
Conquering residual light absorption in the transmissive states of organic electrochromic materials
Anna M.
Österholm
 a,
Linda
Nhon
a,
D. Eric
Shen
a,
Abigail M.
Dejneka
a,
Aimée L.
Tomlinson
b and
John R.
Reynolds
*a
a,
Linda
Nhon
a,
D. Eric
Shen
a,
Abigail M.
Dejneka
a,
Aimée L.
Tomlinson
b and
John R.
Reynolds
*a
aSchool of Chemistry and Biochemistry, School of Materials Science and Engineering, Center for Organic Photonics and Electronics, Georgia Tech Polymer Network, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: reynolds@chemistry.gatech.edu
bDepartment of Chemistry/Biochemistry, University of North Georgia, Dahlonega, Georgia 30597, USA
First published on 12th October 2021
Abstract
In this short review, we provide an overview of our efforts in developing a family of anodically coloring electrochromic (EC) molecules that are fully transparent and colorless in the charge neutral state, and that can rapidly switch to a vibrantly colored state upon oxidation. We employ molecules with reduced conjugation lengths to center the neutral state absorption of the electrochrome in the ultraviolet, as desired for highly transparent and colorless materials. Oxidation creates radical cations that absorb light in the visible and near infrared regions of the electromagnetic spectrum, thus providing a host of accessible colors. Combining a density functional theory (DFT) computational approach fed back to the synthetic effort, target molecules are proposed, synthesized and studied, directing us to develop a complete color palette based on these high contrast ACE molecules. Utilizing pendant phosphonic acid binding substituents in concert with high surface area mesoporous indium tin oxide (ITO) electrodes, the electrochromes can be distributed throughout the oxide film, bringing high extent of light absorption and color density.
1. Electrochromism: a case for mechanism and materials
Electrochromic (EC) molecules and materials, which interconvert from light absorbing colored states to highly transmissive and in some instances transparent states, are finding utility in absorption/transmission (window-type) and reflective (mirror- and display-type) electrochromic devices (ECDs).1 With applications ranging from architectural windows, to automotive mirrors, information communicating displays, and dynamic camouflage, a variety of approaches have been explored that encompass different fundamental mechanisms, device types, and materials. In a fundamental sense, two EC mechanisms are recognized based on cathodic and anodic coloration. In cathodic coloration, a colored material is (typically) converted to a transmissive state upon electrochemical oxidation, and subsequently back to the colored state when reduced. Another type of cathodic coloration is when a colorless material is converted to the colored state upon electrochemical reduction, and then back to the colorless state upon re-oxidation. In contrast, an anodically coloring system utilizes either a colorless electrochrome that is switched to the colored state when electrochemically oxidized, or a colored electrochrome that is switched to a transmissive state when it is electrochemically reduced.In ECDs incorporating molecular electrochromes, two redox-active molecules are typically dissolved in a gel electrolyte. One of the redox couples has a high extinction coefficient in the colored state and is responsible for the color change that is observed in the ECD. The most well-studied of these systems, as well as the most successful commercial devices, incorporate various viologen derivatives and can be found in electrochromic mirrors and in windows in aircrafts. Viologens convert from transparent dicationic species to deeply colored cation radicals, thus giving high optical contrast through light absorption/transmission.2 The other redox couple typically has a low extinction coefficient, but is highly redox active and undergoes an oxidation process when the viologen is reduced, and vice versa. The switching process of this type of ECD relies on mass transport as the two molecules need to diffuse to their respective electrode surfaces for electron transfer to occur in order to change the oxidation state and light absorption characteristics of the chromophore.
Electrochromes can also be affixed to an electrode surface as adsorbed species or deposited films, where they can be switched rapidly between their colored and transparent states. For this device type, transition metal oxides have found especially strong use due to their ability to switch between transparent and deeply absorbing states.3 Tungsten trioxide has to date received the most attention and development as it is especially stable to light and heat, and has long-lived switching characteristics, as needed for architectural applications. Another example of thin film EC materials are inorganic–organic hybrids, exemplified by metallo-supramolecular polymers, where the color change arises from metal-to-ligand or ligand-to-metal charge transfer processes. Here, the color can be tuned to some extent by changing either the identity of the metal or of the ligand.4–6 A third example of thin film EC materials, is all-organic and conjugated electrochromic polymers (ECPs) that have found their niche as they provide a high degree of color control, they can be cathodically or anodically coloring, and as a result of their synthetic versatility complete color palettes have already been developed.7,8 These accessible colors, especially those based on dioxythiophenes, are vibrant with rapid switching times (sub-second for cm2 devices) due to their inherent semiconducting nature and their ability to readily swell with electrolyte.
Also, to be considered in the area of light modulation, is changing the reflectivity at an electrode allowing light to either pass through the device or to fully reflect. This approach is exemplified by reversible metal electrodeposition where solubilized metal ions are reduced to form metallic films. In one instance using Ag deposition, devices have been developed with transparent default states, mirror reflectivity when the Ag is deposited on flat indium tin oxide (ITO), and a black state when the Ag is deposited on a rough ITO particle-modified electrode.9 These metallic films are color neutral, as desired for switchable windows, and have the ability to fully block and reflect all the impinging light with transmission less than 0.1%, as well as the ability to control heat, which leads to significant possibilities in windows with both visual and thermal control.9–16
While it is evident that significant headway has been made in EC material development, systems that bring the full extent of color control with complete transparency remain elusive. To this end, we have pursued a goal of developing a family of anodically coloring, conjugated small molecules that are fully transparent and colorless in the neutral state, and that can rapidly switch to a vibrantly colored state upon oxidation. By reducing the conjugation length, the neutral state absorption can be centered in the ultraviolet (UV), while by tuning the structural design of the molecule, the color attained in the oxidized state can be modulated to span the visible spectrum. Thus far, in the field of conjugated materials, structure–property relationships governing the color of the neutral state have been extensively explored. However, understanding these same relationships in the oxidized state has received little attention. In this short review, we present our developments on a new class of anodically coloring electrochromic (ACE) molecules that attain these goals.17 With this, we show how a new color palette is being developed using these highly transparent-to-colored switching materials with especially high EC contrast.
2. Tuning neutral state color in organic electrochromes
The color of a charge neutral, organic electrochrome originates from the HOMO–LUMO transition of the π-system. Upon electrochemical oxidation/reduction, radical cations/anions and dications/dianions are formed, manifesting themselves by new optical transitions at lower energies relative to the HOMO–LUMO transition, which red-shifts the absorption to longer wavelengths and changes the color of the electrochrome. The neutral state absorption is primarily determined by the molecule's optical gap, the magnitude of which stems from a competition between π-electron confinement (aromaticity) and electron delocalization within the molecule. Heteroatoms (X in Fig. 1a) play an important role in determining the absorption characteristics as they contribute two π-electrons, in addition to those from the π-bonds, and influence both the degree of aromaticity and π-donation. The choice and placement of functional substituents (Y and R in Fig. 1a) can be used to further manipulate the neutral state absorption via steric and electronic effects by altering the equilibrium geometry, bond length alternation, and dihedral angle between subunits (Fig. 1a)2,8,18–21 The rule of thumb is that any structural modification that results in twisting of the molecule will disrupt conjugation, which results in the absorption blue-shifting. Conversely, decreasing the dihedral twist angle will push the neutral state absorption towards lower energies. In addition to the steric and electronic effects that can be induced by functional substituents, the magnitude of the dihedral twist angle can also be modulated through secondary interactions such as hydrogen bonding and attractive sulfur–oxygen interactions.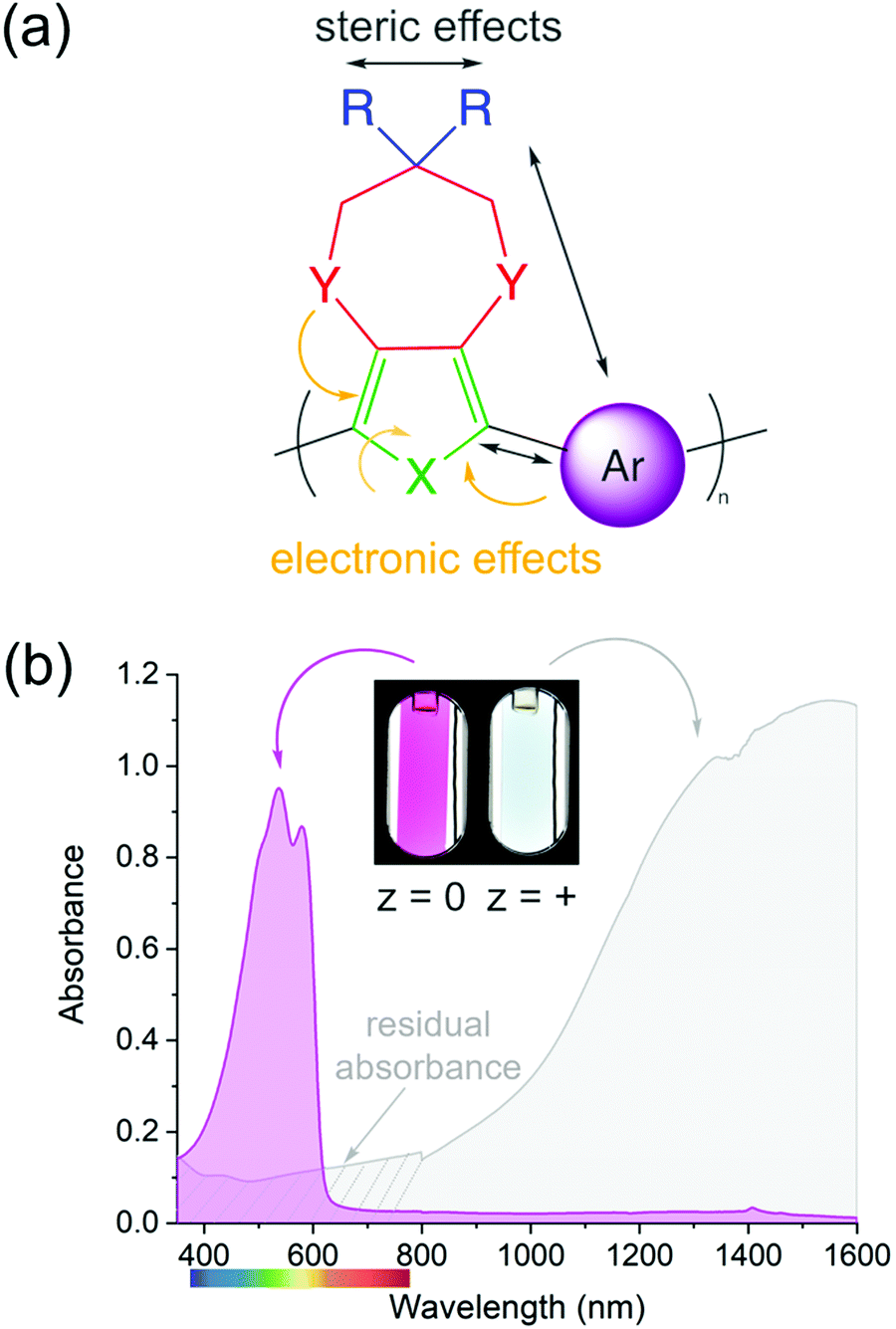 | ||
| Fig. 1 (a) Example of an organic, electrochromic subunit (where n can range from 1 to ∞), (b) absorption spectra and photographs of the charge neutral (magenta) and electrochemically oxidized (grey) forms of a magenta-to-clear switching ECP film. | ||
Among organic electrochromes, extensive structure–property relationships have been developed for conjugated polymers. Because of their extended conjugated backbones, ECPs in their neutral state typically absorb in the visible range (Fig. 1b, magenta spectrum) and an entire color palette of ECPs have already been developed.22 While color tuning of the neutral state is relatively straightforward, the main drawback with cathodically coloring ECPs is that their oxidized, transmissive state has a residual absorption throughout the visible range (as illustrated by the gray spectrum in Fig. 1b), and occasionally absorption bands originating from radical cation and dication states tail into the visible range giving the films a light-blue tint. The latter is particularly challenging to overcome for ECPs with yellow- and orange-colored neutral states that need to absorb high energy blue light when charge neutral, yet the radical cations and dications need to absorb fully in the NIR and beyond.23–28 While the electrochromic contrast in ECPs is high, as evidenced by the photographs in Fig. 1b, and the transmittance of the oxidized state can be enhanced by red-shifting the radical cation/dication bands via optimization of the processing conditions,29–31 decreasing inter-molecular interactions (i.e., fewer redox equilibriums between different charge carriers),32–35 and/or through non-covalent intramolecular interactions (electron donation, sulfur–oxygen interactions, hydrogen bonding, etc.),36–38 the transmissive state will not be perfectly clear. Conversely, because of their shorter conjugation length, small molecules can readily be designed such that their neutral state absorption is centered in the UV, with no absorption in the visible making the neutral state perfectly clear. Upon oxidation/reduction and radical formation, the absorption would then appear in the visible range. While tuning the neutral state absorbance has been thoroughly studied in conjugated systems, it is not well understood whether the same design principles can be applied to predictably control the color of the charged state(s), which often involves absorption from at least two transitions, both of which must be tuned not only for onset of absorption, but also the intensity and the saturation of the hue. In this short review, we highlight how we have utilized a combination of computational and synthetic approaches to establish these design principles to obtain predictable coloration in anodically coloring small molecules.
3. Fundamental approaches and theoretical design for tuning the color of anodically coloring electrochromes
Our starting structural design for an ACE molecule brings together a substituted phenylene ring and an 3,4-alkylenedioxythiophene (XDOT) subunit (Fig. 2). In order to center the neutral absorbance in the UV, the conjugation length is kept short; additionally, the aromaticity of the benzene ring coupled with the steric hindrance between R and Y introduce twisting between the subunits, leading to disruption of the conjugation. The XDOT is an electron-rich subunit that affords the molecule a relatively low oxidation potential to enhance the redox switching and redox stability of the molecule. To further tune steric and electronic effects, this structure also allows us to change the heteroatom and substituents, as well as vary the identity and number of pendant R groups on the benzene ring. Not only can electron withdrawing (e.g. cyano, halogen) and electron donating (e.g. alkoxy) groups be appended to influence electronic effects, steric effects between the subunits can also be exploited by increasing the size of the R group. Using these structural variables, we explored to what extent we could control the complexities of the oxidized spectra shown in Fig. 2, as well as whether we could obtain the desired colorless-to-colored switch through making small changes in substitution pattern.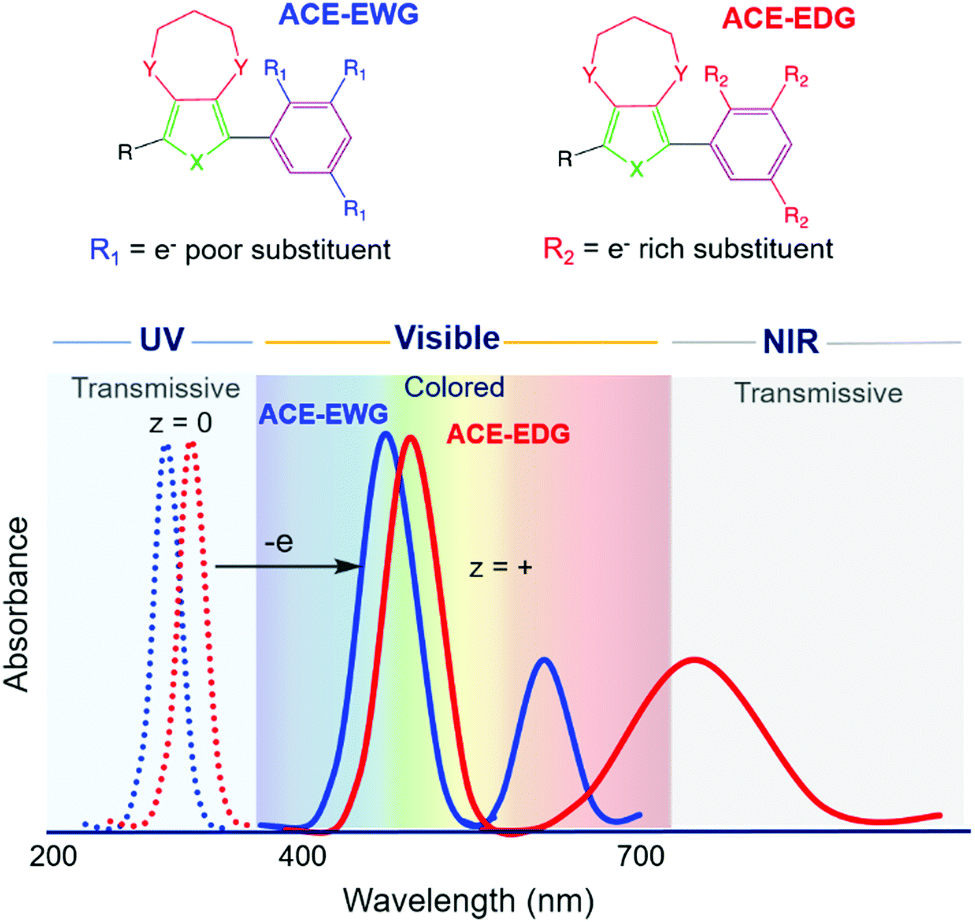 | ||
| Fig. 2 Illustration showing generic chemical structures of anodically coloring electrochromes, ACE-EWG and ACE-EDG and their absorbance profiles. In the neutral state, both molecules absorb in the UV (dashed lines) and in their oxidized states they absorb in the visible range, thereby appearing colored (solid lines). The position of the radical cation absorption can then be blue-shifted using electron withdrawing substituents (solid blue line) or red-shifted (solid red line) using electron donating substituents. | ||
Based on the large number of possible substituents that can be used, in addition to their positions on the arylene ring, we first use density functional theory (DFT) or the extended time-dependent density-functional theory (TDDFT) to screen large sets of systems to determine which ACE molecules and substituents will be the most promising candidates for obtaining specific colors, while also being readily accessible synthetically. In this process, the optimized geometries are frequency verified for the neutral, radical cation, and relevant dication forms, with the 15 lowest lying excited states used to simulate the UV-Vis spectra and excited state transitions. In our experience, we achieve the highest correlation to experimental data by using the mPW1PBE/cc-PVDZ pairing within a dielectric environment that emulates dichloromethane.39 From the simulated results, new ACE molecules can then be prioritized in order of most to least promising for synthesis and further development. Once synthesized, we then evaluate the electrochemical and optical properties of the new ACE molecules to gain an understanding of the color and switching behavior, as well as for continuous benchmarking in an effort to improve the DFT/TDDFT model.
The simulated data combined with spectroelectrochemical results and photographs in Fig. 3 show how changes in electron density impact the color of the radical cation states for two ACE molecules that have the same conjugated scaffold: a thiomethyl-capped 3,4-ethylenedioxythiophene (EDOT) conjugated to methoxy-substituted phenylenes where the extent of substitution modifies the molecule's electron density.17 By constraining the light absorption of the neutral form of these two-ring systems to the UV region, we ensure the molecules are transparent. Here, the mono para-methoxy substituted phenylene molecule (EDOT-MOB) is electron-poor compared to the tri-methoxy substituted analog (EDOT-TMOB).
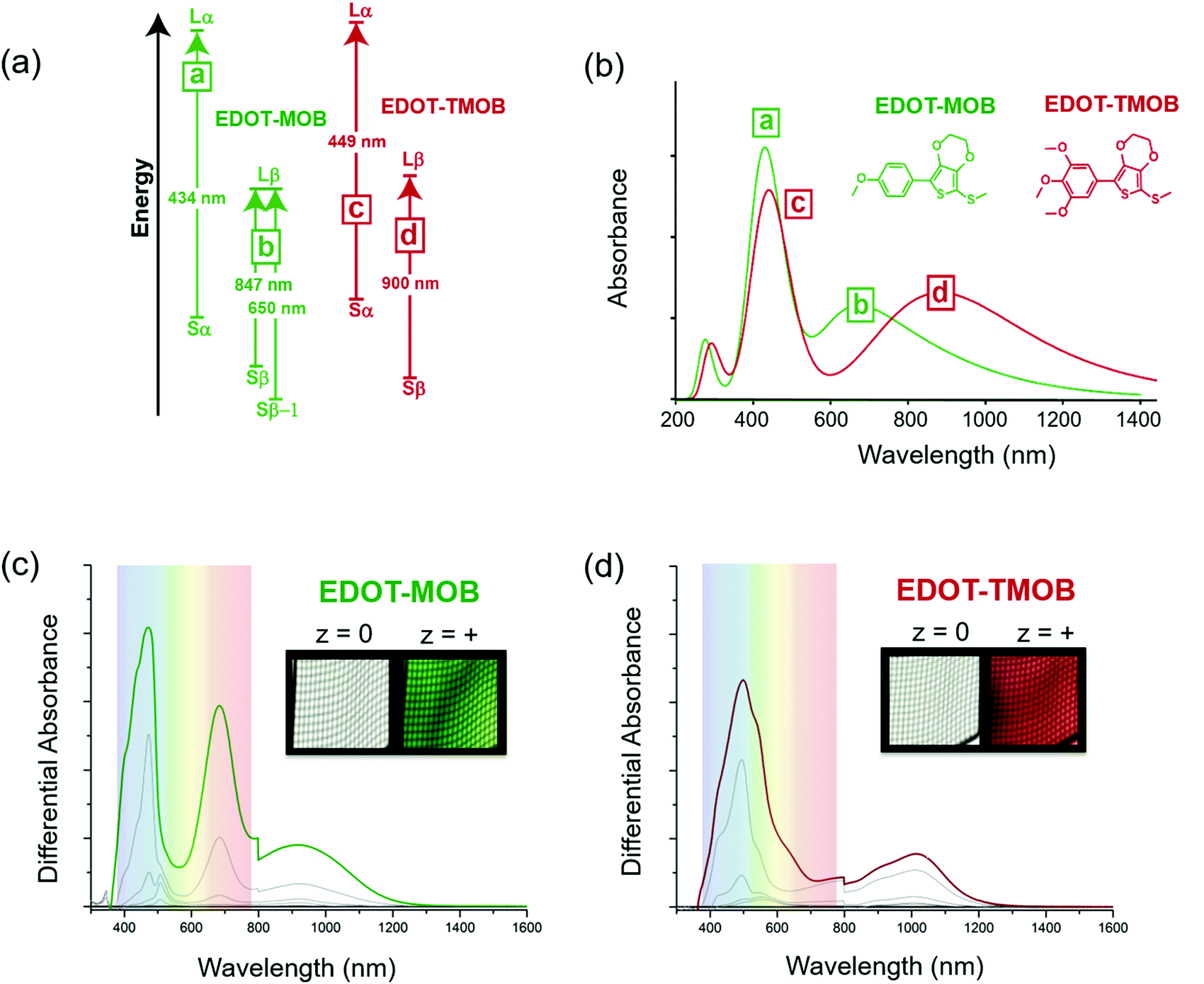 | ||
| Fig. 3 Theoretical and experimental results for the radical cation states of EDOT-MOB in green and the EDOT-TMOB in red. (a) The identity and spin of the energy levels of the most significant electronic transitions (magnitude of the oscillator strength f ≥ 0.1) are given along with the corresponding energy gap in nm. (b) The simulated spectra and chemical structures of EDOT-MOB in green and the EDOT-TMOB in red. Experimentally obtained UV-Vis spectra obtained using an OTTLE cell and photographs of the cell showing the color change as a result of electrochemical oxidation to the radical cation state for (c) EDOT-MOB and (d) EDOT-TMOB. Figure (c) and (d) have been adapted with permission from D. T. Christiansen, et al., J. Am. Chem. Soc., 2019, 141, 3859–3862. Copyright 2019, American Chemical Society. | ||
A detailed analysis of the TDDFT excited-state transitions, associated oscillator strengths, and maximum absorbance (λmax) for EDOT-MOB and EDOT-TMOB, provides insight into the absorption differences between their radical cation states. Three types of transitions are shown in Fig. 3a: a transition from the singly occupied molecular orbital (SOMO) to the lowest unoccupied molecular orbital (LUMO) with electronic spin up (alpha) i.e., Sα → Lα, SOMO to LUMO with electronic spin down (beta) i.e., Sβ → Lβ, and from a lower energy level, Sβ−1 → Lβ. Fig. 3a shows the radical cation of EDOT-MOB having three transitions (a, b), and EDOT-TMOB having two transitions (c, d).
As shown in the simulated spectra in Fig. 3b, the high energy absorption band for both ACE molecules originates from the excited-state electronic transition, Sα → Lα, with a λmax at 434 nm (peak a) for EDOT-MOB and at 449 nm (peak c) for EDOT-TMOB. Based on our calculations, the low-energy absorption for EDOT-TMOB originates from the Sβ → Lβ transition with a λmax at 900 nm (peak d), whereas for EDOT-MOB this absorption arises from the Sβ-1 → Lβ transition with a λmax at 650 nm (peak b). This is the result of differences in the relative oscillator strengths of these transitions. While the band energy diagram in Fig. 3a shows two types of transitions associated with the β spin state (Sβ → Lβ and Sβ−1 → Lβ) for EDOT-MOB, only the transition with the stronger oscillator strength is observed in the spectrum. In EDOT-MOB, the oscillator strength of the Sβ−1 → Lβ (f = 0.2229) is more than twice as high as the Sβ → Lβ (f = 0.0964), which results in the contribution of the latter transition to the radical-cation state spectrum being negligible. Fig. 3b also highlights that the number of methoxy-substituents impacts the low-energy transition much more than the high-energy transition as we only observe a 15 nm difference in λmax for the latter between EDOT-MOB and EDOT-TMOB. Moreover, this observation implies that the β-state transition is mainly responsible for the observed color change.
This finding was confirmed experimentally, as shown in Fig. 3c and d, by comparing the spectra and photographs of these two ACE molecules in an optically transparent thin layer (OTTLE) cell. Both chromophores switch from a perfectly clear transmissive state to a vibrant green radical cation state in the case of EDOT-MOB, and to a vibrant red in the case of EDOT-TMOB. This is a result of both radical cation bands being located in the visible range for EDOT-MOB (λmax at 465 and 671 nm), whereas only one of the two absorption bands is found in the visible range for EDOT-TMOB (λmax at 496 nm), as our simulated data predicted. This demonstrates that increasing electron density indeed facilitates a bathochromic shift in the lower energy excited state transition which, in turn, produces a means for color modification. Surprisingly though, the high energy absorption of the radical cation in these two-ring ACEs is minimally affected by substitution pattern, which is counterintuitive from the design principles established for controlling neutral state absorption. Thus, this is an excellent example of how using DFT/TDDFT provides a powerful tool for guiding synthetic approaches and narrowing down the most promising candidates for further development.
4. Extended conjugation red-shifts the radical cation absorption
Next, we evaluate the impact of expanding from a two-ring to a three-ring electrochrome, where the conjugation remains short enough to center the neutral-state absorption in the UV, while the extra building block affords us with additional ways of tuning steric interactions. In particular, we can access diverse structural conformations affecting planarity and extent of conjugation by varying the types of inter-ring electrostatic forces, specifically oxygen–sulfur (O⋯S) and oxygen–oxygen (O⋯O) interactions. Here, we examined a set of two ACE chromophores, where their structural motif is composed of methoxy-functionalized phenylenes conjugated between two electron-rich 3,4-propylendioxythiophenes (ProDOT) units (ProDOT2-MOB and ProDOT2-TMOB, structures shown in Fig. 4). TDDFT was used to simulate the absorption spectra of the radical cation species and to approximate the van der Waals distances of the O⋯S and O⋯O interactions. Comparing the three-ring systems (Fig. 4) with the two-ring ACE molecules (Fig. 3), the lower energy peaks of these bis-ProDOT derivatives fully absorb in the NIR region due to the extended conjugation length, and increase in electron-rich character from the additional XDOT ring, leaving a single absorption band in the visible region. The incorporation of two additional methoxy groups in ProDOT2-TMOB further red-shifts this low-energy radical cation band. According to our calculations of these three-ring ACEs, the lower energy absorption band is dominated by the Sβ → Lβ excited-state electronic transition. Since this low-energy band is in the NIR, the color saturation and hue of the electrochrome is determined by manipulating the higher-energy radical cation band, which is dominated by the Sα → Lα excited-state transition. Our TDDFT calculations predict that we indeed can fine-tune the high-energy band by manipulating steric interactions in three-ring systems. This finding is shown in Fig. 4 where the simulated spectra show both an intensity increase and a small red-shift in λmax (similar to what we observed for the two-ring systems) of the high-energy band when we decrease steric interaction going from a tri- to a mono-methoxy system. While both electrochromes exhibit favorable O⋯S interactions that increases planarity and the extent of conjugation, the addition of two methoxy groups invokes a repulsive O⋯O interaction that adds torsional strain (∼40° out-of-plane twisting) in ProDOT2-TMOB. This repulsive interaction is sufficient to cause a signficant decrease in absorption intensity, which will affect primarily the color saturation but to some extent also the hue of the electrochrome. Therefore, this simulation suggests that manipulating electron richness mainly influences the low-energy radical cation band, whereas steric interactions (in three-ring systems especially) can be used to fine-tune the high-energy radical cation band. Moving forward, it will be important to examine additional three-ring ACE systems with variations in dihedral angles and van der Waals distances, in order to identify their impact on the simulated UV-Vis spectra, and excited stated electronic transitions.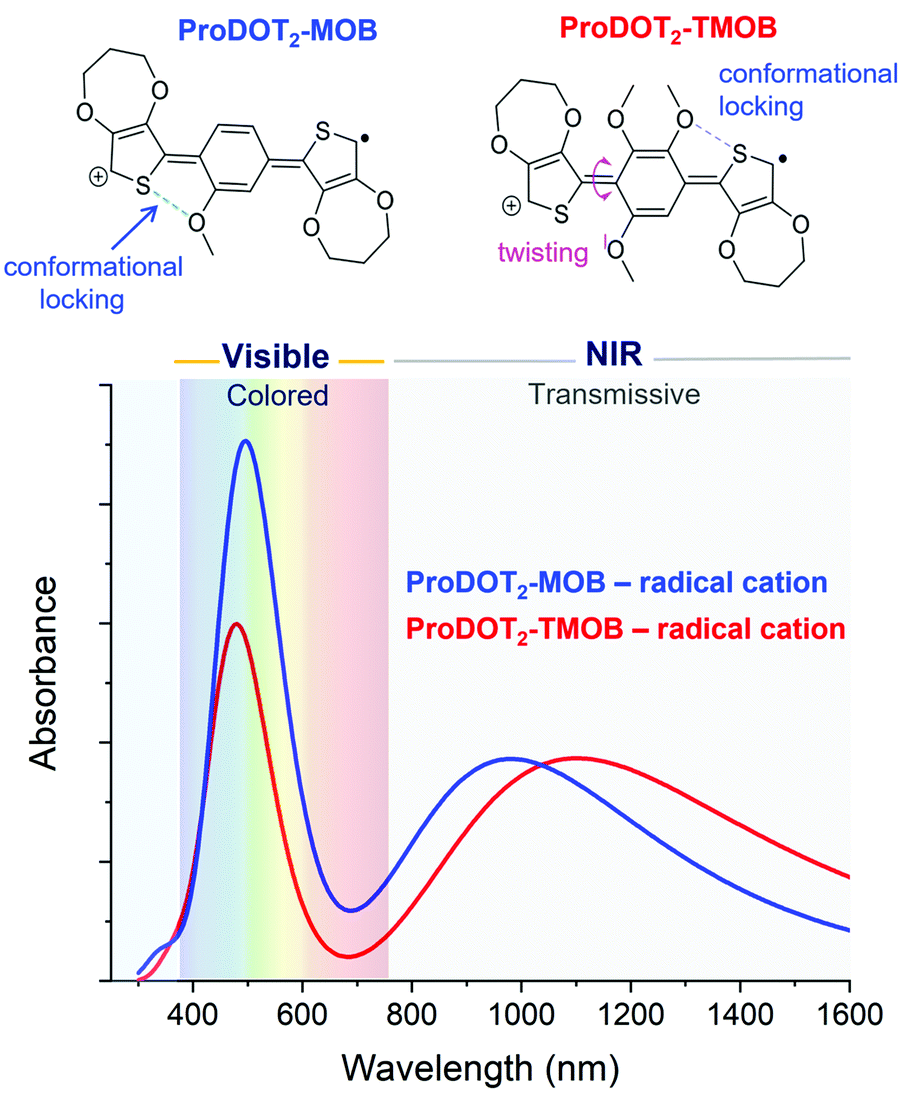 | ||
| Fig. 4 Structure of three-ring ACE systems, ProDOT2-MOB (blue) and ProDOT2-TMOB (red) in their radical cation states. The extended conjugation on these three-ring heteroaryl systems pushes the lower energy band into the NIR region leaving a single absorption band in the visible. Differences between the simulated radical cation spectra of these molecules show the impact on absorbance intensity and λmax of the high-energy absorption bands as a result of differences in the steric interactions. | ||
5. Phosphonate derivative surface adsorption on nanoITO
In the previous sections, we established design principles that provide a fundamental framework for color control in small molecules. In this last section we shift our focus towards the practical challenges that must be overcome to obtain fast, on-demand color switching. While the most commercially successful employment of electrochromic technology, namely rear-view car mirrors and on-demand tinting of airplane windows, makes use of small molecule electrochromes dissolved in the electrolyte,1 we propose that we can enhance the switching rates of the ACE molecules, and make the switching process fully user-controlled, by manipulating electrode–EC molecule interactions using known anchoring approaches. Because small molecules cannot readily be cast as thin films, the redox processes are limited by the rate of mass transport of the molecule to the electrode, and therefore they are inherently slower than the switching of EC films that are in direct contact with an underlying electrode. In the same vein, molecules that have diffused away from the electrode are no longer under user control as illustrated by Fig. 5a. To overcome the mass transport limitations, one strategy is to covalently anchor the electrochromes to the electrode surface and mimic the thin film approach. Surface adsorption of dye molecules gained widespread attention in the early 1990s with dye-sensitized solar cells (DSSCs), which made use of a ruthenium dye anchored onto a high surface area three-dimensional TiO2 network electrode.40 This, in turn, led shortly afterwards to the first anchored electrochrome, where a viologen was adsorbed onto TiO2 and provided a blue-to-white switching electrode.41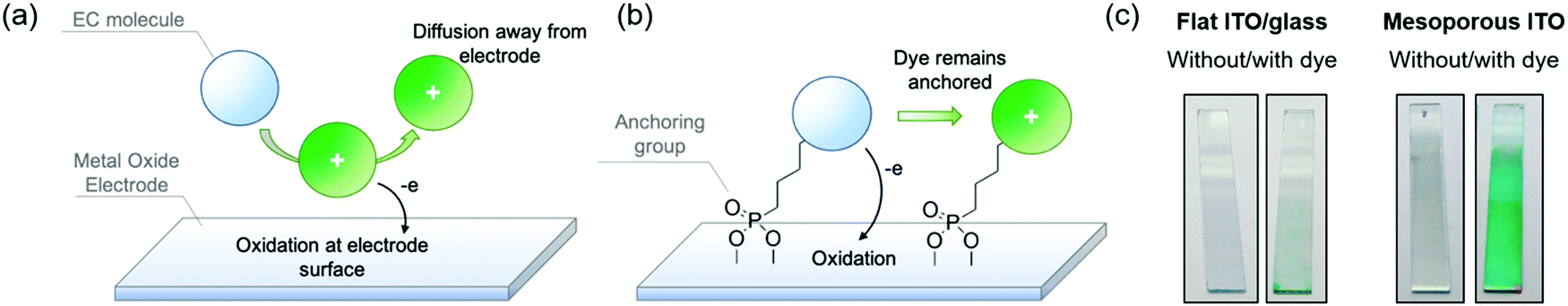 | ||
| Fig. 5 (a) Illustration of an EC molecule dissolved in electrolyte undergoing oxidation at the electrode surface. (b) Illustration of an EC molecule anchored to the electrode surface via phosphonic acid groups undergoing electrochemical oxidation. (c) Photographs of ITO (left pair) and nanoITO electrodes (right pair), uncoated and coated with phosphonic acid-functionalized dye molecules. | ||
To carry out this approach, a functional group that serves as a covalent anchor to the metal oxide electrode must be appended to the electroactive molecule (Fig. 5b). Some of the most popular anchoring groups for attaching molecules onto oxide surfaces are silyl groups, carboxylic acids, and phosphonic acids.42,43 All of these have trade-offs in terms of their ease of synthetic incorporation onto active molecules, reactivity, anchoring strength, and rates of charge transfer between molecule and electrode. For electrochromic applications, the stability of the anchor to the electrode, especially under repeated electrochemical cycling, becomes a critically important property to consider when selecting the appropriate anchoring group. This has led us to focus on functionalizing our ACE molecules with phosphonic acid groups, which offer strong covalent bonding to the electrode, while being synthetically straightforward to attach and incorporate onto a variety of different aromatic systems, making it a versatile approach.44,45
One major challenge with this approach for EC applications specifically is that adsorption occurs only at the surface of the electrode; as a result, the amount of chromophore adsorbed onto smooth ITO/glass electrodes does not offer sufficient coloration for most EC applications as shown in Fig. 5c (left panel). To address this, it is necessary to increase the surface area of the electrode to allow for greater dye adsorption and higher optical density in the colored state. The design logic and choice of electrode should be optimized with certain optical properties in mind. For window-type applications, for example, the electrodes must be as clear and transparent as possible, with minimal haze and minimal coloration. Mesoporous ITO is well-suited for this as a result of its high transmissivity throughout the visible spectrum and haze under 2%, which decreases even further when in contact with electrolyte due to refractive index matching.46 To construct these electrodes, commercially available ITO nanoparticle (nanoITO) dispersions are coated onto ITO/glass, followed by heat treatment >100 °C to remove residual solvent. While increased temperatures and heat treatment under inert atmosphere leads to enhancements in conductivity, we observed that treatment at lower temperatures is sufficient to observe rapid (sub-second to seconds) EC switching, which expands the range of supporting substrates that can be used. The nanoITO coating affords surface areas that are 30–50 times greater than conventional ITO/glass, and can support a high enough chromophore loading to obtain optically vibrant thin films that withstand stresses such as solvent rinsing and electrochemical redox switching (Fig. 5c, right panel).
Applying this to our ACE systems, we synthesized molecules where the phosphonic acid anchor is separated from the chromophore by an alkyl spacer to electronically decouple the anchoring functionality from the electroactive portion of the molecule.47Fig. 6a shows spectra of a hexylthiol-EDOT-phenyl phosphonate ester molecule in solution (EDOT-B-PE, green spectra) along with its phosphonic acid derivative appended onto nanoITO (EDOT-B-PA, purple spectra), in both neutral (colorless) and radical cation (colored) states. Not only are the spectra in excellent agreement with calculations but they also confirm that the alkyl spacer decouples the anchoring group from the chromophore, and that the phosphonate/phosphonic acid group is merely a spectator as far as optical and electronic properties are concerned. Importantly, the spectra of the molecule in solution and adsorbed onto nanoITO have identical peak wavelengths suggesting that these molecules undergo no appreciable intermolecular interactions or aggregation that would be expected to further shift the spectra and potentially move the neutral state absorption into the visible, which would be detrimental. An added benefit of the lack of meaningful intermolecular interactions is that it allows us to more predictably and accurately model the thin film behavior computationally. This affords us a greater degree of control over the properties of the ACE molecules through structural fine-tuning, where spacers as short as a single methyl group are sufficient.
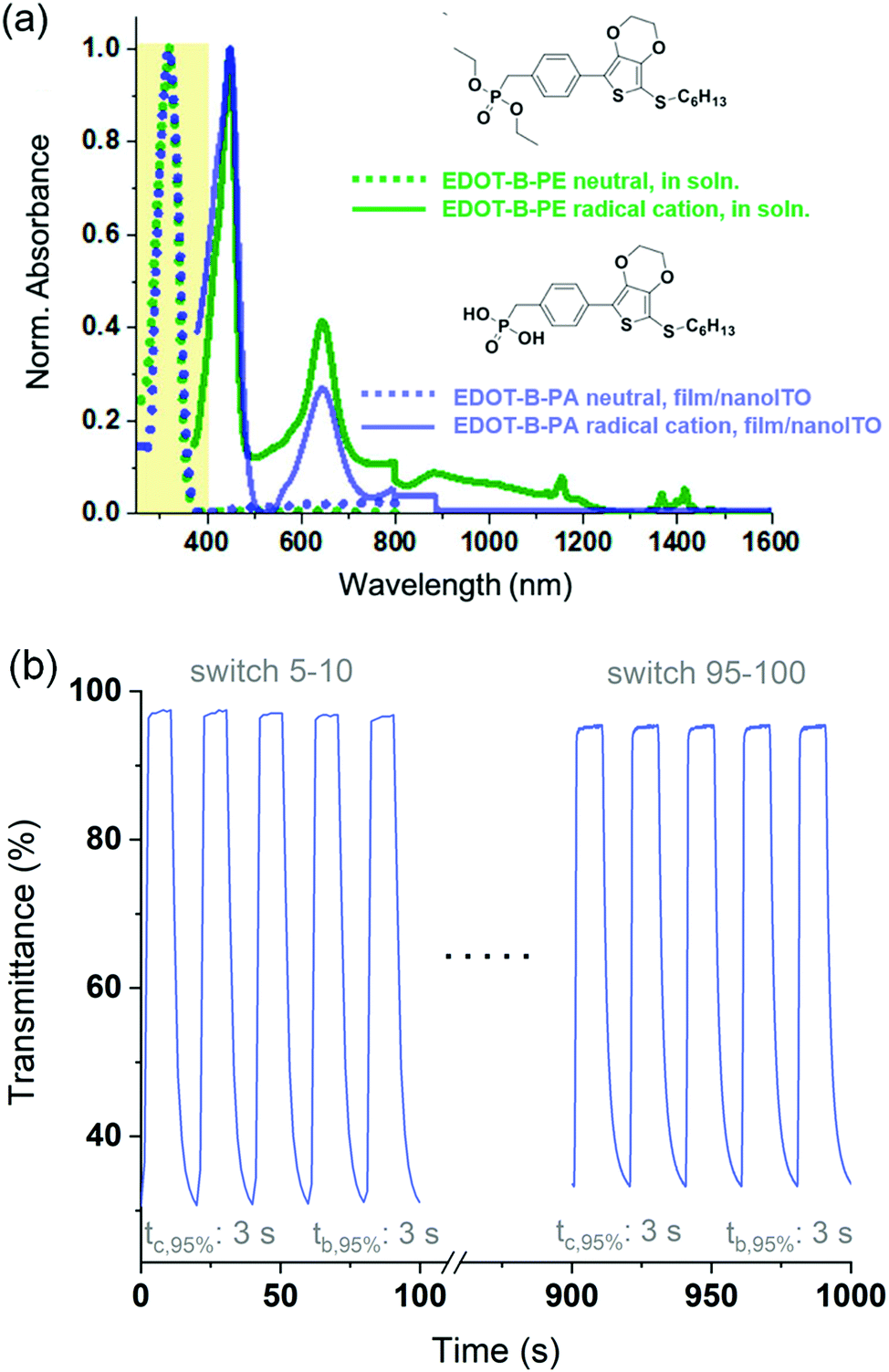 | ||
| Fig. 6 (a) Spectra of the neutral and radical cation states of EDOT-B-PE (dissolved in 0.5 M TBAPF6/dichloromethane, green traces) and EDOT-B-PA adsorbed on a nanoITO electrode and immersed in 0.5 M TBAPF6/dichloromethane (blue traces). Adapted with permission from Nhon, et al. J. Chem. Phys., 2021, 154, 054110. Copyright 2021, AIP. (b) Transmittance at λmax (447 nm) of EDOT-B-PA anchored onto a nanoITO switched between −0.4 and 0.7 V vs. Fc/Fc+ in 0.1 M TBAPF6/propylene carbonate with 10 second intervals for 100 switches (only switches 5–10 and 95–100 are shown here for clarity). The nanoITO electrode was prepared via blade coating of a nanoITO dispersion onto a conventional ITO/glass slide using a speed of 10 mm s−1, and a blade-height of 100 μm. The electrode was then heat treated at 200 °C for 72 hours before soaking it in a 1 mM solution of EDOT-B-PA dissolved in 2-methyltetrahydrofuran. The electrode was then rinsed with acetone and dried at 100 °C for 1 hour prior to use. | ||
Finally, an added benefit of anchoring ACE molecules is that relatively rapid switching times, on par with that of other thin-film EC technologies, can be obtained. This is exemplified in Fig. 6b by EDOT-B-PA anchored on nanoITO where the potential of the electrode is switched between −0.4 V (colorless state, 98%T at λmax) and 0.7 V (colored state, 30%T at λmax) in 10 second intervals for 100 switches. Typically, the switching time of an EC material is reported as the time it takes to reach 95% of a full contrast switch (t95%), as a transmittance change of less than 5% is indistinguishable to the human eye. For EDOT-B-PA the t95% of a ca. 65% contrast switch is just 3 seconds with negligible change observed over the course of 100 switches. This example demonstrates not only the robustness of the phosphonic acid anchor to nanoITO, but also the inherent redox stability of these types XDOT-based molecular electrochromes, and that using this design motif can pave the way for an entirely new color palette of clear-to-colored switching EC materials.
6. Summary and perspective
It is evident that EC systems, using cathodic or anodic coloration, provide numerous routes with which to modulate light intensity, and vary the transmitted wavelengths (i.e., color). Combining organic and inorganic active materials, in conjunction with window-type and display-type device architectures, allow for use in many practical applications where user-controlled color change is desired. As illustrated throughout this short review, the ability to use ACE molecules which only absorb in the UV in their charge neutral form, affords EC materials that are perfectly clear and transmissive in one state. Electrochemical conversion into the radical cation state then transfers light absorption into the visible range where subtle changes in chromophore structure can provide a variety of vibrantly colored states. While the results highlighted in this review focus especially on molecules with red and green radical cation states to illustrate the fundamental concepts, similarly structured molecules have been shown to provide yellow and orange colors,17 whereas blue colored states should be achievable by extending the conjugation, as discussed in conjunction with Fig. 4. We find that a priori determination of the color of the charged states is difficult, without guiding DFT calculations, as subtle spectral changes can lead to large changes in human color perception. Further close collaborative studies between theorists and experimentalists will allow us to fine-tune our color predictions more accurately. The ultimate goal is to provide a color palette of brightly colored electrochromes of all colors that are truly transparent in one state, while further work can be directed to developing secondary colors, along with neutral black and brown colors, via chromophore mixing where the latter may prove important in applications such as electronic paper and switchable eyewear.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge funding of the anodically coloring electrochromes project at the Georgia Institute of Technology and the University of North Georgia by the Air Force Office of Scientific Research (FA9550-21-1-0420).References
- P. M. S. Monk, R. J. Mortimer and D. R. Rosseinsky, Electrochromism: Fundamentals and Applications, VCH Publishers, New York, 1995 Search PubMed.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- C. G. Granqvist, M. A. Arvizu, B. Pehlivan, H. Y. Qu, R. T. Wen and G. A. Niklasson, Electrochim. Acta, 2018, 259, 1170–1182 CrossRef CAS.
- S. Mondal, T. Yoshida, U. Rana, M. K. Bera and M. Higuchi, Sol. Energy Mater. Sol. Cells, 2019, 200, 110000 CrossRef CAS.
- M. Schott, L. Niklaus, J. Clade and U. Posset, Sol. Energy Mater. Sol. Cells, 2019, 200, 110001 CrossRef CAS.
- M. Schott, H. Lorrmann, W. Szczerba, M. Beck and D. G. Kurth, Sol. Energy Mater. Sol. Cells, 2014, 126, 68–73 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS.
- A. M. Österholm, D. E. Shen and J. R. Reynolds, in Conjugated Polymers: Properties, Processing, and Applications, 4th edn, ed. J. R. Reynolds, B. C. Thompson and T. A. Skotheim, CRC Press, Boca Raton, 2019, pp. 201–248 Search PubMed.
- S. Araki, K. Nakamura, K. Kobayashi, A. Tsuboi and N. Kobayashi, Adv. Mater., 2012, 24, 122–126 Search PubMed.
- D. M. Tench, M. A. Cunningham, J. J. Kuo, P. V. Rowell and L. F. Warren Jr., in Electrochromic Materials and Applications, ed. A. Rougier, D. Rauh and G. A. Nazri, The Electrochemical Society, Pennington, New Jersey, 2003, pp. 190–198 Search PubMed.
- A. Tsuboi, K. Nakamura and N. Kobayashi, Chem. Mater., 2014, 26, 6477–6485 CrossRef CAS.
- S. Kimura, K. Nakamura and N. Kobayashi, Sol. Energy Mater. Sol. Cells, 2020, 205, 110247 CrossRef CAS.
- A. Aoki, A. Ito and S. Watanabe, Sol. Energy Mater. Sol. Cells, 2019, 200, 109922 CrossRef CAS.
- S. M. Cho, S. Kim, T. Y. Kim, C. S. Ah, J. Song, S. H. Cheon, J. Y. Kim, H. Ryu, Y. H. Kim, C. S. Hwang and J. I. Lee, Sol. Energy Mater. Sol. Cells, 2018, 179, 161–168 CrossRef CAS.
- X. Tao, D. Liu, J. Yu and H. Cheng, Adv. Opt. Mater., 2021, 9, 2001847 CrossRef CAS.
- M. T. Strand, T. S. Hernandez, M. G. Danner, A. L. Yeang, N. Jarvey, C. J. Barile and M. D. McGehee, Nat. Energy, 2021, 6, 546–554 CrossRef CAS.
- D. T. Christiansen, A. L. Tomlinson and J. R. Reynolds, J. Am. Chem. Soc., 2019, 141, 3859–3862 CrossRef CAS.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
- M. İçli, M. Pamuk, F. Algi, A. M. Önal and A. Cihaner, Chem. Mater., 2010, 22, 4034–4044 CrossRef.
- H. Shin, Y. Kim, T. Bhuvana, J. Lee, X. Yang, C. Park and E. Kim, ACS Appl. Mater. Interfaces, 2011, 4, 185–191 CrossRef.
- Y. Shi, Q. Chen, J. Zheng and C. Xu, Electrochim. Acta, 2020, 341, 136023 CrossRef CAS.
- A. L. Dyer, E. J. Thompson and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2011, 1787–1795 CrossRef CAS.
- E. Oguzhan, H. Bilgili, F. Baycan Koyuncu, E. Ozdemir and S. Koyuncu, Polymer, 2013, 54, 6283–6292 CrossRef CAS.
- M. Içli-Özkut, Z. Öztaş, F. Algi and A. Cihaner, Org. Electron., 2011, 12, 1505–1511 CrossRef.
- T. Dey, M. A. Invernale, Y. Ding, Z. Buyukmumcu and G. A. Sotzing, Macromolecules, 2011, 44, 2415–2417 CrossRef CAS.
- C. M. Amb, J. A. Kerszulis, E. J. Thompson, A. L. Dyer and J. R. Reynolds, Polym. Chem., 2011, 2, 812–814 RSC.
- J. A. Kerszulis, C. M. Amb, A. L. Dyer and J. R. Reynolds, Macromolecules, 2014, 47, 5462–5469 CrossRef CAS.
- K. Cao, D. E. Shen, A. M. Österholm, J. A. Kerszulis and J. R. Reynolds, Macromolecules, 2016, 49, 8498–8507 CrossRef CAS.
- J. Padilla, A. M. Österholm, A. L. Dyer and J. R. Reynolds, Sol. Energy Mater. Sol. Cells, 2015, 140, 54–60 CrossRef CAS.
- E. Poverenov, M. Li, A. Bitler and M. Bendikov, Chem. Mater., 2010, 22, 4019–4025 CrossRef CAS.
- W. T. Neo, Z. Shi, C. M. Cho, S.-J. Chua and J. Xu, ChemPlusChem, 2015, 80, 1298–1305 CrossRef CAS.
- T. Ikeda, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2009, 131, 9158–9159 CrossRef CAS.
- T. Ikeda and M. Higuchi, Langmuir, 2011, 27, 4184–4189 CrossRef CAS.
- R. Shomura, K. Sugiyasu, T. Yasuda, A. Sato and M. Takeuchi, Macromolecules, 2012, 45, 3759–3771 CrossRef CAS.
- B. Karabay, L. C. Pekel and A. Cihaner, Macromolecules, 2015, 48, 1352–1357 CrossRef CAS.
- G. Conboy, H. J. Spencer, E. Angioni, A. L. Kanibolotsky, N. J. Findlay, S. J. Coles, C. Wilson, M. B. Pitak, C. Risko, V. Coropceanu, J.-L. Brédas and P. J. Skabara, Mater. Horiz., 2016, 3, 333–339 RSC.
- J. A. Kerszulis, K. E. Johnson, M. Kuepfert, D. Khoshabo, A. L. Dyer and J. R. Reynolds, J. Mater. Chem. C, 2015, 3, 3211–3218 RSC.
- J. F. Ponder, A. M. Österholm and J. R. Reynolds, Macromolecules, 2016, 49, 2106–2111 CrossRef CAS.
- D. L. Wheeler, L. E. Rainwater, A. R. Green and A. L. Tomlinson, Phys. Chem. Chem. Phys., 2017, 19, 20251–20258 RSC.
- B. O’Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- X. Marguerettaz, R. O’neill and D. Fitzmaurice, J. Am. Chem. Soc., 1994, 116, 2629–2630 CrossRef CAS.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322–6356 CrossRef CAS.
- L. Zhang and J. M. Cole, ACS Appl. Mater. Interfaces, 2015, 7, 3427–3455 CrossRef CAS.
- S. A. Paniagua, E. L. Li and S. R. Marder, Phys. Chem. Chem. Phys., 2014, 16, 2874–2881 RSC.
- R. Stalder, D. Xie, A. Islam, L. Han, J. R. Reynolds and K. S. Schanze, ACS Appl. Mater. Interfaces, 2014, 6, 8715–8722 CrossRef CAS.
- P. G. Hoertz, Z. Chen, C. A. Kent and T. J. Meyer, Inorg. Chem., 2010, 49, 8179–8181 CrossRef CAS.
- L. Nhon, R. Wilkins, J. R. Reynolds and A. Tomlinson, J. Chem. Phys., 2021, 154, 054110 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |