
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the materials space in the smallest particle size range: from heterogeneous catalysis to electrocatalysis and photocatalysis
Juraj
Jašík
 a,
Alessandro
Fortunelli
*b and
Štefan
Vajda
*a
a,
Alessandro
Fortunelli
*b and
Štefan
Vajda
*a
aDepartment of Nanocatalysis J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Science v.v.i, Dolejškova 3, 18223, Prague, Czech Republic. E-mail: stefan.vajda@jh-inst.cas.cz
bCNR-ICCOM, Consiglio Nazionale delle Ricerche, via G. Moruzzi 1, 56124, Pisa, Italy. E-mail: alessandro.fortunelli@cnr.it
First published on 19th April 2022
Abstract
Ultrasmall clusters of subnanometer size can possess unique and even unexpected physical and chemical propensities which make them interesting in various fields of basic science and for potential applications, such as catalysis, photocatalysis, electrocatalysis, and optical and chemical sensors, just to name a few examples. These small particles often offer the tunability of their performance in an atom-by-atom fashion and an economic atom-efficient use of the metal loading. In this paper we review recent progress in the characterization and theory of well-defined subnanometer clusters in catalytic processes, and discuss their optical properties and stability, along with the potential of the size-selected clusters for the understanding of catalytic processes and for the development of new classes of catalysts.
 Juraj Jašík | Juraj Jašík obtained his PhD in Physics of Plasma in 2008 from Comenius University in Bratislava. From 2011 to 2018, he was a member of the group of Jana Roithová at the Department of Organic Chemistry at Charles University in Prague. In 2019, he joined group of Štefan Vajda at the Department of Nanocatalysis at J. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences in Prague. His current research interest focuses on the development and testing of model catalysts based on subnanometer, with atomic precision sized metal clusters supported on solid substrates. |
 Alessandro Fortunelli | Fortunelli obtained his MS in Chemistry (highest honors) in 1983 from University of Pisa and Scuola Normale, which also awarded him a PhD in Theoretical Chemistry. He joined the Italian National Research Council in March 1984, where he is Research Director at the Institute of Chemistry of OrganoMetallic Compounds (ICCOM). He is also Visiting Associate at Caltech, Pasadena (USA). He has developed methods for accelerating computational modelling in molecular and materials sciences, from quantum mechanics to stochastic sampling, and applied these methods to metal nanoclusters/alloys, 2D and nano-materials, amorphous carbon, catalysis (homogeneous, heterogeneous, and electrochemical). |
 Štefan Vajda | Vajda obtained his PhD in Chemistry in 1990 from the Charles University Prague, and in 2003 his Habilitation in Experimental Physics from the Freie Universität Berlin. He joined the Heyrovský Institute of Physical Chemistry in 2019, after a 17 year long tenure at the Argonne National Laboratory, including a one year stay as Detailee at the Department of Energy, Basic Energy Sciences. At the Institute, he currently holds the position of the J. Heyrovský ERA Chair and is the Head of the in 2019 established Department of Nanocatalysis. He has initiated the studies of subnanometer size- and composition selected clusters under realistic conditions of pressure and has developed methods to the testing and characterization of these catalysts under working conditions. His main focus is on the studies of subnanometer and nanometer size clusters in heterogenous catalytic reactions, in collaborations also in electro- and photocatalytic processes. |
Introduction
Designing heterogeneous catalytic systems1 with the desired performance is one of the grand challenges of XXI-century chemistry.2 This challenge has become especially urgent due to the need of transitioning from a linear to a circular economy,3i.e., to an economy based on a sustainable energy cycle, a circular chemicals production and (re-)use, and an environmentally-benign chemistry. Presently available catalysts do not satisfy these societal specifications in all important cases, despite century-long extensive explorations. We thus need to rapidly discover new catalysts/catalytic-sites enabling fundamental industrial processes (from small molecule activation to fine chemistry) under mild conditions, being less energy-consuming, and with a high activity and selectivity.2In this context, we believe that a physical chemistry approach to monitoring catalytic reactions employing a combined, synergic experimental and theoretical approach is key to make real progress not only in understanding but also in controlling and rationally designing new catalysts. In addition, we also believe that the present-day challenges of delivering societally-complying chemical processes can be successfully addressed by exploiting the knowledge drawn from studying highly sophisticated systems, discovered, realized, and characterized in depth: this fundamental knowledge can then open the way to a physical-chemistry-informed rational design of novel nanocatalytic systems. Our focus in this review is on one class of these highly sophisticated systems, that of subnanometer supported cluster systems (reference to larger particles will be introduced as needed). Supported subnanometer clusters present several potential merits and advantages in the catalysis field. As we will see later, their hybrid character, intermediate between organometallic complexes and several-nanometer particles, makes their chemistry in many way unique, i.e., different and tunable at a higher degree than in larger systems. Their economic atom-efficient use of the metal loading is also extremely appealing, especially in view of elemental scarcity and sustainable exploitation of natural resources. Finally, by creating materials in which the metal cluster component is uniquely defined in both size and composition, we streamline the material phase diagram, thus boosting experimental and theoretical characterization, and catalytic testing (coincidence of spatially averaged and local techniques). However, despite these expectations and promises, the actual catalytic potential of ultra-small metal catalysts is presently very far from been exploited, with the lack of fundamental knowledge on structure and properties at this ultra-nano scale being a major reason hindering progress. Subnanometer clusters therefore represent an ideal playground in which a physical chemistry approach to monitoring catalytic reactions can make a breakthrough difference in deriving a fundamental understanding of catalytic processes and simultaneously developing new classes of catalysts. We present selected examples of experimental and theoretical characterization and reaction monitoring, and try to show how theory and experiment, sometimes in a synergistic thrust, sometimes in parallel, complement each other in achieving as atomistically detailed as possible knowledge on the chemical behavior of these systems. While the first era of intense cluster studies was entirely focused on free clusters in vacuum, it was the pioneering studies by Fayet and Wöste et al in the eighties4 and Cox and Kaldor et al. in the nineties5 who built the first bridges towards the interrogation of clusters deposited on surfaces, the first steps in establishing the basics for the expansion into a wide spectrum of research fields and applications of size-selected clusters.
Indeed, this shift of interest from gas-phase to supported small clusters is actually a deja vu. The first pioneering experimental studies of subnanometer metal catalysts in fact dealt with clusters deposited on porous silica or alumina, date back to the years 1970 and 1980, and were conducted by Sinfelt and coworkers at Exxon.6 In these studies, empirical evidence of the potentialities of the field was reported to the best of our knowledge for the first time. Later, these studies were confirmed and the role of subnanometer metal clusters in important reactions was unambiguously demonstrated by EXAFS data showing that in active and efficient nano-catalysts the coordination number of the metals did not exceed 5–6.7
In catalysis, rational design is traditionally associated with “volcano curve” modeling.8 In this approach, the translation of the Sabatier principle9 combined with the Brønsted–Evans–Polanyi (BEP) relationships,10,11 one selects a physical quantity as a “descriptor”, and, assuming that all energy differences and barriers for the given process scale linearly with the descriptor, one searches for the optimal value of the descriptor giving a minimum in the effective barrier (and a corresponding “volcano” maximum in reaction rate).8 Unfortunately, the reaction mechanism of typical industrially-relevant catalytic process is complicated, with many potential rate-determining steps, and can rarely be simplified via a single descriptor volcano curve. In such typical cases, detailed knowledge of atomistic static and dynamic structure of catalytic intermediates and their interconversion as derived from the studies reviewed here seems a pre-condition to make decisive progress to optimal design.
The plan of the review is as follows. We first describe the tools aimed at clarifying the static and dynamic structure of the supported clusters (Section 2), as the basis to draw structure/catalytic-property relationships. Key to the practical use of these strongly under-coordinated species is their stability, addressed in Section 3. Also due to the importance of the field, an increasing number of studies deal with electrochemical rather than gas/solid heterogeneous catalysts, to which Section 4 is dedicated. With a perspective onto photocatalysis, we finally devote Section 5 to the optical properties of subnanometer substrate-stabilized clusters. Section 6 summarizes conclusions. Needless to say that this minireview is not intended to be nor can be all-inclusive. Rather, we deliberately chose to introduce topics such as photocatalysis, electrocatalysis, optical and chemical sensors that are only in their infancy or at a pioneering stage in the subnanometer-cluster field. One of our goals here is in fact to trigger interest on and promote developing new areas that have been scarcely or not at all explored so far, but that we believe are extremely promising in perspective.
Structure and reshaping
Structure
The electronic structure of clusters and the geometry of clusters on a support, including the accompanying cluster–support interaction, and the evolution of these properties as a function of reaction conditions are the factors determining catalytic activity. Clearly, the relative role played by these factors vary with the size of the clusters/particles, with the electronic structure of small clusters especially affected by cluster–support interactions, and adjustable through atomic control of the cluster size and of the hybridization between the electronic states of the adsorbed reactant molecules and of the cluster in order to realize a quantum-controlled catalyst.12–15 Among the various experimental methods to characterize the structure and composition of metal clusters we will first concentrate on microscopy (both electron and tunnel), X-ray and mass spectroscopy as ex situ techniques (Section 2.1), and environmental and scanning microscopy, synchrotron-based, and probe spectroscopy as in situ techniques (Section 2.2).STEM
Aberration-corrected electron microscopy, especially aberration-corrected high-resolution scanning tunnel electron microscope (AC-HR-STEM),16,17 has made important contributions to characterizing supported metal particles down to single atoms. However, accurately determining the structure of supported subnanometer metal clusters in practical catalysts has been a grand challenge. Due to electron-beam induced effects, it is a non-trivial task to keep the atoms and clusters in their pristine configurations and thus provide useful information on the “intact structure” of supported metal atoms and clusters. Electron-beam induced effects and strategies to alleviate them were discussed in ref. 18 and 19.A general view of the characterization of metallic alloy nanoparticles in catalysis using aberration-corrected STEM at atomic resolution is presented in ref. 20, where a clear discussion is provided of the atomic distribution in the metallic nanoparticles affecting the catalytic activity of the nanoalloys, and the capability of high-angle annular darkfield scanning transmission electron microscopy (HAADF-STEM) methods to clarify these features. Down to subnanometer clusters, one of the first applications of AC-HR-STEM with direct atomic-resolution imaging of calcined Au24Pd1 clusters supported on multiwall carbon nanotubes using aberration-corrected STEM was given.21 Theoretical calculations supported the experimental evidence of Au cage structures surrounding the core Pd atom with possible implications for catalytic activity as Pd may act as electron promoter to the surrounding Au atoms when they are involved in catalytic reactions.
Fully exposed Pt3 clusters fabricated on a defective nanodiamond at graphene (ND@G) by the assistance of atomically dispersed Sn promoters were characterized by HAADF-STEM. Results indicate that the size and structure of Pt species are related to the geometric partitioning effect of Sn atoms, indicating that mono-dispersed Sn species facilitate the formation of atomically dispersed Pt clusters. When the atomic ratio Sn/Pt was greater than 1.7, almost all Pt atoms existed in the form of atomically dispersed Pt clusters.22
Fast dynamic STEM combined with a spatio-temporal image denoising algorithm was employed to unveil the dynamic structure of subnanometer Pt clusters anchored to a carbon support.23 The analysis unexpectedly revealed for 30–50 atom Pt clusters a transition from amorphous-like quasi-2D arrangements at room temperature to ordered crystal-like (fcc or hcp) motifs.
For larger particles, size, three-dimensional shape, orientation and atomic arrangement of size-selected gold nanoclusters generated in the gas phase and soft-landed on an amorphous carbon substrate was determined using aberration-corrected STEM.24 The structures of gold nanoclusters containing 309 ± 6 atoms were identified with either Ino-decahedral, cuboctahedral or icosahedral geometries and compared with theoretical modelling. In a further recent development, it has been possible to measure directly the population of competing structural motifs,25 thus providing a quantity which is essential to validate/tune the accuracy of theoretical predictions.
The contribution of theory in this topic has so far concentrated on predicting the relative stability of different isomers and structural families for isolated medium-size and large particles with good success,26–30 although even more sophisticated simulations capturing the effects of electron beam on the cluster structure and thus precisely mimicking the experiment are in principle possible. The field of ultrasmall clusters presents advantages for theory as the limited size of these systems allows one to employ rigorous first-principles modeling, but also specific challenges connected with the need of performing systematic searches via stochastic methods,13 and technical issues such as how to accurately modeling quantum effects31 or cluster/support interaction energetics including e.g. dispersion terms.32 The important role of theory is also to single out energy terms which ultimately determine the cluster structure, such as many-body and directional effects leading e.g. to exotic motifs. To provide simply one example, a new structural family of gold clusters in the size range between 23 and 42 atoms on the MgO(001) surface, the open pyramidal hollow cages, which has no counterpart in gas-phase clusters was proposed by DFT calculations.33
Oxide systems can be investigated as well. For example, organic ligand-free, isolated tantalum oxide complexes (Ta1) and small clusters (Tan>1) on flat silicate supports were prepared by ultra-high vacuum (UHV) techniques followed by oxidation in air. HAADF-STEM analysis of the resulting Tan>1 complexes showed narrow distribution with an size of 7 atoms per cluster, predominantly Ta5+ from X-ray Photoelectron Spectroscopy (XPS) analysis, and binding energies slightly shifted to higher values with respect to the bulk oxide values.34 As another example, preparation of size-controlled tin oxide clusters in mesoporous silica using the dendrimer templating method, employing dendritic phenylazomethine with a tetraphenylmethane core (TPMG4) as the template molecule was reported in ref. 35. HAADF-STEM imaging and XPS analysis showed the presence of both tetravalent Sn(IV) and metastable divalent Sn(II) with a size-dependent composition.
Similar importance bear zeolite supports which are capable of hosting and stabilizing single atoms to well-defined ultrasmall clusters and have been characterized by a variety of techniques,36–48 including zeolites in 2D49 and 1D50 form.
STM
Scanning Tunnel Microscopy (STM) has also been one of the most important tools to achieve information on cluster structures and catalysis.51–61 For this technique, a consolidated synergy between theory and experiment exists, grounded on various decades of combined studies e.g. in the proximal field of ultrathin oxide-on-metal systems.62–65 Functionalized STM tip allows for systematic atomic structure determination with STM of nanoparticles deposited on metal surfaces, with attention to be paid to tip-convolution effects as demonstrated in ref. 66 by a combined experimental/theoretical study of Co clusters (with average diameter 2.5 nm) deposited under ultra-high vacuum onto a thin insulating NaCl film on Au(111) (Fig. 1a).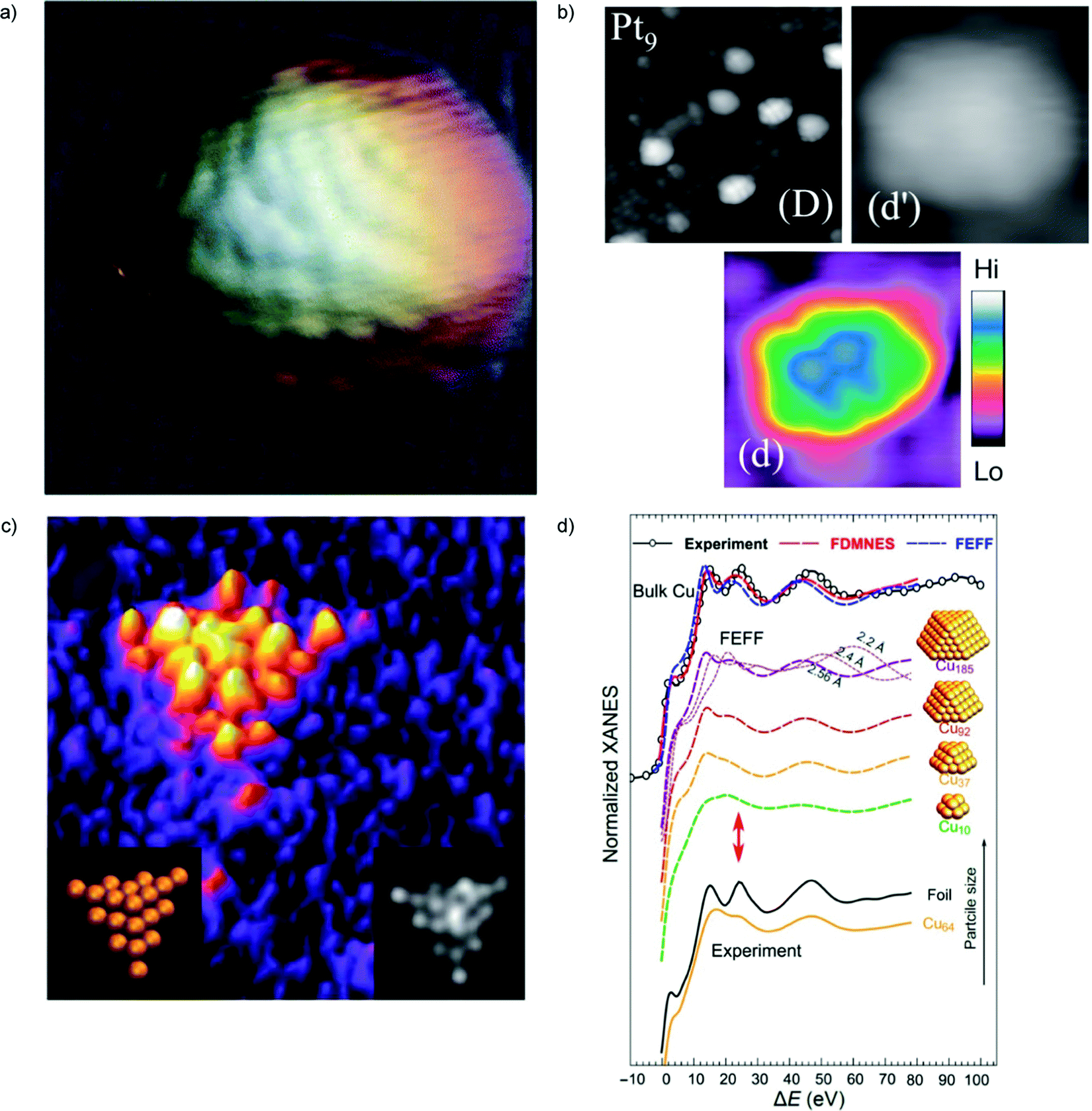 | ||
| Fig. 1 (a) Atomic resolution STM topography image of Co cluster on NaCl–Au(111). Figure size is 7 × 7 nm2. (b) STM images of TiO2(110) surface after deposition of Pt9+ cluster ions. Image (D) is 20 × 20 nm2 and (d), (d′) are 3.5 × 3.5 nm2 views of one cluster on the same surface. A color scale indicates heights for (d′). (c) HAADF-STEM image (2.8 × 2.8 nm2) of a Au20 cluster (left inset shows the orientation of the Au20 cluster; the right inset is the simulated STEM image). (d) Experimental Cu K-edge XANES and theoretical spectra calculated with two ab initio XANES codes (FDMNES and FEFF) for bulk Cu and Cu clusters of different sizes. For the Cu185 cluster, the effect of interatomic distance is also demonstrated based on FEFF simulations. The red arrows mark the feature in XANES spectra that is very sensitive to particle size. (a) is adapted from ref. 66 with permission from the Royal Society of Chemistry, (b) is adapted from ref. 68 with the permission of AIP Publishing, (c) is adapted from ref. 70 with permission from the Royal Society of Chemistry, (d) is adapted with permission from ref. 74. Copyright 2018 American Chemical Society. | ||
Despite the challenges associated with the subnanometer size of the particles, several examples exist in the ultranano cluster field. The morphology of size-selected Ptn (n = 5–13) clusters on a CeO2(111) surface using STM was investigated at room temperature.67 Two-dimensional morphology of Ptn clusters was preferable for n = 5 and a three-dimensional (3D) morphology for n ≥ 6. The preference for a 3D tri-layer structure when n ≥ 10 was observed. The interplay between intra-cluster Pt–Pt and extra-cluster Pt–O interactions (i.e. between the cluster and the CeO2(111) surface) was invoked to rationalize the size-dependent morphology of these Ptn/CeO2(111) clusters.
Atomic-resolution images of size-selected clusters on surfaces enabling the identification of atomic alignments in the clusters on the surface were obtained in ref. 68 (Fig. 1b). The size-selected Ptn (n = 4, 7–10, 15) clusters on TiO2(110) surfaces were scanned using an ultrahigh-vacuum STM with a carbon nanotube tip. The clusters smaller than Pt7 laid flat on the surface with a planar structure. However, a planar-to-three-dimensional transition occurred for Pt8 and both Pt8 and Pt9 had two types of geometric structures.
Sub-molecular resolution in STM imaging was achieved on a 5 nm Ag374 nanoparticle monolayer-protected by tert-butyl benzene thiol.69 The experimental data were compared with simulated topography images from DFT through a pattern recognition algorithm.
Tetrahedral (FCC segment) pyramid structure of Au20 clusters was confirmed using real-space, atomic-resolution images obtained with HAADF-STEM (Fig. 1c). The time-lapse HAADF-STEM imaging shows that the Au20 clusters continuously fluctuate between different structural configurations. The tetrahedral pyramid structure is one of the recurring isomers present in the time-lapse image sequences of the Au20 clusters although lower symmetry structures are more commonly observed.70
In a pioneering experiment, variable-temperature STM was used to study the effect of kinetic cluster energy and rare-gas buffer layers on the deposition process of size-selected silver nanoclusters on a platinum(111) surface.71 At impact energies bellow 1 eV, the clusters landed non-destructively on the substrate, whereas at higher kinetic energies fragmentation and substrate damage were observed. Argon buffer layer on the platinum substrate efficiently dissipated the kinetic energy allowing for soft-landing at elevated impact energy. The opposite case of a hard landing also provides interesting potential for the nanostructuring of surfaces, allowing otherwise thermally unstable structures to be stabilized by the use of defects that were created in the violent deposition process.72
Size-selected Pt cluster ions (size range 5–40 atoms) were deposited on a silicon(111) surface. The surface was characterized using STM under UHV conditions at 77 K. STM images of clusters with different sizes showed that the clusters were flattened and stuck to the surface with a chemical-bond akin to platinum silicide. Platinum atoms occupied preferentially the most reactive sites distributed within a diameter of ∼2 nm on the silicon surface up to a cluster size of 20 atoms, while, above this size, the diameter of the cluster increased with size. The sticking probability of an incoming cluster ion on the surface increased with the cluster size and reached nearly unity at a size larger than 20 atoms.73
X-ray
X-ray and specifically synchrotron techniques represent invaluable tools to furnish information on the structure of cluster catalysts. Small-angle X-ray scattering (SAXS), extended X-ray absorption fine structure (EXAFS), X-ray absorption near-edge structure spectroscopy (XANES), can give precise information about the size and shape of clusters as well as the chemical nature of their metallic components under operating conditions. Agglomeration of the deposited clusters leads to changes in the particle properties and structure that can be different from that in self-standing nanoparticles of similar sizes. In situ studies at different length scales are required to monitor the formation of such complex materials. Combination of SAXS, XANES, ab initio simulations, and machine learning (artificial neural network) techniques was reported in ref. 74. Significant differences between the sizes of particle agglomerates, as probed by SAXS, and the sizes of locally ordered regions, as seen by XANES, were detected. These differences were reported as an evidence for the fractal, grape-cluster-like structure of the agglomerates. From the methodological point of view, it should be noted that this work reports one of the first applications of inverse modeling of experimental spectra (e.g., XANES) to extract atomistic structures. This needs an efficient code to do direct modeling (i.e., a code which, given an atomistic structure, predicts the expected spectrum in – say – less than 1 hour cpu time), which is not the case of first-principles methods, at least so far (Fig. 1d). In multicomponent alloy catalysts, a combination of machine learning and multivariate curve resolution–alternating least square (MCR-ALS) analysis was used to determine the structure of the active site in multicomponent systems.75Another evidence that XANES data are sensitive to size and shape of metallic nanoclusters and neural network can be used to extract structural information from in situ XANES data is provided in ref. 76. Neural network assisted XANES method was used to extract information about the size, shape and interatomic distances in silver clusters, and to monitor their changes during the temperature-controlled particle aggregation showing that particle shape is preserved during the aggregation of Ag clusters in ionic liquid.
A series of size-controlled Pd/hydroxyapatite (HAp) catalysts ranging from single sites (Pd clusters) to nanoparticles (dPd ∼ 30 nm) was prepared and examined in ref. 77. In the subnanosize range, the Pd particles composed of 0D (Pd(IE)), 2D (0.5–2.0 wt% Pd) and 3D (0.5–5.0 wt% Pd) were observed, as identified by XRD, HRTEM, and EXAFS. As it was demonstrated by XPS, in situ diffuse reflectance infrared Fourier-transformed spectroscopy (DRIFTS) of CO, and O2 temperature-programmed desorption (TPD), the electronic structure of the Pd surface can be tailored by varying the particle size. Higher ratios of surface Pd atoms have been found to be positively charged and to form PdOx/Pd interfaces on subnanosized particles.
The geometrical structure of mass-selected Au9 clusters deposited on a silicon substrate prepared by soft-landing conditions was investigated in ref. 78 using grazing incidence small angle X-ray scattering (GISAXS) in combination with a model-based approach. Here the link between structure and observed spectroscopic features is ensured by the use of a simplified (analytic) model, allowing one to obtain the probable morphological information.
Subnanometer Rh clusters derived from Rh-doped SrTiO3 were produced and monitored by in situ XRD and XAFS measurements.79 The in situ DRIFTS measurements were analyzed and suggested that the high CO selectivity in CO2 hydrogenation was attributed to the lower CO binding strength brought about by strong interactions (e.g., charge transfer) between Rh atoms and the defected oxide support see Fig. 2.
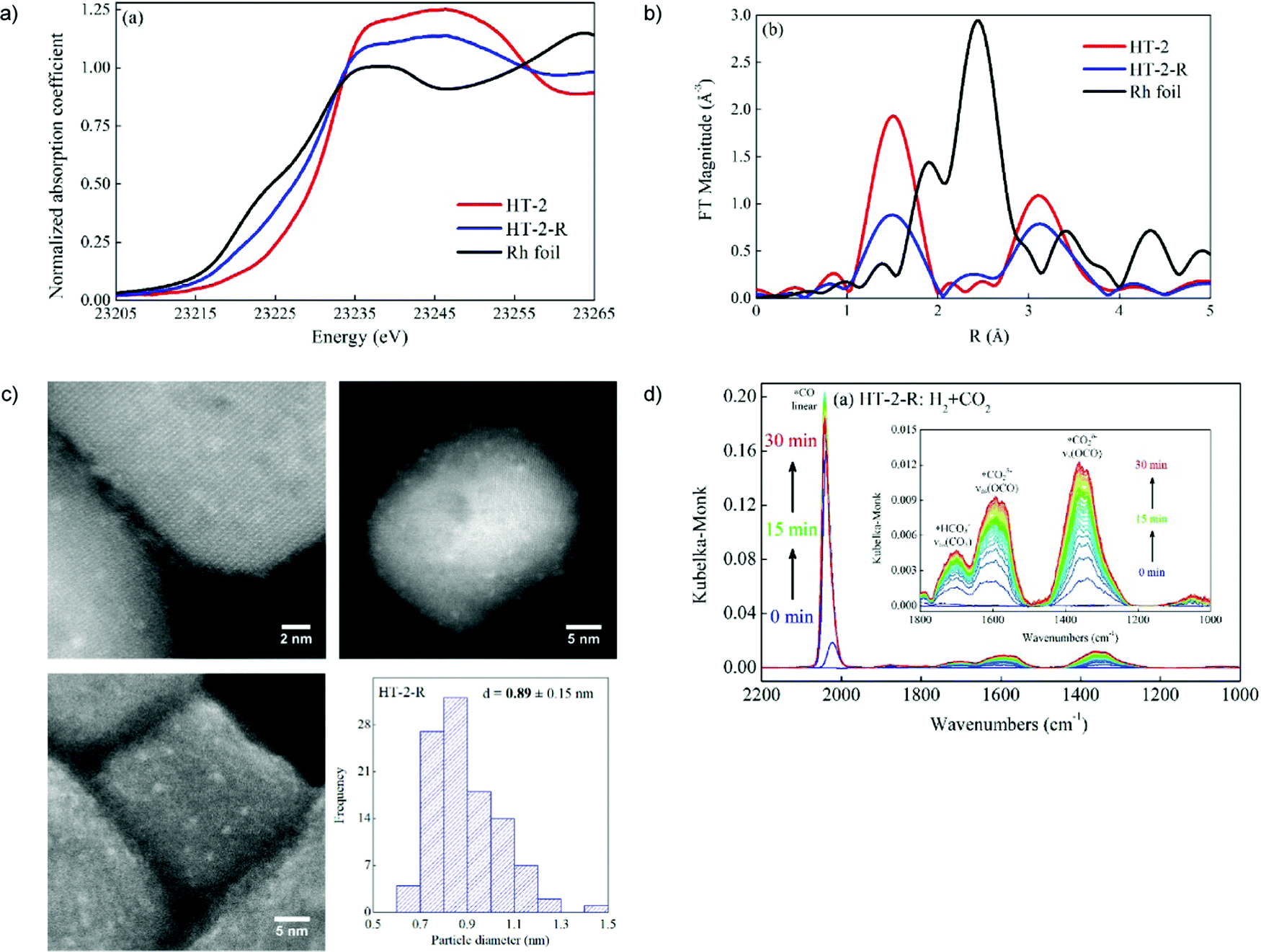 | ||
| Fig. 2 In situ XAFS data of Rh foil, the HT-2 and HT-2-R samples: (a) XANES region, and (b) Fourier transform magnitudes of the k2-weighted EXAFS data. (c) Representative annular dark-field (ADF-)STEM images (top), and high-resolution SEM images (bottom left), and the size distribution of Rh nanoparticles (bottom right) of the HT-2-R sample. (d) In situ DRIFT spectra of CO2 hydrogenation after H2 reduction over HT-2-R. Adapted from ref. 79, Copyright 2018, with permission from Elsevier. | ||
From the preparation point of view, an approach to synthesize atomically dispersed uniform clusters via a cage-separated precursor preselection and pyrolysis strategy was reported in ref. 80 and illustrated in the production of uniform Ru3 clusters stabilized by nitrogen species (Ru3/CN) using well-defined Ru3(CO)12 separated as a precursor by suitable molecular-scale cages of zeolitic imidazolate frameworks (ZIFs). The structure of the Ru3/CN clusters was characterized by HAADF-STEM and XAFS and their oxidation activity of 2-aminobenzyl alcohol investigated via first-principles calculations. Zeolites,36–48 which have been pioneeringly explored by Meurig as a support for subnanometer clusters via decomposition of carbonyl complex precursors,81 including zeolites in 2D49 and 1D50 form, represent another support which is of current considerable interest, despite the great challenges of their structural characterization.
Link with akin systems: ligand-protected metal clusters
The field of supported size-selected metal catalysts presents specifics and advantages that make them unique (see the introduction). However, structural characterization still present problems and challenges, as discussed above, and has not yet reached the targeted ideal, absolute precision. For these and also for synergistic reasons, we believe that the study of these materials would strongly benefit by a closer interaction and comparison with the techniques, results and materials obtained in the related sub-field of size- and composition-selected ligand-protected metal clusters (also called monolayer-protected metal clusters, MPC). The MPC field has been initiated more than 2 decades ago,82 but has witnessed an explosive growth more recently since the first compositionally unique crystalline samples were achieved and their atomistic structure determined via X-ray.83 The crystallization and X-ray structure determination of a p-mercaptobenzoic acid (p-MBA)-protected gold nanoparticle, which comprises 102 gold atoms and 44 p-MBAs was reported in ref. 83. The central gold atoms were packed in a Marks decahedron, surrounded by additional layers of gold atoms in unanticipated geometries. The p-MBAs formed a rigid surface layer due to interaction not only with the gold but also with one another. Since then, nearly 200 distinct MPC compounds have been characterized. The appeal of these materials comes from the possibility of obtaining atomically precise stoichiometry and well-defined crystal structures. Knowledge of stoichiometry and static structure together with the use of advanced experimental and theoretical characterization tools has allowed researchers to achieve an extraordinary accuracy in fundamental studies cross-validating experiment and theory,84 and an in-depth understanding of a wealth of physical phenomena, including e.g. optical absorption and plasmonics.85 We believe that this knowledge can be usefully brought about into the subnanometer size- and composition-selected supported catalyst field, apart from the appealing possibility of using MPC both as precursors for preparing supported species86 and to create hybrid systems combining supported and MPC species e.g. as internal standards in characterization studies (the latter possibility is so far unexplored, to the best of our knowledge). Hereafter and later in the optics section we thus briefly recall a few studies in the MPC field to trigger interest and possible scientific common investigations.Size-dependent optical properties of ligand-protected gold nanoparticles have thus been studied extensively,87 with the catalytic properties more recently also investigated.88,89 Ideal model catalysts should be atomically precise AuNPs with well-defined structures and uncoordinated surface sites as in situ active centers, however, the need to remove ligands can introduce uncertainties in both the structures and sizes of ligand-protected AuNPs for catalytic applications. The discovery of the highly stable tetrahedral (Td) Au20 cluster in the gas phase has inspired exploratory efforts toward its large-scale solution syntheses. In order to keep the tetrahedral structure of this gold pyramid, which has the bulk atomic arrangement with all its atoms on the surface, phosphine ligands were selected.90
An all-amidinate-protected gold nanocluster [Au28(Ph-form)12](OTf)2 (Ph-form = N,N′-diphenylformamidinate) (Au28) has been synthesized and structurally resolved. Single crystal X-ray diffraction reveals that Au28 has a compact Au4@Au24 tetrahedral core–shell structure of T symmetry, which is fully protected by 12 bridging formamidinate ligands. This cluster is quite robust as indicated by the fact that it can stay intact in solution at 80 °C for 6 days. A superatomic electron configuration of 1S21P62S21D4 has been clarified by DFT computations, and the strong gold–ligand binding and geometric shell closure account for the superior stability of Au28.91
Mass spectroscopy (MS), XPS, HR-TEM, FT-IR, UV-vis, and ultrafast spectroscopy techniques are commonly used in this field to unveil composition, morphology, and optical properties of the clusters, see e.g. ref. 92 It is notable that progresses in this field have enabled investigation of monolayer-protected clusters (MPC) up to fairly large sizes. For example, the properties of the ∼300 kDa gold–hexanethiolate compound, extracted from the Faradaurate series of smaller and larger homologues were measured by mass spectrometry, optical spectroscopy, electron microscopy, X-ray scattering, and diffraction.93 A monocrystalline metallic core of ∼3.1 nm minimum dimension supports a clear plasmonic optical response, along with a diffuse exterior shell.
The recent rapid evolution of a multitude of characterization techniques including in realistic reaction environment and often in combination with state-of-the art computational capabilities, allow now tackle the structure–function relationships down at a single atomic level, thus paving the road to the atomic level understanding of and tailoring the properties of matter at the (sub)nanometer scale.
Dynamics, reshaping
Clearly, the atomistic structure of a catalyst is not a static feature, and cluster dynamics and restructuring (reshaping) is an important topic for a deeper understanding of the catalytic event. Especially for ultrasmall systems, fluxional behavior has long been advanced as an important feature13 and evidence that dynamics plays a crucial role in many reactive processes and under many conditions has now accumulated. In the following, we will review this past experience, trying wherever possible to frame it into a set of basic concepts.In particular, we would like to recall the concept of ligand/cluster/support catalytic complex. The theoretical understanding of catalysis by size-selected subnanometer (or ultranano) metal clusters deposited onto an oxide surface is based on three founding concepts: (1) the fluxional character of ultrananoclusters and the consequent need for systematically sampling the potential energy surface of the system, (2) the influence of the underlying support (including possible defects) in terms of geometric relationships, electrostatic and electronic interactions, and the possible active role of the support in the reaction mechanisms, (3) the evolution of the energetics, structure and dynamics of ligand adsorption as a function of coverage, including many-body interactions among ligands. These three concepts can be synthetized into a single one: the formation of ligand/cluster/support catalytically active complex. These species are aggregates involving the metal and support atoms and those of the intermediates produced by the catalytic process, and form in situ with a stoichiometry depending upon the given reaction mechanism and reaction conditions, and are the real actors of the catalytic process. It is, therefore, crucial for assessing efficiency but also the robustness of the catalyst (e.g., in terms of surface diffusion and cluster disaggregation) to single out which is the catalytically active species and which are its features.13
The first aspect, i.e., a tool for systematically sampling the potential energy surface of ultrasmall catalytic systems using a first-principles theoretical approach to heterogeneous catalysis, was provided in ref. 94. There, propylene partial oxidation by Ag3 supported on MgO(100) was studied theoretically. Computational sampling of the reaction path was achieved via a global search in the phase space of structures and stoichiometry combined with filtering, which takes into account the given experimental conditions (catalytically relevant temperature and reactant pressure), and corresponds to an incremental exploration of the disconnectivity diagram of the system. An alternative approach, dubbed global activity sampling method, that directly and globally optimizes the configuration of the supported cluster corresponding to the transition states for the catalytic reaction without requiring prior knowledge on stable cluster structures, was later reported with the example of C–H bond dissociation on Pt7 and Pt8 supported clusters and by an explicit sampling of cluster configurations at C–H dissociation transition states.95
As for the second aspect, i.e., the influence of support, in ref. 96, it was also shown how the presence of an oxide support induces extreme change of the potential energy landscape of the system with respect to the gas phase, favoring configurations that interact positively with the electrostatic field generated by the surface.97
Finally, the reaction energy barriers for the various mechanisms are crucial in the competition between thermodynamically and kinetically favored reaction products. The co-adsorption of O2 and other ligands on small metal clusters have strong cooperative effects (many-body or synergic effects).
STEM
The atom-precise and fully scalable synthesis of platinum clusters on a milligram scale from tiara-like platinum complexes with various ring numbers (n = 5–13) was shown in ref. 98. Atomic resolution STEM showed, unexpectedly, that a Pt10/Ketjenblack catalyst retained its original size features after 2 hours of hydrogenation reaction without obvious aggregation and decomposition. In contrast, in reactions under oxidative conditions, cluster degradation were more pronounced. Formation of single atoms was the mechanism leading to cluster disaggregation and reaggregation.In another study, attention concentrated on the possibility of stabilizing metallic copper clusters under oxidizing reaction conditions by adjusting the atomicity of subnanometer copper clusters.99 By a combination of the XPS and single molecule spectroscopy (SERS) data under the different atmospheres, it was concluded that Cu5 clusters interact weakly with oxygen species and are easier to reduce and more difficult to oxidize than the Cu8 analogues. In contrast, Cu20 nanoparticles were less reducible, while they could be easily reoxidized at 25 °C. DFT simulations supported these findings.
The effect of the size of Cu clusters on the binding strengths of reactants and reaction intermediates, as well as the activation barriers, for the elementary reaction steps underlying CO2 hydrogenation was studied,100 typically finding a linear scaling relationship of reaction barriers with CO and O adsorption energies. Medium-sized Cu19 clusters were predicted to exhibit the highest CO2 hydrogenation activity, ascribed to a compromise between moderate CO2 saturation coverage and a low CO2 dissociation barrier, but all clusters below 55 Cu atoms were predicted to exhibit strong variations of CO and O adsorption energy while the reactivity of larger clusters and nanoparticles was associated with surface atoms with low coordination number.
STEM and XPS characterization before and after reaction to assess the stability of single Pt atoms and Pt clusters on a Si wafer support and the question of heterogeneity versus homogeneity in the catalytic process was addressed in ref. 101. XPS before and after catalysis showed changes in the oxidation states of the clusters, aggregation, and a loss of metal into solution. Characterization of the corresponding Pt clusters supported on TEM grids by means of HAADF-STEM, however, revealed stable clusters before and after exposure to reaction conditions, which was explained by the difference in the support between the silicate TEM grids and the wafers, resulting in different cluster–support interactions.
STM was used to study a Pt1–6/Fe3O4 model catalyst exposed to CO, H2, O2, and mixtures thereof at 550 K. The proposed mechanism foresees that CO extracts lattice oxygen atoms at the cluster perimeter to form CO2, creating holes in the metal oxide surface. H2 and O2 dissociate on the metal clusters and spill over onto the support, the former creating surface hydroxyl groups, ultimately leading to the desorption of water, while the latter reacting with Fe from the support to create new Fe3O4(001) islands. The presence of the Pt is crucial because it catalyzes reactions that already occur on the bare iron oxide surface, but only at higher temperatures102 (Fig. 7b).
The stability of pinned size-selected palladium clusters under reaction conditions (catalytic oxidation of cyclohexane) was investigated by STM measurement both before and after reaction. PdN clusters (N = 10, 20, 55 and 120) were found to be rather stable against sintering under reaction conditions, in a temperature range of 25–300 °C and at a pressure of 800 torr103 (Fig. 3).
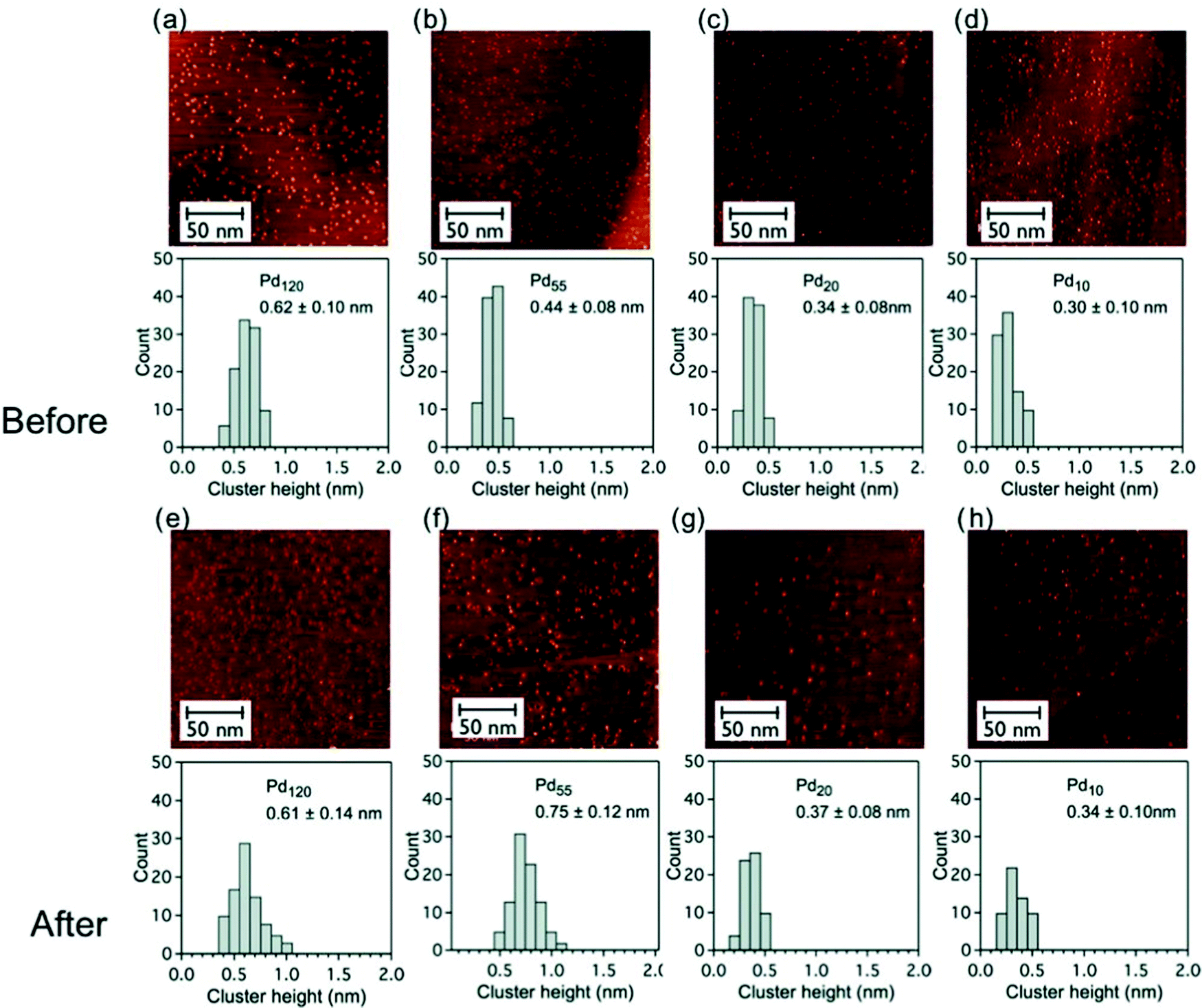 | ||
Fig. 3 Constant current STM images (200 × 200 nm2, 0.4 nA, 0.4 V) of graphite-supported size-selected PdN clusters (N = 120, 55, 20 and 10, low coverage) and histograms of the cluster height distributions before and after catalytic reaction at reactant ratio of C6H12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 = 1:10; (a–d) as-deposited clusters before catalytic measurements and (e–h) after reaction catalytic measurements. The mean height of the cluster is given in each case. Adapted from ref. 103 with permission from Taylor & Francis. :O2 = 1:10; (a–d) as-deposited clusters before catalytic measurements and (e–h) after reaction catalytic measurements. The mean height of the cluster is given in each case. Adapted from ref. 103 with permission from Taylor & Francis. | ||
Sintering of supported Au nanoparticles is significantly different under reaction conditions compared to a vacuum environment.104 The sintering in UHV of Au particles with a size of 2–5 nm occurs above 600 K as it was shown by STM. CO exposure has no apparent effect on the morphology of Au/TiO2(110), whereas significant changes occur after exposure to O2 or CO–O2 at or near room temperature. The Au particle density was greatly reduced in CO–O2, atmosphere because of sintering. Indeed, in ref. 96, it was theoretically demonstrated that the tendency of ultrasmall Au clusters to disaggregation was highly enhanced by the simultaneous adsorption of CO and O2 species, an effect that was mitigated by alloying Au with Ag. An interesting comparison of the catalytic activities reported for platinum group metals and Au catalysts of different sizes in CO oxidation was given in ref. 105. In terms of CO oxidation activity, Au NPs showed remarkable catalytic efficiency at low temperatures, superior to that of the single-atom Au catalyst. Single Pt atoms are superior to Pt clusters at high temperatures above ∼100 °C. Nevertheless, although NPs show catalytic activity for CO oxidation, single Pt atoms do not show activity below 100 °C.105 In their pioneering works, Goodman and coworkers demonstrated the role of Au particle size or the role of gold in dispersing single Pd atoms in highly efficient CO oxidation104,106,107 or the acetylation of ethylene.108
Thermal stability of atomic platinum clusters supported on Al2O3/SiO2/Si(100) as a function of the thickness of the alumina film and presence of hydrogen was studied by in situ GISAX in ref. 109. Extremely high thermal stability of Pt7–10 clusters in vacuo as well as in the presence of hydrogen was observed on SiO2/Si(100) coated with six cycles of Al2O3 film prepared by an atomic layer deposition technique.
Ligation and decomposition of 1,6-hexanedithiol on Cu clusters have been studied using TPD and XPS. Copper cluster anions were size selected and soft landed into a frozen matrix of 1,6-hexandithiol on HOPG maintained at 100 K. After warming up to 298 K, the sample was characterized by a combination of TPD and XPS. Two different binding motifs leading to different decomposition products were observed: the dangling motif is with one sulfur atom binding to a copper cluster, and the bidentate motif is with both sulfur atoms binding to a copper cluster. XPS measurements at varied temperatures confirmed the formation of dithiol ligated copper clusters through Cu–S bond formation, and the decomposition of them via C–S bond scission, consistently with TPD.110
Subnanometer (d = 0.8 ± 0.2 nm) gold particles homogeneously dispersed on amino-functionalized silica catalyze Glaser-type alkyne coupling, providing corresponding 1,3-diynes under mild conditions. The catalyst can be recycled at least five times, giving consistently high isolated yields and maintaining the size and distribution of gold clusters111 (Fig. 5d).
Environmental TEM
Environmental TEM is a technique in which TEM characterization is performed under gaseous pressures up to realistic values, currently typical upper limit being few tens torr but current efforts aim at reaching 1 bar.112E-TEM has already provided information valuable in catalytic studies, and seems especially powerful when combined with concurrent synchrotron characterization techniques, and also in the future with a proper selection of adsorbate gases going beyond typical hydrogen, oxygen and CO molecules. For example, in one of the first in situ studies of nanoparticle dynamical reshaping, the combined use of in situ E-HRTEM, GISAXS, SEM, TPD, and theoretical simulations, led to the idea that the shape can depend on the reaction dynamics/conditions,113 in particular for model size-selected silver large nanoparticle catalysts deposited on amorphous alumina films in the partial oxidation of propylene (Fig. 5b). Notably, the calculated surface energies resulted via the Wulff construction in particle shapes resembling the shapes experimentally observed under realistic reaction conditions of partially oxidized silver clusters, implying that the flow of reactants corresponded to an intermediate oxidation state of the surfaces, a concept now generalized as “steady-state Wulff construction under reaction conditions”.114
A mechanism of oxidation/reduction processes of Pd nanoparticles supported on various substrates, including CeO2, g/d-Al2O3, ZrO2 and SiO2 was studied by means of environmental TEM.115 The observations showed that Pd nanoparticles were transformed to PdO during an exposure to ∼1 mbar of O2 at 400 °C via the formation of surface oxide, and subsequent nucleation and growth of PdO and planar front transformation (Fig. 4).
 | ||
| Fig. 4 High resolution ETEM observation of Pd to PdO transformation at oxygen partial pressure of ∼1 mbar and temperature of 400 °C. (a) Initial Pd nanoparticle supported on CeO2. (b) Transition period corresponding to the formation of surface oxide. (c) Transition period corresponding to growth PdO. (d) Final stage of transformation corresponding to the formation of PdO. Adapted with permission from ref. 115. | ||
The evolution and stabilization of subnanometric Pt species confined in MCM-22 zeolite has been studied by in situ TEM under oxidation–reduction and reaction conditions in ref. 46. Dynamic and reversible transformation between single atoms, clusters and nanoparticles has been observed under CO + O2 reaction conditions at different temperature. The subnanometric Pt clusters can be stabilized in MCM-22 crystallites during NO reduction with CO and H2.
X-ray
Combined in situ grazing incidence small angle X-ray scattering (GISAXS), grazing incidence X-ray absorption near-edge structure (GIXANES) and temperature programmed reaction (TPRx) experiment under a total pressure of 800 mbar was used to monitor the sintering resistance, oxidation state and catalytic performance of the Co4 and Co27 clusters.116 The GISAXS data showed no evidence of agglomeration of the clusters during the course of the reaction. The XANES spectra of the clusters are indicative of Co(II), whereas there is no XANES evidence of Co(III). The spectra of the as-prepared samples suggest that Co clusters occupy various sites on the amorphous substrates.Platinum–titania catalyst was studied by in situ electron microscopy, in situ X-ray photoemission spectroscopy and in situ X-ray diffraction, aided by density functional theory calculations, providing a real time view on how strong metal–support interaction occurs. The migration of reduced titanium oxide, limited in thickness, and the formation of an alloy are competing mechanisms during high temperature reduction. Subsequent exposure to oxygen segregates the titanium from the alloy, and a thicker titania overlayer forms.117
An in situ characterization of Cu nanoassemblies formed transiently from size-selected subnanometer Cu4 clusters supported on amorphous OH-terminated alumina during conversion of CO2 into methanol and hydrocarbons under near-atmospheric pressure reveals that the clusters self-assemble into 2D nanoassemblies at higher temperatures which then disintegrate upon cooling down to room temperature. DFT calculations suggest a formation mechanism of these nanoassemblies by hydrogen-bond bridges between the clusters and H2O molecules, which keep the building blocks together while preventing their coalescence118 (Fig. 5a).
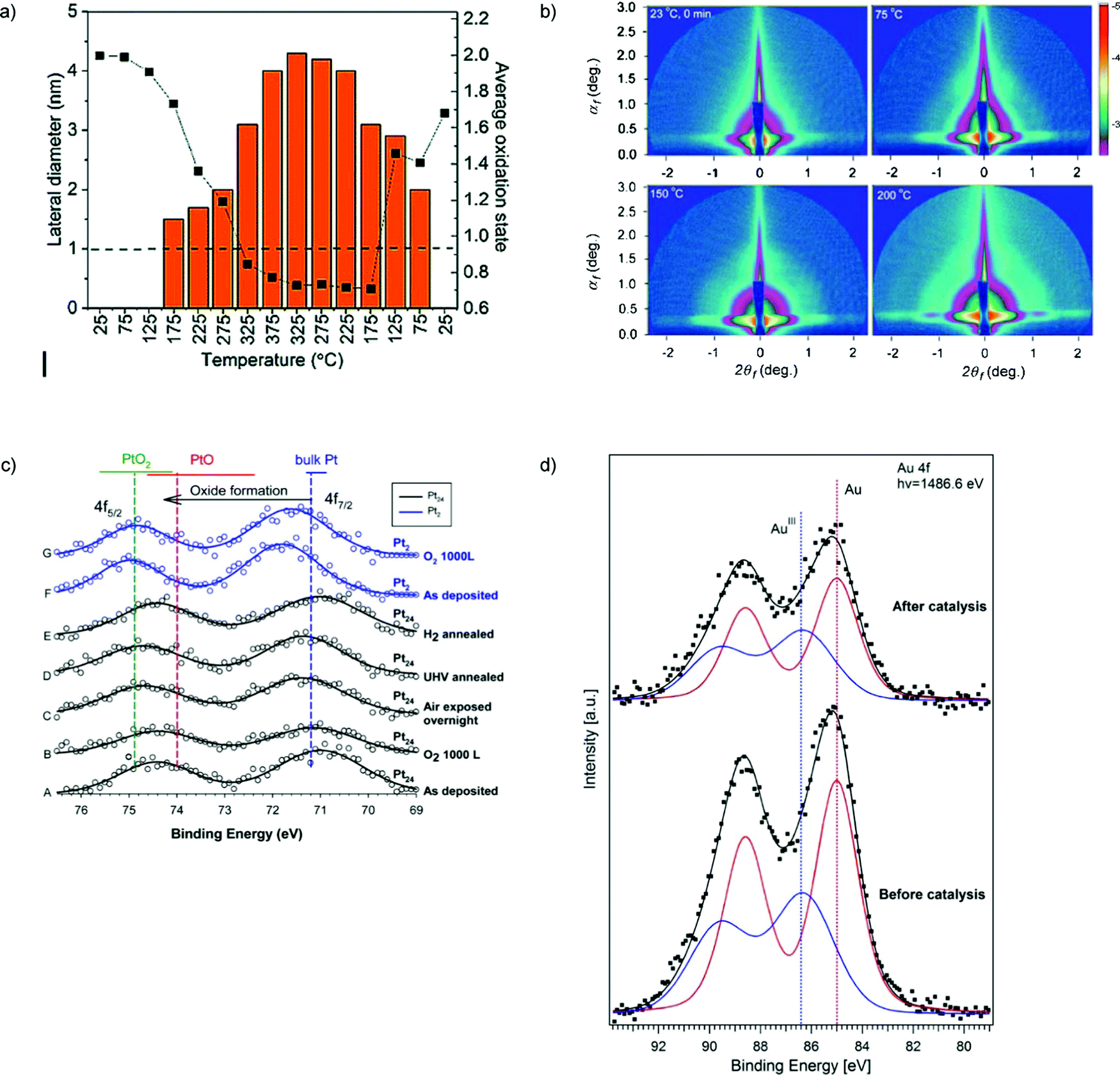 | ||
| Fig. 5 (a) In situ characterization for the Cu4 cluster sample under CO2, H2, and He in a 1:3:1 ratio, supplied at a rate of 5 sccm with 1000 ppm oxygen present. Plot of the change in the oxidation state of copper with the evolution of the particle size of the assembly. The dashed line drawn at 1 nm indicates the cut-off below which the used GISAXS geometry did not allow the resolution of the particle size. The estimated fitting error in the cluster composition obtained from XANES is between 5–10% (relative). (b) GISAXS images on ∼9 nm silver particles. Change of the scattering pattern during ramping the temperature from 23 to 200 °C. (c) (A–E) Pt 4f XPS for Pt24/SiO2/Si: (A) as-prepared in UHV; (B) with 1000 L RT O2 exposure; (C) after overnight air exposure; (D) after annealing to 150 °C in UHV; (E) after annealing at 150 °C in 10−4 torr of H2; (F and G) Pt 4f XPS for Pt2/SiO2/Si, as deposited and after O2 exposure. Horizontal bars show the range of 4f7/2 BEs reported for bulk Pt and oxides. Vertical lines show mean BE values. (d) XPS spectra of subnanometer (d = 0.8 ± 0.2 nm) gold particles supported on 3-aminopropyl-functionalized silicate SBA-15 before and after its use in catalysis. (a) is adapted from ref. 118 with permission from the Royal Society of Chemistry, (b) is adapted from ref. 113, Copyright 2010, with permission from Elsevier, (c) is adapted with permission from ref. 121, Copyright 2016 American Chemical Society, (d) is adapted with permission from ref. 111, Copyright 2017 American Chemical Society. | ||
XPS and STM were used for investigation of structures of Agn cluster anions (n = 3–16) deposited on sputter-damaged HOPG surfaces using a soft-landing technique at room temperature.119 Results suggested that clusters stayed as individual entities with minor agglomeration upon deposition. The deposited Ag clusters were oxidized using atomic oxygen and attempts were made to reduce them with CO, however, such a reduction was not found. The inertness of the small oxidized Ag nanoparticles on HOPG has been attributed to Ag carbonate formation.120
A series of size-selected Ptn/SiO2 samples was analyzed using XPS, low energy ion scattering, GISAXS, and XANES in ref. 121. The oxidation state and morphology were characterized both as-deposited in UHV, and after air/O2 exposure and annealing in H2. The clusters were found to be stable during deposition and upon air exposure, but to sinter when heated above ∼150 °C (Fig. 5c).
A growth of Cu clusters on the TiO2 support, including sintering and oxidation, was modeled using DFT.122Ab initio thermodynamics calculations showed that O2 readily oxidized most clusters, while H2O was a mild oxidant, and CO2 did not oxidize any clusters at relevant temperatures.
In a pioneering study, aberration-corrected STEM was used to analyze several iron oxide–supported Au cluster catalyst samples, ranging from those with little or no activity to others with high activities. High catalytic activity for carbon monoxide oxidation was correlated with the presence of bilayer clusters that are ∼0.5 nanometer in diameter and contain only ∼10 gold atoms123 (Fig. 6c and d).
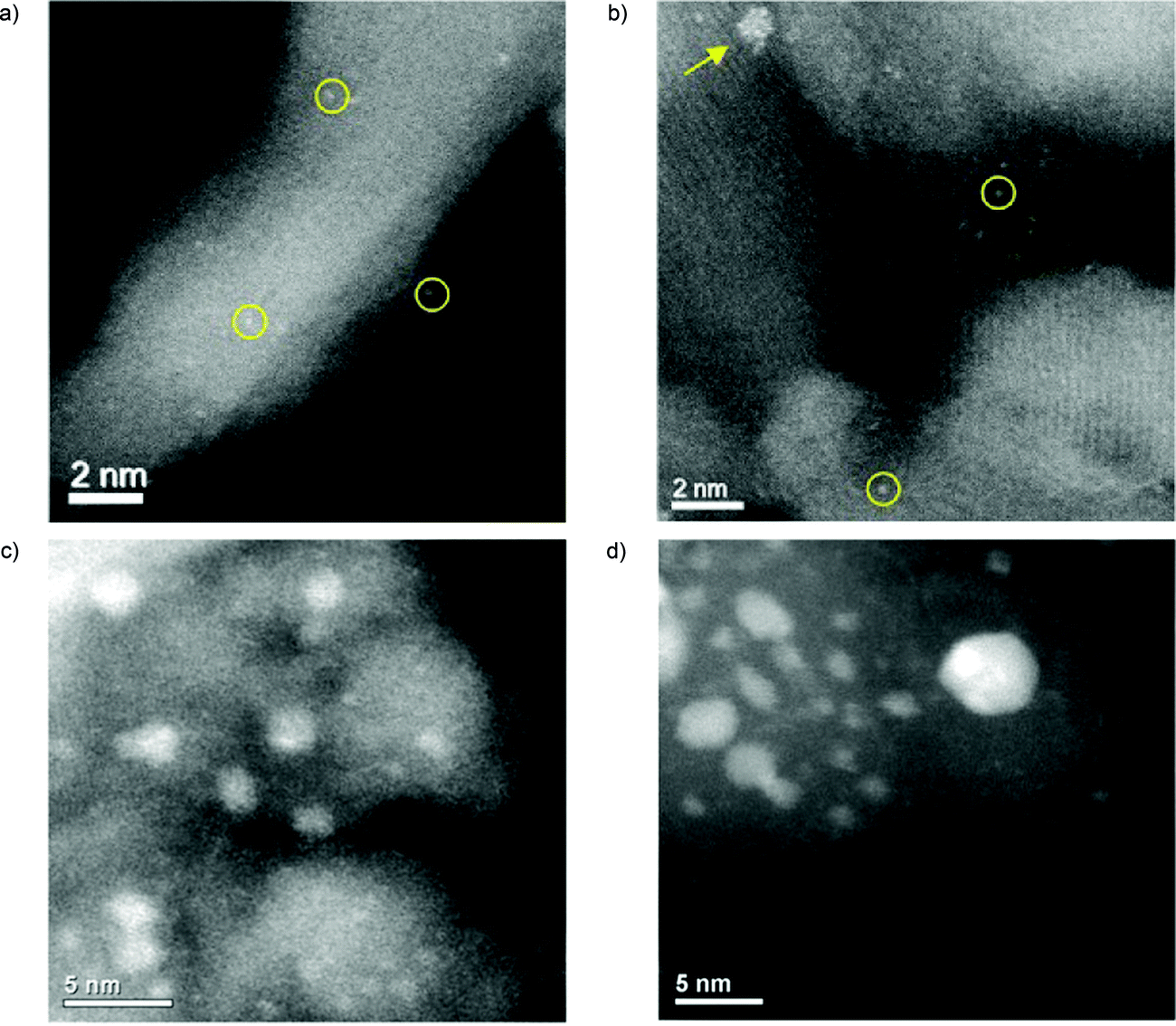 | ||
| Fig. 6 Representative STEM images of Pt/γ-Al2O3 samples through the catalysis experiments. 0.3 wt% Pt/Al2O3 catalyst after calcination (a), and after the full experiment (b) average size of the clusters (such as that pointed by the arrow): 1.0 ± 0.3 nm. Single Ptm+ atoms (m ≥ 2), resulting from the standard impregnation–calcination procedure of SAC preparation, are poorly active. However, they gradually and irreversibly convert into highly active ∼1 nm sized Ptδ+ clusters (δ < 2) throughout the heating–cooling reaction cycles, even under highly oxidizing conditions favorable to atomic dispersion. Aberration-corrected STEM-HAADF images of the active 2.9 atomic% Au/FeOx catalyst 2 calcined for 3 hours at 400 °C (c), and 550 °C (d). The heat-treatment procedures have substantially decreased the population of subnanometer Au clusters relative to the highly active, dried catalyst, while at the same time they have increased the population of particles in the 1 to 3 nm range. (a and b) are adapted with permission from ref. 213, Copyright 2019 American Chemical Society, (c and d) are adapted with permission of American Association for the Advancement of Science, from ref. 123. | ||
Temperature-programmed reaction of CO with O2 catalyzed by Pdn clusters (n = 1, 2, 4, 7, 10, 16, 20, and 25) deposited on rutile TiO2(110) was studied in combination with XPS.124 The Pd 3d binding energy varied non-monotonically with cluster size and the changes correlated with strong size variations in CO oxidation activity. Low activity was correlated with higher-than-expected Pd 3d binding energy, which is attributed to a particularly stable valence electronic structure; electron transfer from the TiO2 support to the Pd clusters also occurs. Ion scattering showed that small clusters form single-layer islands on the surface and that formation of a second layer begins to occur for clusters larger than Pd10.
A liquid-phase-synthesis method to prepare isolated gold atoms supported on functionalized carbon nanotubes was presented and their performance in the oxidation of thiophenol with O2 was studied.125 It was shown that the single gold atoms are not active, but they aggregate under reaction conditions into gold clusters of low atomicity that exhibit a high catalytic activity. When clusters grow into larger nanoparticles, catalyst activity drops to zero.
Paramagnetic clusters possessing a well-defined structure of six equivalent Ag nuclei were prepared by reduction of oxidized silver–sodium containing zeolite LTA using hydrogen. An absorption of O2 and/or C2H4 was inspected by means of continuous wave and pulse electron paramagnetic resonance (EPR) and in situ EXAFS. The unpaired spin density distribution was strongly affected by adsorption of C2H4 at room temperature, and the initially symmetric structure of the reduced Ag+6 cluster turned less symmetric or less compact, lifting the equivalence of the silver atoms. The multiple-shell fitting distinguished a shell that is split into two components of slightly different distances within the limit of EXAFS. The overall coordination number is 3.90 ± 0.07 for the reduced silver cluster consisting of six Ag atoms. The EXAFS results showed that the Ag–Ag coordination number does not change on C2H4 adsorption.126
EXAFS spectroscopy of titania-supported Pd-doped Au nanoparticle catalysts, in the as-prepared and post-catalysis state, shown that the distribution of Pd in Au depends on the chemical history of the samples: as-prepared samples contain highly-dispersed Pd close to the nanoparticle surface, whereas Pd is segregated as larger moieties in the post-catalysis samples. Pd restructures among Au as a result of exposure to the reaction conditions.127
An interesting phenomenon has been unveiled in ref. 128 and 129, i.e., the possibility that clusters change their adhesion to the support as a function of environmental conditions, such as temperature. In the theoretical paper,128 it was shown how increasing temperature of an amorphous silica support leads to de-hydration of the silica surface, in agreement with experimental evidence,130,131 with the consequent formation of stronger adsorption sites for deposited species. In particular, if ultrasmall Au6 clusters are deposited onto such silica support, an orientation transition from clusters singly pinned to the support and therefore perpendicular to it at low T, to clusters doubly anchored to the support and its stronger adsorption sites and therefore parallel to it at higher T is predicted, in excellent agreement with GISAXS measurements on the same system.129
Subnanometer AgxAu3−x/MgO(100) clusters96 transform, after ligand adsorption into reaction complexes which catalyze CO oxidation through a variety of different mechanisms, occurring via both Langmuir–Hinshelwood and Eley–Rideal paths and in some cases directly involving the oxide support. It was concluded that the reaction path for the reaction of an adsorbed O adatom with incoming CO would follow a Langmuir–Hinshelwood mechanism vs. an Eley–Rideal one depending on whether CO adsorption energy on a neighboring site on the cluster was large enough to overcome the loss of the free-energy of gas-phase CO and create a [–CO, –O] co-adsorbed species or whether adsorption was unfavorable with respect to a reactive scattering mechanism of CO “picking up” the O adatom from the cluster (that presented a small energy barrier). This conclusion was claimed to be general: a Eley–Rideal mechanism (e.g., gas-phase CO picking up an O adatom or gas-phase O2 reacting with adsorbed CO to produce CO2 plus an O adatom) should hold in all cases in which (roughly) the absolute values of the entropic free-energy of the gas-phase molecule minus the adsorption energy is larger than the energy barrier for the scattering event (i.e., roughly, the transition state of the adsorption process is higher than the transition state of the scattering event). This claim was supported by a later study of CO oxidation on Ag9Pt2–3 clusters supported on amorphous alumina in which it has been shown that reaction rates predicted along the above assumption are consistent with experiment,132 and in general by the need of assuming adsorption barriers to reconcile predicted and observed catalytic rates for small inert molecule activation at high temperature, see e.g. N2 activation in ref. 133. Notably, the ligand/cluster/support catalytic complex in the case of Ag3 was singled out as a carbonate Ag3(CO3) species. In this case, the formation of the carbonate does not poison the catalyst, on the contrary, it is beneficial for stabilizing the cluster i.e., increasing the barriers to disaggregation and diffusion as demonstrated in successive work.134–136 The alloyed Ag2Au1 cluster was proposed as the best catalyst in terms of efficiency and robustness.96 It is interesting to underline that in successive work it has been shown how the Ag3(CO3) species, which are the ligand/cluster/support catalytic complex for CO oxidation, transform into Ag3(CO3)(NO3)2 species under NO oxidation conditions, which are the ligand/cluster/support catalytic complexes for this reaction, still remaining able to act as a good catalyst,135 a process resembling the catalyst preparation of “real-world” catalyst.
Clearly, dynamics effects can be very important to determine reaction kinetics and therefore to achieve a predictive modeling which can then be turned into rational design. It should be noted that the classical implementation of the Sabatier principle as “volcano curve” modeling8 typically focuses on descriptors based on electronic energy, usually neglecting entropic contributions or assuming they are constant with respect to a reference catalyst. However, as recalled above for the case of CO oxidation on AuxAg3−x and Ag9Pt2–3 clusters, recent research has demonstrated that entropic effects can give substantial contributions to overall reaction free-energy barriers, e.g., stemming from adsorption free-energy barriers of gas-phase molecules at high temperature (such as CO132). A quantitatively predictive modeling must take into account such effects, as done in several recent theoretical work, see e.g. ref. 133, 137 and 138. For example, a recent claim suggests that Brønsted–Evans–Polanyi (BEP) relationships can be broken through fluxional dynamics of the catalyst: indeed, subnanometer gas-phase and graphene-deposited Ptn cluster catalysts were shown to exhibit poor correlation between binding energies of the intermediates, O, OH, and OOH, involved in the scaling relationships for oxygen reduction reaction.138 An advanced view of the issues facing quantitative modeling of entropic phenomena has been recently presented and we refer to this work for more discussion.139
IR and UV-vis
Traditionally, IR and UV-vis spectroscopies have given an important contribution in catalytic studies.140 To quote one clear example, kinetic, isotopic, and infrared studies on well-defined dispersed Pt clusters, in combination with first-principle theoretical methods, on model cluster surfaces, were performed to probe the mechanism and structural requirements for CO oxidation catalysis at conditions typical of its industrial practice.141 In the next section we will report several examples of a more recent implementation of these studies that correspond to operando modality.Cluster stability
The issue of stability is one of the most important in catalysis science. To address the increasingly stricter environmental and sustainability requirements that modern society poses to the field, catalytic processes should be as atom-economical as possible and leach out as little as possible of their active species, which are often precious and sometimes potentially poisonous metals. The issues of catalyst disaggregation and diffusion leading to sintering are thus crucial for the future of the field. It is clear that, also under this respect, the catalyst support cannot be considered as a passive element, but plays a decisive role in maintaining the catalysis in a highly dispersed, intrinsically meta-stable state. Very many groups have therefore investigated the topic of catalyst stability and how to monitor it using physical chemistry techniques. Here we will present first the results of our own investigations, and then complete with reviewing other illustrative cases.For subnanometer cluster catalysts, we start by recalling the concept of ligand/cluster/support catalytic complex.13 As anticipated above, both ligands and support and their interaction, are the factors determining the adhesion of the complex and its energy barriers to diffusion. We have already recalled as an example that the Ag3(CO3) species, generated during COox on Ag3/MgO(100) catalysts, exploits its electrostatic interaction with the substrate to greatly increase its diffusion barriers.96 A further major result in this respect when dealing with oxide, charge separated supports is that, especially at low temperature where e.g. oxide support is hydroxylated,130,131 the structure of the support likely corresponds to an amorphous one. In detail, one of the first in situ studies showing that amorphous support is essential for the stabilization of subnanometer clusters in HUNC is presented in ref. 142. Subnanometer Pt clusters on a crystalline ultrathin alumina film can easily diffuse on the support and sinter already at room temperature, whereas, once deposited onto a similarly ultrathin but amorphous alumina substrate, the ultrasmall Pt clusters are stable up to fairly high temperatures. Even more importantly, in this work, it was demonstrated that the crystalline alumina support transform into an amorphous one under COox realistic reaction conditions, thus bridging the gap between UHV and real-world catalytic systems (Fig. 7a).
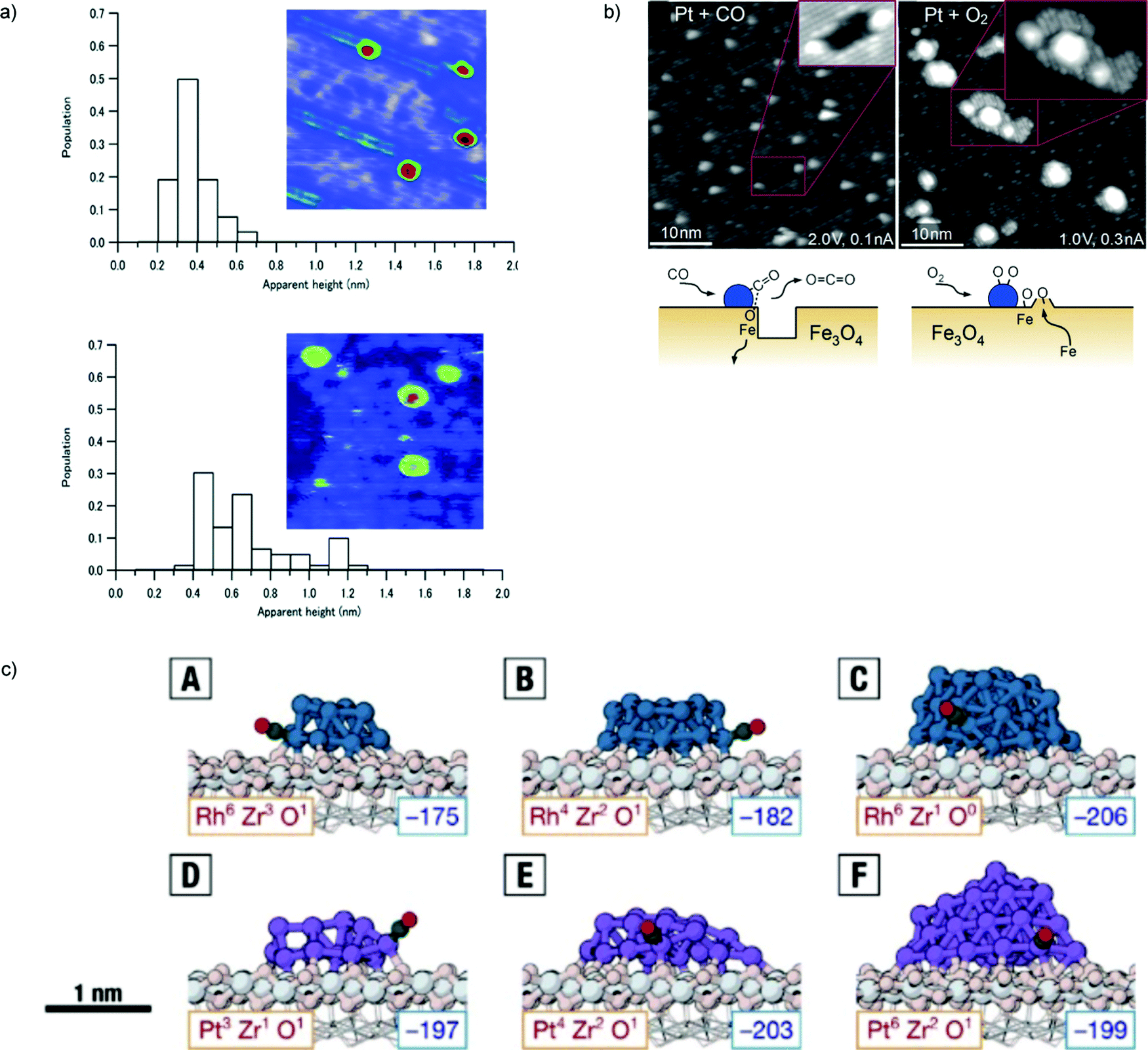 | ||
| Fig. 7 (a) Histograms of apparent height and STM topographic images (24 × 24 nm2, 3.5 Vs, 100 pA) of as-deposited size-selected Pt10 clusters under vacuum at 27 °C (upper part) and after CO and O2 exposure at 27 °C (lower part). (b) STM images acquired following exposure of the Pt/Fe3O4(001) model catalyst to 1 × 10−7 mbar of CO (left), or O2 (right) at 550 K. Following exposure to CO (60 min), approximately 50% of the Pt clusters sit at the edge of or inside monolayer holes in the Fe3O4(001) terrace. The rows (rotated by 90°) of the next Feoct–O layer can be seen inside the holes (inset). The illustration shows how CO extracts Olattice atoms from the cluster perimeter and how CO2 desorbs from the surface. Undercoordinated Fe atoms diffuse into the Fe3O4 bulk. Following exposure to O2 (20 min), all Pt clusters reside on top of islands with a step height of 2 Å. On the larger islands, the rows (rotated by 90°) of the next Fe3O4(001) layer are clearly visible. The islands result from the spillover of oxygen atoms onto the Fe3O4 support, which react with Fe that diffuses out from the bulk. c) Most favorable adsorption of a CO molecule onto the perimeter sites of (A) Rh13/ZrO2, (B) Rh19/ZrO2, (C) Rh43/ZrO2, (D) Pt13/ZrO2, (E) Pt19/ZrO2, and (F) Pt43/ZrO2. The corresponding adsorption energies (in kJ mol−1) are given in the bottom right corners of the panels. The coordination of the corresponding perimeter sites in the absence of CO is given in the bottom left corner of the panels. Color scheme: Rh in teal, Pt in purple, Zr in light gray, oxide O in pink, and CO in green and red. The fixed layers of the supporting oxide are shown in wireframe. (a) is adapted from ref. 142 with permission from the Royal Society of Chemistry, (b) is adapted with permission from ref. 102, Copyright 2015 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, (c) is adapted with permission from ref. 160. | ||
Concerning diffusion, we have conducted a series of systematic studies of this phenomenon143 as the main effect controlling aggregation, Ostwald ripening and sintering. For model systems, an excellent agreement was achieved between the predicted kinetics of diffusion and aggregation and the experimentally observed one as a function of temperature,144 thus validating the theoretical approach.
A competing pathways theory for sintering, which, at the atomistic level, describes the found size-specific sintering behavior, was proposed in ref. 145.
The analogy between homogeneous catalysis and heterogeneous catalysis by very small, subnanometer-size metal clusters, as proposed in ref. 134, seems promising to widen the spectrum of heterogeneous catalyst design and realize synergistic investigations with accumulated knowledge on homogeneous systems. The ligand/cluster/support complexes generated in situ under reaction conditions may be rather different from the original preparation, but it is their properties, and how to tune them, that must be the focus of operative research. Indeed, the ligand/cluster/support catalytic complex generated by CO oxidation over silver trimers deposited on the regular MgO(100) surface – i.e. a Ag3/carbonate or Ag3(CO3)/MgO(100) species – can be used as a catalyst in a different reaction.135 The Ag3(CO3)/MgO(100) species, however, transforms under NOox conditions into an even more complex aggregate, a mixed carbonate/double-nitrite Ag3(CO3)(NO2)2/MgO(100) species, which can then act as an efficient catalyst of NOox. Under NOox reaction conditions a different ligand/cluster/support catalytic complex is formed with respect to the original COox one. It is such diversity of the catalytic chemistry of ultranano metal clusters deposited on oxide substrates, the formation of many different ligand/cluster/support aggregates, and the vast amount of combinatorial possibilities thus opening, which makes detailed physico-chemical studies, including stochastic structural and stoichiometric computational searches, indispensable in order to cope with such a multiform diversity.
Dopant atoms may act as anchoring centers
The high activity and stability of Ag–Pt sub-nanometer clusters, as heterogeneous catalysts in the CO → CO2 reaction (COox), are ascribed to a synergic role of Ag and Pt in ultranano-aggregates.132 It is notable that Pt anchors the clusters to the support and binds and activates two CO molecules, while Ag binds and activates O2, and Ag/Pt surface proximity disfavors poisoning by CO or oxidized species. We recall once more that this Ag9Pt2–3 case study has been important to elucidate the important role of entropic effects (such as adsorption energy barrier) on catalysis, a role particularly crucial in the activation of small inert molecules, that require high temperatures to favor un-coverage of catalytic sites as well as bond breaking, however thus facing the issue of overcoming entropic barriers.The interaction between single metal atoms and support, as well as surface properties of supports, control the catalytic behavior of SACs. A systematic investigation of the effects of supports on CO oxidation by single Pt (Pt1) atoms dispersed on different metal oxides (highly reducible Fe2O3, reducible ZnO, and irreducible γ-Al2O3) found that Pt1 atoms on three metal oxides are active for CO oxidation, and the chemical properties of supports determine the catalytic performance of Pt1 single-atom catalysts (SACs). Both the presence of –OH groups on support surfaces and the addition of H2O significantly modify CO oxidation on three SACs and reduce the effects of supports on their catalytic performances.146 Note however that it has recently been claimed that dopant atoms can increase the reducibility of irreducible supports.147,148
Doping, alloying and support effects on stability and catalyst performance
Modification of ultrasmall clusters by doping, overcoating, changing size and composition with atomic precision or through cluster support interactions provides high-fidelity tuning knobs to control not only their stability, but their activity and, importantly, their selectivity as well.75,116,124,132,149–157Investigation of atomically dispersed Pt/TiO2 catalysts by pre-/postreaction scanning transmission electron microscopy and operando X-ray absorption spectroscopy–mass spectrometry under gas-phase ethanol dehydrogenation conditions under ultraviolet–visible light irradiation shows that the general effect of the reaction on initially fully oxidized (Pt IV) single-atom catalysts (SACs) is Pt reduction and aggregation. This combined phenomenon can be inhibited in two manners. First, when it is supported on high-surface-area anatase, the Pt SAC retains an intermediate oxidation state, and its clustering is limited to Pt dimers and trimers. Second, with the same support, a mild reducing pretreatment generates, through the formation of Pt–Ti bonds, near-neutral and ultradispersed Pt species (from single atoms to nanometric clusters) with the highest stability and activity in photocatalytic hydrogen evolution.158 Platinum complexes supported in zeolite KLTL remained site isolated in the zeolite pores after oxidation and catalysis of CO oxidation44 (Fig. 8a).
 | ||
| Fig. 8 STEM images showing site-isolated Pt atoms in KLTL zeolite in the (a) oxidized and (b) as-prepared samples. White features in dashed blue circles indicate Pt atoms. Magnified views (c–e) of the highlighted regions in (b), containing one Pt atom each at A/B sites in (c), at C/E sites in (d), and at D sites in (e). Simulations (f and g) of the LTL zeolite in the110 direction superimposed on the magnified views in (c–e), showing Pt atoms (green) at A/B sites in (f), at C/E sites in (g) (purple), and at D sites in (h) (red). Pt atoms are located right at the edge of the 12-membered rings of site D; between the two 12-membered rings of sites C/E; and in the center of three 12-membered rings of sites A/B. Adapted with permission from ref. 44, Copyright 2014 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The importance of dispersion effects on the cluster/support interaction was studied in ref. 32 where the energetics of an Au adatom and AuN clusters (N = 2–6) supported on pristine and reduced MgO(100) surfaces was analyzed using an all-electron full-potential density functional theory approach. For AuN clusters (N > 1), dispersion effects start to make an increasingly important contribution to the adhesion energy.
Dispersion corrected density functional theory (DFT-D) was used to study the effect of commonly used support materials (MgO, C, CeO2) on small gold particles with up to 19 atoms.159 The preferred cluster shape and morphology is highly dependent on the support material due to different adsorption strength and structural mismatch between the cluster and the surface material. Depending on the support material, the gold clusters exhibit a positive or negative polarization, which ultimately has strong implications on the catalytic activity of such particles.
A DFT-based genetic approach to obtain globally optimized nanostructures for Rh and Pt clusters on a ZrO2 support was employed in ref. 160 and demonstrated that even an inert support can intricately influence the reactivity of interfacial sites. The analysis of the obtained structures showed that Rh clusters had more compact shapes, whereas Pt preferred elongated and low-symmetry structures. It was found that metal–oxide perimeter sites are structurally different, presenting varying Pt and Rh coordinations and CO adsorption energies. The presence of a support always destabilizes CO adsorption at the cluster edge, but the magnitude of destabilization varies substantially from site to site (Fig. 7c).
Small Cu clusters deposited on alumina support are more susceptible to oxidation than those in gas phase.161 This result was explained in terms of strong cluster–support interactions, which differ significantly for the oxidized and elemental clusters. The stable cluster phases also feature novel oxygen stoichiometries. The results suggest that one can tune the degree of oxidation of Cu catalysts by optimizing not just their size, but also the support they are deposited on. Effect of the morphology of the support on nanocatalysts performance in case of CO2 methanation catalyzed by copper clusters was also pronounced in ref. 162. The Cu4 supported by nanostructured zirconia (NS ZrOx) was found to be about 30% more active compared to Cu4 supported by atomic layer deposited zirconia (ALD ZrOx), whereas Cu12/NS ZrOx was 100% more active compared to Cu12/ALD ZrOx. However, the activity of Cu12/NS ZrOx dropped during the repeated cycle, most likely due to the possible migration of the clusters into the pores and voids with more limited access to reactants.
The strong effect of SiO2 and MgO support on the activity of ethylene hydrogenation catalyzed by size-selected platinum clusters (8–20 atoms per cluster) was observed.163 Predicted trends based on the acidic (SiO2) or basic (MgO) properties of the support are subtle, with the influence of the support also being a function of the cluster size. Pt13 had the lowest hydrogenation activity of all sizes on amorphous SiO2, while being the most active size on MgO. Adding Sn to size-selected Pt clusters deposited on amorphous SiO2 also dramatically enhances the thermal stability of the clusters against sintering. The binding energy of Pt clusters to the support increases by >1 eV upon adding Sn, and Sn forms strong polar covalent bonds with Pt within the clusters and quenches all of the unpaired spins which tends to stabilize the Pt core of the clusters against sintering.150
An operando generated stable Ni–In kinetic phase was discovered that selectively converts CO2 to methanol (CTM) at low pressure compared to the state-of-the-art materials. The catalytic nature of a well-known methanation catalyst, nickel, has been tuned with the introduction of inactive indium, which enhances the CTM process. The remarkable change in the mechanistic pathways toward methanol production has been mapped by operando diffuse reflectance infrared Fourier transform spectroscopy analysis, corroborated by first-principles calculations. The ordered arrangement and pronounced electronegativity difference between metals are attributed to the complete shift in mechanism.164
A novel operando XAFS technique was developed called the operando polarization-dependent total reflection fluorescence (PTRF)-XAFS technique, which can provide information on the valence state (XANES) and three-dimensional (3D) structure (EXAFS) of active metal species dispersed on a well-defined single-crystal surface during catalytic reactions. The technique was applied to a Pt/α-Al2O3(0001) model catalyst during the CO oxidation reaction. Pt clusters that favor an icosahedral Pt55 structure were formed on the α-Al2O3(0001) surface after Pt deposition at room temperature, while they were converted to larger cuboctahedral clusters (Pt147) under the CO oxidation reaction at 493 K. The turnover frequency (TOF) of the CO oxidation activity at 493 K was also estimated to be 0.06 s−1 from simultaneous QMS and XAFS measurements.165
High flux density X-ray beam on the catalyst used for in situ and operando studies can affect the catalyst structure. It was shown that the high flux density causes the reduction of the highly dispersed RhOx/Al2O3 catalyst, even at room temperature. Additionally, exposure to the higher flux density X-rays during in situ reduction results in significant agglomeration of the Rh clusters. The final size of the Rh nanoparticles reduced at 310 °C is equivalent to that of particles formed after the reduction at 600–650 °C in the absence of the beam. Significant beam-induced reduction and agglomeration is also shown for Ni supported on beta zeolite during in situ reduction at an intermediate-flux-density, indicating that beam-induced changes in heterogeneous catalysts could be common at intermediate- and high-flux-density beamlines.166
Zeolites and MOFs
In zeolite-like systems, ultra-small cluster catalysts have been deposited since a while.81 Zeolite-confined noble metals have unique confinement effects on a molecular scale, and thus enables spatially confined catalysis akin to enzyme catalysis.47 Most of the currently available modeling tools are introduced and illustrated on the most challenging problems in zeolite science in review.48The combined experimental–theoretical approach, encompassing advanced molecular dynamics simulations and in situ micro-spectroscopy, gives a profound insight into the role of water in the catalytic process at the molecular and single-particle level in the methanol-to-olefins (MTO) process over H-SAPO-34.167
Mordenite micropores provide a perfect confined environment for the highly selective stabilization of trinuclear copper–oxo clusters that exhibit a high reactivity towards activation of carbon–hydrogen bonds in methane and its subsequent transformation to methanol.43 Structure determination of the copper–oxo active site was determined by DFT calculations and verified by XAS analysis. The reactivity data point to a uniform nature of the Cu sites in the Cu-MOR materials and, therefore, Cu K-edge spectra may yield direct information of the structure of the active clusters. In situ spectroscopy methods (XANES, EXAFS and UV-vis) were used to monitor the formation of the cluster during activation under O2 and its interaction with CH4 under reaction conditions.
Subnanometric hybrid Pd-M(OH)2 (M = Ni, Co) clusters encapsulated within purely siliceous zeolites were synthesized via a hydrothermal synthesis method.40 The hybrid bimetallic nanocatalysts exhibit superior thermal stability at 600 °C–700 °C because the bimetallic hybrid clusters are well confined within the zeolite matrix. The subnanometric sizes of the hybrid bimetallic Pd and Ni(OH)2 clusters were confirmed using STEM and EXAFS analyses.
Metal–organic frameworks (MOFs) have a big potential as platforms for the development of heterogeneous single-site catalysts.168 Single atoms and few-atom clusters of platinum were uniformly installed on the zirconia nodes of a MOF NU-1000 via targeted vapor-phase synthesis.169In situ IR spectroscopy revealed the presence of both single atoms and few-atom clusters that depended upon synthesis conditions. Operando X-ray absorption spectroscopy and X-ray pair distribution analyses revealed changes in chemical bonding environment and cluster size stability while on stream. Density functional theory calculations elucidated a favorable reaction pathway for ethylene hydrogenation with the novel catalyst (Fig. 11b).
Ceria
Silver single atoms and small clusters supported on a model Ce21O42 nanoparticle have been studied computationally at a DFT+U level.170 It was found that silver atoms, trimers and tetramers are oxidized upon interaction with the (100) and (111) nanofacets of the ceria particle and adsorption of silver species at (100) facet is ∼0.5–1.0 eV stronger than to (111) one. Ag3 are adsorbed as upright triangles; Ag4 prefer tetrahedral configuration. Denucleation of silver clusters to atomic species at (100) facet is the most favored.Effect of ligands on electronic properties
Ligands can also strongly affect redox properties of clusters as it was shown in theoretical study.171 Attaching phosphine ligands to simple metal, noble metal, semiconducting, metal–oxide, and metal–chalcogen clusters is shown to severely reduce ionization energies in all classes of clusters. Ligation is shown to be an outstanding strategy for the formation of multiple electron donors.Clusters in electrochemical applications
An overview of studies performed under both realistic reaction conditions aimed at the interrogation, characterization, and understanding of the performance of supported size-selected clusters in heterogeneous and electrochemical reactions, which address the effects of cluster size, cluster composition, cluster–support interactions, and reaction conditions, the key parameters for the understanding and control of catalyst functionality, was provided in ref. 172. Several ultrasmall systems have been studied in this context. Without pretending to be exhaustive, we will discuss a few cases below which further underline the importance of a multi-probe characterization of subnanometer size clusters along with the atomic-precise tunability of their performance in various electrochemical and electrophotochemical processes, such as oxygen or hydrogen evolution reaction, CO2 reduction or batteries.Pd4, Pd6, or Pd17 clusters soft-landed on a ultrananocrystalline diamond (UNCD) Si-coated electrode show stable electrochemical potentials over several cycles, and synchrotron studies of the electrodes show no evidence for evolution or dissolution of either the electrode material or the clusters173 under for the oxygen evolving reaction (OER). Even under the harsh experimental conditions (basic, high potential) typically employed for water oxidation catalysts, UNCD exhibits a very wide potential electrochemical working window and only minor evidence of deterioration, and with Pd clusters decorated, these electrodes reached the highest reported rates of oxygen formation based on metal loading.
Arrays of discrete iridium clusters with identical metal atom number (Ir2, Ir4, or Ir8) were deposited in submonolayer coverage on conductive oxide supports, and the electrochemical properties and activity of each was evaluated.174 Exceptional electroactivity for OER was observed for all cluster samples in acidic electrolyte. Reproducible cluster-size-dependent trends in redox behavior were observed and rationalized via first-principles modeling of the individual discrete-size clusters.
Oxygen reduction (ORR), as well as ethanol oxidation on subnanometer size-selected Pt clusters showed high activity as well as non-linear dependency of activity with cluster size, discussing activity correlated to the electronic structure of the Pt clusters.175
The possibility of realizing low overpotential electrocatalytic conversion of CO2 using subnanometer copper clusters deposited on glassy carbon was demonstrated in ref. 176 where the electrochemical behavior (in the presence of N2 or CO2) of size-controlled naked Cu5 and Cu20 nanoclusters was studied. Cu20 nanoclusters cause anodic redox processes stirring at much lower potential with respect to Cu5 nanoclusters, which behave relatively similar to much larger Cu particles. However, Cu5 nanoclusters coordinate effectively CO2 in solution, in comparison to Cu20 nanoclusters and larger Cu particles. This effect, rather than the redox behavior, is evidently connected to the ability of Cu5 nanoclusters to reduce CO2 under cathodic conditions at low overpotential and indicate the tunability of electrochemical performance within the subnanometer size range.
Size-selected cobalt clusters (Co27) deposited on hematite (Fe2O3) anodes were tested for water oxidation activity in ref. 177. The Co27 clusters are stable to dissolution even in the harsh water oxidation electrochemical environment. The turnover rates for water oxidation are comparable or higher than those reported for Pd- and Co-based materials or for hematite. Annealing of the samples (with the clusters already deposited) significantly improved the electrocatalytic and the photoelectrocatalytic activity of the Co27/Fe2O3 material, another control knob in the design of high-performance subnanometer cluster-based materials.
In the study described, subnanometer sized palladium clusters accommodated into porous ceria were synthesized and showed highly efficient electrocatalytic activity and stability for the hydrogen evolution reaction (HER).178 The synthesized Pd subnanoclusters have a size of about 0.5 nm as it was shown by characterizations from high-resolution transmission electron microscopy (HRTEM) and extended X-ray absorption fine structure (EXAFS). Density functional theory suggested that the active centers are the subunit of Pd clusters rather than CeO2 support based on the free energy of absorption for hydrogen atoms (ΔGH*). Compared to the free Pd nanoclusters, the electron transfer between Pd and O atoms in Pd NC@CeO2 can effectively modulate the adsorption state of H* on the subunit of the Pd cluster, and this cojoint effect can effectively promote the HER catalytic activity of the composite system.
The degradation of size-selected Pt nanoclusters was studied under electrochemical conditions by HAADF-STEM and confirmed by transmission electron microscopy (TEM).179 It was demonstrated that even extremely small Pt clusters exhibit a remarkable stability under electrochemical conditions. Furthermore, applying mixed cluster samples of Pt22 and Pt68, no preferential dissolution of Pt22 by Ostwald ripening – usually held responsible to be the main mechanism for activity loss in Pt fuel cell catalysts – was observed.
Another direction of great interest for future developments is the Hydrogen Evolution Reaction (HER). Indeed, it has been demonstrated that subnanometric metal clusters can be used as photocatalysts for H2 production in the presence of holes or electrons scavengers by water splitting.180
Copper–platinum (Cu–Pt) dual sites were alloyed with palladium nanorings (Pd NRs) containing 1.5 atom% Pt, using atomically dispersed Cu on ultrathin Pd NRs as seeds. The ultrafine structure of atomically dispersed Cu–Pt dual sites was confirmed with EXAFS measurements. The Pd/Cu–Pt NRs exhibit excellent HER properties in acidic solution, also supported by DFT calculations181 (Fig. 9c).
 | ||
| Fig. 9 (a) Snapshots of the structures and v-DoS CO2 (l-CO2), chemisorbed CO2 (b-CO2), *COOH, *CO, and *CO32− from o-QM-MD simulations and 2PT analysis. The color codes are Cu in orange, O in red, C in silver, and H in white. The hydrogen bonds are shown as dashed red lines. In bottom part, v-DoS from o-QM-MD is shown as a solid black line, the experimental frequencies are shown as a red dashed line, and the vibrational frequencies from v-QM optimization are shown as solid blue lines for comparison. (l-CO2 and b-CO2 are the same from v-QM, because b-CO2 is unstable in vacuum and automatically converts to l-CO2 after optimization.) (b) The simulated potential-dependent v-DoS of *HOC-COH at +0.21 V, −0.33 V, and −0.76 V. Inset shows the blue shift of the 1191 cm−1 peak (a feature of C–O stretch of C-OH) as a function of applied potential. (c) Characterization of Pd/Cu NRs. From left to right: TEM image; HAADF-STEM image and the corresponding STEM-EDX elemental mapping; atomic resolution HAADF-STEM image. (a) and (b) are adapted with permission from ref. 187, (c) is adapted with permission from ref. 181, Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Indications for the theoretical design of HER subnanometer catalysts were drawn in ref. 182. Ag–Au subnanometer clusters as catalysts were proposed for the HER and investigated computationally, by conducting an exhaustive first-principles study of the HER process. Interestingly, the hydrogen-saturated resting state under standard conditions was determined as Ag12AuH11, from which HER reaction energies and barriers were predicted also including solvent effects using both implicit and explicit models. It was found that Ag12Au is a good candidate as a HER catalyst, with good stability and an overall reaction energy barrier of 0.89 eV.
A method for the electrodeposition of an isolated single Pt atom or small cluster, up to 9 atoms, on a bismuth ultramicroelectrode (UME) was demonstrated in ref. 183. The electrochemical characterization was made via the hydrogen evolution reaction (HER) that occurs readily on the electrodeposited Pt but not on Bi. Taking the potential at a certain current density as a measure of the relative rate of the HER, it was found that the potential shifted positively as the size increased, with single atoms showing the largest overpotentials compared to bulk Pt.
It should be underlined that a development of great potential in terms of characterization under electrochemical conditions is represented by electrochemical scanning tunneling microscopy (EC-STM),184 naturally in combination with standard electrochemical measurements such as cyclic voltammetry (CV). To the best of our knowledge, this technique has not yet been applied to nanocatalyst systems, but it is to be expected that its recent progresses will allow researchers to achieve this soon.185
Another development that has already achieved very interesting progress and is extremely promising in perspective is related to operando characterization modality. Recent experimental techniques have in fact been developed that allow one to realize operando identification of reaction intermediates using surface infrared, Raman, and XPS spectroscopy. To provide an interpretation of these experimental studies and target for additional studies, theoretical calculations are needed, for example generating vibrational spectra of the various candidate intermediate that can be directly compared with the reported experimental spectroscopy data.186 One recent finding is that explicit consideration of solvent, electrode–electrolyte interface, and applied potential effects in theoretical/computational modeling is indispensable to achieve quantitative agreement and correct interpretation of experiment. Spectroscopic features (peak positions and widths) can depend substantially on both the applied potential in terms of charge residing on the surface, and on interfacial atomistic structures e.g. the network of hydrogen bonds surrounding the adsorbate species under consideration. An example of such operando theoretical spectroscopy, in this case fully based on quantum mechanics molecular dynamics, can be found in ref. 187, where the possible reactive intermediates in carbon dioxide reduction over the Cu(100) surface were characterized via vibrational spectroscopy and important ones identified in the experimental spectra, uniquely different from those predicted neglecting potential and solvent effects (Fig. 9a and b). Experimentally, the determination of XPS core-level shifts under electrochemical conditions is difficult, due to the perturbing background generated by the environment, but progress is continuously being made since the pioneering studies in this field.188 XPS prediction also faces technical issues especially in finite-state simulations due to the hole localization in periodic systems, to overcome which a local fragment similarity (LFS) procedure has been recently proposed.186 In this procedure, accurate and predictive final-state XPS simulations on fragment species are used as a database to complement and correct lower-level simulations on the real systems in the initial-state approximation. Finally, electrochemical scanning tunneling microscopy stands as a particularly promising approach to monitor with atomic-scale precision the catalytic activity of subnanometer species, such as e.g. single iron atoms anchored to carbon vacancies in graphene.189
Lastly, size-selected subnanometer silver and iridium clusters have been also found as efficient electrocatalysts in Li–O2 batteries190 and helped to identify the composition and structure of the active species in iridium-based Li–O2 batteries,191 respectively.
Optical properties of clusters
Optical spectroscopy is potentially a powerful tool for characterization and manipulation (photocatalysis) of nanostructured catalysts. Indeed, the optical properties of larger metal nanoparticles, both pure and alloyed have been the subject of a wealth of studies in the past decades,87,192 with the interest on these properties stemming from their technological applications, which range from plasmonic materials, to sensors and single molecule spectroscopy (SERS). The delocalized electrons of metal nanoparticles are in fact strongly responsive to electromagnetic external fields, to which they react giving rise to coherent and collective excitations and correspondingly strong absorption peaks in the optical region of the spectrum, called surface plasmon resonances (SPR).193 In the present review, it is worthwhile underlining that applications of optical properties include photo-catalytic studies.158,194 It can be added that, when the structure of the clusters is atomically precise as in the subtopic of monolayer-protected clusters, systematic investigations of optical absorption and ultra-fast response, including plasmonic effects, have been conducted in the last decade, providing a mass of often well rationalized data and interesting perspectives, as will also be recalled later.85 This may lead one to expect a similar importance of the topic of chiro-optics also in the subnanometer supported cluster field. However, the technical difficulties associated with weak signals and the need of disentangling response of the catalyst, the reactants/products, and the support have so far hindered the full exploitation of the potentialities of this technique. In the following we will briefly review what has been achieved already together with some appealing perspectives, recalling that one of our goals in this review is to trigger interest on and promote developing new promising areas.A systematic study of optical absorption spectra from the ultraviolet to the visible for size selected neutral Agn clusters (n = 5–120) embedded in solid Ne was published in ref. 195. With increasing size of the Agn clusters, several highly correlated electron excitations gradually develop into a single surface plasmon with energy between 3.9 and 4.1 eV. The plasmon energy is highest for clusters with atom numbers fully filling states with the lowest radial quantum number (e.g. 1s, 1p, 1d,…). TDDFT calculations for clusters with several candidate geometrical structures embedded in Ne show excellent agreement with the experimental data, demonstrating that the absorption bands depend only weakly on the exact structure of the cluster (Fig. 11c).
Surface cavity ring-down (s-CRD) spectroscopy under ultrahigh vacuum (UHV) conditions was used to measure extinction spectra of size-selected, supported Ag20 and Ag55 clusters.196 The reaction with benzenethiol shifted the localized surface plasmon resonance observed around 3.2 eV to lower energies. This was attributed to an increased dielectric function of the surrounding medium as well as to a reduction of the free-electron density inside the silver clusters. Time-resolved study of that shift revealed that two thirds of the overall redshift can be attributed to an increased dielectric constant of the surrounding medium, whereas a reduction of the free-electron density accounts for one third, in agreement with previous theoretical TDDFT predictions.197
The oxidation reaction of supported, size-selected small silver clusters between Ag9 and Ag55 was monitored using surface second harmonic generation spectroscopy (s-SHG).198 A rapid decline of the SH-intensity was attributed to the formation of silver–oxygen-bonds. A double-exponential character of the time evolution of the SH-intensity for all investigated cluster sizes was observed. The two independent pathways of SH-intensity loss were attributed to a surface- and an interface-oxidation of supported clusters, respectively.
In addition to simple absorption, more sophisticated phenomena such as photoluminescence have also been studied. Intense fluorescence of Au20 in He matrix held at temperature of 6 K was reported in ref. 199, related with the HOMO–LUMO diabatic bandgap of the cluster. Since Au20 has only one stable isomer (tetrahedral) and due to low temperature, Au20 shows a very rich absorption fine structure reminiscent of well defined molecule-like quantum levels, which are fairly well reproduced both in absorption and intrinsic fluorescence by DFT and TDDFT calculations (Fig. 11d).
Ag clusters confined within Na-exchanged Linde type A zeolites were studied by X-ray absorption and steady-state and time-resolved photoluminescence spectroscopies in a coordinated effort to reveal the photophysical properties and link them to the precise cluster structure.200 At room temperature, the emission of the hydrated sample decays with 3.4 ns from a state with the same multiplicity as the ground state. Upon dehydration, the entire excited-state dynamics speeds up to 1.2 ps. The microsecond-scale lifetimes observed at 77 K suggest the occurrence of two main decay processes for the initially populated singlet state: intersystem crossing and charge transfer. The intersystem crossing yields the formation of a long-lived (409 μs) triplet state 3P from the 1P state located at lower energy from which luminescence occurs. There is evidence that electron transfer is followed by electron–hole recombination or back electron transfer, yielding a relaxed singlet excited state. The dependence of luminescence lifetime on the water content indicates a possible way to speed or delay the electron–hole recombination in a controlled way (Fig. 11a).
An ultrafast preparation of subnanometer silver particles with high abundance, uniformity (7 Å), and stability into the cages of nanosized zeolite crystals was presented in ref. 41. The particles were prepared using UV excitation of a water suspension of nanozeolite containing photoactive vanadate clusters in the presence of ethanol (as an electron donor) and silver precursor. The subnanometer Ag clusters exhibit exceptional photocatalytic activity and selectivity in the reforming of formic acid to H2 and CO2 under visible light (Fig. 10b).
 | ||
| Fig. 10 (a) Individual component maps of rotatory strength (ICM-RS) and rotatory strength density (RSD) plots as analysis tools of chiro-optical linear response spectra deriving from time-dependent density functional theory (TDDFT) simulations to analyze circular dichroism spectra of selected (Ag–Au)30(SR)18 monolayer-protected metal nanoclusters. (b) Illustration of the Photocatalytic Process Leading to the Formation of Silver Clusters in the Supercage of FAU-Type zeolite (I) silver photoreduction by excitation of the vanadate clusters (VCs) with λ < 300 nm (D); (II) photoreduction Ag0 formed in the stage (I) by excitation of silver clusters with λ > 320 nm (D). (a) is adapted with permission from ref. 209, Copyright 2018 American Chemical Society, (b) is adapted with permission from ref. 41, Copyright 2018 American Chemical Society. | ||
 | ||
| Fig. 11 (a) Ag clusters (AgCLs) confined within Na-exchanged Linde type A zeolites show dependence of luminescence lifetime on the water content indicating a possible way to speed or delay the electron–hole recombination in a controlled way. Proposed simplified kinetic scheme of the processes observed upon excitation of hydrated (A) and dehydrated (B) samples. The processes depicted in black and red are observed at RT and 77 K, respectively. (b) Single atoms and few-atom clusters of platinum exhibit high catalytic activity for ethylene hydrogenation while exhibiting resistance to sintering up to 200 °C. In situ IR spectroscopy reveals the presence of both single atoms and few-atom clusters that depend upon synthesis conditions. IR spectra of CO adsorbed on LT-Pt-AIM (left) and HT-Pt-AIM (right). (c) Measured optical absorption spectra (red) of monodisperse Agn (n = 1–120) in Ne at 6 K. The uppermost spectrum was calculated for bulk Ag and redshifted by 0.17 eV in order to account for the dielectric shift of the Ne matrix. (d) Au20/Ne absorption spectrum (blue) (broad band illumination, spectral detection). Excitation spectrum (black) (monochromatic excitation, monochromatic detection at the main fluorescence line of 739.2 nm). Fluorescence spectrum (red) (excitation at 266 nm, spectral detection). (a) is adapted with permission from ref. 200, Copyright 2019 American Chemical Society, (b) is adapted with permission from ref. 169, Copyright 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, (c) is adapted from ref. 195 with permission from the Royal Society of Chemistry, (d) is adapted from ref. 199 with the permission of AIP Publishing. | ||
A charge engineering strategy to improve the photo-oxidation activity and stability of Au25(PPh3)10(SC3H6Si(OC2H5)3)5Cl2 nanocluster (AuNC) taking advantage of Z-scheme AuNC/ultrathin BiOCl nanosheets (2D-BiOCl) heterojunction for visible light oxidative self-coupling of amines was reported. The enhanced photocatalytic activity of AuNC/2D-BiOCl was predominantly attributed to the formation of Z-scheme heterojunction, which effectively accelerated the transfer and separation of photo-generated carriers. Especially, the consumption of holes in AuNC by injected electrons from 2D-BiOCl can inhibit the self-oxidation of AuNC, thus improve its stability.201
High-energy-resolution X-ray absorption spectroscopy detection of ethylene and CO ligands adsorbed on catalytically active iridium centers isolated on zeolite HY and on MgO supports, supported by density functional theory and FEFF X-ray absorption near-edge modelling, together with IR spectra, was reported in ref. 45. The high-energy-resolution X-ray absorption spectra near the iridium LIII (2p3/2) edge provided clearly ascribable, distinctive signatures of the ethylene and CO ligands and illustrated effects of supports and other ligands. The X-ray absorption technique was more sensitive than conventional IR spectroscopy for characterizing surface intermediates, and it should be applicable to samples having low metal loadings and in reactive atmospheres.
A recent first-principles DFT and TDDFT study investigated the optical response of small (M/M′)6 and (M)3(M′)3 six-atom clusters (unary and equimolar binary hexamers of group IB metals: Cu, Ag, and Au), both in the gas-phase and supported on the MgO(100) surface, chosen as a model of a simple oxide substrate.202 UV-vis spectra were predicted and analyzed in detail in terms of ground- and excited-state wave functions and the involved Kohn–Sham orbitals, to rationalize origin and features of optical absorption in these systems. The interaction with the surface, in particular with the electric field generated by the charge-separated substrate, was found to induce a fragmentation and a shift toward lower energies of the absorption peaks, which was in tune with available experimental information and can be beneficial in terms of applications of these systems as photoenhancers or promoters. The orientation of the transition dipole moment with respect to the support (whether parallel or perpendicular) was also analyzed as a key parameter to translate the present result to non-inert supports to tune and optimize oscillator strengths and interaction with substrate excitations. These indications have been later confirmed in simulations on larger Ag20 and Ag20 clusters deposited on MgO(100), in which modeling was also extended to include defects on the oxide substrate such as oxygen vacancies and/or substitutional cation replacements. Interestingly, it was found that defect excitations can couple with cluster one and that the simultaneous presence of vacancies and cation doping may give rise to fine effects on transition dipole moment orientation.203 Finally, an exploratory study of optical absorption and photo-decay processes occurring in Ag3(XCO3)(C2H4)2(O) ligand/cluster/support catalytic complexes (with X = H or F) after visible light absorption was recently conducted.136 Although a “frailty” of bicarbonate and fluorocarbonate groups to optical adsorption was derived from this study, leading to C2H4 or CO2 detachment or Ag3 disaggregation, from the methodological point of view a palette of computational tools was tuned, enabling one to explore the promising field of subnanometer photo-catalysis.
It should be stressed that decisive progress has been recently made in this topic. Methods have in fact been recently developed to predict photo-induced processes in complex systems such as ligand-protected84 and oxide-supported subnanometer clusters,204,205 with an accuracy quantitatively validated against experiment, thus opening a pathway for predictive modeling photo-induced and photo-chemical reactivity in these materials. Indeed, in ref. 204, a rutile TiO2(110) surface decorated with Ag5 subnanometer clusters and its photon-absorption spectrum were investigated, predicting the formation of surface polarons on TiO2 that has then been confirmed experimentally. Similar phenomena were predicted in ref. 205 on a rutile TiO2(110) surface either bare or decorated with Cu5 subnanometer clusters. It was also predicted that electron donation from the Ag5 or Cu5 cluster enhances overall absorption and extends it somewhat into the visible region, with perspective applications in CO2 photoactivation.206
Indirect nanoplasmonic sensing (INPS) coupled with mass spectrometry was used to study CO and oxygen adsorption as well as CO oxidation, on Pt nanoparticles, in the Torr pressure range. A highly sensitive probe that can detect gas adsorption of a few hundredths of a monolayer, even with a very low number density of Pt particles was obtained. A similarity was observed between the sign and the evolution of the localized surface plasmon resonance (LSPR) peak shift and the work function measurements for CO and oxygen chemisorption.207
We believe that one important step in achieving a deeper understanding of optical phenomena is undoubtedly the development of analysis tools which allow one to decompose the various physical chemical effects at play into a few basic ingredients and single out descriptors able to orient research and design of optimal systems. For the sake of brevity, we here mention only two recent developments that, due to their general character, can be applied to a wide range of phenomena, also after needed methodological extensions. An Individual Component Maps of Oscillatory Strength (ICM-OS) analysis tool of TDDFT simulated spectra have first been introduced and applied in the field of monolayer-protected clusters (MPC) to investigate general phenomena such as coupling of aromatic conjugation and Ag doping, constructive/destructive interference due to long-range off-diagonal single-particle contributions, etc.208 This allowed one to clarify the basic principles of optical spectroscopy of aromatic monolayer-protected alloyed (Ag–Au) clusters and demonstrated how these are uniquely different from those of aliphatic systems. Analogously, Individual Component Maps of Rotatory Strength (ICM-RS) and Rotatory Strength Density (RSD) plots of TDDFT simulated spectra were also introduced as analysis tools of chiral linear response spectra.209 ICM-OS, ICM-RS and RSD allow one to visualize the origin of chiro-optical response in momentum or real space, including signed contributions and therefore highlighting cancellation terms that are ubiquitous in both absorption and chirality phenomena, and should be especially useful in analyzing the spectra of complex systems, to derive insight and understanding, and eventually rational design, in chiro-optical studies of complex systems209 (Fig. 10a), as explored more recently in ref. 210 where the rotatory strength of chiral gold nanowires was designed and brought from negligible to very large values following the leads of the ICM-RS analysis.
We don’t have room to report all the detailed studies conducted on the optical response of MPC. Here, the unique stoichiometry and reasonably well-defined structure have allowed in-depth (and unprecedented in the metal cluster field) investigations of structure/optical-properties relationships, at the same time producing very interesting systems and tracing a path for future studies in parallel fields of heterogeneous and electrochemical catalysis. We refer the reader to recent excellent reviews such as ref. 85, 211 and 212.
The power of operando spectroscopies not only in the electrochemical but also in heterogeneous catalysis context was highlighted in ref. 213 where, in addition to systematic high-resolution scanning transmission electron microscopy, a simultaneous monitoring of the Pt dispersion, oxidation state, and CO oxidation activity by operando fast XAS and DRIFTS, both combined with MS was reported. Poorly active single Ptm+ atoms (m ≥ 2) were converted gradually and irreversibly into highly active ∼1 nm sized Ptδ+ clusters (δ < 2) throughout the heating–cooling reaction cycles. As a final note, we stress that it is expected that such operando modalities (including other spectroscopies such as IR, Raman, and synchrotron ones such as XANES, EXAFS, etc.) will develop quickly in a next future and will provide essential information to achieve the goal of a physical-chemistry-informed rational design of nanocatalytic systems (Fig. 6a and b).
Summary and outlook
Modern society requires a transition to green chemistry, thus green catalysis. Presently available catalysts and catalytic processes, developed in a world in which environmental and energy issues were relatively minor, are for the vast majority not “green”. A societal sustainable growth requires developing atom-economical (optimal use of precious elements), stable (non-leaching undesired toxic compounds or endangered elements), efficient (low-energy spent in high-throughput), selective (low product separation costs) catalysts and catalytic processes, which in turn translates into finding novel catalysts and catalytic sites with all the desired features and performance. In this scenario, as promising new materials we focus our attention on metal and metal/oxide clusters at the subnanometer scale, in the size range from 1 to few tens of atoms, deposited on a proper stabilizing substrate. Several reasons justify our choice and make these materials especially appealing to find novel active sites: the unusual structural motifs encountered at this length scale, the strong quantum mechanical confinement effects, the fact that all metal atoms are at the surface and therefore potentially active for adsorbing and activating reactants, the fact that they can be prepared mass-selected and therefore with a precise definition of size and composition. Then, we review the physico-chemical studies that have been conducted on these systems aimed at clarifying and precisely characterizing their static and dynamic structure (Section 2), and the crucial topic of stability (Section 3). The reasoning behind our choice is our belief that a most promising path to the development of new systems is a physical-chemistry-informed rational design, in which the detailed knowledge achieved from monitoring, characterizing, and testing nanocatalyst materials at work enables researchers to understand the limits and problems but also the potentialities of these systems and to find the way to overcome issue and design drastically improved materials as desired. Separate sections have been dedicated to electrochemical systems (Section 4), due to their specific features and issues, and to optical properties (Section 5), in view of the increasingly important field of photocatalysis. Throughout this review, we tried to emphasize the potential (and often realized) synergy between theory and experiment, and how they can complement each other to achieve at least in part the desired in-depth knowledge and understanding. We have occasionally mentioned topics where further progress is desirable, and is indeed actively pursued by several groups.To conclude, let us summarize the three main messages and the perspective challenges that can be drawn from the above review. The main messages are that, first, we have shown that the field of subnanometer size- and composition-selected clusters indeed hold great promises in the catalysis field, and that subnanometer clusters can indeed be very active systems. Second, we have reported clear cases of the extreme dependence of such activity on support, size and composition, implying then that it requires a higher degree of knowledge to be able to guide researchers in the discovery of new, active and selective systems, so far often found via a trial-and-error approach. Third, the results achieved so far and here extensively reviewed have proved that a physico-chemical approach combining advanced characterization, modelling and controlled synthesis can in principle provide the required degree of knowledge. On the side of perspective challenges, it is clear that this agenda has not yet been fulfilled. Despite the amount of information and sophisticated results so far accumulated, there is yet basically no subnanometer systems for which an ultra-precise knowledge of static and dynamic structure, and of reaction intermediates and their energetics, has been fully achieved. In this respect, we believe that a promising strategy could be to focus the attention of several groups on the most promising systems singled out thus far and to concentrate on these systems a combined effort of the most advanced characterization and modelling tools, also pursuing the development of e.g. new microscopy characterization techniques that could overcome the issues of the perturbing effect of the electron beam,214 together with new approaches, such as possibly the use of MPC internal standards for increasing the accuracy of the techniques, and many others. An additional challenge is to pursue in a more systematic way the new areas that have been scarcely explored so far, such as electrochemical applications and especially the chiro-optical properties of ultra-nanoclusters. We believe we have provided enough evidence that these actually represent exciting areas of research and possible applications, and that e.g. tools to properly model them are rapidly being developed.
We hope that the present excursus, by necessity only partial, will be useful to motivate a larger number of researchers and further studies along this most promising line of research.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
JJ and SV acknowledge support from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 810310, which corresponds to the J. Heyrovsky Chair project (“ERA Chair at J. Heyrovský Institute of Physical Chemistry AS CR – The institutional approach towards ERA”). The funders had no role in the preparation of the article. AF and SV are also grateful to CNR (National Research Council in Italy) and CAS (Czech Academy of Sciences) for support via the bilateral project “Subnanometer Cluster Catalysts for Green Chemistry (SNaGC)”. They also gratefully acknowledge the contribution of the International Research Network IRN on Nanoalloys (CNRS).References
- G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Handbook of Heterogeneous Catalysis, 2nd edition. Second, Completely Revised and Enlarged ed, Wiley-VCH, Weinheim, 2008, vol. 8, p. 4270 Search PubMed.
- E. Keinan and I. Schechter, Chemistry for the 21st Century, 2008, p. 308 Search PubMed.
- T. Keijer, V. Bakker and J. C. Slootweg, Circular chemistry to enable a circular economy, Nat. Chem., 2019, 11(3), 190–195 CrossRef CAS PubMed.
- P. Fayet, F. Granzer, G. Hegenbart, E. Moisar, B. Pischel and L. Wöste, Latent-image generation by deposition of monodisperse silver clusters, Phys. Rev. Lett., 1985, 55(27), 3002–3004 CrossRef CAS PubMed.
- D. M. Cox, W. Eberhardt, P. Fayet, Z. Fu, B. Kessler, R. D. Sherwood, D. Sondericker and A. Kaldor, Electronic structure of deposited monosized metal-clusters, Z. Phys. D: At., Mol. Clusters, 1991, 20(1), 385–386 CrossRef CAS.
- J. H. Sinfelt, Structure of bimetallic clusters, Acc. Chem. Res., 1987, 20(4), 134–139 CrossRef CAS.
- D. Bazin, C. Mottet and G. Tréglia, New opportunities to understand heterogeneous catalysis processes on nanoscale bimetallic particles through synchrotron radiation and theoretical studies, Appl. Catal., A, 2000, 200(1), 47–54 CrossRef CAS.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1(1), 37–46 CrossRef CAS PubMed.
- P. Sabatier, Hydrogénations et déshydrogénations par catalyse, Ber. Dtsch. Chem. Ges., 1911, 44(3), 1984–2001 CrossRef CAS.
- J. N. Bronsted, Acid and basic catalysis, Chem. Rev., 1928, 5(3), 231–338 CrossRef CAS.
- M. G. Evans and M. Polanyi, Inertia and driving force of chemical reactions, Trans. Faraday Soc., 1938, 34(0), 11–24 RSC.
- Y. Watanabe, Atomically precise cluster catalysis towards quantum controlled catalysts, Sci. Technol. Adv. Mater., 2014, 15(6), 063501 CrossRef PubMed.
- F. R. Negreiros, G. Barcaro, L. Sementa and A. Fortunelli, Concepts in theoretical heterogeneous ultrananocatalysis, C. R. Chim, 2014, 17(7–8), 625–633 CrossRef CAS.
- A. Sápi, T. Rajkumar, J. Kiss, Á. Kukovecz, Z. Kónya and G. A. Somorjai, Metallic nanoparticles in heterogeneous catalysis, Catal. Lett., 2021, 151(8), 2153–2175 CrossRef.
- B. Q. Yin and Z. X. Luo, Coinage metal clusters: From superatom chemistry to genetic materials, Coord. Chem. Rev., 2021, 429, 213643 CrossRef CAS.
- N. P. Young, Z. Y. Li, Y. Chen, S. Palomba, M. Di Vece and R. E. Palmer, Weighing supported nanoparticles: Size-selected clusters as mass standards in nanometrology, Phys. Rev. Lett., 2008, 101(24), 246103 CrossRef CAS PubMed.
- L. Piccolo, Z. Y. Li, I. Demiroglu, F. Moyon, Z. Konuspayeva, G. Berhault, P. Afanasiev, W. Lefebvre, J. Yuan and R. L. Johnston, Understanding and controlling the structure and segregation behaviour of AuRh nanocatalysts, Sci. Rep., 2016, 6(1), 35226 CrossRef CAS PubMed.
- J. Liu, Challenges in determining structure of supported subnano metal clusters, Microsc. Microanal., 2019, 25(S2), 1640–1641 CrossRef.
- D. M. Foster, T. Pavloudis, J. Kioseoglou and R. E. Palmer, Atomic-resolution imaging of surface and core melting in individual size-selected Au nanoclusters on carbon, Nat. Commun., 2019, 10(1), 2583 CrossRef CAS PubMed.
- A. Londoño-Calderon, A. Ponce, U. Santiago, S. Mejia and M. José-Yacamán, Controlling the number of atoms on catalytic metallic clusters, in Morphological, Compositional, and Shape Control of Materials for Catalysis, ed. P. Fornasiero and M. Cargnello, Elsevier, 2017, vol. 177, pp. 185–220 Search PubMed.
- A. Bruma, F. R. Negreiros, S. Xie, T. Tsukuda, R. L. Johnston, A. Fortunelli and Z. Y. Li, Direct atomic imaging and density functional theory study of the Au24Pd1 cluster catalyst, Nanoscale, 2013, 5(20), 9620–9625 RSC.
- X. Chen, M. Peng, X. Cai, Y. Chen, Z. Jia, Y. Deng, B. Mei, Z. Jiang, D. Xiao, X. Wen, N. Wang, H. Liu and D. Ma, Regulating coordination number in atomically dispersed Pt species on defect-rich graphene for n-butane dehydrogenation reaction, Nat. Commun., 2021, 12(1), 2664 CrossRef CAS PubMed.
- T. R. Henninen, M. Bon, F. Wang, D. Passerone and R. Erni, The structure of sub-nm platinum clusters at elevated temperatures, Angew. Chem., Int. Ed., 2020, 59(2), 839–845 CrossRef CAS PubMed.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Three-dimensional atomic-scale structure of size-selected gold nanoclusters, Nature, 2008, 451(7174), 46–48 CrossRef CAS PubMed.
- D. M. Foster, R. Ferrando and R. E. Palmer, Experimental determination of the energy difference between competing isomers of deposited, size-selected gold nanoclusters, Nat. Commun., 2018, 9(1), 1323 CrossRef CAS PubMed.
- F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti and C. Mottet, Crossover among structural motifs in transition and noble-metal clusters, J. Chem. Phys., 2002, 116(9), 3856–3863 CrossRef CAS.
- A. Rapallo, G. Rossi, R. Ferrando, A. Fortunelli, B. C. Curley, L. D. Lloyd, G. M. Tarbuck and R. L. Johnston, Global optimization of bimetallic cluster structures. I. Size-mismatched Ag–Cu, Ag–Ni, and Au–Cu systems, J. Chem. Phys., 2005, 122(19), 194308 CrossRef PubMed.
- G. Rossi, R. Ferrando, A. Rapallo, A. Fortunelli, B. C. Curley, L. D. Lloyd and R. L. Johnston, Global optimization of bimetallic cluster structures. II. Size-matched Ag–Pd, Ag–Au, and Pd–Pt systems, J. Chem. Phys., 2005, 122(19), 194309 CrossRef PubMed.
- R. Ferrando, G. Rossi, A. C. Levi, Z. Kuntova, F. Nita, A. Jelea, C. Mottet, G. Barcaro, A. Fortunelli and J. Goniakowski, Structures of metal nanoparticles adsorbed on MgO(001). I. Ag and Au, J. Chem. Phys., 2009, 130(17), 174702 CrossRef PubMed.
- J. Goniakowski, A. Jelea, C. Mottet, G. Barcaro, A. Fortunelli, Z. Kuntova, F. Nita, A. C. Levi, G. Rossi and R. Ferrando, Structures of metal nanoparticles adsorbed on MgO(001). II. Pt and Pd, J. Chem. Phys., 2009, 130(17), 174703 CrossRef PubMed.
- R. Ferrando, A. Fortunelli and G. Rossi, Quantum effects on the structure of pure and binary metallic nanoclusters, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(8), 085449 CrossRef.
- L. O. Paz-Borbón, G. Barcaro, A. Fortunelli and S. V. Levchenko, AuN clusters (N = 1–6) supported on MgO(100) surfaces: Effect of exact exchange and dispersion interactions on adhesion energies, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(15), 155409 CrossRef.
- R. Ferrando, G. Barcaro and A. Fortunelli, Surface-supported gold cages, Phys. Rev. Lett., 2009, 102(21), 216102 CrossRef CAS PubMed.
- G. Zwaschka, M. Rondelli, M. Krause, M. D. Rotzer, M. N. Hedhili, U. Heiz, J. M. Basset, F. F. Schweinberger and V. D'Elia, Supported sub-nanometer Ta oxide clusters as model catalysts for the selective epoxidation of cyclooctene, New J. Chem., 2018, 42(4), 3035–3041 RSC.
- Y. Inomata, K. Albrecht and K. Yamamoto, Size-dependent oxidation state and CO oxidation activity of tin oxide clusters, ACS Catal., 2017, 8(1), 451–456 CrossRef.
- J. Weitkamp, Zeolites and catalysis, Solid State Ionics, 2000, 131(1), 175–188 CrossRef CAS.
- A. Baldansuren, H. Dilger, R.-A. Eichel, J. A. van Bokhoven and E. Roduner, Interaction and reaction of ethylene and oxygen on six-atom silver clusters supported on LTA zeolite, J. Phys. Chem. C, 2009, 113(45), 19623–19632 CrossRef CAS.
- B. M. Weckhuysen and J. Yu, Recent advances in zeolite chemistry and catalysis, Chem. Soc. Rev., 2015, 44(20), 7022–7024 RSC.
- Y. Li and J. Yu, Emerging applications of zeolites in catalysis, separation and host–guest assembly, Nat. Rev. Mater., 2021, 6(12), 1156–1174 CrossRef CAS.
- Q. Sun, N. Wang, Q. Bing, R. Si, J. Liu, R. Bai, P. Zhang, M. Jia and J. Yu, Subnanometric hybrid Pd–M(OH)2, M = Ni, Co, clusters in zeolites as highly efficient nanocatalysts for hydrogen generation, Chem, 2017, 3(3), 477–493 CAS.
- M. El-Roz, I. Telegeiev, N. E. Mordvinova, O. I. Lebedev, N. Barrier, A. Behilil, M. Zaarour, L. Lakiss and V. Valtchev, Uniform generation of sub-nanometer silver clusters in zeolite cages exhibiting high photocatalytic activity under visible light, ACS Appl. Mater. Interfaces, 2018, 10(34), 28702–28708 CrossRef CAS PubMed.
- P. Serna, A. Rodriguez-Fernandez, S. Yacob, C. Kliewer, M. Moliner and A. Corma, Single-site vs. cluster catalysis in high temperature oxidations, Angew. Chem., Int. Ed., 2021, 60(29), 15954 CrossRef CAS PubMed.
- S. Grundner, M. A. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Single-site trinuclear copper oxygen clusters in mordenite for selective conversion of methane to methanol, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C. Y. Chen, N. D. Browning and B. C. Gates, A single-site platinum CO oxidation catalyst in zeolite KLTL: Microscopic and spectroscopic determination of the locations of the platinum atoms, Angew. Chem., Int. Ed., 2014, 53(34), 8904–8907 CrossRef CAS PubMed.
- A. S. Hoffman, D. Sokaras, S. Zhang, L. M. Debefve, C. Y. Fang, A. Gallo, T. Kroll, D. A. Dixon, S. R. Bare and B. C. Gates, High-energy-resolution X-ray absorption spectroscopy for identification of reactive surface species on supported single-site iridium catalysts, Chem. – Eur. J., 2017, 23(59), 14760–14768 CrossRef CAS PubMed.
- L. Liu, D. N. Zakharov, R. Arenal, P. Concepcion, E. A. Stach and A. Corma, Evolution and stabilization of subnanometric metal species in confined space by in situ TEM, Nat. Commun., 2018, 9(1), 574 CrossRef PubMed.
- S. M. Wu, X. Y. Yang and C. Janiak, Confinement effects in zeolite-confined noble metals, Angew. Chem., Int. Ed., 2019, 58(36), 12340–12354 CrossRef CAS PubMed.
- V. Van Speybroeck, K. Hemelsoet, L. Joos, M. Waroquier, R. G. Bell and C. R. Catlow, Advances in theory and their application within the field of zeolite chemistry, Chem. Soc. Rev., 2015, 44(20), 7044–7111 RSC.
- J. A. Boscoboinik, X. Yu, B. Yang, F. D. Fischer, R. Włodarczyk, M. Sierka, S. Shaikhutdinov, J. Sauer and H.-J. Freund, Modeling zeolites with metal-supported two-dimensional aluminosilicate films, Angew. Chem., Int. Ed., 2012, 51(24), 6005–6008 CrossRef CAS PubMed.
- A. Korde, B. Min, E. Kapaca, O. Knio, I. Nezam, Z. Wang, J. Leisen, X. Yin, X. Zhang, D. S. Sholl, X. Zou, T. Willhammar, C. W. Jones and S. Nair, Single-walled zeolitic nanotubes, Science, 2022, 375(6576), 62–66 CrossRef CAS PubMed.
- A. Piednoir, E. Perrot, S. Granjeaud, A. Humbert, C. Chapon and C. R. Henry, Atomic resolution on small three-dimensional metal clusters by STM, Surf. Sci., 1997, 391(1–3), 19–26 CrossRef CAS.
- O. Dulub, W. Hebenstreit and U. Diebold, Imaging cluster surfaces with atomic resolution: the strong metal–support interaction state of Pt supported on TiO2(110), Phys. Rev. Lett., 2000, 84(16), 3646–3649 CrossRef CAS PubMed.
- J. Sauer and H. J. Freund, Models in catalysis, Catal. Lett., 2015, 145(1), 109–125 CrossRef CAS.
- J. Frenken and I. Groot, Live observations of catalysts using high-pressure scanning probe microscopy, in Operando Research in Heterogeneous Catalysis, J. Frenken and I. Groot, Springer, Cham, Cham, 2017, vol. 114, pp. 1–30 Search PubMed.
- M. S. Chen, A. K. Santra and D. W. Goodman, Structure of thin SiO2 films grown on Mo(112), Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(15), 155404 CrossRef.
- J. Weissenrieder, S. Kaya, J. L. Lu, H. J. Gao, S. Shaikhutdinov, H. J. Freund, M. Sierka, T. K. Todorova and J. Sauer, Atomic structure of a thin silica film on a Mo(112) substrate: A two-dimensional network of SiO4 tetrahedra, Phys. Rev. Lett., 2005, 95(7), 076103 CrossRef CAS PubMed.
- H.-J. Freund, Metal-supported ultrathin oxide film systems as designable catalysts and catalyst supports, Surf. Sci., 2007, 601(6), 1438–1442 CrossRef CAS.
- S. Shaikhutdinov and H.-J. Freund, Ultrathin oxide films on metal supports: Structure–reactivity relations, Annu. Rev. Phys. Chem., 2012, 63(1), 619–633 CrossRef CAS PubMed.
- Y. N. Sun, Z. H. Qin, M. Lewandowski, E. Carrasco, M. Sterrer, S. Shaikhutdinov and H. J. Freund, Monolayer iron oxide film on platinum promotes low temperature CO oxidation, J. Catal., 2009, 266(2), 359–368 CrossRef CAS.
- F. Kraushofer, N. Resch, M. Eder, A. Rafsanjani-Abbasi, S. Tobisch, Z. Jakub, G. Franceschi, M. Riva, M. Meier, M. Schmid, U. Diebold and G. S. Parkinson, Surface reduction state determines stabilization and incorporation of Rh on α-Fe2O3(1
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 02), Adv. Mater. Interfaces, 2021, 8(8), 2001908 CrossRef CAS.
02), Adv. Mater. Interfaces, 2021, 8(8), 2001908 CrossRef CAS. - J. Hulva, M. Meier, R. Bliem, Z. Jakub, F. Kraushofer, M. Schmid, U. Diebold, C. Franchini and G. S. Parkinson, Unraveling CO adsorption on model single-atom catalysts, Science, 2021, 371(6527), 375–379 CrossRef CAS PubMed.
- S. Surnev, A. Fortunelli and F. P. Netzer, Structure–property relationship and chemical aspects of oxide–metal hybrid nanostructures, Chem. Rev., 2013, 113(6), 4314–4372 CrossRef CAS PubMed.
- H.-J. Freund and G. Pacchioni, Oxide ultra-thin films on metals: new materials for the design of supported metal catalysts, Chem. Soc. Rev., 2008, 37(10), 2224–2242 RSC.
- L. Giordano, G. Pacchioni, J. Goniakowski, N. Nilius, E. D. L. Rienks and H.-J. Freund, Charging of metal adatoms on ultrathin oxide films: Au and Pd on FeOP(111), Phys. Rev. Lett., 2008, 101(2), 026102 CrossRef PubMed.
- G. Kresse, M. Schmid, E. Napetschnig, M. Shishkin, L. Köhler and P. Varga, Structure of the ultrathin aluminum oxide film on NiAl(110), Science, 2005, 308(5727), 1440–1442 CrossRef CAS PubMed.
- K. Schouteden, K. Lauwaet, E. Janssens, G. Barcaro, A. Fortunelli, C. Van Haesendonck and P. Lievens, Probing the atomic structure of metallic nanoclusters with the tip of a scanning tunneling microscope, Nanoscale, 2014, 6(4), 2170–2176 RSC.
- S. M. F. Shahed, A. Beniya, H. Hirata and Y. Watanabe, Morphology of size-selected Ptn clusters on CeO2(111), J. Chem. Phys., 2018, 148(11), 114702 CrossRef PubMed.
- N. Isomura, X. Wu and Y. Watanabe, Atomic-resolution imaging of size-selected platinum clusters on TiO(2)(110) surfaces, J. Chem. Phys., 2009, 131(16), 164707 CrossRef PubMed.
- Q. Zhou, S. Kaappa, S. Malola, H. Lu, D. Guan, Y. Li, H. Wang, Z. Xie, Z. Ma, H. Hakkinen, N. Zheng, X. Yang and L. Zheng, Real-space imaging with pattern recognition of a ligand-protected Ag374 nanocluster at sub-molecular resolution, Nat. Commun., 2018, 9(1), 2948 CrossRef PubMed.
- Z. W. Wang and R. E. Palmer, Direct atomic imaging and dynamical fluctuations of the tetrahedral Au(20) cluster, Nanoscale, 2012, 4(16), 4947–4949 RSC.
- K. Bromann, C. Felix, H. Brune, W. Harbich, R. Monot, J. Buttet and K. Kern, Controlled deposition of size-selected silver nanoclusters, Science, 1996, 274(5289), 956–958 CrossRef CAS PubMed.
- P. Milani and M. Sowwan, Cluster Beam Deposition of Functional Nanomaterials and Devices, Elsevier, New York, 2015 Search PubMed.
- H. Yasumatsu, T. Hayakawa, S. Koizumi and T. Kondow, Unisized two-dimensional platinum clusters on silicon(111)-7x7 surface observed with scanning tunneling microscope, J. Chem. Phys., 2005, 123(12), 124709 CrossRef PubMed.
- J. Timoshenko, A. Halder, B. Yang, S. Seifert, M. J. Pellin, S. Vajda and A. I. Frenkel, Subnanometer substructures in nanoassemblies formed from clusters under a reactive atmosphere revealed using machine learning, J. Phys. Chem. C, 2018, 122(37), 21686–21693 CrossRef CAS.
- Y. Liu, A. Halder, S. Seifert, N. Marcella, S. Vajda and A. I. Frenkel, Probing active sites in CuxPdy cluster catalysts by machine-learning-assisted X-ray absorption spectroscopy, ACS Appl. Mater. Interfaces, 2021, 13(45), 53363–53374 CrossRef CAS PubMed.
- J. Timoshenko, S. Roese, H. Hövel and A. I. Frenkel, Silver clusters shape determination from in-situ XANES data, Radiat. Phys. Chem., 2020, 175, 108049 CrossRef CAS.
- P. Tian, L. Ouyang, X. Xu, C. Ao, X. Xu, R. Si, X. Shen, M. Lin, J. Xu and Y.-F. Han, The origin of palladium particle size effects in the direct synthesis of H2O2: Is smaller better?, J. Catal., 2017, 349, 30–40 CrossRef CAS.
- D. Chekrygina, A. Rothkirch, I. Baev, F. Kielgast, P. Pandit, W. Wurth and M. Martins, Towards the geometric structure of small supported Au9 clusters on Si, Sci. Rep., 2018, 8(1), 12371 CrossRef CAS PubMed.
- B. H. Yan, Q. Y. Wu, J. J. Cen, J. Timoshenko, A. I. Frenkel, D. Su, X. Y. Chen, J. B. Paris, E. Stach, A. Orlov and J. G. G. Chen, Highly active subnanometer Rh clusters derived from Rh-doped SrTiO3 for CO2 reduction, Appl. Catal., B, 2018, 237, 1003–1011 CrossRef CAS.
- S. Ji, Y. Chen, Q. Fu, Y. Chen, J. Dong, W. Chen, Z. Li, Y. Wang, L. Gu, W. He, C. Chen, Q. Peng, Y. Huang, X. Duan, D. Wang, C. Draxl and Y. Li, Confined pyrolysis within metal–organic frameworks to form uniform Ru3 clusters for efficient oxidation of alcohols, J. Am. Chem. Soc., 2017, 139(29), 9795–9798 CrossRef CAS PubMed.
- J. M. Thomas, R. D. Adams, E. M. Boswell, B. Captain, H. Gronbeck and R. Raja, Synthesis, characterization, electronic structure and catalytic performance of bimetallic and trimetallic nanoparticles containing tin, Faraday Discuss., 2008, 138, 301–315 RSC , discussion 317–335, 433–434.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Nanocrystal gold molecules, Adv. Mater., 1996, 8(5), 428–433 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Structure of a thiol monolayer-protected gold nanoparticle at 1.1 A resolution, Science, 2007, 318(5849), 430–433 CrossRef CAS PubMed.
- M. Medves, L. Sementa, D. Toffoli, G. Fronzoni, K. R. Krishnadas, T. Burgi, S. Bonacchi, T. Dainese, F. Maran, A. Fortunelli and M. Stener, Predictive optical photoabsorption of Ag24Au(DMBT)18 (-) via efficient TDDFT simulations, J. Chem. Phys., 2021, 155(8), 084103 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Atomically precise colloidal metal nanoclusters and nanoparticles: Fundamentals and opportunities, Chem. Rev., 2016, 116(18), 10346–10413 CrossRef CAS PubMed.
- C. García, S. Pollitt, M. van der Linden, V. Truttmann, C. Rameshan, R. Rameshan, E. Pittenauer, G. Allmaier, P. Kregsamer, M. Stöger-Pollach, N. Barrabés and G. Rupprechter, Support effect on the reactivity and stability of Au25(SR)18 and Au144(SR)60 nanoclusters in liquid phase cyclohexane oxidation, Catal. Today, 2019, 336, 174–185 CrossRef.
- G. Barcaro, L. Sementa, A. Fortunelli and M. Stener, Optical properties of nanoalloys, Phys. Chem. Chem. Phys., 2015, 17(42), 27952–27967 RSC.
- G. Li and R. Jin, Atomically precise gold nanoclusters as new model catalysts, Acc. Chem. Res., 2013, 46(8), 1749–1758 CrossRef CAS PubMed.
- V. Sudheeshkumar, K. O. Sulaiman and R. W. J. Scott, Activation of atom-precise clusters for catalysis, Nanoscale Adv., 2020, 2(1), 55–69 RSC.
- Q. F. Zhang, X. Chen and L. S. Wang, Toward solution syntheses of the tetrahedral Au20 pyramid and atomically precise gold nanoclusters with uncoordinated sites, Acc. Chem. Res., 2018, 51(9), 2159–2168 CrossRef CAS PubMed.
- S. F. Yuan, R. L. He, X. S. Han, J. Q. Wang, Z. J. Guan and Q. M. Wang, Robust gold nanocluster protected with amidinates for electrocatalytic CO2 reduction, Angew. Chem., Int. Ed., 2021, 60(26), 14345–14349 CrossRef CAS PubMed.
- Z. Zhuang, Q. Yang and W. Chen, One-step rapid and facile synthesis of subnanometer-sized Pd6(C12H25S)11 clusters with ultra-high catalytic activity for 4-nitrophenol reduction, ACS Sustainable Chem. Eng., 2019, 7(3), 2916–2923 CrossRef CAS.
- C. Kumara, M. M. Hoque, X. Zuo, D. A. Cullen, R. L. Whetten and A. Dass, Isolation of a 300 kDa, Au approximately 1400 gold compound, the standard 3.6 nm capstone to a series of plasmonic nanocrystals protected by aliphatic-like thiolates, J. Phys. Chem. Lett., 2018, 9(23), 6825–6832 CrossRef CAS PubMed.
- F. R. Negreiros, E. Apra, G. Barcaro, L. Sementa, S. Vajda and A. Fortunelli, A first-principles theoretical approach to heterogeneous nanocatalysis, Nanoscale, 2012, 4(4), 1208–1219 RSC.
- G. Sun, J. T. Fuller, A. N. Alexandrova and P. Sautet, Global activity search uncovers reaction induced concomitant catalyst restructuring for alkane dissociation on model Pt catalysts, ACS Catal., 2021, 11(3), 1877–1885 CrossRef CAS.
- F. R. Negreiros, L. Sementa, G. Barcaro, S. Vajda, E. Apra and A. Fortunelli, CO oxidation by subnanometer AgxAu3-x supported clusters via density functional theory simulations, ACS Catal., 2012, 2(9), 1860–1864 CrossRef CAS.
- L. M. Molina and B. Hammer, Some recent theoretical advances in the understanding of the catalytic activity of Au, Appl. Catal., A, 2005, 291(1–2), 21–31 CrossRef CAS.
- T. Imaoka, Y. Akanuma, N. Haruta, S. Tsuchiya, K. Ishihara, T. Okayasu, W. J. Chun, M. Takahashi and K. Yamamoto, Platinum clusters with precise numbers of atoms for preparative-scale catalysis, Nat. Commun., 2017, 8(1), 688 CrossRef PubMed.
- P. Concepción, M. Boronat, S. García-García, E. Fernández and A. Corma, Enhanced stability of Cu clusters of low atomicity against oxidation. Effect on the catalytic redox process, ACS Catal., 2017, 7(5), 3560–3568 CrossRef.
- X. Zhang, J. X. Liu, B. Zijlstra, I. A. W. Filot, Z. Y. Zhou, S. G. Sun and E. J. M. Hensen, Optimum Cu nanoparticle catalysts for CO2 hydrogenation towards methanol, Nano Energy, 2018, 43, 200–209 CrossRef CAS.
- M. Rondelli, G. Zwaschka, M. Krause, M. D. Rotzer, M. N. Hedhili, M. P. Hoger, V. D'Elia, F. F. Schweinberger, J. M. Basset and U. Heiz, Exploring the potential of different-sized supported subnanometer Pt clusters as catalysts for wet chemical applications, ACS Catal., 2017, 7(6), 4152–4162 CrossRef CAS.
- R. Bliem, J. van der Hoeven, A. Zavodny, O. Gamba, J. Pavelec, P. E. de Jongh, M. Schmid, U. Diebold and G. S. Parkinson, An atomic-scale view of CO and H2 oxidation on a Pt/Fe3 O4 model catalyst, Angew. Chem., Int. Ed., 2015, 54(47), 13999–14002 CrossRef CAS PubMed.
- V. Habibpour, C. R. Yin, G. Kwon, S. Vajda and R. E. Palmer, Catalytic oxidation of cyclohexane by size-selected palladium clusters pinned on graphite, J. Exp. Nanosci., 2013, 8(7–8), 993–1003 CrossRef CAS.
- M. Chen and D. W. Goodman, Catalytically active gold on ordered titania supports, Chem. Soc. Rev., 2008, 37(9), 1860–1870 RSC.
- A. Beniya and S. Higashi, Towards dense single-atom catalysts for future automotive applications, Nat. Catal., 2019, 2(7), 590–602 CrossRef.
- M. Chen, Y. Cai, Z. Yan and D. W. Goodman, On the origin of the unique properties of supported Au nanoparticles, J. Am. Chem. Soc., 2006, 128(19), 6341–6346 CrossRef CAS PubMed.
- M. Valden, X. Lai and D. W. Goodman, Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties, Science, 1998, 281(5383), 1647–1650 CrossRef CAS PubMed.
- M. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, The promotional effect of gold in catalysis by palladium–gold, Science, 2005, 310(5746), 291–293 CrossRef CAS PubMed.
- R. E. Winans, S. Vajda, G. E. Ballentine, J. W. Elam, B. D. Lee, M. J. Pelline, S. Seifert, G. Y. Tikhonov and N. A. Tomczyk, Reactivity of supported platinum nanoclusters studied by in situ GISAXS: Clusters stability under hydrogen, Top. Catal., 2006, 39(3–4), 145–149 CrossRef CAS.
- L. J. Wang, N. Blando, Z. Hicks, M. Denchy, X. Tang, H. Bleuel, M. S. Zhang, G. Gantefor and K. H. Bowen, Combined TPD and XPS study of ligation and decomposition of 1,6-hexanedithiol on size-selected copper clusters supported on HOPG, J. Phys. Chem. C, 2018, 122(4), 2173–2183 CrossRef CAS.
- B. Vilhanova, J. Vaclavik, L. Artiglia, M. Ranocchiari, A. Togni and J. A. van Bokhoven, Subnanometer gold clusters on amino-functionalized silica: An efficient catalyst for the synthesis of 1,3-diynes by oxidative alkyne coupling, ACS Catal., 2017, 7(5), 3414–3418 CrossRef CAS.
- J. Nelayah, A. Chmielewski, D. Alloyeau, H. Amara, J. Creuze and C. Ricolleau, Revealing the surface energetics and reactivity of bimetallic copper-gold catalyst nanoparticles by in situ environmental TEM, Microsc. Microanal., 2019, 25(S1), 33–34 CrossRef.
- L. M. Molina, S. Lee, K. Sell, G. Barcaro, A. Fortunelli, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin, I. Barke, V. von Oeynhausen, Y. Lei, R. J. Meyer, J. A. Alonso, A. F. Rodriguez, A. Kleibert, S. Giorgio, C. R. Henry, K. H. Meiwes-Broer and S. Vajda, Size-dependent selectivity and activity of silver nanoclusters in the partial oxidation of propylene to propylene oxide and acrolein: A joint experimental and theoretical study, Catal. Today, 2011, 160(1), 116–130 CrossRef CAS.
- J. Fuller, A. Fortunelli, W. A. Goddard III and Q. An, Reaction mechanism and kinetics for ammonia synthesis on the Fe(211) reconstructed surface, Phys. Chem. Chem. Phys., 2019, 21(21), 11444–11454 RSC.
- L. Kovarik, Z. Wei, C. H. F. Peden and J. Szanyi, Environmental TEM study of oxidation processes of catalytic nanoparticles, Microsc. Microanal., 2019, 25(S2), 1446–1447 CrossRef.
- S. Lee, A. Halder, G. A. Ferguson, S. Seifert, R. E. Winans, D. Teschner, R. Schlogl, V. Papaefthimiou, J. Greeley, L. A. Curtiss and S. Vajda, Subnanometer cobalt oxide clusters as selective low temperature oxidative dehydrogenation catalysts, Nat. Commun., 2019, 10(1), 954 CrossRef PubMed.
- A. Beck, X. Huang, L. Artiglia, M. Zabilskiy, X. Wang, P. Rzepka, D. Palagin, M. G. Willinger and J. A. van Bokhoven, The dynamics of overlayer formation on catalyst nanoparticles and strong metal–support interaction, Nat. Commun., 2020, 11(1), 3220 CrossRef CAS PubMed.
- A. Halder, J. Kioseoglou, B. Yang, K. L. Kolipaka, S. Seifert, J. Ilavsky, M. Pellin, M. Sowwan, P. Grammatikopoulos and S. Vajda, Nanoassemblies of ultrasmall clusters with remarkable activity in carbon dioxide conversion into C1 fuels, Nanoscale, 2019, 11(11), 4683–4687 RSC.
- R. Dietsche, D. C. Lim, M. Bubek, I. Lopez-Salido, G. Ganteför and Y. D. Kim, Comparison of electronic structures of mass-selected Ag clusters and thermally grown Ag islands on sputter-damaged graphite surfaces, Appl. Phys. A: Mater. Sci. Process., 2007, 90(3), 395–398 CrossRef.
- S. H. Jeong, D. C. Lim, J. H. Boo, S. B. Lee, H. N. Hwang, C. C. Hwang and Y. D. Kim, Interaction of silver with oxygen on sputtered pyrolytic graphite, Appl. Catal., A, 2007, 320, 152–158 CrossRef CAS.
- Y. Dai, T. J. Gorey, S. L. Anderson, S. Lee, S. Lee, S. Seifert and R. E. Winans, Inherent size effects on XANES of nanometer metal clusters: Size selected platinum clusters on silica, J. Phys. Chem. C, 2017, 121(1), 361–374 CrossRef CAS.
- S. K. Iyemperumal, T. G. Fenton, S. L. Gillingham, A. D. Carl, R. L. Grimm, G. H. Li and N. A. Deskins, The stability and oxidation of supported atomic-size Cu catalysts in reactive environments, J. Chem. Phys., 2019, 151(5), 054702 CrossRef.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Identification of active gold nanoclusters on iron oxide supports for CO oxidation, Science, 2008, 321(5894), 1331–1335 CrossRef CAS PubMed.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Electronic structure controls reactivity of size-selected Pd clusters adsorbed on TiO2 surfaces, Science, 2009, 326(5954), 826–829 CrossRef CAS PubMed.
- A. Corma, P. Concepcion, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadas, M. A. Lopez-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Exceptional oxidation activity with size-controlled supported gold clusters of low atomicity, Nat. Chem., 2013, 5(9), 775–781 CrossRef CAS PubMed.
- A. Baldansuren, H. Dilger, R. A. Eichel, J. A. van Bokhoven and E. Roduner, Interaction and reaction of ethylene and oxygen on six-atom silver clusters supported on LTA zeolite, J. Phys. Chem. C, 2009, 113(45), 19623–19632 CrossRef CAS.
- A. Boubnov, J. Timoshenko, C. J. Wrasman, A. S. Hoffman, M. Cargnello, A. I. Frenkel and S. R. Bare, Insight into restructuring of Pd–Au nanoparticles using EXAFS, Radiat. Phys. Chem., 2020, 175, 108304 CrossRef CAS.
- G. Barcaro, L. Sementa, F. R. Negreiros and A. Fortunelli, Influence of temperature and H2 adsorption on the structure of silica-supported gold subnanometer clusters, Comput. Theor. Chem., 2013, 1021, 222–228 CrossRef CAS.
- S. Vajda, R. E. Winans, J. W. Elam, B. D. Lee, M. J. Pellin, S. Seifert, G. Y. Tikhonov and N. A. Tomczyk, Supported gold clusters and cluster-based nanomaterials: characterization, stability and growth studies by in situ GISAXS under vacuum conditions and in the presence of hydrogen, Top. Catal., 2006, 39(3-4), 161–166 CrossRef CAS.
- D. W. Sindorf and G. E. Maciel, Solid-state NMR-studies of the reactions of silica surfaces with polyfunctional chloromethylsilanes and ethoxymethylsilanes, J. Am. Chem. Soc., 1983, 105(12), 3767–3776 CrossRef CAS.
- A. W. Ott, J. W. Klaus, J. M. Johnson and S. M. George, Al3O3 thin film growth on Si(100) using binary reaction sequence chemistry, Thin Solid Films, 1997, 292(1-2), 135–144 CrossRef CAS.
- F. R. Negreiros, A. Halder, C. Yin, A. Singh, G. Barcaro, L. Sementa, E. C. Tyo, M. J. Pellin, S. Bartling, K. H. Meiwes-Broer, S. Seifert, P. Sen, S. Nigam, C. Majumder, N. Fukui, H. Yasumatsu, S. Vajda and A. Fortunelli, Bimetallic Ag–Pt sub-nanometer supported clusters as highly efficient and robust oxidation catalysts, Angew. Chem., Int. Ed., 2018, 57(5), 1209–1213 CrossRef CAS PubMed.
- J. Qian, Q. An, A. Fortunelli, R. J. Nielsen and W. A. Goddard, 3rd, Reaction mechanism and kinetics for ammonia synthesis on the Fe(111) surface, J. Am. Chem. Soc., 2018, 140(20), 6288–6297 CrossRef CAS PubMed.
- L. Sementa, G. Barcaro and A. Fortunelli, Analogy between homogeneous and heterogeneous catalysis by subnanometer metal clusters: Ethylene oxidation on Ag trimers supported on MgO(100), Inorg. Chim. Acta, 2015, 431, 150–155 CrossRef CAS.
- L. Sementa, G. Barcaro, F. R. Negreiros and A. Fortunelli, Ligand/cluster/support catalytic complexes in heterogeneous ultrananocatalysis: NO oxidation on Ag3/MgO(100), Phys. Chem. Chem. Phys., 2014, 16(48), 26570–26577 RSC.
- L. Sementa, M. Monti, D. Toffoli, A. Posada-Amarillas, M. Stener and A. Fortunelli, Theoretical investigation of photoinduced processes in subnanometer oxide-supported metal catalysts, J. Phys. Chem. C, 2021, 125(3), 2022–2032 CrossRef CAS.
- M. Jørgensen and H. Grönbeck, Perspectives on computational catalysis for metal nanoparticles, ACS Catal., 2019, 9(10), 8872–8881 CrossRef.
- B. Zandkarimi and A. N. Alexandrova, Dynamics of subnanometer Pt clusters can break the scaling relationships in catalysis, J. Phys. Chem. Lett., 2019, 10(3), 460–467 CrossRef PubMed.
- G. Collinge, S. F. Yuk, M.-T. Nguyen, M.-S. Lee, V.-A. Glezakou and R. Rousseau, Effect of collective dynamics and anharmonicity on entropy in heterogenous catalysis: Building the case for advanced molecular simulations, ACS Catal., 2020, 10(16), 9236–9260 CrossRef CAS.
- A. Zecchina and S. Califano, The Development of Catalysis, 2017 Search PubMed.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, Chemisorption of CO and mechanism of CO oxidation on supported platinum nanoclusters, J. Am. Chem. Soc., 2011, 133(12), 4498–4517 CrossRef CAS PubMed.
- C. Yin, F. R. Negreiros, G. Barcaro, A. Beniya, L. Sementa, E. C. Tyo, S. Bartling, K.-H. Meiwes-Broer, S. Seifert, H. Hirata, N. Isomura, S. Nigam, C. Majumder, Y. Watanabe, A. Fortunelli and S. Vajda, Alumina-supported sub-nanometer Pt10 clusters: amorphization and role of the support material in a highly active CO oxidation catalyst, J. Mater. Chem. A, 2017, 5(10), 4923–4931 RSC.
- R. Ferrando and A. Fortunelli, Diffusion of adatoms and small clusters on magnesium oxide surfaces, J. Condens. Matter Phys., 2009, 21(26), 264001 CrossRef PubMed.
- G. Barcaro, A. Fortunelli, F. Nita and R. Ferrando, Diffusion of palladium clusters on magnesium oxide, Phys. Rev. Lett., 2005, 95(24), 246103 CrossRef CAS PubMed.
- B. Zandkarimi, P. Poths and A. N. Alexandrova, When fluxionality beats size selection: Acceleration of Ostwald ripening of sub-nano clusters, Angew. Chem., Int. Ed., 2021, 60(21), 11973–11982 CrossRef CAS PubMed.
- Y. Lou and J. Y. Liu, CO oxidation on metal oxide supported single Pt atoms: The role of the support, Ind. Eng. Chem. Res., 2017, 56(24), 6916–6925 CrossRef CAS.
- S. Kouva, K. Honkala, L. Lefferts and J. Kanervo, Review: Monoclinic zirconia, its surface sites and their interaction with carbon monoxide, Catal. Sci. Technol., 2015, 5(7), 3473–3490 RSC.
- V. Korpelin, M. M. Melander and K. Honkala, Reducing the irreducible: Dispersed metal atoms facilitate reduction of irreducible oxides, J. Phys. Chem. C, 2022, 126(2), 933–945 CrossRef CAS.
- M. A. Ha, E. T. Baxter, A. C. Cass, S. L. Anderson and A. N. Alexandrova, Boron switch for selectivity of catalytic dehydrogenation on size-selected Pt clusters on Al2O3, J. Am. Chem. Soc., 2017, 139(33), 11568–11575 CrossRef CAS PubMed.
- B. Zandkarimi, T. J. Gorey, G. Li, J. Munarriz, S. L. Anderson and A. N. Alexandrova, Alloying with Sn suppresses sintering of size-selected subnano Pt clusters on SiO2 with and without adsorbates, Chem. Mater., 2020, 32(19), 8595–8605 CrossRef CAS.
- E. T. Baxter, M. A. Ha, A. C. Cass, A. N. Alexandrova and S. L. Anderson, Ethylene dehydrogenation on Pt-4, Pt-7, Pt-8 clusters on Al2O3: Strong cluster size dependence linked to preferred catalyst morphologies, ACS Catal., 2017, 7(5), 3322–3335 CrossRef CAS.
- E. Jimenez-Izal, H. Zhai, J.-Y. Liu and A. N. Alexandrova, Nanoalloying MgO-deposited Pt clusters with Si to control the selectivity of alkane dehydrogenation, ACS Catal., 2018, 8(9), 8346–8356 CrossRef CAS.
- S. Valtera, J. Jašík, M. Vaidulych, J. E. Olszówka, M. Zlámalová, H. Tarábková, L. Kavan and Š. Vajda, Atom by atom built subnanometer copper cluster catalyst for the highly selective oxidative dehydrogenation of cyclohexene, J. Chem. Phys., 2022, 156(11), 114302 CrossRef CAS PubMed.
- A. Halder, M.-A. Ha, H. Zhai, B. Yang, M. J. Pellin, S. Seifert, A. N. Alexandrova and S. Vajda, Oxidative dehydrogenation of cyclohexane by Cu vs. Pd clusters: Selectivity control by specific cluster dynamics, ChemCatChem, 2020, 12(5), 1307–1315 CrossRef CAS.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Selective propene epoxidation on immobilized Au6–10 clusters: The effect of hydrogen and water on activity and selectivity, Angew. Chem., Int. Ed., 2009, 48(8), 1467–1471 CrossRef CAS PubMed.
- T. J. Gorey, B. Zandkarimi, G. Li, E. T. Baxter, A. N. Alexandrova and S. L. Anderson, Preparation of size- and composition-controlled PtnSnx/SiO2 (n = 4, 7, 24) bimetallic model catalysts with atomic layer deposition, J. Phys. Chem. C, 2019, 123(26), 16194 CrossRef CAS.
- M. Xue, M. Nakayama, P. Liu and M. G. White, Electronic interactions of size-selected oxide clusters on metallic and thin film oxide supports, J. Phys. Chem. C, 2017, 121(40), 22234–22247 CrossRef CAS.
- L. Piccolo, P. Afanasiev, F. Morfin, T. Len, C. Dessal, J. L. Rousset, M. Aouine, F. Bourgain, A. Aguilar-Tapia, O. Proux, Y. Chen, L. Soler and J. Llorca, Operando X-ray absorption spectroscopy investigation of photocatalytic hydrogen evolution over ultradispersed Pt/TiO2 catalysts, ACS Catal., 2020, 10(21), 12696–12705 CrossRef CAS.
- J. Engel, S. Francis and A. Roldan, The influence of support materials on the structural and electronic properties of gold nanoparticles – a DFT study, Phys. Chem. Chem. Phys., 2019, 21(35), 19011–19025 RSC.
- A. S. Bazhenov and K. Honkala, Globally optimized equilibrium shapes of zirconia-supported Rh and Pt nanoclusters: Insights into site assembly and reactivity, J. Phys. Chem. C, 2019, 123(12), 7209–7216 CrossRef CAS.
- N. Mammen, L. Spanu, E. C. Tyo, B. Yang, A. Halder, S. Seifert, M. J. Pellin, S. Vajda and S. Narasimhan, Reversing size-dependent trends in the oxidation of copper clusters through support effects, Eur. J. Inorg. Chem., 2018, 16–22 CrossRef CAS.
- A. Halder, C. Lenardi, J. Timoshenko, A. Mravak, B. Yang, L. K. Kolipaka, C. Piazzoni, S. Seifert, V. Bonačić-Koutecký, A. I. Frenkel, P. Milani and S. Vajda, CO2 methanation on Cu-cluster decorated zirconia supports with different morphology: A combined experimental in situ GIXANES/GISAXS, ex situ XPS and theoretical DFT study, ACS Catal., 2021, 11(10), 6210–6224 CrossRef CAS.
- A. S. Crampton, M. D. Rötzer, U. Landman and U. Heiz, Can support acidity predict sub-nanometer catalyst activity trends?, ACS Catal., 2017, 7(10), 6738–6744 CrossRef CAS.
- A. Cherevotan, J. Raj, L. Dheer, S. Roy, S. Sarkar, R. Das, C. P. Vinod, S. Xu, P. Wells, U. V. Waghmare and S. C. Peter, Operando generated ordered heterogeneous catalyst for the selective conversion of CO2 to methanol, ACS Energy Lett., 2021, 6(2), 509–516 CrossRef CAS.
- B. Lu, D. Kido, Y. Sato, H. Xu, W.-J. Chun, K. Asakura and S. Takakusagi, Development of operando polarization-dependent total reflection fluorescence X-ray absorption fine structure technique for three-dimensional structure determination of active metal species on a model catalyst surface under working conditions, J. Phys. Chem. C, 2021, 125(22), 12424–12432 CrossRef CAS.
- M. Albrahim, C. Thompson, D. Leshchev, A. Shrotri, R. R. Unocic, J. Hong, A. S. Hoffman, M. J. Meloni, R. C. Runnebaum, S. R. Bare, E. Stavitski and A. M. Karim, Reduction and agglomeration of supported metal clusters induced by high-flux X-ray absorption spectroscopy measurements, J. Phys. Chem. C, 2021, 125(20), 11048–11057 CrossRef CAS.
- K. De Wispelaere, C. S. Wondergem, B. Ensing, K. Hemelsoet, E. J. Meijer, B. M. Weckhuysen, V. Van Speybroeck and J. Ruiz-Martinez, Insight into the effect of water on the methanol-to-olefins conversion in H-SAPO-34 from molecular simulations and in situ microspectroscopy, ACS Catal., 2016, 6(3), 1991–2002 CrossRef CAS.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepulveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, I. X. F. X. Llabres, V. Van Speybroeck and J. Gascon, Metal–organic and covalent organic frameworks as single-site catalysts, Chem. Soc. Rev., 2017, 46(11), 3134–3184 RSC.
- I. S. Kim, Z. Li, J. Zheng, A. E. Platero-Prats, A. Mavrandonakis, S. Pellizzeri, M. Ferrandon, A. Vjunov, L. C. Gallington, T. E. Webber, N. A. Vermeulen, R. L. Penn, R. B. Getman, C. J. Cramer, K. W. Chapman, D. M. Camaioni, J. L. Fulton, J. A. Lercher, O. K. Farha, J. T. Hupp and A. B. F. Martinson, Sinter-resistant platinum catalyst supported by metal–organic framework, Angew. Chem., Int. Ed., 2018, 57(4), 909–913 CrossRef CAS PubMed.
- V. A. Nasluzov, E. A. Ivanova-Shor, A. M. Shor and K. M. Neyman, Silver atom, trimer and tetramer species supported on a ceria nanoparticle: A density functional study, Surf. Sci., 2019, 681, 38–46 CrossRef CAS.
- A. C. Reber, D. Bista, V. Chauhan and S. N. Khanna, Transforming redox properties of clusters using phosphine ligands, J. Phys. Chem. C, 2019, 123(14), 8983–8989 CrossRef CAS.
- A. Halder, L. A. Curtiss, A. Fortunelli and S. Vajda, Perspective: Size selected clusters for catalysis and electrochemistry, J. Chem. Phys., 2018, 148(11), 110901 CrossRef PubMed.
- G. Kwon, G. A. Ferguson, C. J. Heard, E. C. Tyo, C. Yin, J. DeBartolo, S. Seifert, R. E. Winans, A. J. Kropf, J. Greeley, R. L. Johnston, L. A. Curtiss, M. J. Pellin and S. Vajda, Size-dependent subnanometer Pd cluster (Pd4, Pd6, and Pd17) water oxidation electrocatalysis, ACS Nano, 2013, 7(7), 5808–5817 CrossRef CAS PubMed.
- A. Halder, C. Liu, Z. Liu, J. D. Emery, M. J. Pellin, L. A. Curtiss, P. Zapol, S. Vajda and A. B. F. Martinson, Water oxidation catalysis via size-selected iridium clusters, J. Phys. Chem. C, 2018, 122(18), 9965–9972 CrossRef CAS.
- A. von Weber and S. L. Anderson, Electrocatalysis by mass-selected Ptn clusters, Acc. Chem. Res., 2016, 49(11), 2632–2639 CrossRef CAS PubMed.
- R. Passalacqua, S. Parathoner, G. Centi, A. Halder, E. C. Tyo, B. Yang, S. Seifert and S. Vajda, Electrochemical behaviour of naked sub-nanometre sized copper clusters and effect of CO2, Catal. Sci. Technol., 2016, 6(18), 6977–6985 RSC.
- M. J. Pellin, S. C. Riha, E. C. Tyo, G. Kwon, J. A. Libera, J. W. Elam, S. Seifert, S. Lee and S. Vajda, Water oxidation by size-selected Co27 clusters supported on Fe2 O3, ChemSusChem, 2016, 9(20), 3005–3011 CrossRef CAS PubMed.
- X. H. Gao, G. T. Yu, L. R. Zheng, C. M. Zhang, H. Li, T. Wang, P. D. An, M. Liu, X. Q. Qiu, W. Chen and W. Chen, Strong electron coupling from the sub-nanometer Pd clusters confined in porous ceria nanorods for highly efficient electrochemical hydrogen evolution reaction, ACS Appl. Energy Mater., 2019, 2(2), 966–973 CrossRef CAS.
- J. Quinson, M. Roefzaad, D. Deiana, T. W. Hansen, J. B. Wagner, M. Nesselberger, A. S. Crampton, C. J. Ridge, F. F. Schweinberger, U. Heiz and M. Arenz, Electrochemical stability of subnanometer Pt clusters, Electrochim. Acta, 2018, 277, 211–217 CrossRef CAS.
- Y. Attia and M. Samer, Metal clusters: New era of hydrogen production, Renewable Sustainable Energy Rev., 2017, 79, 878–892 CrossRef CAS.
- T. Chao, X. Luo, W. Chen, B. Jiang, J. Ge, Y. Lin, G. Wu, X. Wang, Y. Hu, Z. Zhuang, Y. Wu, X. Hong and Y. Li, Atomically dispersed copper-platinum dual sites alloyed with palladium nanorings catalyze the hydrogen evolution reaction, Angew. Chem., Int. Ed., 2017, 56(50), 16047–16051 CrossRef CAS PubMed.
- L. Chang, D. Cheng, L. Sementa and A. Fortunelli, Hydrogen evolution reaction (HER) on Au@Ag ultrananoclusters as electro-catalysts, Nanoscale, 2018, 10(37), 17730–17737 RSC.
- M. Zhou, J. E. Dick and A. J. Bard, Electrodeposition of isolated platinum atoms and clusters on bismuth-characterization and electrocatalysis, J. Am. Chem. Soc., 2017, 139(48), 17677–17682 CrossRef CAS PubMed.
- M. Wilms, M. Kruft, G. Bermes and K. Wandelt, A new and sophisticated electrochemical scanning tunneling microscope design for the investigation of potentiodynamic processes, Rev. Sci. Instrum., 1999, 70(9), 3641–3650 CrossRef CAS.
- B. Madry, K. Wandelt and M. Nowicki, Sulfate structures on copper deposits on Au(111): In situ STM investigations, Electrochim. Acta, 2016, 217, 249–261 CrossRef CAS.
- H. Yang, F. R. Negreiros, Q. Sun, M. Xie, L. Sementa, M. Stener, Y. Ye, A. Fortunelli, W. A. Goddard, 3rd and T. Cheng, Predictions of chemical shifts for reactive intermediates in CO2 reduction under operando conditions, ACS Appl. Mater. Interfaces, 2021, 13(27), 31554–31560 CrossRef CAS PubMed.
- T. Cheng, A. Fortunelli and W. A. Goddard, 3rd, Reaction intermediates during operando electrocatalysis identified from full solvent quantum mechanics molecular dynamics, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(16), 7718–7722 CrossRef CAS PubMed.
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Nørskov, A. Nilsson and H. Ogasawara, Direct observation of the oxygenated species during oxygen reduction on a platinum fuel cell cathode, Nat. Commun., 2013, 4(1), 2817 CrossRef.
- T. Kosmala, A. Baby, M. Lunardon, D. Perilli, H. Liu, C. Durante, C. Di Valentin, S. Agnoli and G. Granozzi, Operando visualization of the hydrogen evolution reaction with atomic-scale precision at different metal–graphene interfaces, Nat. Catal., 2021, 4(10), 850–859 CrossRef CAS.
- J. Lu, L. Cheng, K. C. Lau, E. Tyo, X. Luo, J. Wen, D. Miller, R. S. Assary, H.-H. Wang, P. Redfern, H. Wu, J.-B. Park, Y.-K. Sun, S. Vajda, K. Amine and L. A. Curtiss, Effect of the size-selective silver clusters on lithium peroxide morphology in lithium–oxygen batteries, Nat. Commun., 2014, 5(1), 4895 CrossRef CAS PubMed.
- A. Halder, A. T. Ngo, X. Luo, H.-H. Wang, J. G. Wen, P. Abbasi, M. Asadi, C. Zhang, D. Miller, D. Zhang, J. Lu, P. C. Redfern, K. C. Lau, R. Amine, R. S. Assary, Y. J. Lee, A. Salehi-Khojin, S. Vajda, K. Amine and L. A. Curtiss, In situ formed Ir3Li nanoparticles as active cathode material in Li–oxygen batteries, J. Phys. Chem. A, 2019, 123(46), 10047–10056 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Gold nanoparticles: Assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology, Chem. Rev., 2004, 104(1), 293–346 CrossRef CAS PubMed.
- J. Z. Zhang and C. Noguez, Plasmonic optical properties and applications of metal nanostructures, Plasmonics, 2008, 3(4), 127–150 CrossRef CAS.
- M. Murdoch, G. I. N. Waterhouse, M. A. Nadeem, J. B. Metson, M. A. Keane, R. F. Howe, J. Llorca and H. Idriss, The effect of gold loading and particle size on photocatalytic hydrogen production from ethanol over Au/TiO2 nanoparticles, Nat. Chem., 2011, 3(6), 489–492 CrossRef CAS PubMed.
- C. Yu, R. Schira, H. Brune, B. von Issendorff, F. Rabilloud and W. Harbich, Optical properties of size selected neutral Ag clusters: Electronic shell structures and the surface plasmon resonance, Nanoscale, 2018, 10(44), 20821–20827 RSC.
- T. Lunskens, A. von Weber, M. Jakob, T. Lelaidier, A. Kartouzian and U. Heiz, Effect of thiol-ligands on the optical response of supported silver clusters, J. Phys. Chem. C, 2017, 121(17), 9331–9336 CrossRef.
- G. Barcaro, M. Broyer, N. Durante, A. Fortunelli and M. Stener, Alloying effects on the optical properties of Ag–Au nanoclusters from TDDFT calculations, J. Phys. Chem. C, 2011, 115(49), 24085–24091 CrossRef CAS.
- T. Lünskens, C. A. Walenta, P. Heister, A. Kartouzian and U. Heiz, Surface oxidation of supported, size-selected silver clusters, J. Cluster Sci., 2017, 28(6), 3185–3192 CrossRef.
- C. Yu, W. Harbich, L. Sementa, L. Ghiringhelli, E. Apra, M. Stener, A. Fortunelli and H. Brune, Intense fluorescence of Au20, J. Chem. Phys., 2017, 147(7), 074301 CrossRef PubMed.
- E. Fron, S. Aghakhani, W. Baekelant, D. Grandjean, E. Coutino-Gonzalez, M. Van der Auweraer, M. B. J. Roeffaers, P. Lievens and J. Hofkens, Structural and photophysical characterization of Ag clusters in LTA zeolites, J. Phys. Chem. C, 2019, 123(16), 10630–10638 CrossRef CAS.
- H. Chen, L. Peng, Y. Bian, X. Shen, J. Li, H.-C. Yao, S.-Q. Zang and Z. Li, Exerting charge transfer to stabilize Au nanoclusters for enhanced photocatalytic performance toward selective oxidation of amines, Appl. Catal., B, 2021, 284, 119704 CrossRef CAS.
- J. C. Luque-Ceballos, L. Sementa, E. Apra, A. Fortunelli and A. Posada-Amarillas, TDDFT study of the optical spectra of free and supported binary coinage metal hexamers: Effect of doping and support, J. Phys. Chem. C, 2018, 122(40), 23143–23152 CrossRef CAS.
- L. Sementa, M. Stener and A. Fortunelli, Optical activity of metal nanoclusters deposited on regular and doped oxide supports from first-principles simulations, Molecules, 2021, 26(22), 6961 CrossRef CAS PubMed.
- P. López-Caballero, J. M. Ramallo-López, L. J. Giovanetti, D. Buceta, S. Miret-Artés, M. A. López-Quintela, F. G. Requejo and M. P. de Lara-Castells, Exploring the properties of Ag5–TiO2 interfaces: stable surface polaron formation, UV-Vis optical response, and CO2 photoactivation, J. Mater. Chem. A, 2020, 8(14), 6842–6853 RSC.
- M. Pilar de Lara-Castells, A. W. Hauser, J. M. Ramallo-Lopez, D. Buceta, L. J. Giovanetti, M. A. Lopez-Quintela and F. G. Requejo, Increasing the optical response of TiO2 and extending it into the visible region through surface activation with highly stable Cu5 clusters, J. Mater. Chem. A, 2019, 7(13), 7489–7500 RSC.
- P. Lopez-Caballero, A. W. Hauser and M. Pilar de Lara-Castells, Exploring the catalytic properties of unsupported and TiO2-supported Cu5 clusters: CO2 Decomposition to CO and CO2 photoactivation, J. Phys. Chem. C, 2019, 123(37), 23064–23074 CrossRef CAS PubMed.
- B. Demirdjian, I. Ozerov, F. Bedu, A. Ranguis and C. R. Henry, CO and O2 adsorption and CO oxidation on Pt nanoparticles by indirect nanoplasmonic sensing, ACS Omega, 2021, 6(20), 13398–13405 CrossRef CAS PubMed.
- S. Theivendran, L. Chang, A. Mukherjee, L. Sementa, M. Stener, A. Fortunelli and A. Dass, Principles of optical spectroscopy of aromatic alloy nanomolecules: Au36-xAgx(SPh-tBu)(24), J. Phys. Chem. C, 2018, 122(8), 4524–4531 CrossRef CAS.
- L. Chang, O. Baseggio, L. Sementa, D. Cheng, G. Fronzoni, D. Toffoli, E. Apra, M. Stener and A. Fortunelli, Individual component map of rotatory strength and rotatory strength density plots as analysis tools of circular dichroism spectra of complex systems, J. Chem. Theory Comput., 2018, 14(7), 3703–3714 CrossRef CAS PubMed.
- D. Toffoli, M. Medves, G. Fronzoni, E. Coccia, M. Stener, L. Sementa and A. Fortunelli, Plasmonic circular dichroism in chiral gold nanowire dimers, Molecules, 2022, 27(1), 93 CrossRef CAS PubMed.
- S. Knoppe and T. Burgi, Chirality in thiolate-protected gold clusters, Acc. Chem. Res., 2014, 47(4), 1318–1326 CrossRef CAS PubMed.
- N. A. Sakthivel and A. Dass, Aromatic thiolate-protected series of gold nanomolecules and a contrary structural trend in size evolution, Acc. Chem. Res., 2018, 51(8), 1774–1783 CrossRef CAS PubMed.
- C. Dessal, T. Len, F. Morfin, J. L. Rousset, M. Aouine, P. Afanasiev and L. Piccolo, Dynamics of single Pt atoms on alumina during CO oxidation monitored by operando X-ray and infrared spectroscopies, ACS Catal., 2019, 9(6), 5752–5759 CrossRef CAS.
- D. Nicholls, J. Wells, A. Stevens, Y. Zheng, J. Castagna and N. D. Browning, Sub-sampled imaging for STEM: Maximising image speed, resolution and precision through reconstruction parameter refinement, Ultramicroscopy, 2022, 233, 113451 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2022 |