
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning of the elastic modulus of a soft polythiophene through molecular doping†
Sepideh
Zokaei‡
*a,
Donghyun
Kim‡
 b,
Emmy
Järsvall‡
a,
Abigail M.
Fenton
c,
Albree R.
Weisen
c,
Sandra
Hultmark
a,
Phong H.
Nguyen
d,
Amanda M.
Matheson
e,
Anja
Lund
a,
Renee
Kroon
abf,
Michael L.
Chabinyc
e,
Enrique D.
Gomez
cg,
Igor
Zozoulenko
bf and
Christian
Müller
*ah
b,
Emmy
Järsvall‡
a,
Abigail M.
Fenton
c,
Albree R.
Weisen
c,
Sandra
Hultmark
a,
Phong H.
Nguyen
d,
Amanda M.
Matheson
e,
Anja
Lund
a,
Renee
Kroon
abf,
Michael L.
Chabinyc
e,
Enrique D.
Gomez
cg,
Igor
Zozoulenko
bf and
Christian
Müller
*ah
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Göteborg 41296, Sweden. E-mail: zokaei@chalmers.se; christian.muller@chalmers.se
bLaboratory of Organic Electronics, Linköping University, Norrköping 60174, Sweden
cDepartment of Chemical Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
dDepartment of Chemical Engineering, University of California, Santa Barbara, California 93106, USA
eMaterials Department, University of California, Santa Barbara, California 93106, USA
fWallenberg Wood Science Center, Linköping University, Norrköping 60174, Sweden
gDepartment of Materials Science and Engineering, The Pennsylvania State University, University Park, Pennsylvania 16802, USA
hWallenberg Wood Science Center, Chalmers University of Technology, Göteborg 41296, Sweden
First published on 17th November 2021
Abstract
Molecular doping of a polythiophene with oligoethylene glycol side chains is found to strongly modulate not only the electrical but also the mechanical properties of the polymer. An oxidation level of up to 18% results in an electrical conductivity of more than 52 S cm−1 and at the same time significantly enhances the elastic modulus from 8 to more than 200 MPa and toughness from 0.5 to 5.1 MJ m−3. These changes arise because molecular doping strongly influences the glass transition temperature Tg and the degree of π-stacking of the polymer, as indicated by both X-ray diffraction and molecular dynamics simulations. Surprisingly, a comparison of doped materials containing mono- or dianions reveals that – for a comparable oxidation level – the presence of multivalent counterions has little effect on the stiffness. Evidently, molecular doping is a powerful tool that can be used for the design of mechanically robust conducting materials, which may find use within the field of flexible and stretchable electronics.
New conceptsMolecular doping is widely used to control the electrical properties of conjugated polymers, for applications ranging from flexible and wearable electronics to bioelectronics. At the same time, the doped materials must display suitable mechanical properties such as ductility or toughness, which is typically achieved through chemical design of the polymer. Molecular dopants are thought to have a relatively minor impact on the mechanical properties at best, and in some cases are known to result in a brittle material. In this work, a different perspective is established: molecular doping can be used as a tool to not only control the electrical but also mechanical properties of conjugated polymers. The structural changes that occur as a result of doping can be used to tune the stiffness and toughness by a large amount, resulting in electrically conducting materials that feature an elastic modulus comparable to that of commodity polymers such as polyethylene. |
Introduction
Conjugated polymers receive considerable attention for numerous applications from wearable electronics to soft robotics that require well-adjusted mechanical properties. Materials with widely different mechanical behavior from highly stretchable to tough can be realized by chemical design, e.g. through the incorporation of flexible spacers, the use of hydrogen-bonding moieties or copolymerization with non-conjugated, insulating blocks.1–4 Moreover, conjugated polymers can be blended with insulating polymers or be modified through the addition of additives that act as crosslinkers or have a plasticizing effect.2Molecular dopants are additives that are widely used to modulate the electrical properties of conjugated polymers. Most conjugated polymers are relatively stiff and feature a high elastic modulus of several 100 MPa to several GPa at room temperature3–6 due to a high glass transition temperature Tg and/or a high degree of crystalline order. Molecular doping of stiff conjugated polymers does not tend to strongly alter their mechanical properties (Table 1).7–9 As a result, doping is typically not considered as a tool that allows to adjust the elastic modulus of conjugated polymers.
| Polymer | Dopant | mol% dopant | wt% dopant | E neat (MPa) | E doped (MPa) | η | Ref. |
|---|---|---|---|---|---|---|---|
| a Poly(ethylene oxide) (PEO). | |||||||
| P3HT | Mo(tfd-COCF3)3 | 9 | 30 | 340 | 270 | −0.1 | 7 |
| P3HT | EBSA | 9 | 16 | 900 | 345 | −0.4 | 8 |
| P3HT | F4TCNQ | 4 | 6 | 340 | 560 | 0.2 | 7 |
3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 P3HT:PEOa :7 P3HT:PEOa |
F4TCNQ | 20 | 29 | 80 | 188 | 0.4 | 10 |
| P3HT | FeCl3 | 20 | 20 | 230 | 900 | 0.6 | 9 |
| P3OT | FeCl3 | 18 | 15 | 60 | 470 | 0.9 | 11 |
| P3OT | I2 | 7 | 9 | 60 | 80 | 0.1 | 11 |
| P3DDT | FeCl3 | 20 | 14 | 50 | 300 | 0.8 | 9 |
| P3DDT | F4TCNQ | 7 | 8 | 45 | 80 | 0.2 | This work |
| p(g42T-T) | F4TCNQ | 30 | 35 | 8 | 232 | 1.5 | This work |
| p(g42T-T) | F2TCNQ | 20 | 21 | 8 | 377 | 1.7 | This work |
To compare the doping-induced changes in elastic modulus that have been observed for different polymers we here define a figure of merit η = log(Edoped/Eneat), which considers the ratio of the elastic modulus of the doped material Edoped and the neat polymer Eneat. There are only few studies that investigate how the elastic modulus of conjugated polymers changes with molecular doping and values are limited to η ≤ 0.9 for unaligned material (Table 1). A comparison of the few existing reports allows us to predict what type of changes in stiffness can be expected upon doping, as discussed in more detail below: doping of stiff conjugated polymers can lead to a slight decrease in modulus while doping of soft materials tends to increase the modulus.
For stiff conjugated polymers the impact of doping on the mechanical properties appears to be dominated by a plasticization type effect. For example, poly(3-hexylthiophene) (P3HT) with a regioregularity of more than 97% and Tg ≈ 23 °C, was found to have a modulus of about 340 MPa at room temperature, which slightly decreased to 270 MPa upon sequential doping with 9 mol% Mo(tfd-COCF3)3.7 It is feasible that the ingression of the dopant reduced the direct interactions between adjacent polymer chains, resulting in a slight reduction in stiffness but similar Tg ≈ 21 °C. A similar plasticization type impact of doping has been observed in the case of P3HT (regioregularity = 95%) doped with 9 mol% of a latent dopant based on ethylbenzene sulfonic acid (EBSA) caped with a 2-nitrobenzyl moiety that is released upon heating, which resulted in a decrease in Tg from 30 to 15 °C and modulus from 900 to 345 MPa, i.e. a low figure of merit of η ≈ −0.4 (Table 1).8 Further, a diketopyrrolopyrrole (DPP) based copolymer (E = 374 MPa)12 displayed a reduction in Tg from 55 to 27 °C upon doping with 1 wt% of 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane (F4TCNQ) (see Fig. 1 for chemical structure), resulting in a more stretchable material as evidenced by a higher crack onset strain.13 A reduction in stiffness upon molecular doping has also been reported for stretch-aligned polymer films or fibers composed of polyacetylene,5 poly(2,5-dimethoxy-p-phenylenevinylene),14 poly(2,5-thienylene vinylene)15 and P3HT.7
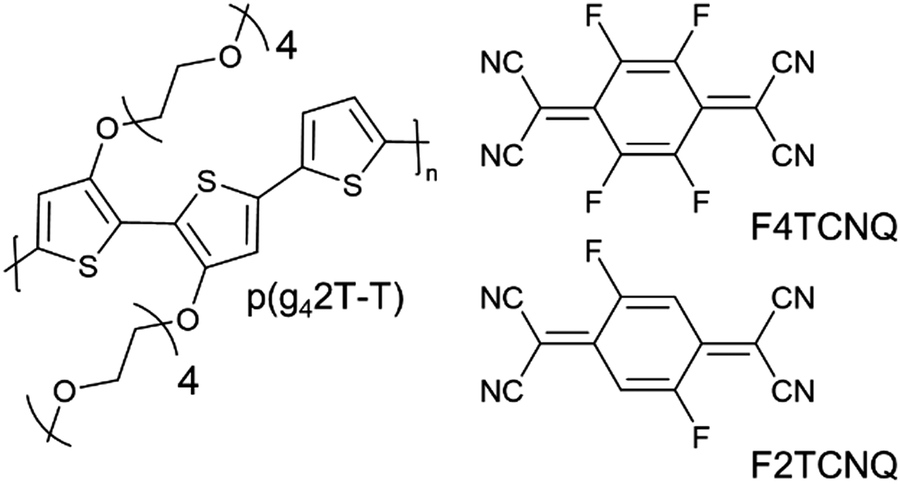 | ||
| Fig. 1 Chemical structures of and p(g42T-T), F4TCNQ and F2TCNQ. | ||
Molecular doping of conjugated polymers with a lower stiffness can have an adverse effect on the elastic modulus (Table 1). Poly(3-octylthiophene) (P3OT) and poly(3-dodecylthiophene) (P3DDT) prepared by oxidative polymerization feature a low regio-regularity of only 75% and hence a low modulus of 60 and 50 MPa at room temperature,9,11 presumably due to a lower crystallinity. Moulton and Smith have argued that molecular doping of these relatively soft materials leads to an increase in π-stacking, which results in a considerably higher elastic modulus.9 As a result, the modulus of unaligned P3OT was found to increase 8-fold to 470 MPa upon doping with 18 mol% FeCl3, resulting in the highest reported figure of merit of η ≈ 0.9.11 Therefore, it can be anticipated that soft conjugated polymers display a more substantial change in modulus upon doping, which may allow to use molecular doping as a tool to not only modulate the electrical but also the mechanical properties.
To explore this hypothesis, we set out to study the impact of molecular doping on the mechanical properties of a soft conjugated polymer. We chose to focus on a polythiophene with tetraethylene glycol side chains, p(g42T-T) (see Fig. 1 for chemical structure), which belongs to a class of polar conjugated polymers that currently receive widespread attention for a myriad of applications from bioelectronics16,17 to thermoelectrics18,19 and energy storage.20,21 p(g42T-T) is very soft due to a low crystallinity and Tg ≈ −46 °C,22 resulting in a low shear storage modulus of only 8 MPa, as we will show in this paper, and therefore doping can be expected to lead to a considerable increase in stiffness (cf. discussion above). Further, the polymer has a low ionization energy of IE0 ≈ 4.7 eV22 and hence can be doped with both F4TCNQ (electron affinity EA0 ≈ 5.2 eV) as well as the anion of F4TCNQ (EA− ≈ 4.7 eV).23 The presence of F4TCNQ dianions opens up the possibility to study the impact of multivalent counterions on the mechanical properties, which has been proposed to lead to ionic type crosslinking when phytic acid24 or MgSO425 are added to the conjugated polymer-based material. We find that doping leads to enhanced π-stacking as well as an increase in Tg. The presence of mono- or dianions, however, which can be readily created through doping with F4TCNQ, are found to have no impact on the modulus, while monoanions improve the ductility and toughness of the material. The electrical and mechanical properties are found to correlate with the oxidation level. An electrical conductivity of up to 52 S cm−1 upon doping with F4TCNQ is accompanied by a 29-fold change in elastic modulus from 8 to 232 MPa, yielding a figure of merit of η ≈ 1.5. An even higher increase to 377 MPa is observed when the dopant 2,5-difluoro-7,7,8,8-tetracyanoquinodimethane (F2TCNQ) is used, which yields a value of η ≈ 1.7.
Results and discussion
In a first set of experiments, we compared the thermo-mechanical properties of neat and strongly doped p(g42T-T). Doping was achieved by processing the polymer and dopant F4TCNQ from the same solution, a 2:1 mixture of chloroform (CHCl3) and acetonitrile (AcN), which was drop cast at 40 °C to obtain 30 to 80 μm thick films (see Experimental for details). The doped material had a uniform appearance, which is in stark contrast to the granular texture of bulk samples of P3HT co-processed with F4TCNQ.8
Neat p(g42T-T) was characterized with oscillatory shear rheometry at 0.16 Hz because the polymer is soft and yields at low strains, which prevented us from characterizing free-standing samples over a wide range of temperatures. The shear storage modulus G′ decreases from a value of about 109 Pa at −80 °C to 108 Pa at −40 °C; storage moduli for glassy polymers are around 1 GPa.4 Thus, we assign this drop in storage modulus to the onset of main-chain relaxation, possibly accompanied by relaxation of part of the side chains. The shear loss modulus G′′ shows a peak at −62 °C with a broad shoulder at higher temperatures (Fig. 2a). We here assign the peak in G′′ to the Tg. We also determined the Tg with differential scanning calorimetry (DSC) using a cooling rate q = −10 °C min−1 (Fig. S1, ESI†) and with dynamic mechanical analysis (DMA) using the glass fiber mesh method and a higher frequency of 1 Hz (Fig. S2a, ESI† and Table 2), which yielded values of Tg ≈ −59 °C and −46 °C, respectively. Fast scanning calorimetry (FSC) was used to study the influence of the cooling rate q, ranging from −0.1 to −1000 K s−1, on the fictive temperature (equivalent to Tg for q = −0.17 K s−1). The dependence of the fictive temperature on q could be described with the Williams–Landel–Ferry (WLF) equation (see Fig. S1, ESI†), which is consistent with an α-relaxation process, i.e. the main-chain relaxation. We would like to point out that relaxation of the side chains is likely frozen in at significantly lower temperatures as reported for polymethacrylates with oligoethylene glycol side chains, which feature a β-relaxation temperature below −100 °C.26 To rule out that the chain length of p(g42T-T) strongly influences the Tg we also studied a low-molecular weight fraction collected through fractionation of the as-synthesized polymer with acetone. DMA of the acetone fraction of p(g42T-T) revealed a Tg ≈ −51 °C, which is only marginally lower than the Tg ≈ −46 °C of p(g42T-T) with Mn ≈ 24 kg mol−1 (Fig. S2, ESI†). We therefore conclude that the chain length does not strongly influence the Tg of p(g42T-T) for the studied range of molecular weights.
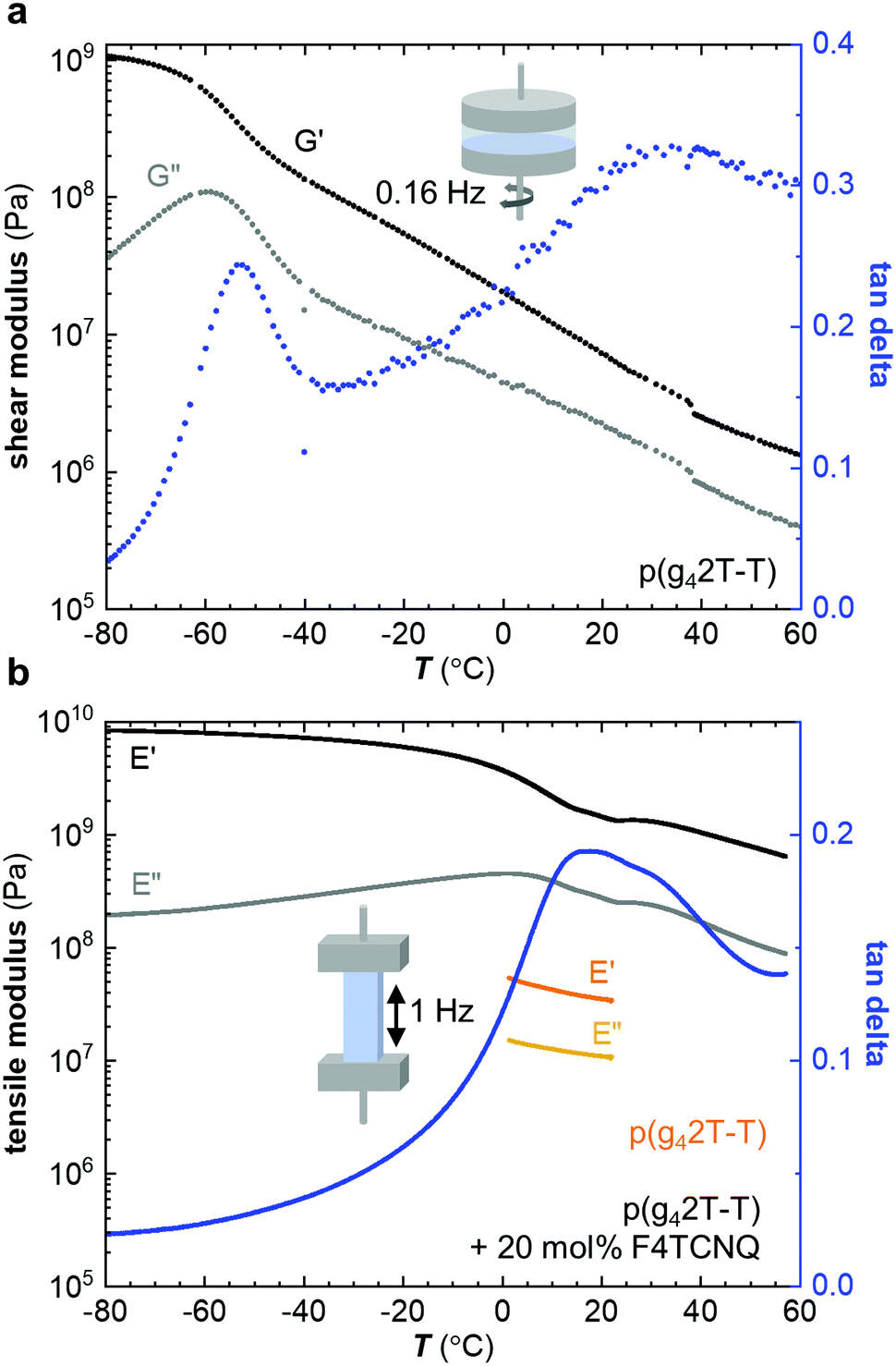 | ||
| Fig. 2 (a) Shear storage and loss modulus, G′ and G′′, and tanδ = G′′/G′ of p(g42T-T) as a function of temperature; (b) tensile storage and loss modulus, E′ and E′′, and tanδ = E′′/E′ of neat p(g42T-T) (orange/yellow) and p(g42T-T) doped with 20 mol% F4TCNQ (black/grey/blue) recorded as a function of temperature; neat p(g42T-T) was only analyzed by cooling from 22 °C to 0 °C because it was difficult to keep the material intact over a wider temperature range. | ||
Co-processing of p(g42T-T) with 20 mol% F4TCNQ resulted in a stiff solid and hence we chose to characterize the doped material with DMA in tensile mode at 1 Hz. The tensile storage modulus E′ has a very high value of 8.4 × 109 Pa in the glassy state at −80 °C and gradually drops to 1.4 × 109 Pa at 20 °C, which is a more than 40-fold increase compared to the neat polymer (η ≈ 1.6), for which we measured a tensile storage modulus of only 34 × 106 Pa at 20 °C and 1 Hz (Fig. 2b). The value measured for the neat polymer is in agreement with the shear storage modulus at 20 °C when assuming a Poisson's ratio of υ = 0.5 so that E′ = 2(1 + ν) × G′ = 3G′. The tensile loss modulus E′′ of p(g42T-T) doped with 20 mol% F4TCNQ features a prominent peak at 3 °C, which we assign to the Tg (Table 2).
We carried out transmission wide-angle X-ray scattering (WAXS) to compare the crystalline order of neat and doped p(g42T-T) bulk samples. The WAXS diffractogram of neat p(g42T-T) features distinct h00 diffraction peaks (h = 1–3; q100 = 0.36 Å−1) due to lamellar stacking (Fig. 3a). Instead of a π-stacking peak there is a broad amorphous halo at q = 1.6 Å−1, which indicates that the backbones of the polymer are disordered. The WAXS diffractogram of p(g42T-T) co-processed with 20 mol% F4TCNQ is remarkably different. The h00 diffraction peaks are now situated at a lower scattering vector (h = 1–2; q100 = 0.30 Å−1), which is commonly observed for polythiophenes doped with F4TCNQ and arises because the dopant is located in the side-chain layers and hence the lattice expands along the side-chain direction.27 Furthermore, a prominent peak can now be discerned at q010 = 1.84 Å−1 (Fig. 3a), which we assign to π-stacking of the p(g42T-T) backbone.
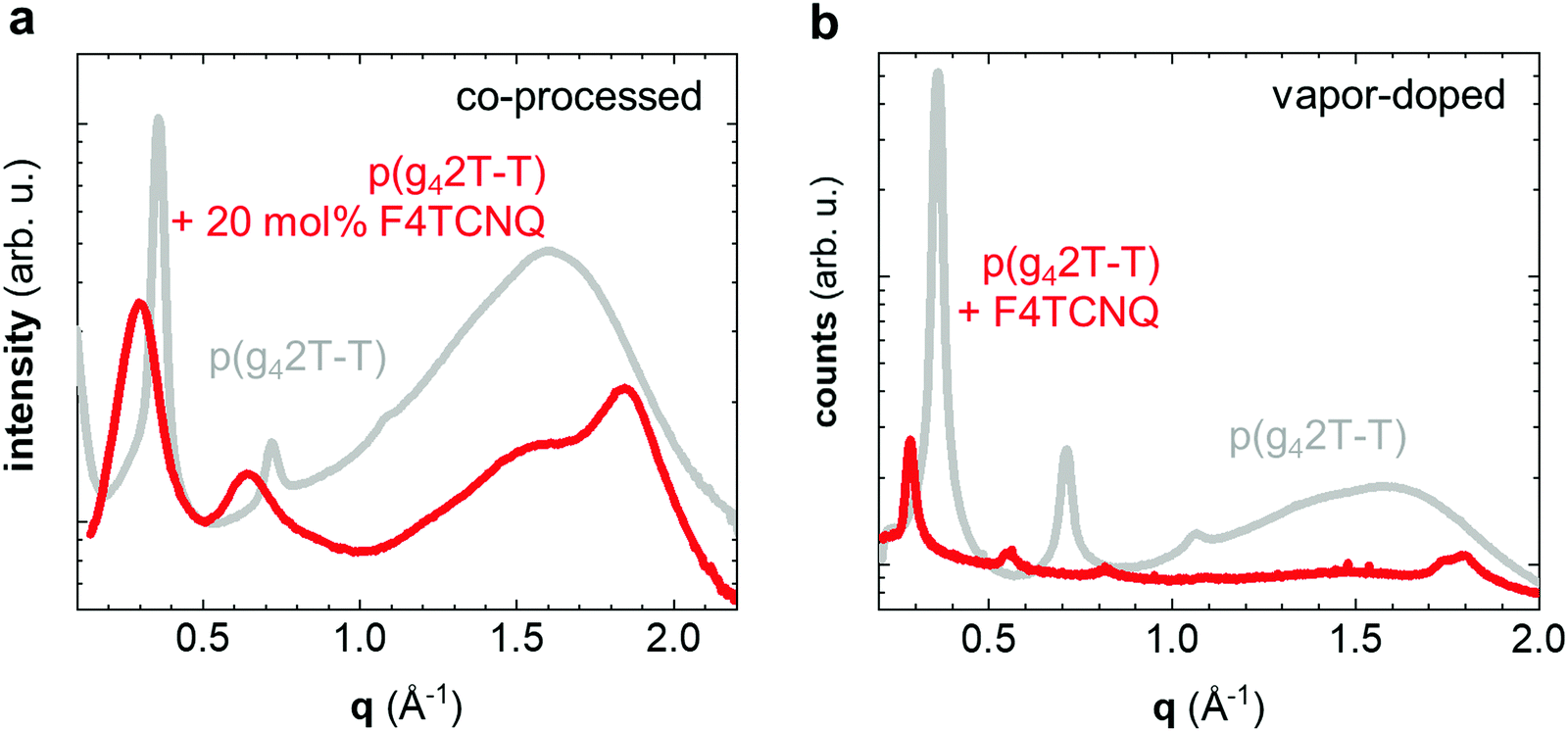 | ||
| Fig. 3 X-ray diffractograms of neat p(g42T-T) (grey) and p(g42T-T) doped with 20 mol% F4TCNQ (red) recorded for (a) co-processed bulk samples using transmission WAXS and (b) vapor-doped thin films using GIWAXS. | ||
The doping process can strongly influence the nanostructure of conjugated polymers.28,29 To separate the impact of doping and processing (e.g. through a change in the solubility of the polymer upon doping) we also vapor-doped thin films of p(g42T-T) with F4TCNQ, which we analyzed with grazing-incidence wide-angle X-ray scattering (GIWAXS). A diffractogram produced by radially integrating a GIWAXS pattern of neat p(g42T-T) over all azimuthal angles is comparable to transmission WAXS measurements on bulk samples, with distinct h00 diffraction peaks (h = 1–3; q100 = 0.37 Å−1) and a broad halo at q = 1.6 Å−1 (Fig. 3b). Vapor doping with F4TCNQ results in a shift in h00 diffraction peaks to lower scattering vectors (h = 1–4; q100 = 0.29 Å−1; Fig. 3b), which retain their preferential out-of-plane orientation (Fig. S3, ESI†). In addition, two in-plane diffraction peaks emerge at 1.74 Å−1 and 1.8 Å−1 (Fig. 3b and Fig. S3b, ESI†), which we assign to two distinct π-stacking motives. Evidently, vapor-doping of p(g42T-T) significantly alters the nanostructure of the polymer, which suggests that the observed structural changes are indeed a result of molecular doping and not merely related to changes in processing conditions. The increase in π-stacking upon doping is consistent with the observed increase in Tg and E′ (see Table 1 and Fig. 2). The large number of crystallites that have developed hinder main-chain relaxation of the remaining amorphous fraction, for which the higher Tg is observed, and at the same time lead to reinforcement of the material, especially at T > Tg.
Molecular dynamics (MD) simulations allowed us to gain detailed insight into the structural changes that occur as a result of molecular doping. A computational box was filled with 200 oligomers, each comprising four g42T-T repeat units with full-length side chains. Oxidation levels Oox, defined as the number of charges per thiophene ring, were created by placing the same integer number of charges N onto each oligomer so that Oox = N/12 resulting in Oox ≈ 0, 8.3, 16.7 and 33.3% for a charge of N = +1, +2 and +4, respectively (Table S1 and Fig. S4a, ESI†). The appropriate number of F4TCNQ anions were added to the simulation box to achieve overall charge neutrality (details of the simulations are described in the Method section). Representative snapshots of the nanostructure for different oxidation levels are shown in Fig. 4a. We then calculated the radial distribution function gt–t(r) of the distance r between the center of mass of thiophene rings that are part of different oligomers (Fig. 4b). For neutral oligomers gt–t(r) is featureless, which is consistent with the high degree of disorder of the polymer backbones inferred from X-ray diffractograms (cf.Fig. 3). In contrast, for the case of oligomers with +1 and +2 charges (Oox ≈ 8.3 and 16.7%), gt–t(r) exhibits a pronounced peak at about 4 Å, which arises due to π-stacking of neighboring chains. With further increase of the doping level to +4 charges (Oox ≈ 33.3%) the oligomers are unable to π-stack, as evidenced by the absence of the peak in gt–t(r). Note that the presence of π-stacking at intermediate doping levels (Oox ≈ 8.3 and 16.7%) and its absence for the neat and highly doped oligomers (Oox ≈ 0 and 33.3%) can also be seen in the MD simulation snapshots (Fig. 4a and Fig. S4b, ESI†). The MD simulations are consistent with our X-ray analysis (Fig. 3), which showed that doped p(g42T-T) forms π-stacks.
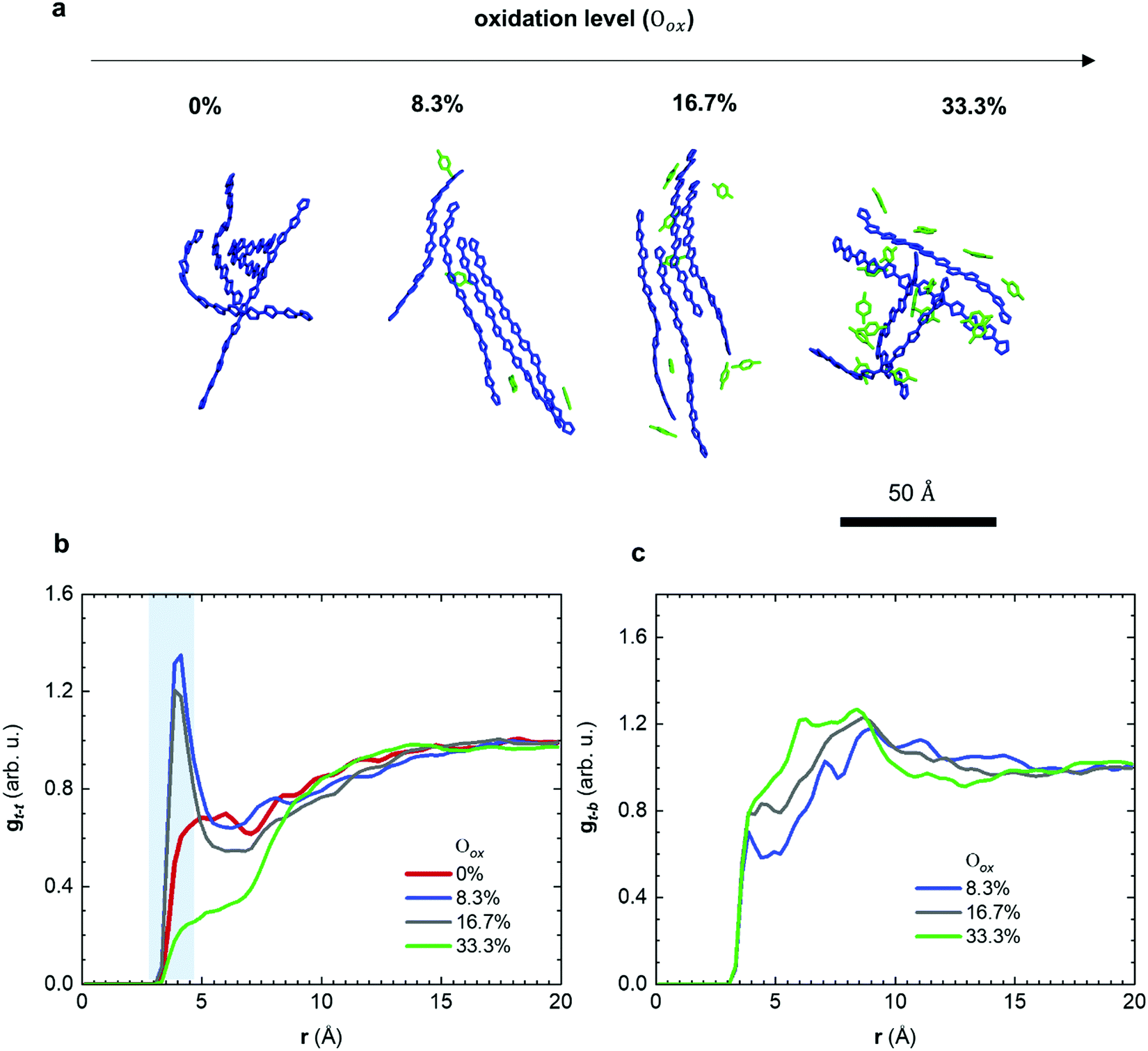 | ||
| Fig. 4 (a) Snapshots of equilibrated nanostructures obtained from molecular dynamics (MD) simulations of p(g42T-T) oligomers (blue) and F4TCNQ anions (green); the tetraethylene glycol side chains of the oligomers and the cyano groups of the anions are omitted; (b) radial distribution function gt–t(r) after MD equilibration of the distance r between the center of mass of thiophene rings that are part of different p(g42T-T) oligomers; and (c) gt–b(r) of the distance r between the center of mass of thiophene rings and the center of mass of the benzene ring of F4TCNQ anions. | ||
The observed trend in the evolution of π-stacking with the doping level can be understood as follows: for Oox ≈ 8.3 to 16.7% the counterions help to bring oligomer chains together, which promotes π-stacking and increases planarity. Note that planarity is also increased because of the change of the character of the bond alternation in the thiophene rings from aromatic to quinoid with the increase of the oxidation level (see Fig. S5, ESI†). In addition, π-stacking enables polarons to delocalize across adjacent chains, which according to previous reports promotes the pronounced π-stacking that occurs when doping regio-random P3HT with F4TCNQ.30,31 With a further increase of the doping level to Oox ≈ 33.3%, Coulomb repulsion between adjacent chains becomes dominant and the excess F4TCNQ is disrupting the microstructure of the film, which prevents π-stacking. The theoretical oxidation level of Oox ≈ 16.7% corresponds to p(g42T-T) doped with 20 mol% F4TCNQ, which has an Oox ≈ 16.8% (Table S2, ESI†).
We also calculated the radial distribution function gt–b(r) of the distance r between the center of mass of thiophene rings and the center of mass of the benzene ring of F4TCNQ anions (Fig. 4c). For all studied doping levels, we observe a sharp onset in gt–b(r) around 3.5 Å, which is comparable to a donor–acceptor distance of 3 to 5 Å predicted by Spano et al. for P3HT and F4TCNQ.32,33 We also carried out MD simulations where we mimicked tensile deformation of the neat and doped material, using a strain rate of 109 s−1, which yields a Young's modulus of almost 4 GPa with only a minor dependence on Oox ranging from 0 to 33.3% (Fig. S6, ESI†). This value is comparable to the storage modulus of 5–9 GPa determined with DMA below −20 °C for p(g42T-T) doped with 20 mol% F4TCNQ (Oox ≈ 16.8%; see Fig. 2).
In a further set of experiments, we studied the impact of the charge of the counterion on the mechanical properties. Each F4TCNQ molecule can undergo two electron transfer processes with polymers that have an IE0 ≤ 4.7 eV, resulting in the formation of F4TCNQ dianions with a charge of −2.23 Dianion formation is most pronounced for low dopant concentrations of 3 and 6 mol% F4TCNQ, as evidenced by a distinct FTIR absorption peak at ϑCN = 2131 cm−1 (Fig. 5a and Fig. S7, ESI†). We estimated the oxidation level using FTIR absorption spectra recorded for spin-coated films of p(g42T-T) co-processed with the dopant (Fig. S7 and S8, ESI†). The anion and dianion of F4TCNQ give rise to distinct absorption peaks at ϑCN that correspond to the cyano stretch vibration. We assumed that at low oxidation levels each dopant molecule undergoes an electron transfer with the polymer and compared the relative intensity of the ϑCN absorption peaks with corresponding FTIR signals recorded for solutions of the lithium and dilithium salt of F4TCNQ.23 A dopant concentration of 3 mol% F4TCNQ gives rise to an ionization efficiency of ηion ≈ 187%, i.e. most dopant molecules generate two polarons, and hence Oox ≈ 5.7% (Table S2, ESI†). We also included samples doped with F2TCNQ (EA0 ≈ 5.1 eV), which can only undergo one electron transfer process with p(g42T-T) per dopant molecule due to a higher EA0 ≈ 5.1 eV and EA− ≈ 4.5 eV (cf.Fig. 5a). For a dopant concentration of 6 mol% F2TCNQ we estimate Oox ≈ 6.4%, assuming that each dopant undergoes one electron transfer with the polymer, i.e. ηion ≈ 100% (Table S2, ESI†). As a result, we are able to carry out a direct comparison of the mechanical properties of doped p(g42T-T) with a similar oxidation level but compensated with counterions that have charge −1 (F2TCNQ anions) or −2 (F4TCNQ dianions).
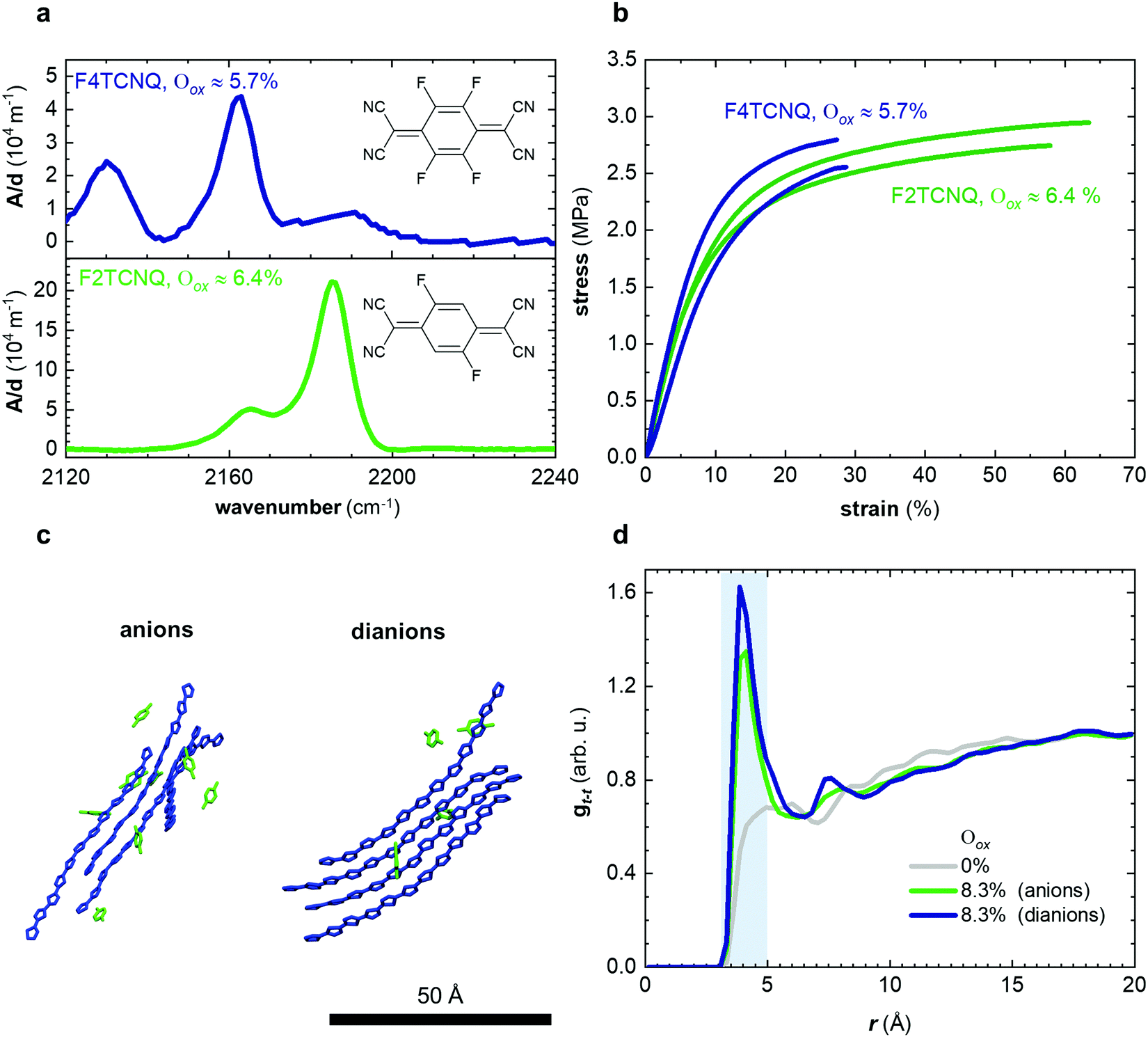 | ||
| Fig. 5 (a) Transmission FTIR absorbance spectra, with the absorbance A normalized by the film thickness d, of p(g42T-T) doped with 3 mol% F4TCNQ (blue; Oox ≈ 5.7%) and 6 mol% F2TCNQ (green; Oox ≈ 6.4%); (b) stress–strain curves recorded at room temperature by tensile deformation of free-standing samples of p(g42T-T) doped with 3 mol% F4TCNQ (blue) and 6 mol% F2TCNQ (green); (c) snapshots from equilibrated MD simulations of p(g42T-T) oligomers with a charge of +1 (Oox ≈ 8.3%) neutralized with F4TCNQ anions (left) and F4TCNQ dianions (right); (d) radial distribution function gt–t(r) after MD equilibration of the distance r between the center of mass of thiophene rings of different oligomers for neutral oligomers (grey) and oligomers with charge +1 neutralized with F4TCNQ anions (green) and F4TCNQ dianions (blue). | ||
We used tensile deformation of free-standing samples at room temperature to analyze the mechanical properties of p(g42T-T). For low oxidation levels the low stiffness made it challenging to both mount samples in our DMA instrument and to ensure their integrity over a wide range of temperatures (see Methods for details). Tensile deformation yielded a comparable Young's modulus of Edoped ≈ (31 ± 2) MPa and (24 ± 4) MPa (Fig. 5b and Table S2, ESI†), which indicates that the charge of the counterion does not influence the stiffness of the doped polymer. WAXS diffractograms recorded for these samples feature a clear π-stacking peak at q010 ≈ 1.84 Å−1 (Fig. S9, ESI†). Moreover, MD simulations of oligomers with charge +1 (Oox ≈ 8.3%) but neutralized with either F4TCNQ anions or dianions yield a comparable radial distribution function between the center of mass of thiophene rings of different oligomers with a distinct peak in gt–t(r) at 4 Å (Fig. 5c, d; note that for the MD simulations we used the same dopant, i.e. F4TCNQ). Doping with F2TCNQ and F4TCNQ appears to enhance the order of the polymer to a similar degree, which suggests that the observed increase in Young's modulus can be explained by changes in the conformation of the polymer and π-stacking. We therefore conclude that the presence of dianions does not lead to ionic type crosslinking of p(g42T-T) in the solid state since the stiffness of the polymer is not affected by the charge of the counterions. However, p(g42T-T) doped with F2TCNQ displays a significantly larger strain at break of εb ≈ (50 ± 10)% as compared to F4TCNQ doped material with εb ≈ (30 ± 5)% (Table S2, ESI†). It appears that the presence of more numerous monoanions instead of dianions has a positive impact on the toughness with values of about 0.8 MJ m−3 and 0.5 MJ m−3 in case of p(g42T-T) doped with 6 mol% F2TCNQ and 3 mol% F4TCNQ, respectively.
In a further set of experiments, we compared the impact of the oxidation level on both the mechanical and electrical properties of doped p(g42T-T). We used tensile deformation at room temperature because we were able to carry out this measurement for a wide range of Oox from 0 to 18.2% (see Methods for details). UV-vis-IR spectra confirm the high oxidation level of the here studied samples doped with F4TCNQ or F2TCNQ, as evidenced by the disappearance of the neat polymer absorption with increasing Oox and the emergence of strong polaronic absorption peaks in the infrared part of the spectrum (Fig. S7 and S8, ESI†).34
The neat, undoped polymer features a low Young's modulus of Eneat ≈ (8 ± 2) MPa, which is three times lower than the value inferred from oscillatory shear rheometry (Table 2), likely due to the low employed tensile deformation rate of 5 mN min−1. The Young's modulus increases with Oox, first gradually to Edoped ≈ (24 ± 4) MPa at Oox ≈ 5.7%, and then more strongly reaching a value of Edoped ≈ (232 ± 16) MPa at Oox ≈ 18.2% (Fig. 6a and b), which yields a figure of merit η ≈ 1.5 (cf.Table 1). The toughness shows minimal increase for Oox < 10% but then increases rapidly to 5.2 MJ m3 at Oox ≈ 18.2% (Fig. S10c, ESI†). The electrical conductivity displays a similar trend as the Young's modulus with Oox and reaches a value of σ ≈ (52 ± 3) S cm−1 for Oox ≈ 18.2% (Fig. 6b). Doping with F2TCNQ results in a comparable trend even though Oox only reaches 13.5% (estimated by comparing the intensity of the ϑCN absorption peak for different amounts of dopant; Fig. S8, ESI†), yielding a lower conductivity of σ ≈ (20 ± 3) S cm−1 but, strikingly, a higher Young's modulus of Edoped ≈ (377 ± 85) MPa and hence η ≈ 1.7 (Fig. S10, ESI†). The close to linear correlation between σ and Edoped (Fig. 6c) is akin to the interplay of electrical and mechanical properties that has been observed for uniaxially aligned conjugated polymer tapes and fibers.9,35,36 Transmission WAXS diffractograms reveal that the intensity of the q010 diffraction due to π-stacking increases with Oox (Fig. S9, ESI†). Since π-stacking aids hopping of charges between neighboring polymer chains as well as the transmission of mechanical force, σ and Edoped increase in tandem with Oox.
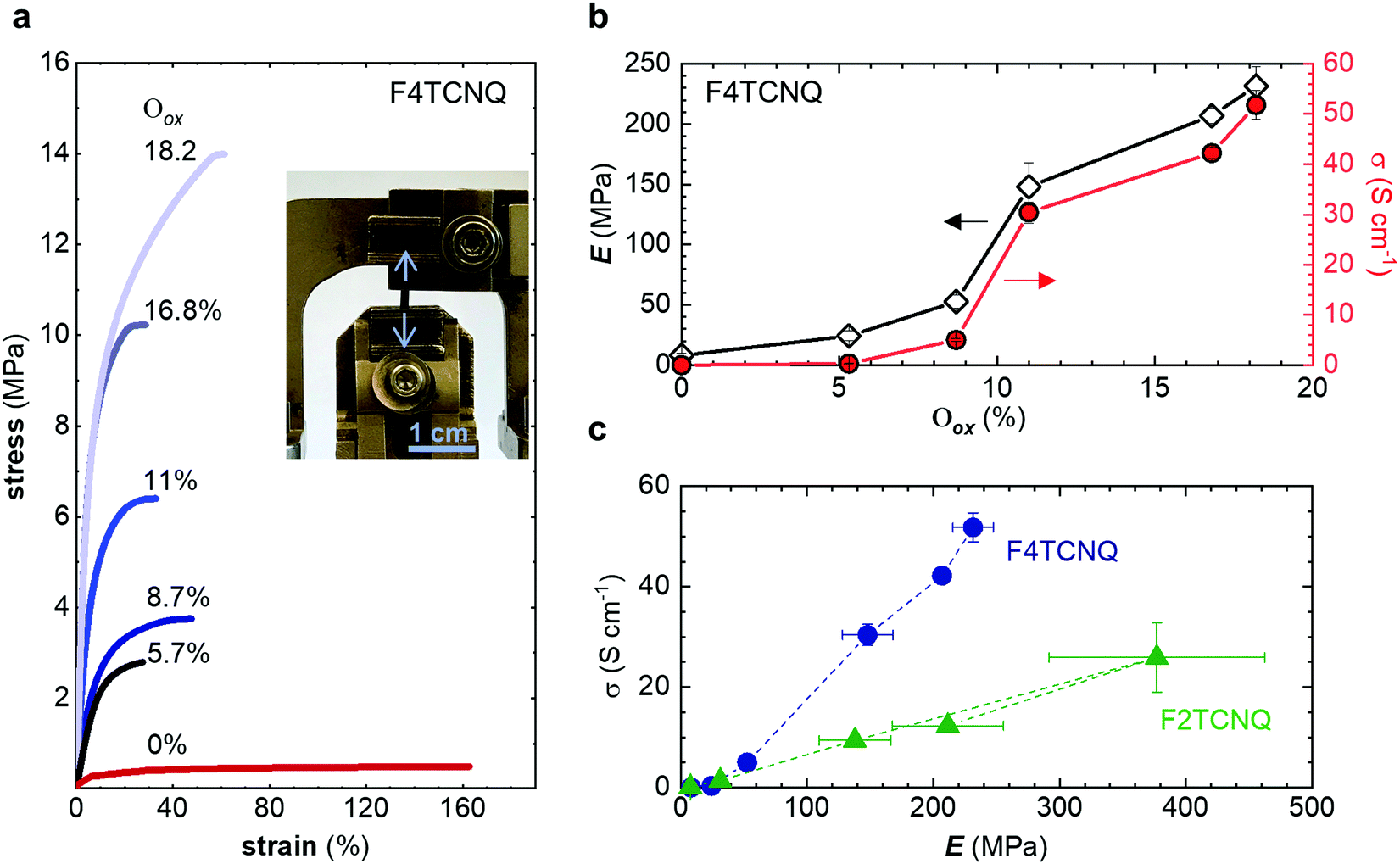 | ||
| Fig. 6 (a) Stress–strain curves recorded at room temperature by tensile deformation of free-standing samples of neat p(g42T-T) (red) and the polymer doped with F4TCNQ (blue) resulting in an oxidation level per thiophene ring Oox ranging from 5.7 to 18.2%; inset: Photograph of a doped polymer sample clamped in a DMA instrument prior to tensile deformation; (b) Young's modulus E (black) and conductivity σ (red) of p(g42T-T) doped with F4TCNQ; (c) σ vs. E of p(g42T-T) doped with F4TCNQ (blue) and F2TCNQ (green). | ||
Finally, we explored if an increase in stiffness can also be achieved with dopants other than F4TCNQ and F2TCNQ. We therefore doped p(g42T-T) with the redox dopants Magic Blue37 and DDQ as well as the acid dopants PDSA and TFSI38 (see Table 3 for chemical structures). In particular for 10 mol% Magic Blue we observe a considerable increase in Young's modulus to Edoped ≈ (148 ± 20) MPa, corresponding to η ≈ 1.3. Intriguingly, the two acid dopants only cause a minor increase in stiffness despite a relatively high electrical conductivity, e.g. σ ≈ (11 ± 2) S cm−1 in case of TFSI. We have previously observed that 10 mol% of acid dopant lead to considerable π-stacking of p(g42T-T).38 Intriguingly, p(g42T-T) doped with 18 mol% TFSI features a Tg ≈ −49 °C (Fig. S11, ESI†), which is much lower than the value observed for p(g42T-T) doped with 20 mol% F4TCNQ (see Table 1). Hence, the use of acid dopants may allow to create conducting materials that remain relatively soft. We also studied whether the type of side chain influences to which extent doping changes the modulus. Regioregular P3DDT features a relatively low Young's modulus of Eneat ≈ (45 ± 6) MPa (cf.Table 1), which increases to Edoped ≈ (80 ± 2) MPa upon sequentially doping with a saturated solution of F4TCNQ in AcN, for 3 days, corresponding to a figure of merit of only η ≈ 0.2 (gravimetric analysis indicates the uptake of 7 mol% F4TCNQ; σ ≈ (5 ± 1) × 10−3 S cm−1).
| Dopant | Structure | mol% dopant | E doped (MPa) | η | σ (S cm−1) |
|---|---|---|---|---|---|
| F4TCNQ |
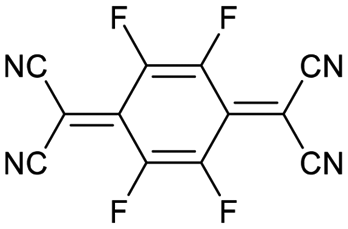
|
10 | 148 ± 20 | 1.3 | 30 ± 2 |
| F2TCNQ |
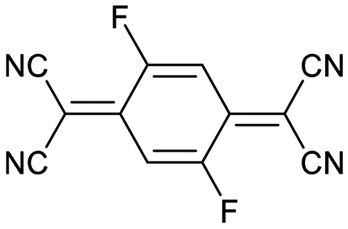
|
10 | 138 ± 28 | 1.2 | 9 ± 1 |
| Magic Blue |
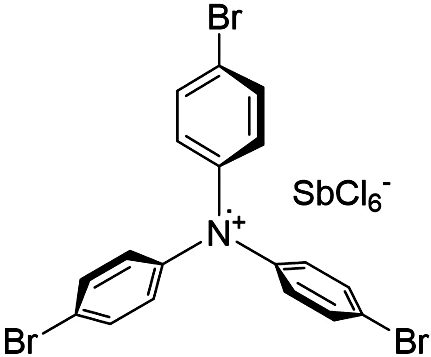
|
10 | 138 ± 24 | 1.2 | 56 ± 4 |
| DDQ |
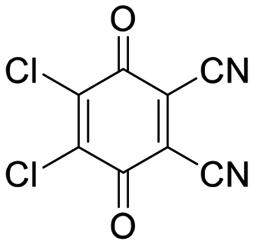
|
10 | 73 ± 11 | 1 | 17 ± 2 |
| PDSA |
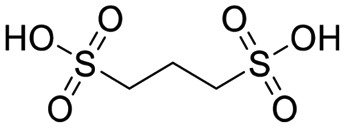
|
10 | 18 ± 5 | 0.3 | 14 ± 10 |
| TFSI |
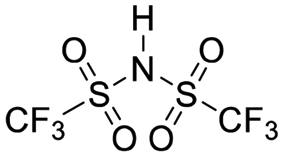
|
10 | 41 ± 17 | 0.7 | 11 ± 2 |
Conclusions
The polymer p(g42T-T) with tetraethylene glycol side chains is very soft with a Young's modulus of only 8 MPa at room temperature due to a low degree of crystallinity and a low Tg ≈ −46 °C, measured with DMA. Molecular doping with F4TCNQ or F2TCNQ strongly enhances the degree of π-stacking of the polymer and increases the Tg to 3 °C in case of an oxidation level Oox ≈ 16.8%. As a result, the Young's modulus increases ∼29-fold to 232 MPa for p(g42T-T) doped with F4TCNQ (Oox ≈ 18.2%). Our findings are corroborated by molecular dynamics simulations. A comparison of less strongly doped samples with Oox ≈ 5.7%, where doping with F4TCNQ mostly yields dianions, indicated that the charge of the counterions (i.e. −1 of anions or −2 of dianions) does not affect the stiffness of the doped polymer, suggesting that dianions do not lead to ionic type crosslinks. However, the choice of dopant influences the ductility and toughness of the doped polymer. Doping of p(g42T-T) with F2TCNQ results in an up to 47-fold increase in Young's modulus to 377 MPa, which corresponds to the strongest relative increase reported for any conjugated polymer. Evidently, molecular doping is a powerful tool that can not only be used to adjust the electrical but also the mechanical properties of conjugated polymers, which may spur the field of flexible and stretchable electronics.Methods
Materials
The synthesis of p(g42T-T) (Mn ≈ 24 kg mol−1; PDI ≈ 3.3) is described elsewhere.39 F4TCNQ and F2TCNQ were purchased from Tokyo Chemical Industry (TCI). Regioregular P3DDT (Mw ≈ 60 kg mol−1), DDQ, Magic Blue, and PDSA (70% aqueous solution) were purchased from Sigma Aldrich. TFSI was purchased from Acros Organics. All dopants were used as received. Chloroform (CHCl3; purity 99.8%) and acetonitrile (AcN; purity 99.95%) were purchased from Fisher Scientific and VWR, respectively.Sample preparation
Co-processed samples were prepared by adding solutions of the dopant in AcN (6 g L−1 for PDSA and 2 g L−1 for the rest of the dopants) to solutions of p(g42T-T) in CHCl3 (3 to 20 g L−1 to achieve different polymer:dopant ratios) and P3DDT in CHCl3 (6 g L−1) together with further AcN to ensure a solvent ratio of 2:1 CHCl3:AcN. The dopant mol% is calculated per thiophene ring of the conjugated polymers. Thin films for spectroscopy were spin-coated at a speed of 1000–5000 rpm for 60 s onto glass slides for UV-vis spectroscopy or CaF2 substrates for FTIR spectroscopy to achieve a film thickness of 35 to 190 nm. Thin films for vapor doping were spin-coated at 1000 rpm for 40 s onto silicon substrates using a solution of p(g42T-T) in chlorobenzene (6 g L−1), followed by annealing for 10 minutes at 120 °C and drying under vacuum. Vapor doping was performed in a nitrogen atmosphere by exposing the p(g42T-T) films to F4TCNQ vapor for 15 minutes. Free-standing samples with a thickness of 30 to 80 μm for mechanical testing were drop-cast at 30 °C onto glass slides followed by removal from the substrate with a sharp blade. Neat p(g42T-T) was frozen in liquid nitrogen prior to the removal of the polymer film from the substrate. Glass fiber supported samples were made through coating glass mesh strands cut at 45° with p(g42T-T) (chlorobenzene, 10 g L−1), acetone fraction p(g42T-T) (CHCl3, 10 g L−1) or a mixture of p(g42T-T) + 3 mol% F4TCNQ, followed by drying at 30 °C under vacuum for 24 hours. The sample for shear rheometry was prepared in a nitrogen glovebox by heating 10 mg polymer up to 200 °C for 45 minutes and pulling vacuum to ensure no bubbles were present in the sample, followed by compressing it using about 1 N of force, and allowing it to cool. The sample diameter was 3 mm and disposable aluminum parallel plates were used.
The thickness of thin and thick films was measured with a KLA Alphastep Tencor D-100 profilometer and a micro-caliper, respectively.
Differential scanning calorimetry (DSC)
DSC measurements were carried out under nitrogen at a flow rate of 60 mL min−1 with a Mettler Toledo DSC2 equipped with a Gas controller GC 200 system at a heating rate of 10 °C min−1.Fast scanning calorimetry (FSC)
Measurements were conducted under nitrogen with a Mettler Toledo Flash DSC 1. A small amount of the polymer was transferred directly to the FSC chip sensor. The sample was first heated to 150 °C to delete the thermal history and then cooled down to −50 °C with different cooling rates ranging from −0.1 K s−1 to −1000 K s−1. Finally, the sample was heated with 600 K s−1. The fictive temperature was calculated using Moynihan's area matching method or by extrapolation if the fictive temperature was below the onset of devitrification.40X-Ray scattering
Transmission wide-angle X-ray scattering (WAXS) was carried out with a Mat:Nordic instrument from SAXSLAB equipped with a Rigaku 003+ high brilliance micro focus Cu Kα-radiation source (wavelength = 1.5406 Å) and a Pilatus 300 K detector placed at a distance of 88.6 mm from the sample. Grazing incidence wide angle X-ray scattering (GIWAXS) measurements were carried out at the Stanford Synchrotron Radiation Lightsource Experimental Station 11-3 using a sample-to-detector distance of 315 mm and an incidence angle of 0.15°.UV-vis absorption spectroscopy
UV-vis-NIR spectra were recorded with a PerkinElmer Lambda 1050 spectrophotometer.Fourier transform infrared spectroscopy (FTIR)
Infrared absorption measurements were performed with a PerkinElmer FT-IR Spectrometer ‘Frontier’ on thin p(g42T-T):F4TCNQ films coated on CaF2.Electrical characterization
The electrical resistivity was measured on fresh films with a 4-point probe setup from Jandel Engineering (cylindrical probe head, RM3000) using co-linear tungsten carbide electrodes with an equidistant spacing of 1 mm. The in-line 4-point probe for films gives a measure of the sheet resistance Rs = π/ln2⋅V/I, where V and I are the voltage and current and π/ln2 is a geometrical correction factor. The conductivity was calculated according to σ = 1/(dRs), where d is the film thickness.
Oscillatory shear rheometry
Measurements were carried out with a Rheometric Scientific ARES LS strain-controlled rheometer using a 3 mm aluminum parallel plate geometry, a strain of 0.2%, which was in the linear regime, and a frequency of 0.16 Hz. The temperature was increased from −80 °C to 180 °C at 5 °C min−1. The sample preparation and measurement were carried out in inert nitrogen atmosphere.Mechanical testing
Dynamic mechanical analysis (DMA) and tensile testing were performed using a Q800 dynamic mechanical analyzer from TA Instruments. To support neat p(g42T-T) and polymer doped with 3 mol% and 6 mol% dopant during mounting, samples were fixated in a paper frame that was cut prior to tensile testing; all other samples were mounted without any support. DMA was carried out at a frequency of 1 Hz while ramping the temperature from −80 °C to 60 °C at a rate of 3 °C min−1. A preload force of 0.003 N–0.009 N and a dynamic strain with a maximum value of 0.03%–0.05% was used for samples supported by a glass fiber mesh. A pre-load force of 0.01 N, a gauge length of 5.1 mm–5.6 mm and a dynamic strain with a maximum value of 0.3% was used for free-standing doped p(g42T-T). DMA of free-standing neat p(g42T-T) was performed at 1 Hz by cooling from 22 °C to 0 °C at a rate of −3 °C min−1 with a pre-load force of 0.01 N, a gauge length of 4.3 mm and a dynamic strain with a maximum value of 0.02%. Tensile testing was performed in a controlled force mode with a force rate of 0.005 N min−1 using a gauge length of 3.8 mm–7 mm.Molecular dynamics (MD) simulations
A parallel MD simulator, LAMMPS package was used to perform all-atom MD simulations with the general AMBER force field (GAFF) as implemented in moltemplate code.41 The Lennard–Jones and Coulombic interactions were cutoff at 1.1 nm, and a k-mean scheme of particle–particle particle-mesh was used for long range Coulombic interactions as implemented in the LAMMPS package. All MD simulations were carried out with a 1.0 fs time step. The initial structure and partial atomic charges of molecules for MD simulations were obtained from geometry optimization and electrostatic potential (ESP) calculation, respectively, using density functional theory (DFT) with the ωB97XD functional and the 6-31G(d) basis set as implemented in Gaussian (Fig. S4, ESI†). 200 oligomer chains consisting of four g42T-T repeat units, with a charge of 0, +1, +2 or +4, were placed in a rectangular computational box of 20 × 20 × 20 nm3 together with F4TCNQ anions or dianions to achieve charge neutrality (see Table S1, ESI†). The solid-state nanostructure was modelled by the following procedure: (1) initial equilibration at 800 K in an isochoric-isothermal (NVT) ensemble for 2 ns and then in an isothermal–isobaric (NPT) ensemble at 0 atm for 5 ns using the Nose–Hoover thermostat and barostat, while allowing the computational box size to decrease, (2) equilibration at 800 K in a microcanonical ensemble for 1 ns using temperature control by a Langevin thermostat and then in a NPT ensemble at 0 atm for 1 ns, and (3) a cooling step from 800 to 300 K at a rate of 0.5 K ps−1 in a NPT ensemble at 0 atm followed by equilibration in a NPT ensemble for 5 ns.Author contributions
S. Z. and C. M. planned the study and wrote the manuscript. R. K. synthesized the polymer. E. J. carried out optical spectroscopy and determined the oxidation level. A. F., A. W. and E. D. G. carried out oscillatory shear rheometry measurements. S. Z. and A. L. carried out the thermomechanical analysis and WAXS characterization. S. H. carried out fast scanning calorimetry measurements. P. N. and M. C. carried out the GIWAXS characterization. D. K. and I. Z. carried out molecular dynamics simulations. All authors read and commented on the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge financial support from the Swedish Research Council (grants no. 2018-03824 and 2016-05990) and the Knut and Alice Wallenberg Foundation through a Wallenberg Academy Fellowship Prolongation grant as well as the project “Mastering Morphology of Solution-Borne Electronics”. A. F., A. W. and E. D. G. acknowledge support from the U.S. National Science Foundation under award DMR-1921854. Research at UCSB was sponsored by the U.S. Army Research Office and accomplished under cooperative agreement W911NF-19-2-0026 for the Institute for Collaborative Biotechnologies. This work was carried out in part at the Chalmers materials analysis laboratory (CMAL). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at NSC and HPC2N.References
- N. Matsuhisa, X. Chen, Z. Bao and T. Someya, Chem. Soc. Rev., 2019, 48, 2946–2966 RSC.
- J. Onorato, V. Pakhnyuk and C. K. Luscombe, Polym. J., 2017, 49, 41–60 CrossRef CAS.
- S. E. Root, S. Savagatrup, A. D. Printz, D. Rodriquez and D. J. Lipomi, Chem. Rev., 2017, 117, 6467–6499 CrossRef CAS PubMed.
- R. Xie, R. H. Colby and E. D. Gomez, Adv. Electron. Mater., 2018, 4, 1700356 CrossRef.
- Y. Cao, P. Smith and A. J. Heeger, Polymer, 1991, 32, 1210–1218 CrossRef CAS.
- B. Roth, S. Savagatrup, N. V. de los Santos, O. Hagemann, J. E. Carlé, M. Helgesen, F. Livi, E. Bundgaard, R. R. Søndergaard, F. C. Krebs and D. J. Lipomi, Chem. Mater., 2016, 28, 2363–2373 CrossRef CAS.
- J. Hynynen, E. Järsvall, R. Kroon, Y. Zhang, S. Barlow, S. R. Marder, M. Kemerink, A. Lund and C. Müller, ACS Macro Lett., 2019, 8, 70–76 CrossRef CAS PubMed.
- R. Kroon, A. I. Hofmann, L. Yu, A. Lund and C. Müller, Chem. Mater., 2019, 31, 2770–2777 CrossRef CAS PubMed.
- J. Moulton and P. Smith, Polymer, 1992, 33, 2340–2347 CrossRef CAS.
- D. Kiefer, L. Yu, E. Fransson, A. Gómez, D. Primetzhofer, A. Amassian, M. Campoy-Quiles and C. Müller, Adv. Sci., 2017, 4, 1600203 CrossRef.
- J. Moulton and P. Smith, Synth. Met., 1991, 40, 13–22 CrossRef CAS.
- Y. Zheng, G. J. N. Wang, J. Kang, M. Nikolka, H. C. Wu, H. Tran, S. Zhang, H. Yan, H. Chen and P. Y. Yuen, Adv. Funct. Mater., 2019, 29, 1905340 CrossRef CAS.
- J. Mun, J. Kang, Y. Zheng, S. Luo, Y. Wu, H. Gong, J.-C. Lai, H.-C. Wu, G. Xue, J. B.-H. Tok and Z. Bao, Adv. Electron. Mater., 2020, 6, 2000251 CrossRef CAS.
- S. Tokito, P. Smith and A. J. Heeger, Polymer, 1991, 32, 464–470 CrossRef CAS.
- S. Tokito, P. Smith and A. J. Heeger, Synth. Met., 1990, 36, 183–194 CrossRef CAS.
- M. Moser, T. C. Hidalgo, J. Surgailis, J. Gladisch, S. Ghosh, R. Sheelamanthula, Q. Thiburce, A. Giovannitti, A. Salleo, N. Gasparini, A. Wadsworth, I. Zozoulenko, M. Berggren, E. Stavrinidou, S. Inal and I. McCulloch, Adv. Mater., 2020, 32, 2002748 CrossRef CAS.
- C. B. Nielsen, A. Giovannitti, D.-T. Sbircea, E. Bandiello, M. R. Niazi, D. A. Hanifi, M. Sessolo, A. Amassian, G. G. Malliaras, J. Rivnay and I. Mcculloch, J. Am. Chem. Soc., 2016, 138, 10252–10259 CrossRef CAS.
- D. Kiefer, A. Giovannitti, H. Sun, T. Biskup, A. Hofmann, M. Koopmans, C. Cendra, S. Weber, L. J. Anton Koster, E. Olsson, J. Rivnay, S. Fabiano, I. McCulloch and C. Müller, ACS Energy Lett., 2018, 3, 278–285 CrossRef CAS PubMed.
- J. Liu, G. Ye, H. G. O. Potgieser, M. Koopmans, S. Sami, M. I. Nugraha, D. R. Villalva, H. Sun, J. Dong, X. Yang, X. Qiu, C. Yao, G. Portale, S. Fabiano, T. D. Anthopoulos, D. Baran, R. W. A. Havenith, R. C. Chiechi and L. J. A. Koster, Adv. Mater., 2021, 33, 2006694 CrossRef CAS PubMed.
- D. Moia, A. Giovannitti, A. A. Szumska, I. P. Maria, E. Rezasoltani, M. Sachs, M. Schnurr, P. R. F. Barnes, I. McCulloch and J. Nelson, Energy Environ. Sci., 2019, 12, 1349–1357 RSC.
- A. V. Volkov, H. Sun, R. Kroon, T.-P. Ruoko, C. Che, J. Edberg, C. Müller, S. Fabiano and X. Crispin, ACS Appl. Energy Mater., 2019, 2, 5350–5355 CrossRef CAS.
- S. Zokaei, R. Kroon, J. Gladisch, B. D. Paulsen, W. Sohn, A. I. Hofmann, G. Persson, A. Stamm, P.-O. Syrén, E. Olsson, J. Rivnay, E. Stavrinidou, A. Lund and C. Müller, Adv. Sci., 2021, 8, 2002778 CrossRef CAS PubMed.
- D. Kiefer, R. Kroon, A. I. Hofmann, H. Sun, X. Liu, A. Giovannitti, D. Stegerer, A. Cano, J. Hynynen, L. Yu, Y. Zhang, D. Nai, T. F. Harrelson, M. Sommer, A. J. Moule, M. Kemerink, S. R. Marder, I. McCulloch, M. Fahlman, S. Fabiano and C. Müller, Nat. Mater., 2019, 18, 149–155 CrossRef CAS.
- L. Pan, A. Chortos, G. Yu, Y. Wang, S. Isaacson, R. Allen, Y. Shi, R. Dauskardt and Z. Bao, Nat. Commun., 2014, 5, 3002 CrossRef.
- S. Ghosh, J. Rasmusson and O. Inganäs, Adv. Mater., 1998, 10, 1097–1099 CrossRef CAS.
- O. Vassiliadou, V. Chrysostomou, S. Pispas, P. A. Klonos and A. Kyritsis, Soft Matter, 2021, 17, 1284–1298 RSC.
- A. Hamidi-Sakr, L. Biniek, J.-L. Bantignies, D. Maurin, L. Herrmann, N. Leclerc, P. Lévêque, V. Vijayakumar, N. Zimmermann and M. Brinkmann, Adv. Funct. Mater., 2017, 27, 1700173 CrossRef.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef CAS PubMed.
- S. N. Patel, A. M. Glaudell, K. A. Peterson, E. M. Thomas, K. A. O’Hara, E. Lim and M. L. Chabinyc, Sci. Adv., 2017, 3, e1700434 CrossRef PubMed.
- P. Y. Yee, D. T. Scholes, B. J. Schwartz and S. H. Tolbert, J. Phys. Chem. Lett., 2019, 10, 4929–4934 CrossRef CAS PubMed.
- E. Lim, A. M. Glaudell, R. Miller and M. L. Chabinyc, Adv. Electron. Mater., 2019, 5, 1800915 CrossRef.
- R. Ghosh, A. R. Chew, J. Onorato, V. Pakhnyuk, C. K. Luscombe, A. Salleo and F. C. Spano, J. Phys. Chem. C, 2018, 122, 18048–18060 CrossRef CAS.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1702654 CrossRef.
- I. Sahalianov, J. Hynynen, S. Barlow, S. R. Marder, C. Müller and I. Zozoulenko, J. Phys. Chem. B, 2020, 124, 11280–11293 CrossRef CAS.
- A. Lund, N. M. van der Velden, N.-K. Persson, M. M. Hamedi and C. Müller, Mater. Sci. Eng., R, 2018, 126, 1–29 CrossRef.
- R. Sarabia-Riquelme, R. Andrews, J. E. Anthony and M. C. Weisenberger, J. Mater. Chem. C, 2020, 8, 11618–11630 RSC.
- A. I. Hofmann, R. Kroon, S. Zokaei, E. Järsvall, C. Malacrida, S. Ludwigs, T. Biskup and C. Müller, Adv. Electron. Mater., 2020, 6, 2000249 CrossRef CAS.
- A. I. Hofmann, R. Kroon, L. Yu and C. Müller, J. Mater. Chem. C, 2018, 6, 6905–6910 RSC.
- R. Kroon, D. Kiefer, D. Stegerer, L. Yu, M. Sommer and C. Müller, Adv. Mater., 2017, 29, 1700930 CrossRef.
- S. Gao, Y. P. Koh and S. L. Simon, Macromolecules, 2013, 46, 562–570 CrossRef CAS.
- A. Jewett, Moltemplate, http://www.moltemplate.org.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1mh01079d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |