
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen-bonded architectures of multivalent calix[4]arenes†
Maria Chiara
Gullo
,
Luciano
Marchiò
 ,
Alessandro
Casnati
and
Laura
Baldini
*
,
Alessandro
Casnati
and
Laura
Baldini
*
Department of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: laura.baldini@unipr.it
First published on 6th May 2022
Abstract
A small family of novel halogen-bonded crystalline supramolecular architectures of calixarenes was obtained by the co-crystallization of cone (1) and 1,3-alternate tetrakis(3-iodopropargyloxy)calix[4]arene (4) as tetradentate halogen donors with different multidentate acceptors. Particularly interesting is the interpenetrated diamondoid network of 4 with DABCO, which represents the first example of a 2D network of calixarene macrocycles where halogen bonding is the key interaction for self-organization.
Introduction
Crystalline supramolecular infinite networks of calixarenes are attractive structures in the field of functional materials. Thanks to the combination of an aromatic cavity available for the complexation of small guests with the ease of functionalization of the phenol moieties, the calixarene macrocycle provides a versatile scaffold for the synthesis of hierarchically porous materials.1 The insertion of appropriate ligating groups on the calixarene rims may result in macrocycle-containing tectons able to self-assemble in structures characterized by two levels of porosity, one associated with the macrocycle and one with the network.2 The non-covalent bond most frequently exploited for the synthesis of this kind of architecture is the metal–ligand interaction, and a number of calixarene-based coordination polymers have been reported in recent decades,3 with promising adsorption,2,4,5 catalytic6–8 and nonlinear optical properties.9On the other hand, for the formation of purely organic calixarene architectures, hydrogen bonding has been the interaction of choice for several years.10 Thanks to its strength and directionality, several hydrogen bonded 1D,11–13 2D14,15 or 3D13,16 infinite networks of calixarenes have been reported. These are based either on the self-assembly of calixarenes functionalized with self-complementary hydrogen bonding groups, such as amides17 or carboxylic acids,13 or take advantage of an external multidentate hydrogen bonding donor or acceptor moiety to bridge complementarily functionalized macrocycles.12,15
In recent years, also halogen bonds (XBs) have been frequently exploited in crystal engineering,18 thanks to some favorable properties,19 such as their high directionality, strength tunability and resistance to solvent polarity.20 Despite these advantages, however, examples of supramolecular networks of calixarenes assembled through XBs are extremely scarce. A few reports describe the crystal structures of upper rim iodo- and bromocalix[4]arenes21 and thiacalix[4]arenes,22–24 where the halogen atoms exchange XBs with the aromatic rings (π acceptors) or halogen and S atoms of neighboring calixarenes. Only one example takes advantage of an external bifunctional XB donor (a 1,4-diiodotetrafluorobenzene) to co-crystallize a calixarene functionalized at the lower rim with two pyridine units, working as a bifunctional acceptor.25 In this case, due to the presence of additional π–π interactions between the external faces of two phenol rings of the macrocycle and the aromatic ring of the XB donor, a 2D network of calixarenes is formed.
With the aim of obtaining robust and possibly porous supramolecular networks of calixarene macrocycles assembled through XBs, we have recently reported the synthesis of tetrakis(3-iodopropargyloxy)calix[4]arene (1, Fig. 1) in the cone geometry as a tetravalent XB donor.26 The iodoalkyne group is well-known to give strong XB interactions27,28 thanks to the increased electronegative character of the sp hybridized C atom linked to the iodine.29 As expected, also when linked to the calixarene, this moiety confirmed its excellent XB donor ability. When crystallized in the absence of external XB acceptors, 1 self-assembles in a compact structure where all the iodine atoms are engaged in XBs with the alkyne C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds of adjacent molecules (Fig. 2, top). On the contrary, in presence of 4,4′-bipyridine (BIPY), a divalent XB acceptor, the four iodine atoms form XBs with the BIPY nitrogen atoms giving rise to a 1D network of calixarenes alternated to couples of BIPYs (Fig. 2, bottom). To the best of our knowledge, this is the first example of a crystalline network of calixarene macrocycles where XB is the key interaction for self-assembly.30
C triple bonds of adjacent molecules (Fig. 2, top). On the contrary, in presence of 4,4′-bipyridine (BIPY), a divalent XB acceptor, the four iodine atoms form XBs with the BIPY nitrogen atoms giving rise to a 1D network of calixarenes alternated to couples of BIPYs (Fig. 2, bottom). To the best of our knowledge, this is the first example of a crystalline network of calixarene macrocycles where XB is the key interaction for self-assembly.30
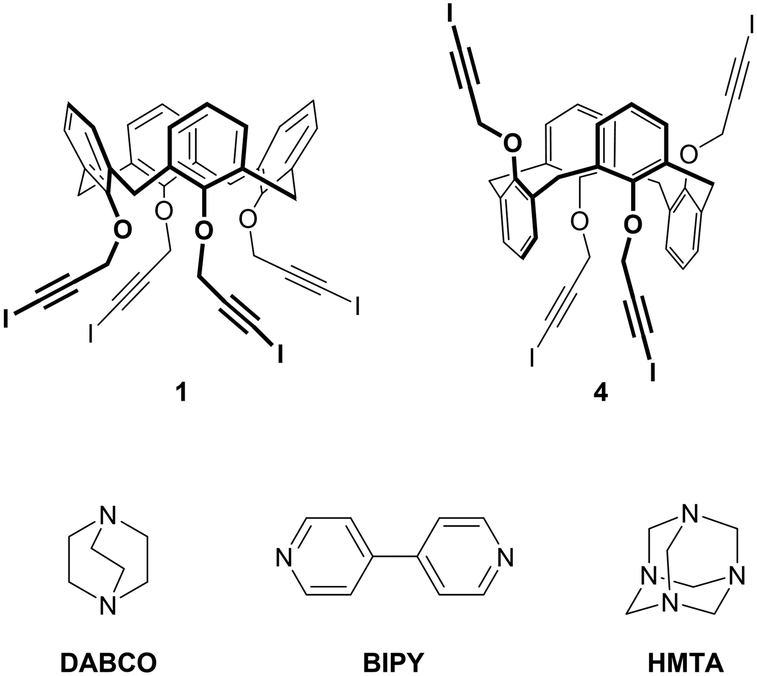 | ||
| Fig. 1 Chemical structure of tetravalent XB donors 1 and 4 and divalent XB acceptors BIPY and DABCO and tetravalent XB acceptor HMTA. | ||
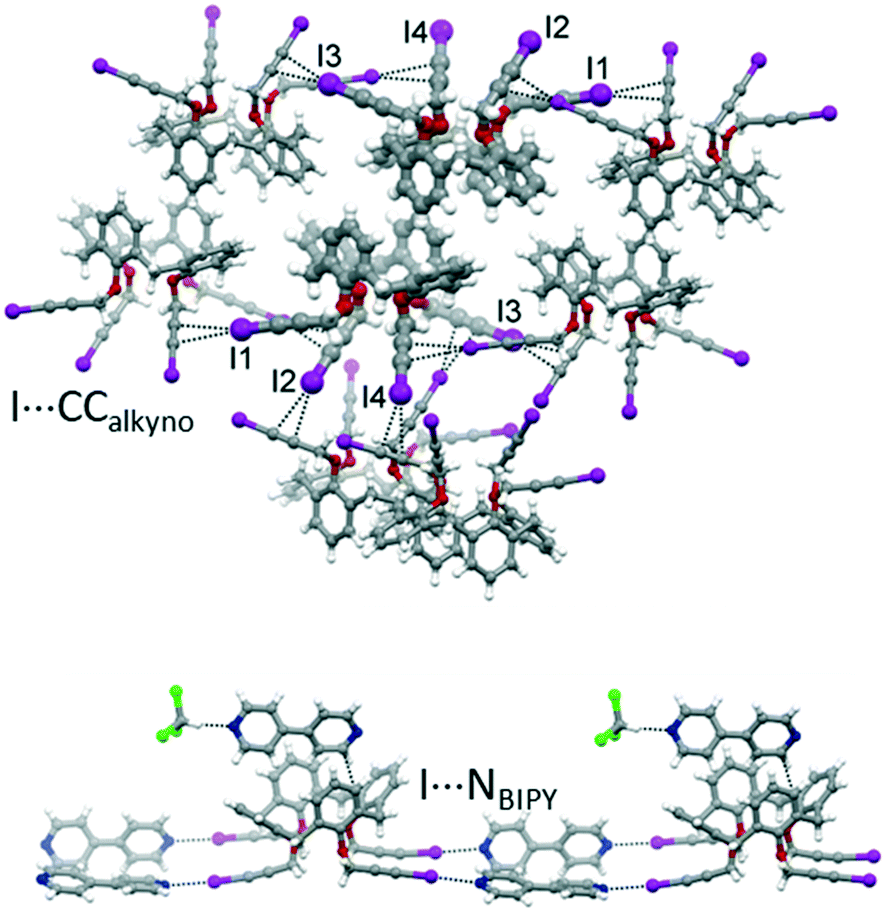 | ||
| Fig. 2 Portions of the crystal packing of 1 (top) and 1-BIPY (bottom) highlighting intermolecular interactions.26 | ||
Even if pleased with the obtained results, we were not entirely satisfied, because in this structure the potentialities of the tetravalent calixarene to form 2D or 3D networks were not fully exploited. The four iodoalkyne struts at the lower rim are not, in fact, pointing in four different directions, but the vicinal iodoalkyne groups are held parallel to each other by attractive CH–π interactions exchanged by the two BIPYs that are edge-to-face oriented. The overall result, on the calixarene side, is a linear synthon with two couples of XB donor groups pointing in opposite directions.
With the aim of obtaining 2D or 3D calixarene networks, we synthesized calixarene 4 (Fig. 1), which is the 1,3-alternate isomer of 1. We anticipated that having the four iodoalkyne groups projected in two opposite directions (two above and two below the plane defined by the methylene bridges of the macrocycle) this compound would be more prone to give multidimensional self-assembled frameworks. As XB acceptors we selected the difunctional (linear) and tetrafunctional (tetrahedral) amines 1,4-diazabicyclo[2.2.2]octane (DABCO) and hexamethylenetetramine (HMTA), respectively. Compared to BIPY, these compounds present the advantage of a N(sp3) nucleophilic site (a more effective XB acceptor than the N(sp2) of BIPY) and, lacking aromatic groups, should avoid undesired π–π or CH–π interactions. DABCO is a stronger Lewis base than HMTA31 and more frequently used as an XB acceptor;32–34 HMTA, on the other side, is a very versatile synthon, being able to function as a mono-, bi-, tri- or tetradentate acceptor.35
We report herein the crystal structures of solventless 4, of a THF solvate of 4 and of the co-crystals of 4 with DABCO and of 1 with HMTA. In all the structures, the supramolecular architecture is directed by XB interactions between all the iodine atoms of the calixarene and the best XB acceptors available. Importantly, the 4-DABCO co-crystal represents the first 2D halogen-bonded network of a calix[4]arene with an external XB donor.
Experimental
Materials and methods
Dry solvents were prepared according to standard procedures, distilled before use and stored over 3 or 4 Å molecular sieves. Analytical TLC was performed using prepared plates of silica gel (Merck 60 F-254 on aluminum). Merck silica gel 60 (70–230 mesh) was used for flash chromatography. 1H and 13C NMR spectra were recorded on Bruker AV300 or AV400 spectrometer. All chemical shifts are reported in part per million (ppm) using the residual peak of the deuterated solvent, whose values are referred to tetramethylsilane (TMS, δTMS = 0), as internal standard. All 13C NMR spectra were performed with proton decoupling. Mass spectra were recorded in ESI mode on a single quadrupole instrument SQ Detector, Waters (capillary voltage 3.7 kV, cone voltage 30–160 eV, extractor voltage 3 eV, source block temperature 80 °C, desolvation temperature 150 °C, cone and desolvation gas (N2) flow rates 1.6 and 8 L min−1, respectively). Melting points were determined with a Gallenkamp apparatus.All reagents were commercially purchased and used without purification. Cone tetrakis(3-iodopropargyloxy)calix[4]arene 1,26 1,3-di(propargyloxy)calix[4]arene 236 and N-iodomorpholine·HI37 were synthesized according to literature.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, v/v) had revealed the complete conversion of calixarene 2, the reaction was quenched with 1 M HCl (15 mL). The resulting white precipitate was collected by filtration and recrystallized from CH2Cl2/methanol obtaining a mixture of 1,3-alternate derivative 3 and its partial cone isomer, from which 3 was isolated by column chromatography (CH2Cl2/hexane 75:25, v/v) in 32% yield (214 mg, 0.37 mmol). M.p. 183–184 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.16 (d, J = 7.5 Hz, 8H, ArH); 6.84 (t, J = 7.5 Hz, 4H, ArH); 4.13 (d, J = 2.4 Hz, 8H, OCH2CCH); 3.80 (s, 8H, ArCH2Ar); 2.47 (t, J = 2.4 Hz, 4H, OCH2CCH). 13C NMR (100 MHz, CDCl3) δ (ppm): 155.3 (OCAr); 134.2 (CAr); 130.4 (CAr); 123.1 (CAr); 80.4 (OCH2CCH); 74.6 (OCH2CCH); 58.9 (OCH2CCH); 37.0 (ArCH2Ar). ESI-MS: m/z calcd for C40H32O4Na [(3 + Na)+] 599.2, found 599.4; calcd for C40H32O4K [(3 + K)+] 615.2, found 615.4.
:4, v/v) had revealed the complete conversion of calixarene 3, 5 mL of a saturated solution of NH4Cl and 10 mL of CH2Cl2 were added. The target compound precipitated as a whitish solid, which was isolated by filtration in 38% yield (155 mg, 0.14 mmol). M.p. 210 °C (dec.). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.04 (d, J = 7.5 Hz, 8H, ArH); 6.76 (t, J = 7.5 Hz, 4H, ArH); 4.28 (s, 8H, ArOCH2CCI); 3.76 (s, 8H, ArCH2Ar). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 156.1 (OCAr); 134.7 (CAr); 130.3 (CAr); 123.2 (CAr); 90.9 (OCH2CCI); 60.7 (OCH2CCI); 37.0 (ArCH2Ar); 13.8 (OCH2CCI).
:3, v/v) had revealed the complete conversion of calixarene 2, the reaction was quenched with 1 M HCl (15 mL). The resulting white precipitate was collected by filtration and recrystallized from CH2Cl2/methanol obtaining a mixture of 1,3-alternate derivative 3 and its partial cone isomer, from which 3 was isolated by column chromatography (CH2Cl2/hexane 75:25, v/v) in 32% yield (214 mg, 0.37 mmol). M.p. 183–184 °C. 1H NMR (300 MHz, CDCl3) δ (ppm): 7.16 (d, J = 7.5 Hz, 8H, ArH); 6.84 (t, J = 7.5 Hz, 4H, ArH); 4.13 (d, J = 2.4 Hz, 8H, OCH2CCH); 3.80 (s, 8H, ArCH2Ar); 2.47 (t, J = 2.4 Hz, 4H, OCH2CCH). 13C NMR (100 MHz, CDCl3) δ (ppm): 155.3 (OCAr); 134.2 (CAr); 130.4 (CAr); 123.1 (CAr); 80.4 (OCH2CCH); 74.6 (OCH2CCH); 58.9 (OCH2CCH); 37.0 (ArCH2Ar). ESI-MS: m/z calcd for C40H32O4Na [(3 + Na)+] 599.2, found 599.4; calcd for C40H32O4K [(3 + K)+] 615.2, found 615.4.
:4, v/v) had revealed the complete conversion of calixarene 3, 5 mL of a saturated solution of NH4Cl and 10 mL of CH2Cl2 were added. The target compound precipitated as a whitish solid, which was isolated by filtration in 38% yield (155 mg, 0.14 mmol). M.p. 210 °C (dec.). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.04 (d, J = 7.5 Hz, 8H, ArH); 6.76 (t, J = 7.5 Hz, 4H, ArH); 4.28 (s, 8H, ArOCH2CCI); 3.76 (s, 8H, ArCH2Ar). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 156.1 (OCAr); 134.7 (CAr); 130.3 (CAr); 123.2 (CAr); 90.9 (OCH2CCI); 60.7 (OCH2CCI); 37.0 (ArCH2Ar); 13.8 (OCH2CCI).
Single-crystal structure determination
Single crystal data were collected with a Bruker D8 Photon II diffractometer, with Mo (λ = 0.71073 Å) or Cu (λ = 1.54178 Å) X-ray source. The intensity data were integrated from several series of exposure frames covering the sphere of reciprocal space.38 Absorption correction was applied using the program SADABS39 for 4·4THF, 4·4DABCO·THF, and 1·2HMTA·3/2CH2Cl2. Crystals of 4 were non-merohedral twins and the absorption correction was applied with the TWINABS program.40 The structures were solved with the ShelXT41 structure solution program using intrinsic phasing and refined with the ShelXL42 refinement package using least squares minimisation. Graphical material was prepared with the Mercury program.43 A summary of data collection and structure refinement for 4, 4·4THF, 4·4DABCO·THF, and 1·2HMTA·3/2CH2Cl2 is reported in Table 1. CCDC 2143352–2143355 contains the ESI† crystallographic data for this paper.| 4 | 4·4DABCO·THF | 4·4THF | 1·2HMTA·3/2CH2Cl2 | |
|---|---|---|---|---|
| Empirical formula | C40H28I4O4 | C56H60I4N4O5 | C56H60I4O8 | C54H56Cl4I4N8O4 |
| Formula weight | 1080.22 | 1376.68 | 1368.64 | 1530.46 |
| Temperature/K | 200 | 200 | 200 | 100 |
| Crystal system | Monoclinic | Orthorhombic | Triclinic | Triclinic |
| Space group | C2/c | Fddd |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 12.8824(8) | 12.9348(2) | 13.2357(16) | 13.1998(18) |
| b/Å | 19.0447(11) | 22.8426(4) | 15.3241(18) | 14.997(2) |
| c/Å | 15.1918(11) | 39.6053(6) | 15.3955(19) | 30.958(4) |
| α/° | 90 | 90 | 112.406(2) | 100.754(4) |
| β/° | 98.548(5) | 90 | 101.238(2) | 90.152(4) |
| γ/° | 90 | 90 | 99.482(2) | 105.402(4) |
| Volume/Å3 | 3685.8(4) | 11702.0(3) | 2731.4(6) | 5795.8(14) |
| Z | 4 | 8 | 2 | 4 |
| ρ calc g cm−3 | 1.947 | 1.563 | 1.664 | 1.754 |
| μ/mm−1 | 26.874 | 2.177 | 2.333 | 2.386 |
| F(000) | 2048.0 | 5408.0 | 1344.0 | 2992.0 |
| Crystal size/mm3 | 0.09 × 0.06 × 0.05 | 0.19 × 0.15 × 0.15 | 0.28 × 0.23 × 0.16 | 0.16 × 0.14 × 0.08 |
| Radiation (Å) | CuKα (λ = 1.54178) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2Θ range (°) | 8.35 to 133.16 | 4.756 to 51.454 | 2.98 to 53.632 | 4.024 to 51.542 |
| Reflections collected | 2999 | 17013 |
34973 |
137466 |
| N Indep (Rint) | 2999 (0.1373) | 2779 (0.0281) | 11670 (0.0328) |
22020 (0.0762) |
| Data/restraints/parameters | 2999/67/206 | 2779/0/157 | 11670/110/705 |
22020/19/1343 |
| Goodness-of-fit on F2 | 1.072 | 1.067 | 1.048 | 1.077 |
| R 1 [I ≥ 2σ(I)] | 0.0990 | 0.0246 | 0.0402 | 0.0743 |
| wR2 [I ≥ 2σ(I)] | 0.2093 | 0.0543 | 0.0922 | 0.1778 |
| Δρmin/max (e Å−3) | 1.11/−0.74 | 0.51/−0.47 | 1.76/−1.55 | 3.58/−1.88 |
Results and discussion
Synthesis
1,3-Alternate tetrakis(3-iodopropargyloxy)calix[4]arene 4 was synthesized in two steps from 1,3-di(propargyloxy)calix[4]arene 236 (Scheme 1). First, compound 2 was alkylated with propargylbromide in presence of Cs2CO3 as a base obtaining a mixture of the 1,3-alternate and partial cone isomers of tetrakis(propargyloxy)calix[4]arene from which the desired compound 3 was isolated in 32% yield by column chromatography. This strategy was preferred to the direct alkylation of parent calix[4]arene44 with excess propargylbromide due to an easier purification of the product. Subsequently, the iodoalkyne derivative 4 was obtained by reaction of 3 with N-iodomorpholine hydrogen iodide in 38% yield.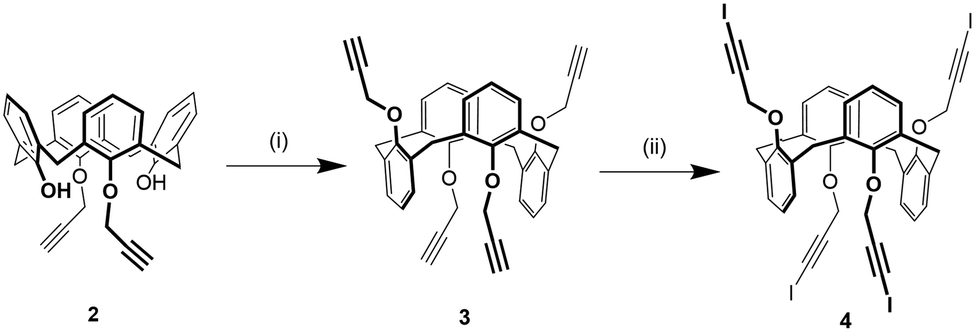 | ||
| Scheme 1 Synthesis of 1,3-alternate tetrakis(3-iodopropargyloxy)calix[4]arene 4: (i) propargyl bromide, Cs2CO3, dry acetone, reflux, 12 h, 32%; (ii) N-iodomorpholine hydrogen iodide, CuI, dry THF, 12 h, 38%. | ||
Solid state self-assembly
1,3-Alternate iodopropargyl calixarene 4 resulted insoluble in all the common organic solvents except THF, dioxane, pyridine and DMSO. The reason for this insolubility, which was not observed for the cone isomer 1, was attributed to multiple intermolecular XB interactions that can possibly form between the eight potential XB acceptors (the electron-rich π clouds of the CC triple bonds and of the aromatic rings) and the four excellent XB donor groups (the iodoalkyne moieties). Moreover, the iodine atoms are positioned at the end of linear struts that are connected to the oxygen atoms of the calixarene through a rotationally unhindered methylene group and are therefore able to explore an ample area to install XB interactions. Only solvents that are good XB acceptors are therefore able to break the intermolecular XBs between the 4 molecules and dissolve the compound.
Two batches of single crystals of calixarene 4 have been obtained, one from a DMSO solution that was left standing at room temperature for a couple of months and the other by slow evaporation of a THF solution. X-ray diffraction analysis of a crystal from each batch revealed two different structures, the first consisting of the sole calixarene 4, the other of a THF solvate.
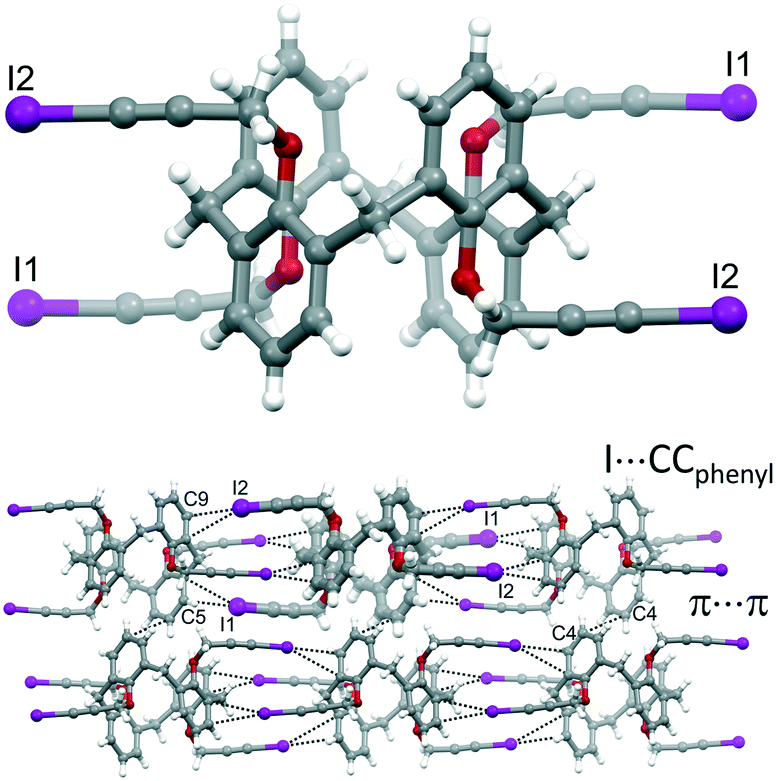 | ||
| Fig. 3 Molecular structure of 4 (top), together with portion of the crystal packing highlighting intermolecular interactions (bottom). | ||
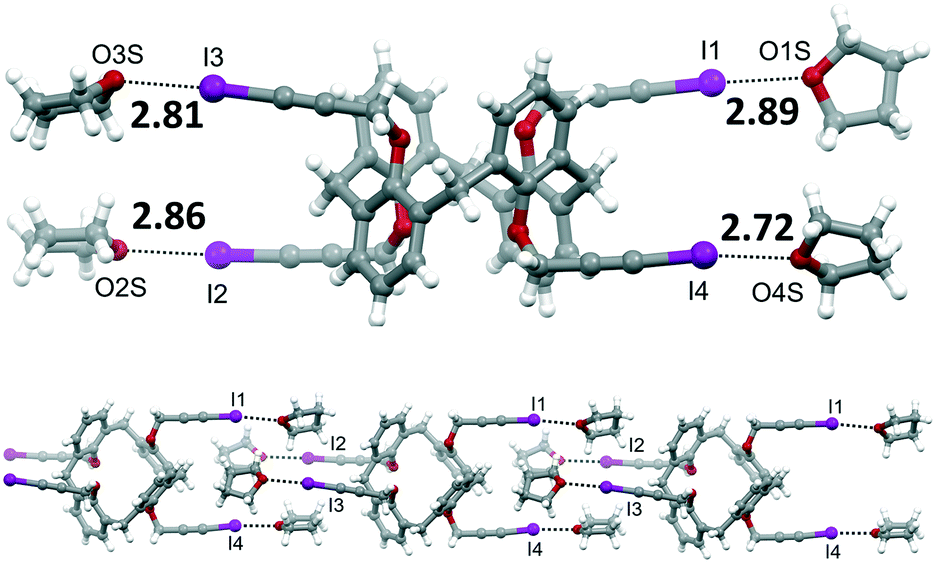 | ||
| Fig. 4 Molecular structure of 4·4THF, with XB distances reported in Å (top). Portion of the supramolecular chain formed by 4·4THF (bottom). | ||
We then explored the ability of iodopropargylcalixarenes 1 and 4 to form self-assembled networks in presence of suitable multidentate XB acceptors by attempting to grow co-crystals of both calixarenes with DABCO and HMTA and of 4 with BIPY (see Table 2). While the high solubility of 1 in most organic solvents allowed us to try several crystallization solvent mixtures, for 4 we were limited to the use of THF, a very competitive XB acceptor. Co-crystals suitable for X-ray diffraction studies were only obtained for 1 with HMTA and for 4 with DABCO. When trying to grow crystals from a mixture of 1 and DABCO in CH2Cl2/hexane, we only obtained crystals of 1 having a structure identical to the one previously reported (Fig. 2, top),26 and no crystalline material was collected from a mixture of 4 and HMTA in THF. The crystals grown from the sample containing 4 and BIPY, instead, consisted of the 4·4THF solvate, with no inclusion of BIPY in the structure. This can be viewed as a confirmation of the lower XB acceptor ability of the sp2 N atoms of bipyridine with respect to the sp3 nitrogens of DABCO. Only the latter, in fact, are able to successfully compete with the THF solvent for halogen bonding with the iodine atoms.
| Entry | XB donor | External XB acceptor | Solvent | Crystal composition | XBs formed | XB lengtha | R | C–I⋯A |
|---|---|---|---|---|---|---|---|---|
| a Ranges are indicated when multiple interactions are present. b R is the normalized interaction distance, defined, according to Lommerse et al.,47 as R = d(I⋯A)/(rI + rA) where A represents the XB acceptor (C, N or O). c Reported in ref. 26. d Calculated considering the shortest I⋯C distance for each XB. | ||||||||
| 1c | 1 | — | CH2Cl2 | 1 | I⋯π cloud of CC |
3.40–3.60d | 0.92–0.98 | 164.1–171.4 |
| 2c | 1 | BIPY | CHCl3 | 1·3BIPY·CHCl3 | I⋯N of BIPY | 2.82–2.85 | 0.80–0.81 | 171.3–177.2 |
| 3 | 1 | DABCO | CH2Cl2 | 1 | I⋯π cloud of CC (same as 1) |
|||
| 4 | 1 | HMTA | CH2Cl2/hexane | 1·2HMTA·3/2CH2Cl2 | I⋯N of HMTA | 2.78–2.92 | 0.79–0.83 | 170.7–179.2 |
| 5 | 4 | — | DMSO | 4 | I⋯π cloud of Ar | 3.27–3.34d | 0.89–0.91 | 165.5–168.9 |
| 6 | 4 | — | THF | 4·4THF | I⋯O of THF | 2.72–2.89 | 0.78 | 170.7–176.6 |
| 7 | 4 | BIPY | THF | 4·4THF | I⋯O of THF (same as 6) | |||
| 8 | 4 | DABCO | THF | 4·4DABCO·THF | I⋯N of DABCO | 2.74 | 0.78 | 178.8 |
| 9 | 4 | HMTA | THF | No crystals | — | |||
:1, mol mol−1) in CH2Cl2/hexane (1:1, v/v). The asymmetric unit comprises two calixarene moieties, four HMTA and three disordered CH2Cl2 molecules of crystallization, 1·2HMTA·3/2CH2Cl2. All of the iodine atoms of 1 are involved in XBs with the nitrogen atoms of HMTA. On the opposite, HMTA employs only two out of four nitrogen atoms to interact with the calixarenes. Interestingly, adjacent calixarenes engage XBs using two iodopropargyloxy moieties on the same couple of HMTA. Consequently, and according to the arrangement of the nitrogen atoms in HTMA at the vertices of an ideal tetrahedron, supramolecular zig-zag chains can be identified, when viewing the calixarenes from the top (Fig. 5). The iodine⋯nitrogen distances are in the 2.78–2.92 Å range, which is significantly shorter than the van der Waals radii sum (approximately 3.53 Å). Furthermore, it is instructive to analyze the geometric parameters pertaining to the XBs within a single chain. Indeed, the two HMTA molecules interact differently with the 3-iodopropargyloxy systems. On one side, the XBs are shorter (∼2.78 Å), the angle I⋯HMTAcentroid⋯I is significantly smaller than 109°, and the C–I⋯N angle deviates from the linearity (∼171°). The other almost parallel motif is characterized by a longer XB distance (∼2.92 Å), by a greater I⋯HMTAcentroid⋯I angle (∼118°) and by a more linear XB geometry, with C–I⋯N of ∼178°. The crystal packing is reported in Fig. S3,† showing a relatively compact structure, in which the dichloromethane molecules of crystallization occupy 13% of the unit cell volume.
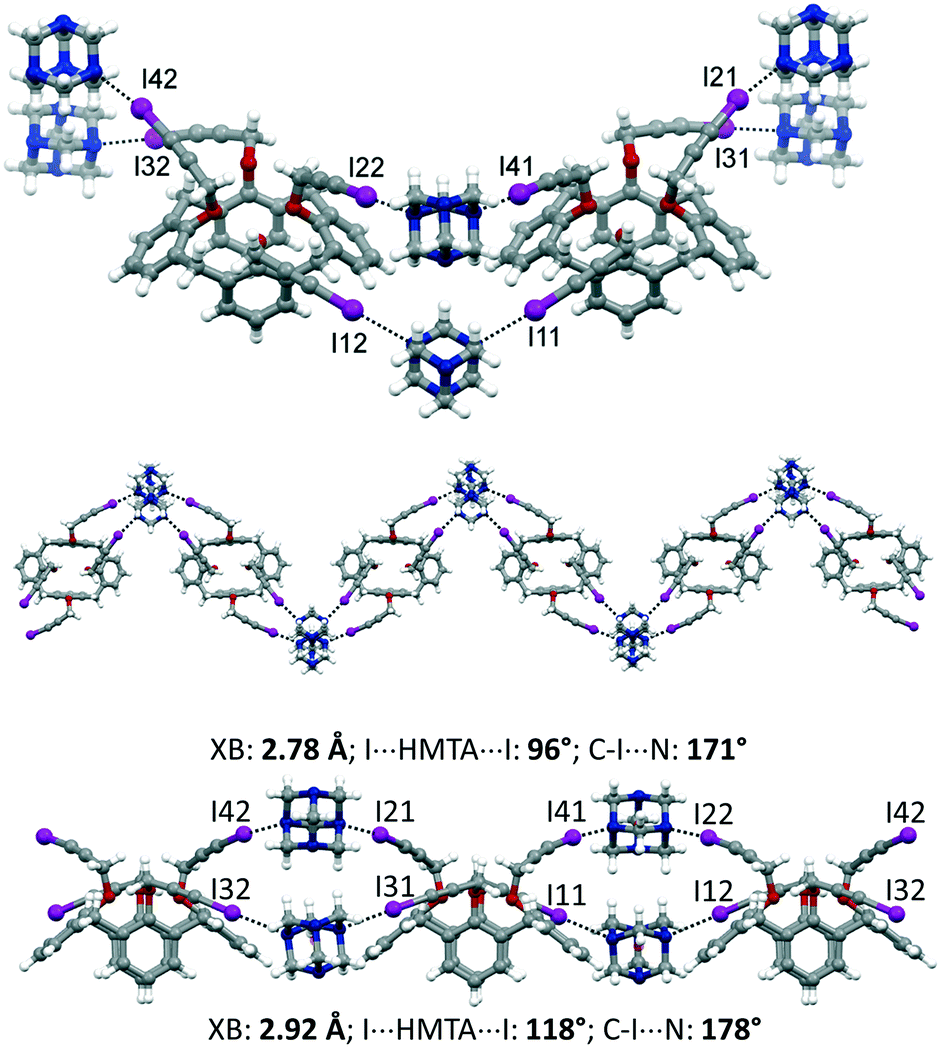 | ||
| Fig. 5 Molecular structure of 1·2HMTA·3/2CH2Cl2 (top). Zig-zag supramolecular chain (middle and bottom), with relevant geometric parameters. | ||
:2, mol mol−1) in THF. The asymmetric unit is represented by 1/4 of the calixarene, a DABCO molecule and 1/4 of disordered THF, with an overall molecular stoichiometry of 4·4DABCO·THF (Fig. 6).
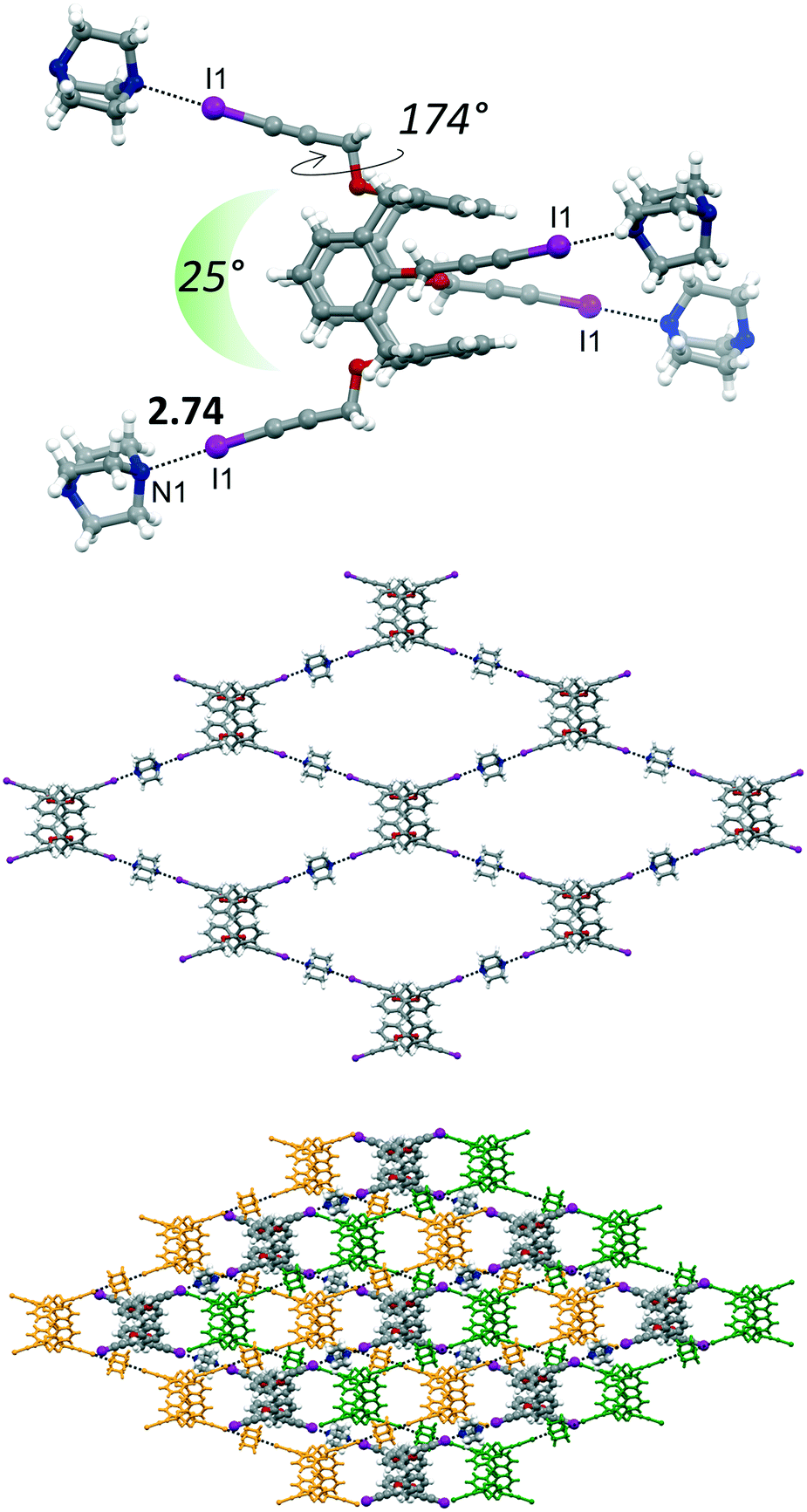 | ||
| Fig. 6 Molecular structure of 4·4DABCO·THF, with XB distance reported in Å (top), single diamondoid network (middle), interpenetrated diamondoid networks (bottom). Solvent of crystallization was omitted for clarity. | ||
The main structural difference, which is then reflected in the supramolecular arrangement, between 4 and 4·4THF with 4·4DABCO·THF is in the peripheral orientation of the iodopropargyl groups with respect to the corresponding phenyl rings. In 4·4DABCO·THF the four iodopropargyloxy struts point in four divergent directions in a conformation suitable to expand the interactions above and below the plane defined by the C atoms of the four methylene bridges. The torsion angle associated to this arrangement is close to 180° in 4·4DABCO·THF, whereas it varies in the 65–87° range for the other compounds. Moreover, in 4·4DABCO·THF the angle formed by two alternate linear C–I vectors is approximately 25° (Fig. 6, top). This overall calixarene arrangement, together with the divergent orientation of the N lone pairs of DABCO, results in the formation of a 2D diamondoid network. The N1⋯I1 distance is 2.74 Å, pointing to the presence of a strong XB. The crystal packing shows then the presence of a structural layer formed by n-interpenetrated networks, which are parallel to the ab crystallographic plane (Fig. S4†). Embedded between the calixarene frameworks is located the THF of crystallization. Interestingly, even after applying moderate vacuum (P ∼ 1 mbar) the THF could still be identified by the difference Fourier map, suggesting a relative capability of the framework in trapping the solvent molecule. The volume occupied by THF is approximately 16% of the unit cell volume.
Conclusions
Together with the previously reported structures of cone tetraiodopropargyloxy calix[4]arene 1 and of the co-crystal 1-BIPY, these results confirm the excellent XB ability of the iodoalkyne strut.29 Systematically, in all the structures, all the iodine atoms are engaged in XB interactions with the same type of acceptor, even when more than one nucleophilic sites are present in the crystallization environment. However, as seen from Table 2, the outcome of the crystallization experiments, when two or three different XB acceptors are available, was in some cases surprising. DMSO, which was reported to be an effective XB acceptor for iodoethynyl moieties,45,46 when used as crystallization solvent, was not halogen-bonded to calixarene 4, which, instead, self-assembles at the solid state through I⋯π XBs. Calixarene 1 forms XBs with BIPY and HMTA, but not with DABCO, which is the most Lewis basic of the three (Table 2, entries 2–4). On the contrary, 1,3-alternate 4 produces a halogen-bonded network only with DABCO (Table 2, entries 7–9). These observations confirm that, due to the moderate strength of XB interactions, the resulting architectures are driven by a subtle interplay of other different factors, which include the spatial orientation and the rotational freedom of the XB donors, the magnitude of secondary supramolecular interactions and the competition by a nucleophilic solvent.34 Nonetheless, the reported crystal structures demonstrate the effectiveness of XBs as key interactions for the self-assembly of supramolecular networks of calixarenes. The dimensionality of these architectures, however, proved hard to predict, and despite the multivalency of both the calixarenes and the XB acceptors, in all but one co-crystals, the main self-assembled structures are 1D chains of alternating calixarenes and XB acceptors. Only for 4-DABCO, the combination of the 1,3-alternate conformation of the macrocycle with the valence and geometry of DABCO allowed to obtain the first example of a 2D halogen-bonded calix[4]arene architecture. Yet, we believe our results provide additional insights into the design of crystalline supramolecular networks of calixarene macrocycles, and further studies are currently underway in our laboratories towards the synthesis of robust 3D supramolecular networks where the macrocycle aromatic cavity could be exploited for host-guest properties. In particular, we plan to extend this approach also to the calix[6]- and calix[8]arene scaffolds, whose higher valency and larger size could facilitate the synthesis of halogen-bonded architectures of higher dimensionality and improved complexation ability.Author contributions
M. C. Gullo: methodology and investigation; L. Marchiò: methodology, visualization, writing – original draft; A. Casnati: supervision, writing – review & editing, funding acquisition; L. Baldini: conceptualization, supervision, writing – original draft.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the Italian Ministry of University and Research projects (COMP- HUB initiative, Departments of Excellence Program and PRIN 2017E44A9P). The “Centro Interfacoltà di Misure” (CIM) of the University of Parma is acknowledged for use of their NMR and mass spectrometers. Chiesi Farmaceutici SpA is acknowledged for the support of the D8 Venture X-ray equipment.Notes and references
- H. Zhang, R. Zou and Y. Zhao, Coord. Chem. Rev., 2015, 292, 74–90 CrossRef CAS.
- S. P. Bew, A. D. Burrows, T. Düren, M. F. Mahon, P. Z. Moghadam, V. M. Sebestyen and S. Thurston, Chem. Commun., 2012, 48, 4824–4826 RSC.
- A. Ovsyannikov, S. Solovieva, I. Antipin and S. Ferlay, Coord. Chem. Rev., 2017, 352, 151–186 CrossRef CAS.
- Z. Zhang, A. Drapailo, Y. Matvieiev, L. Wojtas and M. J. Zaworotko, Chem. Commun., 2013, 49, 8353–8355 RSC.
- E. Lee, Y. Kim, J. Heo and K. M. Park, Cryst. Growth Des., 2015, 15, 3556–3560 CrossRef CAS.
- L. L. Liu, J. Chen, C. X. Yu, W. X. Lv, H. Y. Yu, X. Q. Cui and L. Liu, Dalton Trans., 2017, 46, 178–185 RSC.
- C.-X. Yu, F.-L. Hu, M.-Y. Liu, C.-W. Zhang, Y.-H. Lv, S.-K. Mao and L.-L. Liu, Cryst. Growth Des., 2017, 17, 5441–5448 CrossRef CAS.
- L.-J. Yue, Y.-Y. Liu, G.-H. Xu and J.-F. Ma, New J. Chem., 2019, 43, 15871–15878 RSC.
- Y. J. Liu, J. S. Huang, S. S. Y. Chui, C. H. Li, J. L. Zuo, N. Zhu and C. M. Che, Inorg. Chem., 2008, 47, 11514–11518 CrossRef CAS PubMed.
- M. J. Hardie, Hydrogen Bonded Network Structures Constructed from Molecular Hosts, Springer, Berlin, Heidelberg, 2004, pp. 139–174 Search PubMed.
- W. Wang, S. Gong, Y. Chen and J. Ma, New J. Chem., 2005, 29, 1390–1392 RSC.
- W. Jaunky, M. W. Hosseini, J. M. Planeix, A. De Cian, N. Kyritsakas and J. Fischer, Chem. Commun., 1999, 2313–2314 RSC.
- Z. Xiao, W. Yang, F. Yan, L. Ji, W. Li and W. Wang, CrystEngComm, 2019, 21, 439–448 RSC.
- L. Baldini, F. Sansone, C. Massera, A. Casnati, F. Ugozzoli and R. Ungaro, Inorg. Chim. Acta, 2007, 360, 970–980 CrossRef CAS.
- Y. Liu and M. D. Ward, Cryst. Growth Des., 2009, 9, 3859–3861 CrossRef CAS.
- Y. Li, W. Yang, Y. Chen and S. Gong, CrystEngComm, 2011, 13, 259–268 RSC.
- L. Baldini, F. Sansone, A. Casnati, F. Ugozzoli and R. Ungaro, J. Supramol. Chem., 2002, 2, 219–226 CrossRef CAS.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- C. C. Robertson, R. N. Perutz, L. Brammer and C. A. Hunter, Chem. Sci., 2014, 5, 4179–4183 RSC.
- S. R. Kennedy, M. U. Main, C. R. Pulham, I. Ling and S. J. Dalgarno, CrystEngComm, 2019, 21, 786–790 RSC.
- M. Yamada, Y. Ootashiro, Y. Kondo and F. Hamada, Tetrahedron Lett., 2013, 54, 1510–1514 CrossRef CAS.
- M. Yamada, R. Kanazawa and F. Hamada, CrystEngComm, 2014, 16, 2605–2614 RSC.
- M. Yamada and F. Hamada, Cryst. Growth Des., 2015, 15, 1889–1897 CrossRef CAS.
- M. T. Messina, P. Metrangolo, S. Pappalardo, M. F. Parisi, T. Pilati and G. Resnati, Chem. – Eur. J., 2000, 6, 3495–3500 CrossRef CAS.
- M. C. Gullo, L. Baldini, A. Casnati and L. Marchiò, Cryst. Growth Des., 2020, 20, 3611–3616 CrossRef CAS.
- L. Turunen, N. K. Beyeh, F. Pan, A. Valkonen and K. Rissanen, Chem. Commun., 2014, 50, 15920–15923 RSC.
- C. B. Aakeröy, T. K. Wijethunga, J. Desper and M. Đaković, Cryst. Growth Des., 2015, 15, 3853–3861 CrossRef.
- C. B. Aakeröy, M. Baldrighi, J. Desper, P. Metrangolo and G. Resnati, Chem. – Eur. J., 2013, 19, 16240–16247 CrossRef PubMed.
- On the contrary, few examples of halogen bonded networks of resorcinarene cavitands have been reported, see for example: C. B. Aakeröy, A. Rajbanshi, P. Metrangolo, G. Resnati, M. F. Parisi, J. Desper and T. Pilati, CrystEngComm, 2012, 14, 6366–6368 RSC and ref. 27 and 34.
- K. Raatikainen and K. Rissanen, CrystEngComm, 2011, 13, 6972–6977 RSC.
- C. Perkins, S. Libri, H. Adams and L. Brammer, CrystEngComm, 2012, 14, 3033–3038 RSC.
- L. Catalano, S. Pérez-Estrada, G. Terraneo, T. Pilati, G. Resnati, P. Metrangolo and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2015, 137, 15386–15389 CrossRef CAS PubMed.
- L. Turunen, F. Pan, N. K. Beyeh, J. F. Trant, R. H. A. Ras and K. Rissanen, Cryst. Growth Des., 2018, 18, 513–520 CrossRef CAS.
- G. Anyfanti, A. Bauzá, L. Gentiluomo, J. Rodrigues, G. Portalone, A. Frontera, K. Rissanen and R. Puttreddy, Front. Chem., 2021, 165 Search PubMed.
- W. Xu, J. J. Vittal and R. J. Puddephatt, Can. J. Chem., 2011, 74, 766–774 CrossRef.
- J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018–8021 CrossRef CAS PubMed.
- Bruker, SMART, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- G. M. Sheldrick, TWINABS_2012/1, University of Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- W. Verboom, S. Datta, Z. Asfari, D. N. Reinhoudt and S. Harkema, J. Org. Chem., 1992, 57, 5394–5398 CrossRef CAS.
- J. Lieffrig, O. Jeannin and M. Fourmigué, J. Am. Chem. Soc., 2013, 135, 6200–6210 CrossRef CAS PubMed.
- L. Turunen, F. Pan, N. K. Beyeh, M. Cetina, J. F. Trant, R. H. A. Ras and K. Rissanen, CrystEngComm, 2017, 19, 5223–5229 RSC.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, J. Am. Chem. Soc., 1996, 118, 3108–3116 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2143352–2143355. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ce00193d |
| This journal is © The Royal Society of Chemistry 2022 |