
Accelerating hydrazine-assisted hydrogen production kinetics with Mn dopant modulated CoS2 nanowire arrays†
Junrong
Hou‡
ab,
Xianyun
Peng‡
*cd,
Jiaqiang
Sun
 e,
Shusheng
Zhang
f,
Qian
Liu
g,
Xinzhong
Wang
*a,
Jun
Luo
b and
Xijun
Liu
*h
e,
Shusheng
Zhang
f,
Qian
Liu
g,
Xinzhong
Wang
*a,
Jun
Luo
b and
Xijun
Liu
*h
aInformation Technology Research Institute, Shenzhen Institute of Information Technology, Shenzhen 518172, China. E-mail: wangxz@sziit.com.cn
bInstitute for New Energy Materials & Low-Carbon Technologies and Tianjin Key Lab of Photoelectric Materials & Devices, School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China
cInstitute of Zhejiang University – Quzhou, Quzhou 324000, China. E-mail: xianyunpeng@zju.edu.cn
dKey Laboratory of Biomass Chemical Engineering of Ministry of Education, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China
eState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan 030001, China
fCollege of Chemistry, Zhengzhou University, Zhengzhou 450000, China
gInstitute for Advanced Study, Chengdu University, Chengdu 610106, Sichuan, China
hMOE Key Laboratory of New Processing Technology for Non-Ferrous Metals and Materials, and Guangxi Key Laboratory of Processing for Non-Ferrous Metals and Featured Materials, School of Resource, Environments & Materials, Guangxi University, Nanning 530004, China. E-mail: xjliu@tjut.edu.cn
First published on 5th May 2022
Abstract
Electrochemical H2 production from water splitting is an environmentally sustainable technique but remains a great challenge due to the sluggish anodic oxygen evolution reaction (OER). Replacing the OER with the thermodynamically more favorable electrocatalytic oxidation process is an effective strategy for highly efficient H2 generation. Herein, Mn-doped CoS2 has predicted an excellent bifunctional electrocatalyst for the hydrogen evolution reaction (HER) and the hydrazine oxidation reaction (HzOR). With the introduction of Mn, the Gibbs free energy of the adsorbed H* and the potential rate-limiting step (the dehydrogenation of *NH2NH2 to *NHNH2) for the HzOR process of the catalyst can be significantly reduced. As expected, the Mn-CoS2 catalyst exhibited excellent catalytic activity and robust long-term stability for the HER and HzOR. In detail, the Mn-CoS2 catalyst only acquired potentials of 46 and 77 mV versus the reversible hydrogen electrode for achieving a current density of 10 mA cm−2 for the cathodic HER and anodic HzOR, respectively. In addition, the Mn-CoS2 electrode only needs a cell voltage of 447 mV to output 200 mA cm−2 in the overall hydrazine splitting system as well as exhibits a robust long-term H2 production. This work provides theoretical guidance for the design of advanced bifunctional electrocatalysts and promotes high efficiency and energy-saving H2 production technology.
Introduction
With the increasing depletion of fossil fuels, serious energy shortage, and environmental problems, clean and renewable energy sources have attracted much attention.1,2 Hydrogen energy is deemed as one of the most potential alternatives for a sustainable energy system due to its highest energy density (142 MJ kg−1).3–5 Nevertheless, the current industrial method for H2 production, natural gas reforming, is an energy-intensive and non-clean process. It consumes considerable amounts of fossil fuel and produces a vast amount of carbon dioxide.6 Electrochemical hydrogen (H2) production with high purity via the hydrogen evolution reaction (HER) on the water-splitting cathodic electrode has been emerging as a sustainable, cost-effective, and viable way.7–13 Currently, the major obstacles that restricted the development of H2 production are the lack of high-activity and low-cost electrocatalysts with low overpotential requirements in HER processes.14 Although H2 can be obtained based on the state-of-the-art pH-universal Pt-based electrocatalysts, their wide commercial application is severely limited by their scarcity and high cost.15–17 Therefore, the development of low-cost metal-free or abundant transition metal electrocatalysts toward high-efficient H2 generation to meet the global hydrogen economy is highly desirable yet key challenging.15,18Ongoing research efforts have been devoted to developing cost-effective electrocatalysts with high activity for the HER process, many potential nanomaterials (such as transition metal phosphides,19–22 sulfides,23–26 and nitrides27,28) have been widely put forward and investigated in detail. Among these catalysts, Co-based nanomaterials, especially for the cobalt disulfide (CoS2), have been regarded as promising HER electrocatalysts due to their high conductivity and unique configuration. Despite achieving significant progress, the HER catalytic activities of Co-based catalysts are also still far less than the Pt-based catalyst. In this regard, more attention has been paid to the modulation of the electronic structure and the optimization of chemical compositions, aiming to adjust the energy band structure as well as the electronic conductivity and thermodynamic H* adsorption/desorption state. The doping strategy with transition metal ions has been proved to play a significant role in optimizing the Gibbs free energy of the adsorbed H (ΔGH*) for hydrogen adsorption of HER electrocatalysts.29–35 It has been demonstrated that the HER electrocatalytic activity of Co-based catalysts can be efficiently triggered by varied transition-metal dopants.36,37 In particular, the volcano plot based on a density functional theory (DFT) revealed that elemental Mn is the best candidate dopant to tune the adsorption behavior of H atoms on adjacent Co atoms, the dopant itself, and consequently the HER activity.38
Additionally, the anodic oxygen evolution reaction (OER) is identified as the bottleneck of water splitting. The OER suffered from a high overpotential and sluggish kinetics, which greatly limit the practical application of H2 production.39 Therefore, apart from optimizing catalysts, the electrochemical H2 production can be improved and accelerated by replacing the OER with a thermodynamically more favorable electrocatalytic oxidation process. For instance, the oxidation of hydrazine, 5-hydroxymethylfurfural (HMF),40 and urea41 have been coupled with cathodic H2O reduction to effectively reduce energy consumption toward high-purity H2 generation. Among them, hydrazine (N2H4) oxidation is the most potential candidate due to its much lower oxidation potential of −0.33 V versus reversible hydrogen electrode (vs. RHE) than that of the OER (1.23 V vs. RHE).42–44 Moreover, inert N2 is the anodic oxidation product of N2H4, which can well solve the potential security issues of O2 + H2 mixture gases in membrane-free overall water splitting. In addition, with the assistance of Earth-abundant electrocatalysts, substituted anodic hydrazine oxidation could reduce the cost and overpotential of the entire water electrolysis, resulting in high-efficiency H2 production.
Herein, taking the representative CoS2 as a model electrocatalyst for the HER, we adopted a doping strategy with an Mn atom to accelerate H2 production with the assistance of anodic N2H4 oxidation by rationally modulating the electronic structure. DFT calculations demonstrated that Mn atoms can optimize the Gibbs free energy and enhance the intrinsic activity and tremendously accelerate the kinetic process. Inspired by the theoretical analysis, we successfully prepared Mn-doped CoS2 nanotubes supported on nickel foam (Mn-CoS2). As expected, the Mn-CoS2 electrocatalyst exhibited excellent electrochemical performance with a much lower overpotential of 46 mV at a current density of 10 mA cm−1 and a small Tafel slope of 63.1 mV dec−1 for the HER in 0.1 M KOH aqueous solution. Meanwhile, as for the anodic hydrazine oxidation reaction (HzOR), Mn-CoS2 only needs a small working potential of 77 mV vs. RHE to generate a current density of 10 mA cm−1.
Experimental methods
Computational details
All the geometric optimization and single-point energy calculations were performed using spin-polarized density-functional theory (DFT) implemented in the DMol3 code. Exchange–correlation functions are taken as generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE). The band structure and the corresponding DOS result were conducted by DS-PAW package, and the Device Studio program was used for performing visualization, modeling and simulation45,46 The double numerical plus polarization (DNP) was chosen as the atomic orbital basis set, and the smearing was set to 0.005 Ha to achieve precise electronic convergence. The convergence tolerances of energy, maximum force, and displacement were set to 2 × 10−5 Ha, 0.004 Ha Å−1, and 0.005 Å, respectively, and a basis set a cut-off of 4.4 Å was employed to ensure high-quality computational results. The reciprocal space was sampled using a 3 × 3 × 1 k-point grid for geometry optimizations. A 2 × 2 × 1 supercell was used to calculate the electronic property and hydrogen evolution activity. For Mn-doped CoS2, we used one Mn atom to replace one Co atom. To prevent interaction between two neighboring surfaces, a vacuum slab of 15 Å was employed in the (001) direction of CoS2 for the calculations of hydrogen evolution activity. The free energy diagram for the HER was obtained by calculating the change of the free energy with a hydrogen atom adsorbed on the surface. The activity of the hydrogen evolution reaction was reflected by Gibb's free energy change (ΔGH*) and the values of ΔGH* are obtained using the following formula:| ΔGH* = ΔEH* + ΔEZPE − TΔS |
| ΔEH* = Esurface/H* − Esurface − 1/2EH2 |
The oxidation of hydrazine to nitrogen and hydrogen occurs in the following six consecutive elementary steps:
(1)
| * + N2H4 → *N2H4 |
(2)
| *N2H4 → *N2H3 + H+ + e− |
(3)
| *N2H3 → *N2H2 + H+ + e− |
(4)
| *N2H2 → *N2H + H+ + e− |
(5)
| *N2H → *N2 + H+ + e− |
(6)
| *N2 → * + N2 |
Clean nickel foam
Nickel foam with a three-dimensional skeleton was selected as a substrate for the growth of Mn-CoS2 nanotubes. Before use, nickel foam was carefully cleaned with a dilute hydrochloric acid aqueous solution in an ultrasound bath for several minutes to remove the surface oxide layer. Subsequently, the nickel foam was washed successively with deionized water, acetone, and absolute ethanol. Finally, the cleaned nickel foam was obtained by drying at 60 °C overnight under vacuum conditions.Synthesis of Mn-CoS2
Mn-CoS2 was prepared by a facile hydrothermal reaction and sulfuration method. Typically, Co(NO3)2·6H2O (1.5 mM), NH4F (3 mM), urea (7.5 mM), and MnCl2·4H2O (0.075 mM) were dissolved in 40 mL of deionized water and magnetically stirred for 60 min to form a homogeneous solution. After that, the obtained solution was transferred into a 50 mL Teflon-lined stainless steel autoclave with one piece of clean nickel foam (approximately 1 cm × 2 cm), which was sealed and maintained at 110 °C for 5 h in an oven and was naturally cooled down to room temperature. After cooling to room temperature, the nickel foam with the growth precursor was taken out, washed with water and ethanol, and dried in an oven at 60 °C for 12 h under vacuum conditions.After that, the as-prepared precursor was put into a ceramic boat and placed in a tube furnace. Another ceramic boat containing 0.5 g of sublimed sulfur was put on the upstream side. After vacuum treatment and filling with high-purity Ar, the furnace was heated to 300 °C for 2 h with a heating rate of 1 °C min−1 with a continuous Ar flow with a flow rate of 100 sccm. After the tube furnace was naturally cooled to ambient temperature, Mn-CoS2 was obtained. For comparison, CoS2 was also prepared by a similar approach except without the addition of MnCl2·4H2O. The areal loadings of the CoS2 and Mn-CoS2 catalyst on the nickel foam were confirmed to be 2.7 and 2.9 mg cm−2, respectively. The Mn-CoS2 catalyst with various Mn contents was optimized by adjusting the additive amount of precursor. All the chemicals were directly used after purchase without further purification.
Materials characterization
The crystallographic structure was analyzed by X-ray diffraction (XRD) using an X-ray diffractometer (SmartLab 9 kW) with Cu Kα radiation (λ = 0.154598 nm) at a scan rate of 10° min−1 in the 2θ range from 30° to 70°. The morphology and structure of the prepared sample were characterized using a scanning electron microscope (SEM, Verios 460L) operated at 20 kV and a transmission electron microscope (TEM, Talos F200X) operated at 200 kV equipped with an energy-dispersive X-ray spectrometer (EDS). XPS (ESCALAB 250Xi from Thermo Scientific) was employed for elemental mapping using a monochromatic Al Kα radiation source at a background pressure of 10−9 Torr. The binding energy calibration was performed by referencing the C 1s main peak at 284.8 eV. Inductively coupled plasma atomic emission spectroscopy (ICP-OES) analysis was recorded on a Thermo iCAP RQ instrument.Electrochemical measurements
Electrochemical measurements were carried out with a typical three-electrode system by using an electrochemical workstation (CHI Instruments 760E) at room temperature. A graphite rod and Ag/AgCl electrode were used as the counter and reference electrodes, respectively. The as-prepared catalysts were indirectly used as working electrodes. 0.1 M KOH and 1 M KOH containing 0.5 M N2H4 aqueous solution were used as electrolytes for the HER and HzOR, respectively.Firstly, the HER and HzOR activity of the catalysts was investigated by linear scan voltammogram (LSV) with a scan rate of 10 mA s−1. For comparison, the commercial noble metal Pt/C catalyst was measured under the same conditions. A stability test was performed using the chronopotentiometry–time and continuous cyclic voltammetry measurement technology in 0.1 M KOH. For hydrazine-assisted water electrolysis, a symmetrical full electrolyzer was assembled by directly using Mn-CoS2 as the cathode and anode. LSV was measured at a scan rate of 10 mV s−1 in 1.0 M KOH with 0.5 M N2H4 or 1 M H2SO4 with 0.5 M N2H4. For comparison, the commercial Pt/C catalyst coated nickel foam was measured under the same conditions. All the electrocatalytic current density was normalized to the geometric area of the nickel foam (1 × 1 cm2) and all potentials were given versus reversible hydrogen electrode according to the equation: ERHE = EAg/AgCl + 0.0591pH + 0.197.
Electrochemical impedance spectra (EIS) were recorded at an open-circuit potential in a frequency ranging from 100 kHz to 10 mHz with an AC voltage amplitude of 5 mV. To determine the corresponding electrochemical surface area (ECSA), the double-layer capacitance (Cdl) of all the materials was measured in a 0.1 M KOH aqueous solution. A potential range where no apparent Faradaic process occurred was determined first using cyclic voltammetry (CV) with different scan rates of 2, 4, 6, and 8 mV s−1. The value of Cdl was then obtained from the linear curve versus scan rate.
Results and discussion
To clarify the underlying origins between the catalytic activity and the hybrid structure, DFT calculations were applied to predict both HER and HzOR processes. Firstly, the stable (001) surface of the cubic CoS2 model was built. As shown in Fig. 1a, the structural analysis reveals that CoS2 has a unique crystal structure, where each Co is coordinated with six S atoms to form a slightly distorted octahedron, and the distorted octahedrons are connected by S–S covalent bonds. In the case of the Mn-CoS2 catalyst, one Mn atom instead of one Co atom of CoS2(001) is the theoretical calculation model (Fig. S1†).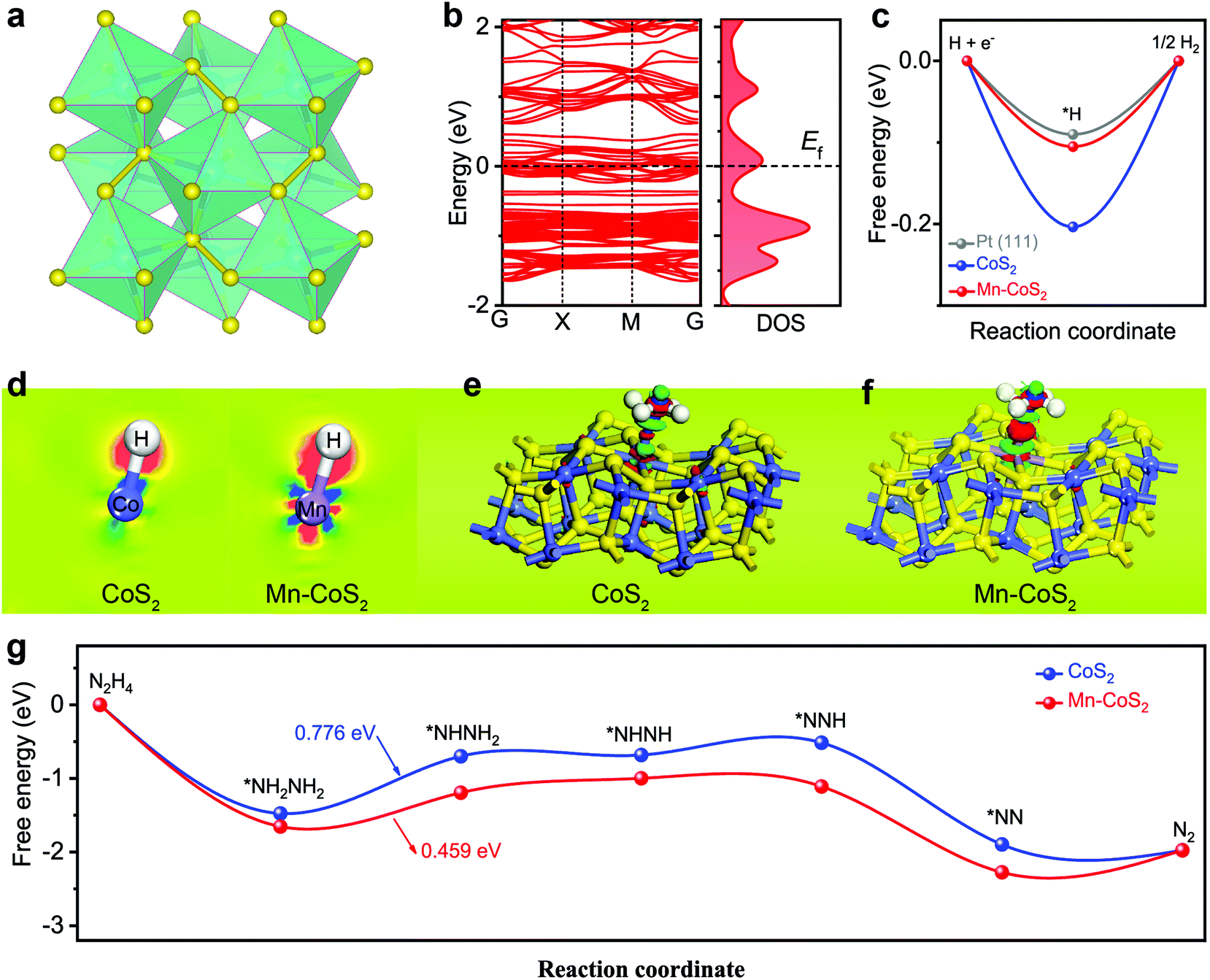 | ||
| Fig. 1 DFT calculation results: (a) crystal structure of CoS2; (b) the calculated electronic band structure and the corresponding DOS results of Mn-CoS2; (c) the free energy profiles of HER pathways for CoS2 and Mn-CoS2; (d) the charge density difference contour plot of Co–H and Mn–H regions, the red and blue colors represent the accumulation and depletion of electrons, respectively; (e and f) the side-view of the three-dimensional charge density difference of the H-adsorbed configuration at different sites over the catalyst surface, the pink and green region representing charge depletion and accumulation in the space, respectively; (g) the free energy profiles of the HzOR reaction pathways for CoS2 and Mn-CoS2. | ||
Firstly, the electronic band structures of CoS2 and Mn-CoS2 are investigated. As shown in Fig. 1b, Mn-CoS2 maintains the metallic nature as pristine CoS2 (Fig. S2†), which would favor the electron transfer process in the electrochemical catalysis process.47 It is well known that the free energy changes of hydrogen adsorption (ΔGH*) are an effective descriptor for evaluating the HER activity of electrocatalysts. The catalysts with ΔGH* close to zero are considered promising candidates for the HER. As shown in Fig. 1c, the pristine CoS2 exhibits a ΔGH* value of −2.03 eV, indicating the strong adsorption of H* on the CoS2 surface with a bond length (Co–H) of 1.471 Å (Fig. S3†). After Mn doping, the bond length of Mn–H is extended to 1.552 Å, resulting in the ΔGH* value of Mn-CoS2 decreasing to −1.05 eV, which is much close to Pt(111), indicating its excellent thermoneutrality for the HER process.
To further analyze the change in the electron distribution of the Co–H region and Mn–H region for CoS2 and Mn-CoS2, the charge density difference (CDD) of the catalyst absorbed H* was studied as shown in Fig. 1d, where the red and blue colors represent the accumulation and depletion of electrons, respectively. As the CDD contour plots shown in Fig. 1d, much more electrons from the Mn atom are transferred to the adsorbed H* in Mn-CoS2 than that of Co in CoS2. In detail, the electrons around the H atom in Mn-CoS2 are much more than that in CoS2 due to the increased electron transfer, and the Mn site in Mn-CoS2 is a relatively electron-rich state compared with the Co site in CoS2, which is probably due to the electronic interaction between Mn and the surrounding atoms (Fig. S4†). These results indicate that the modulation of the electronic structure caused by Mn doping would synergistically promote the water adsorption strength and optimize the adsorption free energy of H*, which can facilitate the electrochemical HER process.31
Apart from the investigation of HER activity, the HzOR process over CoS2 and Mn-CoS2 surfaces was also explored using first-principles calculations. Firstly, the atomic structure adsorbed N2H4 molecule over the Co site of Mn-CoS2 and Co site of CoS2 was optimized. As shown in Fig. S5,† Mn-CoS2 displays the strongest N2H4 adsorption with a more negative binding energy of −1.65 eV than that of CoS2 (−1.48 eV). Furthermore, the charge density difference was also calculated. As the charge density difference contour plot is shown in Fig. 1e and f, it can be seen that much more electrons were transferred from the Mn site to adsorbed N2H4 and the charge redistribution was dominantly restricted around the Mn site, proving the strong adsorption of N2H4 on Mn-CoS2. This electron localization behavior of the Mn-CoS2 catalyst upon Mn doping could contribute to the enhanced catalytic activity.43,48,49
The HzOR pathway on the catalyst is further studied. As shown in Fig. 1g, it can be found that adsorption of N2H4 on the Mn site of the Mn-CoS2 surface is exothermic by −1.65 eV, lower than that of the Co site of the CoS2 surface (−1.48 eV), indicating that the electron-deficient Mn species on Mn-CoS2 would facilitate the adsorption of the N2H4 molecule.50,51 Moreover, it has been revealed that the dehydrogenation process of *NH2NH2 to *NHNH2 is the potential rate-limiting step (PLS) of the HzOR on the CoS2 and Mn-CoS2 surfaces, and the PDS barrier of Mn-CoS2 (0.459 eV) is lower than that of CoS2 (0.776 eV). Therefore, the Mn-CoS2 catalyst is preferred for propelling the electrochemical HzOR process than CoS2.
Motivated by the above promising prediction, we successfully synthesized Mn-doped CoS2 nanotube supported nickel foam (Mn-CoS2/NF). Fig. 2a illustrates the synthesis schematics of the Mn-CoS2/NF. Before the growth of Mn-CoS2, the commercial NF was cleaned with acid to remove the oxide layer. Then, the cleaned NF serves as the three-dimensional skeleton for the uniform growth of Mn-Co(OH)2 nanoarrays by a facile hydrothermal synthesis at 200 °C for 6 h. Finally, the Mn-CoS2 nanotubes were obtained by the sulfuration of Mn-Co(OH)2 with sublimed sulfur as a S source at 250 °C for 2 h (please see the Experimental section for more details). For comparison, CoS2 nanoarrays were also fabricated by a similar method except without the addition of a Mn precursor.
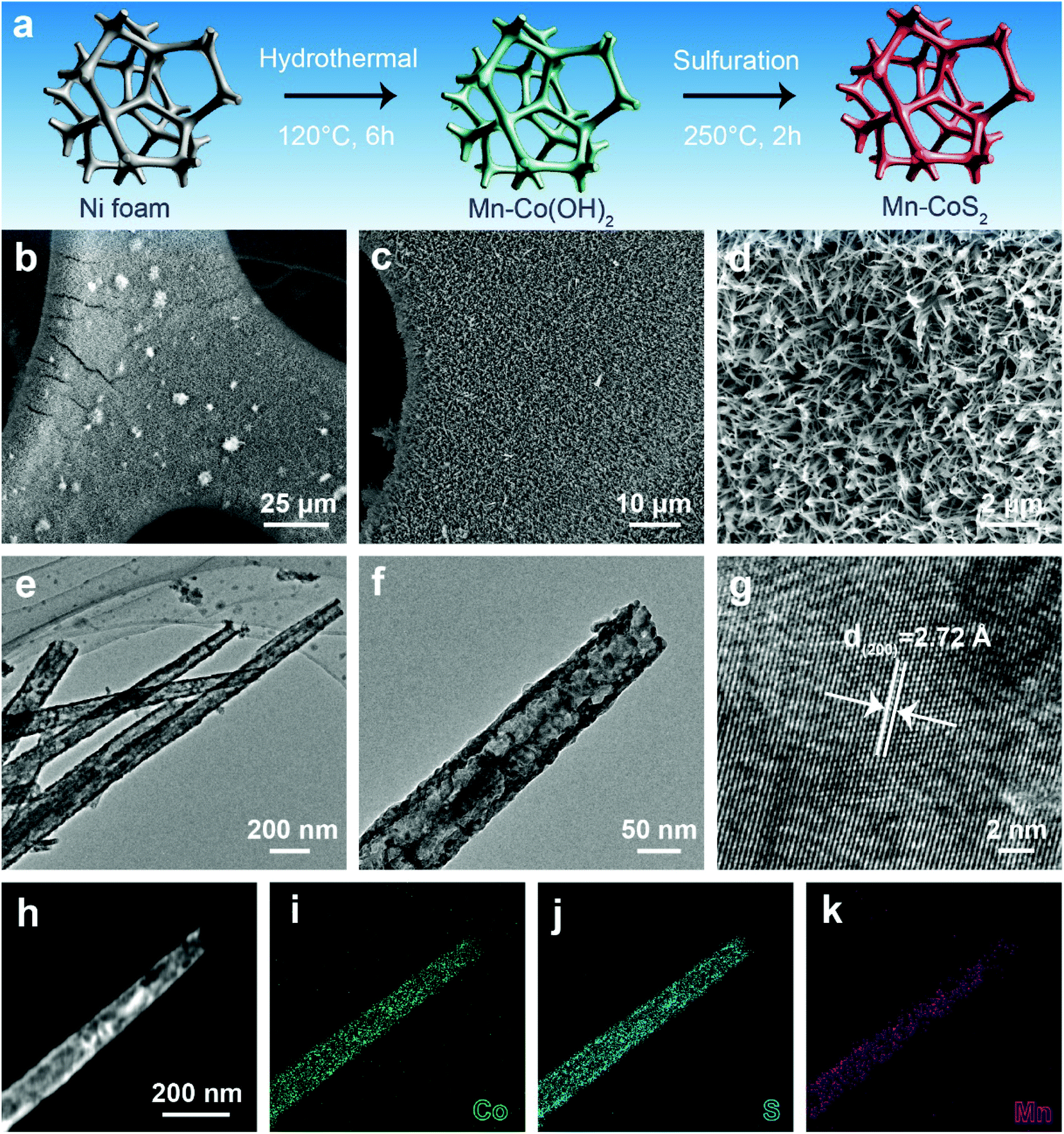 | ||
| Fig. 2 Morphology and structural characterization of Mn-CoS2: (a) schematic illustration of the synthesized process of Mn-CoS2; (b–d) SEM images; (e and f) low-magnification TEM images; (g) high-magnification TEM image; (h–k) HAADF-STEM image and the corresponding EDS mapping. | ||
Scanning electron microscopy (SEM) images show that the whole surface of NF is completely covered by interconnected Mn-CoS2 nanoarrays (Fig. 2b–d). The as-prepared Mn-CoS2 sample displays a nanotube morphology with a few hundred microns in length and a diameter of about 100 nanometers, as confirmed by transmission electron microscopy (TEM) (Fig. 2e and f). A high-resolution TEM image shows the visible lattice fringes with an equal interplanar distance of 2.72 Å that corresponds to the (200) plane of cubic CoS2, revealing the high crystallization feature of the Mn-CoS2 nanotubes (Fig. 2g). Furthermore, this nanotube structure is further explored by the elemental mapping images of the energy-dispersive X-ray spectroscopy (EDS) analysis. As shown in Fig. 2h–k, the Mn signals uniformly overlap with the Co and S signals in the as-prepared Mn-CoS2 catalyst.
The crystallographic structures of the as-prepared CoS2 and Mn-CoS2 catalysts are also characterized by X-ray diffraction (XRD). As the XRD pattern is shown in Fig. 3a, the as-prepared CoS2 and Mn-CoS2 exhibit similar XRD diffraction peak positions. In detail, the diffraction peaks at 32.2°, 36.2°, 39.78°, 46.31°, 54.88°, 57.53°, 60.22°, and 62.6°, which correspond to the (200), (210), (211), (220), (311), (222), (023), and (321) planes of cubic CoS2 (JCPDS no. 89-1492). In detail, it can be observed that the XRD diffraction peaks are shifted to a high angle relative to that of CoS2 (Fig. S6†), which is caused by the incorporation of Mn atoms with a smaller atomic radius into the lattice of CoS2 by replacing partial of Co atoms, confirming the successfully doping of Mn atoms into CoS2.44 Importantly, no Mn-related phase could be detected within the XRD detection range, indicating that a Mn-source was completely inserted in the CoS2 lattice. The Mn-doping level is determined to be 2.1wt% by inductively coupled plasma mass spectrometry (ICP-MS) analysis. Furthermore, X-ray photoelectron spectroscopy (XPS) measurements are employed to investigate the chemical state of the as-prepared catalysts. As shown in Fig. 3b and c, it can be found that CoS2 and Mn-CoS2 have similar high-resolution Co 2p and S 2p spectra, revealing that Mn doping does not affect the valence states of Co and S. In detail, as for the Co 2p spectra of Mn-CoS2, the core-level peaks at binding energies of 778.7 eV, 782.3 eV, 793.6, eV and 798.6 eV correspond to Co3+ 2p3/2, Co2+ 2p3/2, Co3+ 2p1/2, and Co2+ 2p1/2, respectively.26,52 Concerning the S region of Mn-CoS2 (Fig. 3c), the signals at 162.6 and 163.9 eV are attributed to the binding energies of S 2p3/2 and S 2p1/2, respectively, which belong to the typical metal–sulfur bonds.26,52 The peak at the binding energy of 168.7 eV could represent the S–O bond arising from surface air exposure.53 Impressively, the Co2+ 3d3/2 doublet of Mn-CoS2 is shifted by ≈1.6 eV toward a higher binding energy relative to that of pristine CoS2, whereas S 2p3/2 is shifted by ≈0.3 eV toward a higher binding energy. These shifts might be attributed to that the Mn dopant can provide more electrons to S than Co after Mn doping. Moreover, the high-resolution Mn 2p XPS spectrum (Fig. 2c) displays two representative XPS characteristic peaks at 643.1 and 653.4 eV corresponding to the Mn 2p3/2 and Mn 2p1/2 with an Mn4+ oxidation state in Mn-CoS2, respectively. Based on the above XRD and XPS analysis, it can be concluded that the Mn-CoS2 catalyst was successfully synthesized.
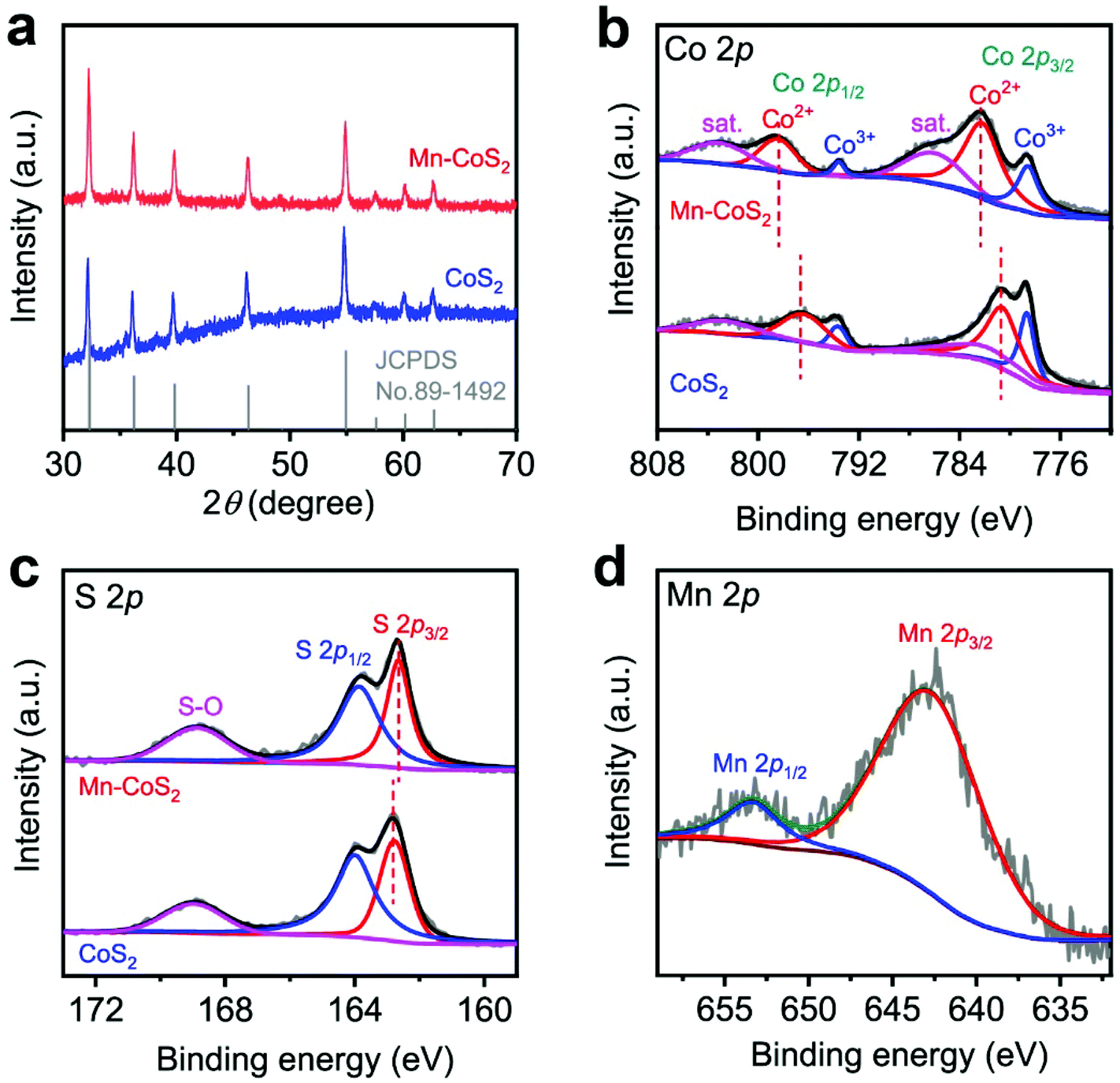 | ||
| Fig. 3 Structural characterization of the as-prepared CoS2 and Mn-CoS2: (a) XRD patterns; high-resolution XPS (b) Co 2p, (c) S 2p, and (d) Mn 2p spectra. | ||
To demonstrate the facilitation of catalytic reactions that arise from Mn-doping, the as-prepared Mn-CoS2 catalyst was evaluated for the HER with a three-electrode configuration in 0.1 M KOH electrolytes, where Mn-CoS2 supported on NF was directly used as the working electrode, and the reference electrode and counter electrode were the Ag/AgCl electrode (filled with saturated KCl) and the carbon rod, respectively. Firstly, electrochemical HER catalytic activity was evaluated using linear scan voltammogram (LSV) with a scan rate of 10 mA s−1. As the polarization curves are shown in Fig. 4a, the Mn-CoS2 catalyst exhibits a negligible onset potential of 7 mV vs. RHE at a current density of −1 mA cm−2 near the thermodynamic potential of the HER. Moreover, Mn-CoS2 achieved a much lower overpotential of 46 mV at a current density of −10 mA cm−2, which is much better than that of pristine CoS2 (228 mV) and comparable to those of 20 wt% commercial Pt/C catalysts (Fig. 4b), Mn-CoS2 with different Mn contents (Fig. S7†), and previously reported HER catalysts (Table S1†). Furthermore, Tafel plots were derived from the polarization curves to provide a deeper insight into the detailed mechanism of the HER of the as-prepared Mn-CoS2 catalyst. As shown in Fig. 4c, a Tafel slope of 63.1 mV dec−1 for the Mn-CoS2 catalyst is close to that of Pt/C (38.2 mV dec−1).
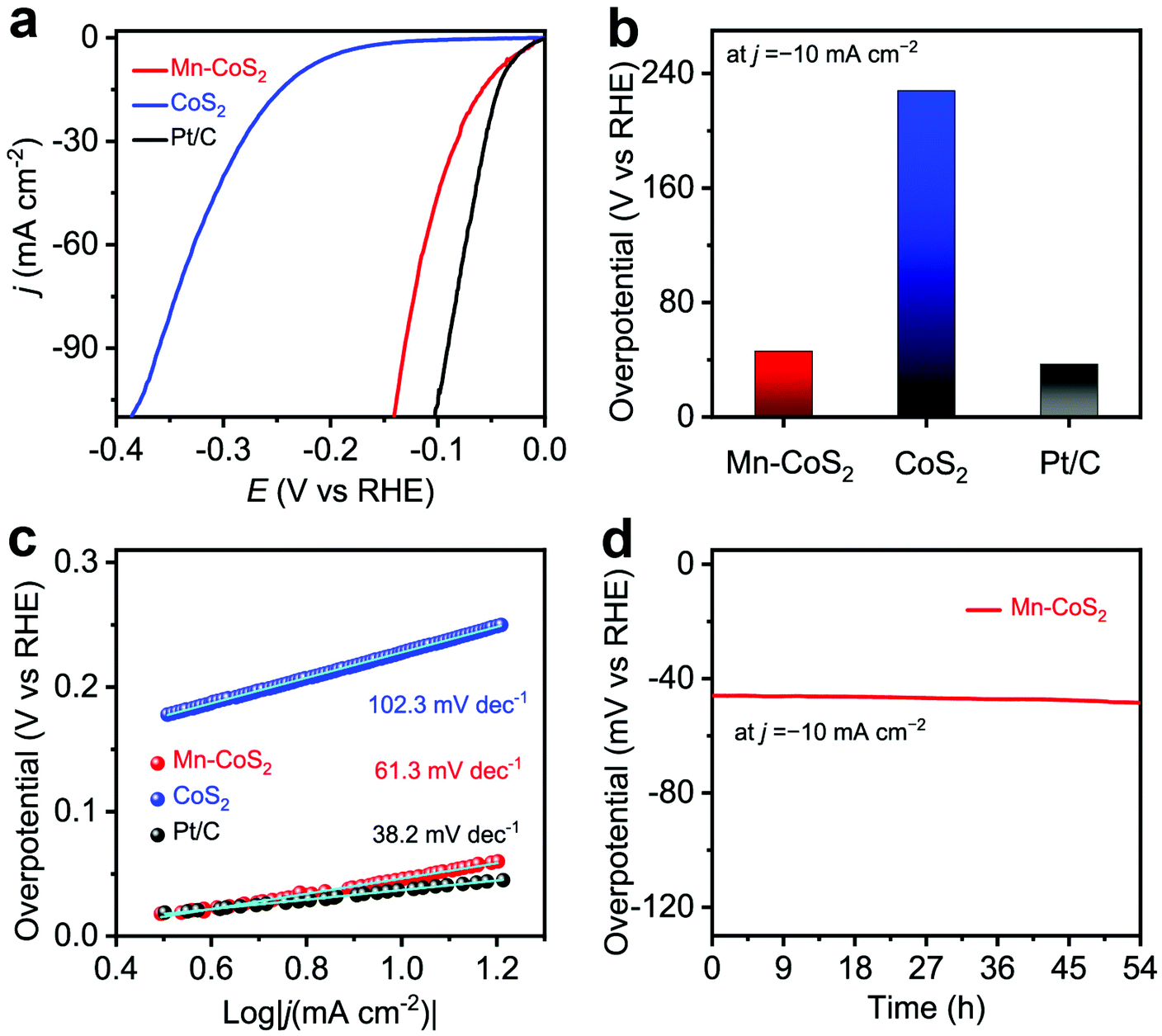 | ||
| Fig. 4 HER performance measurements in 0.1 M KOH aqueous solution: (a) polarization curves of Mn-CoS2 in comparison with CoS2 and commercial Pt/C (20 wt%); (b) the activity of the Mn-CoS2 catalyst at a current density of −10 mA cm−2 with respect to the reference catalysts. (c) Tafel plots derived from the corresponding polarization curves. (d) The time-dependent overpotential curve of the Mn-CoS2 electrode obtained at a constant current density of −10 mA cm−2 for 54 h under alkaline conditions. | ||
Furthermore, to gain insights into the interfacial charge transfer kinetics on the catalytic surface during the HER process, electrochemical impedance spectroscopy (EIS) was conducted. Fig. S8† shows the Nyquist plots of the CoS2 and Mn-CoS2 electrodes with a frequency from 100 kHz to 10 mHz. It can be found that the Nyquist plots present a typical semicircle in the high-frequency region and a straight line in the low-frequency region profiles, which can be ascribed to the charge-transfer resistance of the electrochemical H+ reaction at the electrode–electrolyte interface and the diffusion-controlled impedance, respectively.54,55 Obviously, the Mn-CoS2 electrode exhibits an enhanced electron transfer rate and faster catalytic kinetics during the HER process, eventually leading to the enhanced activity for the HER. To explore the enhanced HER activity of Mn-CoS2 relative to CoS2, electrochemical double-layer capacitances (Cdl) were measured. As shown in Fig. S9,† the cyclic voltammograms (CVs) of Mn-CoS2 and CoS2 at scan rates of 2, 4, 6, and 8 mV s−1, respectively. The Mn-CoS2 electrode achieved a Cdl value of 82.5 mF cm−2, which is 4.12 times larger than that of CoS2 (20.5 mF cm−2), implying a higher surface area and more exposed active sites for Mn-CoS2, which is beneficial for enhancing the HER activity.56,57
To assess the durability of the Mn-CoS2 catalyst, the chronopotentiometry–time measurement technology was employed. As shown in Fig. 4d, it can be seen that the Mn-CoS2 electrode can maintain a high stable overpotential at a current density of −10 mA cm−2 for 54 hours with a negligible increase (Fig. 3e). Moreover, the long-term cycling stability of Mn-CoS2 was investigated by performing continuous cyclic voltammetry (CV) at a scan rate of 100 mV s−1 in 0.1 M KOH. As shown in Fig. S10,† the HER polarization curves showed a negligible difference between the curves at the initial and after 1000 cycles, implying the superior stability of Mn-CoS2 in the long-term electrochemical process. Besides, the Mn-CoS2 catalyst after the long-term measurement was further characterized by XRD, SEM, and TEM. As shown in Fig. S11,† the crystalline structure and morphology were well preserved after the long-term electrolysis, demonstrating the robust stability of Mn-CoS2 in alkaline electrolytes.
Next, the electrochemical HzOR performances of the as-prepared catalyst were evaluated in 1.0 M KOH + 0.5 M N2H4 aqueous solutions. Meanwhile, the OER performance was also tested in a 1.0 M KOH aqueous solution. Firstly, the HzOR and OER catalytic activity was measured (Fig. 5a and Fig. S12†). As shown, the Mn-CoS2 electrode exhibits a rapid increase in the HzOR activity in the alkaline electrolyte than that of the acidic one. The comparative polarization curves of the HzOR and OER suggest a dramatic improvement of activity after the addition of N2H4, indicating a great thermodynamic advantage of the HzOR. In detail, it only needs a working potential of 77 mV vs. RHE to achieve a current density of 10 mA cm−2, which is much lower than that of the CoS2 electrode (102 mV vs. RHE). In particular, the Mn-CoS2 electrode possesses large current densities of 100 and 200 mA cm−2 and only requires the potentials of 160 and 188 mV vs. RHE, which is much larger than CoS2 (216 and 282 mV vs. RHE) and even better than the commercial Pt/C catalyst (161 and 262 mV vs. RHE), indicating that the outstanding HzOR activity of the Mn-CoS2 electrode makes it a superior HzOR electrocatalyst, which is highly comparable to previously reported HzOR electrocatalysts (Table S2†). Fast charge transfer kinetics is further demonstrated by a small Tafel slope of 47.1 mV dec−1, which is much smaller than that of CoS2 (58 mV dec−1) and much closer to that of Pt/C (41.5 mV dec−1). In addition, the current density of 10 mA cm−2 just changes slightly during a long-term test for 40 h (Fig. 5d) and the HzOR polarization curves showed a slight change between the curves at the initial and after 1000 cycles (Fig. S13†), demonstrating the high stability of the Mn-CoS2 electrode in the HzOR for practical applications.
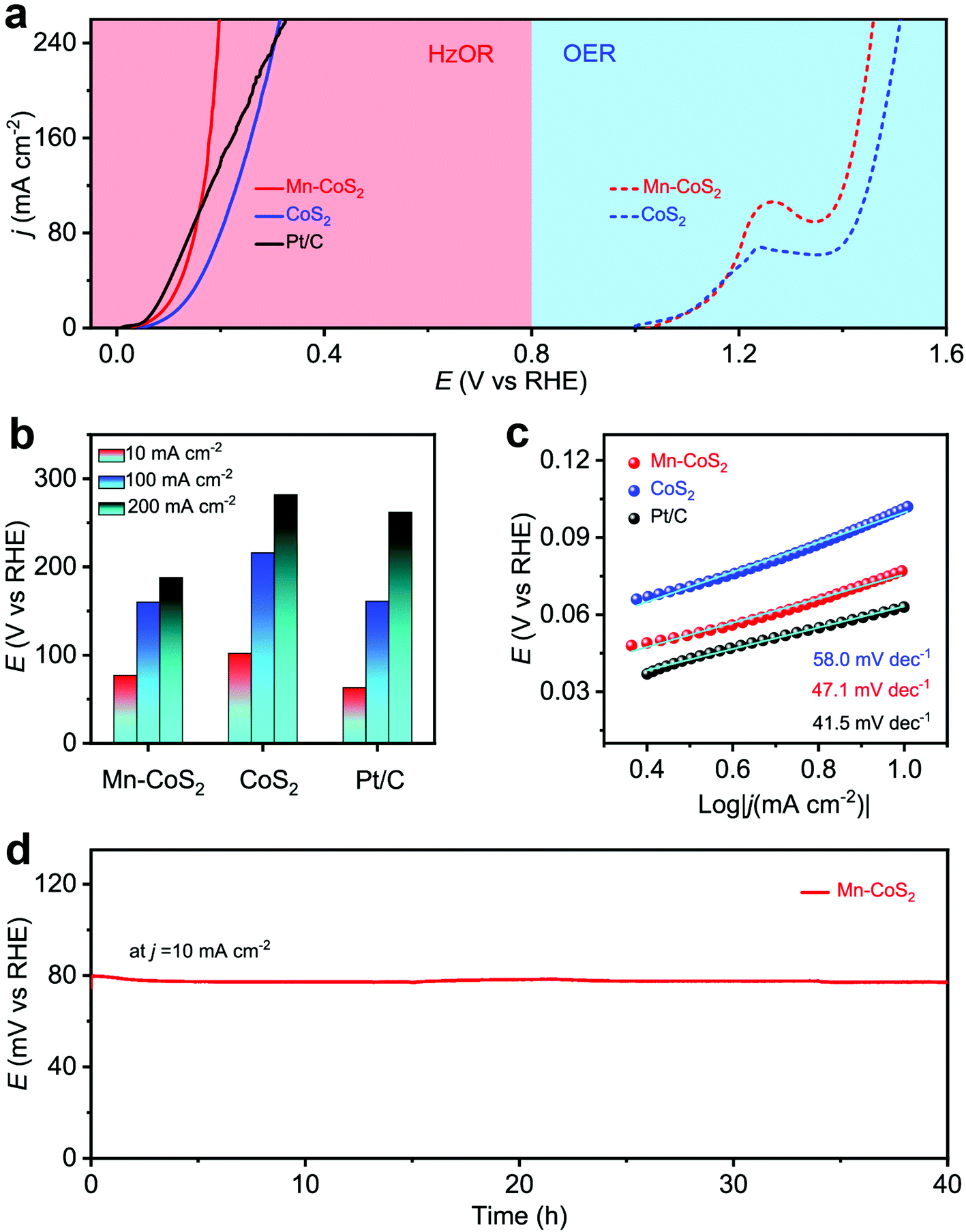 | ||
| Fig. 5 HzOR and OER measurements: (a) HzOR polarization curves of Mn-CoS2 in comparison with CoS2 and commercial Pt/C (20 wt%) in 1 M KOH + 0.5 M N2H4 aqueous solutions and the OER–HzOR polarization curves in 1.0 M KOH aqueous solutions; (b) the activity of the Mn-CoS2 catalyst at various current densities with respect to the reference catalysts; (c) HzOR Tafel plots derived from the corresponding HzOR polarization curves; (d) the time-dependent working potential curve of the Mn-CoS2 electrode obtained at a constant current density of 10 mA cm−2 for 40 h. | ||
Considering the intrinsic activities of the Mn-CoS2 catalyst, we envisioned that it could realize energy-saving H2 production by utilizing Mn-CoS2 as a bifunctional electrocatalyst toward the HER and HzOR. Fig. 6a shows the comparative LSV curves of overall hydrazine splitting (OHzS), in which significantly enhanced energy efficiency can be intuitively seen using hydrazine oxidation-assisted H2 production. Specifically, the OHzS cell using Mn-CoS2 as both the anode and cathode only demands 111, 329, and 447 mV to drive current densities of 10, 100, and 200 mA cm−2, respectively (Fig. 6b), which are much lower than the overall water-splitting theoretical value of 1230 mV. By contrast, CoS2 (405, 933, and 1227 mV) and commercial Pt/C (86, 319, and 467 mV) catalysts require a higher voltage to reach up to the current densities of 10, 100, and 200 mA cm−2 (Fig. 6b). Besides, the bifunctional Mn-CoS2 catalyst in the OHzS system can maintain higher voltage stability at various current densities (10 and 50 mA cm−2) during a continuous 24 h test (Fig. 6c). Moreover, the OHzS stability test of the Mn-CoS2 electrode was also performed for 24 h at a cell potential of 111 mV. As shown in Fig. 6, the current density retention can be determined to be 96.5%. The Mn-CoS2 electrode is highly comparable to previously reported state-of-the-art OHzS electrocatalysts (Table S3†). These results demonstrate that the Earth-abundant Mn-CoS2 catalyst as a bifunctional electrode for the HER and HzOR can efficiently save energy for H2 production by water-assisted electrolysis.
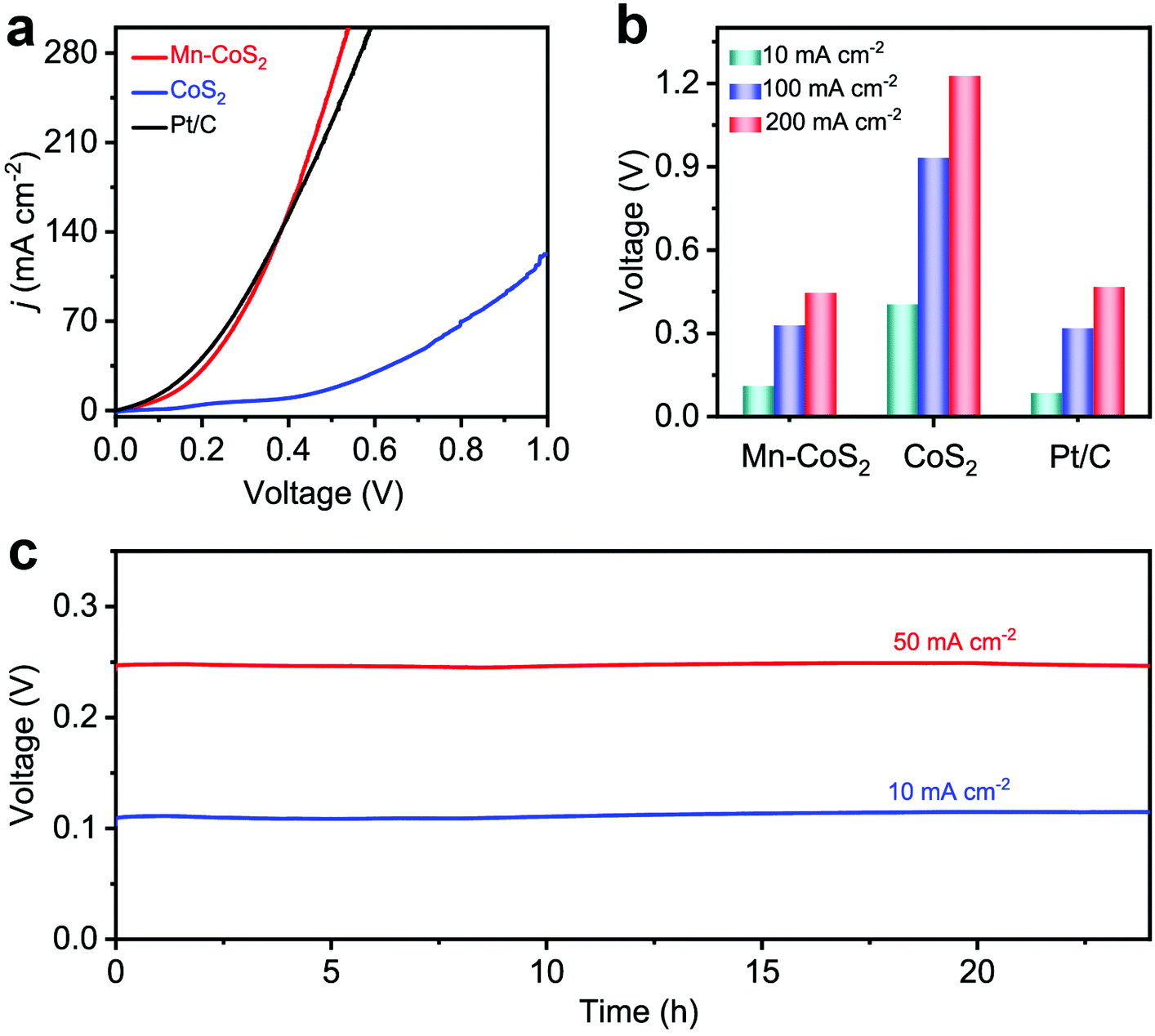 | ||
| Fig. 6 OHzS measurements: (a) OHzS polarization curves of Mn-CoS2 in comparison with CoS2 and commercial Pt/C (20 wt%) in 1 M KOH + 0.5 M N2H4 aqueous solutions; (b) the OHzS activity of the catalysts at various current densities; (c) the time-dependent working potential curve of the Mn-CoS2 electrode for OHzS obtained at various current densities for 24 h. | ||
Conclusions
In summary, we designed and prepared highly active Mn-doped CoS2 nanotubes supported nickel foam for boosting the electrochemical H2 evolution reaction by replacing the sluggish OER with a thermodynamically more favorable HzOR. DFT calculations manifest that the incorporation of Mn into the CoS2 lattice realizes the modulation of the electronic structure and charge distribution on the catalyst, which significantly decrease the hydrogen adsorption Gibbs free energy for the HER and the potential rate-limiting step for the HzOR process. The experimental results demonstrated that the Mn-CoS2 catalyst exhibited excellent catalytic activity, which can achieve 10 mA cm−2 at a overpotential of 46 mV vs. RHE for the HER and 77 mV vs. RHE for the anodic HzOR, respectively. In addition, the Mn-CoS2 electrode only needs a cell voltage of only 447 mV to output 200 mA cm−1 in the OHzS system and exhibits robust long-term H2 production. It is expected that this work can extend the pathway of the design for high-efficiency electrochemical H2 evolution catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22075211, 21601136, 51971157, 62005173, and 51621003), the Zhejiang Provincial Natural Science Foundation of China (LR19B060002), the Research Funds of Institute of Zhejiang University-Quzhou. The Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (2016), the Guangdong Third Generation Semiconductor Engineering Technology Development Center (2020GCZX007), the Science, Technology, and Innovation Commission of Shenzhen Municipality (RCBS20200714114818140), the China Postdoctoral Science Foundation (2019M663118), and the School level Scientific Research Project of Shenzhen Institute of Information Technology (PT2019E002). We are grateful for the HZWTECH for providing computation facilities.Notes and references
- N. Armaroli and V. Balzani, The Future of Energy Supply: Challenges and Opportunities, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, The Role of Hydrogen and Fuel Cells in the Global Energy System, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- I. P. Jain, Hydrogen the Fuel for 21st Century, Int. J. Hydrogen Energy, 2009, 34, 7368–7378 CrossRef CAS.
- C.-J. Winter, Hydrogen Energy - Abundant, Efficient, Clean: A Debate over the Energy-system-of-Change, Int. J. Hydrogen Energy, 2009, 34, S1–S52 CrossRef CAS.
- J. A. Turner, Sustainable Hydrogen Production, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Combining Theory and Experiment in Electrocatalysis: Insights into Materials Design, Science, 2017, 355, eaad4998 CrossRef PubMed.
- R. Abbasi, B. P. Setzler, S. Lin, J. Wang, Y. Zhao, H. Xu, B. Pivovar, B. Tian, X. Chen, G. Wu and Y. Yan, A Roadmap to Low-Cost Hydrogen with Hydroxide Exchange Membrane Electrolyzers, Adv. Mater., 2019, 31, 1805876 CrossRef PubMed.
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Water Photolysis at 12.3% Efficiency via Perovskite Photovoltaics and Earth-Abundant Catalysts, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Earth-Abundant Catalysts for Electrochemical and Photoelectrochemical Water Splitting, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Design of Active and Stable Co-Mo-Sx Chalcogels as pH-Universal Catalysts for the Hydrogen Evolution Reaction, Nat. Mater., 2016, 15, 197–203 CrossRef CAS PubMed.
- J. Yang, J. K. Cooper, F. M. Toma, K. A. Walczak, M. Favaro, J. W. Beeman, L. H. Hess, C. Wang, C. Zhu, S. Gul, J. Yano, C. Kisielowski, A. Schwartzberg and I. D. Sharp, A Multifunctional Biphasic Water Splitting Catalyst Tailored for Integration with High-Performance Semiconductor Photoanodes, Nat. Mater., 2017, 16, 335–341 CrossRef CAS PubMed.
- X. Li, X. Hao, A. Abudula and G. Guan, Nanostructured Catalysts for Electrochemical Water Splitting: Current State and Prospects, J. Mater. Chem. A, 2016, 4, 11973–12000 RSC.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. B. Chorkendorff and J. K. Norskov, Computational High-Throughput Screening of Electrocatalytic Materials for Hydrogen Evolution, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- Y. Jiao, Y. Zheng, K. Davey and S.-Z. Qiao, Activity Origin and Catalyst Design Principles for Electrocatalytic Hydrogen Evolution on Heteroatom-Doped Graphene, Nat. Energy, 2016, 1, 16130 CrossRef CAS.
- P. Chen, T. Zhou, M. Zhang, Y. Tong, C. Zhong, N. Zhang, L. Zhang, C. Wu and Y. Xie, 3D Nitrogen-Anion-Decorated Nickel Sulfides for Highly Efficient Overall Water Splitting, Adv. Mater., 2017, 29, 1701584 CrossRef PubMed.
- Z. Pu, S. Wei, Z. Chen and S. Mu, Flexible Molybdenum Phosphide Nanosheet Array Electrodes for Hydrogen Evolution Reaction in a Wide pH Range, Appl. Catal., B, 2016, 196, 193–198 CrossRef CAS.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Advancing the Electrochemistry of the Hydrogen-Evolution Reaction Through Combining Experiment and Theory, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- J. Yin, Q. Fan, Y. Li, F. Cheng, P. Zhou, P. Xi and S. Sun, Ni-C-N Nanosheets as Catalyst for Hydrogen Evolution Reaction, J. Am. Chem. Soc., 2016, 138, 14546–14549 CrossRef CAS PubMed.
- H. Liu, J. Guan, S. Yang, Y. Yu, R. Shao, Z. Zhang, M. Dou, F. Wang and Q. Xu, Metal-Organic-Framework-Derived Co2P Nanoparticle/Multi-doped Porous Carbon as a Trifunctional Electrocatalyst, Adv. Mater., 2020, 32, 2003649 CrossRef CAS PubMed.
- T. Liu, D. Liu, F. Qu, D. Wang, L. Zhang, R. Ge, S. Hao, Y. Ma, G. Du, A. M. Asiri, L. Chen and X. Sun, Enhanced Electrocatalysis for Energy-Efficient Hydrogen Production over CoP Catalyst with Nonelectroactive Zn as A Promoter, Adv. Energy Mater., 2017, 7, 1700020 CrossRef.
- X. Wang, Y. V. Kolen'ko, X. Q. Bao, K. Kovnir and L. Liu, One-Step Synthesis of Self-Supported Nickel Phosphide Nanosheet Array Cathodes for Efficient Electrocatalytic Hydrogen Generation, Angew. Chem., Int. Ed., 2015, 54, 8188–8192 CrossRef CAS PubMed.
- L. Yang, R. Liu and L. Jiao, Electronic Redistribution: Construction and Modulation of Interface Engineering on CoP for Enhancing Overall Water Splitting, Adv. Funct. Mater., 2020, 30, 1909618 CrossRef CAS.
- P. Chen, T. Zhou, M. Chen, Y. Tong, N. Zhang, X. Peng, W. Chu, X. Wu, C. Wu and Y. Xie, Enhanced Catalytic Activity in Nitrogen-Anion Modified Metallic Cobalt Disulfide Porous Nanowire Arrays for Hydrogen Evolution, ACS Catal., 2017, 7, 7405–7411 CrossRef CAS.
- J. Hao, W. Yang, Z. Peng, C. Zhang, Z. Huang and W. Shi, A Nitrogen Doping Method for CoS2 Electrocatalysts with Enhanced Water Oxidation Performance, ACS Catal., 2017, 7, 4214–4220 CrossRef CAS.
- R. Miao, B. Dutta, S. Sahoo, J. He, W. Zhong, S. A. Cetegen, T. Jiang, S. P. Alpay and S. L. Suib, Mesoporous Iron Sulfide for Highly Efficient Electrocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2017, 139, 13604–13607 CrossRef CAS PubMed.
- J. Zhang, W. Xiao, P. Xi, S. Xi, Y. Du, D. Gao and J. Ding, Activating and Optimizing Activity of CoS2 for Hydrogen Evolution Reaction Through the Synergic Effect of N Dopants and S Vacancies, ACS Energy Lett., 2017, 2, 1022–1028 CrossRef CAS.
- Z. Chen, Y. Song, J. Cai, X. Zheng, D. Han, Y. Wu, Y. Zang, S. Niu, Y. Liu, J. Zhu, X. Liu and G. Wang, Tailoring the d-band Centers Enables Co4N Nanosheets to be Highly Active for Hydrogen Evolution Catalysis, Angew. Chem., Int. Ed., 2018, 57, 5076–5080 CrossRef CAS PubMed.
- N. Yao, P. Li, Z. Zhou, Y. Zhao, G. Cheng, S. Chen and W. Luo, Synergistically Tuning Water and Hydrogen Binding Abilities over Co4N by Cr Doping for Exceptional Alkaline Hydrogen Evolution Electrocatalysis, Adv. Energy Mater., 2019, 9, 1902449 CrossRef CAS.
- Y. Men, P. Li, F. Yang, G. Cheng, S. Chen and W. Luo, Nitrogen-Doped CoP as Robust Electrocatalyst for High-Efficiency pH-universal Hydrogen Evolution Reaction, Appl. Catal., B, 2019, 253, 21–27 CrossRef CAS.
- Y. Men, P. Li, J. Zhou, S. Chen and W. Luo, Trends in Alkaline Hydrogen Evolution Activity on Cobalt Phosphide Electrocatalysts Doped with Transition Metals, Cell Rep. Phys. Sci., 2020, 1, 100136 CrossRef CAS.
- Y. Men, P. Li, J. Zhou, G. Cheng, S. Chen and W. Luo, Tailoring the Electronic Structure of Co2P by N Doping for boosting Hydrogen Evolution Reaction at All pH Values, ACS Catal., 2019, 9, 3744–3752 CrossRef CAS.
- Y. Men, Y. Tan, P. Li, X. Cao, S. Jia, J. Wang, S. Chen and W. Luo, Tailoring the 3d-Orbital Electron Filling Degree of Metal Center to Boost Alkaline Hydrogen Evolution Electrocatalysis, Appl. Catal., B, 2021, 284, 119718 CrossRef CAS.
- Y. Li, J. He, W. Cheng, H. Su, C. Li, H. Zhang, M. Liu, W. Zhou, X. Chen and Q. Liu, High Mass-Specific Reactivity of a Defect-Enriched Ru Electrocatalyst for Hydrogen Evolution in Harsh Alkaline and Acidic Media, Sci. China Mater., 2021, 64, 2467–2476 CrossRef CAS.
- Y. Pan, C. Zhang, Y. Lin, Z. Liu, M. Wang and C. Chen, Electrocatalyst Engineering and Structure-Activity Relationship in Hydrogen Evolution Reaction: From Nanostructures to Single Atoms, Sci. China Mater., 2020, 63, 921–948 CrossRef CAS.
- H. Yang, C. Wang, F. Hu, Y. Zhang, H. Lu and Q. Wang, Atomic-Scale Pt Clusters Decorated on Porous α-Ni(OH)2 Nanowires as Highly Efficient Electrocatalyst for Hydrogen Evolution Reaction, Sci. China Mater., 2017, 60, 1121–1128 CrossRef CAS.
- J. T. Ren, Y. Yao and Z. Y. Yuan, Fabrication Strategies of Porous Precious-Metal-Free Bifunctional Electrocatalysts for Overall Water Splitting: Recent advances, Green Energy Environ., 2021, 6, 620–643 CrossRef.
- X. W. Lv, W. S. Xu, W. W. Tian, H. Y. Wang and Z. Y. Yuan, Activity Promotion of Core and Shell in Multifunctional Core-Shell Co2P@NC Electrocatalyst by Secondary Metal Doping for Water Electrolysis and Zn-Air Batteries, Small, 2021, 17, 2101856 CrossRef CAS PubMed.
- J. Zhang, Y. Liu, C. Sun, P. Xi, S. Peng, D. Gao and D. Xue, Accelerated Hydrogen Evolution Reaction in CoS2 by Transition-Metal Doping, ACS Energy Lett., 2018, 3, 779–786 CrossRef CAS.
- C. C. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Electrochemical Oxidation of 5-Hydroxymethylfurfural on Nickel Nitride/Carbon Nanosheets: Reaction Pathway Determined by In Situ Sum Frequency Generation Vibrational Spectroscopy, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- B. Zhu, Z. Liang and R. Zou, Designing Advanced Catalysts for Energy Conversion Based on Urea Oxidation Reaction, Small, 2020, 16, 1906133 CrossRef CAS PubMed.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li and G. Zhang, Realizing the Synergy of Interface Engineering and Chemical Substitution for Ni3N Enables Its Bifunctionality Toward Hydrazine Oxidation Assisted Energy-Saving Hydrogen Production, Adv. Funct. Mater., 2021, 31, 2103673 CrossRef CAS.
- F. Sun, J. Qin, Z. Wang, M. Yu, X. Wu, X. Sun and J. Qiu, Energy-Saving Hydrogen Production by Chlorine-Free Hybrid Seawater Splitting Coupling Hydrazine Degradation, Nat. Commun., 2021, 12, 4182 CrossRef CAS PubMed.
- J. Y. Zhang, H. Wang, Y. Tian, Y. Yan, Q. Xue, T. He, H. Liu, C. Wang, Y. Chen and B. Y. Xia, Anodic Hydrazine Oxidation Assists Energy-Efficient Hydrogen Evolution over A Bifunctional Cobalt Perselenide Nanosheet Electrode, Angew. Chem., Int. Ed., 2018, 57, 7649–7653 CrossRef CAS PubMed.
- Hongzhiwei Technology, Device Studio, Version 2021B, China, 2021. Available online: https://iresearch.net.cn/cloudSoftware (accessed on 24 Feb. 2022).
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- M. Wang, W. Zhang, F. Zhang, Z. Zhang, B. Tang, J. Li and X. Wang, Theoretical Expectation and Experimental Implementation of In Situ Al-doped CoS2 Nanowires on Dealloying-Derived Nanoporous Intermetallic Substrate as An Efficient Electrocatalyst for Boosting Hydrogen Production, ACS Catal., 2019, 9, 1489–1502 CrossRef CAS.
- Y. Liu, J. Zhang, Y. Li, Q. Qian, Z. Li, Y. Zhu and G. Zhang, Manipulating Dehydrogenation Kinetics Through Dual-Doping Co3N Electrode Enables Highly Efficient Hydrazine Oxidation Assisting Self-Powered H2 Production, Nat. Commun., 2020, 11, 1853 CrossRef CAS PubMed.
- X. Peng, Y. Mi, X. Liu, J. Sun, Y. Qiu, S. Zhang, X. Ke, X. Wang and J. Luo, Self-Driven Dual Hydrogen Production System Based on Bifunctional Single-Atomic Rh Catalyst, J. Mater. Chem. A, 2022, 10, 6134–6145 RSC.
- Q. Qian, J. Zhang, J. Li, Y. Li, X. Jin, Y. Zhu, Y. Liu, Z. Li, A. El-Harairy, C. Xiao, G. Zhang and Y. Xie, Artificial Heterointerfaces Achieve Delicate Reaction Kinetics Towards Hydrogen Evolution and Hydrazine Oxidation Catalysis, Angew. Chem., Int. Ed., 2021, 60, 5984–5993 CrossRef CAS PubMed.
- L. Zhou, M. Shao, C. Zhang, J. Zhao, S. He, D. Rao, M. Wei, D. G. Evans and X. Duan, Hierarchical CoNi-sulfide Nanosheet Arrays Derived from Layered Double Hydroxides Toward Efficient Hydrazine Electrooxidation, Adv. Mater., 2017, 29, 1604080 CrossRef PubMed.
- Y. Li, Z. Mao, Q. Wang, D. Li, R. Wang, B. He, Y. Gong and H. Wang, Hollow Nanosheet Array of Phosphorus-Anion-Decorated Cobalt Disulfide as An Efficient Electrocatalyst for Overall Water Splitting, Chem. Eng. J., 2020, 390, 124556 CrossRef CAS.
- X. Cui, Z. Xie and Y. Wang, Novel CoS2 Embedded Carbon Nanocages by Direct Sulfurizing Metal-Organic Frameworks for Dye-Sensitized Solar Cells, Nanoscale, 2016, 8, 11984–11992 RSC.
- X. Tao, J. Wang, Z. Ying, Q. Cai, G. Zheng, Y. Gan, H. Huang, Y. Xia, C. Liang, W. Zhang and Y. Cui, Strong Sulfur Binding with Conducting Magneli-Phase TinO2n−1 Nanomaterials for Improving Lithium-Sulfur Batteries, Nano Lett., 2014, 14, 5288–5294 CrossRef CAS PubMed.
- W. Zhang, X. Zhou, X. Tao, H. Huang, Y. Gan and C. Wang, In Situ Construction of Carbon Nano-Interconnects between the LiFePO4 Grains Using Ultra Low-Cost Asphalt, Electrochim. Acta, 2010, 55, 2592–2596 CrossRef CAS.
- J. Tian, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Self-Supported Cu3P Nanowire Arrays as An Integrated High-Performance Three-Dimensional Cathode for Generating Hydrogen from Water, Angew. Chem., Int. Ed., 2014, 53, 9577–9581 CrossRef CAS PubMed.
- X. Peng, S. Zhao, Y. Mi, L. Han, X. Liu, D. Qi, J. Sun, Y. Liu, H. Bao, L. Zhuo, H. L. Xin, J. Luo and X. Sun, Trifunctional Single-Atomic Ru Sites Enable Efficient Overall Water Splitting and Oxygen Reduction in Acidic Media, Small, 2020, 16, 2002888 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The optimized structural model, electronic band structure, DOS, charge density difference, adsorption energy, XRD pattern, EIS, CV, ECSA, LSV, and i–t curves. See DOI: https://doi.org/10.1039/d2qi00083k |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2022 |