
Recent applications of covalent chemistries in protein–protein interaction inhibitors
Alexandria M.
Chan†
 a,
Christopher C.
Goodis†
b,
Elie G.
Pommier
b and
Steven
Fletcher
*ac
a,
Christopher C.
Goodis†
b,
Elie G.
Pommier
b and
Steven
Fletcher
*ac
aUniversity of Maryland School of Pharmacy, Department of Pharmaceutical Sciences, 20 N. Pine St, Baltimore, MD 21201, USA. E-mail: steven.fletcher@rx.umaryland.edu
bUniversity of Maryland School of Pharmacy, PharmD Program, 20 N. Pine St, Baltimore, MD 21201, USA
cUniversity of Maryland Greenebaum Cancer Center, 20 S. Greene St, Baltimore, MD 21201, USA
First published on 3rd June 2022
Abstract
Protein–protein interactions (PPIs) are large, often featureless domains whose modulations by small-molecules are challenging. Whilst there are some notable successes, such as the BCL-2 inhibitor venetoclax, the requirement for larger ligands to achieve the desired level of potency and selectivity may result in poor “drug-like” properties. Covalent chemistry is presently enjoying a renaissance. In particular, targeted covalent inhibition (TCI), in which a weakly electrophilic “warhead” is installed onto a protein ligand scaffold, is a powerful strategy to develop potent inhibitors of PPIs that are smaller/more drug-like yet have enhanced affinities by virtue of the reinforcing effect on the existing non-covalent interactions by the resulting protein–ligand covalent bond. Furthermore, the covalent bond delivers sustained inhibition, which may translate into significantly reduced therapeutic dosing. Herein, we discuss recent applications of a spectrum of TCIs, as well as covalent screening strategies, in the discovery of more effective inhibitors of PPIs using the HDM2 and BCL-2 protein families as case studies.
Introduction
Protein–protein interactions (PPIs) are fundamental to the regulation of signalling pathways in cells, and their interrogations with small-molecules may lead to improved understanding of particular biological events.1–3 Moreover, aberrant PPIs, which may include those between endogenous proteins, as well as bacterial proteins, or both, represent therapeutic targets for the development of new drugs to treat a range of diseases, including cancers, neurodegenerative diseases and bacterial infections.4 Targeting PPIs is non-trivial owing to their expansive interfaces and to a lack of well-defined binding sites/cavities. Although there are typically “hot-spots” – clusters of residues at the PPI interface that are responsible for the majority of the binding free energy – often, large molecules are required to effectively compete with the native protein.1–3 However, notwithstanding the success of the BCL-2 inhibitor venetoclax (molecular weight = 868),5 increasingly larger molecules may battle to penetrate the cell membrane, and exhibit systematically worsening pharmacokinetics through increased opportunities for metabolism.6,7 The challenge in the development of small-molecule PPI inhibitors is managing the size, whilst also achieving sufficient potency and selectivity. With a recent reboot of covalent drug discovery, one possible strategy to circumvent this dilemma is targeted covalent inhibition, or TCI, which is the introduction of a weakly electrophilic “warhead” into an existing protein ligand scaffold.8,9 These warheads react most typically with cysteines, as with the acrylamide and 2-chloroacetamide warheads,9–12 but there are emerging warheads that react with different amino acid residues, such as aryl sulfonyl fluorides that react with lysines, tyrosines or histidines.13 Weakly electrophilic warheads are preferred to mitigate indiscriminate reactions: the ligand first engages the protein through non-covalent interactions, and then, due to the resulting increase in effective molarity – provided the warhead is in proximity to the targeted side chain – the chemical reaction occurs, which, in turn, underpins the initial non-covalent interaction. The net result is irreversible inhibition that, in addition to greater potency through the reinforcement of the non-covalent interactions by virtue of the ligand now chemically fixed at the binding site, also results in a sustained duration of inhibition, i.e., a greater residency time.9 These specific advantages offered by TCI may help mitigate the large molecular mass requirement to develop a highly potent and selective inhibitor with desirable pharmacologic properties, and advance to the clinic. Our group has a special interest in targeting the human double minute 2 (HDM2) and B-cell lymphoma 2 (BCL-2) families of proteins towards the discovery of new anti-cancer therapeutics.14–22 In recent years, covalent inhibitors of these protein families have emerged. Thus, the focus of this review is the application of TCI, as well as covalent chemistry screening strategies, in the discovery of potent inhibitors of HDM2 and, within the BCL-2 family, MCL-1 and BFL-1.HDM2
p53, also regarded as the “guardian of the genome”, is a tumour suppressor, and its role is the regulation of the cell-cycle and apoptosis;23 mutation or deletion of the p53 gene is a hallmark of cancer.24 HDM2 (or MDM2) binds the transactivation domain (TAD) of p53, inhibiting its ability to activate transcription.25 In addition to this negative regulatory role, HDM2 targets p53 for destruction by the proteasome through its role as an E3 ubiquitin ligase.26 Over-expressed HDM2 captures – thereby “neutralizes” – p53, causing the dysregulation of cell growth, which may develop into cancer.27 Indeed, elevated levels of HDM2 are found in a range of haematological malignancies and solid tumours.27 For this reason, targeting the p53/HDM2 PPI to develop small-molecule therapeutics has become a major focus of research.28Central to the interaction between p53 and HDM2 is a deep hydrophobic cleft on HDM2 that is engaged by three key amino acids (Phe19, Trp23 and Leu26) projected from one face of the α-helical TAD of p53 (PDB ID: 1YCR).29 Around 20 years of research have been devoted to disrupting this PPI through fashioning small-molecules that recapitulate the p53 recognition profile, and some such HDM2 inhibitors are presently in clinical trials.27,28,30,31 Crucially, however, HDM2 inhibitors are yet to advance to the clinic, and different modes of inhibition, such as with TCI, may provide the winning strategy to prolonged/sustained activation of p53.
HDM2 – irreversible inhibition, NASA warhead: terminal amine, tyrosine
Hamachi's group recently introduced an N-acyl-N-alkyl sulfonamide, or “NASA” group as an electrophilic warhead that was reported to acylate a noncatalytic lysine residue of Hsp90 (Scheme 1).32,33 With this data in hand, the group then wanted to determine if they could extend their technology to develop a more potent inhibitor of the HDM2/p53 PPI based on the existing family of inhibitors termed “nutlins”30 by targeting nucleophilic residues near the nutlin binding site.34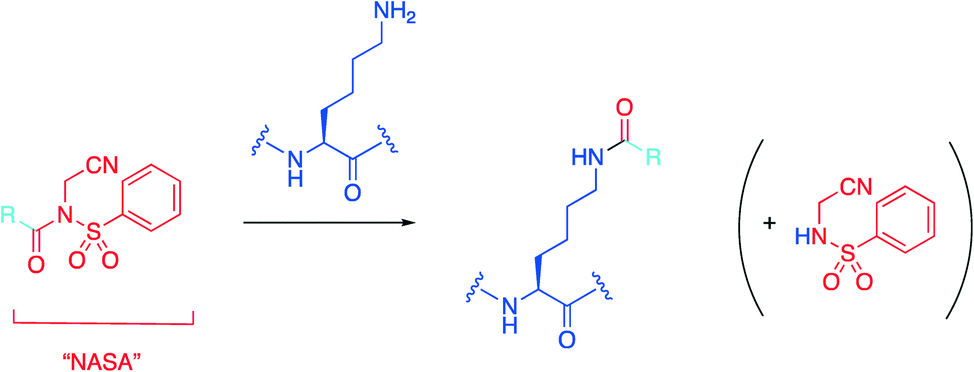 | ||
| Scheme 1 The NASA warhead is grafted onto an existing protein ligand represented by R, which then irreversibly acylates the ε-amino group of a lysine residue. | ||
Upon inspection of the co-crystal structure of HDM2 and Nutlin-3a (PDB ID: 4HG7), the authors noted that the ε-amino group of Lys51 is about 11 Å away from the 2-oxopiperazine of the ligand (Fig. 1). Accordingly, they prepared a panel of inhibitors based on (±)-nutlin-3 that incorporated the NASA group via various linkers grafted through the piperazine moiety. Lead compound 1 exhibited a a two-fold reduction in binding with a Kd of 570 nM relative to (±)-nutlin-3 (Kd = 263 nM), indicating this modification was not significantly detrimental. Mass spectrometry (MS)-based peptide mapping revealed that 1 modified the N-terminal α-amino group of HDM2 and also the Tyr67 side chain, rather than the ε-amino group of Lys51 as designed. However, the authors rationalized these findings by noting that the distances of these functional groups from Nutlin-3a in the co-crystal structure are somewhat similar to that with the ε-amino group in Lys51, and that α-amino groups typically have a lower pKa (pKa = 8) than that of lysine side chains (pKa = 10), and are thus more likely to be unprotonated and nucleophilic. In addition, these covalent modifications were confirmed in HEK293T cells, and the expected activation of the p53 pathway was observed in osteosarcoma SJSA1 cells.
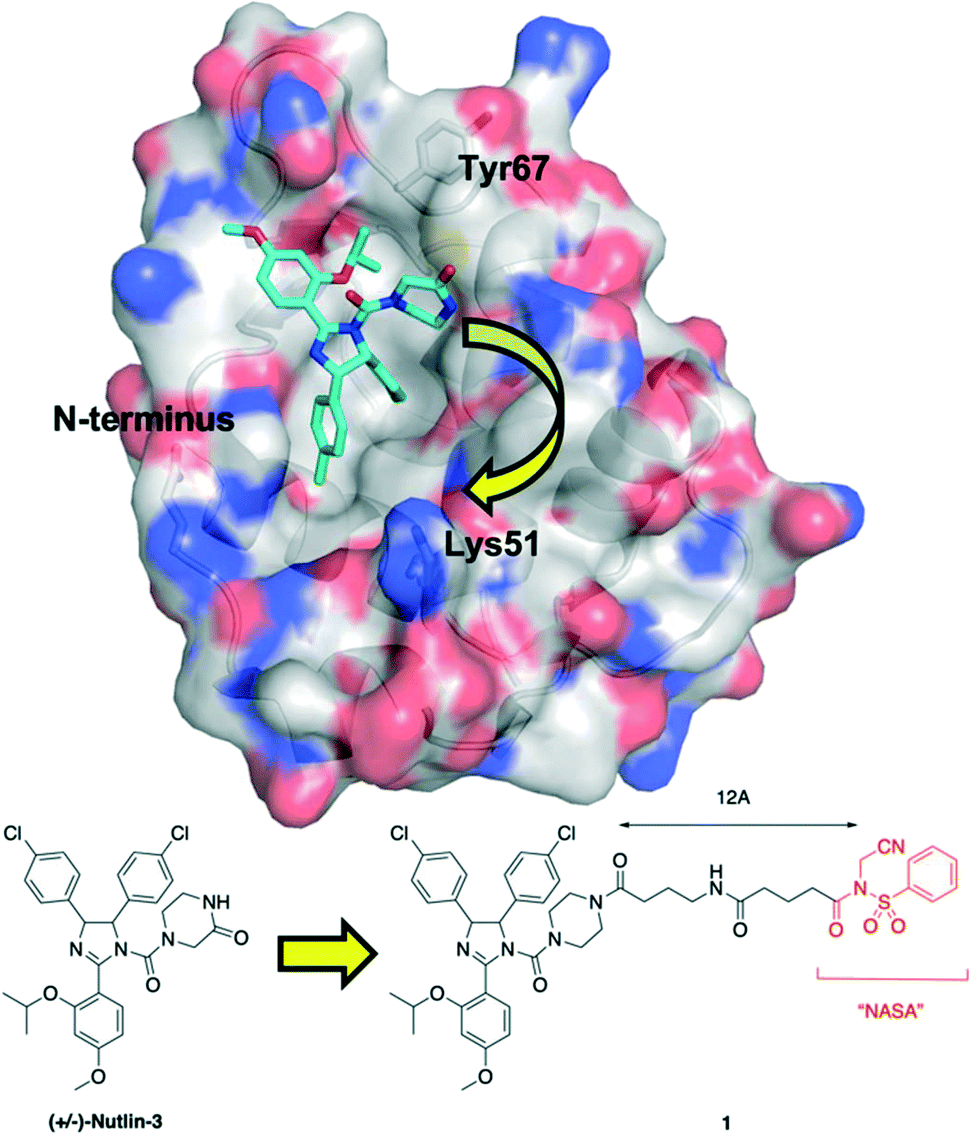 | ||
| Fig. 1 Tethering the NASA warhead onto the HDM2 inhibitor (±)-Nutlin-3. Crystal structure (PDB ID: 4HG7) of HDM2 (surfaced, coloured by atom type) bound to Nutlin-3a (cyan, coloured by atom type). Whilst the ε-amino group of Lys51 was the intended target for the NASA warhead, the chemical reaction instead occurred through acylation of the N-terminal α-amino group and the hydroxyl of Tyr67. | ||
Finally, the cytotoxic effects of covalent inhibitor 1 were compared with the parent, non-covalent inhibitor (±)-nutlin-3 under washout conditions, in which cells were exposed to either compound for 1 h, followed by thorough washing, then incubation for 23 h; this was repeated a total of 5 times. Cell viability, as determined by a WST-8 assay, indicated compound 1 had a greater cytotoxic effect on wild type-p53 MCF7 cells than (±)-nutlin-3, which was even more pronounced with SJSA1 cells. Conversely, neither compound had any impact on the viability of mutant-p53 HeLa and A431 cells. Collectively, these data demonstrate that a TCI-approach to the disruption of the HDM2/p53 PPI led to a sustained activation of the p53 pathway, which has significant implications not only in the field of oncology but, more generally, in the development of drugs to target PPIs, as these findings suggest therapeutic windows may be widened through reduced dosing and frequency of dosing.
BCL-2 family
The BCL-2 family of proteins regulates the intrinsic apoptosis pathway through forming heterodimeric or homo-oligomeric structures in the mitochondrial outer membrane (MOM), and these PPIs determine the cellular fate.35–37 The family is divided into anti-apoptotic proteins, which include MCL-1, BCL-xL and BFL-1, and pro-apoptotic proteins that are further classified as (i) “BH3-only” proteins, which contain the BCL-2 homology 3 killer domain and include BIM, PUMA and NOXA, BAX and BAK, and (ii) the multidomain proteins BAX and BAK. When the multidomain pro-apoptotic proteins engage the BH3-only proteins, a conformational change occurs in the former, which subsequently homo-oligomerize, penetrate the MOM and initiate the apoptosis cascade. Under times of stress, this is the fate of the cell. Conversely, anti-apoptotic proteins seize and “neutralize” BH3-only and multidomain pro-apoptotic proteins, thereby averting mitochondrial apoptosis. Cancer cells capitalize on this mechanism by over-expressing the anti-apoptotic BCL-2 proteins.35–37The pro-apoptotic proteins project key residues from one face of their α-helical BH3 domains into specific sub-pockets p1–p4 that constitute the BH3-binding groove on the surfaces of the anti-apoptotic proteins.38 Since many cancer cells overexpress the anti-apoptotic BCL-2 proteins, around two decades of intense medicinal chemistry research have been dedicated towards the discovery of selective inhibitors of BCL-xL, BCL-2 and, MCL-1 by targeting these sub-pockets, thereby competing with the pro-apoptotic proteins and ultimately reactivating apoptosis.39,40 Indeed, the development of the BCL-2 inhibitor venetoclax5 is the most notable and successful example, gaining FDA approval for the treatment of BCL-2-dependent cancers, which include acute myeloid leukaemia and chronic lymphocytic leukaemia.41 MCL-1 is receiving considerable attention right now as a cancer target in its own right, particularly in haematological cancers, but also as a major contributor to chemoresistance,42–48 while BFL-1 is emerging as a potential target in lymphoma and melanoma, among other cancers.49–51 Whilst there are several clinical trials on-going with MCL-1 inhibitors,45 there are currently no drugs that have advanced to the clinic for MCL-1 or BFL-1.
MCL-1 – reversible covalent inhibition, iminoboronate: lysine
Due to their high nucleophilicity, cysteines are often the focus in TCI with small-molecules.8,9 However, the presence of surface cysteines at PPIs is fairly rare. Akin to Hamachi's goals,34 Akçay and colleagues52 at AstraZeneca wanted to expand the scope of targetable amino acid side chains in TCI and were inspired by the application of ortho-carbonyl aryl boronic acids to covalently bind to several surface lysines of a range of proteins, including lysozyme, cytochrome c and myoglobin, forming an iminoboronate.53Scheme 1 illustrates the general principle of this iminoboronate chemistry in which the resulting imine – formed by reaction of a primary amine with an ortho-carbonyl aryl boronic acid – is stabilized by the establishment of an additional coordinate/dative covalent bond via the donation of the imine nitrogen's lone pair of electrons into the vacant p orbital of the boronic acid, which is only possible with this ortho relationship (Scheme 2). Importantly, this chemistry is reversible, likely mitigating toxicity that may be observed with an analogous irreversible inhibitor.53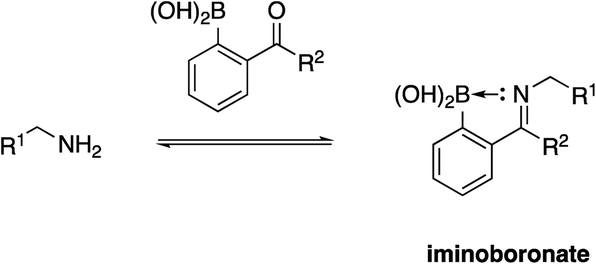 | ||
| Scheme 2 Formation of an iminoboronate with a stabilizing coordinate/dative covalent bond. | ||
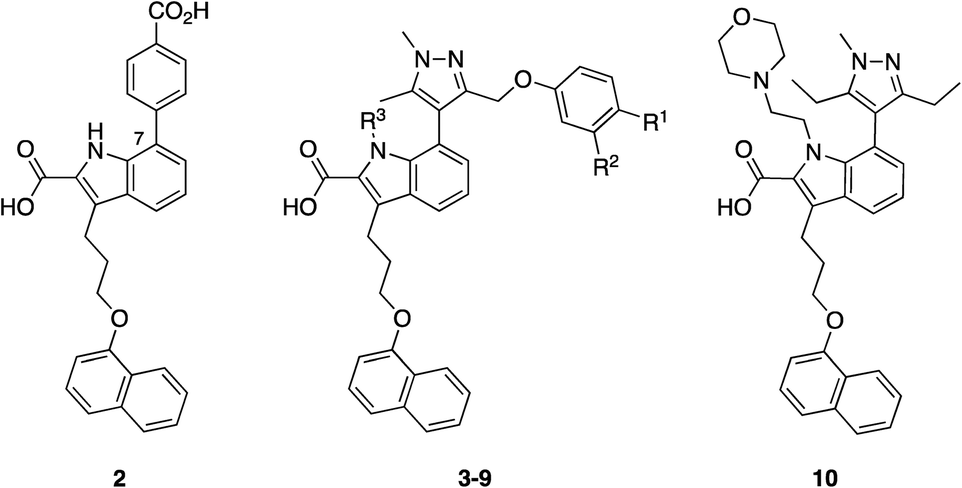
Initially, the AstraZeneca team conducted dilution studies of some control 2-carbonyl aryl boronic acids and a lysine derivative under physiological pH in D2O, and 1H NMR spectroscopy indicated the preference for the iminoboronate at high concentrations, which dissociated upon dilution. More universally, these findings imply that a less-exposed lysine, such as that in a well-defined pocket like an active site, might persist for longer as the covalent adduct with a concomitant greater increase in potency.52 With this data in hand, they then embarked upon modifying an MCL-1 inhibitor to incorporate this reactive ortho-carbonyl aryl boronic acid warhead. Inspection of a crystal structure of 2 with MCL-1 (PDB ID: 3WIX; Fig. 2) suggested that building off the 7 position of the indole core might permit the entrapment of the ε-amino group of Lys234, located near the p1 pocket. Various proposals were evaluated in silico for their abilities to retain binding to the BH3-binding groove as well as placing the warhead within 3 Å of Lys234, and lead compounds included a pyrazole substituent with a phenol linker that also housed the ortho-carbony aryl boronic acid warhead.
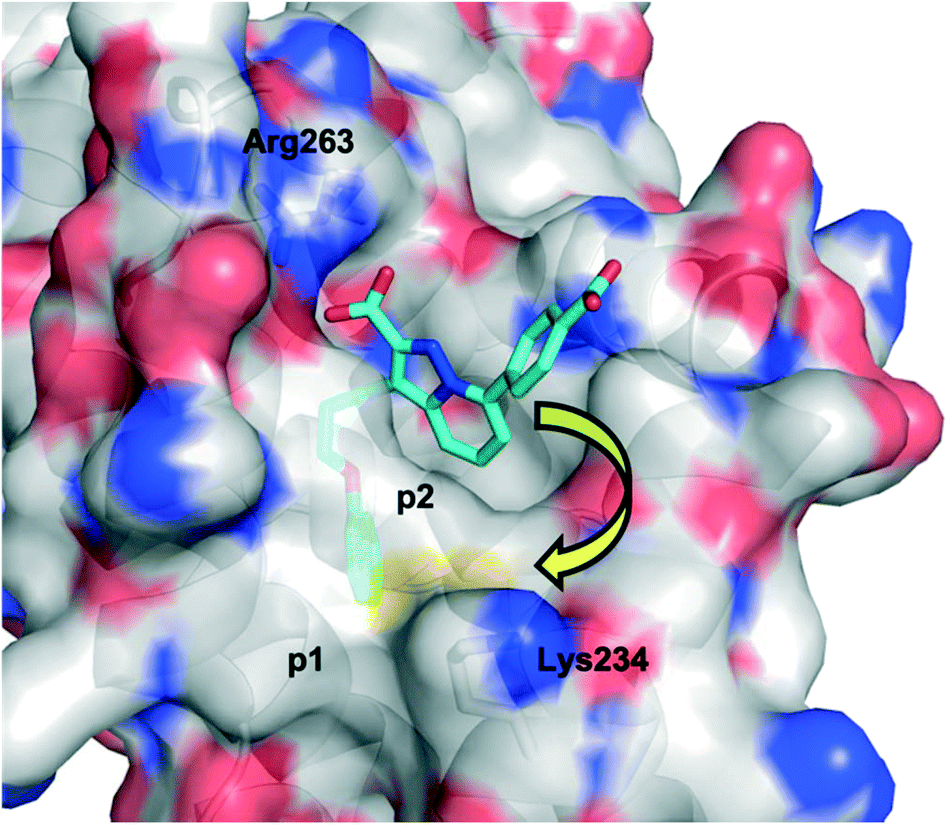 | ||
| Fig. 2 The PPI between MCL-1 (surfaced, coloured by atom type) and small-molecule 2 (cyan, coloured by atom type), highlighting the proximity to the surface residue Lys234 (PDB ID: 3WIX). | ||
Accordingly, a panel of ortho-carbonyl aryl boronic acid-modified derivatives of indole 2 (in which the benzoic acid motif was replaced with a pyrazole) were synthesized and evaluated in a time-resolved fluorescence resonance energy transfer (TR-FRET)-based binding assay (Table 1). Relative to the noncovalent control compound 8, the introduction of one of the constituent components of the warhead, i.e., an aldehyde, ketone or boronic acid, resulted in improved inhibition of MCL-1 most markedly with the aldehyde 5 (IC50 59.5 nM vs. 383 nM). Pleasingly, the combination of either an aldehyde and a boronic acid (3) or a methyl ketone and a boronic acid (4), which would now permit the formation of an iminoboronate, resulted in an even greater improvement in potency to 3.4 and 4.7 nM, respectively. Likewise, a similar trend was observed in an MCL-1-dependent multiple myeloma (MOLP-8) cellular caspase-3/7 activity assay with N-methylated indole 9 demonstrating a six-fold enhancement in caspase activation – hence increased activation of apoptosis – over the more polar analogue 3, which itself was more than 24-fold better than the noncovalent control 8. Significantly, lead compounds 3 and 9 demonstrated on-target activity, proving active in additional MCL-1-dependent cells (L-363, LP-1 and NCI-H929), whilst inactive in the MCL-1-independent cell lines KMS-12-PE and MM.1S. Immunoprecipitation assays with MCL-1 and BAK indicated that compound 3 effected disruption of the MCL-1–BAK PPI. Liquid chromatography-mass spectrometry (LC-MS) studies revealed a new peak that corresponded to the addition of 3 to MCL-1 with the loss of water, consistent with the formation of an imine, hence the iminoboronate. Additionally, the formation of this peak was much faster than the corresponding peak formed by reaction of ketone 4 with MCL-1, reflecting the relative reactivities of aldehydes vs. ketones. The authors presumed they would need to first reduce the imine with sodium cyanoborohydride to enable its detection (as the corresponding amine) given its reversible nature, but the imine proved sufficiently stable to be observed, which may be due to incomplete unfolding of MCL-1 under the conditions of the experiment. Surface plasmon resonance (SPR) demonstrated the reversibility of the binding and the observed time dependency suggested a covalent mechanism of inhibition. Lastly, site-directed mutagenesis of Lys234 to alanine resulted in a significant reduction in the TR-FRET IC50 for compound 4 (reference compound 10 confirmed this mutation did not interfere with MCL-1 inhibition), and MS experiments did not reveal any adduct formation. Taken together, these findings support the conclusion that the ortho-carbonyl aryl boronic acid warhead attacks Lys234 in a reversible, covalent manner.
|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | TR-FRET | MOLP-8 |
| IC50 (nM) | EC50 (μM) | ||||
| 3 | B(OH)2 | CHO | H | 3.4 | 0.46 |
| 4 | B(OH)2 | COCH3 | H | 4.7 | 1.17 |
| 5 | H | CHO | H | 59.5 | >11 |
| 6 | H | COCH3 | H | 237 | 2.70 |
| 7 | B(OH)2 | H | H | 162 | >11 |
| 8 | H | H | H | 383 | >11 |
| 9 | B(OH)2 | CHO | Me | 4.2 | 0.075 |
| 10 | — | — | — | 5.96 | 0.34 |
BFL-1 – irreversible inhibition, acrylamide: cysteine
The Walensky laboratory is one of the forerunners of stapled peptide chemistry, in which non-interacting residues on one face of an α-helix – most typically the i and i + 3/4 or i and i + 7 positions – are synthetically modified then conjugated together, often through a ring closing metathesis reaction between two terminal alkenes.54 This so-called “staple” rigidifies the peptide into an α-helix, and those that are specifically stabilized α-helices of BCL-2 domains have been dubbed SAHBs.55 Huhn and Guerra et al. noticed the apposition of cysteines at the binding interface in the crystal structure of BFL-1 and the BH3 domain of NOXA (PDB ID: 3MQP), one of BFL-1's pro-apoptotic partners: Cys55, at the edge of the canonical groove of BFL-1, and Cys25, towards the N-terminus of NOXA-BH3 (Fig. 3). It was hypothesized that a disulfide bond may be fashionable between the two.56 Indeed, dithiothreitol reduction followed by glutathione disulfide oxidation resulted in a mass shift of BFL-1 when incubated with the NOXA SAHBA (the subscript A indicates their stapled peptides were constrained through the classical “A” position55) as determined by gel electrophoresis. This was confirmed by the reported labelling of BFL-1 with a fluorescein isothiocyanate (FITC)-tagged NOXA SAHBA in a FITC scan. Lastly, selectivity for BFL-1 was provided by a lack of mass shift nor FITC labelling upon incubation of FITC-NOXA SAHBA with the anti-apoptotic sister proteins MCL-1 nor BCL-xL, both of which contain surface cysteines (Cys286 and Cys151, respectively), although neither of these are located in the BH3 binding groove.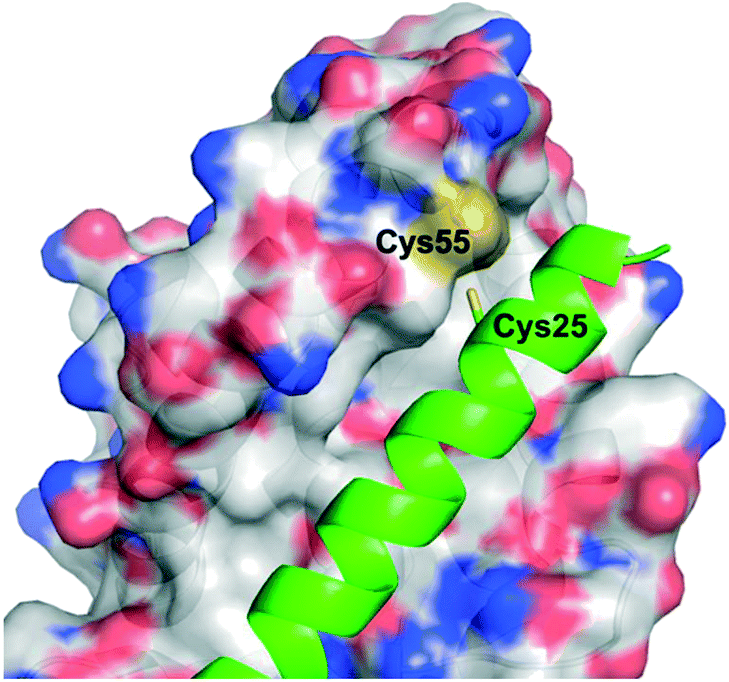 | ||
| Fig. 3 The PPI between BFL-1 (surfaced, coloured by atom type) and the NOXA BH3 peptide (helix, green, coloured by atom type) highlighting the juxtaposition of cysteines on each binding partner (PDB ID: 3MQP). | ||
The reversibility of disulfide bond formation in vivo is a key reason this type of covalent bond formation in late-stage drug discovery is typically avoided, and is more commonly employed in early-stage, fragment-based drug design (see next section).57,58 Thus, the authors shifted their focus to replacing the cysteine thiol of NOXA Cys25 with alternative, acrylamide-based electrophilic warheads, which are more “drug-like”. Careful inspection of a BFL-1/NOXA-BH3 crystal structure revealed that NOXA Leu21 (and Trp147 of BIM) was even closer to BFL-1 Cys55 than was NOXA Cys25. Therefore, the authors made warhead-charged SAHBAs of the MCL-1/BFL-1 dual-selective NOXA BH3 peptide and the pan-active BIM BH3 peptide by replacing Leu21 or Trp147 at the N-terminus of NOXA or BIM, respectively, with a range of Michael acceptor acrylamide derivatives. One of the best warheads was the N-acryloyl-D-nipecotic acid (11, Fig. 4), thus NOXA and BIM SAHBAs carrying this warhead, dubbed NOXA SAHBA-3 and BIM SAHBA-3, respectively, were advanced to further studies. BFL-1 constructs lacking Cys55 did not react with either SAHBAs, nor did BFL-1 constructs containing Cys4 and/or Cys19 (other surface cysteines) indicating selectivity for the cysteine located at the edge of the canonical BH3-binding groove (Cys55). Moreover, as with the NOXA Cys25 SAHBA, incubation of these SAHBAs with MCL-1 and BCL-xL (which, it may be recalled, present surface cysteines but away from the BH3 binding groove) did not result in a chemical reaction, providing further evidence for their selectivity. Pulldown assays in which there was an equimolar mixture of the reactive SAHBA-3 or the unreactive SAHBA and differentially tagged BCL-xL, MCL-1 and BFL-1 also revealed a preference for BFL-1 targeting with the reactive SAHBAs over the unreactive parent SAHBAs. Finally, the reactive BIM SAHBA-3 demonstrated a time-dependent cytotoxic effect in the BFL-1-expressing human melanoma cell line A375P manifested as an increase in apoptosis over time, which is consistent with a covalent mechanism of action. A particularly significant finding in this research was the observation that the installation of an electrophilic warhead into the BIM SAHBA resulted in the transformation of a pan-BCL-2 inhibitor into a selective BFL-1 inhibitor. With co-crystal structures in hand, a similar strategy may be possible with small-molecule pan-BCL-2 inhibitors.
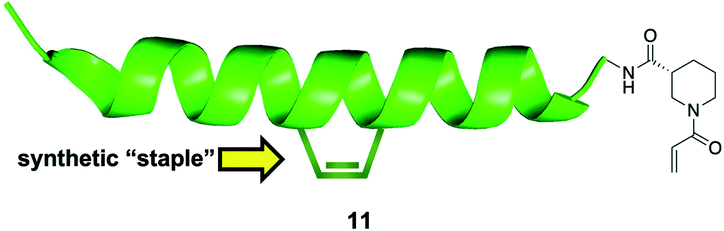 | ||
| Fig. 4 A “stapled peptide” inhibitor of BFL-1 functionalized with an acrylamide-derived D-isonipecotic acid warhead at its N-terminus. | ||
BFL-1 – disulfide tethering: cysteine
Harvey et al. employed a disulfide tethering57,58 screen – also referred to as disulfide trapping – through incubation of a 1600-member, thiol-containing small-molecule library with a construct of BFL-1 lacking the C-terminal α9 helix. In addition, the two cysteines not located in the BH3-binding grove (Cys4 and Cys19) were mutated to serines.59 Quantification of the degree of tethering was then determined by intact MS. This led to the discovery of a subset of 31 compounds, many of which exhibited a (hetero)aromatic ring connected to a thiol via four to six bonds. These hits were then verified by performing a fluorescence polarization competition assay (FPCA) with a fluorescein-labelled BID BH3 peptide and BFL-1. All compounds effectively competed with the BID BH3 ligand, corroborating the intact MS findings, but inhibitor 4E14 (Fig. 5) emerged as the lead compound. Additionally, the selectivity of disulfide bond formation in the BH3-binding groove with Cys55 was demonstrated with a construct of BFL-1 that lacked Cys55, but retained Cys4 and Cys19; no binding was observed in the FPCA assay. Furthermore, no binding was detected between 4E14 and BCL-xL nor MCL-1, confirming the small-molecule's selectivity for BFL-1. This is especially noteworthy because, as already noted in the previous section, BCL-xL and MCL-1 both present surface cysteines, albeit not in the canonical BH3-binding groove. Hydrogen–deuterium exchange mass spectrometry (HXMS) revealed that Cys55 and residues in the p1 and p2 pockets were most protected from H/D-exchange in the presence of 4E14, suggesting that the small-molecule resided there. Although the team were unsuccessful in determining the binding mode of 4E14 experimentally, a solved crystal structure of BFL-1 incubated with 4E14 revealed that the small-molecule (which was not visualized) caused conformational changes in the BH3-binding groove. Taking the crystal structure data together with the HXMS data as well as molecular dynamics simulations, the authors arrived at a model structure for the binding of 4E14 to BFL-1, which is shown in Fig. 6. A mixed disulfide bond is proposed with Cys55, the indole is predicted to bind in the p2 pocket, and the indole NH is engaged in a hydrogen bond with Asp78. Presumably, the authors have moved away from the disulfide lead and introduced more drug-like warheads, such as an acrylamide, as in the previous section, as well as engaged in structure–activity relationship studies towards the optimization of 4E14.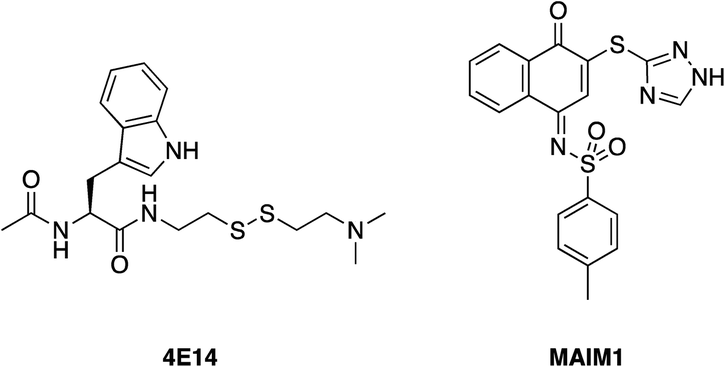 | ||
| Fig. 5 4E14 – an irreversible BFL-1 inhibitor discovered by a disulfide tethering screen; MAIM1 – an allosteric, covalent inhibitor of MCL-1, also discovered by an in vitro screen. Both inhibitors react with surface cysteines. | ||
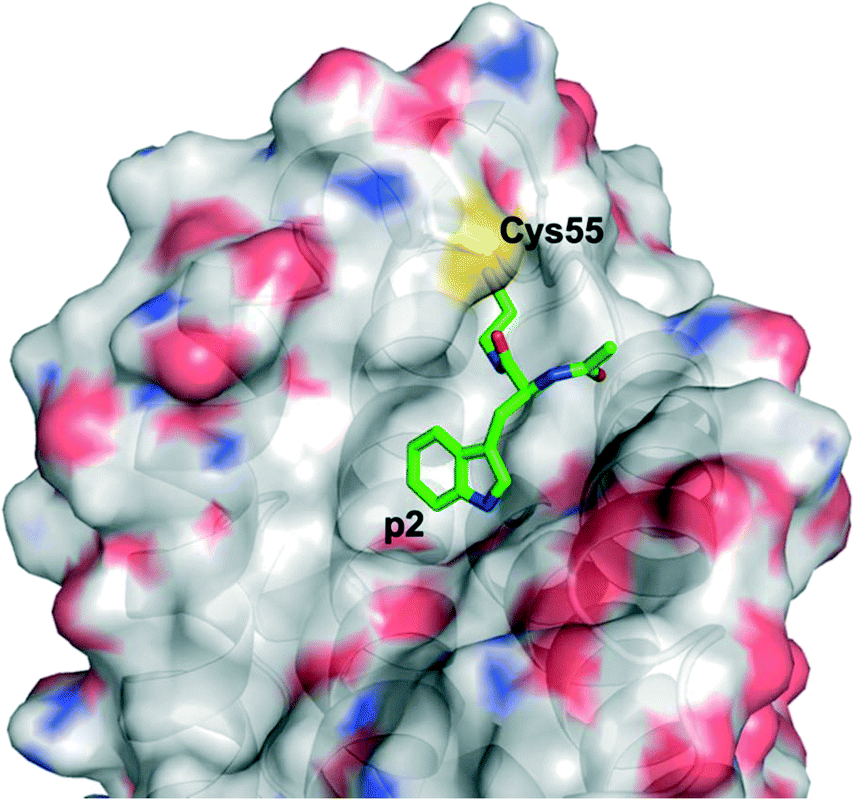 | ||
| Fig. 6 A model for the binding interaction of 4E14 with BFL-1 (PyMOL .pse file provided by Dr. Walensky): protein surfaced and coloured by atom type; 4E14 ligand is green and coloured by atom type. Note the omitted (displaced) 2-(dimethylamino)ethane-1-thiol motif indicating the disulfide exchange with Cys55 has taken place. | ||
MCL-1 – allosteric inhibition, naphthquinone: cysteine
Lee and co-workers established a novel screening platform to discover small-molecule MCL-1 inhibitors, and uncovered several irreversible binders.60 Their assay focused on the competitive displacement of an MCL-1 SAHBA from MCL-1. One of their best hits was MAIM1 (“MCL-1 allosteric inhibitor molecule 1”, Fig. 5), so termed because they would later discover its mechanism of action to be allosteric. MAIM1 disrupted the interaction between FITC-labelled BID BH3 and MCL-1 with an IC50 of 450 nM in a standard FPCA. MAIM1 is a naphthoquinone arylsulfonamide that belongs to the PAINs family of molecules, and can covalently modify cysteines.61 Indeed, dilution experiments suggested that MAIM1's mechanism of action was irreversible inhibition. MS studies revealed covalent modification of Cys286, which, as stated earlier, is not located within the canonical BH3 binding groove, and is in fact found on the opposite face of the protein (Fig. 7). The authors conducted C286S mutagenesis of MCL-1, which essentially abolished the inhibitory effect of MAIM1. Interestingly, MAIM1 was an ineffective disruptor of the FITC BID-BH3–BCL-xL PPI, despite the anti-apoptotic BCL-xL protein also exhibiting a surface cysteine (Cys151). Lastly, alkylation of Cys286 with iodoacetamide did not result in inhibition of the FITC BID-BH3MCL-1 PPI. Taken together, these data confirm the selectivity for Cys286 on MCL-1 over Cys151 on BCL-xL, and also that a significant mass attached to Cys286 – such as covalent alkylation by MAIM1 – is required to effect perturbation of the canonical PPI.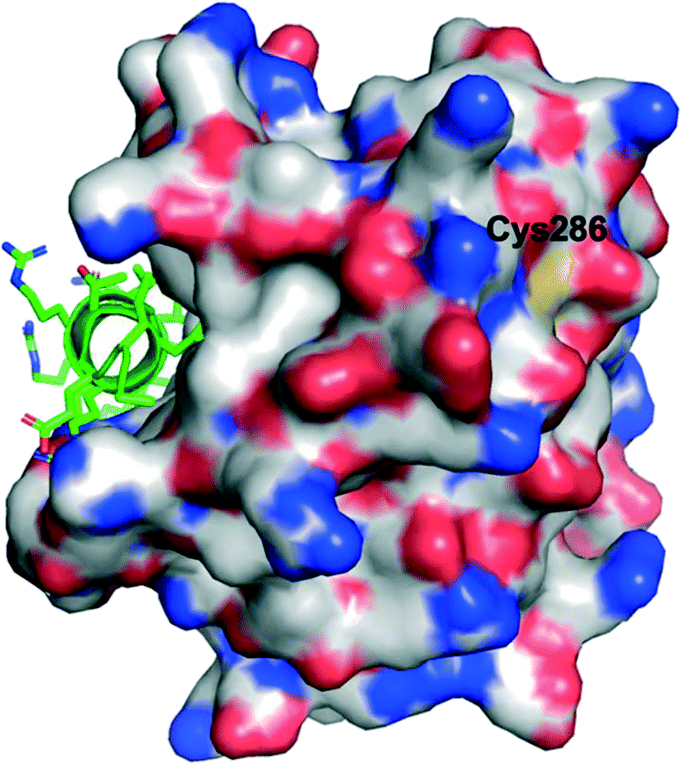 | ||
| Fig. 7 The PPI between MCL-1 (surfaced, coloured by atom type) and an MCL-1 SAHBA that binds in the canonical BH3-binding groove (helix, green, coloured by atom type), highlighting the distal location of Cys286 (PDB ID: 3MV8). | ||
HXMS studies of MCL-1 with BID BH3 peptide revealed that, in the absence of MAIM1, residues lining the BH3-binding groove were protected from deuterium exchange by the presence of BID BH3. Likewise, deuterium exchange of the BID BH3 peptide was heavily abrogated in the presence of MCL-1. These findings are consistent with the shielding effects offered by each protein/peptide by virtue of their established PPI. However, in the presence of MAIM1, the protective effects of deuterium exchange on the BID BH3 peptide were impaired whilst minimal protection of exchange was observed with MCL-1, but this was only in the vicinity of Cys286 (distant from the BH3 binding groove), providing further support that MAIM1 is an allosteric inhibitor of MCL-1. Finally, a C286W mutant emulated the allosteric inhibition manifested by MAIM1.
In summary, the authors described that, in addition to uncovering a new mode of inhibition of this oncoprotein, the covalent allosteric effect results in compromising – rather than completely abolishing – the ability of MCL-1 to recognize its pro-apoptotic counterparts, i.e. partial inhibition as opposed to complete inhibition of MCL-1. Indeed, this may result in the most viable path forward towards the development of safe and effective MCL-1 inhibitors given that MCL-1 deletion in mouse models caused cardiomyopathy, mitochondrial dysfunction, loss of hematopoietic stem cells, and also resulted in embryonic lethality.62–65 Furthermore, several clinical trials of small-molecule MCL-1 inhibitors that target the canonical BH3-binding groove have either been terminated or suspended due to safety concerns, including at least one fatality (https://www.fiercebiotech.com/biotech/amid-amgen-s-similar-struggles-astrazeneca-slams-brakes-mcl-1-blood-cancer-drug).
Conclusions
PPIs are challenging drug targets. Whilst there have been some successes, such as with the BCL-2 inhibitor venetoclax that is now in the clinic, achieving a balance between potency, selectivity and “drug-like” properties continues to hamper PPI drug development. Capitalizing on the recent resurgence of covalent inhibitors in drug discovery, TCI has now been extended to the development of drugs to inhibit PPIs. In fact, we consider the important examples discussed in this review may lay the foundation for a paradigm shift in the inhibition of PPIs. The reinforcing effect on the non-covalent interactions and the prolonged residence time both afforded by covalent inhibition may have significant impact on molecular size with smaller, more “drug-like” molecules exhibiting comparable, or better, potency to larger, “non-traditional” architectures. Moreover, TCI may ultimately result in reduced dosing for the required therapeutic effect, thereby addressing drugs with narrow therapeutic windows. Further work needs to be conducted in this area, but we believe the rebooting of covalent chemistry in drug discovery is leading to a regeneration of medicinal chemistry and will directly tackle the high drug attrition rate, especially in the context of the more challenging PPIs, not only in terms of their disruption but also their stabilization.66Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Maryland School of Pharmacy for support of our work in this area, and we acknowledge the T32 training grant (NIH/NIGMS T32 GM066706) to AMC and CCG.Notes and references
- S. Fletcher and A. D. Hamilton, Curr. Opin. Chem. Biol., 2005, 9, 632–638 CrossRef CAS PubMed.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS PubMed.
- L. Mabonga and A. P. Kappo, Biophys. Rev., 2019, 11, 559–581 CrossRef CAS PubMed.
- Y. H. Jeon, J. Y. Lee and S. Kim, Bioorg. Med. Chem., 2012, 20, 1893–1901 CrossRef CAS PubMed.
- A. J. Souers, et al. , Nat. Med., 2013, 19, 202–208 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- L. Z. Benet, C. M. Hosey, O. Ursu and T. I. Oprea, Adv. Drug Delivery Rev., 2016, 101, 89–98 CrossRef CAS PubMed.
- J. Singh, R. C. Petter, T. A. Baillie and A. Whitty, Nat. Rev. Drug Discovery, 2011, 10, 307–317 CrossRef CAS PubMed.
- T. A. Baillie, Angew. Chem., Int. Ed., 2016, 55, 13408–13421 CrossRef CAS PubMed.
- M. Gehringer and S. A. Laufer, J. Med. Chem., 2019, 62, 5673–5724 CrossRef CAS PubMed.
- R. Lonsdale and R. A. Ward, Chem. Soc. Rev., 2018, 47, 3816–3830 RSC.
- T. Zhang, J. M. Hatcher, M. Teng, N. S. Gray and M. Kostic, Cell Chem. Biol., 2019, 26, 1486–1500 CrossRef CAS PubMed.
- L. Gambini, et al. , J. Med. Chem., 2019, 62, 5616–5627 CrossRef CAS PubMed.
- M. E. Lanning, et al. , Org. Biomol. Chem., 2015, 13, 8642–8646 RSC.
- X. Cao, et al. , Mol. Cancer, 2013, 12, 42 CrossRef CAS PubMed.
- L. Chen, et al. , Org. Biomol. Chem., 2016, 14, 5505–5510 RSC.
- I. L. Conlon, et al. , ChemMedChem, 2020, 15, 1691–1698 CrossRef CAS PubMed.
- I. L. Conlon, et al. , Bioorg. Med. Chem. Lett., 2018, 28, 1949–1953 CrossRef CAS PubMed.
- B. Drennen, et al. , ChemMedChem, 2016, 11, 827–833 CrossRef CAS PubMed.
- E. Whiting, et al. , Bioorg. Med. Chem. Lett., 2018, 28, 523–528 CrossRef CAS PubMed.
- K.-Y. Jung, et al. , Org. Lett., 2013, 15, 3234–3237 CrossRef CAS PubMed.
- M. E. Lanning, et al. , Eur. J. Med. Chem., 2016, 113, 273–292 CrossRef CAS PubMed.
- S. L. Harris and A. J. Levine, Oncogene, 2005, 24, 2899–2908 CrossRef CAS PubMed.
- P. Hainaut and M. Hollstein, Adv. Cancer Res., 2000, 77, 81–137 CrossRef CAS PubMed.
- G. L. Bond, W. Hu and A. J. Levine, Curr. Cancer Drug Targets, 2005, 5, 3–8 CrossRef CAS PubMed.
- M. H. Kubbutat, S. N. Jones and K. H. Vousden, Nature, 1997, 387, 299–303 CrossRef CAS PubMed.
- V. Tisato, R. Voltan, A. Gonelli, P. Secchiero and G. Zauli, J. Hematol. Oncol., 2017, 10, 133 CrossRef PubMed.
- M. Konopleva, et al. , Leukemia, 2020, 34, 2858–2874 CrossRef PubMed.
- P. H. Kussie, et al. , Science, 1996, 274, 948–995 CrossRef CAS PubMed.
- L. T. Vassilev, et al. , Science, 2004, 303, 844–848 CrossRef CAS PubMed.
- K. H. Khoo, K. K. Hoe, C. S. Verma and D. P. Lane, Nat. Rev. Drug Discovery, 2014, 13, 217–236 CrossRef CAS PubMed.
- T. Tamura, et al. , Nat. Commun., 2018, 9, 1870 CrossRef PubMed.
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- T. Ueda, et al. , J. Am. Chem. Soc., 2021, 143, 4766–4774 CrossRef CAS PubMed.
- P. E. Czabotar, G. Lessene, A. Strasser and J. M. Adams, Nat. Rev. Mol. Cell Biol., 2014, 15(1), 1 CrossRef PubMed.
- J. Kale, E. J. Osterlund and D. W. Andrews, Cell Death Differ., 2018, 25, 65–80 CrossRef CAS PubMed.
- J. M. Adams and S. Cory, Cell Death Differ., 2018, 25, 27–36 CrossRef CAS PubMed.
- M. Sattler, et al. , Science, 1997, 275, 983–986 CrossRef CAS PubMed.
- J. L. Yap, L. Chen, M. E. Lanning and S. Fletcher, J. Med. Chem., 2017, 60, 821–838 CrossRef CAS PubMed.
- S. T. Diepstraten, M. A. Anderson, P. E. Czabotar, G. Lessene, A. Strasser and G. L. Kelly, Nat. Rev. Cancer, 2022, 22, 45–64 CrossRef CAS PubMed.
- D. A. Pollyea, et al. , Am. J. Hematol., 2021, 96, 208–217 CrossRef CAS PubMed.
- S. Fletcher, MCL-1 inhibitors - where are we now (2019)?, Expert Opin. Ther. Pat., 2019, 29, 909–919 CrossRef CAS PubMed.
- A. W. Hird and A. E. Tron, Pharmacol. Ther., 2019, 198, 59–67 CrossRef CAS PubMed.
- A. Negi and P. V. Murphy, Eur. J. Med. Chem., 2021, 210, 113038 CrossRef CAS PubMed.
- H. Wang, M. Guo, H. Wei and Y. Chen, J. Hematol. Oncol., 2021, 14, 67 CrossRef CAS PubMed.
- A. Bolomsky, et al. , J. Hematol. Oncol., 2020, 13, 173 CrossRef PubMed.
- X. Wu, Q. Luo and Z. Liu, Cell Death Dis., 2020, 11(7), 7 Search PubMed.
- K. H. Lin, et al. , Sci. Rep., 2016, 6, 27696 CrossRef CAS PubMed.
- S. Boiko, et al. , Blood, 2021, 137, 2947–2957 CrossRef CAS PubMed.
- S. Boiko, et al. , Blood, 2021, 137, 2947–2957 CrossRef CAS PubMed.
- X. Li, J. Dou, Q. You and Z. Jiang, Eur. J. Med. Chem., 2021, 220, 113539 CrossRef CAS PubMed.
- G. Akçay, et al. , Nat. Chem. Biol., 2016, 12, 931–936 CrossRef PubMed.
- C.-U. Lee and T. N. Grossmann, Angew. Chem., Int. Ed., 2012, 51, 8699–8700 CrossRef CAS PubMed.
- R. A. Hillman, J. W. Nadraws and M. A. Bertucci, Curr. Top. Med. Chem., 2018, 18, 611–624 CrossRef CAS PubMed.
- L. D. Walensky, et al. , Science, 2004, 305, 1466–1470 CrossRef CAS PubMed.
- A. J. Huhn, R. M. Guerra, E. P. Harvey, G. H. Bird and L. D. Walensky, Cell Chem. Biol., 2016, 23, 1123–1134 CrossRef CAS PubMed.
- D. A. Erlanson, et al. , Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9367–9372 CrossRef CAS PubMed.
- S. G. Kathman and A. V. Statsyuk, Med. Chem. Commun., 2016, 7, 576–585 RSC.
- E. P. Harvey, et al. , Cell Chem. Biol., 2020, 27, 647–656 CrossRef CAS PubMed.
- S. Lee, et al. , Nat. Struct. Mol. Biol., 2016, 23, 600–607 CrossRef CAS PubMed.
- J. B. Baell and J. W. M. Nissink, ACS Chem. Biol., 2018, 13, 36–44 CrossRef CAS PubMed.
- X. Wang, et al. , Genes Dev., 2013, 27, 1351–1364 CrossRef CAS PubMed.
- B. Vick, et al. , Hepatology, 2009, 49, 627–636 CrossRef CAS PubMed.
- J. T. Opferman, et al. , Science, 2005, 307, 1101–1104 CrossRef CAS PubMed.
- J. L. Rinkenberger, S. Horning, B. Klocke, K. Roth and S. J. Korsmeyer, Genes Dev., 2000, 14, 23–27 CrossRef CAS PubMed.
- P. J. Cossar, J. Am. Chem. Soc., 2021, 143, 8454–8464 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |