez, B. Zhang and Q. Xu, J. Am. Chem. Soc., 2019, 141, 13143 CrossRef CAS PubMed.
- S. L. Bayliss, A. D. Chepelianskii, A. Sepe, B. J. Walker, B. Ehrler, M. J. Bruzek, J. E. Anthony and N. C. Greenham, Phys. Rev. Lett., 2014, 112, 238701 CrossRef PubMed.
- E. Vauthey, ChemPhysChem, 2012, 13, 2001 CrossRef CAS PubMed.
- N. Banerji, A. Fürstenberg, S. Bhosale, A. L. Sisson, N. Sakai, S. Matile and E. Vauthey, J. Phys. Chem. B, 2008, 112, 8912 CrossRef CAS PubMed.
- A. K. Pal, K. Bhattacharyya and A. Datta, J. Chem. Theory Comput., 2019, 15, 5014 CrossRef CAS.
- E. A. Buchanan, Z. Havlas and J. Michl, Adv. Quantum Chem., 2017, 75, 175 CrossRef CAS.
- K. E. Brown, W. A. Salamant, L. E. Shoer, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2014, 5, 2588 CrossRef CAS PubMed.
- R. E. Cook, B. T. Phelan, R. J. Kamire, M. B. Majewski, R. M. Young and M. R. Wasielewski, J. Phys. Chem. A, 2017, 121, 1607 CrossRef CAS.
- C. B. Dover, J. K. Gallaher, L. Frazer, P. C. Tapping, A. J. Petty II, M. J. Crossley, J. E. Anthony, T. W. Kee and T. W. Schmidt, Nat. Chem., 2018, 10, 305 CrossRef CAS.
- Y. Gao, H. Liu, S. Zhang, Q. Gu, Y. Shen, Y. Ge and B. Yang, Phys. Chem. Chem. Phys., 2018, 20, 12129 RSC.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schartz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749 CrossRef CAS PubMed.
- N. Renaud and F. C. Grozema, J. Phys. Chem. Lett., 2015, 6, 360 CrossRef CAS PubMed.
- S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 14701 CrossRef CAS PubMed.
- W. E. Ford and P. V. Kamat, J. Phys. Chem., 1987, 91, 6373 CrossRef CAS.
- A. K. Le, J. A. Bender and S. T. Roberts, J. Phys. Chem. Lett., 2016, 7, 4922 CrossRef CAS PubMed.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120 CrossRef CAS PubMed.
- X. Zhao, Y. J. Bae, M. Chen, S. M. Harvey, C. Lin, J. Zhou, R. D. Schaller, R. M. Young and M. R. Wasielewski, J. Chem. Phys., 2020, 153, 244306 CrossRef CAS PubMed.
- E. A. Margulies, J. L. Logsdon, C. E. Miller, L. Ma, E. Simonoff, R. M. Young, G. C. Schatz and M. R. Wasielewski, J. Am. Chem. Soc., 2017, 139, 663 CrossRef CAS PubMed.
- K. Tajima, K. Matsuo, H. Yamada, S. Seki, N. Fukui and H. Shinokubo, Angew. Chem., Int. Ed., 2021, 25, 14060 CrossRef.
- S. Kumar, K. Yoshida, Y. Hattori, T. Higashino, H. Imahori and S. Seki, Chem. Sci., 2022, 13, 1593 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2171334 and 2171335. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc04187a |
| ‡ D. B. and A. K. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |