
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances and perspectives of actinide chemistry from ex situ high pressure and high temperature chemical studies
Gabriel L.
Murphy
 *,
Philip
Kegler
and
Evgeny V.
Alekseev
*
*,
Philip
Kegler
and
Evgeny V.
Alekseev
*
Institute of Energy and Climate Research, Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: g.murphy@fz-juelich.de; e.alekseev@fz-juelich.de
First published on 20th April 2022
Abstract
High pressure high temperature (HP/HT) studies of actinide compounds allow the chemistry and bonding of among the most exotic elements in the periodic table to be examined under the conditions often only found in the severest environments of nature. Peering into this realm of physical extremity, chemists have extracted detailed knowledge of the fundamental chemistry of actinide elements and how they contribute to bonding, structure formation and intricate properties in compounds under such conditions. The last decade has resulted in some of the most significant contributions to actinide chemical science and this holds true for ex situ chemical studies of actinides resulting from HP/HT conditions of over 1 GPa and elevated temperature. Often conducted in tandem with ab initio calculations, HP/HT studies of actinides have further helped guide and develop theoretical modelling approaches and uncovered associated difficulties. Accordingly, this perspective article is devoted to reviewing the latest advancements made in actinide HP/HT ex situ chemical studies over the last decade, the state-of-the-art, challenges and discussing potential future directions of the science. The discussion is given with emphasis on thorium and uranium compounds due to the prevalence of their investigation but also highlights some of the latest advancements in high pressure chemical studies of transuranium compounds. The perspective also describes technical aspects involved in HP/HT investigation of actinide compounds.
Introduction
Since the dawn of the nuclear age, the chemistry of actinide elements and the compounds they form has captivated the attention of society, particularly regarding the deliberate or unintentional applications of their nuclear properties. Transcending this, considerable efforts have been made by chemists to understand the nature of chemical bonding within actinide materials and the associated properties that are derived therein. Pertinent to the chemistry of the actinides is the pronounced relativistic effects which their core electrons present and the occurrence of considerable spin–orbit coupling which vary significantly between individual elements. Although the theoretical framework for relativistic and heavy electron systems has been available since the early days of quantum theory,1 accounting for these effects in ab initio wave-function based calculations has only recently become somewhat accessible.2 However, as contemporary studies have highlighted3 limitations still remain in executing correct standalone calculations without the aid of experimental data to benchmark obtained results.4–9 Consequently, to correctly understand chemical bonding in actinide compounds requires controlled experimental approaches that can both directly probe and examine their limitations in a systematic form. Ex situ HP/HT experimental methods have subsequently formed a powerful means to achieve this and advance fundamental chemical insight. Typically, published studies of actinides under high pressure and temperature are relatively few compared to those of ambient pressure elevated temperature studies, a consequence of the radiological hazard combined with operating costs and difficulties associated with HP/HT apparatuses. Nevertheless, those that are available have often divulged significant insight into the fundamental chemistry of actinides, pertinent in the context of basic scientific endeavour and as applied functional materials for both nuclear and non-nuclear applications.In the early days of actinide high pressure science, studies were initially conducted using ex situ belt-type HP/HT apparatuses, which although can be considered relatively primitive by today's standards, hinted at the flexibility and peculiarity of actinide bonding, and could maintain conditions above 4 GPa and over 1000 °C. For instance, in 1966 Young10 and shortly after in 1967 Hoekstra and Marshall11 using such apparatuses with post powder diffraction measurements concluded the formation of NiUO4 polymorphs could only be achieved through pressure induced overlap of atomic orbitals resulting in a distorted structure of which was later confirmed by single crystal X-ray diffraction methods.3,12
The advent of the diamond anvil cell (DAC) by Van Valkenburg and co-workers13 heralded a new era in high-pressure science, of which the actinides were not excluded. It meant smaller sample amounts could be examined in situ at greater pressures reducing radiological risk whilst dramatically increasing the operating range compared to that offered by piston cylinder or other multi-anvil devices. Accordingly, it resulted in a decline of studies using ex situ HP/HT apparatuses in place of DAC related in situ studies. The latter studies in the past were generally focused on the metallic form of actinides but also the binary oxides, due to their applications and relevance to the nuclear fuel cycle. Such studies led to the acquisition of the high-pressure phases of UO2 and ThO2.14 The discovery of heavy fermion systems in the late 1970s, invigorated fundamental interest by physicists of placing actinides under pressure in exploring the superconductive phases and condensed electron systems of the notable compounds URu2Si2, and UGe2 among others.15,16
The turn of the century coincided with new trends in actinide chemical investigation where the utilisation of precise high-resolution measurements of compound arising from a multitude of conditions such as high temperature, hydrothermal, solvothermal and supercritical water conditions.17–21 This resulted in one of greatest volumes of published literature for actinides of any period. These inorganic chemistry studies were conducted with the purpose of building a fundamental understanding of actinide chemistry behind the nuclear fuel cycle and particularly nuclear waste materials. However, a notable absentee of the conditions examined in this period was that of pressures above 1 GPa and under elevated temperatures. This limitation of conditions examined within previous studies, and by extension associated knowledge of actinide chemistry therein, is well defined observed within a p–T diagram as is presented in Fig. 1. From the figure, it is apparent that the breadth of field for HP/HT conditions that actinides can be examined greatly exceeds that of those produced by high temperature, hydrothermal, solvothermal or supercritical water conditions. Although high temperature and hydrothermal methods can involve some elevated pressure, such as using sealed quartz tubes, it is dwarfed by what HP/HT techniques can access. Nevertheless, this realm of heightened pressure and temperature is greatly understudied.
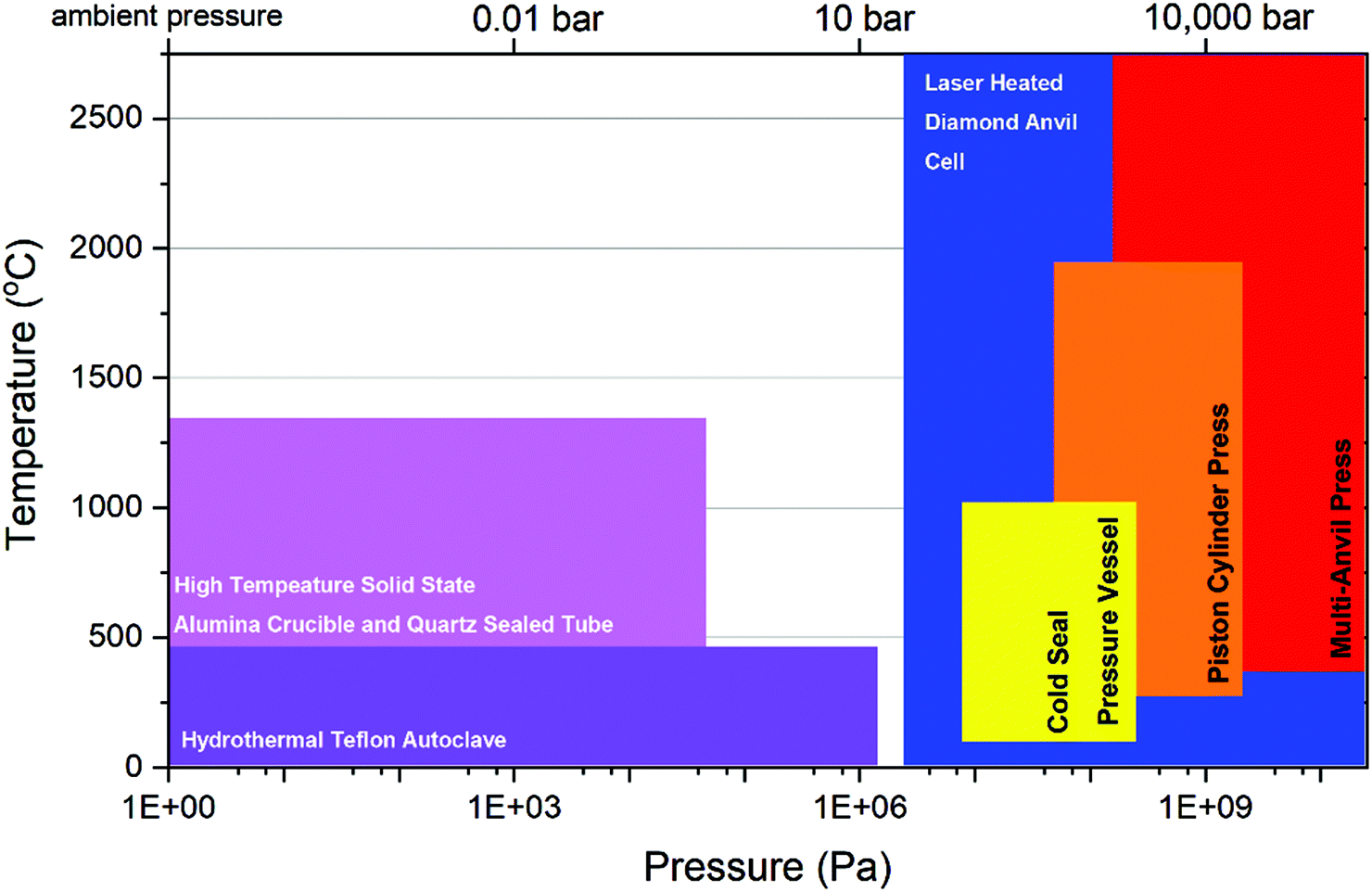 | ||
| Fig. 1 Typical experimental methods used for examining the chemistry of actinides with respect to temperatures and pressures they can access. | ||
Outside of the laboratory, actinides can be exposed to HP/HT conditions in a variety of real and engineering systems, in addition to nature. Terrestrially in nature, uranium and thorium are most abundant of the actinides (22 and 80 ppb for bulk silicate Earth) and are typically found within the Earth's crust (2.7 and 9.6 ppm) which extends to a depth of 70 km corresponding to pressures and temperatures of over 600 °C and >1–2 GPa.22,23 Actinide occurrence within the Earth extends even further to within the Earth's mantle at depths of 2890 km where pressures and temperatures of over 135–140 GPa and 3650 °C can be encountered.23 It is here actinides, particularly uranium and thorium, provide a key proportion of the radiogenic input into the Earth’ internal heat budget.24 Anthropogenically, actinides occur within and around applications of the nuclear fuel cycle and weapons technology. Within a light water reactor (LRW) the primary coolant circuit typically operates at temperatures of 315 °C with pressures around 10 MPa, whereas in future designs such as the Supercritical Water Reactor (SCWR), as part of GEN IV reactors, considers approximate pressures and temperatures of 25 MPa and 370 °C.25,26 Within the centre of LWR UO2 fuels, HP/HT conditions can be more realised as the centreline fuel pellet temperature is held typically below 1400 °C27 and it has been estimated the internal pressures generated during He bubble formation can reach GPa level.28 Within spent nuclear fuel (SNF) near-surface geological repositories, such as the planned Swedish Forsmark SNF repository, maximum pressures and temperatures of a few MPa and around 100 °C of these near surface facilities are predicted.29 In deep borehole designs (DBD), which have been examined due favourable safety aspects, account for pressures of over 150 MPa and significantly higher temperatures than that of near surface facility types.30
As a waste stream from fuel enrichment, depleted uranium (DU) has found considerable recycled usage in military munitions due its high density and pyrophoricity. Depending on firing velocity, projectile design and target composition pressures and temperatures of over several MPa and above 600 °C can be reached in which oxidation of the DU metal rapidly occurs resulting in binary and ternary oxide formation such as U3O8, UO3, FeUO4 and Fe2UO6 from primary impact.31,32 The synthesis of FeUO4 is known to require elevated pressure. During the detonation of a nuclear weapon, depending on the design, efficiency and assembly of the warhead, extreme pressures and temperatures can be encountered. For instance, temperatures and pressures of over 7700 °C and above 8 GPa are suspected to have occurred during the Trinity test detonation of the ‘gadget’ in 1945.33 From these extreme conditions, the peculiar and infamous mineral phase trinitite with some An-content was recovered, which formed through the combination of these extreme conditions with the nuclear debris and surrounding sand.34 Although this description of where actinides can potentially encounter HP/HT conditions only scratches the surface of scenarios, it is evident w.r.t. Fig. 1, minimum hydrothermal and better yet, HP/HT pressure devices are necessary to appropriately uncover and explore the chemical properties that can occur in these often extreme places.
The Fukushima Daiichi nuclear reactor incident of 2011 led to the public perception of nuclear energy sinking to one of its lowest points. To ameliorate this, significant efforts were made by industry and governments to ensure the safety of reactor and nuclear systems, whilst behind this, actinide researchers began focusing on understanding the behaviour of actinides when exposed to extreme environments, for instance what is potentially encountered in an accident scenario. This culminated in the re-emergence of HP/HT experimental work, particularly of ex situ method, on actinides systems towards understanding their chemistry when placed in extreme scenarios and understanding changes to their bonding and properties therein. Subsequently, the previous decade afforded some of most significant output of studies relating to ex situ HP/HT investigation of actinides, particularly the early members, Th and U. These studies have shed significant light on their fundamental chemistry as compounds but also on the nature of their bonding. Considering this and that there are no current review or perspective articles available to this topic, the present perspective article is devoted to discussing the advances in actinide chemical science through HP/HT ex situ studies that use pressures greater than 1 GPa and under elevated temperatures over the last decade. Focus will be given primarily to the early actinides, Th and U, due to the significant steps made in their research, particularly on the future direction of HP/HT ex situ science, but discussion also on recent advances on the later actinides. Note unless specially stated otherwise, e.g. in situ DAC related studies, ex situ HP/HT studies will be often referred to as HP/HT studies throughout the perspective article.
High pressure/high temperature ex situ technical aspects
Placing actinides under HP/HT conditions in a laboratory setting is not a trivial task and despite many apparatuses available globally, few are licensed to run actinides due the radiological risk. Of the HP/HT actinide synthesis studies produced over the previous decade, the Voggenreiter LP 1000-540/50, a combined piston cylinder/multi anvil press (Fig. 2) located at the Institute of Reactor Safety and Nuclear Waste Management (IEK-6) Forschungszentrum Jülich Germany, has produced the majority of those available. | ||
| Fig. 2 The Voggenreiter LP 1000-540/50 HP/HT apparatus present in the laboratories of the Institute of Reactor Safety and Nuclear Waste Management (IEK-6), Forschungszentrum Jülich, Germany. | ||
The Voggenreiter LP 1000-540/50 uses an end loaded piston cylinder module allowing pressures up to 4.5 GPa and temperatures up to 1800 °C, which can be easily replaced by a Walker-type multi anvil module allowing conditions up to 24 GPa and 2300 °C.35 The combination of classical piston cylinder and multi anvil techniques with today's fully computerised temperature and pressure control,36 allow precise experimental conditions to be maintained for the synthesis of comparably large quantities (depending on experimental pressure of up to hundreds of milligrams) of new HP/HT materials, allowing for the ex situ measurements of the products in standard in house analytical devices like powder and single crystal diffraction. The complex assemblies for both modules, which are designed for the generation of hydrothermal pressure conditions, are internally heated (graphite or LaCrO3 heater) and temperature control is conducted using an in situ thermocouple. A comparable setup was used by Huppertz, Hinteregger and co-workers37–40 for their high-pressure research on 3d and 4f elements. Hydrothermal conditions (p < 0.25 GPa, t < 850 °C) with sample quantities of up to 1 gram can be achieved using a cold seal vessel41 (here water is used as pressure media in an externally heated pressure vessel, often made out of inconel®). The sample itself is introduced into a capsule that is made from a noble metal (mostly gold) which is placed inside the pressure vessel, this is then closed using a water cooled, cone shaped seal. This method was used in synthesis of a uranyl aluminoborate complex.41
Thorium HP/HT studies
Interest in thorium chemistry has typically been overshadowed by that of its more societally relevant sister, uranium. The further absence of 5f electrons and preference to remain tetravalent typically leads to a perception that thorium is less exotic and more mundane chemically than uranium. However as contemporary investigations have demonstrated, this prosaic notion is incorrect. The spherical nature of the Th(IV) cation allows it adopt to a broad range of coordination environments (6–15) allowing the formation of higher dimensional structures (at greater degree than that of uranium as example) which possess a range of chemical properties leading to pertinent applications in areas such as ion-exchange and catalysis.42–44 Traditional studies of thorium compounds with high pressure or HP/HT techniques have typically focused on its binary oxide form in addition to hydrides and carbides.45–48 The latter two examples were examined particularly in the context of potential superconducting materials. Many contemporary studies relating to thorium under HP/HT conditions have largely shifted focused towards utilising its flexible coordination chemistry to examine novel structure formation with a variety of inorganic ligand donors such as borate, tungstate and molybdate among others. Such works have added significant insight into the fundamental chemistry of thorium and its behaviour under HP/HT conditions. The following sections will briefly review recent success in the HP/HT studies of thorium compounds over the previous decade.Thorium molybdates
A pertinent example that illustrates the structural flexibility of thorium and how it can be manipulated through HP/HT conditions is exemplified by ThMo2O8.49 At ambient pressure, using specific high temperature conditions ThMo2O8 can adopt three different structural types, monoclinic (SG P21/c), orthorhombic (SG Pbca) and hexagonal (SG P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ).49 When exposed to conditions of over 3.5 GPa and 1000 °C an orthorhombic variant (SG Pna21) can be generated culminating in a reduction of molar volume by 20% compared to that of the ambient pressure monoclinic variant. Fig. 3 compares the general structures of the monoclinic and HP/HT orthorhombic variants.
).49 When exposed to conditions of over 3.5 GPa and 1000 °C an orthorhombic variant (SG Pna21) can be generated culminating in a reduction of molar volume by 20% compared to that of the ambient pressure monoclinic variant. Fig. 3 compares the general structures of the monoclinic and HP/HT orthorhombic variants.
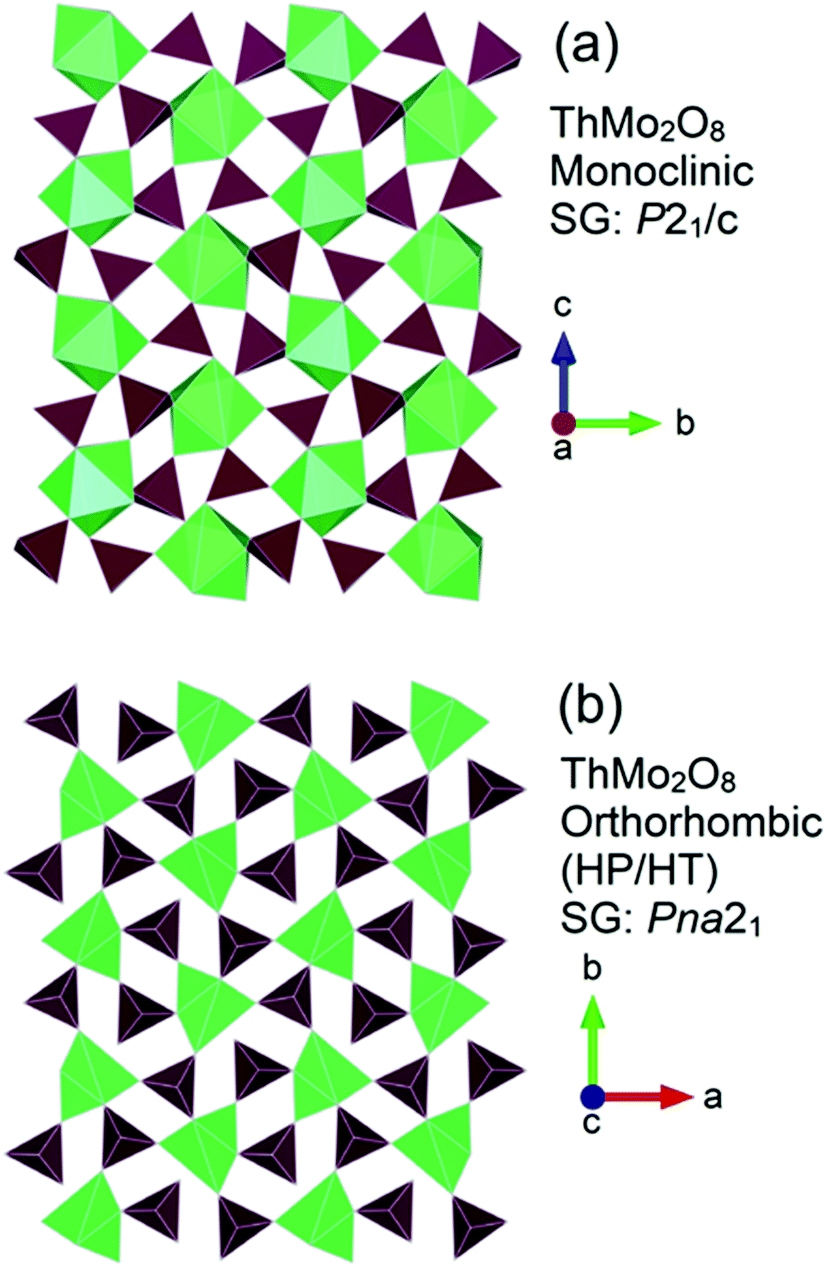 | ||
| Fig. 3 General structure representations of the ThMo2O8 polymorphs of (a) the ambient pressure high temperature synthesised monoclinic (SG P21/c) and (b) the HP/HT orthorhombic (SG Pna21). Note Th and Mo polyhedra are represented by green and maroon polygons respectively. Figure adapted from ref. 49. | ||
This significant reduction of volume in ThMo2O8 is attributed to the extreme flexibility of the Th and Mo polyhedra. At ambient pressure, in the monoclinic form, like polyhedra are found approximately 6 Å apart in a linked geometry of Th-centres connected via MoO42− units in an approximately linear arrangement of Th and Mo cations (Fig. 4). Upon transformation to the HP/HT variant, the linear motif is lost. Instead, Th and Mo are now independently aligned and the distance between them has been reduced to 4 Å (Fig. 4).
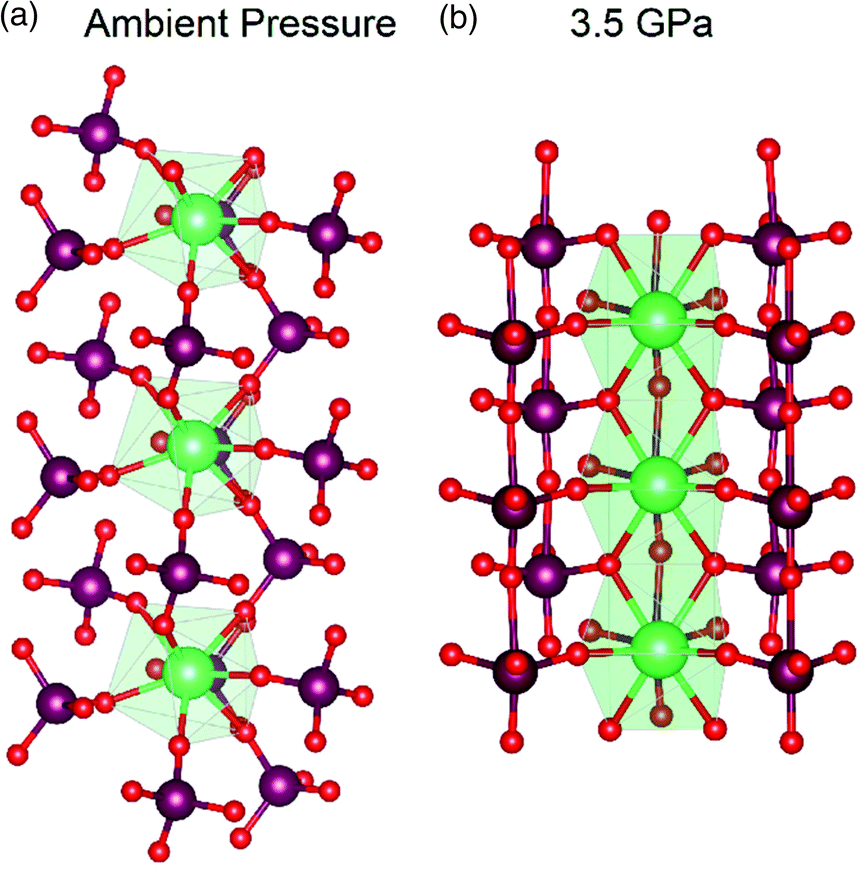 | ||
| Fig. 4 Comparison of the coordination motifs between the Th and Mo polyhedra at (a) ambient pressure synthesised monoclinic variant and (b) HP/HT orthorhombic variant of ThMo2O8. The figure illustrates the reduced distance between Th polyhedra between the ambient and higher-pressure variants. Note Th polyhedra, Mo and O atoms are represented by green polygons, maroon and red spheres respectively. Figure adapted from ref. 49. | ||
Between the three ambient pressure polymorphs of ThMo2O8 monoclinic, orthorhombic and hexagonal in addition to the HP/HT orthorhombic, the Th cation respectively adopts oxygen coordination environments of ThO8, ThO8, ThO6/ThO6/ThO9 and ThO11. When contrasted against the broader family AB2O8 structures (A = Zr, Hf and Th, B = Mo, W), the non-actinide equivalents have less structural diversity compared to ThMo2O8, only reported so far to occur with cubic structure types. This lack of diversity can be traced to the largely restrictive coordination environments of the Zr and Hf cations which typically can only adopt coordination environments of 6–8, Th can take on coordination environments from 6–15.
Thorium tellurates/tellurites
The oxyanions of tellurium, tellurate, Te(IV), and tellurite, Te(VI), have been utilised and explored extensively in inorganic synthesis due to their diverse chemical behaviour and that they can exist in a variety of geometries and coordination environments, TeOx (x = 3–6). The further presence of lone-pair second-order Jahn–Teller distorting electrons on the Te(IV) cation leads to non-centrosymmetric structure (NSC) formation allowing desirable materials with second harmonic generation (SHG) and non-linear optical (NLO) properties to be obtained. Comparatively, tellurites can exist in a greater range of coordination geometries than tellurates where, as building units, their geometric flexibility leads to a variety structure being generated from 0D clusters, 1D chains, 2D layers to 3D frameworks with desirable functional properties. Utilisation of tellurate and tellurite chemistry with uranium and thorium has been somewhat focused on the former actinide with a variety of studies under conditions such as high temperature, hydrothermal, solvothermal, HP/HT and even many examples seen in minerals.20,50 In contrast there are significantly less studies on thorium tellurate and tellurite compounds, no known related minerals and only one study under HP/HT conditions.51 Nevertheless, the study by Xiao et al.51 examined the formation of thorium–tellurium compounds through HP/HT conditions of which Th2Te3O11 was uncovered, a mixed thorium tellurite–tellurate compound (Fig. 5). Unlike previous studies on thorium–tellurium compounds under ambient pressure conditions none have produced structures containing mixed Te cations. The occurrence of Te(IV) and Te(VI) in Th2Te3O11 further invokes the formation of mixed Te coordination's of Te(IV)O3, Te(IV)O4, and Te(VI)O6. This multivalent and multicoordination possessing Te actinide compound was argued to be a consequence of the HP/HT conditions used, coupled with the flexibility of Te and Th coordination chemistry. The same study, further uncovered the first alkaline thorium tellurates of Na4Th2(Te(VI)O15) and K2Th(Te(VI)O4)3 in addition to the high pressure form of Th(Te(IV)O3)(SO4). The latter structure, compared to its lower pressure polymorph,52 through HP/HT conditions involves an apparent total reconstructive phase transformation involving the Th coordination environment increasing from 8 to 9 and also dimensionality of the structure moving from 2D to 3D.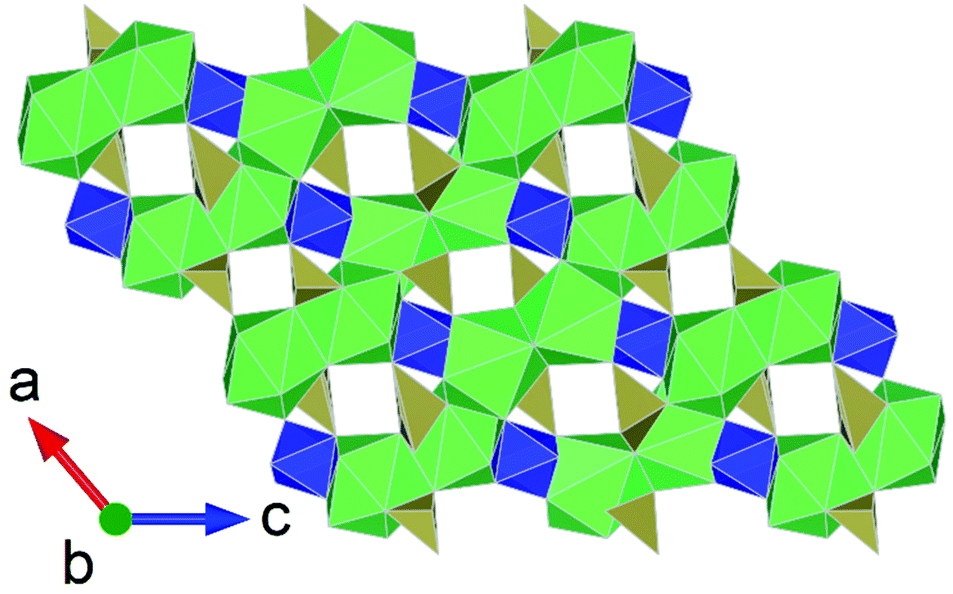 | ||
| Fig. 5 Structural representation of Th2Te3O11 highlighting coordination motif of different Te valences adapted from ref. 51. Note: Th(IV), Te(IV) and Te(VI) polyhedra are presented as green, tan and blue polygons. | ||
Thorium arsenates/arsenites
Similar to tellurates and tellurites, actinide arsenic compounds have been investigated extensively due to their diversity in structure formation, a consequence of the As(III) and As(VI) cations being able to adopt multiple coordination environments and also being able to polymerise. The chemistry of arsenic is further of interest due to its toxicity and has accordingly garnered interest in the context of environmental safety.53 Subsequently, studies have focused on examining the valence chemistry of arsenic compounds particularly with respect to forming mixed valent compounds and how actinides can coerce this. The first example of an actinide mixed valent arsenic compound was uncovered by Yu et al.54 through applying HP/HT conditions to Th(NO3)4·5H2O–As2O3–CsNO3 leading to the formation of Th(As(III)4As(V)4O18). Aside to this study some thorium arsenates/arsenites compounds had been reported from high temperature synthesis55,56 but none with mixed arsenic valences. The structure of Th(As(III)4As(V)4O18) consists of (AsIII4AsV4O18)4− layers that are interconnected via Th4+ cations leading to a 3D framework structure. In ambient pressure studies of thorium arsenite/arsenate compounds, AsOx, units typically act as singular bridging units between Th4+ cations. Accordingly, the application of pressure through HP/HT synthesis is suggested to coerce polymerisation of AsOx units and can be considered a tool in aiding this for synthesis design (Fig. 6).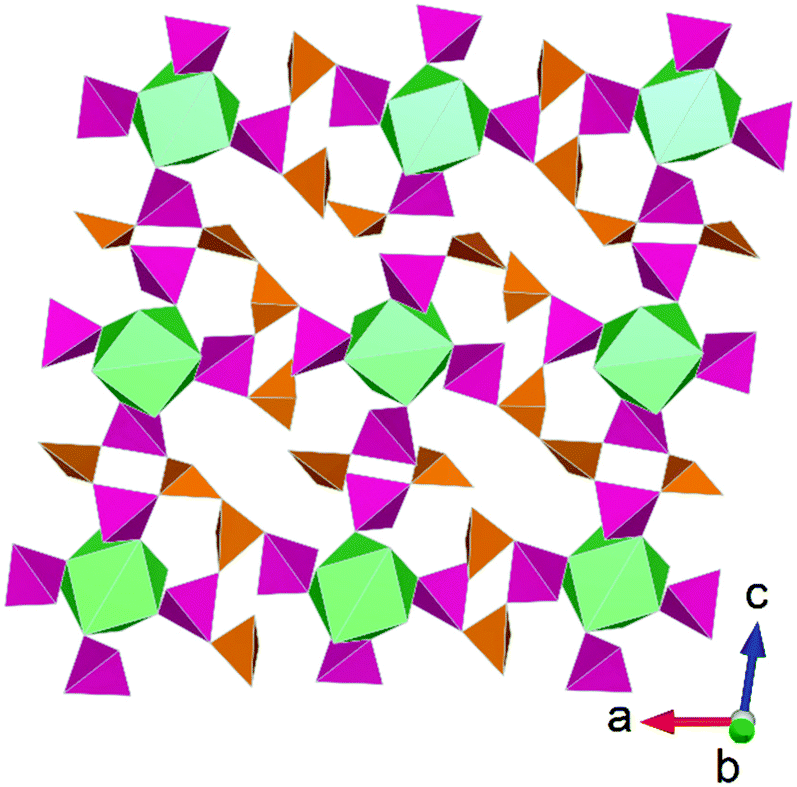 | ||
| Fig. 6 Structural representation of Th(As(III)4As(V)4O18) highlighting coordination motif of different As valences in the layer plane. Note that Th polyhedra (green) are coordinated firstly by As(III)O4 (orange) forming discrete clusters that are linked by As(V)O3 polyhedra (pink). Figure adapted from ref. 54. | ||
Thorium borates
Interest in actinide borate chemistry has a rich history, traditionally examined in the context of understanding interactions in glass-waste form matrices and immobilisation.17 Contemporary studies have shifted their focus towards advanced functional material development. This shift is related to the flexibility in structure formation that borate units often provide in facilitating diverse actinide structure formation that typically features pores which can be used for ion-exchange applications,57 notably involving thorium.44 A number of HP/HT studies involving uranium borates have been conducted (discussed below), but only so far one study involving thorium borates under such conditions is reported.37 The study by Hinteregger et al.37 examined the formation of AnB4O8 (An = Th, U) under conditions of 5.5 GPa and 1100 °C for the thorium variant (uranium discussed below). The structure of ThB4O8 was found to bare considerable resemblance to that of rare-Earth borates synthesised from HP/HT conditions, namely being built from tetrahedral BO4 groups as 2D layered structures.58,59 However, in the case of ThB4O8 it is found to be a 3D framework structure consisting of discrete channels (Fig. 7). As suggested by Hinteregger et al.37 the occurrence of Th(IV) and U(IV) within the AnB4O8 structure implies it should be possible for transuranium variants to be established, for instance containing Pu(IV) (or as surrogate Ce(IV)). However, there are yet to be any reports of this.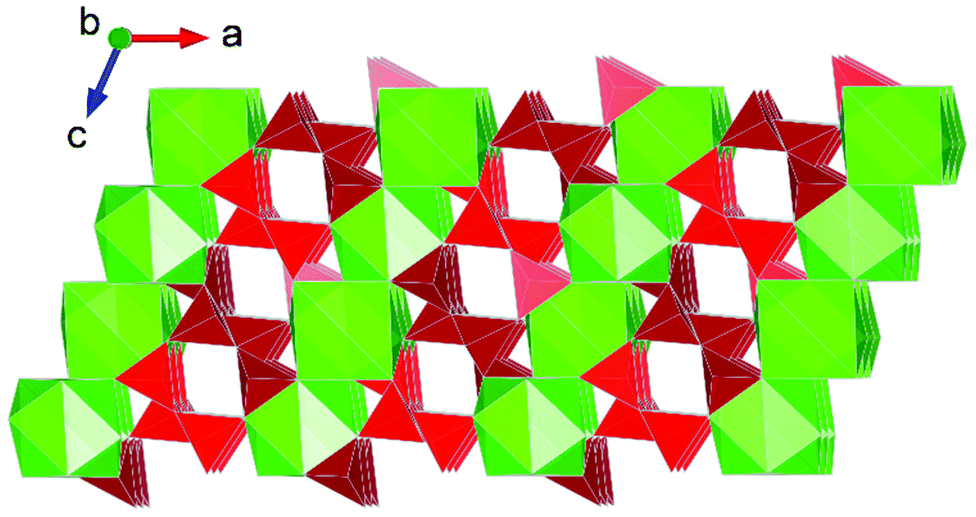 | ||
| Fig. 7 Structural representation of the monoclinic channel structure of AnB4O8 (An = Th, U), (Th variant depicted). Note Th and B polyhedra are represented by green and red polygons respectively. Figure adapted from ref. 37. | ||
Uranium HP/HT studies
As the primary actinide element utilised within the nuclear fuel cycle, uranium has endured sustained interest in which publications pertaining to actinides are most associated with, and this holds further true for studies involving HP/HT conditions. Chemically, uranium can adopt a range of valence states from II–VI but of which the tetravalent ([Rn]5f2), pentavalent ([Rn]5f1) and hexavalent ([Rn]5f0) are most relevant for applications and solid-state materials.60 The U(V) state is traditionally considered a rarer intermediate between U(IV) and (VI), due to the unfavourable occupation of an unpaired single 5f electron in the 5f lowest unoccupied molecular orbital and thus will rapidly oxidise, reduce or disproportionate to retain a stable electronic configuration, particularly in aqueous state. However, contemporary investigations often by solid state and organometallic chemists had shed light on the ability to isolate U(V) in solid compounds, through intricate synthesis methods including the use of pressure.20 Compared to U(IV), U(VI) has an exceedingly higher solubility, considered greatly more reactive and accordingly is often studied in the context of environmental radiological contamination. Ubiquitous to their chemistry particularly in the solid state, the U(V) and U(VI) cations can be hosted within the oxo collinear uranyl group, UO22+, but which is restricted for U(IV). The uranyl, a part of the general actinyl group, is pertinent to the chemistry of uranium and actinides and directly influences the structure dimensionality and properties of compounds. Its oxo nature typically leads to the formation of lower dimensional 2D layer or 1D chain structures as opposed to the spherical non-uranyl containing U(IV) and Th(IV) cations which adopt more higher dimensional structures. This subsequent preference in structure type formation based on uranium valence state provides then a basis for accurate and controlled synthesis design. Traditional studies involving HP/HT conditions of uranium compounds, like thorium, largely focused on examining the pressure dependence of binary oxides like UO2 and also metallic using DACs in addition to some on mixed oxides as mentioned in the introduction previously.10,11 However contemporary investigations on uranium materials under HP/HT conditions have expanded to broader classes of materials such as those of oxo-salt and mixed oxide compounds, particularly to utilise the relationship between uranium valence and structure type. Many of these HP/HT studies have become conducted in the context of alteration phases arising from SNF, advanced functional material development and fundamental explorative research of uranium chemistry. Accordingly, the next sections will briefly review and highlight successes of some of these contemporary investigations.Ternary uranium oxides
Interest in ternary uranium oxide compounds, AxUyOx, (A = di- or trivalent cation) extends to the days of the Manhattan Project era with the early works of Zacharisen examining phases such as SrUO4 and MgUO4.61,62 Indeed, these simple ternary uranium oxides known as the monouranates of form the AUO4 have received the considerable interest in published literature and are still topical.4–6,60,63,64 As previously mentioned, the AUO4 monouranates were some of the first actinide compounds to be examined using HP/HT methods in the case of the polymorphs of NiUO4 (α and β) examined by Young, Hoekstra and Marshall10,11 in the 1960s. These studies suggested the inability of the NiUO4 polymorphs to form under ambient pressure high temperature conditions was a consequence of the size mismatch between the small Ni cation and that of the large uranium cation. Many of these traditional studies were often conducted with a sense of serendipity regarding acquiring structures and compounds from crudely designed synthesis experiments. Many contemporary investigations apply more systematic methods in synthesis design and analysis to better understand structure–property relationships. This was exemplified in a study re-examining the formation of structures from the Ni–U–O system, mono-, NiUO4, di-, NiU2O6, and triuranates, NiU3O10, and particularly uncovering the need for HP/HP conditions for the synthesis of the monouranate variants.12 The study using high-resolution measurements identified, consistent with the postulates of Brisi and Young, that HP/HT conditions are necessary to overcome the size mismatch between the Ni and U cations in the synthesis of the NiUO4 polymorphs. However further to this, structure formation can be achieved via excess U addition in place of pressure, which can bridge the size mismatch of the cations allowing the formation of the di and triuranates. This relationship between Ni–U–O structures, U/Ni ratio and pressure is graphically depicted in Fig. 8. W.r.t. the figure note the decreasing separation between Ni polyhedra with increasing pressure and reduced requirement for a higher H/Ni ratio to overcome this.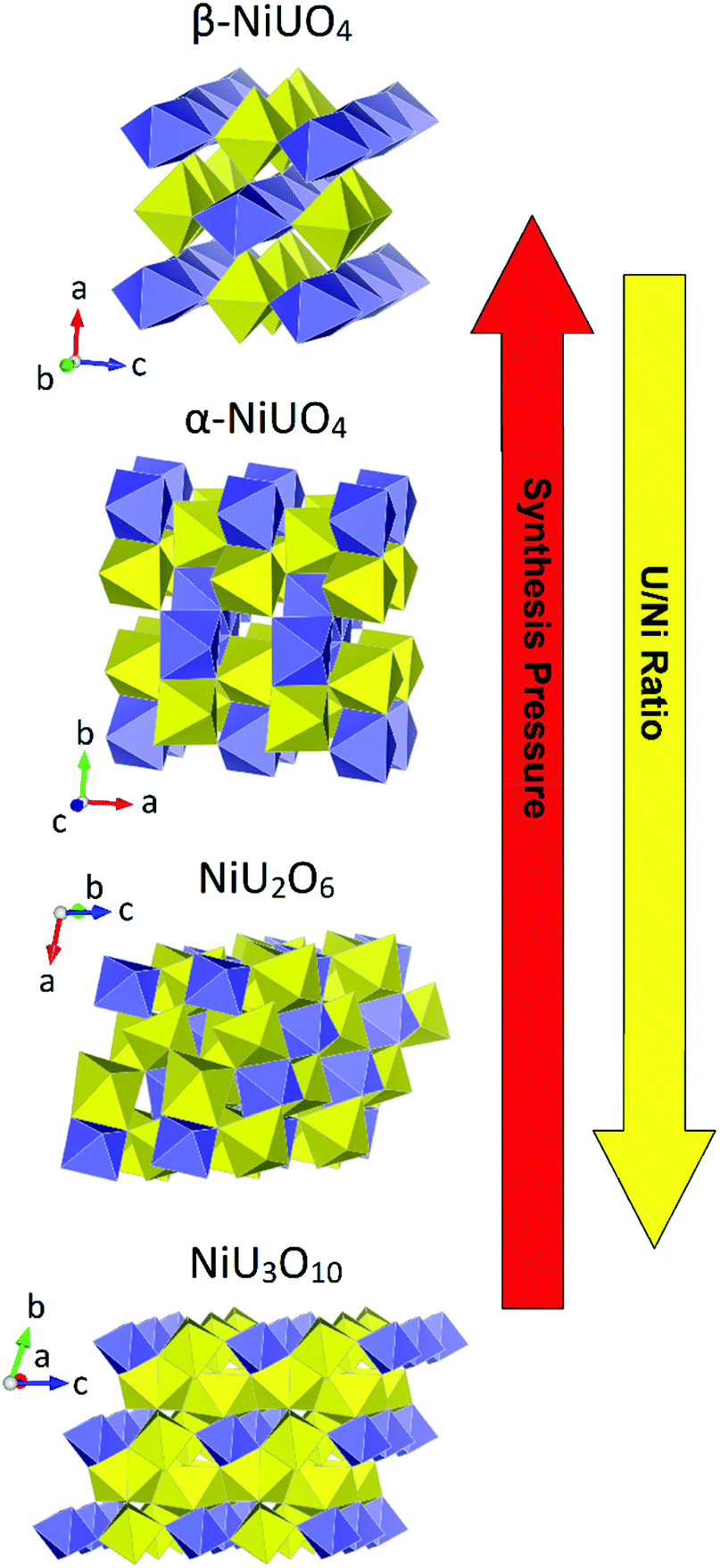 | ||
| Fig. 8 Structural representations of mono-, di- and triuranates of the Ni–U–O system emphasising the relationship between the structure type and synthesis conditions. Note uranium and nickel polyhedra are given in yellow and purple respectively. Structures and conditions adapted from ref. 12. | ||
The structures of the NiUO4 polymorphs, α and β, form respectively form rutile type structures of respectively 2D chain/layer and 1D chains structures which can be related to α-PbO2 and TiO2.12 A pertinent follow up study3 found that by relating the rutile relationships present in the NiUO4 polymorphs with the broader family AUO4 monouranates which also possess rutile structures, subtle distortions and tilting in respective UO6 and AO6 polyhedra could be modulated through pressure, temperature or cation size. The study found that the AUO4 structures in space group Ibmm, A = Mn, Co, Mg and β-Ni which are related to TiO2, can be potentially described in the higher symmetry space group Cmmm that β-CdUO4 possesses, but the size mismatch between the U and A cations leads to structural distortions and symmetry lowering. However interestingly by applying temperature through in situ synchrotron powder diffraction heating measurements, the higher symmetry non-distorted space group Cmmm structure could be obtained. Conversely by applying pressure through ab initio DFT + U simulations the lower symmetry distorted space group Ibmm structure could be obtained. Fig. 9 illustrates the difference in distortion of polyhedra and tilt between the distorted and non-distorted space group Ibmm and Cmmm structures respectively in addition to the relationship between pressure and temperature. The study gives a beautiful insight into the role temperature and pressure have upon modulating atomic position towards structure formation. The study3 further saliently highlighted limitations in ab initio calculations using DFT + U for actinide metal oxides, often considered the gold standard for such computational work. The investigation found issues in applying the Hubbard U energy term which consistently resulted in the overestimation of the structural energy and consequently incorrect prediction of stability within the AUO4 compounds. The authors pointed towards the need to be better represent d and f states in calculations such as instead applying Wannier projections. Such insight is highly pertinent in the context of calculations relating to modern nuclear fuels which often use transition metals in their fuel blends.65
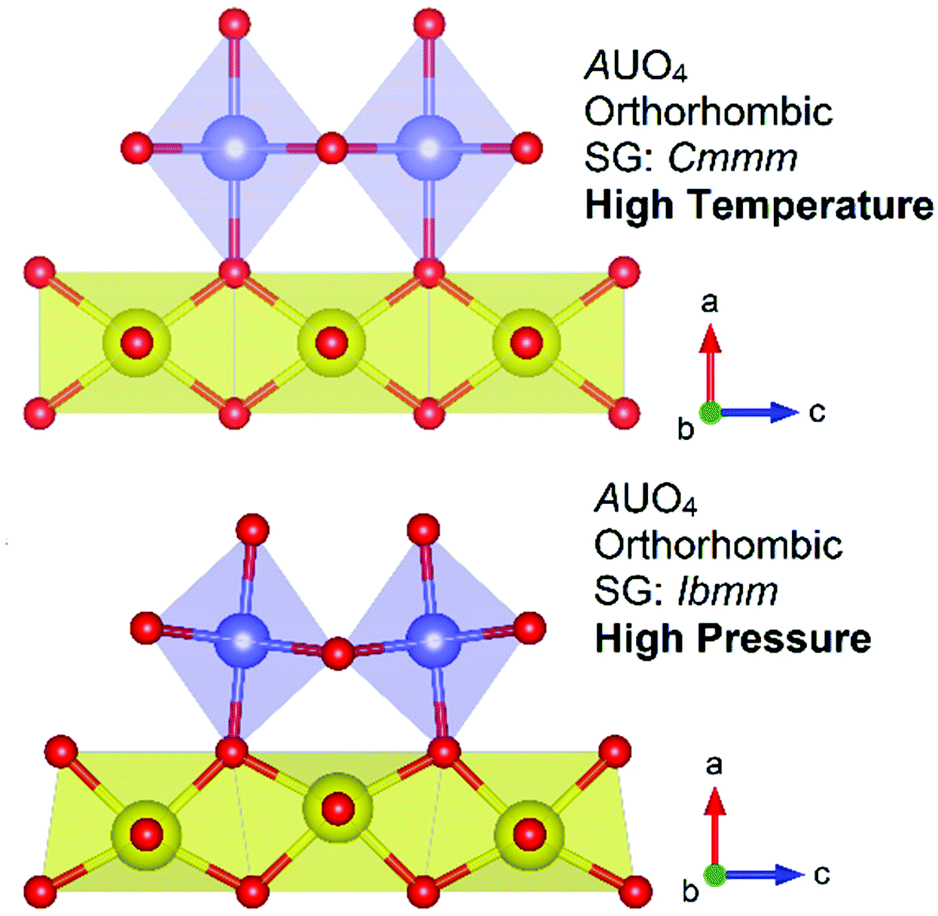 | ||
| Fig. 9 Structural representations of the AUO4 orthorhombic structures in space groups Cmmm and Ibmm where it has been shown3 high temperatures favour the Cmmm structure non-distorted structure and high pressures favour the distorted Ibmm structure. Note the tilt in the AO6 polyhedra (purple) and distortion of UO6 polyhedra (yellow) w.r.t. oxygen atoms (red spheres). | ||
Uranium molybdates
Uranium molybdates have been explored extensively for similar reasons to that of thorium counterparts, namely their contribution to desirable physical properties but also due to the pertinent occurrence of Mo in SNF.66 However, unlike the spherical Th4+ cation, the oxo-uranyl group possessing UO22+ cation often leads to lower dimensional structure formation forming 1D chains or 2D layered structures over 3D framework. This was highlighted in so far the only study67 of uranium molybdates synthesised under HP/HT conditions, which reported the compounds K2[UO2(Mo2O7)2], K2[(UO2)2(Mo(VI)4Mo(IV)(OH)2)O16], K3[(UO2)6(OH)2(MoO4)6(MoO3OH)] and K5[(UO2)10MoO5O11OH]·H2O from 900 °C and 4 GPa. Three of the four structures were found to adopt 3D framework structures with K2[UO2(Mo2O7)2] being the exception adopting a 2D layered structure. Salient to this structure is the occurrence of oligomerised molybdate unit, (Mo2O7)2−, which at time of writing has only observed in that structure and yet to be observed in potassium uranium molybdates resulting from more ambient pressure synthesis conditions (Fig. 10). It was highlighted by the study at time of publication that proportionally w.r.t. to other studies, HP/HT methods generally lead to a greater proportion of structures adopting 3D framework structures as opposed to high temperature and hydrothermal methods which use considerably lower pressures. The authors argued that both pressure and a high ratio of U/Mo units leads to the condensation of polyhedra resulting in the increased likelihood of oligomerisation of molybdate units, for instance with the occurrence of Mo2O72− dimers and higher dimensionality of formed structure. Although, the study's sample size of known HP/HT derived uranium molybdates is relatively small, the postulate is well consistent with that which was similarly observed in thorium molybdates, ternary uranium oxides and in non-actinide molybdate based studies.68 Consequently, the study points towards the ability of HP/HT methods to generate higher dimensional compounds and coerce oligomerisation of functional groups.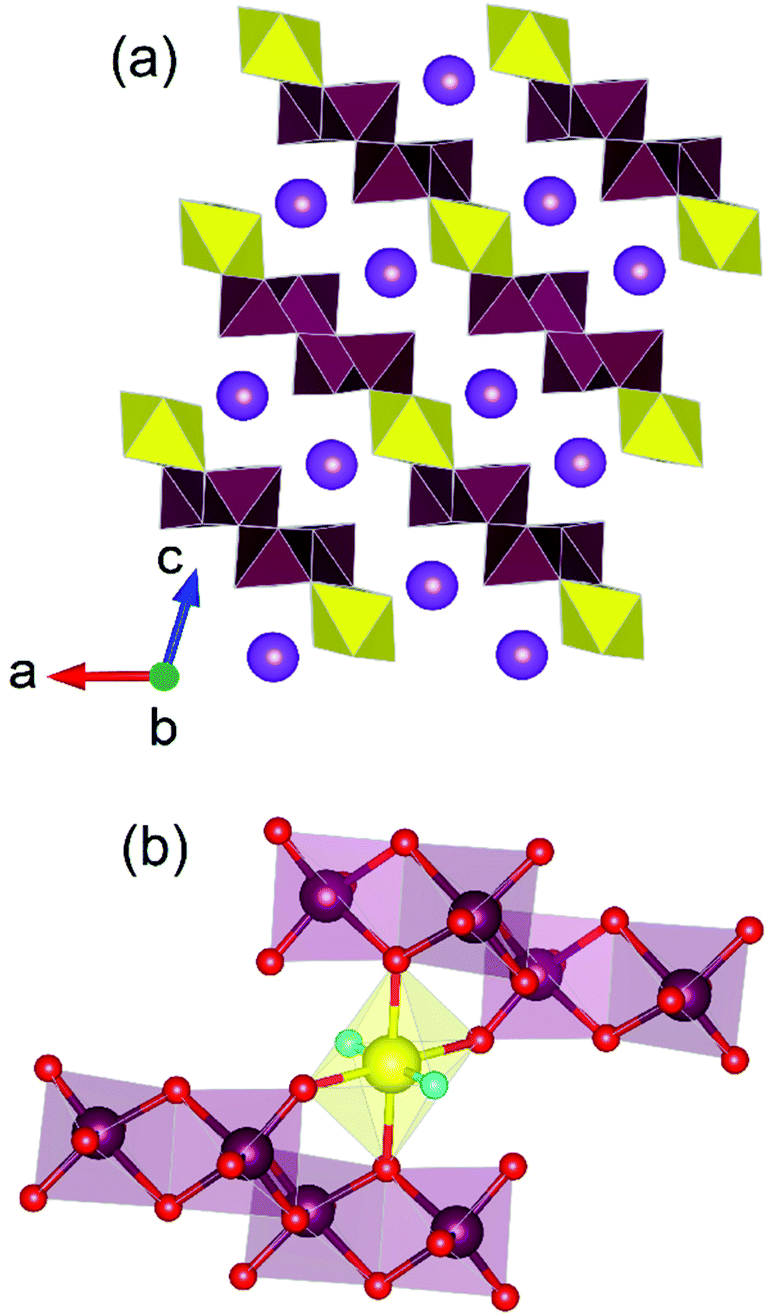 | ||
| Fig. 10 (a) Structural layer motif of K2[UO2(Mo2O7)2] highlighting the occurrence of (b) the (Mo2O7)2− unit. Note maroon and yellow polyhedra respectively represent Mo and U polyhedra whereas purple, maroon, yellow, red and aqua spheres respectively represent K, Mo, U, O and uranyl-O atoms respectively. Figure adapted from ref. 67. | ||
Uranium tellurates/tellurites
Formation of uranium tellurates and tellurites was explored by Xiao et al.69 under HP/HT conditions of 3.5 GPa and 1100 °C resulting in the structures Na2[UO2(Te(VI)2O8], Na2[UO2(Te(VI)2O5)], Na[UO2Te(IV)2O5OH] and Na[UO2Te(IV)6O13OH]. Interestingly the structure of Na[UO2Te(IV)6O13OH] was found to be isotypical to the mineral cliffordite UO2(Te3O7),50 and crystallises in the centrosymmetric space group of Pa. Due to the oxo-bonding nature of the uranyl group, uranyl based compounds typically adopt NSC structures.70 So far, there are no further studies of uranium tellurates and tellurites under HP/HT conditions.
Uranium arsenates/arsenites
As previously described for thorium arsenates and arsenites, the uranium counterparts have been studied extensively using ambient pressure methods owing to the rich chemistry and diverse chemical behaviour that can be extracted from combing the chemistry of arsenic and uranium. Under high temperature ambient pressure and HP/HT conditions Yu et al.71 examined the formation of caesium uranium arsenates with conditions of 900 °C and 3 GPa. The investigation led to four novel uranyl arsenates Cs2[(UO2)(As2O7)], α-Cs[(UO2)(HAs2O7)], β-Cs[(UO2)(HAs2O7)] and Cs[(UO2)(HAs2O7)]·0.17H2O of which the latter three are the first examples of uranyl arsenates to be obtained under HP/HT conditions. However, there are yet to be any studies of uranyl arensites resulting from HP/HT conditions. The study further supports the conclusions made on potassium uranyl molybdates under HP/HT67 conditions previously discussed, that the application of pressure seems to assist in the transformation from 2D layered motif of Cs2[(UO2)(As2O7)] to that of framework structures of the high HP/HT synthesised phases. Accordingly, the investigation supports the narrative of HP/HT conditions being a driver towards increasing dimensionality of compounds.Uranium silicates
The elucidation of interactions and structure formation between uranium and silicon is a highly topical and long-examined area of actinide chemistry and mineralogy due to the strong association the elements have with SNF alteration phases and geochemistry.72,73 It is known that uranium provides approximately 25% of the total energy generated within the Earth through its radioactive decay, and accordingly geochemists have investigated its speciation within Earth's layers.24 It is suspected that approximately 50% of terrestrial uranium is found within the lower mantle, where temperatures over 2000 °C and pressures above 50 GPa are experienced. Under these conditions, coffinite, USiO4, is expected to decompose into UO2 and SiO2 and incorporation of the uranium oxide into region prevalent silicate perovskites including davemaoite CaSiO3,74 has been of strong topical interest. A pertinent investigation by Gréaux et al.75,76 using HP/HT conditions provided by a laser heated DAC (LH-DAC) and using X-ray diffraction and spectroscopy, was able to show under conditions of up 54 GPa and 2400 °C, uranium incorporation into the CaSiO3 indeed occurs. The authors conclude consequently that Al rich CaSiO3 is the only candidate mineral able to host uranium providing key radiogenic heat (and also suspected thorium) in the Earth's mantle. Although the study is in situ based, it emphasises the importance of applying HP/HT conditions to actinide relevant minerals towards understanding geological behaviour of the Earth.Uranium borates
Of the actinide system studies that have been examined in the previous decade, uranium borates have arguably received the most attention and a large body of literature is available for them under a variety of conditions.17,77,78 The ability for borate units to occur as seemingly an innumerable amount of fundamental building blocks through BO3 triangle and BO4 combinations that can also polymerise, gives a large breadth of investigation for chemists particularly using HP/HT conditions. Furthermore, as opposed to thorium borate studies, the chemistry of uranium allows even a greater diversity of structures to be obtained due to the greater accessibility of valence states of the latter actinide. In the previously discussed study by Hinteregger et al.37 which examined the formation of AnB4O8 (An = Th, U) structures, additional to the synthesis of thorium borate, they were also able to achieve synthesis of the uranium in its tetravalent state, despite beginning with hexavalent uranium nitrate as starting reagent. Compared to the thorium counterpart which used pressures and temperatures of 5.5 GPa and 1100 °C, a higher pressure of 10 GPa was employed. Although such a result is surprising considering the lack of reducing conditions used in their synthesis, a growing body evidence points towards the flexibility of uranium valence in facility structure formation based on size requirements. For example, in isostructural monouranates, α-NiUO4, CrUO4 and FeUO4, the adoption of the pentavalent uranium in the Cr and Fe members and hexavalent in Ni (synthesised under HP/HT conditions),12 is argued to be based on structure and cation size ratio requirements, which is facilitated by the flexibility of the uranium valence to conform.3 Similar is expected to occur in UB4O8, where the larger tetravalent uranium cation is needed over the smaller hexavalent of which the combination of extreme pressure and temperature through HP/HT synthesis facilitates its formation.A number of studies in the early 2010's utilising supercritical water conditions79,80 demonstrated the exotic and unique chemistry that can be obtained from elevated pressures and temperatures on uranyl borates. Inspired by these successes, researchers began examining uranyl borates under HP/HT conditions. A pertinent study by Wu et al.79 examined the formation of uranyl borates at temperatures between 650 and 1200 °C and pressures between 0.2 and 2.5 GPa. The study unveiled three potassium uranyl borates which exhibited firstly a U/B ratio greater than 1 (rare for uranium borates) in addition to novel coordination geometries of uranyl centres that have not been encountered in ambient pressure studies. Two of the structures from the study, K12[(UO2)19(UO4)(B2O5)2(BO3)6(BO2OH)O10]·nH2O, and K4[(UO2)5(BO3)2O4]·H2O, are presented in Fig. 11 which illustrates the starkly different layer configurations compared to somewhat similar structural configurations. The study eloquently demonstrated the novel coordination types and structural architectures which HP/HT conditions can provide for in uranyl borate synthesis. A later study by Hao et al.81 added to this narrative, in using systematic methods to examine the formation of uranyl borates under high temperature, hydrothermal and HP/HT synthesis conditions. The study highlighted the variety of different uranyl borate layer topologies that can be obtained, in which the topology of the borate units is markedly contrasted, from different synthesis methods.
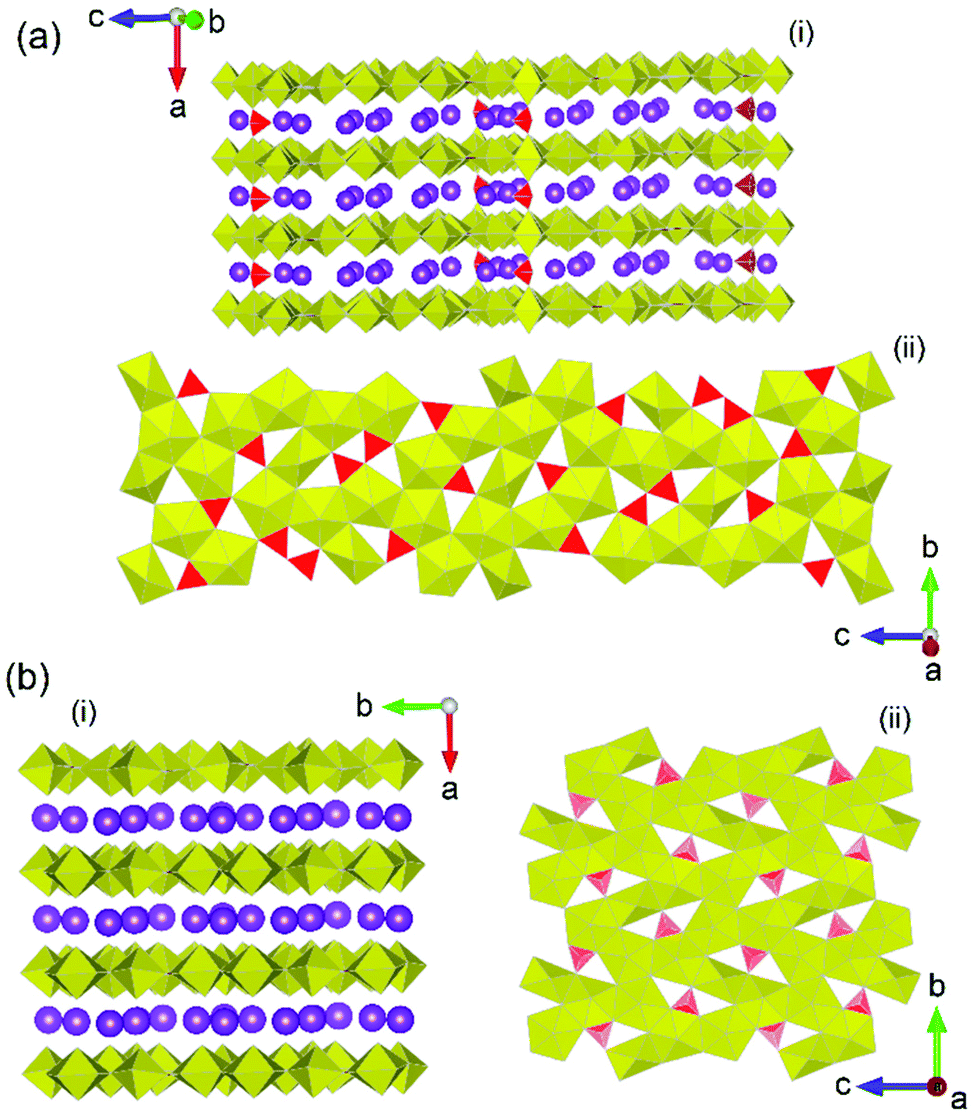 | ||
| Fig. 11 Structural representations of (a) orthorhombic K12[(UO2)19(UO4)(B2O5)2(BO3)6(BO2OH)O10]·nH2O in (i) general and (ii) layer format, and (b) monoclinic K4[(UO2)5(BO3)2O4]·H2O in (i) general and (ii) layer formats. Yellow and red polyhedra represent U and B polyhedra respectively whereas purple spheres represent K atoms. Figures adapted from ref. 79. | ||
A topical sub-field of uranium borate chemistry is that of mixed anionic borate materials such as aluminoborates or borophosphates.41,82 Both types of uranium borates have been studied extensively in the context of nuclear waste form glasses but also because of the similar flexible bonding nature of phosphate and aluminate groups that are highly compatible with borate groups. This leads to an even greater degree of permutations and combinations that can be obtained when combined with uranium chemistry. The uranyl aluminoborate UO2[B3Al4O11(OH)] featuring a highly complex 3D structure was achieved by Wu et al.41 using a cold seal vessel with conditions of 0.2 GPa and 650 °C. Pertinent to the structure is the occurrence of multi-oxo components of uranyl in addition to AlO6 and AlO5 groups that gives rise to its unique structure which deviates sharply from ambient pressure studies. Hinteregger et al.38 examined the formation uranyl borophosphates under HP/HT conditions of 7.0 GPa and 700 °C. Similar to their previous study37 on uranium borates, they were able to synthesis a tetravalent uranium borophosphate despite starting with hexavalent uranium nitrate. The structure, U2[BO4][PO4], interestingly was found to be isotypic to Th2[BO4][PO4] which was obtained from high temperature ambient pressure conditions.83 It is suspected the reason for the reduction of the uranium during synthesis is again related to cation size requirement in structure formation, of which the flexibility of uranium redox chemistry can accommodate this through HP/HT conditions.
Uranium sulfates
There is so far only one such study of uranium sulfates under HP/HT conditions.84 As is often the result of fundamental investigations involving extreme conditions, the study describes the serendipitous synthesis of a HP/HT uranium sulfate phase, Na4[(UO2)(SO4)3], from a synthesis at 3.5 GPa and 1000 °C that was intended to examine the formation of actinide oxo-chalcogenates. Pertinently, the structure possesses the uranyl group, which is typically linear with only small angular deviations,85 with a pronounced bend away from 180° to 165.6(12)° (Fig. 12). Significant efforts have been made in general uranium and actinide chemistry to understand the behaviour of the uranyl/actinyl group regarding reactivity such as in cation–cation interactions (CCIs) or particularly examining limitations in bending and its deformation. To probe the latter phenomena, chemists have often used scaffolding methods often with organometallic ligands to induce bending or deformation that is steric or electronic in origin.86 At present, the maximum uranyl bend reported is approximately 161°.87,88 In the case of Na4[(UO2)(SO4)3], the authors argue that bending originates from the physical pressure applied to the structure rather than an electronic or steric effect. The latter effects arise from large steric groups pushing the uranyl, of which sulphate groups do not possess such strength. Rather, the sulphate groups are strong equatorial donors, which can weaken the uranyl moiety, leading to its bending when placed under HP/HT conditions during structure formation and subsequent quenching. The authors used ab initio calculations to support these conclusions. The study is significant, as it offers a new pathway towards understanding the fundamental chemistry of the uranyl group, of which was only previously examined using organic and organometallic ligands.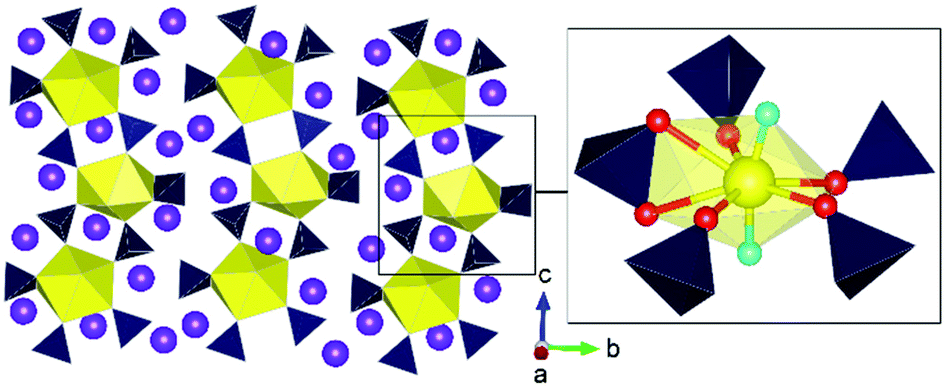 | ||
| Fig. 12 Structure of Na4[(UO2)(SO4)3] synthesised from HP/HT conditions illustrating bending of the uranyl group in the inset. Note Yellow and dark polyhedra represent U and S polyhedra respectively whereas purple, yellow, red and aqua blue spheres represent K, U, O and uranyl O atoms respectively. Figure adapted from ref. 84. | ||
Transuranics
Despite the significant advances made in the chemistry of uranium and thorium under HP/HT conditions, little progress has been made for transuranium element containing compounds. A consequence of the heightened radiological danger associated with the elements coupled with the unavailability of HP/HT apparatuses and laboratories that are licensed and can accommodate them. Despite this, the last decade has unveiled considerable new insight into the chemistry of transuranium compounds, divulging significant chemical insight particularly under conditions of elevated pressure via pertinent studies involving DACs. Accordingly, some of these will be mentioned briefly considering their salience to actinide chemistry.Plutonium metals have been examined extensively at higher pressures (and also temperatures) due to the complexity in their electronic structure which leads to a plethora of allotropes.89 In the last decade several papers have examined the associated pressure and temperature induced transitions. A long-standing question of plutonium chemistry has been the observation of anomalous large softening of the bulk modulus in the metallic form as temperature increases irrespective of the sign of the coefficient of thermal expansion, which is not encountered to such an extent as other metals. However it was recently shown by Harrison90 that the softening of the bulk modulus can be attributed to the compressibility of plutonium's electronic configuration. Such phenomena arises from the unique ability for plutonium to adopt large number of electronic configurations that are non-degenerate.91 This result obtained through statistical thermodynamic modelling, underscores the exotic chemistry of plutonium and that despite 82 years since its discovery, key questions remain behind its HP/HT behaviour even its metallic form.
Transplutonium elements, unlike their lighter actinide sisters, possess unique localisation of 5f electrons at ambient pressures but delocalise and partake in bonding at higher pressures. This results in a rich array of crystallographic structures that form under elevated pressure which exhibit intriguing structural and magnetic properties.92,93 This is well exemplified by curium which has been examined extensively under high pressure conditions over the last decades and several allotropes ideanified.94 Yet, still the story is not over with recent discoveries occurring under elevated pressure conditions such as the predicted occurrence of quasiparticle multiplets at high pressure.95 Further pioneering studies into californium by Heathman et al.96 found in 2013 that californium to be first actinide to exhibit more than one metallic valence (from divalent to trivalent) near ambient conditions from in situ pressure studies. This phenomenon is due to the strong ability for californium ability to delocalise its f electrons under pressure allowing them to adopt different valence states.
It has often been thought close parallels can be made between the electronic characteristics of heavy 4f lanthanide elements with that of the 5f transplutonium and by extension bonding characteristics. However, over the last decade this perception has been rapidly reduced through pioneering transplutonium studies by the group of Albrecht-Schönzart utilising optical spectroscopy measurements under pressure using DACs. A study by Sperling et al.97 examined the compression of curium pyrrolidinedithiocarbamate (pydtc) along with its neodymium analogue using absorption spectroscopy under pressures up to 11 GPa. They showed that the f–f transitions in [Cm(pydtc)4]− (Fig. 13) with its Cm–S bonds are more considerable than that of the Nd analogue and also Cm mellitate with its Cm–O bonds. The study highlighted the complexity of curium bonding, particularly covalent interactions at high pressure and the unexpected further impact it has on the ligands. Similar results were also reported by Sperling et al.98–100 on related americium in addition to similar berkelium and californium compounds by like methods highlighting both the sharp departure in bonding behaviour compared to non-actinide variants. Paucities of information still remain for the transuranics under pressure both from the understanding of the fundamental electronic states to their bonding with compounds. Nevertheless, significant advancements have been made in both areas which have enabled deeper understanding to their core chemical properties and bonding behaviours.
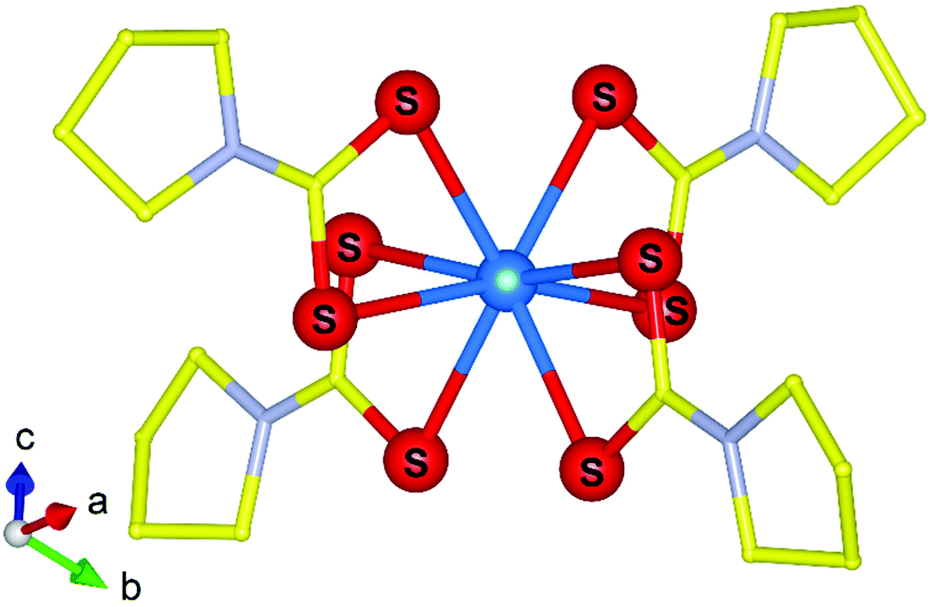 | ||
| Fig. 13 Molecular structure representation of the [Cm(pydtc)4]− anion that was examined by Sperling et al.97 using absorption spectroscopy via DACs. Note Cm, S, N and C atoms are represented by blue, red, silver and yellow spheres respectively. Figure adapted from ref. 97. | ||
Perspectives, challenges, and future directions
From this overview, over the last decade ex situ HP/HT studies of actinide compounds have allowed significant and novel understanding of their physical–chemical properties to be obtained. For instance, it is now more comfortably understood that when considering the results of potassium uranyl molybdates,67 arsenates/arsenites71 and thorium tellurates/tellurites,51 HP/HT conditions, particularly pressure, enable strong control to be obtained over the dimensionality of structure formation. Specifically, higher pressure conditions favour high dimensional structures. Indeed, this seems to extend further with the coerced oligomerisation of building units with the application of pressure and temperature. Another interesting observations seems to be the unexpected flexibility of actinide valence states under pressure, emphasised in uranium studies12,37,38 where despite beginning with oxidised uranium either reduced forms are found after HP/HT conditions are applied or structure formation seems to diverge from what is expected in ambient pressure studies regarding uranium valence.63 Generally, it can be observed that HP/HT conditions provide an open playground in which actinide compounds are able to exhibit flexible and exotic structure formation, through the adoption of multiple polymorphs at elevated pressures, exhibit distinctive bonding and polyhedra distortions and sometimes unexpected valence behaviour.It is also quite evident from the discussion thus far that several thorium, uranium and particularly transuranium chemical systems are underexplored. For instance, thorium compounds of phosphate, tungstate, iodate and selenate/selenite have received broad interest under variable temperature and ambient to low pressure conditions, but there are no reports of these from HP/HT conditions.101–103 Similarly for uranium, compounds of iodate, phosphate, tungstate and selenate/selenite among others have not been reported under HT/HP conditions, despite considerable general interest.104–107 In the case of transuranium compounds, there are yet to be any reports of compounds from HP/HT conditions. Although, the works of Albrecht-Schönzart and co-workers have hinted at the unique bonding characteristics of transuranium compounds under pressure, unfortunately there appears to be no laboratories operating that are equipped for HP/HT experimentation with transuranium elements at this time. This absence is more related to the lack and associated cost of HP/HT infrastructure, rather than transuranium laboratories. Nevertheless, the innovative use of DAC devices in transuranium laboratories has offered a somewhat alterative to this, and it is hoped then that LH-DAC can provide a relatively inexpensive substitute to HP/HT devices and find further application. There is a further general absence of studies pertaining to pressures above 10 GPa for actinide compounds. Although piston cylinder/multi-anvil devices such as that describe previously (Fig. 1), can reach such pressures, it is generally an expensive exercise particularly w.r.t. time. However, as demonstrated by geochemical studies such as the speciation of uranium in the Earth's mantle,74 there is an inherent interest to examine compounds and materials arising under such conditions in quantities larger than what a DAC can produce. Accordingly, despite the advancements in DAC measurements there is an on-going need for devices that can produce larger size samples from HP/HT conditions such as that described and presented in Fig. 1.
It has been previously established that HP/HT conditions are an alternative means to produce good quality single crystals, particularly from materials that have high melting points.108,109 Indeed, actinide materials, particularly those of oxide or silicate such as UO2 and coffinite are notoriously difficult to generate single crystals of.73 The synthesis of coffinite even as a powder is a difficult task,73 often requiring intricate synthesis methods involving elevated pressure via hydrothermal conditions to synthesis it,72,73,110,111 indeed there are no further reports of single crystal growth. In the case of UO2, methods such as arc-melting, floating zone techniques and vapor deposition are required, which many actinide laboratories do not possess.112 Accordingly, it is argued that combined with appropriate fluxes, HP/HT conditions can be used as an alternative means to produce often sort after actinide single crystals such as UO2 and potentially more difficult to obtain, coffinite and related silicate minerals.
A topical area of actinide chemistry recently has been examining the formation and properties of actinide metal organic framework (MOF) compounds. MOFs have received long standing interest due to their notable applications in hydrogen and carbon storage, electrocatalysis, semiconductors, gas separation among others. Placing MOFs under pressure has often been conducted to understand load bearing, deformation, and pressure-induced amorphisation of MOFs but it has also been found they can exhibit extreme compressibility.113 Studies relating to actinide MOFs have largely focused on topics of sequestration and capture of radionuclides in addition to fundamental synthesis studies of MOFs using actinides as building blocks. It is postulated HP/HT conditions would be then fertile ground for MOFs synthesis in utilising the unique chemical properties of actinide bonding under pressure to unlock novel structures and properties, as has been achieved in non-actinide HP/HT studies of MOFs.114,115
Concluding remarks
Overall, HP/HT studies of uranium and thorium compounds have uncovered unique and unexpected structure formation and bonding motifs. Whereas those of transuranium compounds under elevated pressure highlight the starkly different chemical properties of heavy actinide elements compared to relative lanthanides and lighter actinides. From these studies, a good basis of chemical knowledge of actinides under HP/HT conditions has been established. Nevertheless, it is important that this fundamental knowledge finds suitable use and application in areas of geoscience, nuclear waste form/NFC materials and advanced functional development. The latter subject has particularly gained considerable momentum by Chinese research groups, demonstrating profound properties in a variety of materials from ion-conductors, SHG materials to radiation detectors.116–118 However importantly, the developments of the fundamental understanding behind many HP/HT study conclusions began only approximately 10 years ago when a firm basis of actinide chemistry under ambient pressure and medium to high temperature conditions was already established. Accordingly, it can be viewed that HP/HT science of actinides is still a few years behind that of ambient pressure studies. Nevertheless, the renewed interest in actinide chemistry particularly in the context of reinvigoration of nuclear energy and overhauling nuclear waste disposal is expected to drive research back into HP/HT related studies, towards supporting science related to the NFC and fundamental discovery and materials development.Conflicts of interest
There are no conflicts to declare.Acknowledgements
GLM is grateful to funding and support from the German Federal Ministry of Education and Research (BMBF), Project No. 02NUK060. EVA and GLM are grateful to the funding and support from the German Science Foundation, DFG, Project No. AL1527/3-1.References
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1926, 112, 661–677 CAS.
- S. Knecht, H. J. A. Jensen and T. Saue, Nat. Chem., 2019, 11, 40–44 CrossRef CAS PubMed.
- G. L. Murphy, Z. Zhang, R. Tesch, P. M. Kowalski, M. Avdeev, E. Y. Kuo, D. J. Gregg, P. Kegler, E. V. Alekseev and B. J. Kennedy, Inorg. Chem., 2021, 60, 2246–2260 CrossRef CAS PubMed.
- G. L. Murphy, C. H. Wang, Z. M. Zhang, P. M. Kowalski, G. Beridze, M. Avdeev, O. Muransky, H. E. A. Brand, Q. F. Gu and B. J. Kennedy, Inorg. Chem., 2019, 58, 6143–6154 CrossRef CAS PubMed.
- G. L. Murphy, C.-H. Wang, G. Beridze, Z. Zhang, J. A. Kimpton, M. Avdeev, P. M. Kowalski and B. J. Kennedy, Inorg. Chem., 2018, 57, 5948–5958 CrossRef CAS PubMed.
- G. L. Murphy, B. J. Kennedy, J. A. Kimpton, Q. Gu, B. Johannessen, G. Beridze, P. M. Kowalski, D. Bosbach, M. Avdeev and Z. Zhang, Inorg. Chem., 2016, 55, 9329–9334 CrossRef CAS PubMed.
- P. M. Kowalski, G. Beridze, Y. Ji and Y. Li, MRS Adv., 2017, 2, 491–497 CrossRef CAS.
- T. Connor, O. Cheong, T. Bornhake, R. Tesch, M. Sun, Z. He, A. Bukayemsky, V. Vinograd, S. C. Finkeldei and P. M. Kowalski, Front. Chem., 2021, 940 Search PubMed.
- G. Beridze and P. M. Kowalski, J. Phys. Chem. A, 2014, 118, 11797–11810 CrossRef CAS PubMed.
- A. Young, Science, 1966, 153, 1380–1381 CrossRef CAS PubMed.
- H. R. Hoekstra and R. H. Marshall, Some uranium-transition element double oxides, ACS Publications, 1967 Search PubMed.
- G. L. Murphy, P. Kegler, Y. Zhang, Z. Zhang, E. V. Alekseev, M. D. de Jonge and B. J. Kennedy, Inorg. Chem., 2018, 57, 13847–13858 CrossRef CAS PubMed.
- C. Weir, E. Lippincott, A. Van Valkenburg and E. Bunting, J. Res. Natl. Bur. Stand., Sect. A, 1959, 63, 55 CrossRef PubMed.
- M. Idiri, T. Le Bihan, S. Heathman and J. Rebizant, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 014113 CrossRef.
- M. McElfresh, J. Thompson, J. Willis, M. Maple, T. Kohara and M. Torikachvili, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 43 CrossRef CAS PubMed.
- N. Tateiwa, T. Kobayashi, K. Hanazono, K. Amaya, Y. Haga, R. Settai and Y. Onuki, J. Phys.: Condens. Matter, 2001, 13, L17 CrossRef CAS.
- S. A. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Chem. Commun., 2011, 47, 10874–10885 RSC.
- N. N. Krot and M. S. Grigoriev, Usp. Khim., 2004, 73, 94–106 Search PubMed.
- B. I. Kharisov and M. A. Mendez-Rojas, Usp. Khim., 2001, 70, 974–995 Search PubMed.
- C. R. Graves and J. L. Kiplinger, Chem. Commun., 2009, 3831–3853, 10.1039/b902969a.
- J. Drozdzynski, Coord. Chem. Rev., 2005, 249, 2351–2373 CrossRef CAS.
- S. Taylor, Geochim. Cosmochim. Acta, 1964, 28, 1273–1285 CrossRef CAS.
- K. Lodders and B. Fegley, The planetary scientist's companion, Oxford University Press on Demand, 1998 Search PubMed.
- E. R. Oxburgh and R. K. O'Nions, Science, 1987, 237, 1583–1588 CrossRef CAS PubMed.
- R. B. Rebak, Accident-Tolerant Materials for Light Water Reactor Fuels, Elsevier, 2020 Search PubMed.
- M. Ruzickova, T. Schulenberg, D. C. Visser, R. Novotny, A. Kiss, C. Maraczy and A. Toivonen, Prog. Nucl. Energy, 2014, 77, 381–389 CrossRef CAS.
- T. Abe and K. Asakura, Comprehensive Nuclear Materials, ed. R. J. M. Konings, Elsevier, Oxford, 2012, pp. 393–422. DOI:10.1016/B978-0-08-056033-5.00036-7.
- L. Van Brutzel and A. Chartier, Mechanical behaviour of UO2 under irradiation: a molecular dynamics study, MMSNF (The Materials Modelling and Simulation for Nuclear Fuels), 2017 Search PubMed.
- C. Lee, J. Lee, S. Park, S. Kwon, W.-J. Cho and G. Y. Kim, Tunn. Undergr. Space Technol., 2020, 103, 103452 CrossRef.
- A. J. Beswick, F. G. Gibb and K. P. Travis, Proc. Inst. Civ. Eng.: Energy, 2014, 167, 47–66 Search PubMed.
- V. Chazel, P. Gerasimo, V. Debouis, P. Laroche and F. Paquet, Radiat. Prot. Dosim., 2003, 105, 163–166 CrossRef CAS PubMed.
- D. E. Crean, M. C. Stennett, F. R. Livens, D. Grolimund, C. N. Borca and N. C. Hyatt, Environ. Sci.: Processes Impacts, 2020, 22, 1577–1585 RSC.
- G. N. Eby, N. Charnley, D. Pirrie, R. Hermes, J. Smoliga and G. Rollinson, Am. Mineral., 2015, 100, 427–441 CrossRef.
- N. Eby, R. Hermes, N. Charnley and J. A. Smoliga, Geol. Today, 2010, 26, 180–185 CrossRef.
- D. Walker, M. Carpenter and C. Hitch, Am. Mineral., 1990, 75, 1020–1028 Search PubMed.
- J. R. Holloway and B. J. Wood, Simulating the Earth: experimental geochemistry, Springer Science & Business Media, 2012 Search PubMed.
- E. Hinteregger, T. S. Hofer, G. Heymann, L. Perfler, F. Kraus and H. Huppertz, Chem. – Eur. J., 2013, 19, 15985–15992 CrossRef CAS PubMed.
- E. Hinteregger, K. Wurst, L. Perfler, F. Kraus and H. Huppertz, Eur. J. Inorg. Chem., 2013, 5247–5252 CrossRef CAS PubMed.
- H. Huppertz, Chem. Commun., 2011, 47, 131–140 RSC.
- H. Huppertz, Z. fur Krist. – Cryst. Mater., 2004, 219, 330–338 CrossRef CAS.
- S. Wu, O. Beermann, S. Wang, A. Holzheid, W. Depmeier, T. Malcherek, G. Modolo, E. V. Alekseev and T. E. Albrecht-Schmitt, Chem. – Eur. J., 2012, 18, 4166–4169 CrossRef CAS PubMed.
- M. E. Garner, B. F. Parker, S. Hohloch, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2017, 139, 12935–12938 CrossRef CAS PubMed.
- J. L. R. Costa, G. S. Marchetti and M. do Carmo Rangel, Catal. Today, 2002, 77, 205–213 CrossRef.
- S. A. Wang, E. V. Alekseev, D. W. Juan, W. H. Casey, B. L. Phillips, W. Depmeier and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 1057–1060 CrossRef CAS PubMed.
- C. Satterthwaite and I. Toepke, Phys. Rev. Lett., 1970, 25, 741 CrossRef CAS.
- M. Krupka, A. Giorgi, N. Krikorian and E. Szklarz, J. Less-Common Met., 1969, 19, 113–119 CrossRef CAS.
- J.-P. Dancausse, E. Gering, S. Heathman and U. Benedict, High Pressure Res., 1990, 2, 381–389 CrossRef.
- A. Jayaraman, G. Kourouklis and L. Van Uitert, Pramana, 1988, 30, 225–231 CrossRef CAS.
- B. Xiao, P. Kegler, T. M. Gesing, L. Robben, A. Blanca-Romero, P. M. Kowalski, Y. Li, V. Klepov, D. Bosbach and E. V. Alekseev, Chem. – Eur. J., 2016, 22, 946–958 CrossRef CAS PubMed.
- R. V. Gaines, Am. Mineral., 1969, 54, 697–701 CAS.
- B. Xiao, P. Kegler, D. Bosbach and E. V. Alekseev, Inorg. Chem., 2017, 56, 2926–2935 CrossRef CAS PubMed.
- J. Lin, J. N. Cross, J. Diwu, N. A. Meredith and T. E. Albrecht-Schmitt, Inorg. Chem., 2013, 52, 4277–4281 CrossRef CAS PubMed.
- P. Bhattacharya, A. H. Welch, K. G. Stollenwerk, M. J. McLaughlin, J. Bundschuh and G. Panaullah, Journal, 2007, 379, 109–120 CAS.
- N. Yu, V. V. Klepov, P. Kegler, D. Bosbach, T. E. Albrecht-Schmitt and E. V. Alekseev, Inorg. Chem., 2014, 53, 8194–8196 CrossRef CAS PubMed.
- Y. Hao, E. V. Alekseev, V. V. Klepov and N. Yu, Eur. J. Inorg. Chem., 2020, 3187–3193 CrossRef CAS.
- N. Yu, V. V. Klepov, H. Schlenz, D. Bosbach, P. M. Kowalski, Y. Li and E. V. Alekseev, Cryst. Growth Des., 2017, 17, 1339–1346 CrossRef CAS.
- S. Wu, S. Wang, A. Simonetti, F. Chen and T. E. Albrecht-Schmitt, Radiochim. Acta, 2011, 99, 573–579 CrossRef CAS.
- G. Heymann, T. Soltner and H. Huppertz, Solid State Sci., 2006, 8, 821–829 CrossRef CAS.
- A. Haberer, G. Heymann and H. Huppertz, Z. Naturforsch. B, 2007, 62, 759–764 CrossRef CAS.
- G. L. Murphy, Z. Zhang and B. J. Kennedy, Complex Oxides: An Introduction, WORLD SCIENTIFIC, Singapore, 2019, pp. 103–130 Search PubMed.
- W. H. Zachariasen, Acta Crystallogr., 1954, 7, 788–791 CrossRef CAS.
- W. H. Zachariasen, Acta Crystallogr., 1948, 1, 281–285 CrossRef CAS.
- X. F. Guo, E. Tiferet, L. Qi, J. M. Solomon, A. Lanzirotti, M. Newville, M. H. Engelhard, R. K. Kukkadapu, D. Wu, E. S. Ilton, M. Asta, S. R. Sutton, H. W. Xu and A. Navrotsky, Dalton Trans., 2016, 45, 4622–4632 RSC.
- G. Murphy, B. J. Kennedy, B. Johannessen, J. A. Kimpton, M. Avdeev, C. S. Griffith, G. J. Thorogood and Z. M. Zhang, J. Solid State Chem., 2016, 237, 86–92 CrossRef CAS.
- P. Kegler, M. Klinkenberg, A. Bukaemskiy, G. L. Murphy, G. Deissmann, F. Brandt and D. Bosbach, Materials, 2021, 14, 6160 CrossRef CAS PubMed.
- L. Johnson, C. Ferry, C. Poinssot and P. Lovera, J. Nucl. Mater., 2005, 346, 56–65 CrossRef CAS.
- G. L. Murphy, P. Kegler, M. Klinkenberg, S. Wang and E. V. Alekseev, Dalton Trans., 2020, 49, 15843–15853 RSC.
- A. Jayaraman, S. Sharma, S. Wang, S. Shieh, L. Ming and S. W. Cheong, J. Raman Spectrosc., 1996, 27, 485–490 CrossRef CAS.
- B. Xiao, P. Kegler, D. Bosbach and E. V. Alekseev, Inorg. Chem., 2016, 55, 4626–4635 CrossRef CAS PubMed.
- G. L. Murphy, E. M. Langer, O. Walter, Y. Wang, S. Wang and E. V. Alekseev, Inorg. Chem., 2020, 59, 7204–7215 CrossRef CAS PubMed.
- N. Yu, P. Kegler, V. V. Klepov, J. Dellen, H. Schlenz, E. M. Langer, D. Bosbach and E. V. Alekseev, Dalton Trans., 2015, 44, 20735–20744 RSC.
- X. Guo, S. Szenknect, A. Mesbah, N. Clavier, C. Poinssot, S. V. Ushakov, H. Curtius, D. Bosbach, R. C. Ewing and P. C. Burns, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6551–6555 CrossRef CAS PubMed.
- A. Mesbah, S. Szenknect, N. Clavier, J. Lozano-Rodriguez, C. Poinssot, C. Den Auwer, R. C. Ewing and N. Dacheux, Inorg. Chem., 2015, 54, 6687–6696 CrossRef CAS PubMed.
- O. Tschauner, S. Huang, S. Yang, M. Humayun, W. Liu, S. N. Gilbert Corder, H. A. Bechtel, J. Tischler and G. R. Rossman, Science, 2021, 374, 891–894 CrossRef CAS PubMed.
- S. Greaux, L. Gautron, D. Andrault, N. Bolfan-Casanova, N. Guignot and M. A. Bouhifd, Phys. Earth Planet. Inter., 2009, 174, 254–263 CrossRef CAS.
- S. Gréaux, F. Farges, L. Gautron, N. Trcera, A.-M. Flank and P. Lagarde, Am. Mineral., 2012, 97, 100–109 CrossRef.
- M. A. Silver and T. E. Albrecht-Schmitt, Coord. Chem. Rev., 2016, 323, 36–51 CrossRef CAS.
- G. Yuan and D. Xue, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 353–362 CrossRef CAS PubMed.
- S. Wu, S. Wang, M. Polinski, O. Beermann, P. Kegler, T. Malcherek, A. Holzheid, W. Depmeier, D. Bosbach, T. E. Albrecht-Schmitt and E. V. Alekseev, Inorg. Chem., 2013, 52, 5110–5118 CrossRef CAS PubMed.
- J. T. Stritzinger, E. V. Alekseev, M. J. Polinski, J. N. Cross, T. M. Eaton and T. E. Albrecht-Schmitt, Inorg. Chem., 2014, 53, 5294–5299 CrossRef CAS PubMed.
- Y. Hao, P. Kegler, T. E. Albrecht-Schmitt, S. Wang, Q. Dong and E. V. Alekseev, Eur. J. Inorg. Chem., 2020, 407–416 CrossRef CAS.
- Y. Hao, G. L. Murphy, D. Bosbach, G. Modolo, T. E. Albrecht-Schmitt and E. V. Alekseev, Inorg. Chem., 2017, 56, 9311–9320 CrossRef CAS PubMed.
- C. Lipp and P. C. Burns, Can. Mineral., 2011, 49, 1211–1220 CrossRef CAS.
- E. M. Langer, P. Kegler, P. M. Kowalski, S. Wang and E. V. Alekseev, Inorg. Chem., 2021, 60, 8419–8422 CrossRef CAS PubMed.
- R. J. Baker, Chem. – Eur. J., 2012, 18, 16258–16271 CrossRef CAS PubMed.
- T. W. Hayton, Dalton Trans., 2018, 47, 1003–1009 RSC.
- E. A. Pedrick, J. W. Schultz, G. Wu, L. M. Mirica and T. W. Hayton, Inorg. Chem., 2016, 55, 5693–5701 CrossRef CAS PubMed.
- S. Schöne, T. Radoske, J. März, T. Stumpf, M. Patzschke and A. Ikeda-Ohno, Chem. – Eur. J., 2017, 23, 13574–13578 CrossRef PubMed.
- J. C. Martz, F. J. Freibert and D. L. Clark, Nucl. Technol., 2021, 207, S266–S285 CrossRef.
- N. Harrison, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4480–4485 CrossRef CAS PubMed.
- A. Svane, L. Petit, Z. Szotek and W. M. Temmerman, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 115116 CrossRef.
- S. Heathman, R. Haire, T. Le Bihan, A. Lindbaum, M. Idiri, P. Normile, S. Li, R. Ahuja, B. Johansson and G. Lander, Science, 2005, 309, 110–113 CrossRef CAS PubMed.
- A. Lukoyanov, A. Shorikov, V. Bystrushkin, A. Dyachenko, L. Kabirova, Y. Y. Tsiovkin, A. Povzner, V. Dremov, M. Korotin and V. Anisimov, J. Phys.: Condens. Matter, 2010, 22, 495501 CrossRef CAS PubMed.
- K. Moore, G. van der Laan, R. Haire, M. Wall, A. Schwartz and P. Söderlind, Phys. Rev. Lett., 2007, 98, 236402 CrossRef CAS PubMed.
- L. Huang, R. Chen and H. Lu, Phys. Rev. B, 2020, 101, 195123 CrossRef CAS.
- S. Heathman, T. Le Bihan, S. Yagoubi, B. Johansson and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 214111 CrossRef.
- J. M. Sperling, E. J. Warzecha, C. Celis-Barros, D.-C. Sergentu, X. Wang, B. E. Klamm, C. J. Windorff, A. N. Gaiser, F. D. White and D. A. Beery, Nature, 2020, 583, 396–399 CrossRef CAS PubMed.
- J. M. Sperling, E. Warzecha, B. E. Klamm, A. N. Gaiser, C. J. Windorff, M. A. Whitefoot and T. E. Albrecht-Schönzart, Inorg. Chem., 2020, 60, 476–483 CrossRef PubMed.
- J. M. Sperling, N. Beck, B. Scheibe, Z. Bai, J. Brannon, D. Gomez-Martinez, D. Grödler, J. A. Johnson, X. Lin, B. M. Rotermund and T. E. Albrecht-Schönzart, Chem. Commun., 2022, 58, 2200–2203 RSC.
- J. M. Sperling, E. Warzecha, C. J. Windorff, B. E. Klamm, A. N. Gaiser, M. A. Whitefoot, F. D. White, T. N. Poe and T. E. Albrecht-Schönzart, Inorg. Chem., 2020, 59, 10794–10801 CrossRef CAS PubMed.
- B. Xiao, M. Klinkenberg, D. Bosbach, E. V. Suleimanov and E. V. Alekseev, Inorg. Chem., 2015, 54, 5981–5990 CrossRef CAS PubMed.
- E. M. Villa, S. Wang, E. V. Alekseev, W. Depmeier and T. E. Albrecht-Schmitt, Facile routes to ThIV, UIV, and NpIV phosphites and phosphates, 2011, 3749–3754 Search PubMed.
- M. Qie, J. Lin, F. Kong, M. A. Silver, Z. Yue, X. Wang, L. Zhang, H. Bao, T. E. Albrecht-Schmitt and J.-Q. Wang, Inorg. Chem., 2018, 57, 1676–1683 CrossRef CAS PubMed.
- G. L. Murphy, Y. Wang, P. Kegler, Y. Wang, S. Wang and E. V. Alekseev, Chem. Commun., 2021, 57, 496–499 RSC.
- G. L. Murphy, P. Kegler, M. Klinkenberg, A. Wilden, M. Henkes, D. Schneider and E. V. Alekseev, Incorporation of iodine into uranium oxyhydroxide phases, Dalton Trans., 2021, 50, 17257–17264 RSC.
- G. L. Murphy, P. Kegler, E. M. Langer and E. V. Alekseev, Crystals, 2021, 11, 965 CrossRef CAS.
- E. M. Langer, O. Walter, J.-Y. Colle, D. Bosbach and E. V. Alekseev, Inorg. Chem., 2018, 57, 1604–1613 CrossRef CAS PubMed.
- P. Toulemonde, C. Darie, C. Goujon, M. Legendre, T. Mendonca, M. Alvarez-Murga, V. Simonet, P. Bordet, P. Bouvier and J. Kreisel, High Pressure Res., 2009, 29, 600–604 CrossRef CAS.
- Y. Zhang, C. Zang, H. Ma, Z. Liang, L. Zhou, S. Li and X. Jia, Diamond Relat. Mater., 2008, 17, 209–211 CrossRef CAS.
- D. Costin, A. Mesbah, N. Clavier, N. Dacheux, C. Poinssot, S. Szenknect and J. Ravaux, Inorg. Chem., 2011, 50, 11117–11126 CrossRef CAS PubMed.
- H. R. Hoekstra and L. H. Fuchs, Science, 1956, 123, 105–105 CrossRef CAS PubMed.
- A. T. Chapman, G. W. Clark, D. E. Hendrix, C. S. Yust and O. B. Cavin, J. Am. Ceram. Soc., 1970, 53, 46–51 CrossRef CAS.
- S. G. Duyker, V. K. Peterson, G. J. Kearley, A. J. Studer and C. J. Kepert, Nat. Chem., 2016, 8, 270–275 CrossRef CAS PubMed.
- N. Campagnol, T. Van Assche, T. Boudewijns, J. Denayer, K. Binnemans, D. De Vos and J. Fransaer, J. Mater. Chem. A, 2013, 1, 5827–5830 RSC.
- L. Paseta, G. Potier, S. Sorribas and J. Coronas, ACS Sustainable Chem. Eng., 2016, 4, 3780–3785 CrossRef CAS.
- D. Gui, W. Duan, J. Shu, F. Zhai, N. Wang, X. Wang, J. Xie, H. Li, L. Chen, J. Diwu, Z. Chai and S. Wang, CCS Chem., 2019, 1, 197–206 CAS.
- Y. Wang, Y. Li, Z. Bai, C. Xiao, Z. Liu, W. Liu, L. Chen, W. He, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt and S. Wang, Dalton Trans., 2015, 44, 18810–18814 RSC.
- Y. Wang, X. Yin, J. Chen, Y. Wang, Z. Chai and S. Wang, Chem. – Eur. J., 2020, 26, 1900–1905 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |