
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyoxometalates as chemically and structurally versatile components in self-assembled materials†
Yanting
Gao
 ab,
Manjiri
Choudhari
b,
Georgina K.
Such
*a and
Chris
Ritchie
*b
ab,
Manjiri
Choudhari
b,
Georgina K.
Such
*a and
Chris
Ritchie
*b
aSchool of Chemistry, The University of Melbourne, Parkville, Victoria 3010, Australia. E-mail: gsuch@unimelb.edu.au
bSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: chris.ritchie@monash.edu
First published on 22nd December 2021
Abstract
Polyoxometalates (POMs) are anionic molecular metal oxides with expansive diversity in terms of their composition, structure, nuclearity and charge. Within this vast collection of compounds are dominant structural motifs (POM platforms), that are amenable to significant chemical tuning with minimal perturbation of the inorganic oxide molecular structure. Consequently, this enables the systematic investigation of these compounds as inorganic additives within materials whereby structure and charge can be tuned independently i.e. [PW12O40]3−vs. [SiW12O40]4− while also investigating the impact of varying the charge balancing cations on self-assembly. The rich surface chemistry of POMs also supports their functionalisation by organic components to yield so-called inorganic–organic hybrids which will be the key focus of this perspective. We will introduce the modifications possible for each POM platform, as well as discussing the range of nanoparticles, microparticles and surfaces that have been developed using both surfactant and polymer building blocks. We will also illustrate important examples of POM-hybrids alongside their potential utility in applications such as imaging, therapeutic delivery and energy storage.
 From left to right: Yanting Gao, Manjiri Choudhari, Georgina K. Such, and Chris Ritchie | Ms Yanting Gao received her BSc (2017) in Material Chemistry from Sun Yat-sen University, China, and MSc (2019) in Chemistry from the University of Melbourne, Australia. She is currently a PhD student in the groups of A. Prof. Georgina Such and Dr Chris Ritchie. Ms Manjiri Choudhari completed an integrated Master's degree in chemistry from the Indian Institute of Technology, Kharagpur, in (2017) and subsequently joined the group of Dr Chris Ritchie as a PhD student in (2019). A. Prof. Georgina Such and Dr Chris Ritchie run research groups at the University of Melbourne and Monash University respectively, and share a mutual interest in the design and development of stimuli-responsive materials through the utility of diverse chemical building blocks for applications including biomedicine, controlled release and photochromism. |
1 Introduction
Polyoxometalates (POMs) are molecular anions composed of high oxidation state metal ions and oxide ligands. A broad selection of synthetic methodologies are available for their preparation which can involve either one metallic element to yield isopolyoxometalates, or multiple elements, at least one of which must be a metal in the case of heteropolyoxometalates. The expansive chemical diversity of POMs coincides with impressive variation in their size, charge, topology, and structure to provide an extensive library of solution-processable molecular metal oxides.1–5 Examples such as the Keggin ion [XW12O40]n− provide access to variants with analogous connectivity but varying in charge, while also providing a means to tune surface reactivity and electronic structure.6 Being anionic, the molecular metal-oxides require charge balancing counter cations which are often incorrectly assumed to be innocent spectator ions. In reality, they perform important roles such as perturbing solution state reaction equilibria to facilitate the access of otherwise inaccessible reactive intermediates alongside directing intermolecular interactions and consequently extended structure.7 As the majority of POMs are synthesised in aqueous media from simple water-soluble building blocks, the early literature is dominated by POM salts where H+ along with group one counter-cations such as Li+, Na+, K+, Cs+ and less commonly the alkali-earth cations balance the anionic charge of the POMs. Compounds of this type can be crystallised and their solid-state structures determined using single-crystal X-ray diffraction. Several decades ago Corigliano and Pope demonstrated that POMs could be transferred into aprotic, apolar solvents including hexane using appropriate phase transfer agents.8,9 This work signalled the beginning of a new vein of research where POMs could be studied in non-aqueous media, which in turn stimulated the invention of POM-based materials. This development catalysed the investigation of non-covalent interactions such as electrostatic, hydrophobic and solvophobic interactions between POMs and either cationic surfactants or polymers leading to the discovery of intriguing self-assembled structures instead of crystalline solids (Scheme 1). The solubility of these molecular metal oxides in organic solvents also made POMs amenable to chemistries that are inaccessible in aqueous media. Consequently, various methodologies to exploit the versatility of POM surface chemistry have been developed resulting in the establishment of so-called “POM platforms” (Fig. 1). These platforms are based upon the four dominant POM structural classes utilised in the synthesis of hybrid materials i.e. Anderson–Evans (P1), Lindqvist (P2), Keggin (P3) and Dawson (P4) (Fig. 1). Of note is that each of these platforms can be compositionally modified to develop a library of functionalisation protocols that provide methodologies by which a wide range of hybrid materials have been and continue to be produced.10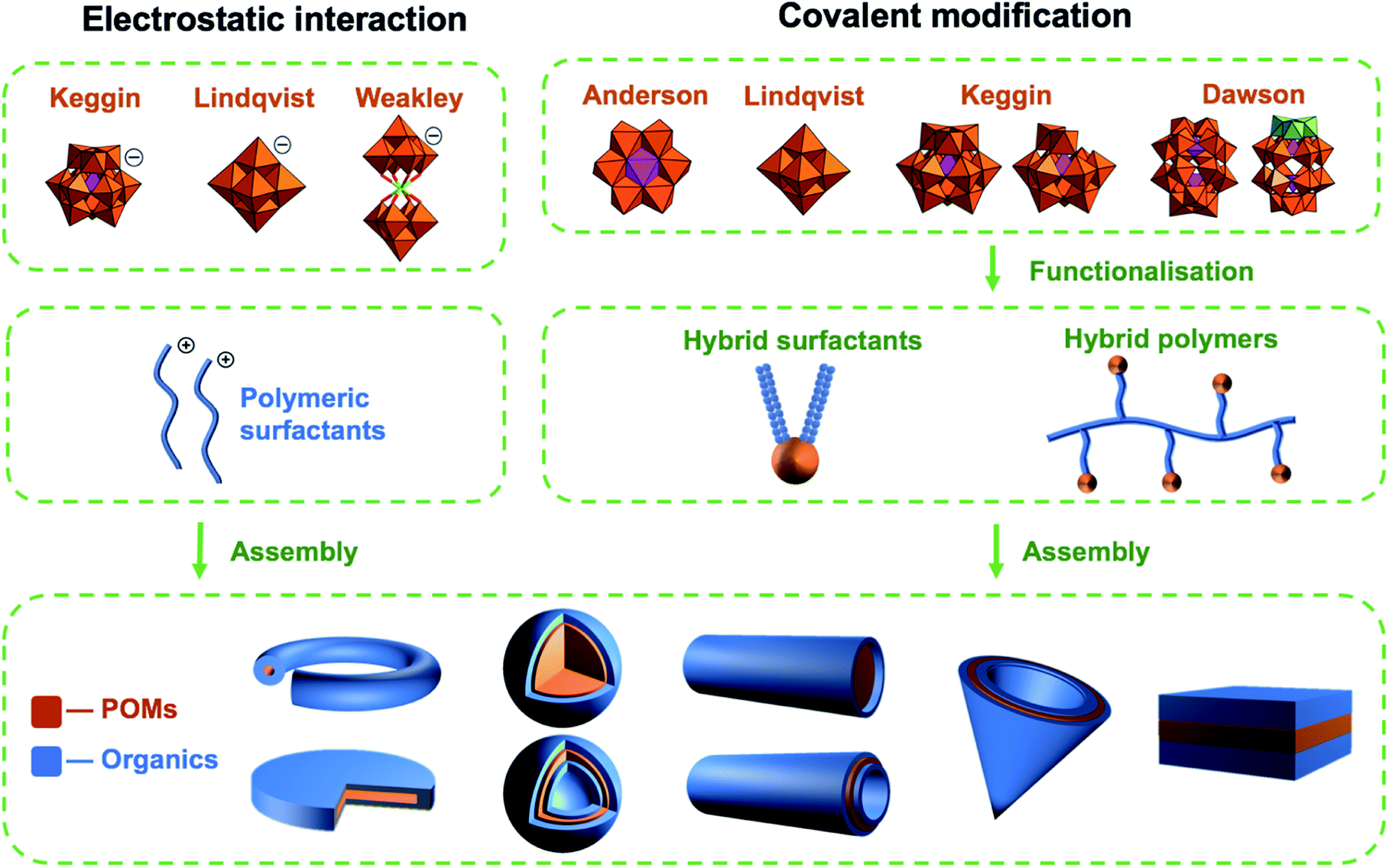 | ||
| Scheme 1 A graphical overview of the most frequently studied POMs, their functionalisation and self-assembled architectures. | ||
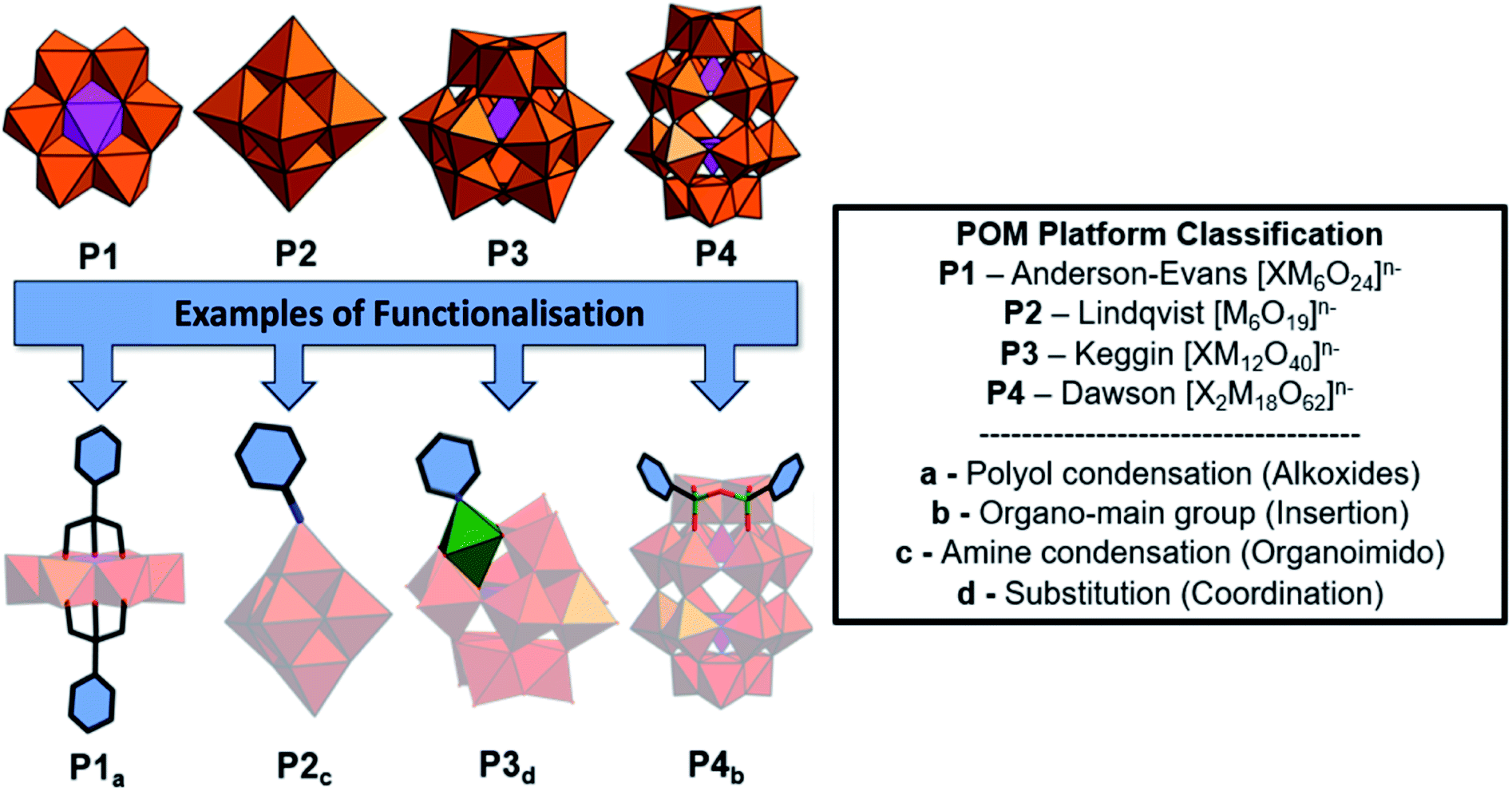 | ||
| Fig. 1 The dominant POM structural classes and most frequently utilised functionalisation methods are provided with graphical examples. The combinations provided are not exclusive e.g.P3 can be functionalised using all four methods through variations in the polynanions chemical composition. We will use a, b, c and d as subscripts to distinguish between each functionalization method. | ||
Extensive documentation of cluster stability alongside a demonstrated tolerance of organic reaction conditions including cross-coupling reactions (Suzuki–Miyaura, Heck, Buchwald–Hartwig) and polymerization approaches such as ring-opening metathesis polymerization, atom transfer radical polymerization, and reversible addition-fragmentation polymerization (ROMP, ATRP, and RAFT) has enhanced the ability to incorporate POMs into advanced materials. This work demonstrates the viability of hybrid POM species as reagents within complex reaction schemes that may be thought to be incompatible with the polyanions (see ESI – Table S1† and references therein). In particular, the use of polymers with controlled structure and composition such as those afforded by controlled polymerisation approaches offers the potential to precisely control the location of the POM within the structure. In addition, there is significant progress in developing advanced polymerisation techniques using robotics or flow chemistry, accelerating the rate at which libraries of hybrid materials can be designed and realised. However, this exciting research area is still in its infancy, with the combination of POMs with stimuli-responsive or cleavable polymers representing some of the most interesting prospects.
In this perspective, we will discuss the different strategies that have been used to design and prepare POM-containing materials with a focus on self-assembled substances that use organic surfactants and polymers (naturally occurring and synthetic). The discussion will highlight the synthesis of the POM hybrids and the assembly of these materials into nanoparticles, microparticles and surfaces. We will begin with a description of the most frequently used POM structural classes and the dominant POM platforms (Fig. 1) that have been developed to date, followed by compounds prepared via self-assembly utilizing non-covalent interactions. Subsequently, we will provide details of covalently functionalized POMs including amphiphilic species and polymerizable POM monomers. The remainder of the perspective will then focus on summarising advances regarding the applications of POM-based polymeric materials in terms of synthesis, structure, and their potential for applications in medicine and energy storage.
Historically, polyoxometalates belonging to the Anderson–Evans (P1), Lindqvist (P2), Keggin (P3) and Dawson (P4) (Fig. 1) structural classes have been most extensively studied, with well-established protocols for the diversification of their compositions and physicochemical properties being investigated. Each type of POM (P1–P4) can be thought of as a POM platform. The Keggin (P3) is probably the most intensely studied of all POM structural classes due to the immense variety of composition, charge, and redox states that are accessible with comparatively minor changes to the molecular structure. Constructed from four {M3O13} triads, the Keggin [XM12O40]n− is assembled by condensation of these building blocks in the presence or absence of an additional central templating heteroatom{X}. Rotational isomerism of the triads results in five possible isomers, all of which have been observed experimentally, with the Td symmetrical alpha isomer being the most prevalent. The tunability of anionic charge through choice of heteroatom, i.e. [α-XW12O40]n−; X, n = PV, 3; SiIV, 4; BIII, 5, with negligible change to the molecular structure has been studied extensively, including the impact of these changes on their self-assembly with surfactants and polymers. Another classical POM is the Wells–Dawson anion (P4) [X2M18O62]n− which has a more elongated structure than the Keggin (Fig. 1) and contains two templating heteroatoms. The anion can also be prepared as various rotational isomers. However, the [α-X2W18O62]6− species dominates the literature due to its stability. The Lindqvist anion (P2) is the most intensely studied isopolyanion and is formed via the condensation of six {MoO6} octahedra to yield [Mo6O19]2− with a μ-6 oxo ligand located at the centre of the cluster, templating the Oh symmetric molecule. The Anderson–Evans anion (P1) is composed of {MoO6} or {WO6} octahedra surrounded by a central heteroatom forming planar arrangement with D3d symmetry. The general formulation of Anderson–Evans can be stated as [Hy(XO6)M6O18]n−, where y = 0–6, n = 2–8, M = addenda atoms (MoVI or WVI), and X = central heteroatom. There are several more relevant plenary (defect-free) POM structural classes that could be described herein, however, we have limited our discussion to these key POM platforms as they represent the majority of work in this area. Beyond the selection of templating heteroatoms within the POM metal-oxide framework, the composition of plenary POMs can be selectively altered using controlled base mediated degradation.11,12 This process provides access to lacunary (defect-containing) species with increased basicity at the vacancy, introducing site-specific reactivity (Fig. 1). The reaction of these species with a variety of elements including transition metals, main group elements, and organic components yields compounds with site-specific functionalization. Depending on the choice of platform and preferred post-synthetic approach, the number of coordination sites or covalent modifications can be controlled providing a means to investigate the impact of these changes on molecular and bulk properties.10
2 Electrostatic interaction
Non-covalent interactions between POMs and small inorganic cations with well-defined point charges are dominated by ionic interactions and contribute to high lattice energies and the facile crystallisation of such materials.7 Replacement of these small cations by comparatively large organic species dramatically alters the nature of the non-covalent intermolecular interactions between oppositely and like-charged ions. Consequently, the stability of such compounds is afforded by the cumulative energetics of numerous interactions (van der Waals, hydrophobic, hydrogen bonding, dipole–dipole etc.), which in many instances results in a very flat energy surface.13 Ultimately this manifests in dynamic behaviour on the nanoscale where structures respond to various inputs such as changes in solvent polarity, electrochemical potential, pH and light.14–162.1 Surfactant encapsulated clusters
Owing to the expansive selection of POMs and surfactants that can be combined, ranging from the small (0.8 nm) dianionic [Mo6O19]2− (P2) to the large highly charged POM [(Mo176O528)(H2O)80]32−, the volume of reported compounds prohibits an exhaustive review.7,17 Selected examples of surfactant encapsulated clusters (SECs) will be discussed with an emphasis on dynamic self-organization.The polar cationic surfactant, dimethyl–dioctadecylammonium (DODA) has proven effective at replacing the alkali cations of polyanions to yield amphiphilic salts.18 As mentioned earlier, the molecular composition of particular classes of POMs such as the Keggin anion can be controlled with relative ease, resulting in a change in net charge but with negligible change to the size and shape of the molecule. This molecular control enabled studies, which focussed on understanding the impact that the change in charge has on the attractive and repulsive intermolecular interactions between the phosphotungstate [PW12O40]3− and silicotungstate [SiW12O40]4− (P3) anions and DODA (Fig. 2).14,19 Interestingly, dissolution of both of these compounds in the same solvent system (chloroform–butanol 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v), resulted in dramatically different self-assembled structures that were studied in both solution and solid states.14,19,20 The most probable explanation for the different assemblies is the number of surfactant molecules associated with the POM surfaces. A common feature of both systems is the regular lamellar structure of the composites with layer spacings of 2.9–3.2 nm. The self-assembled architectures are susceptible to solvent composition, with dynamic interconversion between disk and cone-like structures reported when the solvent system is changed to (chloroform–butanol 2:1 v/v). This process is reversible and has been ascribed to a shift in the thermodynamic equilibria of the various non-covalent interactions.14
:1 v/v), resulted in dramatically different self-assembled structures that were studied in both solution and solid states.14,19,20 The most probable explanation for the different assemblies is the number of surfactant molecules associated with the POM surfaces. A common feature of both systems is the regular lamellar structure of the composites with layer spacings of 2.9–3.2 nm. The self-assembled architectures are susceptible to solvent composition, with dynamic interconversion between disk and cone-like structures reported when the solvent system is changed to (chloroform–butanol 2:1 v/v). This process is reversible and has been ascribed to a shift in the thermodynamic equilibria of the various non-covalent interactions.14
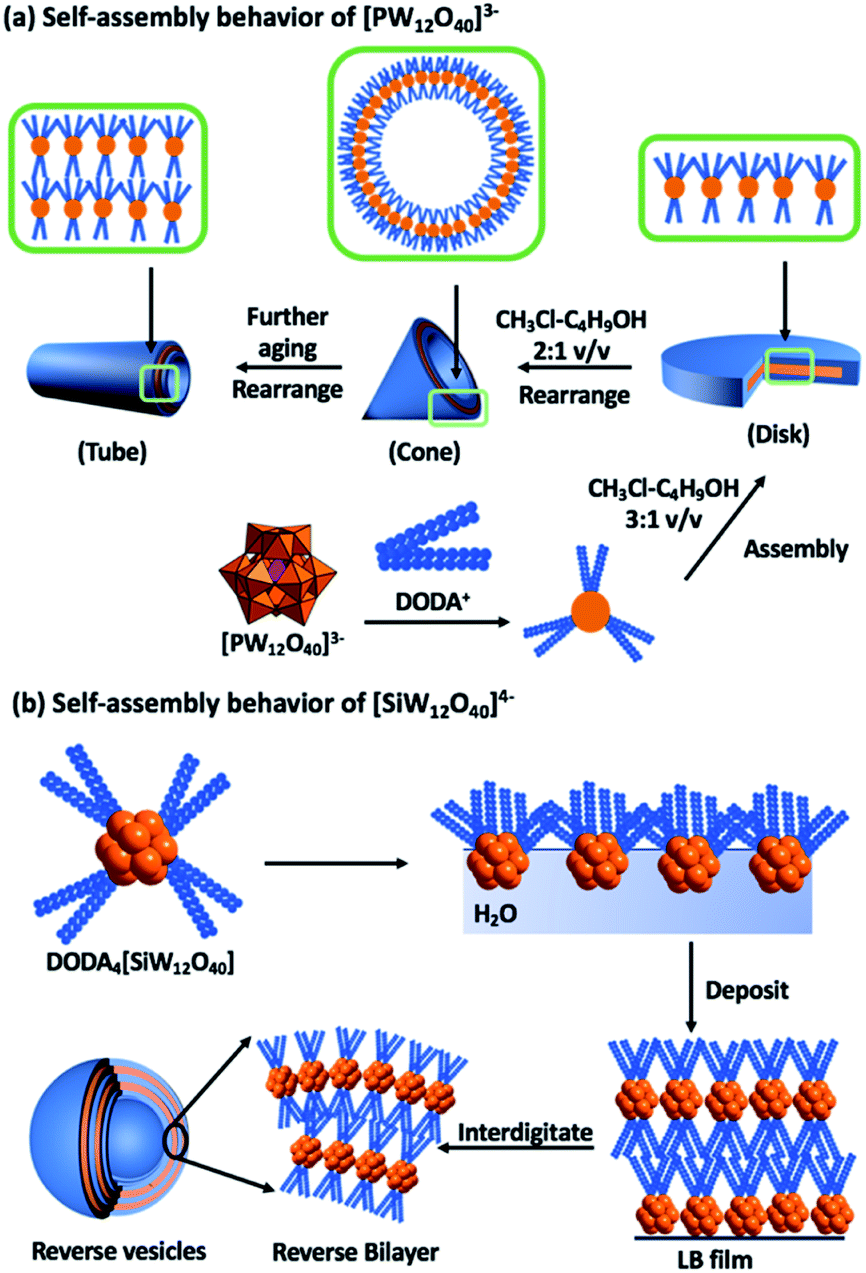 | ||
| Fig. 2 Graphical representation of the dynamic self-assembly behaviour of dimethyl dioctadecylammonium (DODA) – Keggin assemblies and the macroscopic features of the resulting materials. | ||
To establish the SEC self-assembly mechanism, Wu and co-workers carried out a series of experiments using the dendritic surfactant, N-(3,5′-bis[3,5-bis(heptyloxy)benzyloxy])benzyl-N,N,N-trimethylammonium (D). They associated D with (P3) and [P2W15O56]12− (P4) species POMs with varying charges controlled by their heteroatoms. Observations arising from these experiments include the self-assembly of vesicular aggregates with lamellar structures in acetone when the total lateral surface covered by cationic surfactants is less than 1/3 i.e. (P3)4−/5−. An increase in surface coverage for more highly charged species of similar size negates aggregation in solution but favours columnar assemblies on deposition. This leads to the formation of hexagonally packed structures for anions such as the monolacunary derivative D7Na[SiW11O39]. The main driving force for these differences was attributed to the phase separation of hydrophilic POMs and hydrophobic cationic dendrons while reaching an energy minimum through efficient packing.21 In contradiction to the above results, increasing the POM surface coverage by surfactants bearing flexible long-chain hydrocarbons such as DODA does not result in a transition from the lamellar structure due to more orientations of the surfactant that can be effectively arranged around the polyanion20,22 These examples stress the importance of the molecular packing parameter in the structural evolution observed for such self-assembled systems.23
In 2000, Kurth et al. described the self-assembly of an SEC from, (DODA)40(NH4)2[(H2O)n ⊂ Mo132O372(CH3COO)30(H2O)72] (n ≈ 50), which consists of a giant polyoxometalate core covered by DODA cations to form a hydrophobic shell.24 These species form stable and reproducible Langmuir monolayers at the air–water interface as confirmed by X-ray reflectance measurements.25 A subsequent study by Polarz et al. using the same polyanion in addition to (Cat)32[(MoO3)176(H2O)80]; cation – DDMA (didodecyl dimethylammonium)/DOMA(dioctadecyl dimethylammonium) discovered that the POM-surfactant building block could control the symmetry of the resulting self-assembled 3D superstructures. For example, the replacement of the charge-balancing protons of H32[(MoO3)176(H2O)80] and the subsequent ion-pairing with DOMA cations yields disk-shaped 3D structures while the same displacement of ammonium cations from (NH4)42[Mo132O372(CH3COO)30(H2O)72] yields cubic 3D assemblies. These examples, show how different POMs can alter the structure of the resulting assemblies.26
Recently, Yu et al. reported a tri-stimuli responsive supramolecular assembly consisting of ethyl-4′-(trimethylaminohexyloxy)azobenzene (ETAB) and luminescent POM [Eu(W5O18)2]9−. This assembly was found to be responsive to UV, pH, and Cu2+. As reported, the emission intensity of Eu-POM in aqueous media is very low due to outer-sphere quenching, however, when the ETAB cations displace the Na+ ions, the hydration associated with the sodium ions is removed, and the emission intensity of Eu-POM is enhanced due to the increased hydrophobicity of the ETAB(Eu-POM) assemblies. The morphology of the ETAB/Eu-POM assemblies was not impacted by irradiation. However, the mean size of the spherical aggregates was decreased and accompanied by an increase in polydispersity. Upon prolonged UV irradiation, the fluorescence of the assemblies was quenched. This assembly was also found to be pH-responsive with this responsiveness arising from the formation of new hydrogen bonds with the POM surface (W–O⋯H⋯O–H) which were monitored by UV-Vis at a selection of pHs. Finally, the addition of Cu2+ was shown to quench the fluorescence when it coordinated to the cluster surface.16
2.2 POM-based ionic liquids
The definition of an ionic liquid is a salt that exists in the liquid state below 100 °C, with the research topic dominated by the modification and functionalization of organic cations, such as imidazolium and quaternary alkylammonium. Comparatively the anionic component has received less attention with the exploration of POMs as the anions only commencing recently to yield (POM-ILs).In important work, the catalytically active [W2O3(O2)4]2− anion was paired with protic cations (N-dodecylimidazolium and N-hexylimidazolium) and two aprotic imidazolium (1-hexyl-3-methyl-imidazolium and 1-dodecyl-3-methyl-imidazolium) to synthesize four kinds of (POM-ILs). Among them, N-hexylimidazolium peroxotungstate showed the best catalytic performance for the epoxidation of cyclooctene in different solvents. Moreover, this catalyst precipitated at the end of the reaction, making it is easy to separate and reuse.27 In 2013, Li and co-workers paired the cations [(n-C8H17)3NCH3]+ and [(n-C12H25)3NCH3]+ with [PO4(MoO(O2)2)4]3− and [PO4(WO(O2)2)4]3− respectively, to obtain a series of POM-ILs. This hybrid material not only acted as a catalyst but also performed as an extractant for the oxidation of dibenzothiophene (DBT). After reacting with H2O2 to generate active peroxo species Mo(O2), these POM-ILs removed almost 100% of sulfur at 60 °C after 2 h (500 μg mL−1 DBT).28 The group of Ganglin also reported four types of POM-ILs that were used as the catalyst for H2O2-involved oxidation desulfurization. They expanded their previous reported catalyst to a POM-IL system and synthesized 1-butyl 3-methyl imidazolium-based [PW11O39TM(H2O)]n− (TM = MnII, CoII, NiII and ZnII). The CoII mono-substituted POM-IL exhibited the highest catalytic efficiency, removing 99.8% dibenzothiophene (500 ppm) at 50 °C after 1 h.29
In addition to their catalytic performance, the conductivity of POM-ILs has also been studied. Qingyin Wu and co-workers used tetraalkylammonium bromide and tetraalkylphosphonium chloride together with (P3) and (P4) – [PW9V3O40]6−, [P2W16V2O62]8−, [PW10V2O40]8−, to prepare a series of POM-ILs by exchange of the halide anions by the POMs. Findings showed that the phosphonium-based POM-IL was more thermally stable with enhanced ionic conductivity, while the physicochemical properties of the POM-ILs were correlated with the POM size with larger anions possessing higher ionic conductivity, thermal performance, and oxidation properties, along with a lower phase inversion temperature. Both of these performance improvements may be due to their weaker coulombic interactions.30,31 This group also reported a series of hybrid gel electrolytes derived from POM-ILs including the pairing of the 1-(3-sulfonic group)propyl-3-methyl imidazolium (MIMPS) cation with [PW11VO40]4−, [SiW11VO40]5−, [SiMo11VO40]5− and [SiW9V3O40]7− (P3) respectively. All of these hybrids exhibited temperature-dependent conductivity, showing better performance above the phase transition temperature. In their studies, (MIMPS)4[PW11VO40] showed the highest conductivity 8.60 × 10−2 S cm−1 at 83 °C and 80% relative humidity. The chemical composition of POM also affected the redox behaviour of POM-ILs. The Mo-containing POM-ILs showed greater oxidizability than the W-containing ones, and POM-ILs with more vanadium also enhanced the oxidizability.32,33
Recently, as more attention has been paid to POM-ILs, their potential applications in different fields have been gradually explored. In 2014, Streb and co-workers used room-temperature POM-IL composed of [SiW11O39TM(H2O)]n− (TM = CuII, FeIII) (P3) and (CnH2n+1)4N+ (n = 7–8) to protect copper metal disks against acid corrosion. The coating recovered from mechanical damage within one minute without performance loss and can be removed by organic solvent.34 In other work, (CnH2n+1)4N+ (n = 6–8) salts of unsubstituted lacunary (P3) anions [α-SiW11O39]8− were immobilized on commercial porous silica. These materials were used for the filtration of polluted water and showed antimicrobial behaviour against E. coli, P. aeruginosa, and S. aureus.35 Transition-metal functionalized POM-IL was also applied to remove Cd2+ and Pb2+ metal ions from water. [PW11O39TM(H2O)]n− (TM = MnII, FeII, CoII, NiII, CuII, and ZnII) was mixed with tetraoctylammonium to prepare the POM-IL with melting temperatures less than 100 °C, these materials extracted 99% metal ion from the aqueous phase at 80 °C under constant stirring.36
2.3 Additives in polymeric materials
As an extension of the surfactant-based approaches discussed earlier, the loading of POMs as additives into positively charged polymeric building blocks has been a long-standing method for the preparation of hybrid materials. While this strategy readily provides access to hybrid materials, there are also associated drawbacks of this non-covalent association such as the limitation on polymer composition as it needs to include sites for protonation, cargo leakage as a result of anion exchange and poor stability under certain conditions (high ionic strength, high pH and humidity).However, the incorporation of non-specific POMs has been found to offer interesting control over the self-assembly properties of the hybrid materials. Noritaka and co-workers reported a series of micelles and vesicles (SVP-1 to SVP-7) formed from poly(styrene-b-4-vinyl-N-methylpyridinium iodide) (PS-b-P4VMP) and [PW12O40]3− (P3), the morphology of which can be controlled by varying the POM loading. The rationale for this morphology change is the increased hydrophilic volume and interfacial tension that arises from the inclusion of more polyanions.37 Other interesting polymeric architectures synthesized by the same approach were reported by this group. One example included the combination of POM-based supramolecular star polymers (PSPs) formed by the electrostatic interaction between [Mo132O372(CH3COO)30(H2O)72]42− with quaternary-ammonium terminated PS with POM-hybrids based on the synthesis of PS-b-P4VMP with [PW12O40]3− clusters (SVP-6).38 This work demonstrated the particle structure could be tuned based on solvent choice. It was shown that the SVP-6 was hosted within the PSP structure with high methanol concentration, allowing multilayer POM hybrids to be formed. Another study demonstrating morphology control was reported by Xinhua and co-workers in 2018 using the triblock copolymer poly(ethylene oxide)-b-styrene-b-2-(dimethylamino)ethyl methacrylate (PEO-b-PS-b-PDMAEMA) with the morphology being varied between micelles, rods, toroids and vesicles by tuning the molar ratio of POM. In this study, the authors postulated strong interactions between [PMo12O40]3− (P3) and PDMAEMA in the core of the structure which was then followed up by using the luminescent [Eu(W5O18)2]9−, with the Eu3+ centred emission being strongly dependant on the environment of the polyanion and highly sensitive to changes in pH suggesting the interactions between PDMAEMA and [Eu(W5O18)2]9− are of central importance to the nanoparticle structure.39
[Eu(W5O18)2]9− hybrids were again investigated through the formation of core–shell nanoparticles by simply mixing with the cationic copolymer PEO-b-PDMAEMA in an aqueous solution (Fig. 3). Enhanced Eu3+ luminescence (20-fold) was evident on association with the polymer by reducing luminescence quenching by vibronic coupling with outer-sphere water molecules. Unfortunately, the hybrid material was unstable in high ionic strength media due to the weakened electrostatic interactions between the POM and polymer, which limited the application of these compounds in the field of bio-imaging and bio-detection.15 In related work, Li et al. used this method to successfully prepare PS-based star polymers assembled around a core of [Eu(W5O18)2]9−via post-association polymerisation. The extent of polymerisation was shown to impact the POM sensitized emission.40
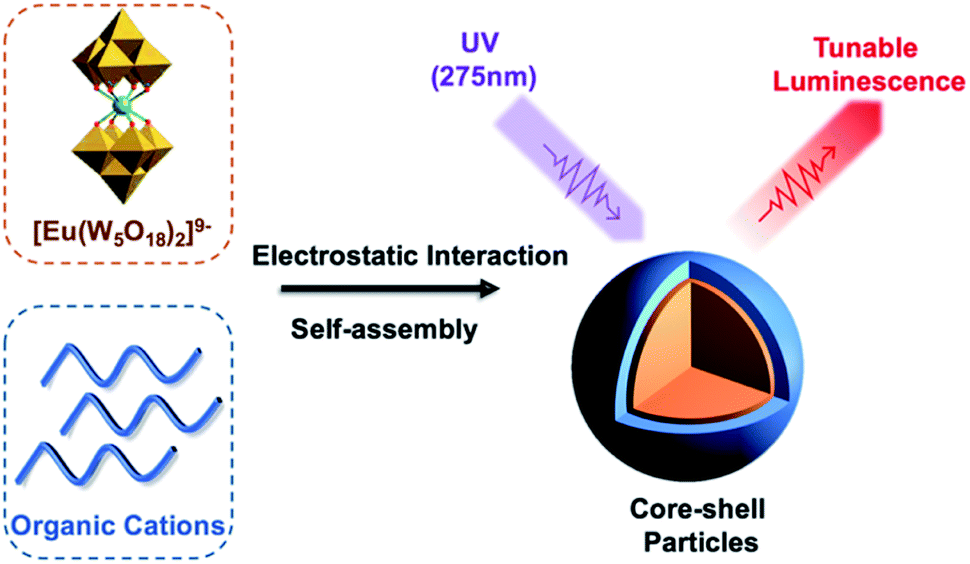 | ||
| Fig. 3 Graphical representation of the Eu-containing core–shell nanoparticles with tunable luminescence. | ||
The group of Haolong Li used [PW12O40]3− and [SiW12O40]4− (P3) as electrostatic crosslinkers to form bicontinuous41 inverse hexagonal42 and lamellar morphologies43 with the positively charged polymer poly(styrene-b-2-vinylpyridinium) (PS-b-P2VP). These nanocomposites exhibited enhanced proton conductivity and mechanical strength as the result of using macroions as functional additives. In another study, the same heteropolyacids (P3) were doped into a perfluorosulfonic acid polymer to investigate their proton conductivity. The activation energy for proton conduction, as measured by pulsed gradient spin echo NMR, was lower than that of the undoped materials, with a practical proton conductivity of 113 mS cm−1 being measured at 100 °C and 80% relative humidity. By comparison, proton conductivity of commercial proton exchange membrane Nafion® 212 was lower than 100 mS cm−1 under the same conditions.44,45 Unfortunately, stability issues were observed due to the leaching from the membrane in the presence of water. This was of course problematic as it severely hampered the performance of the materials but in turn, stimulated research into covalent methodologies to address these issues (see Section 3.2.1.).
3 Covalent systems
3.1 Organic-modified POM platforms
While strategies such as changing the heteroatom(s) tune the net anionic charge of heteropolyoxometalates, and consequently the non-covalent interactions of amphiphilic POM based materials, this approach offers minimal control regarding the spatial separation of hydrophobic or electrostatic interactions within these materials which is controlled by the nature of the cation. An alternative approach is the covalent functionalisation of POM surfaces with hydrophobic moieties with systematic studies being possible through the POM platforms. The choice of platform (Fig. 1) provides a means to control the POM: hydrophobe ratio and orientation, with in some instances asymmetric functionalisation of the POM surface (see ESI; Table S1†).Recently, the Anderson–Evans polyanion (P1) has been extensively derivatized by organic functionalities, with protonation of the six basic μ3 oxo ligands surrounding the central MnIII ion in [H6MnMo6O24]3− enabling the condensation of appropriate triols. Trishydroxylmethylamine is readily attached to both the hexagonal faces of the cluster to yield the bifunctionalised species [MnMo6O18{(OCH2)3CNH2}2]3− (P1a) as reported by Gouzerh.46 The resulting divergent primary amines present an opportunity for post-synthetic functionalisation of the polyanion, with those discussed herein representing only a small cross-section. This approach has also been utilised for other POM platforms such as (P3a) and (P4a) for which the protonation pattern of their surfaces is well-established.47–49
Generic amide formation conditions are well-tolerated by the Mn-Anderson, with the covalent attachment of long-chain aliphatics by Cronin, Wu, and Mialane yielding [MnMo6O18{(OCH2)3NH–CO–(CH2)n−2–CH3}2]3− (n = 16, 18) (P1a) amphiphiles, where the hydrophilic POM is sandwiched between the hydrophobic groups. The DOMA salts of (P1a) showed excellent solubility in chloroform and a relatively low value of transition temperature (Tc) during phase transition.50 The addition of a more polar solvent, i.e., in chloroform:acetonitrile (MeCN) (6:1), increases the interaction between cations, which increases Tc. The tetrabutylammonium (TBA) salts of (P1a) show uniform rod-like morphologies whereas DOMA salts form sea urchin-like morphologies. These are very different from the “onion” or “honeycomb” like morphologies of DOMA SECs described in the previous section.14,19 In further investigations, it was found that TBA salts of (P1a) (Mn–C16) are soluble in both MeCN and a MeCN/water mixed solvent system. In the mixed solvents, (Mn–C16) slowly (∼57 days) assemble into membrane-like vesicles when the amount of MeCN is controlled to between (35–60 v%). The formation of vesicles can be accelerated by the elevation of temperature but has an obvious effect on the size of vesicles. When the alkyl chain length was reduced from 16 to 6, vesicle formation was slower with fewer assemblies observed using dynamic light scattering analysis. This observation was attributed to the greater solubility of the short-chain alkyl derivatives in MeCN.51 Furthermore, reverse vesicle formation of (P1a) (Mn–C6 and Mn–C16) in organic medium i.e. MeCN/toluene mixed solvents were demonstrated. In this case, elevating the temperature accelerates the assembly process, while the alkyl chain length has only a minor influence on the vesicle size. There was also no obvious lag time before the formation of vesicles compared to the initiation of the process as noted for polar solvents (MeCN/water), mentioned above. The size of the reverse vesicles is quite sensitive to the MeCN/toluene ratio; therefore, vesicle size can be controlled by changes in solvent polarity.52
As an extension of these studies, the impact of asymmetric functionalization of (P1) was investigated.53 This required the use of reverse-phase chromatography to overcome the challenge of obtaining analytically pure compounds from statistical mixtures arising from the chemical equivalence of the POM protonation sites. In this approach, functionalization of Mn-Anderson was obtained by reacting [Mo8O26]3−, Mn(OAc)3, and tris(hydroxymethyl)methane derivatives (Fig. 4). The asymmetrically functionalized species (P1a) with (HOCH2)3CNH–CH2–C16H9 and (HOCH2)3–C–C9H17 functional groups resulted in high-aspect-ratio anisotropic nanostructures due to self-recognition of aromatic and aliphatic moieties on the POM surface.54 Later, Mialane et al. presented a different approach where they grafted spiropyran moieties onto [MnMo6O18{(OCH2)3CNH2}2]3− using peptide coupling.55 Following this synthetic route symmetrically and asymmetric functionalized Mn-Anderson can be obtained by the appropriate stoichiometry of POM and spiropyran. The asymmetrically spiropyran functionalized Mn-Anderson has a free reactive primary amine on one face of the polyanion, which was used as a precursor to further construct Mn-Anderson with a long alkyl chain (C16H32O2) on one side and spiropyran moieties on the other side.56 This asymmetric Mn-Anderson self-assembled into vesicles in polar solvents and reverse vesicles in apolar solvents. Additionally, the assembly and disassembly of the nanostructures were shown to be controlled by light with the illumination of the sample by UV light (λ = 365) driving assembly while exposure to visible light promoted disassembly. This phenomenon accompanied the photochemical conversion of the spiropyran to its charge-separated merocyanine form, triggering the aggregation while the thermally activated back-reaction resulted in re-equilibration and re-assembly.
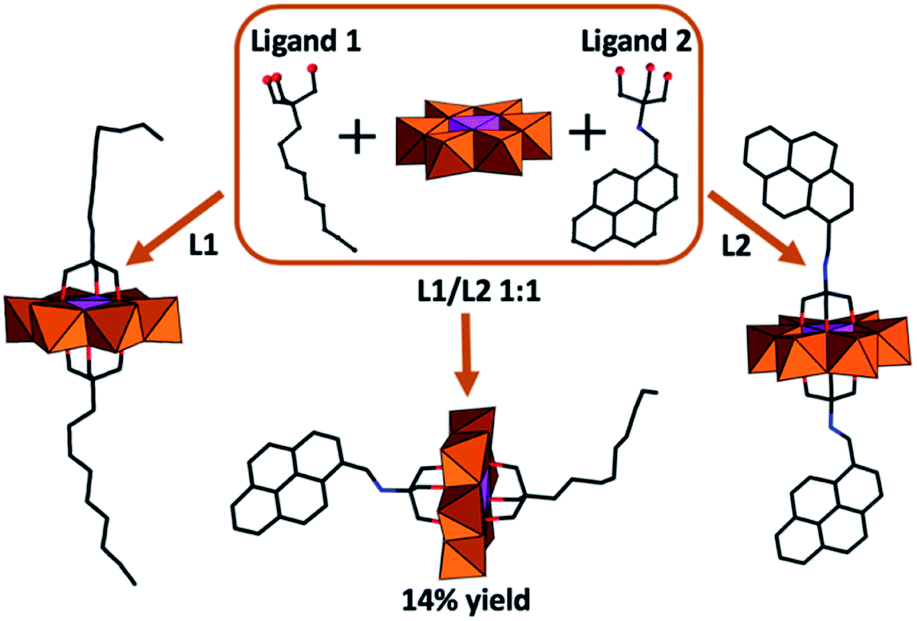 | ||
| Fig. 4 Graphical representation of the symmetrically and asymmetrically functionalised (P1a) obtained from a stoichiometric mixture of ligands and POM precursors. | ||
Unlike Anderson–Evans (P1), the protons associated with heteropolyacids (HPA) such as H3[PW12O40] (P3) were de-localized prohibiting the controlled condensation of alcohols to its surface. By implementing the well-defined acid–base chemistry of [PW12O40]3−, its controlled conversion to the mono-lacunary species [PW11O39]7− resulted in a marked increase in basicity and nuclearity of the oxide ligands located at this site.57 The introduction of main-group organic moieties at lacunary sites of different POMs such as phosphonate or siloxane has been explored by various groups.58–62 In one of the early reports from Thouvenot et al., the introduction of organophosphonic acids (RPO(OH)2) to [γ-SiW10O39]8− resulted in the formation of [γ-SiW10O39(RPO)2]4− (P3b), where R could be H, Et, t-Bu, n-Bu, Ph.59 Similar to this approach, the addition of siloxane to phosphotungstates POMs has also been reported. Addition of (EtO)3SiR with [PW11O39]7− affords disubstituted [PW11O39 (SiR)2O]3− (P3b) where R = Et, (CH2)nCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (n = 0, 1, 4).63
CH2 (n = 0, 1, 4).63
In 2010, Polarz utilised this methodology initially developed by Thouvenot and Proust to covalently graft long-chain alkoxy-silanes into the monolacunary site of [PW11O39]7−.64 This process enabled the synthesis of [PW11O39(SiCn)2]3− (n = 8, 12, 18) (P3b) as MeCN soluble TBA salts, which were subsequently converted to Na+ salts using ion-exchange chromatography. The cation exchange step has a significant impact on the self-assembly properties of the molecular hybrid as the cations are located around the polyanionic headgroup, with hydrophobic quaternary ammonium cations reducing the surfactants amphiphilic character. Liquid crystalline properties were observed for the compounds regardless of cation however the materials' structure was impacted by the nature of the cation and the aliphatic chain length (P3b). (C16, C18) compounds form hexagonally packed cylinders with the lattice constant increasing from (C16) – 4.62 nm to (C18) – 5.37 nm in line with what was expected for the increased hydrocarbon chain length. Dispersion of the sodium salts in water yielded micelles where the hydrodynamic radius of the nanoobjects scale with the alkyl chain length.
Using the same POM platform (P3), the bola-amphiphile (bola = hydrophobic skeleton covalently attached to two hydrophilic groups) H3[PW11O39(SiC11H22NH3Cl)2O] (P3b) was prepared with the terminal methyl groups of the previously discussed compounds being replaced by a primary amine.65 In general terms, the self-organization properties of bolas are sensitive to the nature of the hydrophilic headgroups with asymmetrical bolas tending to form small vesicles with an unsymmetrical monolayer membrane. As an extreme variant [PW11O39(SiC11H22NH3Cl)2O]3− follows this behaviour yielding vesicles with (DH – hydrodynamic diameter) of 16 nm at room temperature on dispersion in water. The small size of the monolayer lipid membrane (MLM) vesicles is attributed to the size disparity between the bola primary ammonium and POM head groups which results in highly curved interfaces with the POM located on the exterior of the assembly. Remarkably, the critical aggregation concentration of the bola was determined as 3 × 10−6 mol L−1, which is several orders of magnitude lower than typical micellar systems. When considering the resurgence of interest in POM based materials as potential host–guest systems, the integrity of the self-assembled structures in highly dilute systems is significant. Finally, the MLM was found to have extremely low permeability for a selection of guests with the successful entrapment and retention of hydrophilic, hydrophobic and ionic species that could be released using a pH trigger. Subsequent studies using the same POM platform have explored the rich coordination and electrochemical properties of the POM head group where grafting of metal ions and electrochemical switching results in tuneable readouts from the self-assembled structures via intermolecular interactions with guest molecules such as Förster Resonance Energy Transfer.
A third POM platform that has been studied in detail is that of the vanadium substituted Dawson polyanion [P2V3W15O62]9− (P4). The trilacunary POM precursor [P2W15O56]12− can be routinely synthesised in gram quantities, while a subsequent stoichiometric reaction with sodium metavanadate yields [P2V3W15O62]9−.66,67 The increased basicity of the bridging oxo ligands within the {V3O13} cap results in the site-specific and reliable condensation of tris(hydroxymethyl) containing organics. Over several years the groups of Cronin and Liu studied different derivatives reporting the impact of solvent polarity, POM: hydrophobe aspect ratio, cations and pH on the self-assembly behaviour of these materials. In one of the initial studies, Cronin and Liu et al. reported nanosized (3.4 nm) hybrids (P4a) starting from [P2V3W15O62]9− (P4) and linear bis(Tris) ligands. These hybrids can self-assemble into vesicles with an average size of 60 nm in a water/acetone mixture (1:1). The vesicle formation of this hybrid is rare due to having large polar head groups like POM, and the organic linker is closely packed, which was not observed previously.68 Further, Cronin and Liu et al. provided systematic studies on hybrids starting from different bis(tris) ligands and (P4). This study investigated the effect of varying tris ligands on the self-assembly process, assembly sizes, stability, thermodynamic properties, and phase behaviour. The hydrophobicity of the organic linker also determines the packing of monolayers of nanohybrids at air/water interfaces, i.e., in the case of a more hydrophobic linker, the monolayer becomes ordered at the interface.69 These investigations provide guiding principles to predict the self-assembly behaviour of this type of (P4a). Therefore a family of hybrid surfactants can be constructed, providing an opportunity to expand POM applications in organic media. In later work, the nanohybrid starting from [P2V3W15O62]9− (P4) with a 15-carbon alkyl chain was synthesized. Instead of two polar heads (POM) and bis tris ligand, this hybrid (P4a) has a single polar head group and a tris organic linker attached to it. This hybrid assemble into vesicles in polar solvents such as acetone and MeCN. The hydrodynamic radius of vesicular structures at pH 1.5 is 80 nm which decreases to 64 nm at pH 5 and sharply decreases to 38 nm at pH 12. The decrease in the vesicular structure's size can be attributed to the deprotonation of polar head groups leading to an increase in total net charge, which increases the repulsion between the polar head group and aggregates. This increased repulsion results in polar head groups and aggregates, leading to the formation of smaller vesicles.70
In a different approach, researchers designed heteroclusters composed of a pair of dissimilar clusters connected by various organic linkers. The two clusters chosen were a [P2V3W15O62]9− (P4) and a polyhedral oligomeric silsesquioxane. It was demonstrated by transmission electron microscope (TEM) analysis that these materials (P4a) could form hybrid cubosomes in a four-stage process.71 Another interesting asymmetric Wells–Dawson hybrid POM was synthesised containing two organophosphate moieties with contrasting properties, one a chelating metal-binding group and the other a long aliphatic chain that facilitated self-assembly.72 The approach to synthesise these materials (P4d) was simple and is thus an attractive strategy to build libraries of hybrid POM materials. These asymmetrical POMs formed micelles in a water–MeCN mixture (9:1 v/v) of approximately 6 nm. The authors demonstrated they could complex Fe2+ to form dimeric structures; however, these structures were less uniform.
In addition to the three platforms mentioned earlier in this section, a few examples are reported in the literature where covalent functionalization of the Lindqist platform (P2) has been reported. Like the Anderson–Evan platform (P1), covalent functionalization of [V6O19]2−/3− (P2) can be achieved using trishydroxylmethylamine to isolate hexavanadate–organic hybrids (P2a). The self-assembly behaviour of these amphiphilic hexavandate hybrids has been studied based on factors such as counter cations, the organic linker attached to the vanadate, and pH. Liu and Wei et al. reported the vanadate hybrid where two C18 alkyl chains were grafted on opposite sides of a vanadate POM via tris linker (P2a). When the TBA counter cations of this hybrid are replaced by H+, Na+ blue luminescence was observed. This vanadate-based hybrid shows amphiphilic properties in the acetone/water mixture by forming vesicles at the water/air interface and reducing surface tension.73 In another report from Liu and Hill et al., a pyrene moiety was grafted on [V6O13{(OCH2)3CNH2}2]2− (P2c) symmetrically as well as asymmetrically. Similar to the previous hybrid, the fluorescent properties depended on the type of counter cations of the hybrids. These hybrids show amphiphilic properties by forming spherical vesicular structures in polar solvents mixture such as H2O/DMSO, H2O/MeCN.74
3.2 Polymerizable POM-containing monomers
The development of organo-functionalized POMs during the past 50 years provided another approach to prepare POM-polymer hybrid materials. In 1975 Pope and co-workers took the lead in the successful synthesis of molybdate complexes of dialkyl and diaryl arsinic acids, [R2AsMo4O14(OH)]2− with R = CH3, C2H5, C6H5, C2H4NH3+ and p-CH2C6H4NH3+ and determined their structures by single-crystal X-ray diffraction analysis.75 After this work, a notable increase in the number of studies focusing on the organic functionalization of POMs was observed, leading to the establishment of the platforms discussed throughout. One of the first POM-containing polymerizable molecules were reported by Judeinstein in 1992 (Section 2.2.1.).76 This work sparked significant interest in the direct incorporation of POMs into polymeric materials using more robust covalent bonds. Over the past decade, advances in polymerisation methodologies and the expansion of viable POM platforms has seen strong growth in this research area including a recent increase in materials displaying responsive and dynamic properties. In this section, POM hybrid materials are roughly divided into four categories according to how the inorganic component is incorporated into the polymeric composite (Fig. 5). Firstly, we will discuss the co-polymerisation of organic monomers with POM crosslinkers followed by the development of polymers with POM as pendants through the use of a polymerizable group and conclude with a discussion of polymers containing POMs as main chain components.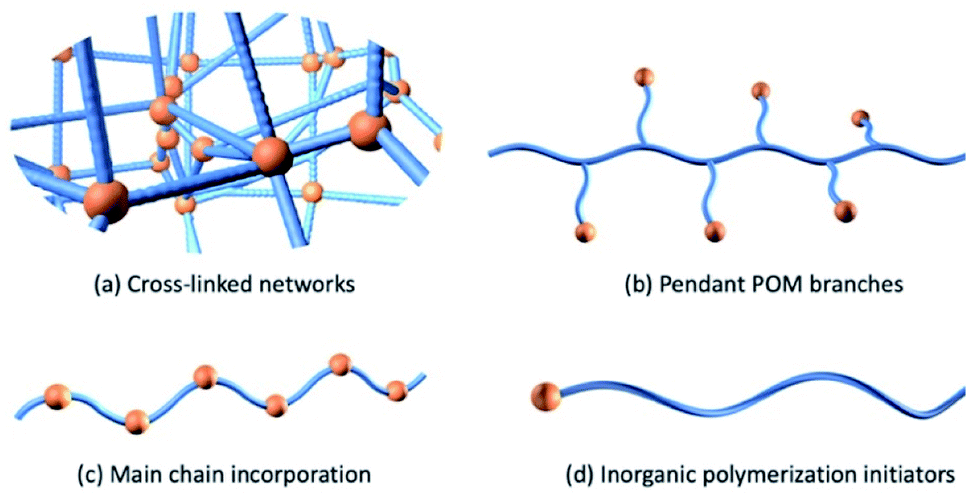 | ||
| Fig. 5 Four main typical strategies for the covalent modification of POM-polymer composites. | ||
Several studies focused on investigating the influence of POMs on the bulk proton conductivity of functionalised polymers. Horan and co-workers prepared a series of proton exchange membranes using the vinyl-functionalized heteropoly acid H4[SiW11O39Si(R)2O] (P3b) (R – vinyl), butyl acrylate and 1,6-hexanediol diacrylate via UV initiated polymerizations.79,80 The HPA exhibited high proton diffusivity due to hydrogen bonding and was incorporated into polymeric membranes as the sole proton-conducting species. The authors showed that the membrane morphology was sensitive to the weight fraction of POM loading. With less than 50 wt% POM, the materials appeared homogeneous however when reaching 80 wt% the polyanions form channels spanning 20–100 nm in width, which was determined by AFM and SEM. Covalently grafted hybrid membranes overcome the issue of water-soluble POM being washed out under high humidity conditions where the covalent linkages are resistant to hydrolysis. The proton conductivities increased with increasing humidity, temperature and POM contents. The best performance 0.40 S cm−1 was observed at 90 °C and 95% relative humidity (RH) with 85 wt% POM loading, which is significantly better than the commonly used perfluorinated ionomer 825 EW 3 M (0.14 S cm−1, 80 °C, 100% RH).
The incorporation of POMs into polymeric networks is also a strategy of interest for the design of heterogeneous catalysts. It is proposed that amphiphilic POMs could lead to the formation of porous structures by influencing the solubility of growing polymers and the trapping of solvent droplets.81 These hybrid catalysts could be used in fixed-bed and membrane reactors, such as fuel cleansers due to their oxidative desulfurization properties and resistance to decomposition under operating conditions. POM platforms such as (P3b) that are functionalised by organo main-group species facilitate the preparation of POM based monomers with multiple sites for potential polymerisation as discussed earlier. The resulting polymerizable POMs have been used to prepare macroporous hybrid catalysts through free radical polymerization with methyl methacrylate. Catalytic tests and recycling experiments showed the hybrid polymer activated hydrogen peroxide for oxygen transfer, such as oxidation of dibenzothiophene by H2O2 in MeCN and MeCN/n-octane.82 In another study [P2W17O61(SiC6H4CH2N3)2O]6− (P4b) was used to control the spatial arrangement of two divergent azido functional groups that were reacted with the microporous benzylamine functionalised resin with a pore size distribution of 47.4 nm using click chemistry. Following preparation, this material was shown to possess catalytic oxidation properties through the installation of the POMs within the resin.83
In conclusion, organically functionalized POMs have been used to generate cross-linked POM hybrid materials. However, the extent of synthetic methodologies explored is limited with free-radical polymerization being the most widely adopted. This has limited the homogeneity of the resulting products and therefore control over the resulting self-assembled architectures. More recently the emphasis has transitioned to other polymerization methods such as living radical polymerization and ROMP, which can control the POM distribution and loading.
CH2)]2− (P2c) were reacted with 4-methylstyrene to yield hybrid polymers bearing Lindqvist pendants by free radical polymerization. The ratio of 4-methylstyrene to POM in the final product was about three to one determined using FT-IR and NMR characterization.84 While the work of Moore was a successful proof of concept, the resulting materials did not exploit the electronic conjugation between the POM and polymer through the imido linkage which involves the electronic coupling of the organic and inorganic moieties due to significant d–p orbital overlap from Mo and N respectively. In 2005, Peng and co-workers successfully synthesized conjugated hybrid polymers with the same POM platform (P2c) on the side chain through one-pot synthesis.85 In this work, the polymerizable POM monomers were prepared by the reaction of the arylamine with the hexamolybdate cluster. After determining the structure of this hybrid monomer by 1H NMR and X-ray single-crystal diffraction, POM-modified poly(phenylene ethynylene) was produced via the Sonogashira cross-coupling reaction. The target products obtained by the hybridization–polymerization method were then utilized to test electrical, optical and electrochemical properties. The hybrid materials with conjugated linkages exhibited significant fluorescent quenching of the conjugated polymer emission due to the photoinduced electron transfer from the polymer backbone to the POMs. Moreover, this hybrid showed amphiphilic behaviour in toluene and self-assembled into reverse vesicles. In polar solvents like DMSO, the POM pendants were charged because of the dissociation of counterions and formed tubular structures. The authors provided a mechanistic hypothesis for the tubular structures, where the solvophobic and rigid rod-like nature of the polymer backbone (phenylene–ethynylene backbone) facilitated preferred aggregation in one dimension to form a tube through pi–pi stacking interactions.86 In their following work, the post-polymerization functionalization approach was employed to successfully prepare rod-coil diblock copolymers with (P2c) pendants attached to the coil block, which was designed to provide separated charge-transporting channels to minimize electron–hole charge recombination. The conjugated polymer, poly(phenylene vinylene), served as the electron donor and the (P2c) in the coil block functioned as the electron acceptor. This hybrid showed negligible fluorescence quenching in solution, but efficient fluorescence quenching in the solid state. As the emission of the hybrid films was blue-shifted by 10 nm, the authors attributed the fluorescence quenching to electron transfer from the conjugated polymer exciton to the POM clusters rather than increased π stacking of poly(phenylene vinylene).87,88
As eluded to earlier, a more appropriate polymerization methodology for controlling the molecular weight and configuration of polymers is ROMP. Wang has successfully used this method for the preparation of well-controlled hybrid polymers with the Dawson tungstovanadate [P2V3W15O62]9− pendant groups (P4a). They used size-exclusion chromatography to demonstrate the formation of well-defined hybrid polymers with high molecular weight (∼775 kDa) and low polydispersity (PDI < 1.16).89 Subsequently, they proved that ROMP was also suitable for preparing block copolymers with POMs covalently grafted in one segment. When dissolved in acrylonitrile the block copolymers formed a hexagonal close-packed structure. The catalytic oxidation of tetrahydrothiophene into tetrahydrothiophene oxide was investigated using both the block copolymer and the poly(POM)5 homopolymer. The results showed that the homopolymer had higher activity than the block copolymer, which may be due to the heterogeneous nature of the hybrid block copolymer system.90 Ruhlmann has used a bottom-up synthetic strategy to prepare POM-porphyrin copolymers bearing four kinds of conjugated spacers with various geometries. [P2V3W15O62]9− (P4) was reacted with diol-amide ligands at 80 °C under microwave irradiation to yield a POM with two pendant electro-polymerizable pyridyl groups (P4a). A novel electropolymerization technique was then used in the presence of zinc β-octaethylporphyrin, resulting in POM–porphyrin copolymer films.91 A further study of their electrochemical behaviour showed that compared to the conjugation group between POM and ligands, the geometry of pyridyl groups carried by monomers had a more noticeable effect on the performance.92
In 2017, Bazzan and co-workers synthesized a new photochromic hybrid polymer using organo-modified [MnMo6O18]3− (P1) via free radical polymerization (PDI ∼ 1.7). Methacrylate modified Anderson–Evan POM (P1a) with spiropyran appended groups were covalently or ionically incorporated into a polymeric matrix, and their solid-state photophysical properties were fully investigated under ambient conditions.93
Another important technique to design polymers with controlled molecular weight and dispersity is reversible deactivation radical polymerizations techniques such as ATRP and RAFT. Recently, we reported the rational design of pH-responsive core–shell nanoparticles that incorporate a [BIIIW11O39CoIII]6− (P3d) cargo. An amphiphilic polymer poly(ethylene glycol) methacrylate-b-poly(2-diethylamino)ethyl methacrylate (PEGMA-b-PDEAEMA) and a statistical copolymer of poly(2-diethylamino)ethyl methacrylate-r-poly(2-(diisopropylamino)ethyl methacrylate)-r-poly(pyridin-4-yl methyl methacrylate) (PDEAEMA-r-PDPAEMA-r-PPyMMA) were synthesized through RAFT polymerization. Post-polymerization modification of pyridyl functionality allows for the formation of kinetically inert coordinate bonds with the (P3d) cargo. Large-scale structural rearrangement was observed in response to acidification. This is a significantly different response to the POM-free materials, which underwent complete nanoparticle disassembly when the pH was reduced below 6.4. Cryo-TEM was also conducted on POM-containing samples at pH 8, 7 and 6 to probe the various states of the particle rearrangement. Most interestingly, at pH 6, the migration of POMs formed the interface between core and shell components, and the samples were markedly less stable with rapid beam damage observed. Based on the data from all analytical techniques, we have attributed this structural rearrangement to the increase in inter- and intra-polymer hydrogen bonding with the polyanion surface and stronger point charge interactions, which ultimately prevents particle disassembly.94
The development of research on organic modification of POM clusters as well as the design and control of molecular weight and morphology have become the key factors affecting the application performance of the POM-polymer hybrids. Thus, the synthetic methodology of polymer hybrids with POM as the pendant sidechain has evolved from free radical polymerization to more controlled polymerization methods, such as RAFT and ROMP. We predict this field will grow in the future as material scientists investigate how different polymeric functionality interacts with POMs incorporated into their structure.
Cross-coupling. Challenges associated with the incorporation of bulky components such as POMs into a polymer backbone are exacerbated in comparison to pendant functionality due to an increase in steric constraints and reactivity challenges. Consequently, most examples of main chain incorporation are achieved by using coupling reactions to realise hybrid linear polymers.
To the best of our knowledge, Peng and co-workers reported the first main-chain POM-containing polymers in 2002.95 In this groundbreaking work, they found a novel and efficient method to prepare and purify bifunctionalized POM monomers [Mo6O17(NAr)2]2− (P2c). They had previously reported a convenient approach to modify the (P2) platform but failed to obtain the bifunctionalized product in high yields.96 Interestingly, the use of the α-octamolybdate ion, [Mo8O26]4−, as the molybdate source generated over 60% bifunctionalized products under the same reaction conditions. The compound bearing two iodo groups was chosen to synthesize linear polymers with 2,5-di(2,2-dimethylpropoxy-1,4-diethynylbenzene) using palladium-catalyzed coupling reactions. Cyclic voltammetry studies showed a reversible reduction wave at −1.19 V versus Ag/Ag+, and the I–V test under 100 mW cm−2 illumination showed 1.12 mA cm−2 short-circuit current and 0.15% power conversion efficiency, both of which were an order of magnitude higher than that of typical single layer polymer device.97
Photo-induced. (P1a) have also been used to design main-chain POM materials. For example, Song and co-workers reported a novel photosensitive organo-POM hybrid bearing coumarins on each face [MnMo6O18{(OCH2)3CNHC12H9O4}2]3− (P1a). Irradiation (365 nm) induced light-driven polymerization, which confirmed spectroscopically and using dynamic light scattering measurements. Illumination with deep UV (254 nm) reversed the polymerisation process revealing the dynamic nature of these compounds.98 Cronin and co-workers designed and synthesized a series of asymmetric and symmetric (P1a) species bearing azide and/or alkyne groups. A Cu-catalyzed alkyne–azide cycloaddition reaction was used to lock these ‘blocks’ together to form oligomers. More importantly, the asymmetric hybrid acted as a monofunctionalized compound until the other site was activated by post-functionalization, allowing the synthesis of monodisperse POM oligomers/polymers with precisely controlled chain lengths and composition. Four examples of monodisperse oligomeric hybrids, from dimers to pentamers, were synthesized to demonstrate the extraordinary control level over the size and structure of POM hybrids.99 Most recently, symmetric (P1a) hybrids bearing two anthracene groups were synthesized, with supramolecular chemistry utilised to assist the photopolymerisation process. Association of the anthracene moieties of two (P1a) hybrids inside the cavity of a single γ-cyclodextrin facilitated the controlled cyclodimerization of the organic components to yield covalent polymers which were visualized and characterized using TEM.100
POM monomers. POM-based monomers of [MnMo6O18{(OCH2)3–CNH2}2]3− (P1a) bearing methacrylate functionality were prepared and co-polymerised with methyl methacrylate to obtain a hybrid polymer with dispersity of 1.63. These materials were produced and incorporated into devices as thin films to investigate their redox properties and their potential function as rewritable multilevel resistive memory. By applying different RESET voltages, the devices encouragingly exhibited rewritable switching properties among three redox states with good endurance and retention.101 The same derivatives were copolymerized with (methacryloyloxy) phenyldimethylsulfoniumtriflate (MAPDST) to yield a photoresponsive material due to the well-documented photoresist properties of MAPDST containing polymers. The resulting hybrid was fabricated into hexamethyldisilazane-treated RCA-cleaned p-type Si wafer pieces to explore its photo-conducting properties, where it was revealed that improved photo-conducting performance was achieved in comparison to the control devices based on MAPDST polymers and POM-poly(methyl methacrylate) separately.102 The authors believe this synergistic effect comes from the combination of redox-active POM and the photogeneration of ions from MAPDST. These select examples are indicative of the versatility of POM platforms as reagents that can be incorporated in polymeric backbones. Also apparent is the scope for further research both with regards to the design of more complex materials and the improvement of functionality that maximises the synergy between polymer and POM.
3.3 Inorganic polymerization initiators
Using the same methodologies discussed throughout, it is possible to install organic functionality on the surface of POMs that facilitates their use as inorganic polymerization initiators. While this approach limits the POM loading on each polymer chain, the benefits of increased control concerning polymer molecular weight and chain configuration can be advantageous including minimising purification. Nevertheless, there are only a few examples of systematically studied POM-based macro initiators, with an emphasis on ATRP and RAFT initiated polymerisation.:POM aspect ratio and their amphiphilic nature.103 Building upon these initial findings, a follow-up study focussed on the impact of heating and resulting changes to particle morphology observed following thermal treatment and sample ageing. A comparison of the average vesicle diameters before and following heating (70 °C – 12 h) revealed a significant temperature effect with elevated temperature resulting in an average 74% increase in vesicle diameter. Furthermore, the heat treatment resulted in defect-free vesicles most likely due to improved dispersion of the hybrid polymers prior to self-assembly. Ageing of the samples at 25 °C over 21 days resulted in a transition of the assembly morphology from vesicles to tubes containing a POM core and PS corona which grew via end-to-end fusion to reach a lower energy state.104 In another recent study, Yu et al. demonstrated that a POM-PS hybrid could be self-assembled in DMF due to the different solubility of the POM and polymer in this solvent. The self-assembled structures changed from micelles to self-assembled vesicles, cylindrical vesicles and then large vesicles based on the concentration of the POM-polymer hybrid (P4a-PS) (Fig. 6).105
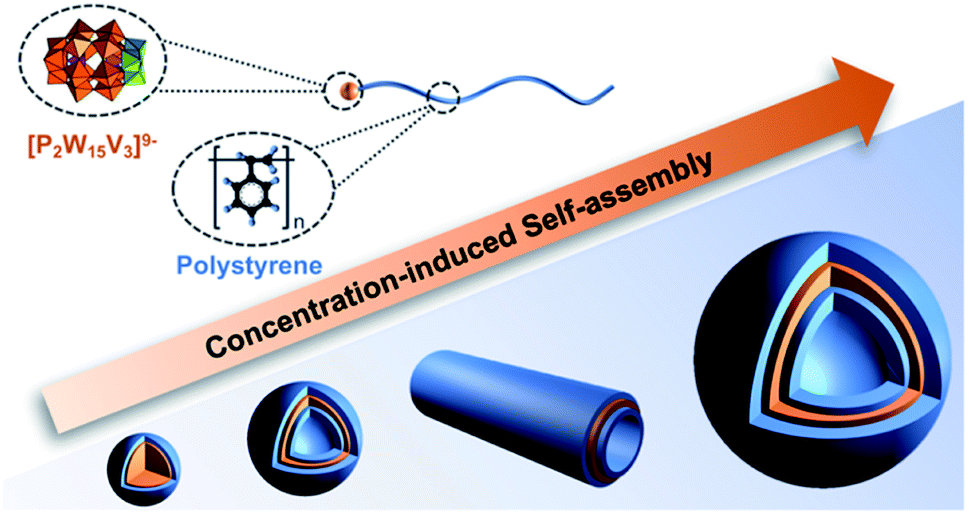 | ||
| Fig. 6 The morphology evolution of concentration-induced POM-PS hybrid assembles. | ||
In the above examples, the targeted linear polymers were obtained using ATRP and a POM-based macroinitiator, with the resulting observation of morphology control suggesting that these compounds may find utility in functional materials. A recurring feature of POM-based ATRP macroinitiators is the higher PDI of the resulting materials compared to analogous POM-free systems. This observation indicates that the POMs are causing side reactions or impeding efficient chain propagation. To the best of our knowledge, the origin of this deviation from typical ATRP behaviour remains to be investigated.
A final notable mention is the function of HPAs as initiators for cationic and ring-opening polymerisation in addition to their previously discussed amphiphilic properties. The [PW11O39(SiR)2]3− (R – C18 alkyl chains) (P3b) amphiphile discussed earlier (see Section 3.1.) can also serve as a source of protons for cationic polymerisation, with the resulting polymer nanoparticles (300–350 nm) having their surfaces decorated by POMs as evidenced by TEM and SEM studies.64 [MnMo6O18{(OCH2)3CCH2OH}2]3− (P1a) was also used to initiate a ring-opening polymerization of ε-caprolactone by two OH groups on the sides. The growth of isothermal crystallization was monitored by a hot stage microscope, and polarized light microscopy was employed to visualize the crystalline morphologies.108 This work did not investigate the polymerization mechanism.
4 Current applications
POMs have demonstrated potential for a range of biomedical applications (Fig. 7); however, they are limited due to non-specific interactions with their biological environment.109 Thus, their encapsulation into nanomaterials can enhance their properties. In one such study, a new platinum(IV)-substituted pro-drug (P3) was synthesised which required reductive elimination by endogenous reductants to release the cancer agent platinum(II)-substituted Keggin-type POM.110 This molecule was not functional until it was converted into its active form within a tumour. In this work, the pro-drug was encapsulated by 1,2 distearoyl-sn-glycero-3-phosphethanolamine-N-[methoxy(polyethylene glycol)] DSPE-PEG2000 carboxylic acid to form cargo loaded nanoparticles. Nanoparticles were synthesised using the film dispersion method and formed uniform nanoparticles with a size of 31.2 ± 6.1 nm by TEM. These particles showed decreased cell viability for HT29 cancer cells compared to the positive control of cisplatin. However, later results showed that the impact on tumour volume of the nanoparticle was not significant compared to the cisplatin control. However, this is still of significant interest as the starting cargo was a pro-drug and thus would likely have reduced off-target effects compared to free drugs.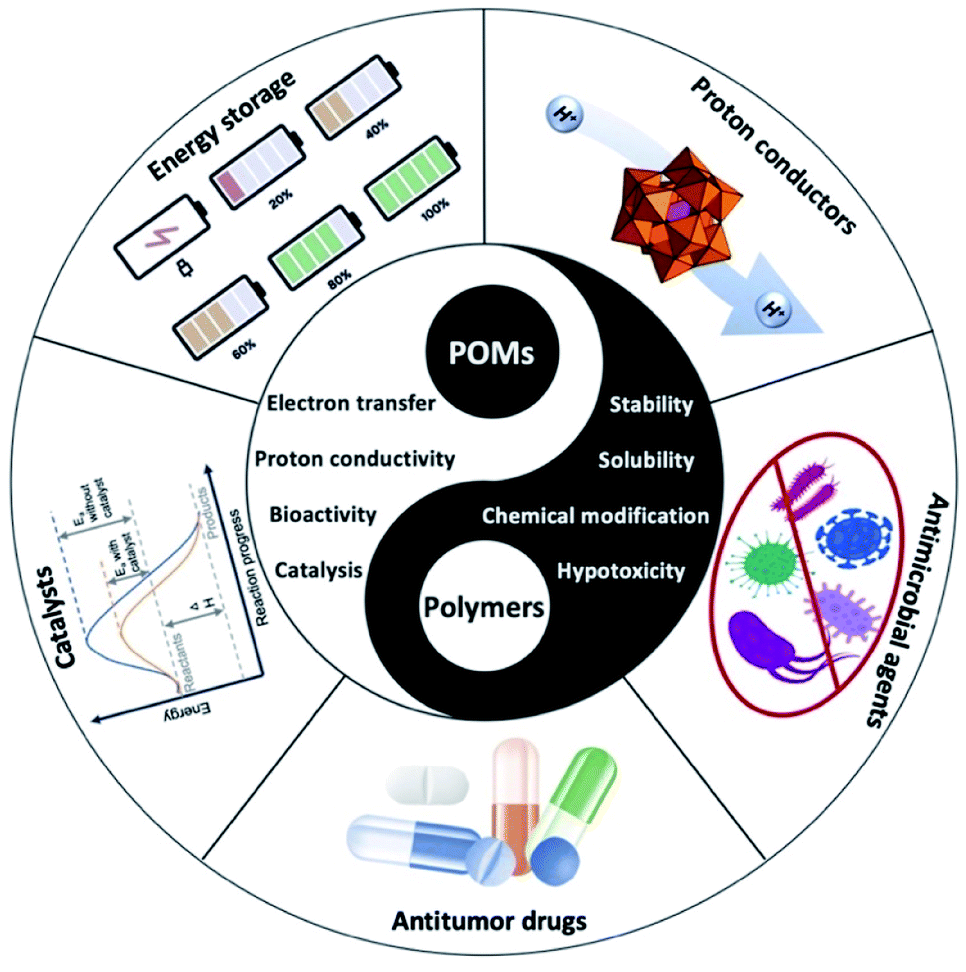 | ||
| Fig. 7 Potential applications for POM containing hybrid materials. | ||
While several POMs have therapeutic activity, they suffer from low stability in biological conditions. Thus, there are several studies to enhance this property using polymer encapsulation. The advantages of nanoparticle encapsulation include limiting non-specific interaction of the POM and allowing targeting towards a certain treatment site. Nanoparticle carriers can also be designed to respond to specific triggers, thus releasing the functional POM cargo when at a target site. One recent example involves the encapsulation of [NaSb9W21O86]18−, [α-P2W18O62]6− and [Mo7O24]6− within chitosan and carboxymethyl chitosan to afford nanoparticles.111 The [NaSb9W21O86]18− and [α-P2W18O62]6− loaded nanoparticles showed enhanced toxicity for HeLa cells (a model cancer cell line) compared to normal cells. In contrast, [Mo7O24]6− showed similar results in both cell lines.
POMs have also generated significant interest as phototheranostics. For example, in recent work, a semi-conducting brush polymer (SBP) with thiol functionalised brushes were synthesised. This polymer was successfully complexed to a molybdenum-based POM cluster through thiol-metal complexation forming nanoparticles of approximately 100 nm.112 Mo-based POMs are of interest as they display near-infrared absorption as a result of glutathione-triggered reduction from MoVI to MoV to yield IVCT bands. Thus they can be used for imaging tumours, and this NIR signal is also enhanced in a tumour environment. The nanoparticles have exhibited better potential for this application than POMs in isolation due to improved retention in a tumour. The nanoparticles were shown to increase in size due to proton-induced hydrogen bonding. It was also demonstrated that SPB@POM nanoparticles performed significantly better at photothermal treatment on U87MG tumours as compared to the SPB control particles, with full survival over 45 days. The particles were also used to perform positron emission tomography.
Due to their charged nature POMs are also of interest for complexing biological materials such as proteins. Recently, Li et al. showed β-sheet nanofibers could be synthesised by the electrostatic complexation of small cationic peptides based on an alternating sequence of lysine and alanine residues with [SiW12O40]4− (P3) in an aqueous solution.113 The maximum β-sheet content was reached with a molar ratio of peptide to POM of approximately 2:1. These nanofibers had antimicrobial activity against E. coli, while the control peptide and POM showed limited activity. The results showed that the addition of the POM facilitated the formation of advanced structures which possibly enhanced the interaction of the hybrid materials with the surface of the E. coli. It was also proved that the stability of the peptide fiber was significantly enhanced in the hybrid materials. POMs containing gadolinium have generated interest as MRI contrast agents as they have a longer rotational correlation time and higher longitudinal relaxation rate than conventional gadolinium complexes due to the chemical structure and larger size. A third-generation dendron was synthesised in recent work with a cationic head group and tri(ethylene glycol) end groups.114 This was shown to interact with [Gd(β2-SiW11O39)2]13− through electrostatic interactions to form hybrid nanoparticles. The in vivo T1-weighted MRI was investigated as compared to a commercial gadolinium complex (gadopentetate dimeglumine) (Gd–DTPA), and it was found that the POM hybrid had an enhancement of approximately 44.9 ± 15.2% whereas only 15.4 ± 10.3% occurred at 30 min in the case of Gd–DTPA. It should be noted that the [Gd(β2-SiW11O39)2]13− POMs took longer to reach maximum enhancement owing to the higher molecular weight and larger size compared to Gd-DTPA.
Another interesting application is catalysis (Fig. 7). Recently, it was shown that two different POMs, [PMo12O40]3− or [SiMo12O40]4− (P3), could be loaded into a trimethylolpropane triacrylate matrix using radical polymerisation induced by irradiation with light.115 The matrix efficiently photodegraded Eosin Y dye using UV light LED@375 nm. It was not as effective as the free POM control, however, it was shown that the composite allowed the dye to be recovered and recycled. Furthermore, EPR showed that POM structure could be regenerated by simple air contact. Another potential application for POM-based materials is in photovoltaic devices or energy storage systems. In such devices, bicontinuous polymer structures are desired to enhance charge transport but remain challenging to produce. Zhang et al. showed that [SiW12O40]4− (P3) could be used to induce a phase transition of a poly(styrene)-b-(2-vinyl pyridine) (PS-b-P2VP) to a stable bicontinuous phase. It was postulated that the POM was able to protonate the P2VP chains, thus forming electrostatic crosslinks between the POM and P2VP. Results showed that the transition to the bicontinuous phase was achieved by increasing the amount of POM (4.3–21.0 wt%). Importantly, it was also shown that this process was generalizable to other POMs. These systems are of interest as potential photovoltaics as they had high conductivity (0.1 mS cm−1 at 298 K) and enhanced materials properties.41 A related study involved the synthesis of POM coated Fe2O4 spheres.116 In this work, Fe3O4 spheres were encapsulated with poly(N-(3-dimethylamino propyl)methacrylamide) (PDMAPMA) using distillation precipitation. The free amino groups were used to chelate Zn2+ which then were used for a coordination polymerisation of the Anderson–Evan POM [MnMo6O18{(OCH2)3CNH2}2]3−. This material showed potential as an energy storage device with large areal capacitance (2.23 mF cm−2 at 0.01 mAcm−2) and stable cycling.
5 Conclusions
In this perspective, we have discussed the varied approaches that have been used to prepare POM containing materials, with a focus on the most widely utilised polyanions. The field of POM synthesis has matured significantly, allowing huge versatility in POM structure and modification approaches. We have classified four key families as POM platforms and have discussed their structure and how they can be modified to demystify these versatile inorganic building blocks for researchers in other fields to better utilise these materials in their research. The field of POM-hybrid materials is still young, with a significant amount of research focusing on well-established polymers and surfactants. However, as illustrated in this perspective, there is huge scope for the design of interesting nanostructures using the combination of these building blocks. We have highlighted recent work in several applications. Our view is that there remains significant scope for the development of smart stimuli-responsive materials that derive their value from an interplay of properties arising from both organic and POM componentry such as electron transport, structural rearrangement, photoresponsivity, response to pH variation. One emerging area of interest is in the area of biomedicine where POMs have shown potential but are limited by stability. There is obvious potential to combine advances in polymer science and POM chemistry to design stimuli-responsive POM-polymer hybrids that protect and release this cargo for a targeted therapeutic outcome.6 Future outlook
As the chemistry of POMs has evolved, the development of approaches for their systematic functionalisation has given rise to a wealth of compositionally and structurally diverse molecular species. Likewise, polymer science has progressed over the same period with the development of more sophisticated polymeric architectures such as multiblock or star polymers as well as greater versatility in the types of functionalities that can be incorporated within the polymer structure. In contrast, the implementation of these techniques in the preparation of smarter POM containing polymer hybrids is still an emerging research area. Looking forward it would be intriguing to determine if the tuneable molecular electronics of POM-based RAFT agents could be used to control the behaviour of RAFT agents to switch on and off polymerisation, by controlling the ability of the monomer to add efficiently to the agent. It would be even more significant if polymerisation could be tuned by light or redox chemistry. The use of different polymeric architectures to build POM-polymer hybrids is an area that has significant potential. Miktoarm polymers (asymmetric branched macromolecules) offer the possibility to devise an infinitely large library of materials with structures intended to instigate the formation of specific supramolecular interactions. The spatially controlled incorporation of POM containing motifs within these assemblies provides an opportunity to direct the formation of elaborate assemblies that utilise the numerous interactions we have highlighted in this article. POM-polymer hybrids have potential as therapeutics if we incorporate POMs that have a specific biological function. Encapsulation within a nanoparticle carrier can help to direct the POM more directly to a specific target location while maintaining stability, and thus offers an important avenue for further investigation. Further directions could involve the use of POMs to drive a catalytic therapeutic cycle, potentially through the use of light or redox potential. There is also scope to investigate the combination of different POMs within a single nanoparticle structure. Recently we have demonstrated the impact that the inclusion of POMs in pH-responsive core–shell nanoparticles has on the disassembly process, with the inclusion of the POM affording increased particle stability at low pH accompanied by a notable structural change. We have continued this approach to elucidate the POM induced stability threshold as we as expanding the study to other responsive componentry including photo-induced processes. In time we foresee the utility of these materials as delivery agents whereby the POM fulfils the role of either a structural mediator or as a relay for electron transfer.Abbreviations
| POMs | Polyoxometalates |
| ROMP | Ring-opening metathesis polymerization |
| ATRP | Atom transfer radical polymerization |
| RAFT | Reversible addition-fragmentation polymerization |
| SECs | Surfactant encapsulated clusters |
| DDMA | Didodecyl dimethylammonium |
| DOMA | Dioctadecyl dimethylammonium |
| DODA | Dimethyl-dioctadecylammonium |
| ETAB | Ethyl-4′-(trimethylaminohexyloxy)azobenzene |
| POM-ILs | Polyoxometalate-based ionic liquids |
| DBT | Dibenzothiophene |
| MIMPS | 1-(3-Sulfonic group)propyl-3-methyl imidazolium |
| PSP | Supramolecular star polymers |
| P4VMP | Poly(4-vinyl-N-methylpyridinium iodide) |
| PEO | Poly(ethylene oxide) |
| PS | Polystyrene |
| PDMAEMA | Poly(2-(dimethylamino)ethyl methacrylate) |
| P2VP | Poly(2-vinylpyridinium) |
| TBA | Tetrabutylammonium |
| MeCN | Acetonitrile |
| HPAs | Heteropolyacids |
| MLM | Monolayer lipid membrane |
| TEM | Transmission electron microscopes |
| DMSO | Dimethyl sulfoxide |
| DMF | N,N-Dimethylformamide |
| TM | Transition metal |
| AFM | Atomic force microscopy |
| SEM | Scanning electron microscope |
| RH | Relative humidity |
| FT-IR | Fourier-transform infrared |
| NMR | Nuclear magnetic resonance |
| PDI | Polydispersity |
| PEGMA | Poly(ethylene glycol)methacrylate |
| PDEAEMA | Poly(2-(diethylamino)ethyl methacrylate) |
| PDPAEMA | Poly(2-(diisopropylamino)ethyl methacrylate) |
| PPyMMA | Poly(pyridin-4-yl methyl methacrylate) |
| MAPDST | (Methacryloyloxy)phenyldimethylsulfoniumtriflate |
| PDEAAm | Moly(N,N-diethylacrylamide) |
| SBP | Semi-conducting brush polymer |
| Gd–DTPA | Gadolinium complex (gadopentetate dimeglumine) |
| PDMAPMA | Poly(N-(3-dimethylamino propyl) methacrylamide) |
Author contributions
All authors contributed to the preparation of this article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. R. and G. S. gratefully acknowledge financial support from the Australian Research Council for projects FT180100610 and DP180100844. M. C. thanks Monash University for support through the Deans scholarship scheme.References
- L. Baker and D. C. Glick, Chem. Rev., 1998, 98, 3–50 CrossRef CAS PubMed.
- J. J. Borrás-Almenar, E. Coronado, A. Müller and M. Pope, Polyoxometalate molecular science, Springer Science & Business Media, 2003 Search PubMed.
- C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34–48 CrossRef.
- J. Xu, H. Volfova, R. J. Mulder, L. Goerigk, G. Bryant, E. Riedle and C. Ritchie, J. Am. Chem. Soc., 2018, 140, 10482–10487 CrossRef CAS PubMed.
- X. Lopez, J. J. Carbo, C. Bo and J. M. Poblet, Chem. Soc. Rev., 2012, 41, 7537–7571 RSC.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed. Engl., 2020, 59, 596–612 CrossRef CAS PubMed.
- D. E. Katsoulis and M. T. Pope, J. Am. Chem. Soc., 1984, 106, 2737–2738 CrossRef CAS.
- F. Corigliano and S. Di Pasquale, Inorg. Chim. Acta, 1975, 12, 99–101 CrossRef.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- C. Rocchiccioli-Deltcheff and R. Thouvenot, Spectrosc. Lett., 1979, 12, 127–138 CrossRef CAS.
- G. B. McGarvey and J. B. Moffat, J. Mol. Catal., 1991, 69, 137–155 CrossRef CAS.
- A. Nisar, Y. Lu and X. Wang, Chem. Mater., 2010, 22, 3511–3518 CrossRef CAS.
- A. Nisar, J. Zhuang and X. Wang, Chem. Mater., 2009, 21, 3745–3751 CrossRef CAS.
- J. Zhang, Y. Liu, Y. Li, H. Zhao and X. Wan, Angew. Chem., Int. Ed. Engl., 2012, 51, 4598–4602 CrossRef CAS PubMed.
- Y. Guo, Y. Gong, Y. Gao, J. Xiao, T. Wang and L. Yu, Langmuir, 2016, 32, 9293–9300 CrossRef CAS PubMed.
- B. Li, W. Li, H. Li and L. Wu, Acc. Chem. Res., 2017, 50, 1391–1399 CrossRef CAS PubMed.
- M. Clemente-Leon, B. Agricole, C. Mingotaud, C. Gomez-Garcia, E. Coronado and P. Delhaes, Langmuir, 1997, 13, 2340–2347 CrossRef CAS.
- A. Nisar, X. Xu, S. Shen, S. Hu and X. Wang, Adv. Funct. Mater., 2009, 19, 860–865 CrossRef CAS.
- H. Li, H. Sun, W. Qi, M. Xu and L. Wu, Angew. Chem., Int. Ed. Engl., 2007, 46, 1300–1303 CrossRef CAS PubMed.
- Y. Yang, Y. Wang, H. Li, W. Li and L. Wu, Chem.–Eur. J., 2010, 16, 8062–8071 CrossRef CAS PubMed.
- Y. Wang, W. Li and L. Wu, Langmuir, 2009, 25, 13194–13200 CrossRef CAS PubMed.
- R. Nagarajan, Langmuir, 2002, 18, 31–38 CrossRef CAS.
- D. Volkmer, A. Du Chesne, D. G. Kurth, H. Schnablegger, P. Lehmann, M. J. Koop and A. Müller, J. Am. Chem. Soc., 2000, 122, 1995–1998 CrossRef CAS.
- D. G. Kurth, P. Lehmann, D. Volkmer, A. Müller and D. Schwahn, J. Chem. Soc., Dalton Trans., 2000, 21, 3989–3998 RSC.
- S. Polarz, B. Smarsly and M. Antonietti, ChemPhysChem, 2001, 7, 457–461 CrossRef.
- Y. Qiao, Z. Hou, H. Li, Y. Hu, B. Feng, X. Wang, L. Hua and Q. Huang, Green Chem., 2009, 11, 1955–1960 RSC.
- W. Zhu, G. Zhu, H. Li, Y. Chao, M. Zhang, D. Du, Q. Wang and Z. Zhao, Fuel Process. Technol., 2013, 106, 70–76 CrossRef CAS.
- Y. Li, Y. Zhang, P. Wu, C. Feng and G. Xue, Catalysts, 2018, 8, 639 CrossRef.
- Y. Li, X. Wu, Q. Wu, H. Ding and W. Yan, Dalton Trans., 2014, 43, 13591–13595 RSC.
- X. Wu, W. Wu, Q. Wu and W. Yan, Langmuir, 2017, 33, 4242–4249 CrossRef CAS PubMed.
- X. Wu, X. Tong, Q. Wu, H. Ding and W. Yan, J. Mater. Chem. A, 2014, 2, 5780–5784 RSC.
- T. Huang, Z. Xie, Q. Wu and W. Yan, Dalton Trans., 2016, 45, 3958–3963 RSC.
- S. Herrmann, M. Kostrzewa, A. Wierschem and C. Streb, Angew. Chem., Int. Ed. Engl., 2014, 53, 13596–13599 CrossRef CAS PubMed.
- A. L. Kubo, L. Kremer, S. Herrmann, S. G. Mitchell, O. M. Bondarenko, A. Kahru and C. Streb, ChemPlusChem, 2017, 82, 867–871 CrossRef CAS PubMed.
- K. Shakeela and G. R. Rao, ACS Appl. Nano Mater., 2018, 1, 4642–4651 CrossRef CAS.
- W. Bu, S. Uchida and N. Mizuno, Angew. Chem., Int. Ed. Engl., 2009, 48, 8281–8284 CrossRef CAS PubMed.
- Q. Zhang, Y. Liao and W. Bu, Langmuir, 2013, 29, 10630–10634 CrossRef CAS PubMed.
- J. Tan, D. Chong, Y. Zhou, R. Wang, X. Wan and J. Zhang, Langmuir, 2018, 34, 8975–8982 CrossRef CAS PubMed.
- D. Li, H. Li and L. Wu, Polym. Chem., 2014, 5, 1930–1937 RSC.
- L. Zhang, T. Cui, X. Cao, C. Zhao, Q. Chen, L. Wu and H. Li, Angew. Chem., Int. Ed. Engl., 2017, 56, 9013–9017 CrossRef CAS PubMed.
- L. Zhai, S. Chai, G. Wang, W. Zhang, H. He and H. Li, Macromol. Rapid Commun., 2020, 41, 2000438 CrossRef CAS PubMed.
- H. He, G. Wang, S. Chai, X. Li, L. Zhai, L. Wu and H. Li, Chin. Chem. Lett., 2021, 32, 2013–2016 CrossRef CAS.
- F. Meng, N. V. Aieta, S. F. Dec, J. L. Horan, D. Williamson, M. H. Frey, P. Pham, J. A. Turner, M. A. Yandrasits, S. J. Hamrock and A. M. Herring, Electrochim. Acta, 2007, 53, 1372–1378 CrossRef CAS.
- S. Y. Lee, D. W. Shin, C. Wang, K. H. Lee, M. D. Guiver and Y. M. Lee, Electrochem. Commun., 2013, 31, 120–124 CrossRef CAS.
- P. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2003, 2406–2412 CrossRef.
- D. Karimian, B. Yadollahi and V. Mirkhani, Microporous Mesoporous Mater., 2017, 247, 23–30 CrossRef CAS.
- C. P. Pradeep, D. L. Long, G. N. Newton, Y. F. Song and L. Cronin, Angew. Chem., 2008, 120, 4460–4463 CrossRef.
- V. Kalyani, V. S. V. Satyanarayana, V. Singh, C. P. Pradeep, S. Ghosh, S. K. Sharma and K. E. Gonsalves, Chem.–Eur. J., 2015, 21, 2250–2258 CrossRef CAS PubMed.
- Y. F. Song, N. McMillan, D. L. Long, J. Thiel, Y. Ding, H. Chen, N. Gadegaard and L. Cronin, Chemistry, 2008, 14, 2349–2354 CrossRef CAS PubMed.
- Y. F. Song, J. Zhang, L. Cronin and T. Liu, J. Am. Chem. Soc., 2008, 130, 14408–14409 CrossRef PubMed.
- J. Zhang, Y. F. Song, L. Cronin and T. Liu, Chemistry, 2010, 16, 11320–11324 CrossRef CAS PubMed.
- C. Allain, S. Favette, L. M. Chamoreau, J. Vaissermann, L. Ruhlmann and B. Hasenknopf, Eur. J. Inorg. Chem., 2008, 2008, 3433–3441 CrossRef.
- M. H. Rosnes, C. Musumeci, C. P. Pradeep, J. S. Mathieson, D. L. Long, Y. F. Song, B. Pignataro, R. Cogdell and L. Cronin, J. Am. Chem. Soc., 2010, 132, 15490–15492 CrossRef CAS PubMed.
- O. Oms, K. Hakouk, R. Dessapt, P. Deniard, S. Jobic, A. Dolbecq, T. Palacin, L. Nadjo, B. Keita, J. Marrot and P. Mialane, Chem. Commun., 2012, 48, 12103–12105 RSC.
- Y. Chu, A. Saad, P. Yin, J. Wu, O. Oms, A. Dolbecq, P. Mialane and T. Liu, Chemistry, 2016, 22, 11756–11762 CrossRef CAS PubMed.
- X. López, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2002, 124, 12574–12582 CrossRef PubMed.
- A. Mazeaud, Y. Dromzee and R. Thouvenot, Inorg. Chem., 2000, 39, 4735–4740 CrossRef CAS PubMed.
- C. R. Mayer, P. Herson and R. Thouvenot, Inorg. Chem., 1999, 38, 6152–6158 CrossRef CAS PubMed.
- D. Agustin, C. Coelho, A. Mazeaud, P. Herson, A. Proust and R. Thouvenot, Z. Anorg. Allg. Chem., 2004, 630, 2049–2053 CrossRef CAS.
- T. Minato, K. Suzuki, K. Yamaguchi and N. Mizuno, Chemistry, 2017, 23, 14213–14220 CrossRef CAS PubMed.
- R. Villanneau, A. B. Djamâa, L. M. Chamoreau, G. Gontard and A. Proust, Eur. J. Inorg. Chem., 2013, 2013, 1815–1820 CrossRef CAS.
- D. Agustin, J. Dallery, C. Coelho, A. Proust and R. Thouvenot, J. Organomet. Chem., 2007, 692, 746–754 CrossRef CAS.
- S. Landsmann, C. Lizandara-Pueyo and S. Polarz, J. Am. Chem. Soc., 2010, 132, 5315–5321 CrossRef CAS PubMed.
- S. Landsmann, M. Luka and S. Polarz, Nat. Commun., 2012, 3, 1299 CrossRef PubMed.
- R. G. Finke, B. Rapko, R. J. Saxton and P. J. Domaille, J. Am. Chem. Soc., 1986, 108, 2947–2960 CrossRef CAS.
- B. J. Hornstein and R. G. Finke, Inorg. Chem., 2002, 41, 2720–2730 CrossRef CAS PubMed.
- C. P. Pradeep, M. F. Misdrahi, F. Y. Li, J. Zhang, L. Xu, D. L. Long, T. Liu and L. Cronin, Angew. Chem., Int. Ed. Engl., 2009, 48, 8309–8313 CrossRef CAS PubMed.
- M. F. Misdrahi, M. Wang, C. P. Pradeep, F. Y. Li, C. Lydon, L. Xu, L. Cronin and T. Liu, Langmuir, 2011, 27, 9193–9202 CrossRef CAS PubMed.
- P. Yin, C. P. Pradeep, B. Zhang, F. Y. Li, C. Lydon, M. H. Rosnes, D. Li, E. Bitterlich, L. Xu, L. Cronin and T. Liu, Chemistry, 2012, 18, 8157–8162 CrossRef CAS PubMed.
- H. K. Liu, L. J. Ren, H. Wu, Y. L. Ma, S. Richter, M. Godehardt, C. Kübel and W. Wang, J. Am. Chem. Soc., 2018, 141, 831–839 CrossRef PubMed.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., Int. Ed. Engl., 2019, 58, 18281–18285 CrossRef CAS PubMed.
- P. Yin, P. Wu, Z. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. Liu and Y. Wei, Angew. Chem., Int. Ed. Engl., 2011, 50, 2521–2525 CrossRef CAS PubMed.
- D. Li, J. Song, P. Yin, S. Simotwo, A. J. Bassler, Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. Liu, J. Am. Chem. Soc., 2011, 133, 14010–14016 CrossRef CAS PubMed.
- K. M. Barkigia, L. M. Rajkovic, M. T. Pope and C. O. Quicksall, J. Am. Chem. Soc., 1975, 97, 4146–4147 CrossRef CAS.
- P. Judeinstein, Chem. Mater., 1992, 4, 4–7 CrossRef CAS.
- C. R. Mayer, R. Thouvenot and T. Lalot, Chem. Mater., 2000, 12, 257–260 CrossRef CAS.
- C. R. Mayer, V. Cabuil, T. Lalot and R. Thouvenot, Angew. Chem., Int. Ed. Engl., 1999, 38, 3672–3675 CrossRef CAS PubMed.
- J. L. Horan, A. Genupur, H. Ren, B. J. Sikora, M. C. Kuo, F. Meng, S. F. Dec, G. M. Haugen, M. A. Yandrasits, S. J. Hamrock, M. H. Frey, A. M. Herring, J. Sikora, M. C. Kuo, F. Meng, A. M. Herring, J. L. Horan, S. F. Dec and M. H. Frey, ChemSusChem, 2009, 3, 226–229 CrossRef PubMed.
- J. L. Horan, A. Lingutla, H. Ren, M. C. Kuo, S. Sachdeva, Y. Yang, S. Seifert, L. F. Greenlee, M. A. Yandrasits, S. J. Hamrock, M. H. Frey and A. M. Herring, J. Phys. Chem. C, 2014, 118, 135–144 CrossRef CAS.
- H. Sun, H. Li and L. Wu, Polymer, 2009, 50, 2113–2122 CrossRef CAS.
- M. Carraro, G. Fiorani, L. Mognon, F. Caneva, M. Gardan, C. Maccato and M. Bonchio, Chemistry, 2012, 18, 13195–13202 CrossRef CAS PubMed.
- Y. Xiao, D. Chen, N. Ma, Z. Hou, M. Hu, C. Wang and W. Wang, RSC Adv., 2013, 3, 21544–21551 RSC.
- A. R. Moore, H. Kwen, A. M. Beatty and E. A. Maatta, Chem. Commun., 2000, 1793–1794 RSC.
- B. Xu, M. Lu, J. Kang, D. Wang, J. Brown and Z. Peng, Chem. Mater., 2005, 17, 2841–2851 CrossRef CAS.
- P. Yin, L. Jin, D. Li and P. Cheng, Chem.–Eur. J., 2012, 18, 6754–6758 CrossRef CAS PubMed.
- S. Chakraborty, A. Keightley, V. Dusevich, Y. Wang and Z. Peng, Chem. Mater., 2010, 22, 3995–4006 CrossRef CAS.
- S. Chakraborty, L. Jin, Y. Li, Y. Liu, T. Dutta, D. M. Zhu, X. Yan, A. Keightley and Z. Peng, Eur. J. Inorg. Chem., 2013, 2013, 1799–1807 CrossRef CAS.
- W. K. Miao, Y. K. Yan, X. L. Wang, Y. Xiao, L. J. Ren, P. Zheng, C. H. Wang, L. X. Ren and W. Wang, ACS Macro Lett., 2014, 3, 211–215 CrossRef CAS.
- W. K. Miao, A. Yi, Y. K. Yan, L. J. Ren, D. Chen, C. H. Wang and W. Wang, Polym. Chem., 2015, 6, 7418–7426 RSC.
- I. Azcarate, I. Ahmed, R. Farha, M. Goldmann, X. Wang, H. Xu, B. Hasenknopf, E. Lacôte and L. Ruhlmann, Dalton Trans., 2013, 42, 12688–12698 RSC.
- Z. Huo, I. Azcarate, R. Farha, M. Goldmann, H. Xu, B. Hasenknopf, E. Lacôte and L. Ruhlmann, J. Solid State Electrochem., 2015, 19, 2611–2621 CrossRef CAS.
- I. Bazzan, P. Bolle, O. Oms, H. Salmi, N. Aubry-Barroca, A. Dolbecq, H. Serier-Brault, R. Dessapt, P. Roger and P. Mialane, J. Mater. Chem. C, 2017, 5, 6343–6351 RSC.
- Y. Gao, J. Xu, C. Zhang, H. Venugopal, S. S. Kermaniyan, G. Such and C. Ritchie, ACS Appl. Nano Mater., 2020, 3, 11247–11253 CrossRef CAS.
- L. Xu, M. Lu, B. Xu, Y. Wei, Z. Peng and D. R. Powell, Angew. Chem., Int. Ed., 2002, 41, 4129–4132 CrossRef CAS PubMed.
- Y. Wei, B. Xu, C. L. Barnes and Z. Peng, J. Am. Chem. Soc., 2001, 123, 4083–4084 CrossRef CAS PubMed.
- M. Lu, B. Xie, J. Kang, F. C. Chen, Y. Yang and Z. Peng, Chem. Mater., 2005, 17, 402–408 CrossRef CAS.
- U. Tong, W. Chen, C. Ritchie, X. Wang and Y. F. Song, Chem.–Eur. J., 2014, 20, 1500–1504 CrossRef CAS PubMed.
- A. Macdonell, N. A. B. Johnson, A. J. Surman and L. Cronin, J. Am. Chem. Soc., 2015, 137, 5662–5665 CrossRef CAS PubMed.
- W. Guan, G. Wang, J. Ding, B. Li and L. Wu, Chem. Commun., 2019, 55, 10788–10791 RSC.
- B. Hu, C. Wang, J. Wang, J. Gao, K. Wang, J. Wu, G. Zhang, W. Cheng, B. Venkateswarlu, M. Wang, P. S. Lee and Q. Zhang, Chem. Sci., 2014, 5, 3404–3408 RSC.
- V. Kalyani, V. S. V. Satyanarayana, A. S. Sarkar, A. Kumar, S. K. Pal, S. Ghosh, K. E. Gonsalves and C. P. Pradeep, RSC Adv., 2015, 5, 36727–36731 RSC.
- Y. Han, Y. Xiao, Z. Zhang, B. Liu, P. Zheng, S. He and W. Wang, Macromolecules, 2009, 42, 6543–6548 CrossRef CAS.
- Y. K. Han, Z. J. Zhang, Y. L. Wang, N. Xia, B. Liu, Y. Xiao, L. X. Jin, P. Zheng and W. Wang, Macromol. Chem. Phys., 2011, 212, 81–87 CrossRef CAS.
- S. J. Yu, Y. K. Han and W. Wang, Polymer, 2019, 162, 73–79 CrossRef CAS.
- J. Lesage de la Haye, P. Beaunier, L. Ruhlmann, B. Hasenknopf, E. Lacôte and J. Rieger, ChemPlusChem, 2014, 79, 250–256 CrossRef CAS PubMed.
- J. Lesage de La Haye, J. M. Guigner, E. Marceau, L. Ruhlmann, B. Hasenknopf, E. Lacôte and J. Rieger, Chem.–Eur. J., 2015, 21, 2948–2953 CrossRef CAS PubMed.
- N. Xia, W. Yu, Y. Wang, Y. Han, P. Zheng, W. Wang, G. Sakaguchi, K. Matsuda, K. Saijo, M. Takenaka and H. Hasegawa, Polymer, 2011, 52, 1772–1780 CrossRef CAS.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed.
- T. Sun, W. Cui, M. Yan, G. Qin, W. Guo, H. Gu, S. Liu and Q. Wu, Adv. Mater., 2016, 28, 7397–7404 CrossRef CAS PubMed.
- M. Croce, S. Conti, C. Maake and G. R. Patzke, Eur. J. Inorg. Chem., 2019, 2019, 348–356 CrossRef CAS.
- Z. Yang, W. Fan, W. Tang, Z. Shen, Y. Dai, J. Song, Z. Wang, Y. Liu, L. Lin, L. Shan, Y. Liu, O. Jacobson, P. Rong, W. Wang and X. Chen, Angew. Chem., Int. Ed. Engl., 2018, 57, 14101–14105 CrossRef CAS PubMed.
- J. Li, Z. Chen, M. Zhou, J. Jing, W. Li, Y. Wang, L. Wu, L. Wang, Y. Wang and M. Lee, Angew. Chem., Int. Ed. Engl., 2016, 55, 2592–2595 CrossRef CAS PubMed.
- S. Zhang, Y. Zheng, S. Yin, J. Sun, B. Li and L. Wu, Chemistry, 2017, 23, 2802–2810 CrossRef CAS PubMed.
- M. Ghali, C. Brahmi, M. Benltifa, F. Dumur, S. Duval, C. Simonnet-Jégat, F. Morlet-Savary, S. Jellali, L. Bousselmi and J. Lalevée, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1538–1549 CrossRef CAS.
- S. Xu, L. You, P. Zhang, Y. Zhang, J. Guo and C. Wang, Chem. Commun., 2013, 49, 2427–2429 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc05879g |
| This journal is © The Royal Society of Chemistry 2022 |