
Ultrafast and efficient energy transfer in a one- and two-photon sensitized rhodamine-BODIPY dyad: a perspective for broadly absorbing photocages†
Marvin
Asido‡
 a,
Carsten
Hamerla‡
a,
Rebekka
Weber‡
b,
Maximiliane
Horz‡
a,
Madhava Shyam
Niraghatam
a,
Alexander
Heckel
*b,
Irene
Burghardt
*a and
Josef
Wachtveitl
*a
a,
Carsten
Hamerla‡
a,
Rebekka
Weber‡
b,
Maximiliane
Horz‡
a,
Madhava Shyam
Niraghatam
a,
Alexander
Heckel
*b,
Irene
Burghardt
*a and
Josef
Wachtveitl
*a
aInstitute of Physical and Theoretical Chemistry, Goethe University Frankfurt, Max-von-Laue Straße 7, 60438 Frankfurt am Main, Germany. E-mail: burghardt@chemie.uni-frankfurt.de; wveitl@theochem.uni-frankfurt.de
bInstitute of Organic Chemistry and Chemical Biology, Goethe University Frankfurt, Max-von-Laue Straße 7, 60438 Frankfurt am Main, Germany. E-mail: heckel@uni-frankfurt.de
First published on 20th December 2021
Abstract
In view of the demand for photoactivatable probes that operate in the visible (VIS) to near infrared (NIR) region of the spectrum, we designed a bichromophoric system based on a rhodamine fluorophore and a BODIPY photocage. Two-photon excited fluorescence (TPEF) measurements and quantum chemical calculations reveal excellent two-photon properties of the employed rhodamine derivative. Excitation of the rhodamine unit via a one- or two-photon process leads to excitation energy transfer (EET) onto the BODIPY part, which is followed by the liberation of the leaving group. Ultrafast transient absorption spectroscopy provides evidence for a highly efficient EET dynamics on a sub-500 femtosecond scale. Complementary quantum dynamical calculations using the multi-layer multiconfiguration time-dependent Hartree (ML-MCTDH) approach highlight the quantum coherent character of the EET transfer. Photorelease of p-nitroaniline (PNA) was investigated by UV/vis absorption spectroscopy by either excitation of the rhodamine or the BODIPY moiety. Even though a quantitative assessment of the PNA yield could not be achieved for this particular BODIPY cage, the present study provides a design principle for a class of photocages that can be broadly activated between 500 and 900 nm.
Introduction
The possibility to control chemical processes with light paved the way for numerous innovations in biological and medical science ranging from high resolution imaging techniques1 to photodynamic therapy2–4 and drug release.5,6 Such applications favor excitation light of the so called “phototherapeutic window” (600–1000 nm) which provides less phototoxicity and deep tissue penetration due to little light scattering and competitive absorption.7 Photolabile Protecting Groups (PPGs) that operate in this area are rare and mainly include BODIPY-, xanthene- or cyanine-derived structures.8,9 However, while demonstrating strong absorption, these compounds often lack efficient photorelease quantum yields.Alternatively, photoactivation in the NIR region can be realized by two-photon absorption (2PA).10,11 The tight spatial focus of 2PA (due to the quadratic dependence on the intensity of the incident light) offers a particular advantage for photorelease reactions in a physiological context since biological signaling molecules can be liberated with very high precision.12,13 Unfortunately, most of the established PPGs show only little 2P response.14 Efforts have been made to convert these compounds into better 2P absorbers by structural modifications, which resulted in the development of a number of 2P activatable photocages including ortho-nitrobenzyl,15,16 coumarinyl,17,18 nitroindolinyl19,20 or quinoline21,22 based chromophores. However, improvement of the 2P cross section σ2P by such structural alterations often has an adverse effect on the photorelease quantum yield Φrel.23 Therefore high 2P action cross sections δu (with δu being the product of σ2P and Φrel)24 are difficult to achieve. The idea to separate the absorption process from the uncaging process was realized by the development of cooperative dyads based on photoinduced electron transfer (PET),25 fluorescence resonance energy transfer (FRET)26 or triplet–triplet energy transfer (TT-ET).27 In 2013 the Blanchard-Desce group introduced a FRET-based tandem system for enhanced two-photon uncaging which was further optimized to reach record two-photon action cross section of estimated 20 GM at 710 nm.28 In 2018 we presented a related system with special focus put on the electronic interactions between the coactive chromophores.29
In this work we investigate the excitation energy transfer (EET) dynamics in a one- and two-photon sensitive uncaging system based on covalently linked rhodamine (RHO) and BODIPY (BPY) chromophores (Fig. 1a). Rhodamine was chosen as 2P donor molecule since this class of fluorophores features high stability and brightness as well as outstanding 2PA properties.30,31 In our experiments, the conformationally rigid X-type rhodamine showed superior 2PA response between 810 and 870 nm compared to other alkylated rhodamines (Fig. S3, ESI†). A styryl-substituted BODIPY photocage with red-shifted absorption served as suitable EET acceptor. The two chromophores were connected via a rigid alkyne linker to prevent electronic interactions due to π-stacking. This dyad system undergoes an ultrafast EET on the sub-500 fs scale with high efficiency, which was shown by ultrafast transient absorption spectroscopy. Given that the transfer dynamics falls into a non-Förster regime, high-dimensional quantum dynamical calculations were carried out which are in agreement with the observed time scale and provide an interpretation of the quantum coherent EET step.
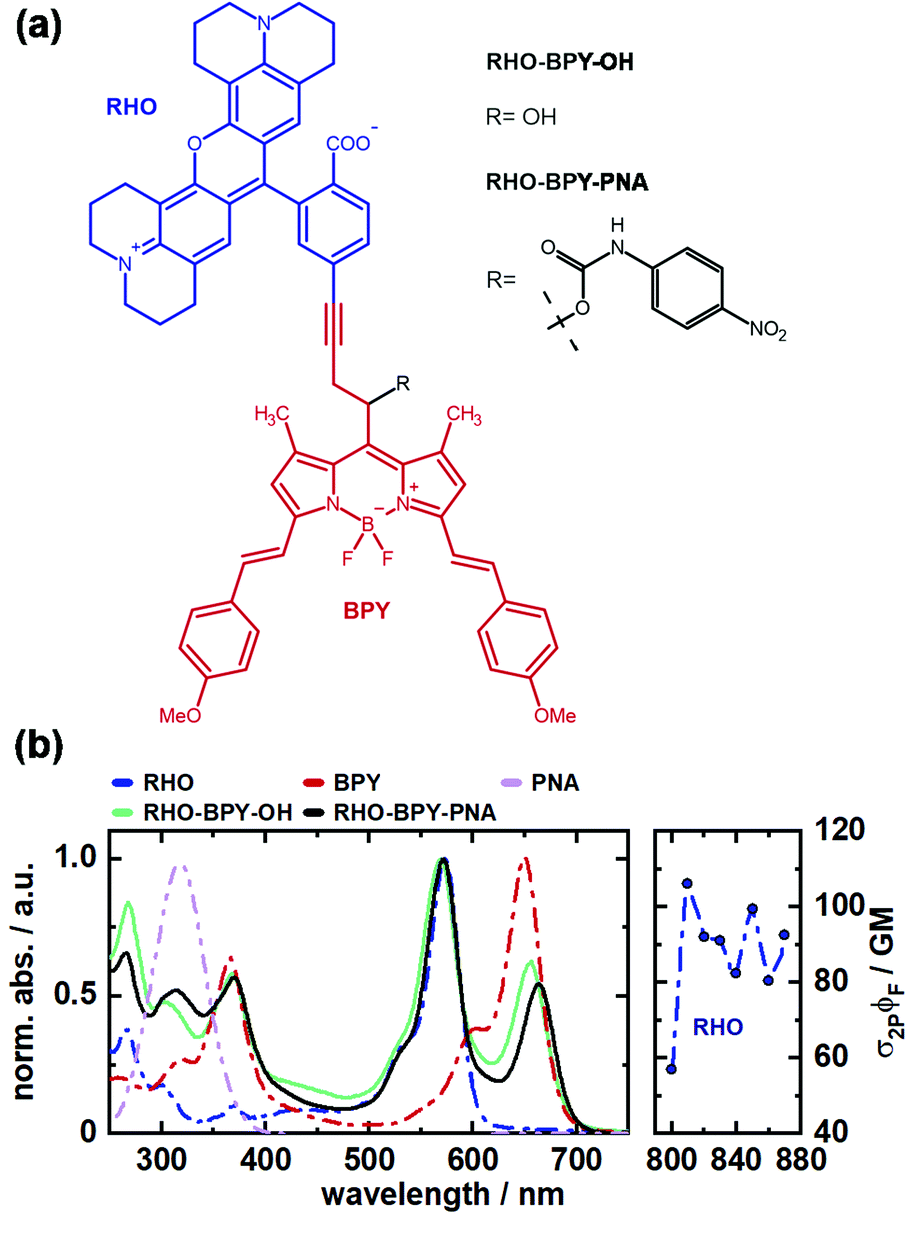 | ||
| Fig. 1 (a) Molecular structure of RHO-BPY-OH and RHO-BPY-PNA with rhodamine part (RHO) labeled blue and BODIPY part (BPY) labeled red. (b) Corresponding 1P absorption spectra of RHO-BPY-OH and RHO-BPY-PNA, as well as their corresponding individual constituents (left) and 2P absorption cross section of RHO (right) in MeOH. | ||
To test the suitability of the dyad for an application as a PPG, p-nitroaniline (PNA) was inserted as model leaving group. Details of the synthesis of the depicted compounds can be found in the ESI.†
The absorption spectra of both, RHO-BPY-OH as well as RHO-BPY-PNA, reflect well the main bands of their individual constituents (Fig. 1b), which absorb from 500–600 nm (RHO) and 570–700 nm (BPY). In the UV region below 400 nm the spectral components are more overlapping. For both dyads the bands at around 250 nm can be attributed to a higher order transition of RHO, whereas the bands at 380 nm mostly originate from BPY. In contrast to RHO-BPY-OH, the dyad with an attached leaving group (PNA) has a broader and slightly red-shifted absorption around 300–320 nm. The spectrum of the sensitizer (RHO) extends into the NIR region by considering 2PA effects. With 2P action cross sections (σ2PΦF) of 80–110 GM in the range of 810–870 nm, RHO shows excellent 2PA abilities similar to the values of other rhodamines found in the literature.32–35 Such high GM values for rhodamines are usually explained by the corresponding S0–S2 transition, which is 1P forbidden due to its symmetry, and in turn strongly 2P active.36,37
Results and discussion
Time-resolved spectroscopy
In order to investigate the ultrafast dynamics of all compounds we employed UV/vis transient absorption spectroscopy. In the initial experiment on RHO-BPY-OH we used pump pulses with a central wavelength of 560 nm to account for an excitation of RHO. The bleaching of the RHO ground state (GSB1) is reflected in the negative signal around 550–620 nm (Fig. 2a). Correspondingly, the excited state of RHO becomes populated, which leads to an excited state absorption (ESA1) with a central wavelength of 470 nm. The rise of these signals is faster than the temporal resolution of this experiment, which is around 75 fs.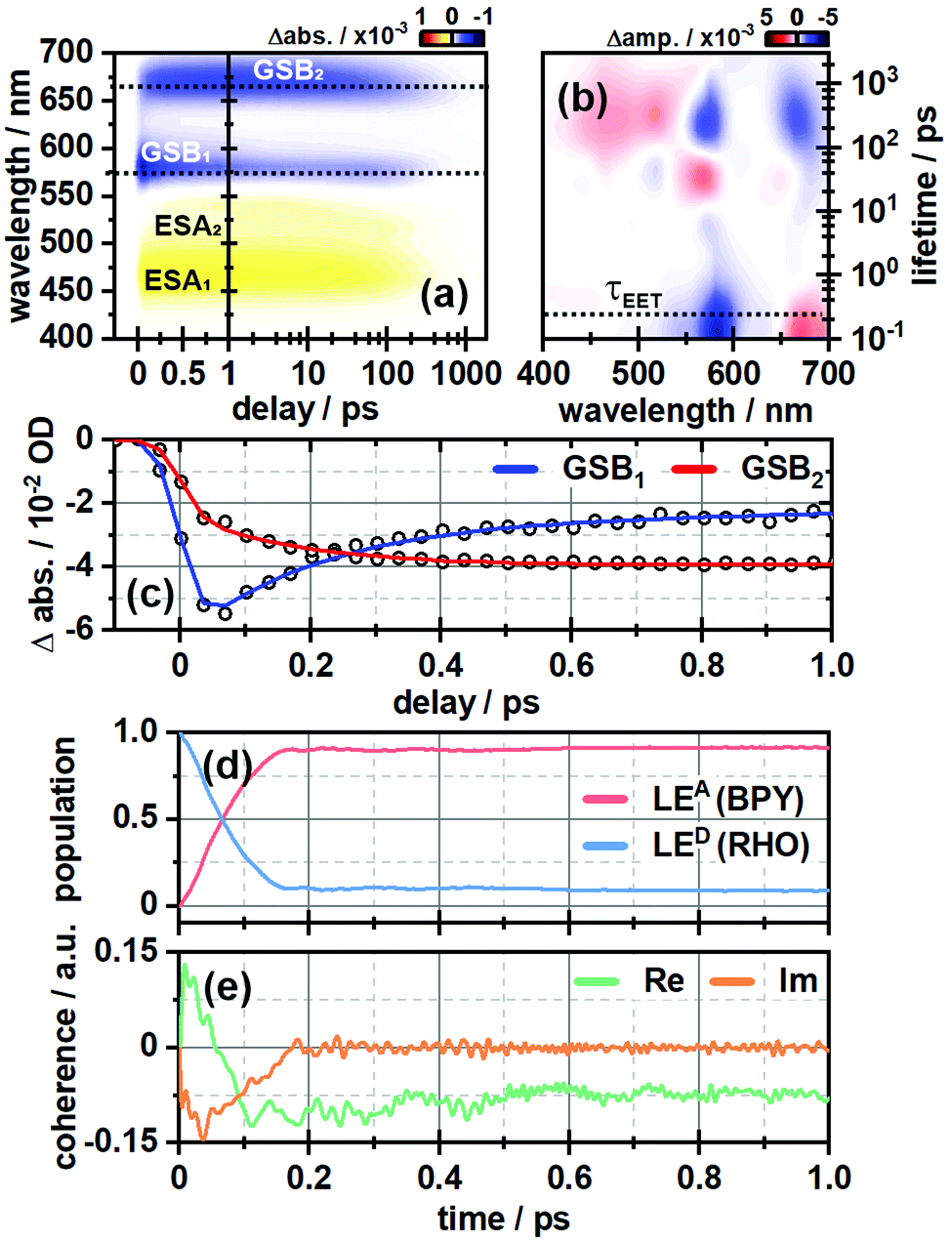 | ||
| Fig. 2 (a) Ultrafast transient absorption dataset of RHO-BPY-OH (in MeOH) and (b) its corresponding lifetime density analysis. The time of energy transfer τEET is highlighted by the dashed line. (c) Transients of GSB1 and GSB2, which are indicative for the EET from RHO to BPY. (d) Population of the locally excited states at the BODIPY (LEA) and the rhodamine moiety (LED) as a function of time derived from quantum dynamical calculations. (e) Corresponding coherences according to eqn (3). | ||
The decay, however, begins to occur on the 200–500 fs timescale, as can be seen in the lifetime density analysis (LDA) of the dataset. Simultaneously one can observe the rise of an additional bleaching signal (GSB2) centered at 670 nm, which corresponds to the BPY ground state absorption. This is easily visualized by comparing the transients at 575 nm and 670 nm (Fig. 2c). The bleaching signal at 575 nm decreases to 50% of its initial value in the first 300 fs after photoexcitation. On the same timescale GSB2 reaches more than 90% of its maximum value. Hence we assume an efficient EET from RHO to BPY, which then levels off once an equilibrium with the locally excited RHO is formed.
In the same manner we can identify a delayed excited state absorption at 525 nm (ESA2) with a similar rise time as GSB2, which indicates an excited state absorption of BPY. Unlike GSB1, ESA1 does not show a corresponding decay component. This can be explained by comparing the excited state dynamics of the isolated RHO and BPY, respectively (Fig. S5, ESI†). Whereas the ESA of RHO is centered around 470 nm (Fig. S5a, ESI†), BPY has a rather broad ESA with two main bands at 470 nm and 570 nm (Fig. S5c, ESI†). Therefore, the ESA1 band of RHO-BPY-OH is actually a superposition of the RHO and BPY excited state absorptions. ESA2 can then be considered as the residual shoulder of the BPY band at 570 nm, which partly overlaps with the GSB1 signal.
In order to determine the possibility of electronic coupling, we also excited RHO-BPY-OH with 655 nm pulses, which should mainly target the BPY part of the molecule. Indeed, in this case the photophysics of RHO-BPY-OH is dominated by the ultrafast dynamics of BPY (Fig. S6, ESI†). However, one can also observe an additional bleaching component of RHO at 575 nm, which occurs on the 10–100 ps timescale. This could stem from a slight spectral overlap of the RHO absorption and the excitation pulses. On the earlier timescale, this band overlaps with (and is compensated by) the red shoulder of the BPY excited state absorption, which is also apparent in the corresponding LDM (Fig. S6b, ESI†).
Computational analysis
To interpret the spectroscopic finding of an ultrafast energy transfer from the donor fragment RHO to the acceptor fragment BPY, electronic structure calculations and a quantum dynamical analysis were performed for the considered dyad. Time-dependent density functional theory (TD-DFT) was employed to study the low-lying singlet excitations of the dyad. For the quantum-dynamical calculations, the multi-layer multiconfiguration time-dependent Hartree (ML-MCTDH) method38–40 was employed, based upon a linear vibronic coupling (LVC) Hamiltonian for two electronic states and 266 vibrational modes. Due to the ultrafast transfer dynamics, conventional FRET rates are not expected to give reliable results.All electronic structure calculations were performed using DFT and TD-DFT as implemented in the Gaussian16 package.41
The ground state geometry was optimized using the B3LYP functional42,43 and 6-31G* basis.44 Other functionals yielded implausible ground state geometries where the carboxylate group of the respective rhodamine fragment formed a lactone type ring with the π-system, leading to a loss of planarity, as discussed in the Fig. S8 (ESI†). The minimum geometry based on B3LYP was confirmed using the Hessian. Solvent effects (here, methanol) were accounted for by the Polarizable Continuum Model (PCM).45,46
Excited state calculations were performed by means of TD-DFT with the long-range corrected CAM-B3LYP functional47 and 6-31G* basis set. Quadratic response calculations using the same functional were carried out with the DALTON program package.48 As detailed in the Table S2 (ESI†), benchmark calculations with other functional and basis set combinations were performed, from which we concluded that CAM-B3LYP/6-31G* yields the most accurate description of the system with respect to state energies and two-photon absorption properties as compared to the experimental findings. This functional was also employed in ref. 49, where 2PA spectra of rhodamine 6G were computed.49
The excited state analysis of the RHO-BPY-OH dyad (Table 1) shows three spectroscopic (one-photon) bright states with excitation energies of 2.14 eV (579 nm), 2.86 eV (434 nm) and 3.43 eV (362 nm) followed by a dark state at 3.51 eV (354 nm). The first bright excited state (S1), is localized on the acceptor BPY fragment whereas the S2 state is localized on the donor RHO fragment. The S3 state is another BPY centered state while the subsequent dark state (S4), at 3.51 eV is again localized at the RHO fragment. The latter state (S4), is the two-photon active state as further explained below. These four excited states S1, S2, S3 and S4 are illustrated in Fig. 3a.
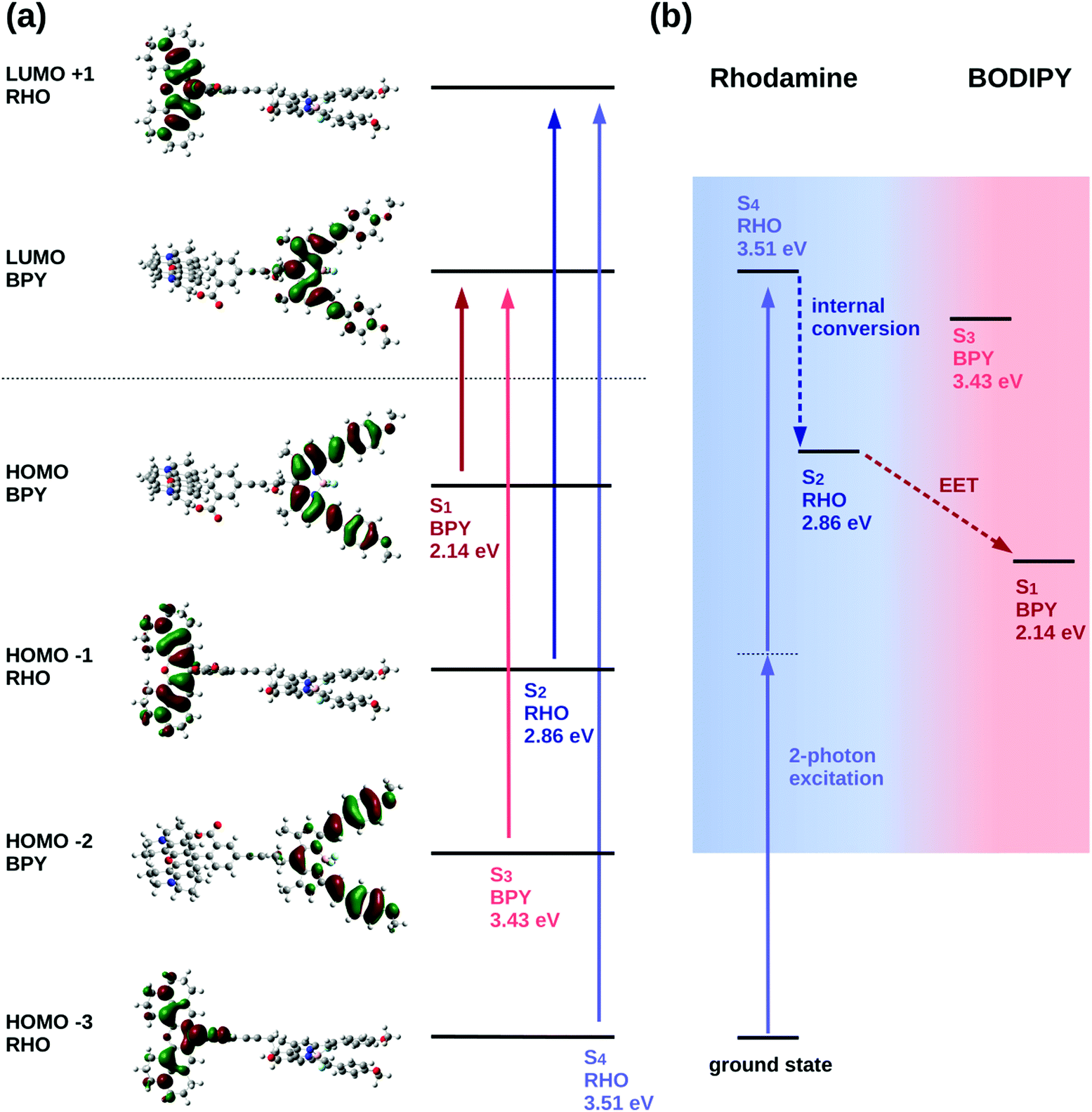 | ||
| Fig. 3 (a) Frontier molecular orbitals involved in the excited electronic states of interest in RHO-BPY-OH. (b) Schematic illustration of the processes triggered by two-photon excitation of RHO-BPY-OH. | ||
For the quadratic response calculations, a smaller model system was used; i.e., a slightly adapted dyad (referred to as “model” system (M) in the following, see ESI† Table S3) consisting of a dimethyl substituted Rho1 moiety and a BPY1 fragment without the ortho-methoxyphenyl substituent linked through an acetylene bond. The excited state analysis of the model system turns out to be very similar to the original RHO-BPY system and exhibits the same orbital transitions contributing to the respective states (Fig. S11 and S12, ESI†). The SM1 state is a bright state at 2.86 eV (433 nm) localized on the BPY1 moiety, the SM2 state is another bright state at 3.06 eV (405 nm) localized on the Rho1 fragment and the subsequent state, the SM3 state, is a dark state at 3.56 eV (348 nm), also localized on the Rho1 fragment.
The quadratic response calculation shows that the SM3 state has the highest two-photon absorption cross-section. That is, the SM3 state has a two-photon cross section σ2P of 175 GM, whereas the SM1 and the SM2 states have two-photon cross-sections of 3 GM and 28 GM, respectively. The SM3 state exhibits the same orbital transitions as the S4 state of RHO-BPY-OH and is, hence, considered the equivalent state in the model system. We therefore conclude that the S4 state of RHO-BPY-OH is the two-photon active state.
From the assignments above, the following picture of the photochemical process results: following excitation by a two-photon pulse the molecule is first directly excited to the S4 state in the case of RHO-BPY-OH (or the SM3 state in case of the model system Rho1-BPY1-OH). This is followed by internal conversion (IC) to the S2/SM2 state within the RHO/Rho1 fragment, and subsequently the energy is transferred via EET to the S1/SM1 state on the BPY/BPY1 fragment. A scheme representing these steps following the initial two-photon excitation is shown in Fig. 3b. The next higher state involving a transition on the BPY fragment is the S3 state of RHO-BPY-OH, also shown in Fig. 3. This state features a negligible admixture of transitions on the RHO fragment, such that there is no indication that the S3 state is involved in the IC step. Indeed, the IC can be understood as an intramolecular process within the RHO fragment, involving the S4 → S2 transition. In the following we assume that the IC and EET steps are sequential, but a more detailed analysis would allow for temporal overlap of these steps.
| System | RHO-BPY-OH | Rho1-BPY1-OH (M) | |
|---|---|---|---|
| S1 | Excitation Energy [eV] | 2.1413 | 2.8611 |
| Oscillator strength f | 1.1307 | 0.5711 | |
| σ 2P [GM] | 3.0 | ||
| S2 | Excitation Energy [eV] | 2.8624 | 3.0616 |
| Oscillator strength f | 1.0555 | 0.9615 | |
| σ 2P [GM] | 27.5 | ||
| S3 | Excitation Energy [eV] | 3.4293 | 3.5648 |
| Oscillator strength f | 1.6060 | 0.3422 | |
| σ 2P [GM] | 175 | ||
| S4 | Excitation Energy [eV] | 3.5065 | 3.7837 |
| Oscillator strength f | 0.0788 | 0.0559 | |
| σ 2P [GM] | 5.1 |
In the quantum dynamical analysis, we focus on the EET step, assuming that the preceding IC happens within tens of femtoseconds. Here, too, we refer to the smaller model system permitting to include the full set of N = 266 vibrational modes; details are given in the ESI† (Sec. Excited state analysis of Rho1-BPY1-OH). An LVC model was parameterized based upon the electronic structure calculations complemented by excited state gradients. The LVC Hamiltonian reads as follows, using mass- and frequency weighted coordinates
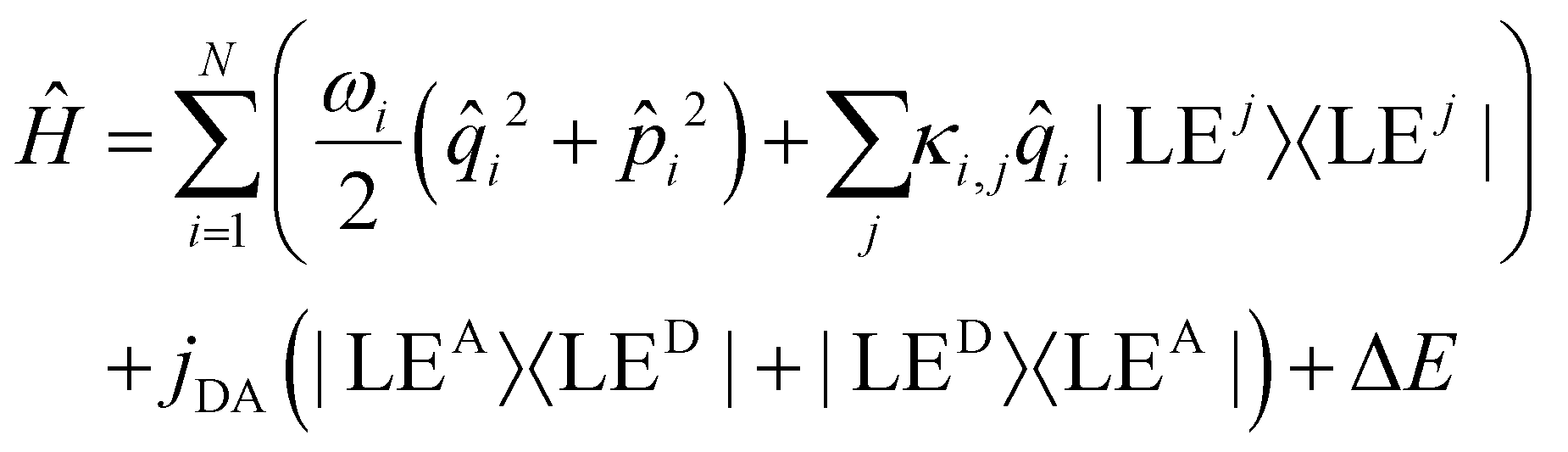 | (1) |
![[q with combining circumflex]](https://www.rsc.org/images/entities/i_char_0071_0302.gif) i being the position operator, the momentum operator
i being the position operator, the momentum operator 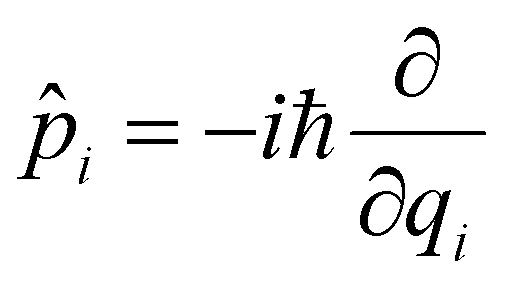 , the vibronic couplings κi,j, Coulombic EET coupling jDA and the electronic offset ΔE.
, the vibronic couplings κi,j, Coulombic EET coupling jDA and the electronic offset ΔE.
The Hamiltonian encompasses two diabatic electronic states, which here correspond to locally excited states (denoted LEA and LED). These locally excited (diabatic) states correspond to a very good approximation to the adiabatic SM1 and SM2 state of the Rho1-BPY1 model dyad obtained by the above electronic structure calculations. Furthermore, the Hamiltonian includes N = 266 normal modes, among which NA = 117 modes are localized on the BPY1 (acceptor) fragment and ND = 149 modes are localized on the Rho1 (donor) fragment, respectively. The vibronic coupling constants {κi,j} are computed by projecting the gradient of the two relevant excited states at the Franck–Condon geometry onto the ground state normal modes. For this purpose, the CAM-B3LYP method had to be consistently used for the ground state geometry optimization and the excited state gradient calculation; thus, a constraint was introduced in order to match the CAM-B3LYP optimized geometry with the B3LYP reference geometry (see Fig. S14, ESI†). The normal-mode frequencies {ωi} were obtained by normal mode analysis for the donor and acceptor fragment respectively. Analysis of the spectral densities (that can be computed from the vibronic couplings, see ESI† eqn (S9)) shows, that the normal modes are localized either on the Rho1 or BPY1 fragment and only couple to one of the locally excited states LEA or LED respectively. The electronic coupling jDA (0.024 eV) was determined using a transition density cube (TDC) procedure,50 implemented in an in-house code as detailed in the ESI.† The coupling derives from the respective transition densities as follows51
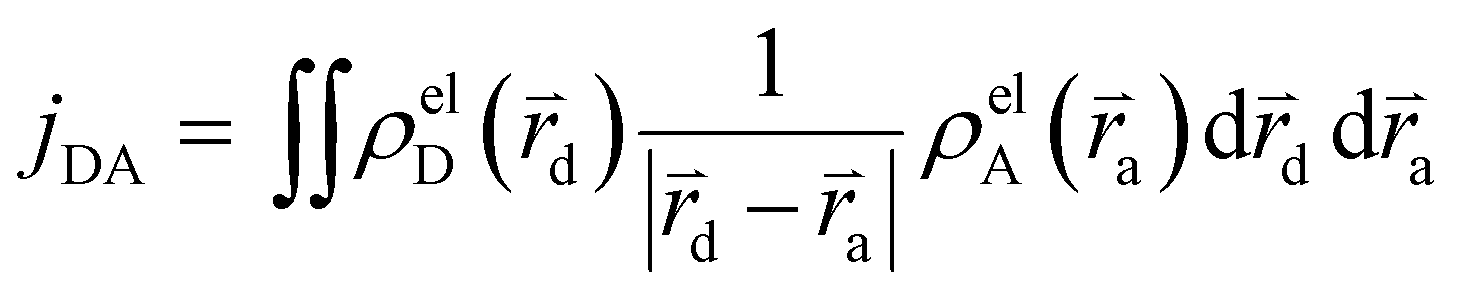 | (2) |
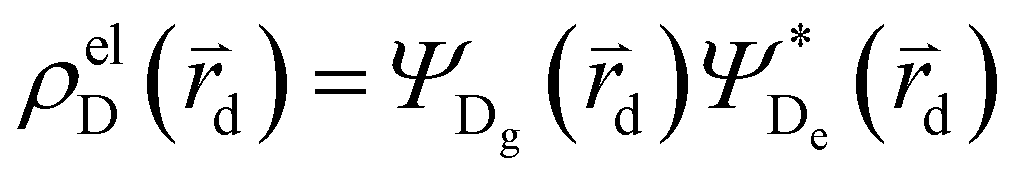 and
and 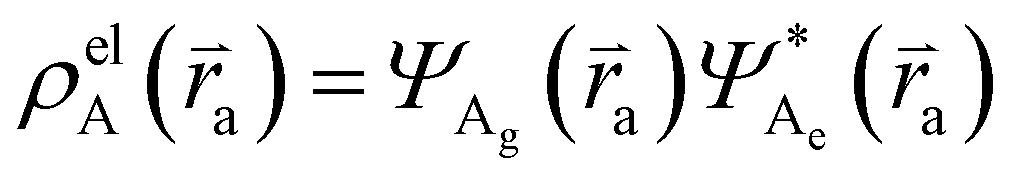 , where
, where 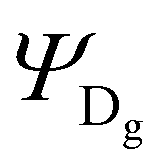 , ΨDe, ΨAg and ΨAe are the donor and acceptor ground (g) and excited state (e) wavefunctions and
, ΨDe, ΨAg and ΨAe are the donor and acceptor ground (g) and excited state (e) wavefunctions and ![[r with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0072_20d1.gif) d and a are the electron coordinates on the donor and acceptor fragment. The energy difference between the two relevant electronic states ΔE (0.25 eV) was taken from the experimental absorption spectrum.
d and a are the electron coordinates on the donor and acceptor fragment. The energy difference between the two relevant electronic states ΔE (0.25 eV) was taken from the experimental absorption spectrum.
Fig. 2d shows the time-evolving diabatic populations of the two relevant states LEA and LED and Fig. 2e depicts the time-evolving diabatic electronic coherence ρDA(t). The latter was obtained by taking the trace over the electronic and vibrational degrees of freedom of the density operator 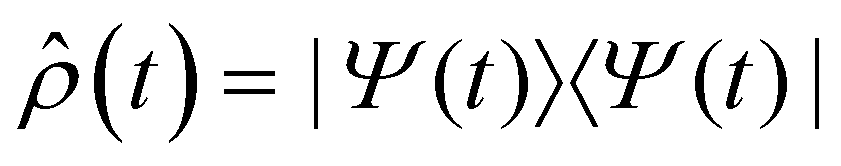 of the full electronic-vibrational system.
of the full electronic-vibrational system.
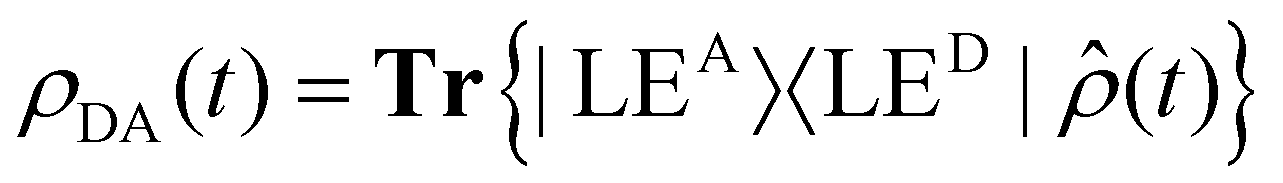 | (3) |
From the quantum dynamical analysis, an initial decay to the LEA state mainly “localized” on the acceptor fragment BPY1, is found on a time scale of 200 fs. Although a bit faster than the experiment this is in good agreement with the time-resolved spectroscopic measurements where the energy transfer occurs on a 300 fs timescale (Fig. 2a–c). The coherence decays on a similar time scale; more precisely, the transient state-to-state population flux (corresponding to the imaginary part of the coherence), decays to zero, while the persistent real part indicates that the system tends towards a coherent superposition involving a non-zero admixture of the LED state (PD ∼ 0.1) to the mainly populated LEA state (PA ∼ 0.9). The equilibrated donor population is possibly underestimated as compared with experiment, one of the reasons being that solvent effects were not explicitly included.
Analysis of photocleavage
In order to investigate the proposed system in terms of photocleavage, we performed illumination experiments with high-power LEDs of 565 nm and 660 nm central wavelength, respectively. Irradiation of RHO-BPY-PNA with 565 nm light (Fig. 4a–c) results in a strong photobleaching at 300–400 nm and 580–700 nm, which correspond to the absorption bands of the BPY scaffold (see Fig. 1b). In contrast, the main absorption band of RHO at around 560 nm remains mostly intact. At the blue end of the spectrum, a new absorption feature at 300 nm arises. This difference band could indeed originate from liberated PNA, which is expected to absorb at 250–380 nm (Fig. 1b) and/or a blue shift due to the formation of a stable (leaving group free) photoproduct (Fig. 4b). The latter argument can be rationalized by the red-shifted absorption of RHO-BPY-PNA compared to the leaving group free reference compound RHO-BPY-OH in the spectral range of 280–350 nm (Fig. 1b).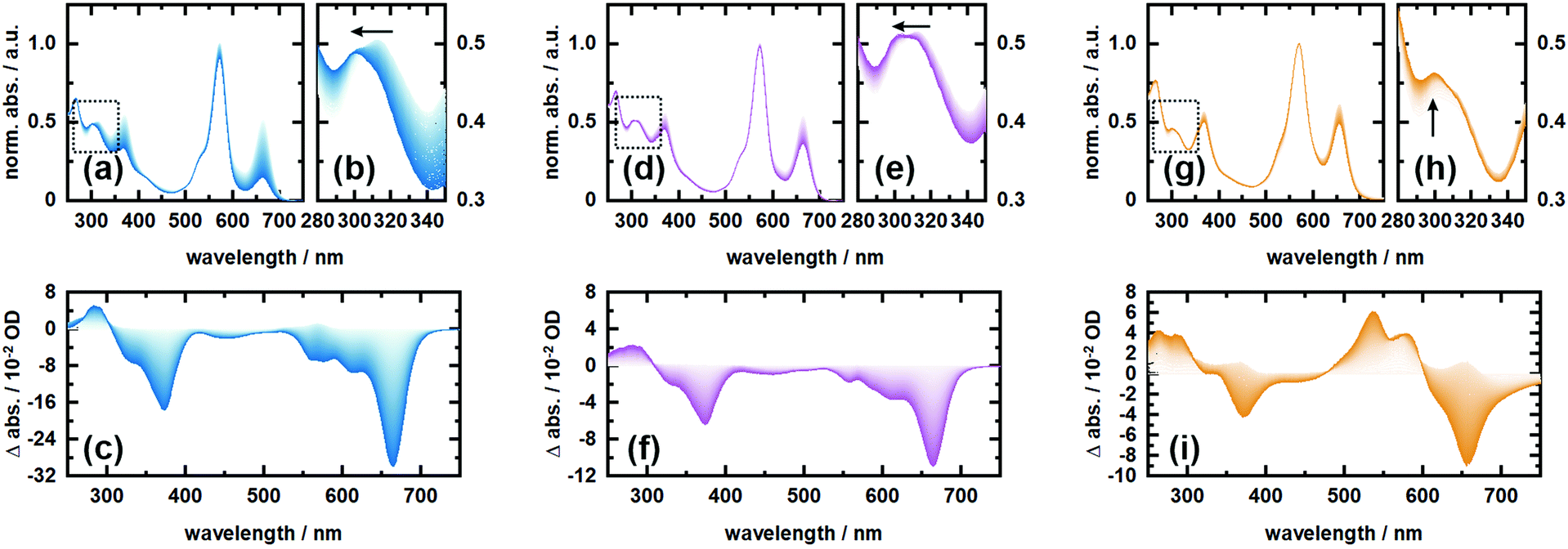 | ||
| Fig. 4 Irradiation experiments of RHO-BPY-PNA (in MeOH) with 565 nm light (a–c) and 660 nm light (d–f), as well as irradiation of RHO-BPY-OH (in MeOH) with 565 nm light (g–i). (a), (d) and (g) depict the respective normalized spectra with a closer zoom in (b), (e) and (h) as indicated by the dashed boxes. (c), (f) and (i) show the respective difference spectra referenced to the initial dark measurement. The spectra were recorded in 30 s intervals in a total timeframe of 6000 s. | ||
Strikingly, the illumination of the same sample with 660 nm light (Fig. 4d–f) leads to the same difference spectrum, albeit with a significantly weaker amplitude compared to the 565 nm illumination. On the one hand this underlines that direct excitation of either the RHO or BPY scaffold leads to the same photochemistry. On the other hand it further underlines the remarkable ability of RHO to collect photons due to its high extinction coefficient, as well as its very efficient energy transfer to BPY, which is reflected in the almost 3-fold signal amplitude upon excitation with 565 nm light. Unfortunately, the direct observation, and therefore a quantitative assessment, of PNA photorelease is challenging due to the strong spectral overlap of the PNA absorption with the relatively strong BPY band in the UV.
Therefore, we also irradiated RHO-BPY-OH with 565 nm light (Fig. 4g–i) as a control to substantiate our observation of photocleavage. The resulting difference spectrum (Fig. 4i) clearly differs from Fig. 4c and f. The two bleaching bands centered at 360 nm and 660 nm, which are again mainly due to BPY contributions, are each accompanied by a rise of a blue shifted absorption at 250–300 nm and 500–600 nm, respectively. Unlike RHO-BPY-PNA (Fig. 4b and e), no spectral blue-shift of the band around 300 nm is observed (Fig. 4h). The shapes of the difference bands in Fig. 4i also indicate a significant spectral overlap between reactant and product, which further underlines a different photochemistry compared to the illumination of RHO-BPY-PNA. Here, we assume that the excitation energy is mainly funneled into the cleavage or a disruption of the conjugation of the styryl-residues, which consequently leads to an intact but blue shifted BPY scaffold due to its reduced π-electron system. This is in contrast to results reported by Peterson et al.,52 which show such a cleavage of a styryl residue only as a result of actual cargo release. However, they investigated a BODIPY system with a different leaving group and no additional rhodamine attached, which makes a direct comparison difficult.
Conclusions
In conclusion, we developed a cooperative dyad based on a rhodamine fluorophore and a BODIPY photocage for one- or two-photon induced uncaging between 500 and 900 nm. This broad spectral window could be advantageous for applications and studies, which demand the usage of different excitation sources. The utilized X-rhodamine derivative displays high two-photon action cross sections (σ2PΦF) of around 100 GM between 810 and 870 nm, covering parts of the phototherapeutic window. Investigation of the energy transfer from rhodamine to BODIPY by ultrafast transient absorption spectroscopy and quantum dynamics simulations revealed a very fast and efficient EET on the sub-500 fs timescale. Photolysis experiments were performed at 565 and 660 nm using p-nitroaniline as a leaving group. It can be deduced from the UV/vis spectra that PNA is released upon illumination. However, the quantification of the uncaging reaction remained challenging due to overlapping bands in the spectrum. Moreover, the employed BODIPY derivative is known for low uncaging quantum yields and photodecomposition, which can partly be compensated by the large extinction coefficient of rhodamine. Future work will therefore focus on identifying variants of the BODIPY moiety which improve its uncaging properties.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank Jan von Cosel and Konstantin Falahati for providing the in-house implementation used to compute the electronic TDC coupling and Francesco Di Maiolo for useful discussions and help with preparing the figures. We gratefully acknowledge funding of the Deutsche Forschungsgemeinschaft (DFG) through the research training group “CLiC” (GRK 1986, Complex Light-Control).Notes and references
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73–76 CrossRef CAS PubMed.
- J. D. Bhawalkar, N. D. Kumar, C.-F. Zhao and P. N. Prasad, J. Clin. Laser Med. Surg., 1997, 15, 201–204 CrossRef CAS PubMed.
- P. Majumdar, R. Nomula and J. Zhao, J. Mater. Chem. C, 2014, 2, 5982–5997 RSC.
- A. M. Durantini, L. E. Greene, R. Lincoln, S. R. Martínez and G. Cosa, J. Am. Chem. Soc., 2016, 138, 1215–1225 CrossRef CAS PubMed.
- M. Matsuzaki, G. C. R. Ellis-Davies, T. Nemoto, Y. Miyashita, M. Iino and H. Kasai, Nat. Neurosci., 2001, 4, 1086–1092 CrossRef CAS PubMed.
- I. Elamri, C. Abdellaoui, J. K. Bains, K. F. Hohmann, S. L. Gande, E. Stirnal, J. Wachtveitl and H. Schwalbe, J. Am. Chem. Soc., 2021, 143, 10596–10603 CrossRef CAS PubMed.
- C. Morville, J. Chaud, F. Bolze and A. Specht, J. Inclusion Phenom. Macrocyclic Chem., 2021, 101, 291–304 CrossRef CAS.
- R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed.
- L. Josa-Culleré and A. Llebaria, ChemPhotoChem, 2021, 5, 298–316 CrossRef.
- W. Denk, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6629–6633 CrossRef CAS PubMed.
- P. Lipp and E. Niggli, J. Physiol., 1998, 508, 801–809 CrossRef CAS PubMed.
- G. C. R. Ellis-Davies, Front. Synaptic Neurosci., 2019, 10, 1–13 Search PubMed.
- G. C. R. Ellis-Davies, Acc. Chem. Res., 2020, 53, 1593–1604 CrossRef CAS PubMed.
- T. Furuta, S. S. H. Wang, J. L. Dantzker, T. M. Dore, W. J. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193–1200 CrossRef CAS PubMed.
- A. Momotake, N. Lindegger, E. Niggli, R. J. Barsotti and G. C. R. Ellis-Davies, Nat. Methods, 2006, 3, 35–40 CrossRef CAS PubMed.
- Y. Becker, S. Roth, M. Scheurer, A. Jakob, D. A. Gacek, P. J. Walla, A. Dreuw, J. Wachtveitl and A. Heckel, Chem. – Eur. J., 2021, 27, 2212–2218 CrossRef CAS PubMed.
- J. P. Olson, H. B. Kwon, K. T. Takasaki, C. Q. Chiu, M. J. Higley, B. L. Sabatini and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2013, 135, 5954–5957 CrossRef CAS PubMed.
- M. Bojtár, A. Kormos, K. Kis-Petik, M. Kellermayer and P. Kele, Org. Lett., 2019, 21, 9410–9414 CrossRef PubMed.
- J. Morrison, P. Wan, J. E. T. Corrie and G. Papageorgiou, Photochem. Photobiol. Sci., 2002, 1, 960–969 CrossRef CAS.
- G. C. R. Ellis-Davies, M. Matsuzaki, M. Paukert, H. Kasai and D. E. Bergles, J. Neurosci., 2007, 27, 6601–6604 CrossRef CAS PubMed.
- Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267–4276 CrossRef CAS PubMed.
- C. Tran, T. Gallavardin, M. Petit, R. Slimi, H. Dhimane, M. Blanchard-Desce, F. C. Acher, D. Ogden and P. I. Dalko, Org. Lett., 2015, 17, 402–405 CrossRef CAS PubMed.
- I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J. B. Baudin, P. Neveu and L. Jullien, Chem. – Eur. J., 2006, 12, 6865–6879 CrossRef CAS PubMed.
- M. Abe, Y. Chitose, S. Jakkampudi, P. T. T. Thuy, Q. Lin, B. T. Van, A. Yamada, R. Oyama, M. Sasaki and C. Katan, Adv. Synth. Catal., 2017, 49, 3337–3346 CAS.
- K. A. Korzycka, P. M. Bennett, E. J. Cueto-Diaz, G. Wicks, M. Drobizhev, M. Blanchard-Desce, A. Rebane and H. L. Anderson, Chem. Sci., 2015, 6, 2419–2426 RSC.
- S. Picard, E. J. Cueto-Diaz, E. Genin, G. Clermont, F. Acher, D. Ogden and M. Blanchard-Desce, Chem. Commun., 2013, 49, 10805–10807 RSC.
- G. Papageorgiou, M. Lukeman, P. Wan and J. E. T. Corrie, Photochem. Photobiol. Sci., 2004, 3, 366–373 CrossRef CAS PubMed.
- E. Cueto Diaz, S. Picard, M. Klausen, V. Hugues, P. Pagano, E. Genin and M. Blanchard-Desce, Chem. – Eur. J., 2016, 22, 10848–10859 CrossRef CAS PubMed.
- C. A. Hammer, K. Falahati, A. Jakob, R. Klimek, I. Burghardt, A. Heckel and J. Wachtveitl, J. Phys. Chem. Lett., 2018, 9, 1448–1453 CrossRef CAS PubMed.
- M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- A. Nag and D. Goswami, J. Photochem. Photobiol., A, 2009, 206, 188–197 CrossRef CAS PubMed.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 CrossRef CAS.
- C. Xu, R. M. Williams, W. Zipfel and W. W. Webb, Bioimaging, 1996, 4, 198–207 CrossRef CAS.
- M. A. Albota, C. Xu and W. W. Webb, Appl. Opt., 1998, 37, 7352–7356 CrossRef CAS PubMed.
- D. A. Oulianov, I. V. Tomov, A. S. Dvornikov and P. M. Rentzepis, Opt. Commun., 2001, 191, 235–243 CrossRef CAS.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, J. Am. Chem. Soc., 2011, 133, 14590–14592 CrossRef CAS PubMed.
- C. B. Milojevich, D. W. Silverstein, L. Jensen and J. P. Camden, J. Phys. Chem. C, 2013, 117, 3046–3054 CrossRef CAS.
- H. Wang, J. Phys. Chem. A, 2015, 119, 7951–7965 CrossRef CAS PubMed.
- M. H. Beck, A. Jäckle, G. A. Worth and H.-D. Meyer, Phys. Rep., 2000, 324, 1–105 CrossRef CAS.
- G. A. Worth, M. H. Beck, A. Jäckle and H.-D. Meyer, MCTDH Package, Version 8.5.5, Heidelberg University, 2016, http://www.mctdh.uni-hd.de Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef.
- S. Miertus and J. Tomasi, Chem. Phys., 1982, 65, 239–245 Search PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenæs, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjærgaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnæs, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, WIREs Comput. Mol. Sci., 2014, 4, 269–284 CrossRef CAS PubMed.
- R. Di Remigio, T. Giovannini, M. Ambrosetti, C. Cappelli and L. Frediani, J. Chem. Theory Comput., 2019, 15, 4056–4068 CrossRef CAS PubMed.
- B. P. Krueger, G. D. Scholes and G. R. Fleming, J. Phys. Chem. B, 1998, 102, 5378–5386 CrossRef CAS.
- H. Tamura, J.-M. Mallet, M. Oheim and I. Burghardt, J. Phys. Chem. C, 2009, 113, 7548–7552 CrossRef CAS.
- J. A. Peterson, C. Wijesooriya, E. J. Gehrmann, K. M. Mahoney, P. P. Goswami, T. R. Albright, A. Syed, A. S. Dutton, E. A. Smith and A. H. Winter, J. Am. Chem. Soc., 2018, 140, 7343–7346 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General procedures; chemical synthesis; experimental details; steady-state absorption/fluorescence experiments; two-photon excitation fluorescence (TPEF) experiments of rhodamine compounds using rhodamine 6G as reference; fluorescence lifetimes of rhodamine compounds; ultrafast transient absorption measurements; electronic structure calculations; excited state analysis; electronic coupling analysis; NMR- & mass spectra of new compounds. See DOI: 10.1039/d1cp04528h |
| ‡ These authors contributed equally. |
| This journal is © the Owner Societies 2022 |