
Advanced approaches of developing targeted covalent drugs
Conghao
Gai
 *a,
Suzannah J.
Harnor
b,
Shihao
Zhang
a,
Céline
Cano
b,
Chunlin
Zhuang
a and
Qingjie
Zhao
*a
*a,
Suzannah J.
Harnor
b,
Shihao
Zhang
a,
Céline
Cano
b,
Chunlin
Zhuang
a and
Qingjie
Zhao
*a
aOrganic Chemistry Group, College of Pharmacy, Naval Medical University, Shanghai, 200433, P. R. China. E-mail: c.gai2@outlook.com
bCancer Research UK Newcastle Drug Discovery Unit, Newcastle University Centre for Cancer, School of Natural and Environmental Sciences, Bedson Building, Newcastle University, Newcastle upon Tyne NE1 7RU, UK
First published on 11th October 2022
Abstract
In recent years, the development of targeted covalent inhibitors has gained popularity around the world. Specific groups (electrophilic warheads) form irreversible bonds with the side chain of nucleophilic amino acid residues, thus changing the function of biological targets such as proteins. Since the first targeted covalent inhibitor was disclosed in the 1990s, great efforts have been made to develop covalent ligands from known reversible leads or drugs by addition of tolerated electrophilic warheads. However, high reactivity and “off-target” toxicity remain challenging issues. This review covers the concept of targeted covalent inhibition to diseases, discusses traditional and interdisciplinary strategies of cysteine-focused covalent drug discovery, and exhibits newly disclosed electrophilic warheads majorly targeting the cysteine residue. Successful applications to address the challenges of designing effective covalent drugs are also introduced.
Drugs that covalently bind to their biological targets have a long history in drug discovery. There is an increased interest in covalent therapeutics in the literature and recent years have witnessed a significant increase in the number of drug candidates with a covalent mechanism of action progressing through clinical trials. Liu et al. has reviewed several protein kinases, RAS proteins, and a number of other enzymes that have been studied extensively as targets for covalent inhibition, claiming that covalent strategy may reduce the risk of idiosyncratic toxicity.1 Dömling et al. also provided a historic overview, including structural aspects and examples on challenging targets and several developed covalent inhibitors from 2011 to 2019.2 The development of small-molecule drugs that covalently inhibit biological targets dates back to 1897 with the discovery of aspirin. Over the past twenty years, targeted drugs have shown increasingly rapid advances, for example, reduced toxicity in healthy tissue, leading to an important strategy for cancer treatment.3 With this in mind, targeted covalent drugs, such as the β-lactam-containing antibiotic penicillin, chemotherapeutic fluorouracil, and osimertinib, rationally designed molecules that irreversibly bind to target proteins are being developed.4 Compared to drugs with reversible binding modes, covalent drugs allow high potency to be routinely achieved in compounds of low molecular mass, along with all the associated beneficial pharmaceutical properties.5 Renato has summarized the pros and cons for covalent inhibitors, especially highlighting that covalent inhibition may be an underused strategy for addressing challenging targets and ‘undruggable’ modalities in human disease, such as odanacatib to cathepsin K.6
To date, much effort has gone into developing good covalent ligands from known reversible leads or drugs by introduction of tolerated electrophilic warheads to the scaffold.7–9 Elena exhibited several types of warheads found in FDA-approved drugs and their targeted moieties. The percentage clearly showed acrylamide as the most popular warhead in FDA-approved TCIs targeting cysteine residues.10 High-throughput screening has also proven to be an excellent approach to the development of novel highly selective covalent ligands targeting specific proteins, such as KRasG12C.11 Additionally, the development of fragment-based drug discovery (FBDD) has succeeded in probing potential binding sites of ‘undruggable’ target proteins, thus inspiring the strategy of building novel targeted covalent inhibitors from covalent fragments. Several new techniques and multidisciplinary ideas including proteolysis targeting chimeras (PROTACs) and peptide drugs currently promote the development of either reversible or irreversible covalent inhibitors also.12,13 The strategy employed depends on prior knowledge and structural tolerance of the scaffold and target when processing a new project.
Although toxicity is still a bottleneck restricting the development of targeted covalent inhibitors (TCIs), a 2009 report revealed that the risk of idiosyncratic toxicities may be mitigated via lower doses of administered drug irrespective of the drug mechanism.14 This review covers the concept of targeted covalent inhibition, traditional and interdisciplinary strategies of covalent drug discovery, and the exhibited covalent binding modes of newly disclosed electrophilic warheads and potential risk mitigation.
1. Targeted covalent inhibitors and binding modes
Remarkably, it was not known until the 1970s that aspirin acts through covalent and irreversible inhibition of cyclooxygenase-1 (COX-1) and −2, the enzymes responsible for the biosynthesis of prostaglandins.15 Despite previous underlying concerns regarding safety, development of covalent inhibitors has made significant contributions to intervention of human health conditions over the past two decades.Covalent inhibitors selectively bind to amino acid residues on the catalytic domain of the target protein, causing loss of function, leading to anti-tumour, anti-inflammatory and anti-viral effects. In this section we review newly disclosed clinical covalent inhibitors and their exhibited irreversible binding adducts with great respect.
1.1 Anti-tumour
Covalent inhibitors for targeted cancer therapy have developed rapidly in the past twenty years, with approximately 30% of TCIs being used for oncology-related targets.2As a traditional druggable target, third-generation covalent inhibitors of EGFR aim to overcome drug resistance of the first- and second-generation EGFR covalent inhibitors. Osimertinib (Tagrisso®) currently dominates the third-generation EGFR covalent inhibitor landscape,18 which initially gained full FDA approval for patients with metastatic EGFR T790M mutant non-small-cell lung cancer (NSCLC) after showing improved efficacy to first- or second-generation EGFR TCIs. Osimertinib subsequently gained approval for first-line treatment of EGFR mutant lung cancer. It binds irreversibly to the cysteine 797 residue at the ATP binding site of EGFR (Fig. 1).16,19
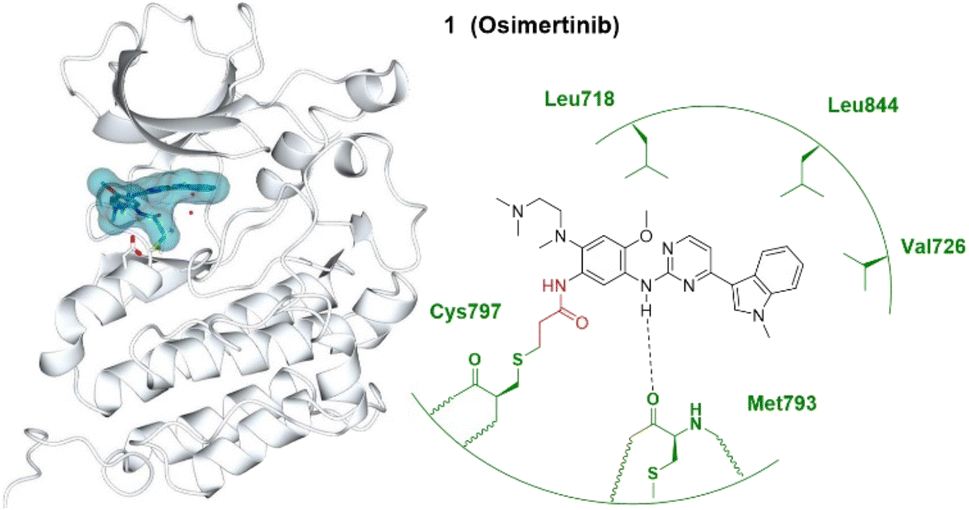 | ||
| Fig. 1 Covalent inhibitor osimertinib 1 and the co-crystal structure of 1 (dark cyan) bound with the Cys797 of the EGFRT790M (pale white) hinge pocket. The electrophilic warhead, acrylamide, is highlighted in red, forming an irreversible bond with the thiol of Cys797 by Michael addition. Image generated in CCP4MG at a resolution of 2.53 Å, PDB 6JX0.17 | ||
In November 2015, the FDA approved nazartinib for the treatment of patients with EGFR T790M-positive tumors. Clinical trials have shown more promising efficacy when compared to osimertinib and markedly wider therapeutic windows than either erlotinib or afatinib (also prescribed for the treatment of NSCLC).20 However, not all third-generation compounds showed superiority to first- and second-generation inhibitors. The clinical development of rociletinib and olmutinib was halted due to their reported off-target toxicity and unfavorable side effects.16 Naquotinib and mavelertinib are in further development to evaluate the efficacy and tolerability of the drug alone or in combination with other inhibitors.
Dacomitinib (Vizimpro®) is the most recently approved covalent inhibitor targeting EGFR.21 The discovery program that led to the identification of dacomitinib began as canertinib (CI-1033). Unfavorable side effects were identified and it became apparent that additional, more potent and more selective pan-ErbB tyrosine kinase inhibitors (TKIs) could be of clinical benefit to patients with EGFR dysregulated cancers.22
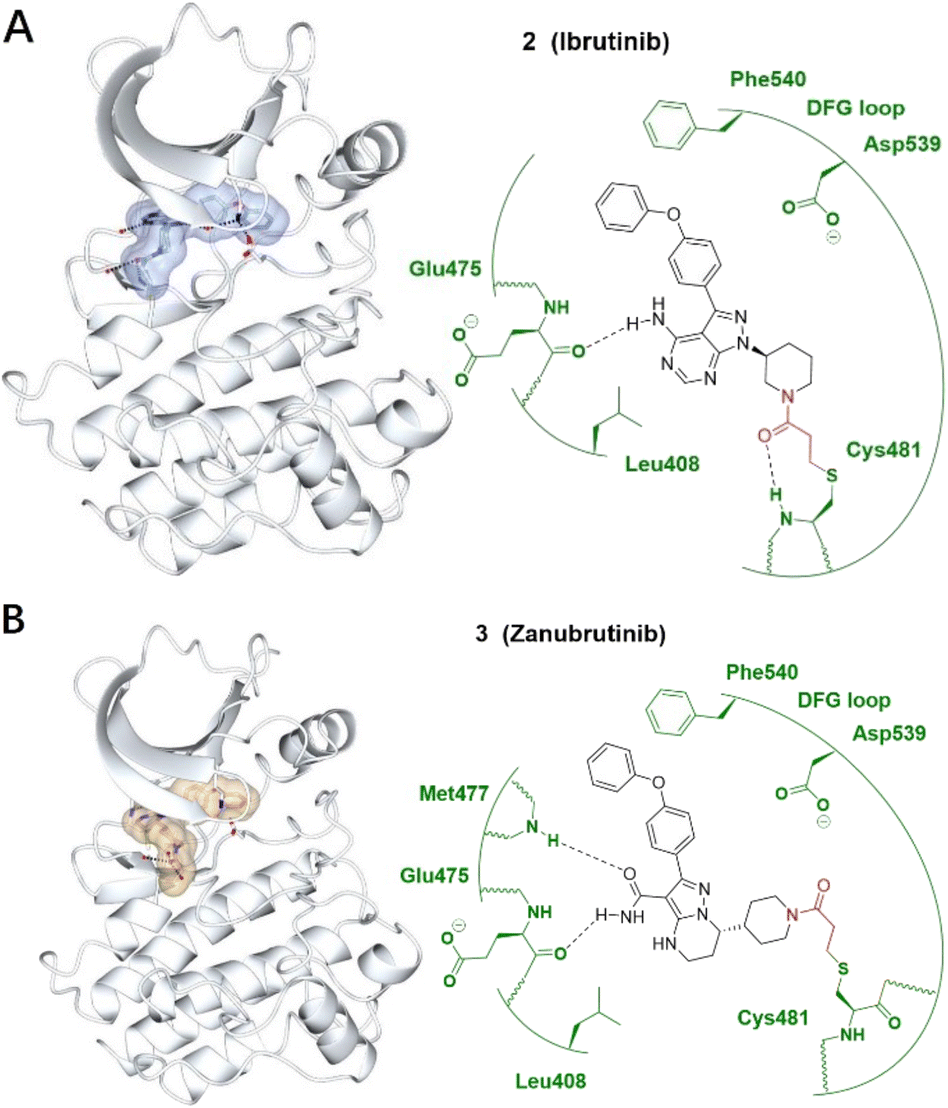 | ||
| Fig. 2 (A) Covalent inhibitor ibrutinib 2 and the co-crystal structure of 2 (ice blue) bound with the Cys481 of the BTK (pale white) hinge pocket. (B) Covalent inhibitor zanubrutinib 3 and the co-crystal structure of 3 (burlywood) bound with the Cys481 of the BTK (pale white) hinge pocket. The electrophilic warhead, acrylamide, is highlighted in red, forming an irreversible bond with the thiol of Cys481 by Michael addition. Image generated in CCP4MG at a resolution of 1.08 Å and 1.25 Å, PDB 5P9J and 6J6M, respectively.23,24 | ||
Although ibrutinib is an effective treatment of B-cell lymphomas and leukaemia, side effects such as infections, anaemia and diarrhoea can be distressing and challenging for patients. Acalabrutinib and zanubrutinib are two second-generation BTK inhibitors which offer better potency and bioavailability as well as reducing off-target toxicity binding to EGFR and other Tec family proteins (Fig. 2B).
Resistance to ibrutinib treatment has been attributed to the selection of cells carrying a pathogenic mutant (C481S), altering BTK or its downstream effector PLCG2. Other BTK variants such as C481F, C481G, C481R and C481Y are reported in some CLL patients but occur at a much lower frequency than C481S.25 These mutations disrupt the binding of covalent BTK inhibitors but hastened the rationale for the design of non-covalent BTK inhibitors.26–28
CDKs have been widely investigated as drug targets for many years.31 THZ1 is a covalent CDK7 inhibitor, targeting a remote cysteine residue (Cys312).32 Research showed that potent CDK12/13 off-target activity of THZ1 obscured the contribution of CDK7 to this phenotype.32 THZ531, a derivative of THZ1 which retains the phenylaminopyrimidine core scaffold, was later developed and used to selectively target CDK12/13 and exhibits approximately 20 times more potent inhibition than CDK7.33
In 2019, Gray et al. published their successful discovery of YKL-5-124, a highly selective CDK7 inhibitor containing an aminopyrazole core.34 Covalent targeting of CDK7 at Cys312 is essential for YKL-5-124 activity as shown in molecular docking studies. This new scaffold was developed from a previously unexplored CDK-targeting scaffold, itself originating from a PAK4 inhibitor (PF-3758309) in work by the Murray group.34
The widespread nature of covalent drug discovery has also resulted in novel antagonists such as SY-1365, a highly potent and selective CDK7 inhibitor which has been in clinical investigations for treatment of ovarian and breast cancers.35 In 2015, the first covalent, irreversible and ATP-competitive CDK2 inhibitor was identified at the Northern Institute for Cancer Research (NICR). NU6102 is a potent and selective ATP-competitive inhibitor of CDK2 in which the sulphonamide is positioned close to a pair of lysine residues. Using this structure, NU6300 was designed and this forms an irreversible interaction with Lys89 of CDK2 (Fig. 3).30 This purine scaffold allows for three hydrogen bonds to be made between the purine ring and the CDK2 protein and an additional hydrogen bonding interaction gained from the vinyl sulfone with Asp86.
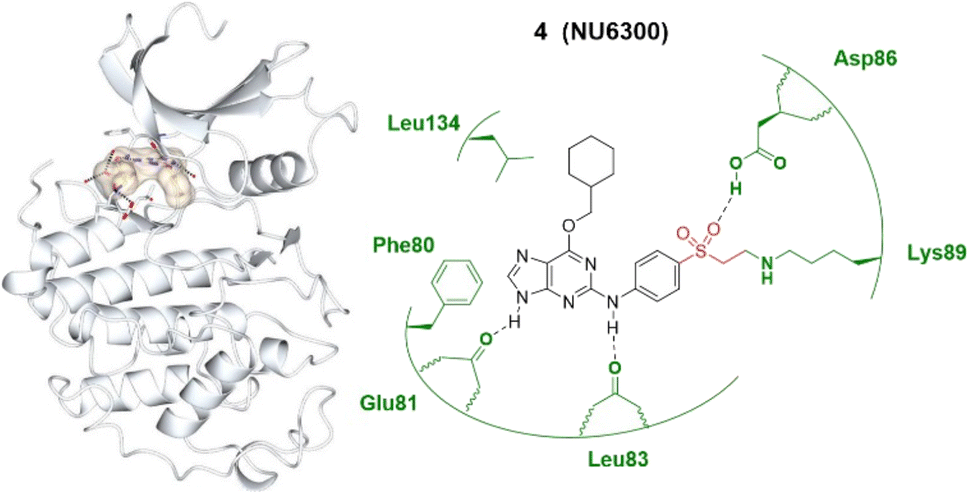 | ||
| Fig. 3 Covalent inhibitor NU6300 4 and the co-crystal structure of 4 (bisque) bound with the Lys89 of the CDK2 (pale white) hinge pocket. The vinylsulfonyl warhead is highlighted in red, forming an irreversible bond with the terminal amino group of Lys89 by Michael addition. Image generated in CCP4MG at a resolution of 2.00 Å, PDB 5CYI.30 | ||
In 2014, London et al. employed a series of covalent reversible cyanoacrylamide-based inhibitors to measure both inhibitory activity in vitro and isoform selectivity to JAK3.38 Forster et al. identified a JAK3-specific cysteine residue Cys909 and a ligand-induced binding pocket where a novel class of covalent reversible JAK3 inhibitors provided picomolar cellular activity and high isoform and kinome selectivity (>400-fold).37 Interestingly, the high-resolution crystal structure of JAK3 in complex with a 1,6-dihydroimidazo[4,5-d]pyrrolo[2,3-b]pyridine core showed the coexistence of the non-covalently and the covalently bound inhibitor 5.37 The presence of both binding modes, also confirmed by electrospray ionisation mass spectrometry (ESI-MS), underlines the highly reversible character of the covalent interactions and an equilibrium state between covalently and non-covalently bound 5 (Fig. 4). This discovery further validates the concept of covalent reversible enzyme inhibition with cyanoacrylamide-modified Michael acceptors.
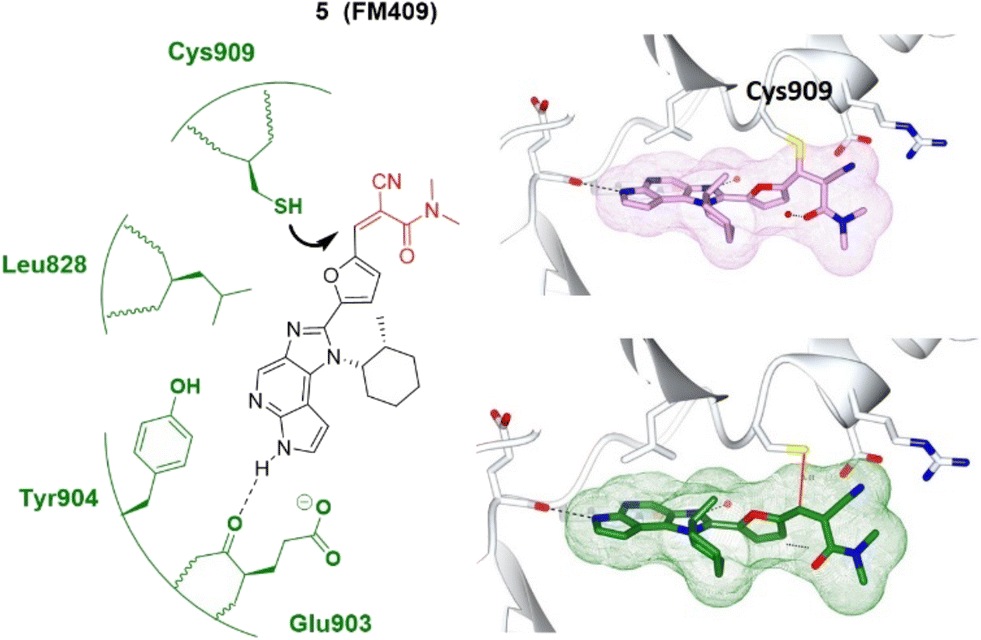 | ||
| Fig. 4 Covalent inhibitor FM409 5 and the co-crystal structure of 5 covalently (plum) and non-covalently (green) bound with the Cys909 of the JAK3 (pale white) hinge pocket, respectively. The electrophilic warhead, cyanoacrylamide, is highlighted in red and forms reversible covalent interactions with the thiol of Cys909 (black arrow). Image generated in CCP4MG at a resolution of 1.60 Å, PDB 5LWN.37 | ||
JAKs also mediate several inflammatory signalling pathways stimulated by interferons, such as IL-2, IL-3 and IL-5, in diseases such as rheumatoid arthritis and asthma. Numerous JAK1 inhibitors have been approved by the FDA, including filgotinib, upadacitinib, solcitinib and tofacitinib.39 JAK1 inhibition appears to be associated with fewer haematological effects compared to other JAK inhibitors.40
JAK3 has been shown to be a selective anti-inflammatory target due to its unique ligand-binding pocket and poorly conserved isoform-specific cysteine (Cys909).41 However, only one potent candidate, PF-06651600, displayed covalent interaction with the targeted cysteine thiol (Cys909) in the catalytic domain of JAK3.42 Keserű et al. employed JAK3 to test a small library of electrophiles including pyrazolopyrimidines that provided reactivity and accessibility information on targeted cysteines, which might be useful for identifying tractable targets for covalent inhibition.43
The KRAS G12C mutation occurs in approximately 13% of NSCLCs and in 1–3% of colorectal cancers and other solid cancers.44 The glycine-to-cysteine mutation at position 12 favours the active form of the KRAS protein, resulting in a predominantly GTP-bound KRAS oncoprotein and enhanced proliferation and survival in tumour cells.45 The mutated cysteine resides next to a pocket (P2) of the switch II region. The P2 pocket is present only in the inactive GDP-bound conformation of KRAS and has been exploited to design covalent inhibitors of KRASG12C.11,46
In 2013, Shokat et al. provided structure-based validation of a new allosteric regulatory site that can be targeted in a KRASG12C mutant-specific manner.11 A series of novel covalent inhibitors which derived from two hits, 6H05 and 2E07, targeted this allosteric pocket, disrupting both switch-I and switch-II regions and the native nucleotide preference to favour GDP over GTP and impairing KRASG12C binding to Raf (Fig. 5A).11 These compounds rely on the mutant cysteine for binding and therefore do not affect the wild-type KRAS protein. Further efforts have shown that ARS-1620/ARS-3248, AMG510 and adagrasib (MRTX849), developed by Wellspring, Amgen, and Mirati Therapeutics, respectively, potently inhibit KRASG12C activity in vitro and in vivo.47–50 As a result, ARS-3248, AMG510, and adagrasib entered phase I clinical trials and have shown promising results.51 Amgen disclosed that AMG510 (sotorasib) specifically and irreversibly inhibits KRASG12C through a unique interaction with its P2 pocket (Fig. 5B).52 This allosteric covalent drug was subsequently approved by the FDA in May 2021 for the treatment of patients with KRAS G12C-mutated locally advanced or metastatic non-small-cell lung cancer (NSCLC) following at least one prior systemic therapy.
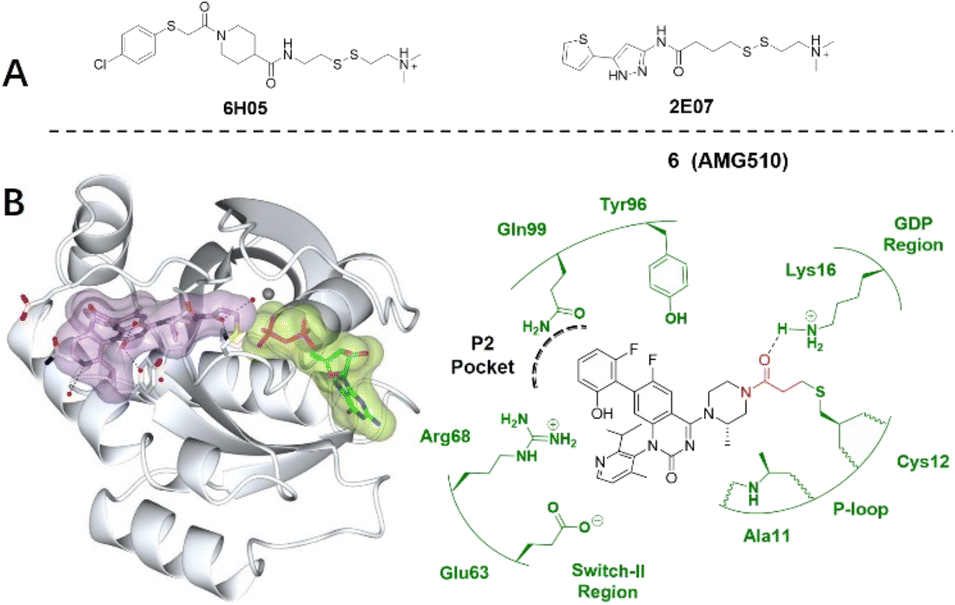 | ||
| Fig. 5 (A) Structures of screen hits, 6H05 and 2E07. (B) Covalent inhibitor AMG510 6 and the co-crystal structure of inhibitor 6 (lilac) and GDP (light green) bound with the Cys12 of KRASG12C (pale white). The allosteric P2 pocket is defined by dash lines. The electrophilic warhead, acrylamide, is highlighted in red, forming an irreversible bond with the thiol of Cys12 by Michael addition. Image generated in CCP4MG at a resolution of 1.65 Å, PDB 6OIM.53 | ||
1.2 Anti-viral
Covalent drugs also exhibit crucial improvements in the treatment of AIDS resistance. Chan et al. report several covalent inhibitors that can completely knock out the activity of the resistant mutant HIV-1 reverse transcriptase (RTY181C) and of the particularly challenging Lys103Asn/Tyr181Cys variant. The co-crystallography shows that the residue of Cys181 forms irreversible bonds with thiol-tolerated electrophilic warheads such as chloromethylamide and acrylamide at high resolutions. Another necessary interaction for potency is a hydrogen bond with O–N between a uracilyl oxygen atom of inhibitors and the backbone nitrogen of Lys103 in both structures (Fig. 6).54 | ||
| Fig. 6 Co-crystal structures of covalent inhibitors JLJ684 7 (brown) and JLJ686 8 (khaki) bound with the Cys181 of HIV-1 RTY181C (pale white), respectively. The electrophilic warheads, acrylamide and chloromethylamide, are highlighted in red, forming irreversible covalent bonds with the thiol of Cys181 by Michael addition and nucleophilic substitution, respectively (black arrow). Image generated in CCP4MG at a resolution of 2.58 Å and 2.40 Å, PDB 5VQV and 5VQX, respectively.54 | ||
Recently, Liu et al. performed a virtual screen of a clinical and investigational drug library, on the basis of the “steric-clashes alleviating receptor” (SCAR) strategy. Nine drugs that might be repurposed as covalent inhibitors of the priming proteases (cathepsin B, cathepsin L, and TMPRSS2) of the spike protein of SARS-CoV-2 were identified.55 Taking their computational studies together, the nucleophilic residues including CatB Cys29 and CatL Cys25 as well as TMPRSS1 Ser353 and TMPRSS2 Ser441 would be potential druggable sites to covalently bind to electrophilic ligands, which might facilitate development of anti-SARS-CoV-2 drugs.
Although Wang and Chen et al. described the rational design of di- and trihaloacetamides as covalent SARS-CoV-2 main protease (Mpro) inhibitors, these new compounds did not significantly inhibit the host cysteine proteases.56 The target specificity promised by co-crystal structures of SARS-CoV-2 Mpro with Jun9-62-2R and Jun9-57-3R showed that both compounds form a covalent adduct with the catalytic Cys145 (Fig. 7).56
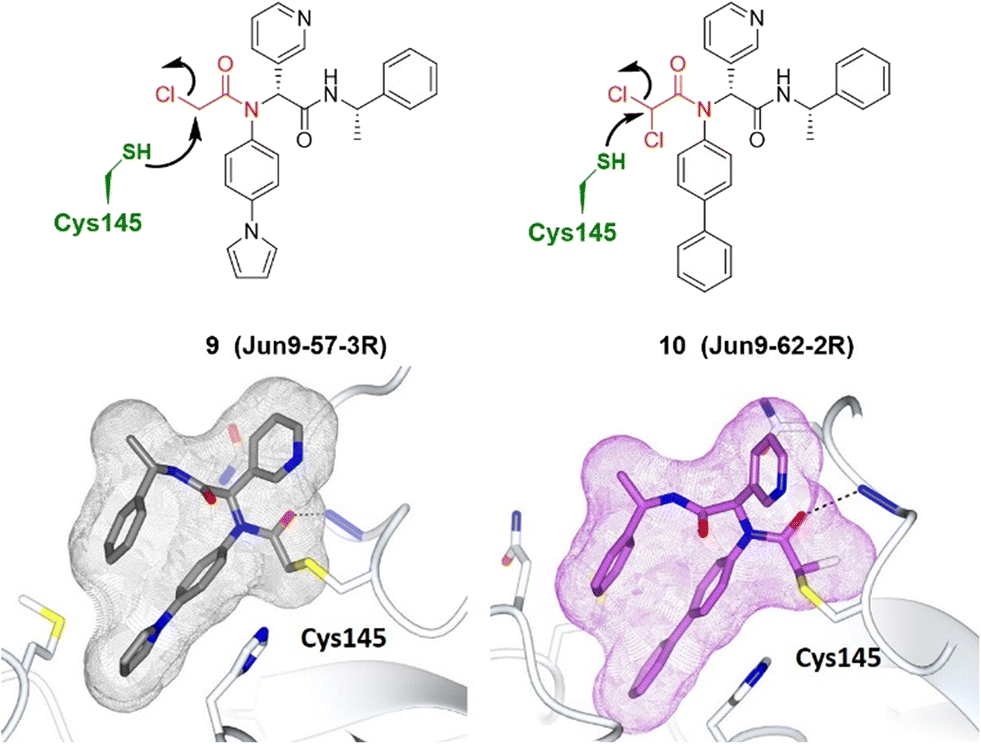 | ||
| Fig. 7 Co-crystal structures of covalent inhibitors Jun9-57-3R 9 (gray) and Jun9-62-2R 10 (purple) bound with the Cys145 of SARS-CoV-2 Mpro (pale white), respectively. The electrophilic warhead, chloroacetamide, is highlighted in red, forming an irreversible bond with the thiol of Cys145 by nucleophilic substitution. Image generated in CCP4MG at a resolution of 2.25 Å and 2.30 Å, PDB 7RN0 and 7RN1, respectively.56 | ||
Su and Kuai et al. reported their discovery of two acrylamide-containing molecules that irreversibly bind to SARS-CoV-2 Mprovia the commercially available covalent DNA-encoded library (DEL) screening platform.57 Time-dependent enzymatic assay showed that the IC50 values after 1 h and 16 h of incubation time were 15.9 μM and 1.9 μM, respectively. The inhibitory effect against SARS-CoV-2 infection was investigated by cellular assay, resulting in an anti-viral EC50 value of 33 μM.
Nirmatrelvir analogues with different warheads and their inhibitory activities were also investigated, suggesting that hydroxymethylketone and ketobenzothiazole warheads can also be employed for treating coronavirus infections equipotent to the nitrile.58
To clearly characterise differences between structures and biochemical activities, herein we exhibit the structures and cellular/enzymatic IC50 values of all drugs mentioned above (Table 1).
| Name | Structure | Protein/targeted residue | Potency (IC50) |
|---|---|---|---|
| a Inhibition of EGFR phosphorylation. Double mutant (DM) cell line: H1975. b Bound to CDK12. c Inhibition of cell proliferation in Jurkat cells. d Competitive pull-down assays in HAP1 cells. e 50% inhibition to pan-CDK2. f Inhibition to CDK7/CycH/MAT1 in leukemia cells. g Measured by the inhibition of IFNγ production in Th1 cells. h Measured by Ba/F3 cells expressing KRASG12C. i Incubation time of 24 h for WT HIV-1 reverse transcriptase bearing the Y181C mutation, which contains Tyr181. | |||
| Osimertinib |
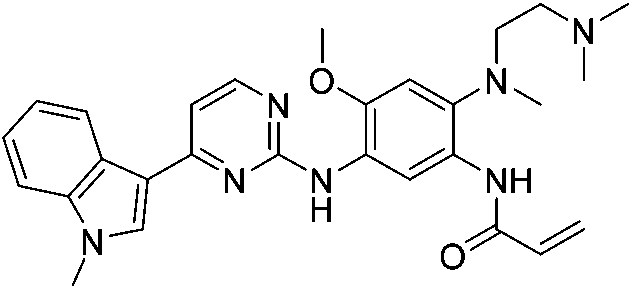
|
EGFRL858R/T790M Cys797 | 15 nMa |
| Nazartinib |
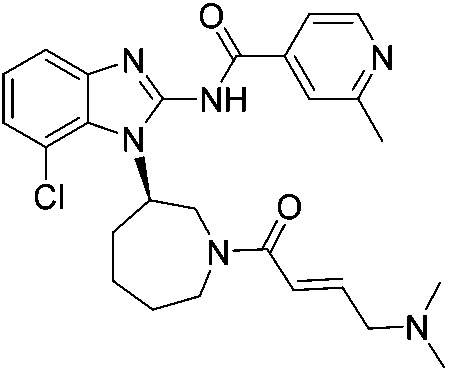
|
EGFRL858R/T790M Cys797 | 31 nMa |
| Dacomitinib |
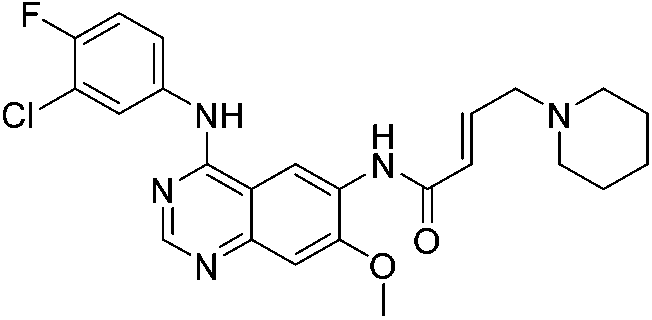
|
EGFRL858R/T790M Cys797 | 0.44 μMa |
| Ibrutinib |
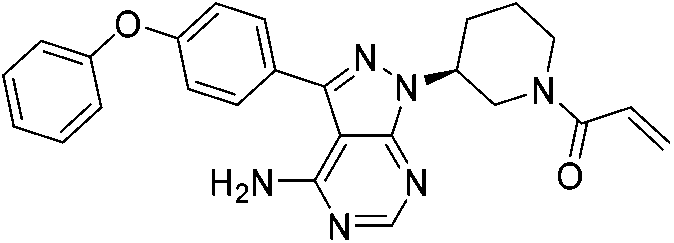
|
BTK Cys481 | 0.5 nM |
| Acalabrutinib |
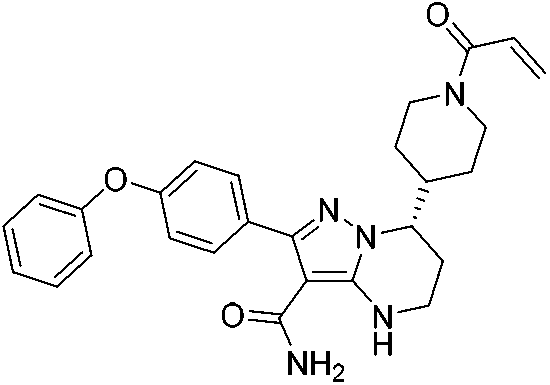
|
BTK Cys481 | 3 nM |
| Zanubrutinib |
|
BTK Cys481 | 0.5 nM |
| THZ1 |
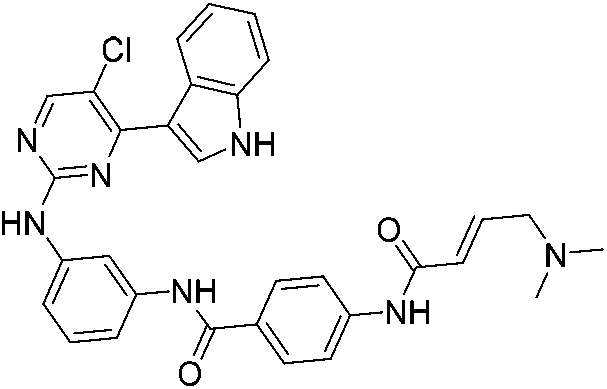
|
CDK7 Cys312 | 0.24 μMd |
| THZ531 |
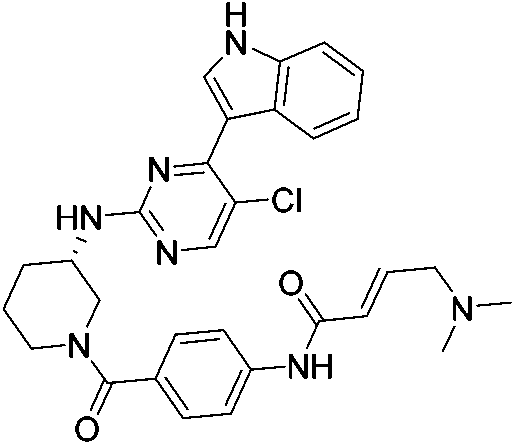
|
CDK12/13 Cys1039 | 50 nMb,c |
| YKL-5-124 |
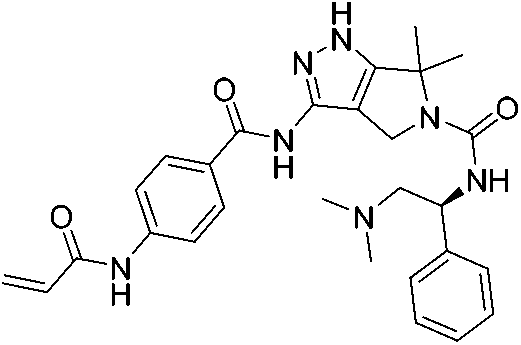
|
CDK7 Cys312 | 53.5 nMd |
| NU6300 |
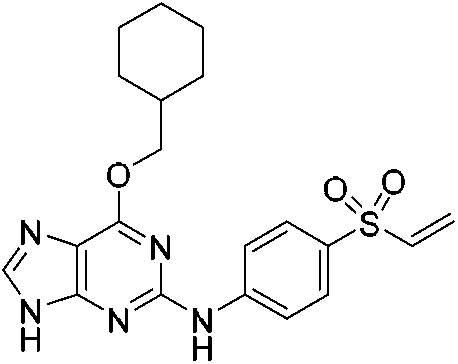
|
CDK2 Lys89 | 0.16 μMe |
| SY-1365 |
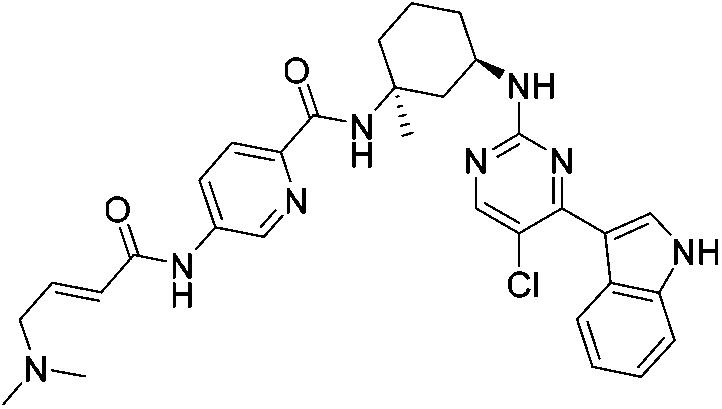
|
CDK7 Cys312 | 20 nMf |
| FM409 |
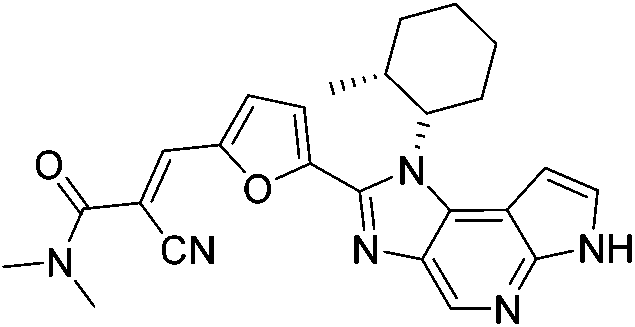
|
JAK3 Cys909 | 17 nM |
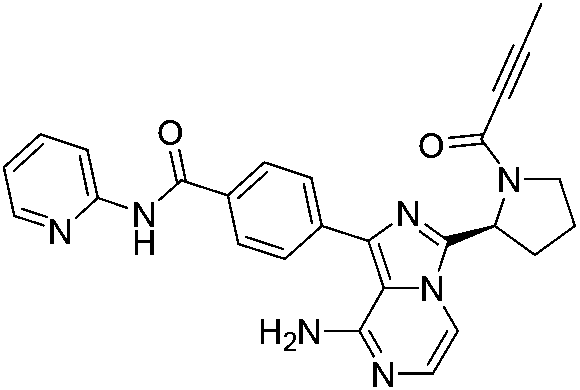
| PF-06651600 |
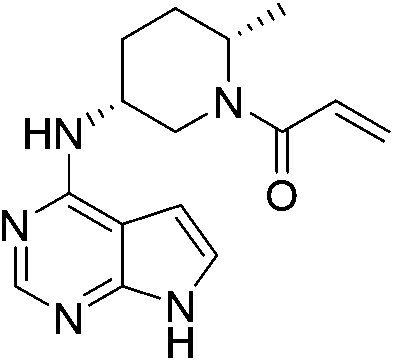
|
JAK3 Cys909 | 48 nMg |
| Sotorasib |
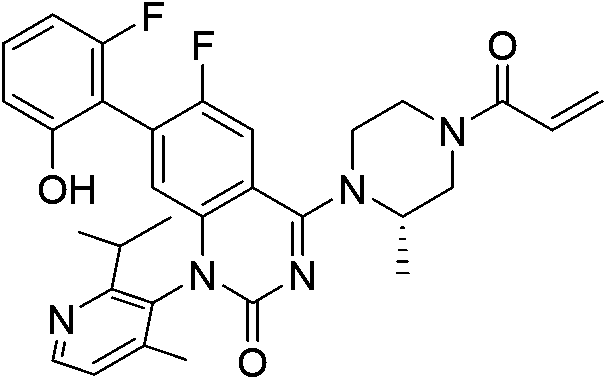
|
KRASG12C Cys12 | 12.4 nMh |
| Adagrasib |
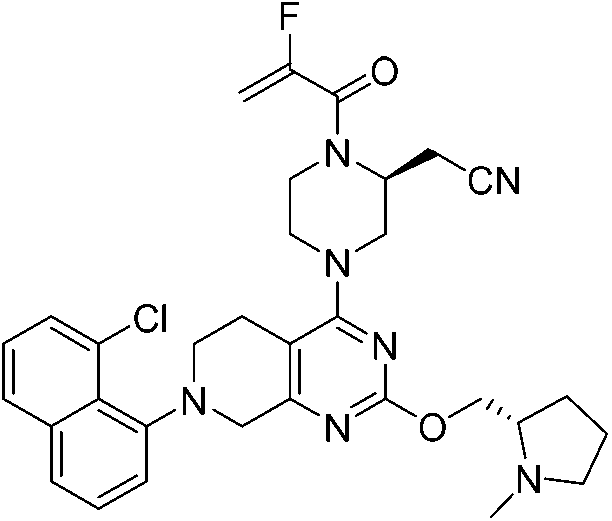
|
KRASG12C Cys12 | 1.3 nMf |
| JLJ684 |
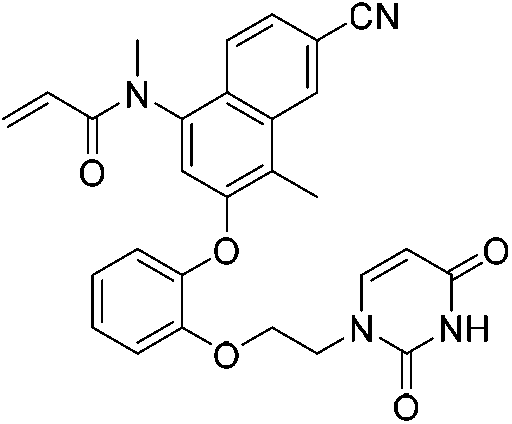
|
HIV-1 RTY181C Cys181 | 0.40 μMi |
| JLJ686 |
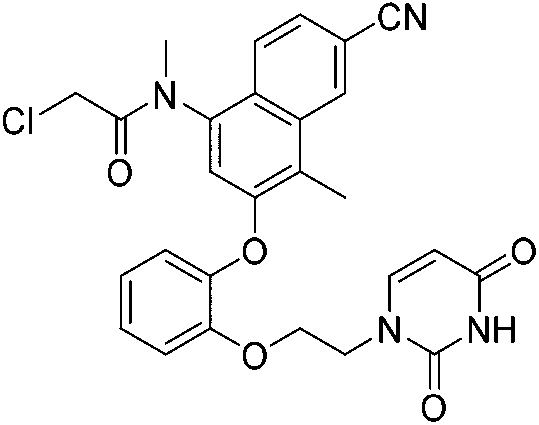
|
HIV-1 RTY181C Cys181 | 0.48 μMi |
| Jun9-57-3R |

|
SARS-CoV-2 Mpro Cys145 | 0.05 μM |
| Jun9-62-2R |
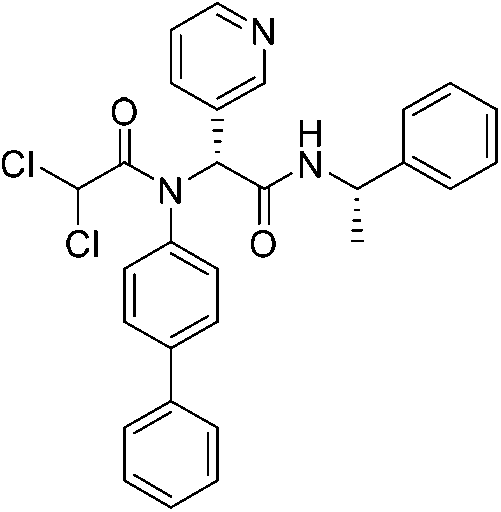
|
SARS-CoV-2 Mpro Cys145 | 0.43 μM |
2. The role of irreversible and reversible covalent inhibition
2.1 Irreversible covalent inhibitors
Designing an irreversible covalent inhibitor fundamentally requires an acceptable covalent bond sharing an electron pair between atoms from the electrophile of a ligand and the nucleophile of a target. Acrylamide is known as the most classic warhead for the irreversible binding mode to cysteine thiol, which has been widely applied in many cases.Besides acrylamide, moieties such as vinyl sulfonamide and propynamide are also popular electrophilic warheads that target cysteine proteases by Michael addition. Martin et al. reported that vinyl sulfonamide reacts more rapidly with the thiol group than the acrylamide.69 Several approved TCIs, including afatinib and neratinib, utilise substituted acrylamides as warheads. Research has demonstrated that they are relatively poor electrophiles to the cysteine residue of targets, which only display good binding affinities if sufficiently close to their target proteins for reaction.69 Interestingly, it was also reported that neratinib could form a reversible covalent adduct with K190 of human serum albumin. This might provide a potential approach to investigate the off-target reactivity and minimise the safety issue of irreversible inhibitors via proteomics-based screening.
Although there are not many commercial drugs containing chloroacetamides, likely due to their long-standing reactivity against targets, they work well as chemical probe screening for the validation of available thiol groups on targets in the early stage of covalent drug discovery. Recently, London et al. employed an electrophile-fragment library (containing chloroacetamides as majority compounds), which successfully disclosed two potentially druggable enzymes, the deubiquitinase OTUB2 and the pyrophosphatase NUDT7.70 Combining the approach with high-throughput crystallography, they observed co-crystal structures of both proteins in complex with covalent probes at high resolutions. A potent NUDT7 inhibitor was subsequently discovered by fragment merging.
Irreversible covalent inhibitors improve the binding affinity, forming adducts with targeted enzymes and causing a prolonged therapeutic effect above clearance. Whilst this is advantageous, it also brings forth undesirable disadvantages, such as over-reactivity or off-target toxicity. Once the molecule tightly binds with plasma proteins, the side effect can be critical. Zhong et al. studied the t1/2 values of several irreversible covalent inhibitors, including osimertinib and ibrutinib, bound to human serum albumin in vitro. It was highlighted that the electrophilicity of the acrylamide β-carbon atom is an important factor in the rate of covalent binding between covalent drugs and plasma proteins.71
2.2 Reversible covalent inhibitors
In contrast, reversible covalent inhibitors mitigate the risk of reactivity and derive from the basis of off-target toxicity of irreversible covalent inhibitors. Some electrophilic groups that are represented by benzonitrile are employed to reversibly bind target cysteine residues and slowly form covalent adducts via Markovnikov addition.72 Since non-covalent inhibitors would result in ‘fake’ complete inhibition as well as proceed in high enough dosing concentrations, Singh et al. insisted that explicit consideration of the time dependence of inhibition is crucial to recognize reversible covalent interactions from reversible inhibitors and assess their absolute or relative activities. Recently, the kinetics of inhibition for reversible inhibitors, particularly the target residence time, has initiated the study of potential novel covalent binding modes for reversible inhibitors. However, reversible covalent inhibitors also face the challenge of resistance, so there is a significant need to find novel electrophilic moieties and more accurate reversible binding modes. Bradshaw et al. presented “residence time by design” that employed some cysteine-reactive cyanoacrylamide electrophiles to modulate and improve the duration of target engagement in vivo.733. Approaches of designing covalent inhibitors
3.1 Phenotypic screening
Phenotypic screening in biological research is widely used to identify small molecules, peptides or RNAi that alter the phenotype of a cell or organism in a desired manner. As a traditional method in drug discovery, phenotypic screening developed rapidly in the 1950s and 1960s due to the in-depth study of enzymes and enzyme kinetics.74 With the development of molecular biology, research around small-molecule drugs moved into the target-based strategy stage. In 2011, Swinney and Anthony published an analysis highlighting 75 new, clinically approved first-in-class drug entities with novel active binding modes between 1999 and 2008. Among these candidates, 50 were small molecules and 25 were biologics. Delving into the discovery of these 50 small molecular drugs, 28 (56%) were obtained through phenotypic screening.75 Recently, this established approach has experienced an upsurge. It has been developed to build a chemogenomic library which would assist in the target identification and mechanism of action induced by drugs.76The biggest advantage of phenotypic screening is to produce the bioactive compound directly on an advanced disease-relevant parameter. Secondly, due to the direct reflex on animal models or complete cells, phenotypic screening barely needs to have a thorough understanding of the action mechanism, which is mainly used towards non-covalent drug development. On the other hand, limitations must be highlighted. The efficacy of drug combinations, by their nature, can be hard to predict nor can screening evaluate new mutations that have not yet been described. In covalent drug discovery, the amount of diversity is typically limited within current covalent libraries; it may also be challenged by the selectivity to a target without the conformational structure available.9
3.2 Screening of covalent leads
Lead compounds refer to chemical compounds which have biological activity against a drug target and can then be used as a starting point to obtain further compounds with improved activity, selectivity or pharmacokinetic parameters. Covalent leads can be identified through extensive screening, rational drug design, existing drugs, high-throughput screening (HTS) and other methods. Screening for leads usually produces compounds with weak binding affinity and inhibitory activity, which then require structure–activity relationship (SAR) studies to identify more active compounds after lead optimisation.High-throughput virtual screening is currently overtaking the more expensive traditional screening to give drug-like compounds in a shorter time period and introducing better tolerated electrophilic groups after calculation. An appropriate algorithm to confirm the irreversible binding mode between compounds and proteins is necessary. However, building a highly efficient and trained model from a large number of diverse compounds is still in development.9
3.3 Covalent optimisation based on reversible lead scaffolds
In covalent drug discovery, the strategy aims to place an electrophilic moiety on the inhibitor, allowing it to undergo attack from a nucleophilic amino acid residue upon binding to the target protein. An irreversible bond is formed that is much stronger than typical reversible interactions. It usually starts from a highly active drug molecule, or lead compound, combined with the rational binding mode at a high resolution, which can be regarded as the key point determining successful irreversible optimisation. Pharmacophores on the lead scaffold take charge of introducing necessary non-covalent interactions and maintaining potent binding affinity to the target protein. On that basis, the tolerated electrophilic moiety could potentially result in irreversible interaction with the target nucleophilic residue. SAR studies around this moiety are also necessary to gain a better understanding of the environment and valid interactions around the binding site.However, the premise of forming an expected irreversible bond requires a suitable distance between the electrophilic moiety and the targeted nucleophilic residue. Therefore, effective co-crystallographic mapping significantly enhances structural information, in turn saving on resource, allowing the gain of better results faster.9
3.4 Covalent fragment screening
The physicochemical properties of molecules to be developed into drugs are important. Generally, Lipinski's rule of 5 is conceived to aid the development of orally bioavailable drugs, but it was not designed to guide the medicinal chemistry development of all small-molecule drugs.77,78Given the advantages of fragment-based drug discovery, covalent fragments refer to small molecules which comply with an adapted form of Lipinski's rules containing the electrophilic warhead. As a result of their small size, the fragments inherently have more scope for further optimisation, allowing potency improvement to be achieved via fragment growing, linking and merging. Screening aims to use these covalent fragments as probes to search for potential irreversible binding sites on the target protein and to develop covalent leads. Numerous popular targets, e.g. KRASG12C, have been done by warhead screening of covalent fragments in recent publications.79–81 This strategy has also been applied to validate novel targets and to generate viable chemical starting points even for targets that are sometimes described as undruggable.82,83 X-ray crystallography is necessary for this strategy, providing the structural information on fragment binding sites and guiding the subsequent hit generation or hit-to-lead studies.
Inspired by fragment-based ligand discovery, Backus and Cravatt et al. reported a quantitative analysis of cysteine-reactive small-molecule fragments screened against thousands of proteins in human proteomes and cells.84 Over 700 cysteines in both druggable proteins and proteins deficient in chemical probes, including BTK and isocitrate dehydrogenases (IDHs), were targeted by fragment electrophiles via isotopic tandem orthogonal proteolysis-activity-based protein profiling (isoTOP-ABPP). Ligandable lysines could also be mapped by this method.85,86
Interestingly, Ding and Li et al. discovered the application of ynamide, a versatile synthon, which can efficiently modify carboxyl residues in situ and in vitro to form covalent inhibition for the first time.87 Since this, we suppose that incorporation of electrophiles with novel structures into fragment-screening probes would be helpful for disclosing innovative warheads used in the development of new types of covalent inhibitors by chemical proteomic approaches.
Herein, we exhibit a recognised flow chart for covalent drug discovery that contains the approaches of covalent drug development from target validation (Fig. 8). It would be helpful for groups who may be interested in starting exploration and the following screenings, etc.
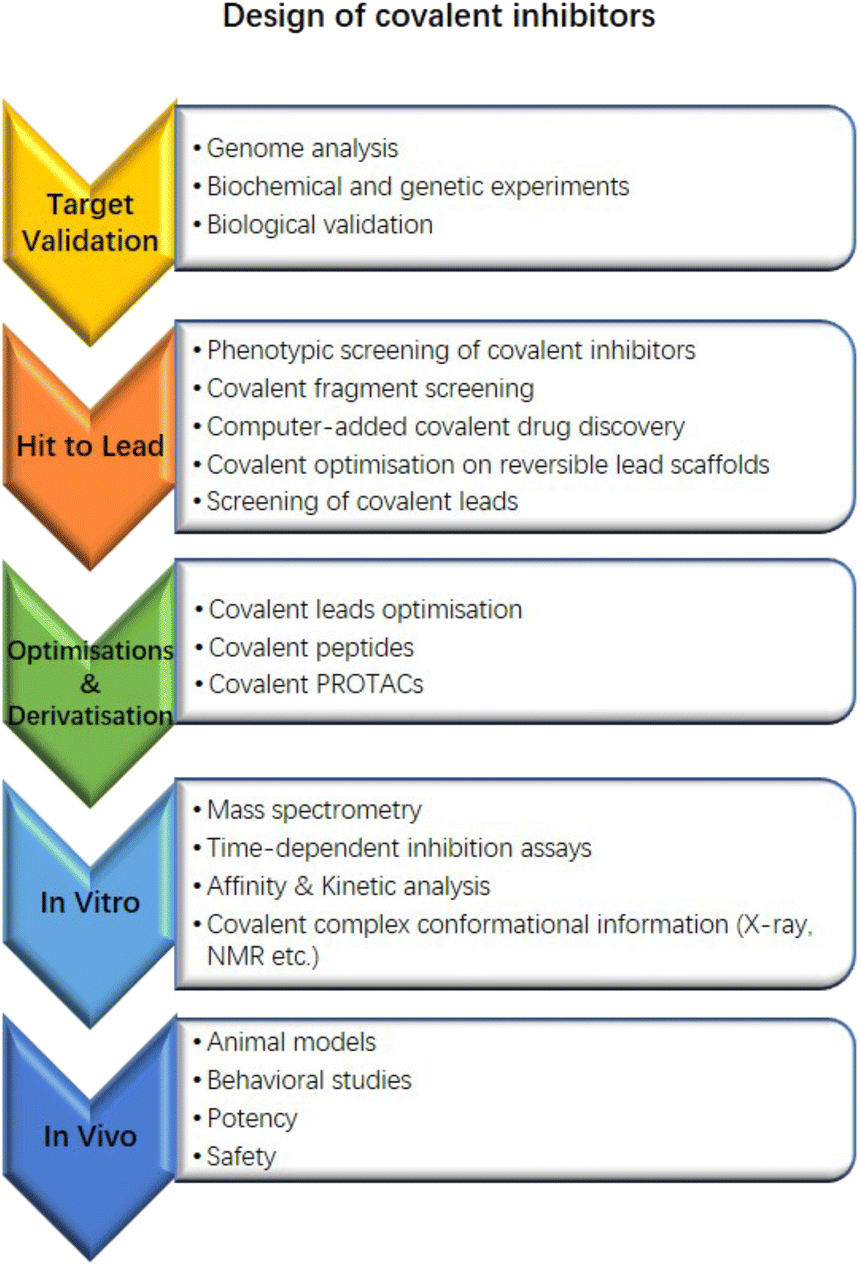 | ||
| Fig. 8 The flow chart of developing covalent drugs and corresponding approaches. | ||
4. Covalent strategy in multidisciplinary applications
4.1 Computer-aided covalent drug discovery
With the rapid development of computer modelling techniques, such as improved algorithms, deep learning and cloud computing resources, which provide more powerful computing capacity, scientists have increasingly been employing artificial intelligence (AI) to manage biological data processing and automatic proteome structural analysis.88,89Computer-aided drug design (CADD) is a highly regarded modern approach, which utilises data from known biomolecular targets and interactions, to drive faster and more successful compound characterisation for the discovery of small-molecule candidates.90 Covalent molecular docking has been also recently implemented in CADD workflows to describe covalent interactions between inhibitors and biological targets.
Proven by many successful cases, CADD works as a useful tool to model covalent interactions between ligands and targets. For example, Blake et al. reported the application of a unique hybrid ligand/structure-based virtual screening using covalent docking to search for irreversible protein splicing inhibitors as potential anti-tuberculosis drugs.91 Dong et al. showed covalent docking for substrate discovery into 14 representative glutathione S-transferase (GST) enzymes with known structures and substrates.92,93
Covalent docking algorithms can provide protocols for the binding mode prediction of covalent ligands. Keserű et al. described a typical virtual screening of compound sets including various warheads, which could promote the design and characterisation of covalent binders.94 London et al. recruited a covalent docking protocol to screen large virtual libraries of electrophilic fragments to identify reversible covalent fragments that target non-conserved cysteine residues in several protein kinases.38 Zhou et al. assessed four popular covalent docking tools, including MOE, and investigated their covalent docking performance through parameter comparison.95
Successful computer-aided covalent drug discovery was performed by Shoichet and Taunton's groups which targeted a challenging enzyme, eukaryotic translation initiation factor 4E (eIF4E). Since there are no cysteines near the eIF4E cap binding site, they developed a covalent docking approach and focused on a noncatalytic lysine (Lys162). As a result, two analogues equipped with the arylsulfonyl fluoride achieved irreversible interactions with the protein surface from cocrystal structures, which are claimed as the first covalent eIF4E inhibitors with cellular activity.96
Although computational chemistry techniques can have an impact on covalent inhibitor design, some crucial aspects such as the accuracy, speed, ligand sampling and protein flexibility should be revisited with improved algorithms to overcome such shortfalls.97,98
4.2 PROTACs
Proteolysis targeting chimeras (PROTACs) were first reported by Craig Crews in 2001 and are heterobifunctional molecules consisting of two active domains and a linker.99 The ligand portion of a PROTAC binds to a target protein meant for degradation, while another portion stretches into the pocket of an E3 ubiquitin ligase that is recruited for ubiquitination of target proteins.Duan et al. divided PROTACs into three types depending on their binding mode to the E3 ligase: irreversible covalent, reversible non-covalent and reversible covalent binding.12 To date, most reported PROTACs bind to target proteins by means of a reversible non-covalent pattern, e.g. the first oral PROTACs (ARV-110 and ARV-471) showed encouraging results in clinical trials for prostate and breast cancer treatment.100,101 However, researchers disclosed that reversible non-covalent PROTACs have poor selectivity and permeability.102,103 For example, the clinical development of the recently reported BRD4 PROTACs have stagnated since safety risks were confirmed.104 Crews et al. reported the development of LC-2, the first PROTAC capable of degrading endogenous KRASG12C. It covalently binds KRASG12C with a MRTX849 warhead and recruits the E3 ligase VHL, inducing rapid and sustained KRASG12C degradation, leading to suppression of MAPK signalling in both homozygous and heterozygous KRASG12C cell lines.105 Alternatively, the irreversible bond can also be set on the E3 ubiquitin ligase which recruits multiple target proteins for ubiquitination and degradation without the need to re-form the E3–PROTAC complex, eliminating the kinetic process of this step. Xue et al. disclosed a range of successful BTK-PROTACs that utilised two different covalent ligands for the BTK binding moiety and recruited two different E3 ligases, pomalidomide and VH032.106 This research highlights the advantage of irreversible covalent chemistry used in targeted protein degradation. However, irreversible binding may reduce the potency of PROTACs due to their decreased catalytic nature according to investigations.107
Reversible covalent PROTACs take advantage of covalent binding, including selectivity and increased potency, while keeping the reversibility which is necessary for catalytic properties of chemical reactions of a PROTAC's efficacy.108
Several reversible covalent moieties were also published in recent years, targeting nucleophilic amino acids through the mechanism of, for example, imine formation and 1,2-addition.109–112 Cyanoacrylamide-containing moieties were recruited as highly potent, selective, reversible covalent ‘warheads’ for designing reversible covalent PROTACs. Gabizon et al. reported that cyanoacrylamide-containing PROTACs exhibited much better potency towards BTK protein than irreversible acrylamide analogues and equivalent non-covalent PROTACs.107,113
Collectively, covalent PROTACs present a highly promising approach for current and future drug discovery and promotion in biology with better degradation activity as well as longer duration of action compared to noncovalent PROTACs. Keys to designing reversible covalent PROTACs are to disclose a reversible covalent E3 recruiter or introduce a reversible covalent ligand binding to the target protein, such as cyanoacrylamide or dimethylated cyanoacrylamide.113–115 Interestingly, although both molecular glue degradation agents and PROTACs ultimately degrade target proteins through the proteasome pathway, rarely found is the combination of covalent chemistry with molecular glues, promising a potential alternative strategy in targeted covalent drug discovery.
4.3 Covalent peptide inhibitors
Some proteins cannot be targeted by small molecules and are thus deemed “undruggable.” Covalent peptide inhibitors merge the advantages of peptidomimetic drugs that can better bind to smooth and flat protein surfaces and covalent chemistry, resulting in better inhibition with lower dosage.Only one of the twenty natural amino acids, cysteine, can form a reversible covalent bond with the thiol residue of another cysteine via the disulfide bridge without enzymatic catalysis. However, its weak stability and redox sensitivity are not permitted in designing covalent peptide inhibitors. Alternatively, medicinal chemists may introduce electrophilic warheads such as sulfonyl fluoride into current amino acids to develop peptide inhibitors that can covalently bind with native amino acid residues, e.g. cysteine, serine and lysine. Herein, we briefly overview some successful cases describing covalent strategy in peptide drug discovery.
Stebbins et al. converted BI-107D1 into a covalent peptide by introducing an acrylamide warhead onto a residue in the peptide. The Lys-acrylamide cross-linked an endogenous cysteine residue next to the binding pocket of the peptide. Cellular studies showed that the covalent peptide was more efficient in inhibiting Siah protein than the non-covalent peptide.116 Similar to covalent small-molecule inhibitors, employing different electrophilic moieties can achieve reaction specificity of covalent peptides and change the preferential binding to target residues. A recent study around BIM peptide to Bcl2A1 disclosed that the acrylamide-modified peptide could bind selectively and irreversibly to Cys55 within the helix-binding groove of Bcl2A1, but not to the other two surface-exposed cysteine residues. When using more reactive chloroacetamide or propiolamide warheads, non-specific targeting was observed.117
A stapled peptide, mSF-SAH, contains an electrophilic aryl sulfonyl fluoride warhead and showed efficient binding and cross-linking of the peptide with the Lys or His residue of MDM2 and MDM4, leading to target inhibition.118–120 Inspired by this work, Spring et al. designed a ‘two- component’ stapled peptide covalently bound to MDM2, which showed an improved dissociation constant over time when compared with the non-covalent peptide.13,121 With the growing applications of sulfur fluoride exchange (SuFEx) click chemistry, follow-up studies proved that aryl sulfonyl fluoride and aryl fluorosulfate are increasingly appreciated as improved warheads for targeting other nucleophilic residues including Lys, Tyr, His, Thr, and Ser in a protein context.122–126
It is known that peptide hydrophilicity is responsible for its poor permeability through physiological barriers and biological membranes. Undesirable physicochemical properties of peptides, such as variable solubility, low bioavailability and limited stability, make their systemic delivery difficult. Most electrophilic warheads contain double/triple bonds and flat, smooth conformational features. Thus, covalent optimisation can introduce several unsaturated structures or heterocyclic conjugations into a scaffold, which modulates the lipid solubility and conformational flexibility of peptide leads. We suppose that the application of covalent mechanisms would promote the development of peptide drugs.
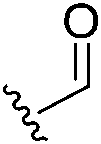
5. Overview of newly disclosed electrophilic warheads to cysteine
As alluded to above, fragment-sized molecules with identical scaffolds, but equipped with diverse electrophilic warheads, can assist in covalent drug discovery through either the initial step of covalent fragment screening or covalent optimisation to reversible lead scaffolds.Although other amino acids, e.g. serine and lysine, are also targeted among the FDA-approved drugs such as bortezomib and voxelotor, cysteine remains the most favourite residue to develop covalent inhibitors due to its high nucleophilicity of thiol under physiological conditions and the good conservative property at functionally important sites.127 Herein, we exhibit recently disclosed warheads to cysteine.
In 2020, Keserű introduced a ligand-based technique for mapping cysteine reactivity and accessibility by screening a set of covalent probes with diverse reactivity, which includes a wide range of known cysteine-sensitive fragment probes taken from recent studies around covalent molecules (Table 2).43
| No. | Structures | Mechanism |
|---|---|---|
| 1 |
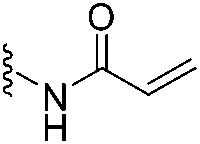
|

|
| 2 |
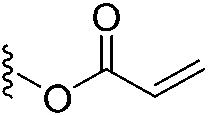
|

|
| 3 |
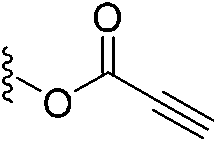
|
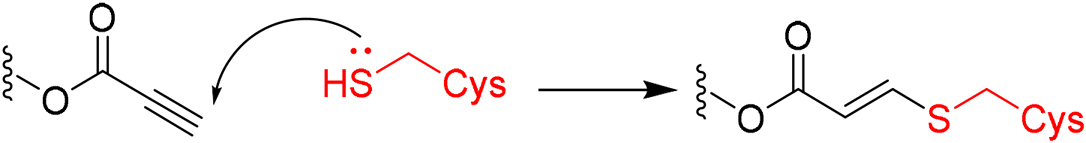
|
| 4 |
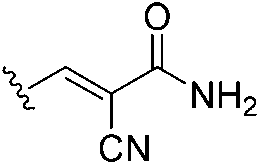
|
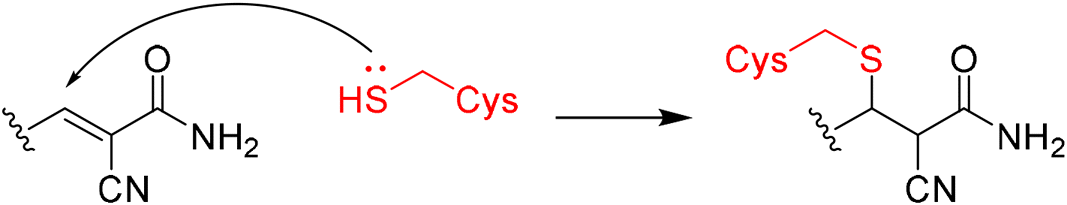
|
| 5 |
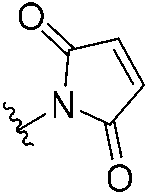
|
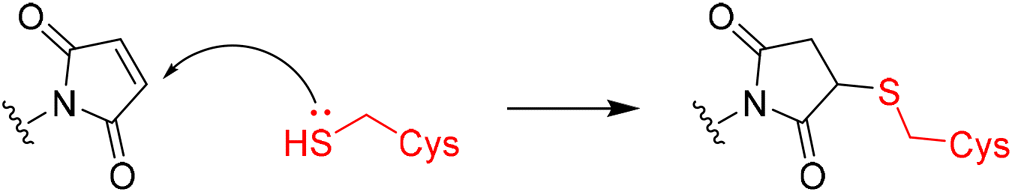
|
| 6 |

|
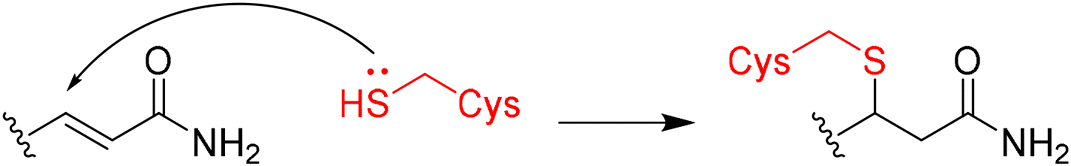
|
| 7 |
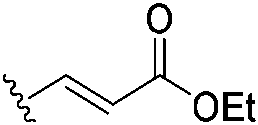
|

|
| 8 |
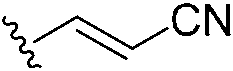
|
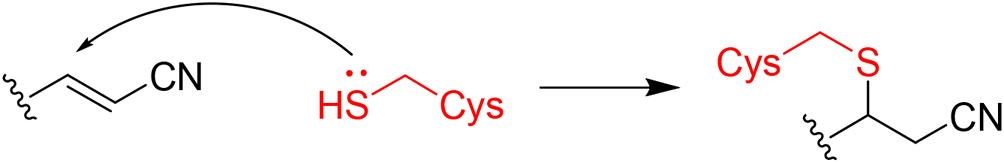
|
| 9 |
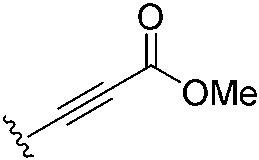
|
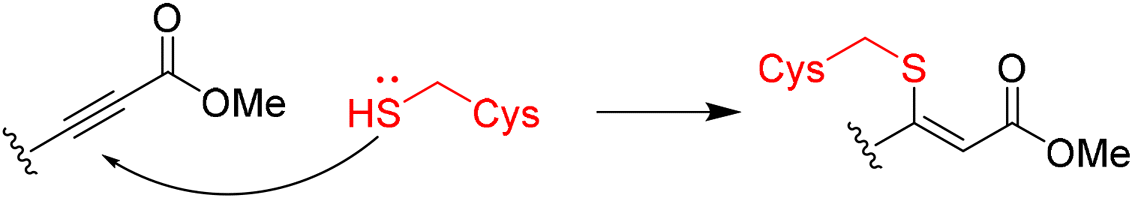
|
| 10 |
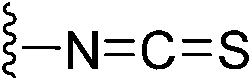
|
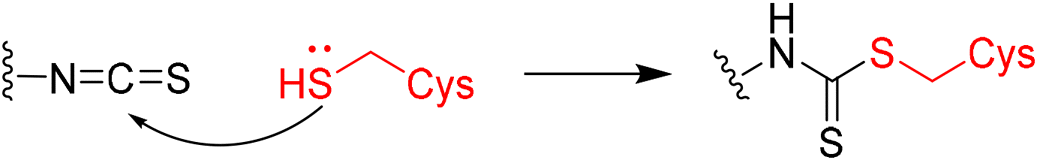
|
| 11 |

|
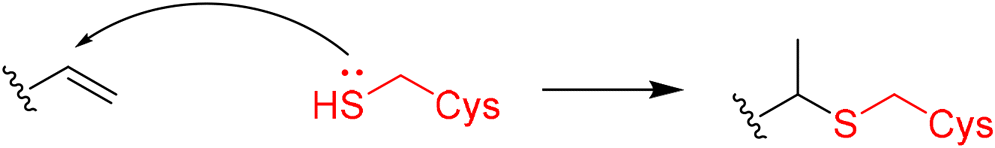
|
| 12 |

|
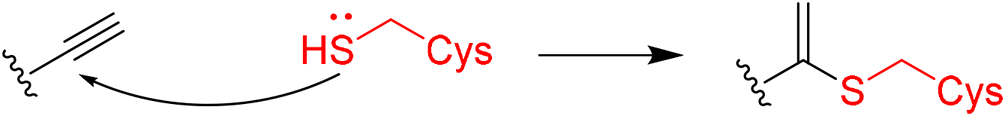
|
| 13 |
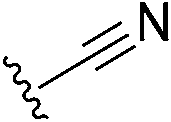
|

|
| 14 |
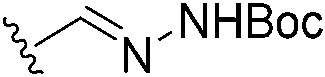
|
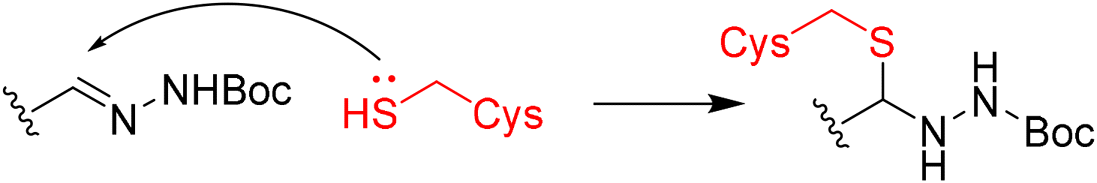
|
| 15 |
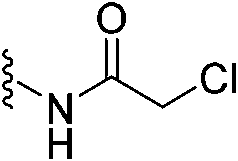
|
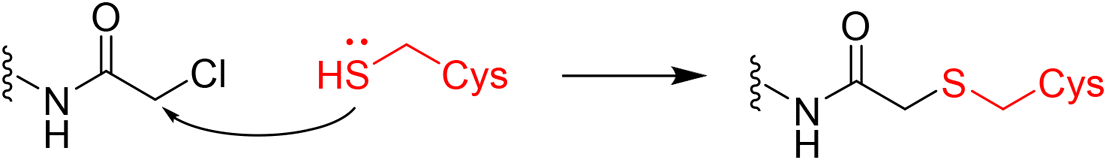
|
| 16 |
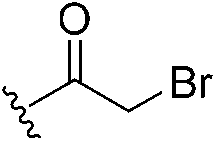
|
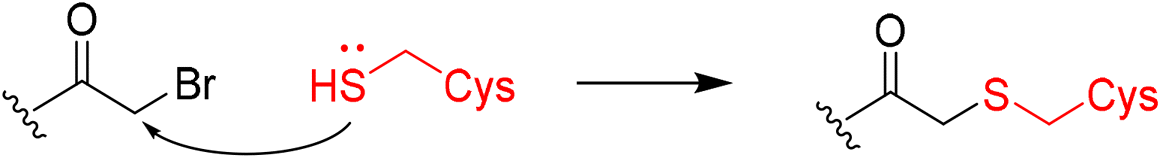
|
| 17 |
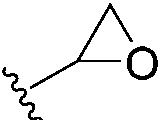
|

|
| 18 |
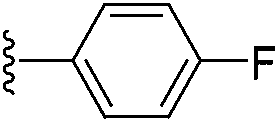
|
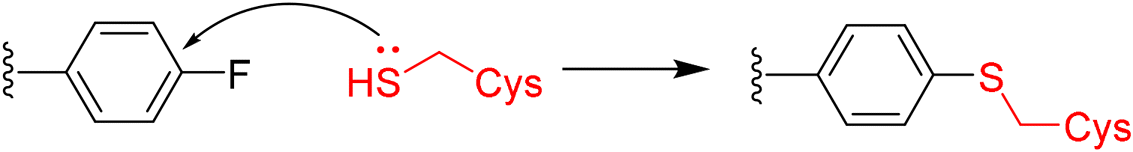
|
| 19 |
|
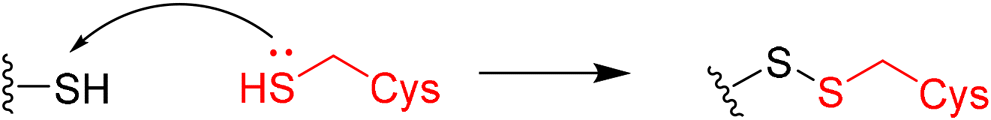
|
| 20 |
|
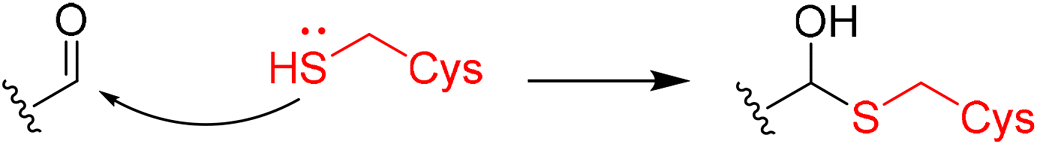
|
Moreover, an electrophilic fragment library based on heterocycles was also designed and characterised in a GSH-based reactivity assay using HPLC-MS or NMR-based kinetic methods.131 These heterocyclic electrophiles could be used to replace aromatic moieties in known non-covalent ligands with minimal influence on key non-covalent interactions, presenting an alternative design strategy in covalent drug discovery (Fig. 9).131 Most experimental data are discrepant from the theoretical reactivity predicted by computational chemistry.132
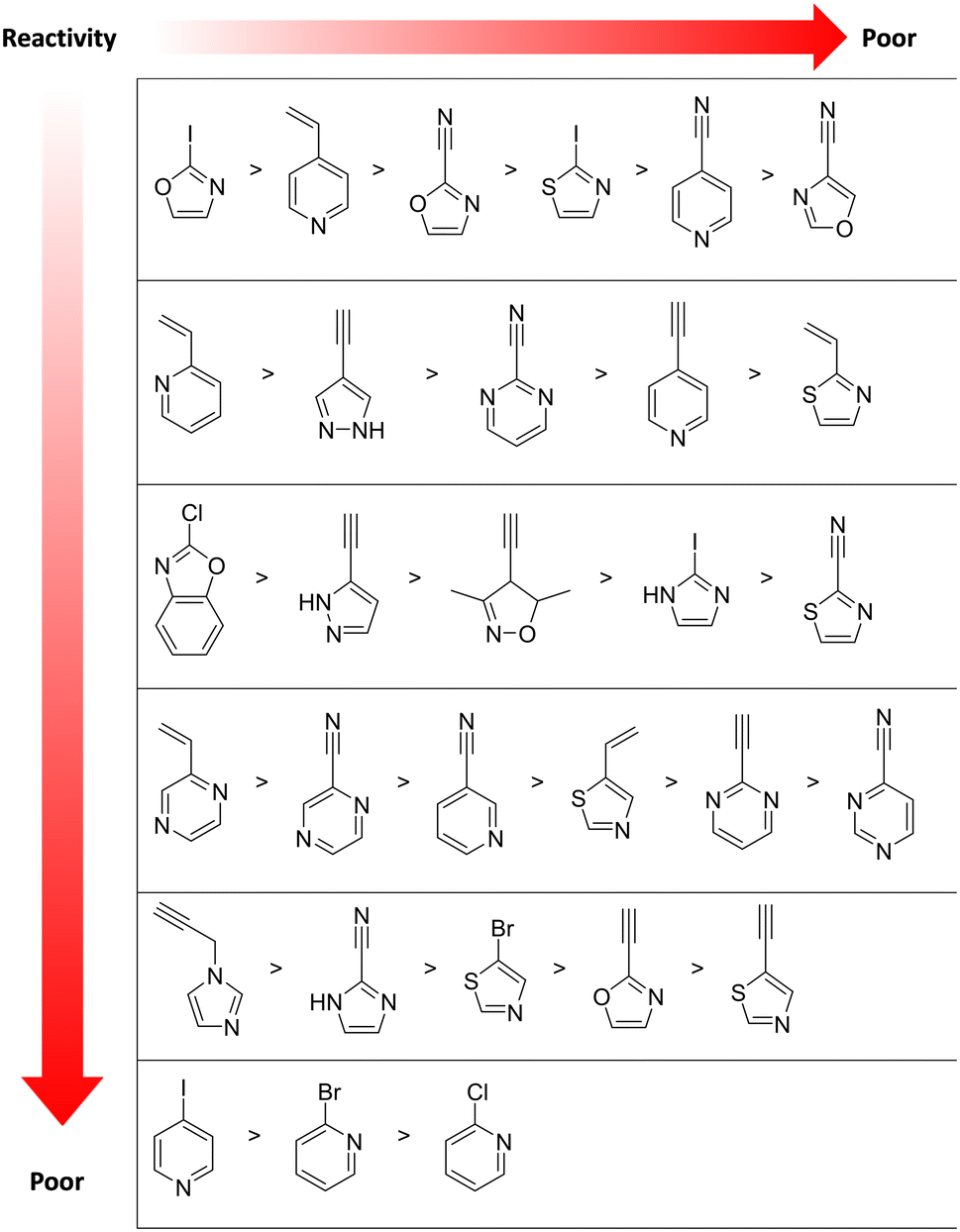 | ||
| Fig. 9 Measured GSH reactivity of diverse heterocycle moieties. The reactivity vector indicates decreasing electrophilicity.131 | ||
Inspired by the acrylamide moiety of osimertinib, Kettle et al. described their studies on newly disclosed alkynyl benzoxazines and dihydroquinazolines capable of cysteine bond formation that are different from commonly employed systems such as acrylamide.133 These electrophilic groups not only have a desirable reactivity, stability, and compatibility profile but also possess potential application as chemical biological probes and warheads in drug molecules. Although iterative optimisation against a particular target was not the focus, the group's efforts successfully demonstrated a great covalent lead discovery to JAK3 on the basis of the utility of these novel chemical probes.133
Notably, Keserű et al. overviewed recent drug discovery applications of covalent fragment libraries and especially exhibited a great table including published electrophilic moieties with their targeting information and corresponding screening methods.134 Reviews that cover a similar field were also published recently by both Gray's and London's groups, which summarised efforts in covalent fragment-based ligand discovery and the benefits of covalent targeting and fragment-based medicinal chemistry.135,136
Conclusions
To date, there are several papers published recently that overviewed the application of covalent strategies in drug discovery.1,2,6,10 In this work, we aimed to provide a brief overview of several classic covalent targets, such as EGFR, JAKs and BTK, in addition to KRASG12C, a newly disclosed cysteine-mutated GTPase protein. The co-crystal structure displayed irreversible binding modes in the ligand–protein complex, which facilitated structure-guided optimisation and rational development in covalent drug discovery. Notably, studies on tri-/difluoroketone-containing inhibitors introduced the tetrahedral adductive conformation with the serine nucleophilic residues at active sites, which may progress to an innovative stage in reversible covalent drug discovery.101Covalent inhibition strategies have been widely utilised in many progressive technologies and tools, e.g. PROTAC and peptidomimetic drugs. Targeted covalent drug design mainly focuses on the optimisation of the non-covalent scaffold followed by the attachment of extended warheads, commonly by Michael addition to the cysteine residue. However, using covalent fragments as chemical biological probes would be a potential technique to validate site-selective proteins and to search for tolerated electrophilic moieties before incorporation into kinase drug scaffolds.
Although many novel electrophilic groups have been disclosed to date, some show greater potency and are better tolerated. A promising systematic cascade of covalent drug discovery is still in development.
Author contributions
The manuscript was written by C. G. and S. Z., checked and polished by S. J. H., and reviewed by C. C. and Q. Z. The outline was proposed by C. Z. C. G. and Q. Z. are joint corresponding authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank all authors for their help and efforts. This work was supported by National Natural Science Foundation of China (82204347), Naval Medical University Young Research Fellowship Grant (2021QN12), Naval Medical University Undergraduates' Innovation and Practice Training Programs (MS2021047, FH2021097).Notes and references
- A. K. Ghosh, I. Samanta, A. Mondal and W. R. Liu, ChemMedChem, 2019, 14, 889–906 CrossRef CAS PubMed.
- F. Sutanto, M. Konstantinidou and A. Dömling, RSC Med. Chem., 2020, 11, 876–884 RSC.
- D. M. K. Keefe and E. H. Bateman, J. Natl. Cancer Inst. Monogr., 2019, 2019, 25–29 CrossRef PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 CrossRef CAS PubMed.
- A. J. Smith, X. Zhang, A. G. Leach and K. N. Houk, J. Med. Chem., 2009, 52, 225–233 CrossRef CAS PubMed.
- R. A. Bauer, Drug Discovery Today, 2015, 20, 1061–1073 CrossRef CAS PubMed.
- L. Petri, A. Egyed, D. Bajusz, T. Imre, A. Hetényi, T. Martinek, P. Ábrányi-Balogh and G. M. Keserű, Eur. J. Med. Chem., 2020, 207, 112836 CrossRef CAS PubMed.
- H. Ma, Y. Wu, W. Zhang, H. Zhang, Z. Miao and C. Zhuang, Chin. Chem. Lett., 2021, 32, 1197–1201 CrossRef CAS.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- E. D. Vita, Future Med. Chem., 2021, 13, 193–210 CrossRef CAS PubMed.
- J. M. Ostrem, U. Peters, M. L. Sos, J. A. Wells and K. M. Shokat, Nature, 2013, 503, 548–551 CrossRef CAS PubMed.
- M. Yuan, Y. Chu and Y. Duan, Front. Chem., 2021, 9, 691093 CrossRef CAS PubMed.
- J. Charoenpattarapreeda, Y. S. Tan, J. Iegre, S. J. Walsh, E. Fowler, R. S. Eapen, Y. Wu, H. F. Sore, C. S. Verma, L. Itzhaki and D. R. Spring, Chem. Commun., 2019, 55, 7914–7917 RSC.
- S. Nakayama, R. Atsumi, H. Takakusa, Y. Kobayashi, A. Kurihara, Y. Nagai, D. Nakai and O. Okazaki, Drug Metab. Dispos., 2009, 37, 1970–1977 CrossRef CAS PubMed.
- G. J. Roth, N. Stanford and P. W. Majerus, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3073–3076 CrossRef CAS PubMed.
- A. Murtuza, A. Bulbul, J. P. Shen, P. Keshavarzian, B. D. Woodward, F. J. Lopez-Diaz, S. M. Lippman and H. Husain, Cancer Res., 2019, 79, 689–698 CrossRef CAS PubMed.
- X. E. Yan, P. Ayaz, S. J. Zhu, P. Zhao, L. Liang, C. H. Zhang, Y. C. Wu, J. L. Li, H. G. Choi, X. Huang, Y. Shan, D. E. Shaw and C. H. Yun, J. Med. Chem., 2020, 63, 8502–8511 CrossRef CAS PubMed.
- A. C. Tan, Y. L. Teh, G. G. Y. Lai and D. S. W. Tan, Expert Rev. Anticancer Ther., 2019, 19, 431–435 CrossRef CAS PubMed.
- S. Butterworth, D. A. E. Cross, M. R. V. Finlay, R. A. Ward and M. J. Waring, MedChemComm, 2017, 8, 820–822 RSC.
- K. Masuzawa, H. Yasuda, J. Hamamoto, S. Nukaga, T. Hirano, I. Kawada, K. Naoki, K. Soejima and T. Betsuyaku, Oncotarget, 2017, 8, 105479–105491 CrossRef PubMed.
- R. Roskoski, Jr., Pharmacol. Res., 2021, 165, 105422–105439 CrossRef PubMed.
- J. E. Reed and J. B. Smaill, in Comprehensive Accounts of Pharmaceutical Research and Development: From Discovery to Late-Stage Process Development, 2016, vol. 1, pp. 207–233, DOI:10.1021/bk-2016-1239.ch008.
- A. T. Bender, A. Gardberg, A. Pereira, T. Johnson, Y. Wu, R. Grenningloh, J. Head, F. Morandi, P. Haselmayer and L. Liu-Bujalski, Mol. Pharmacol., 2017, 91, 208–219 CrossRef CAS PubMed.
- Y. Guo, Y. Liu, N. Hu, D. Yu, C. Zhou, G. Shi, B. Zhang, M. Wei, J. Liu, L. Luo, Z. Tang, H. Song, Y. Guo, X. Liu, D. Su, S. Zhang, X. Song, X. Zhou, Y. Hong, S. Chen, Z. Cheng, S. Young, Q. Wei, H. Wang, Q. Wang, L. Lv, F. Wang, H. Xu, H. Sun, H. Xing, N. Li, W. Zhang, Z. Wang, G. Liu, Z. Sun, D. Zhou, W. Li, L. Liu, L. Wang and Z. Wang, J. Med. Chem., 2019, 62, 7923–7940 CrossRef CAS PubMed.
- H. Y. Estupinan, Q. Wang, A. Berglof, G. C. P. Schaafsma, Y. Shi, L. Zhou, D. K. Mohammad, L. Yu, M. Vihinen, R. Zain and C. I. E. Smith, Leukemia, 2021, 35, 1317–1329 CrossRef CAS PubMed.
- D. Gu, H. Tang, J. Wu, J. Li and Y. Miao, J. Hematol. Oncol., 2021, 14, 40–55 CrossRef CAS PubMed.
- J. J. Crawford, A. R. Johnson, D. L. Misner, L. D. Belmont, G. Castanedo, R. Choy, M. Coraggio, L. Dong, C. Eigenbrot, R. Erickson, N. Ghilardi, J. Hau, A. Katewa, P. B. Kohli, W. Lee, J. W. Lubach, B. S. McKenzie, D. F. Ortwine, L. Schutt, S. Tay, B. Wei, K. Reif, L. Liu, H. Wong and W. B. Young, J. Med. Chem., 2018, 61, 2227–2245 CrossRef CAS PubMed.
- B. Tasso, A. Spallarossa, E. Russo and C. Brullo, Molecules, 2021, 26, 7411–7442 CrossRef CAS PubMed.
- L. Ding, J. Cao, W. Lin, H. Chen, X. Xiong, H. Ao, M. Yu, J. Lin and Q. Cui, Int. J. Mol. Sci., 2020, 21, 2935–2969 CrossRef PubMed.
- E. Anscombe, E. Meschini, R. Mora-Vidal, M. P. Martin, D. Staunton, M. Geitmann, U. H. Danielson, W. A. Stanley, L. Z. Wang, T. Reuillon, B. T. Golding, C. Cano, D. R. Newell, M. E. Noble, S. R. Wedge, J. A. Endicott and R. J. Griffin, Chem. Biol., 2015, 22, 1159–1164 CrossRef CAS PubMed.
- W. Cheng, Z. Yang, S. Wang, Y. Li, H. Wei, X. Tian and Q. Kan, Eur. J. Med. Chem., 2019, 164, 615–639 CrossRef CAS PubMed.
- N. Kwiatkowski, T. Zhang, P. B. Rahl, B. J. Abraham, J. Reddy, S. B. Ficarro, A. Dastur, A. Amzallag, S. Ramaswamy, B. Tesar, C. E. Jenkins, N. M. Hannett, D. McMillin, T. Sanda, T. Sim, N. D. Kim, T. Look, C. S. Mitsiades, A. P. Weng, J. R. Brown, C. H. Benes, J. A. Marto, R. A. Young and N. S. Gray, Nature, 2014, 511, 616–620 CrossRef CAS PubMed.
- B. Jiang, J. Jiang, I. H. Kaltheuner, A. B. Iniguez, K. Anand, F. M. Ferguson, S. B. Ficarro, B. K. A. Seong, A. K. Greifenberg, S. Dust, N. P. Kwiatkowski, J. A. Marto, K. Stegmaier, T. Zhang, M. Geyer and N. S. Gray, Eur. J. Med. Chem., 2021, 221, 113481–113497 CrossRef CAS PubMed.
- C. M. Olson, Y. Liang, A. Leggett, W. D. Park, L. Li, C. E. Mills, S. Z. Elsarrag, S. B. Ficarro, T. Zhang, R. Duster, M. Geyer, T. Sim, J. A. Marto, P. K. Sorger, K. D. Westover, C. Y. Lin, N. Kwiatkowski and N. S. Gray, Cell Chem. Biol., 2019, 26, 792–803 CrossRef CAS PubMed.
- S. Hu, J. J. Marineau, N. Rajagopal, K. B. Hamman, Y. J. Choi, D. R. Schmidt, N. Ke, L. Johannessen, M. J. Bradley, D. A. Orlando, S. R. Alnemy, Y. Ren, S. Ciblat, D. K. Winter, A. Kabro, K. T. Sprott, J. G. Hodgson, C. C. Fritz, J. P. Carulli, E. di Tomaso and E. R. Olson, Cancer Res., 2019, 79, 3479–3491 CrossRef PubMed.
- W. Vainchenker and S. N. Constantinescu, Oncogene, 2013, 32, 2601–2613 CrossRef CAS PubMed.
- M. Forster, A. Chaikuad, S. M. Bauer, J. Holstein, M. B. Robers, C. R. Corona, M. Gehringer, E. Pfaffenrot, K. Ghoreschi, S. Knapp and S. A. Laufer, Cell Chem. Biol., 2016, 23, 1335–1340 CrossRef CAS PubMed.
- N. London, R. M. Miller, S. Krishnan, K. Uchida, J. J. Irwin, O. Eidam, L. Gibold, P. Cimermančič, R. Bonnet, B. K. Shoichet and J. Taunton, Nat. Chem. Biol., 2014, 10, 1066–1072 CrossRef CAS PubMed.
- A. M. Shawky, F. A. Almalki, A. N. Abdalla, A. H. Abdelazeem and A. M. Gouda, Pharmaceutics, 2022, 14, 1001–1064 CrossRef CAS PubMed.
- B. Clarke, M. Yates, M. Adas, K. Bechman and J. Galloway, Rheumatology, 2021, 60, 24–30 CrossRef PubMed.
- A. Abdeldayem, Y. S. Raouf, S. N. Constantinescu, R. Moriggl and P. T. Gunning, Chem. Soc. Rev., 2020, 49, 2617–2687 RSC.
- M. F. Robinson, N. Damjanov, B. Stamenkovic, G. Radunovic, A. Kivitz, L. Cox, Z. Manukyan, C. Banfield, M. Saunders, D. Chandra, M. S. Vincent, J. Mancuso, E. Peeva and J. S. Beebe, Arthritis Rheumatol., 2020, 72, 1621–1631 CrossRef CAS PubMed.
- L. Petri, A. Egyed, D. Bajusz, T. Imre, A. Hetenyi, T. Martinek, P. Abranyi-Balogh and G. M. Keserű, Eur. J. Med. Chem., 2020, 207, 112836–112845 CrossRef CAS PubMed.
- A. Addeo, G. L. Banna and A. Friedlaender, Cancers, 2021, 13, 2541–2556 CrossRef CAS PubMed.
- V. Amodio, G. Mauri, N. M. Reilly, A. Sartore-Bianchi, S. Siena, A. Bardelli and G. Germano, Cancers, 2021, 13, 2638–2671 CrossRef CAS PubMed.
- D. S. Hong, M. G. Fakih, J. H. Strickler, J. Desai, G. A. Durm, G. I. Shapiro, G. S. Falchook, T. J. Price, A. Sacher, C. S. Denlinger, Y. J. Bang, G. K. Dy, J. C. Krauss, Y. Kuboki, J. C. Kuo, A. L. Coveler, K. Park, T. W. Kim, F. Barlesi, P. N. Munster, S. S. Ramalingam, T. F. Burns, F. Meric-Bernstam, H. Henary, J. Ngang, G. Ngarmchamnanrith, J. Kim, B. E. Houk, J. Canon, J. R. Lipford, G. Friberg, P. Lito, R. Govindan and B. T. Li, N. Engl. J. Med., 2020, 383, 1207–1217 CrossRef CAS PubMed.
- M. R. Janes, J. Zhang, L. S. Li, R. Hansen, U. Peters, X. Guo, Y. Chen, A. Babbar, S. J. Firdaus, L. Darjania, J. Feng, J. H. Chen, S. Li, S. Li, Y. O. Long, C. Thach, Y. Liu, A. Zarieh, T. Ely, J. M. Kucharski, L. V. Kessler, T. Wu, K. Yu, Y. Wang, Y. Yao, X. Deng, P. P. Zarrinkar, D. Brehmer, D. Dhanak, M. V. Lorenzi, D. Hu-Lowe, M. P. Patricelli, P. Ren and Y. Liu, Cell, 2018, 172, 578–589 CrossRef CAS PubMed.
- J. Hallin, L. D. Engstrom, L. Hargis, A. Calinisan, R. Aranda, D. M. Briere, N. Sudhakar, V. Bowcut, B. R. Baer, J. A. Ballard, M. R. Burkard, J. B. Fell, J. P. Fischer, G. P. Vigers, Y. Xue, S. Gatto, J. Fernandez-Banet, A. Pavlicek, K. Velastagui, R. C. Chao, J. Barton, M. Pierobon, E. Baldelli, E. F. Patricoin, 3rd, D. P. Cassidy, M. A. Marx, I. I. Rybkin, M. L. Johnson, S. I. Ou, P. Lito, K. P. Papadopoulos, P. A. Janne, P. Olson and J. G. Christensen, Cancer Discovery, 2020, 10, 54–71 CrossRef CAS PubMed.
- C. Sheridan, Nat. Biotechnol., 2019, 37, 1245–1246 CrossRef CAS PubMed.
- Nat. Biotechnol., 2019, 37, 1247 Search PubMed.
- S. Seton-Rogers, Nat. Rev. Cancer, 2020, 20, 3 CrossRef CAS PubMed.
- B. A. Lanman, J. R. Allen, J. G. Allen, A. K. Amegadzie, K. S. Ashton, S. K. Booker, J. J. Chen, N. Chen, M. J. Frohn, G. Goodman, D. J. Kopecky, L. Liu, P. Lopez, J. D. Low, V. Ma, A. E. Minatti, T. T. Nguyen, N. Nishimura, A. J. Pickrell, A. B. Reed, Y. Shin, A. C. Siegmund, N. A. Tamayo, C. M. Tegley, M. C. Walton, H. L. Wang, R. P. Wurz, M. Xue, K. C. Yang, P. Achanta, M. D. Bartberger, J. Canon, L. S. Hollis, J. D. McCarter, C. Mohr, K. Rex, A. Y. Saiki, T. San Miguel, L. P. Volak, K. H. Wang, D. A. Whittington, S. G. Zech, J. R. Lipford and V. J. Cee, J. Med. Chem., 2020, 63, 52–65 CrossRef CAS PubMed.
- J. Canon, K. Rex, A. Y. Saiki, C. Mohr, K. Cooke, D. Bagal, K. Gaida, T. Holt, C. G. Knutson, N. Koppada, B. A. Lanman, J. Werner, A. S. Rapaport, T. San Miguel, R. Ortiz, T. Osgood, J. R. Sun, X. Zhu, J. D. McCarter, L. P. Volak, B. E. Houk, M. G. Fakih, B. H. O'Neil, T. J. Price, G. S. Falchook, J. Desai, J. Kuo, R. Govindan, D. S. Hong, W. Ouyang, H. Henary, T. Arvedson, V. J. Cee and J. R. Lipford, Nature, 2019, 575, 217–223 CrossRef CAS PubMed.
- A. H. Chan, W. G. Lee, K. A. Spasov, J. A. Cisneros, S. N. Kudalkar, Z. O. Petrova, A. B. Buckingham, K. S. Anderson and W. L. Jorgensen, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 9725–9730 CrossRef CAS PubMed.
- Q. Li, Z. Wang, Q. Zheng and S. Liu, Comput. Struct. Biotechnol. J., 2020, 18, 2200–2208 CrossRef CAS PubMed.
- C. Ma, Z. Xia, M. D. Sacco, Y. Hu, J. A. Townsend, X. Meng, J. Choza, H. Tan, J. Jang, M. V. Gongora, X. Zhang, F. Zhang, Y. Xiang, M. T. Marty, Y. Chen and J. Wang, J. Am. Chem. Soc., 2021, 143, 20697–20709 CrossRef CAS PubMed.
- R. Ge, Z. Shen, J. Yin, W. Chen, Q. Zhang, Y. An, D. Tang, A. L. Satz, W. Su and L. Kuai, SLAS Discovery, 2022, 27, 79–85 CrossRef PubMed.
- S. Vankadara, M. D. Dawson, J. Y. Fong, Q. Y. Oh, Q. A. Ang, B. Liu, H. Y. Chang, J. Koh, X. Koh, Q. W. Tan, J. Joy and C. S. B. Chia, ACS Med. Chem. Lett., 2022, 13, 1345–1350 CrossRef CAS PubMed.
- M. R. V. Finlay, M. Anderton, S. Ashton, P. Ballard, P. A. Bethel, M. R. Box, R. H. Bradbury, S. J. Brown, S. Butterworth, A. Campbell, C. Chorley, N. Colclough, D. A. E. Cross, G. S. Currie, M. Grist, L. Hassall, G. B. Hill, D. James, M. James, P. Kemmitt, T. Klinowska, G. Lamont, S. G. Lamont, N. Martin, H. L. McFarland, M. J. Mellor, J. P. Orme, D. Perkins, P. Perkins, G. Richmond, P. Smith, R. A. Ward, M. J. Waring, D. Whittaker, S. Wells and G. L. Wrigley, J. Med. Chem., 2014, 57, 8249–8267 CrossRef CAS PubMed.
- Y. Jia, J. Juarez, J. Li, M. Manuia, M. J. Niederst, C. Tompkins, N. Timple, M.-T. Vaillancourt, A. C. Pferdekamper, E. L. Lockerman, C. Li, J. Anderson, C. Costa, D. Liao, E. Murphy, M. DiDonato, B. Bursulaya, G. Lelais, J. Barretina, M. McNeill, R. Epple, T. H. Marsilje, N. Pathan, J. A. Engelman, P.-Y. Michellys, P. McNamara, J. Harris, S. Bender and S. Kasibhatla, Cancer Res., 2016, 76, 1591–1602 CrossRef CAS PubMed.
- L. Cheng, S. Zhou, S. Zhou, K. Shi, Y. Cheng, M.-C. Cai, K. Ye, L. Lin, Z. Zhang, C. Jia, H. Xiang, J. Zang, M. Zhang, X. Yin, Y. Li, W. Di, G. Zhuang and L. Tan, Cancer Res., 2022 DOI:10.1158/0008-5472.Can-22-0222.
- T. Zhang, N. Kwiatkowski, C. M. Olson, S. E. Dixon-Clarke, B. J. Abraham, A. K. Greifenberg, S. B. Ficarro, J. M. Elkins, Y. Liang, N. M. Hannett, T. Manz, M. Hao, B. Bartkowiak, A. L. Greenleaf, J. A. Marto, M. Geyer, A. N. Bullock, R. A. Young and N. S. Gray, Nat. Chem. Biol., 2016, 12, 876–884 CrossRef CAS PubMed.
- B. N. Marak, J. Dowarah, L. Khiangte and V. P. Singh, Eur. J. Med. Chem., 2020, 203, 112571 CrossRef CAS PubMed.
- J.-B. Telliez, M. E. Dowty, L. Wang, J. Jussif, T. Lin, L. Li, E. Moy, P. Balbo, W. Li, Y. Zhao, K. Crouse, C. Dickinson, P. Symanowicz, M. Hegen, M. E. Banker, F. Vincent, R. Unwalla, S. Liang, A. M. Gilbert, M. F. Brown, M. Hayward, J. Montgomery, X. Yang, J. Bauman, J. I. Trujillo, A. Casimiro-Garcia, F. F. Vajdos, L. Leung, K. F. Geoghegan, A. Quazi, D. Xuan, L. Jones, E. Hett, K. Wright, J. D. Clark and A. Thorarensen, ACS Chem. Biol., 2016, 11, 3442–3451 CrossRef CAS PubMed.
- M. Forster, A. Chaikuad, Silke M. Bauer, J. Holstein, Matthew B. Robers, Cesear R. Corona, M. Gehringer, E. Pfaffenrot, K. Ghoreschi, S. Knapp and Stefan A. Laufer, Cell Chem. Biol., 2016, 23, 1335–1340 CrossRef CAS PubMed.
- C. M. Olson, Y. Liang, A. Leggett, W. D. Park, L. Li, C. E. Mills, S. Z. Elsarrag, S. B. Ficarro, T. Zhang, R. Düster, M. Geyer, T. Sim, J. A. Marto, P. K. Sorger, K. D. Westover, C. Y. Lin, N. Kwiatkowski and N. S. Gray, Cell Chem. Biol., 2019, 26, 792–803.e710 CrossRef CAS PubMed.
- G. Hodgson, L. Johannessen, N. Rajagopal, S. Hu, D. Orlando, M. Eaton, M. McKeown, J. W. Tyner, S. E. Kurtz, K. Sprott, C. Fritz and E. Di Tomaso, Blood, 2017, 130, 2651–2651 Search PubMed.
- J. A. Engelman, K. Zejnullahu, C.-M. Gale, E. Lifshits, A. J. Gonzales, T. Shimamura, F. Zhao, P. W. Vincent, G. N. Naumov, J. E. Bradner, I. W. Althaus, L. Gandhi, G. I. Shapiro, J. M. Nelson, J. V. Heymach, M. Meyerson, K.-K. Wong and P. A. Jänne, Cancer Res., 2007, 67, 11924–11932 CrossRef CAS PubMed.
- J. S. Martin, C. J. MacKenzie, D. Fletcher and I. H. Gilbert, Bioorg. Med. Chem., 2019, 27, 2066–2074 CrossRef CAS PubMed.
- E. Resnick, A. Bradley, J. Gan, A. Douangamath, T. Krojer, R. Sethi, P. P. Geurink, A. Aimon, G. Amitai, D. Bellini, J. Bennett, M. Fairhead, O. Fedorov, R. Gabizon, J. Gan, J. Guo, A. Plotnikov, N. Reznik, G. F. Ruda, L. Díaz-Sáez, V. M. Straub, T. Szommer, S. Velupillai, D. Zaidman, Y. Zhang, A. R. Coker, C. G. Dowson, H. M. Barr, C. Wang, K. V. M. Huber, P. E. Brennan, H. Ovaa, F. von Delft and N. London, J. Am. Chem. Soc., 2019, 141, 8951–8968 CrossRef CAS PubMed.
- X. Liu, D. Feng, M. Zheng, Y. Cui and D. Zhong, Drug Metab. Pharmacokinet., 2020, 35, 456–465 CrossRef CAS PubMed.
- E. Mons, R. Q. Kim, B. R. van Doodewaerd, P. A. van Veelen, M. P. C. Mulder and H. Ovaa, J. Am. Chem. Soc., 2021, 143, 6423–6433 CrossRef CAS PubMed.
- J. M. Bradshaw, J. M. McFarland, V. O. Paavilainen, A. Bisconte, D. Tam, V. T. Phan, S. Romanov, D. Finkle, J. Shu, V. Patel, T. Ton, X. Li, D. G. Loughhead, P. A. Nunn, D. E. Karr, M. E. Gerritsen, J. O. Funk, T. D. Owens, E. Verner, K. A. Brameld, R. J. Hill, D. M. Goldstein and J. Taunton, Nat. Chem. Biol., 2015, 11, 525–531 CrossRef CAS PubMed.
- W. Zheng, N. Thorne and J. C. McKew, Drug Discovery Today, 2013, 18, 1067–1073 CrossRef CAS PubMed.
- A. T. Plowright and L. Drowley, in Platform Technologies in Drug Discovery and Validation, 2017, pp. 263–299, DOI:10.1016/bs.armc.2017.07.001.
- B. Dafniet, N. Cerisier, B. Boezio, A. Clary, P. Ducrot, T. Dorval, A. Gohier, D. Brown, K. Audouze and O. Taboureau, Aust. J. Chem., 2021, 13, 91–103 CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- S. Neidle, in Therapeutic Applications of Quadruplex Nucleic Acids, ed. S. Neidle, Academic Press, Boston, 2012, pp. 151–174, DOI:10.1016/B978-0-12-375138-6.00009-1.
- L. Petri, P. Ábrányi-Balogh, I. Tímea, G. Pálfy, A. Perczel, D. Knez, M. Hrast, M. Gobec, I. Sosič, K. Nyíri, B. G. Vértessy, N. Jänsch, C. Desczyk, F.-J. Meyer-Almes, I. Ogris, S. Golič Grdadolnik, L. G. Iacovino, C. Binda, S. Gobec and G. M. Keserű, ChemBioChem, 2021, 22, 743–753 CrossRef CAS PubMed.
- L. Li, H. Zhao, H. Liao, J. Chen, J. Liu and J. Chen, Bioorg. Chem., 2021, 110, 104825 CrossRef CAS PubMed.
- L. M. McGregor, M. L. Jenkins, C. Kerwin, J. E. Burke and K. M. Shokat, Biochemistry, 2017, 56, 3178–3183 CrossRef CAS PubMed.
- Q. T. N. Tran, D. W. S. Tan, W. S. F. Wong and C. L. L. Chai, Eur. J. Med. Chem., 2020, 204, 112481–112498 CrossRef CAS PubMed.
- S. Fan, L. Yue, W. Wan, Y. Zhang, B. Zhang, C. Otomo, Q. Li, T. Lin, J. Hu, P. Xu, M. Zhu, H. Tao, Z. Chen, L. Li, H. Ding, Z. Yao, J. Lu, Y. Wen, N. Zhang, M. Tan, K. Chen, Y. Xie, T. Otomo, B. Zhou, H. Jiang, Y. Dang and C. Luo, Angew. Chem., Int. Ed., 2021, 60, 26105–26114 CrossRef CAS PubMed.
- K. M. Backus, B. E. Correia, K. M. Lum, S. Forli, B. D. Horning, G. E. González-Páez, S. Chatterjee, B. R. Lanning, J. R. Teijaro, A. J. Olson, D. W. Wolan and B. F. Cravatt, Nature, 2016, 534, 570–574 CrossRef CAS PubMed.
- M. E. Abbasov, M. E. Kavanagh, T.-A. Ichu, M. R. Lazear, Y. Tao, V. M. Crowley, C. W. A. Ende, S. M. Hacker, J. Ho, M. M. Dix, R. Suciu, M. M. Hayward, L. L. Kiessling and B. F. Cravatt, Nat. Chem., 2021, 13, 1081–1092 CrossRef CAS PubMed.
- S. M. Hacker, K. M. Backus, M. R. Lazear, S. Forli, B. E. Correia and B. F. Cravatt, Nat. Chem., 2017, 9, 1181–1190 CrossRef CAS PubMed.
- S. Li, P. Zhang, F. Xu, S. Hu, J. Liu, Y. Tan, Z. Tu, H. Sun, Z.-M. Zhang, Q.-Y. He, P. Sun, K. Ding and Z. Li, J. Med. Chem., 2022, 65, 10408–10418 CrossRef CAS PubMed.
- J. Yang, I. Anishchenko, H. Park, Z. Peng, S. Ovchinnikov and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1496–1503 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Zidek, A. Potapenko, A. Bridgland, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- L. Frye, S. Bhat, K. Akinsanya and R. Abel, Drug Discovery Today: Technol., 2021, 39, 111–117 CrossRef CAS PubMed.
- L. Blake and M. Soliman, Med. Chem. Res., 2013, 23, 2312–2323 CrossRef.
- G. Q. Dong, S. Calhoun, H. Fan, C. Kalyanaraman, M. C. Branch, S. T. Mashiyama, N. London, M. P. Jacobson, P. C. Babbitt, B. K. Shoichet, R. N. Armstrong and A. Sali, J. Chem. Inf. Model., 2014, 54, 1687–1699 CrossRef CAS PubMed.
- H. M. Kumalo, S. Bhakat and M. E. S. Soliman, Molecules, 2015, 20, 1984–2000 CrossRef PubMed.
- A. Scarpino, G. G. Ferenczy and G. M. Keserű, in Protein-Ligand Interactions and Drug Design, ed. F. Ballante, Springer US, New York, NY, 2021, pp. 73–88, DOI:10.1007/978-1-0716-1209-5_4.
- C. Wen, X. Yan, Q. Gu, J. Du, D. Wu, Y. Lu, H. Zhou and J. Xu, Molecules, 2019, 24, 2183–2203 CrossRef CAS PubMed.
- X. Wan, T. Yang, A. Cuesta, X. Pang, T. E. Balius, J. J. Irwin, B. K. Shoichet and J. Taunton, J. Am. Chem. Soc., 2020, 142, 4960–4964 CrossRef CAS PubMed.
- H. M. Kumalo, S. Bhakat and M. E. Soliman, Molecules, 2015, 20, 1984–2000 CrossRef PubMed.
- A. Scarpino, G. G. Ferenczy and G. M. Keserű, Curr. Pharm. Des., 2020, 26, 5684–5699 CrossRef CAS PubMed.
- M. Sakamoto Kathleen, B. Kim Kyung, A. Kumagai, F. Mercurio, M. Crews Craig and J. Deshaies Raymond, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef PubMed.
- J. Y. Xi, R. Y. Zhang, K. Chen, L. Yao, M. Q. Li, R. Jiang, X. Y. Li and L. Fan, Bioorg. Chem., 2022, 125, 105848–105867 CrossRef CAS PubMed.
- L. Dzhekieva, S. A. Adediran, R. Herman, F. Kerff, C. Duez, P. Charlier, E. Sauvage and R. F. Pratt, Biochemistry, 2013, 52, 2128–2138 CrossRef CAS PubMed.
- J. Lu, Y. Qian, M. Altieri, H. Dong, J. Wang, K. Raina, J. Hines, J. D. Winkler, A. P. Crew, K. Coleman and C. M. Crews, Chem. Biol., 2015, 22, 755–763 CrossRef CAS PubMed.
- C. Cecchini, S. Pannilunghi, S. Tardy and L. Scapozza, Front. Chem., 2021, 9, 672267–672267 CrossRef CAS PubMed.
- R. Hu, W. L. Wang, Y. Y. Yang, X. T. Hu, Q. W. Wang, W. Q. Zuo, Y. Xu, Q. Feng and N. Y. Wang, Eur. J. Med. Chem., 2022, 227, 113922–113936 CrossRef CAS PubMed.
- M. J. Bond, L. Chu, D. A. Nalawansha, K. Li and C. M. Crews, ACS Cent. Sci., 2020, 6, 1367–1375 CrossRef CAS PubMed.
- G. Xue, J. Chen, L. Liu, D. Zhou, Y. Zuo, T. Fu and Z. Pan, Chem. Commun., 2020, 56, 1521–1524 RSC.
- R. Gabizon, A. Shraga, P. Gehrtz, E. Livnah, Y. Shorer, N. Gurwicz, L. Avram, T. Unger, H. Aharoni, S. Albeck, A. Brandis, Z. Shulman, B. Z. Katz, Y. Herishanu and N. London, J. Am. Chem. Soc., 2020, 142, 11734–11742 CrossRef CAS PubMed.
- F. Yu, M. Cai, L. Shao and J. Zhang, Front. Chem., 2021, 9, 679120–679134 CrossRef CAS PubMed.
- N. Shindo and A. Ojida, Bioorg. Med. Chem., 2021, 47, 116386–116398 CrossRef CAS PubMed.
- D. Quach, G. Tang, J. Anantharajan, N. Baburajendran, A. Poulsen, J. L. K. Wee, P. Retna, R. Li, B. Liu, D. H. Y. Tee, P. Z. Kwek, J. K. Joy, W. Q. Yang, C. J. Zhang, K. Foo, T. H. Keller and S. Q. Yao, Angew. Chem., Int. Ed., 2021, 60, 17131–17137 CrossRef CAS PubMed.
- W. Dai, B. Zhang, X.-M. Jiang, H. Su, J. Li, Y. Zhao, X. Xie, Z. Jin, J. Peng, F. Liu, C. Li, Y. Li, F. Bai, H. Wang, X. Cheng, X. Cen, S. Hu, X. Yang, J. Wang, X. Liu, G. Xiao, H. Jiang, Z. Rao, L.-K. Zhang, Y. Xu, H. Yang and H. Liu, Science, 2020, 368, 1331–1335 CrossRef CAS PubMed.
- L. Zhang, D. Lin, X. Sun, U. Curth, C. Drosten, L. Sauerhering, S. Becker, K. Rox and R. Hilgenfeld, Science, 2020, 368, 409–412 CrossRef CAS PubMed.
- W. H. Guo, X. Qi, X. Yu, Y. Liu, C. I. Chung, F. Bai, X. Lin, D. Lu, L. Wang, J. Chen, L. H. Su, K. J. Nomie, F. Li, M. C. Wang, X. Shu, J. N. Onuchic, J. A. Woyach, M. L. Wang and J. Wang, Nat. Commun., 2020, 11, 4268–4284 CrossRef PubMed.
- H. Kiely-Collins, G. E. Winter and G. J. L. Bernardes, Cell Chem. Biol., 2021, 28, 952–968 CrossRef CAS PubMed.
- R. Gabizon, A. Shraga, P. Gehrtz, E. Livnah, Y. Shorer, N. Gurwicz, L. Avram, T. Unger, H. Aharoni, S. Albeck, A. Brandis, Z. Shulman, B.-Z. Katz, Y. Herishanu and N. London, J. Am. Chem. Soc., 2020, 142, 11316–11316 CrossRef CAS PubMed.
- J. L. Stebbins, E. Santelli, Y. Feng, S. K. De, A. Purves, K. Motamedchaboki, B. Wu, Z. A. Ronai, R. C. Liddington and M. Pellecchia, Chem. Biol., 2013, 20, 973–982 CrossRef CAS PubMed.
- A. D. de Araujo, J. Lim, A. C. Good, R. T. Skerlj and D. P. Fairlie, ACS Med. Chem. Lett., 2017, 8, 22–26 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 RSC.
- Q. Li and G. Lozano, Clin. Cancer Res., 2013, 19, 34–41 CrossRef CAS PubMed.
- Y. H. Lau, Y. Wu, P. de Andrade, W. R. Galloway and D. R. Spring, Nat. Protoc., 2015, 10, 585–594 CrossRef CAS PubMed.
- A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC.
- W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 CrossRef CAS PubMed.
- D. E. Mortenson, G. J. Brighty, L. Plate, G. Bare, W. Chen, S. Li, H. Wang, B. F. Cravatt, S. Forli, E. T. Powers, K. B. Sharpless, I. A. Wilson and J. W. Kelly, J. Am. Chem. Soc., 2018, 140, 200–210 CrossRef CAS PubMed.
- N. Wang, B. Yang, C. Fu, H. Zhu, F. Zheng, T. Kobayashi, J. Liu, S. Li, C. Ma, P. G. Wang, Q. Wang and L. Wang, J. Am. Chem. Soc., 2018, 140, 4995–4999 CrossRef CAS PubMed.
- L. Gambini, C. Baggio, P. Udompholkul, J. Jossart, A. F. Salem, J. J. P. Perry and M. Pellecchia, J. Med. Chem., 2019, 62, 5616–5627 CrossRef CAS PubMed.
- A. J. Maurais and E. Weerapana, Curr. Opin. Chem. Biol., 2019, 50, 29–36 CrossRef CAS PubMed.
- C. Brullo, C. Villa, B. Tasso, E. Russo and A. Spallarossa, Int. J. Mol. Sci., 2021, 22, 7641–7657 CrossRef CAS PubMed.
- Y. Liu, S. Lv, L. Peng, C. Xie, L. Gao, H. Sun, L. Lin, K. Ding and Z. Li, Biochem. Pharmacol., 2021, 190, 114636–114645 CrossRef CAS PubMed.
- M. Cocco, D. Garella, A. Di Stilo, E. Borretto, L. Stevanato, M. Giorgis, E. Marini, R. Fantozzi, G. Miglio and M. Bertinaria, J. Med. Chem., 2014, 57, 10366–10382 CrossRef CAS PubMed.
- A. Keeley, P. Abranyi-Balogh and G. M. Keseru, MedChemComm, 2019, 10, 263–267 RSC.
- R. M. Oballa, J. F. Truchon, C. I. Bayly, N. Chauret, S. Day, S. Crane and C. Berthelette, Bioorg. Med. Chem. Lett., 2007, 17, 998–1002 CrossRef CAS PubMed.
- K. McAulay, E. A. Hoyt, M. Thomas, M. Schimpl, M. S. Bodnarchuk, H. J. Lewis, D. Barratt, D. Bhavsar, D. M. Robinson, M. J. Deery, D. J. Ogg, G. J. L. Bernardes, R. A. Ward, M. J. Waring and J. G. Kettle, J. Am. Chem. Soc., 2020, 142, 10358–10372 CrossRef CAS PubMed.
- A. Keeley, L. Petri, P. Ábrányi-Balogh and G. M. Keserű, Drug Discovery Today, 2020, 25, 983–996 CrossRef CAS PubMed.
- A. Shraga, E. Resnick, R. Gabizon and N. London, in Annual Reports in Medicinal Chemistry, ed. R. A. Ward and N. P. Grimster, Academic Press, 2021, vol. 56, pp. 243–265 Search PubMed.
- W. Lu, M. Kostic, T. Zhang, J. Che, M. P. Patricelli, L. H. Jones, E. T. Chouchani and N. S. Gray, RSC Chem. Biol., 2021, 2, 354–367 RSC.
| This journal is © The Royal Society of Chemistry 2022 |