
Engineering mesoporous silica nanoparticles towards oral delivery of vancomycin†
John
Ndayishimiye
 a,
Yuxue
Cao
a,
Tushar
Kumeria
b,
Mark A. T.
Blaskovich
c,
James Robert
Falconer
a and
Amirali
Popat
*ad
a,
Yuxue
Cao
a,
Tushar
Kumeria
b,
Mark A. T.
Blaskovich
c,
James Robert
Falconer
a and
Amirali
Popat
*ad
aSchool of Pharmacy, Pharmacy Australia Centre of Excellence, The University of Queensland, Brisbane, Queensland 4102, Australia. E-mail: a.popat@uq.edu.au
bSchool of Materials Science and Engineering, University of New South Wales, New South Wales, Australia
cCentre for Superbug Solutions, Institute for Molecular Bioscience, The University of Queensland, St. Lucia, Queensland 4072, Australia
dMater Research Institute – The University of Queensland, Translational Research Institute, 37 Kent St, Woolloongabba, QLD 4102, Australia
First published on 9th August 2021
Abstract
Vancomycin (Van) is a key antibiotic of choice for the treatment of systemic methicillin resistant Staphylococcus aureus (MRSA) infections. However, due to its poor membrane permeability, it is administered parenterally, adding to the cost and effort of treatment. The poor oral bioavailability of Van is mainly due to its physico-chemical properties that limit its paracellular and transcellular transport across gastrointestinal (GI) epithelium. Herein we report the development of silica nanoparticles (SNPs)-based formulations that are able to enhance the epithelial permeability of Van. We synthesized SNPs of different pore sizes (2 nm and 9 nm) and modified their surface charge and polarity by attaching different functional groups (–NH2, –PO3, and –CH3). Van was loaded within these SNPs at a loading capacity in the range of ca. 18–29 wt%. The Van-loaded SNPs exhibited a controlled release behaviour when compared to un-encapsulated Van which showed rapid release due to its hydrophilic nature. Among Van-loaded SNPs, SNPs with large pores showed a prolonged release compared to SNPs with small pores while SNPs functionalised with -CH3 groups exhibited a slowest release among the functionalised SNPs. Importantly, Van-loaded SNPs, especially the large pore SNPs with negative charge, enhanced the permeability of Van across an epithelial cell monolayer (Caco-2 cell model) by up to 6-fold, with Papp values up to 1.716 × 10−5 cm s−1 (vs. 0.304 × 10−5 cm s−1 for un-encapsulated Van) after 3 h. The enhancement was dependent on both the type of SNPs and their surface functionalisation. The permeation enhancing effect of SNPs was due to its ability to transiently open the tight junctions measured by decrease in transepithelial resistance (TEER) which was reversible after 3 h. All in all, our data highlights the potential of SNPs (especially SNPs with large pores) for oral delivery of Van or other antimicrobial peptides.
1. Introduction
Infectious diseases are among the leading causes of death globally and it is estimated that, based on current trends, infectious diseases would be the leading cause of death by 2050 – more than cancer or cardiovascular diseases.1 Notably, Staphylococcus aureus causes a wide range of life threatening infections, ranging from skin and soft tissue infections to infective endocarditis and bloodstream infections.2 Due to its tendency to develop resistance to conventional antibiotics such as penicillins, cephalosporins and other β-lactam antibiotics, S. aureus has become increasingly challenging from a public health perspective.3,4 These methicillin resistant S. aureus (MRSA) infections can become real medical emergencies and their treatment presents a major challenge to the public and medical community.5Although there are several options for treating invasive MRSA infections, vancomycin‡ (Van) has continued to be the primary course of treatment for the last fifty years.6–8 Van (Fig. S1, ESI†) is a tricyclic glycopeptide that acts by inhibiting the bacterial cell-wall synthesis by binding to the D–Ala–D–Ala terminal group of the growing peptidoglycan peptide chain.9 Although Van has been the drug of choice for MRSA, it must be administered via the intravenous route for S. aureus systemic infections,10 due to its physico-chemical properties and the complexity of GI epithelium.§10,11 (Note that it is given orally for C. difficile infections, but this is essentially topical delivery as the infection is in the gut12,13). Van is a large molecule of 1485.7 Da,14 which is substantially greater than 500 Da maximum suggested by Lipinski for oral drug delivery.15,16 In addition Van is hydrophilic17 and this makes it difficult to partition and permeate through the enterocytes and reach lamina propria for systemic circulation either via paracellular or transcellular routes.11 Moreover, its hydrogen bond donor and hydrogen bond acceptor counts are 20 and 26, respectively,14 and this again greatly violates the Lipinski's guidelines for oral drug delivery (no more than 5 and 10, respectively).15,16
From the drug delivery point of view, an oral Van therapy could provide many benefits compared to Van administration via injection. While it can be dependent on case by case basis (inappropriate for critically ill patients or patients who are unable to absorb oral medications), switching from IV to oral administration would enable the reduction of length of stay in a hospital and improve cost effectiveness.18,19 Moreover, a fear of injections is real and prevalent across populations, sometimes even surpassing 80% prevalence in patients,20 where approximately 20–30% of patients are further classified as suffering from needle phobia.21 In addition, “red man” syndrome and “pain and spasm” syndrome are other adverse effects associated with Van IV infusion.22,23
Different delivery systems and approaches have been used to improve the permeability via GI epithelium and/or oral delivery of macromolecules.11,24,25 These delivery systems include for instance (1) focusing on the colloidal level, where ileal, colonic, and rectal permeation of peptides are enhanced using emulsions with different excipients and compositions,11 (2) using polymer-based carrier systems, where polymeric nanoparticles have been used to overcome the oral physiological barriers,26 (3) using solid lipid nanoparticles to specifically improve the stability of drugs of peptide nature27 and (4) incorporating mucus-penetrating agents28 and/or permeation enhancers29 into the carrier systems in order to enhance the penetration of drugs into intestinal lumen mucus layer and permeate through GI epithelium. Another emerging approach that has been used to improve permeability and oral delivery of biologics is the use of mesoporous silica nanoparticles30–32 where the drug can be entrapped and/or adsorb in the pores of silica nanoparticles (SNPs) either by physisorption (due to weak van der Waals forces) or by electrostatic interactions.33
Regarding Van and SNPs, previous works have mainly focused on for example: (1) Van encapsulation and release and its antimicrobial activity where Van is either physically adsorbed or covalently bonded to SNPs,34,35 (2) localised and sustained release of Van for treatment of infected bone defects, where a composite scaffold based on Van-loaded mesoporous SNPs was fabricated and studied,36 and (3) covalently bonding van on mesoporous SNPs for selective recognition and killing of pathogenic Gram-positive bacteria over macrophage-like cells.37 However, there is no study on the applicability of functionalised SNPs with tunable chemistry for enhancing Van release and permeability with the purpose of improving its oral bioavailability. Therefore, to the best of our knowledge, this is the first work focusing specifically on the possibility of engineering and characterising SNPs with the purpose of controlling its release and improving the oral bioavailability.
SNPs possess many attractive characteristics, such as high surface area, abundant and tuneable porosity, tuneable particle size and morphology, tuneable surface charge and chemistry and good chemical and mechanical stability.32,38,39 Here we thought that we can explore the possibility of tuning parameters such as pore size, and surface chemistry in order to improve the permeability and other characteristics of loaded Van. Starting by the pore size, it has been documented that the pore size of SNPs plays an important role in loading and release of drugs40 although the literature is conflicting. For instance, some literatures say that SNPs with larger pores normally give higher drug loading and faster drug release in comparison to SNPs with small pores, which is often ascribed to a steric hindrance effect.40–44 Other literatures say otherwise.45,46 So, our first hypothesis was whether the change of pore size can have an impact on loading, release behaviour and permeability of Van.
Besides pore size, surface charge is another interesting parameter that should be taken into consideration. Here due to the presence of silanol groups on SNPs surfaces, it is possible to modify the SNPs surfaces with groups of various charges, which would consequently play a role in the release and permeability characteristics of a drug.40,47–49 The impact of SNPs surface charge on the delivery of therapeutics has been studied.32,40,47,49–52 For instance, studying the impact of different functional groups, although their study was not on Van, Lamson et al.30 used SNPs with different sizes and charges for oral delivery of insulin. They discovered that the more negatively charged nanoparticles with smaller size enhanced the permeability of insulin in vitro and in vivo. Our preliminary experiments showed that un-functionalised SNPs are anionic but does the increase of negative charge, by functionalizing SNPs with more anionic phosphonate groups, increase the permeability of Van? What if we functionalize them with cationic groups? Do cationic characteristics of SNPs surfaces have an impact on the permeability of Van? Surface polarity of SNPs is another parameter that we considered while engineering the surface chemistry of the SNPs. SNPs surface's polarity plays a critical role especially when the drug is being delivered transcellularly.40,49,52 It is possible to modify the SNPs surfaces with the introduction of groups with different polarity. In the literature for example, neutral and hydrophilic nanoparticles have shown the ability to permeate through the intestinal mucus layer,53 whereas hydrophobic nanoparticles have been successful in epithelial transport.54 Here, we hypothesized that the functionalisation of SNPs with hydrophobic groups would affect the release behaviour, permeability, and other characteristics of SNPs-loaded Van.
Therefore, based on the above hypotheses and research questions, we synthesized and characterized SNPs with different pore sizes which were surface modified by different functional groups for the purpose of loading Van to improve its controlled release, permeability, and other characteristics. To achieve that, we designed and executed the work as follows: We initiated our work by synthesizing and characterizing SNPs with different pore sizes. Here the SNPs were characterized for their particle size and morphology, surface area, charge, and porosity. We then functionalized the SNPs with different functional groups to achieve different cationic/anionic properties and polarity of SNPs surfaces. Then SNPs (functionalised or un-functionalised) were loaded with Van and characterised. Finally, we performed the cytotoxicity assays for SNPs and carried out the permeability studies for SNPs-loaded Van using widely accepted Caco-2 cell monolayer assay.
2. Experimental
2.1. Chemicals
Vancomycin hydrochloride (#861987), hexadecyltrimethylammonium chloride (CTAC), triethanolamine (TEA), tetraethylorthosilicate (TEOS), 3-(trihydroxysilyl) propyl methyl-phosphonate (THMP), (3-aminopropyl) triethoxysilane (APTES), Dulbecco's modified Eagle medium (DMEM, D6546), Dulbecco's phosphate-buffered saline (PBS), fetal bovine serum (FBS, F9423) and MEM nonessential amino acid solution were purchased from Sigma-Aldrich (Australia). Dimethyl sulfoxide (DMSO), ammonium dihydrogen phosphate, and octadecyl trimethoxysilane (ODMS) were obtained from ThermoFisher Scientific (NJ, USA). Hank's balanced salt solution (HBSS), pen-strep (penicillin 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mL−1 and streptomycin 10000 μg mL−1), L-glutamine, sodium pyruvate (100 mM), trypsin-EDTA, and (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Gibco (Thermo Fisher Scientific, Australia).
000 U mL−1 and streptomycin 10000 μg mL−1), L-glutamine, sodium pyruvate (100 mM), trypsin-EDTA, and (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Gibco (Thermo Fisher Scientific, Australia).
2.2. Synthesis of SNPs with different pore sizes
The synthesis of SNPs is described in Scheme 1A. The synthesis procedure for small pore-sized SNPs was slightly modified from the method reported by Chaudhary et al.55 2 g of CTAC and 0.04 g of TEA were dissolved in 20 mL of Milli-Q water and stirred (400 rpm) for 1 h at 95 °C. After 1 h, TEOS (1.5 mL) was added (dropwise) and the suspension mixture was, under the same conditions, stirred for another hour. The mixture was then cooled, and centrifuged (24650×g, 15 min) to collect the particles as pellets. The particles were washed with ethanol (twice) and oven-dried at 60 °C overnight. Finally, they were crushed and calcined at 550 °C for 5 h. The resulting particles were designated as “SNPs SP”. The synthesis procedure for large pore-sized SNPs was adapted from the method reported by Juère et al.56 CTAC (8 mL) and TEA (360 mg) were dissolved in Milli-Q water (72 mL) and stirred (375 rpm) for 1 h at 60 °C. After 1 h, the pre-mixture of cyclohexane (32 mL) and TEOS (8 mL) was added, and the mixture was aged overnight under similar conditions. The resulting product was centrifuged (24650×g, 10 min) and the pellets were collected, washed twice with ethanol, oven-dried and then calcined. These particles were designated as “SNPs LP”.
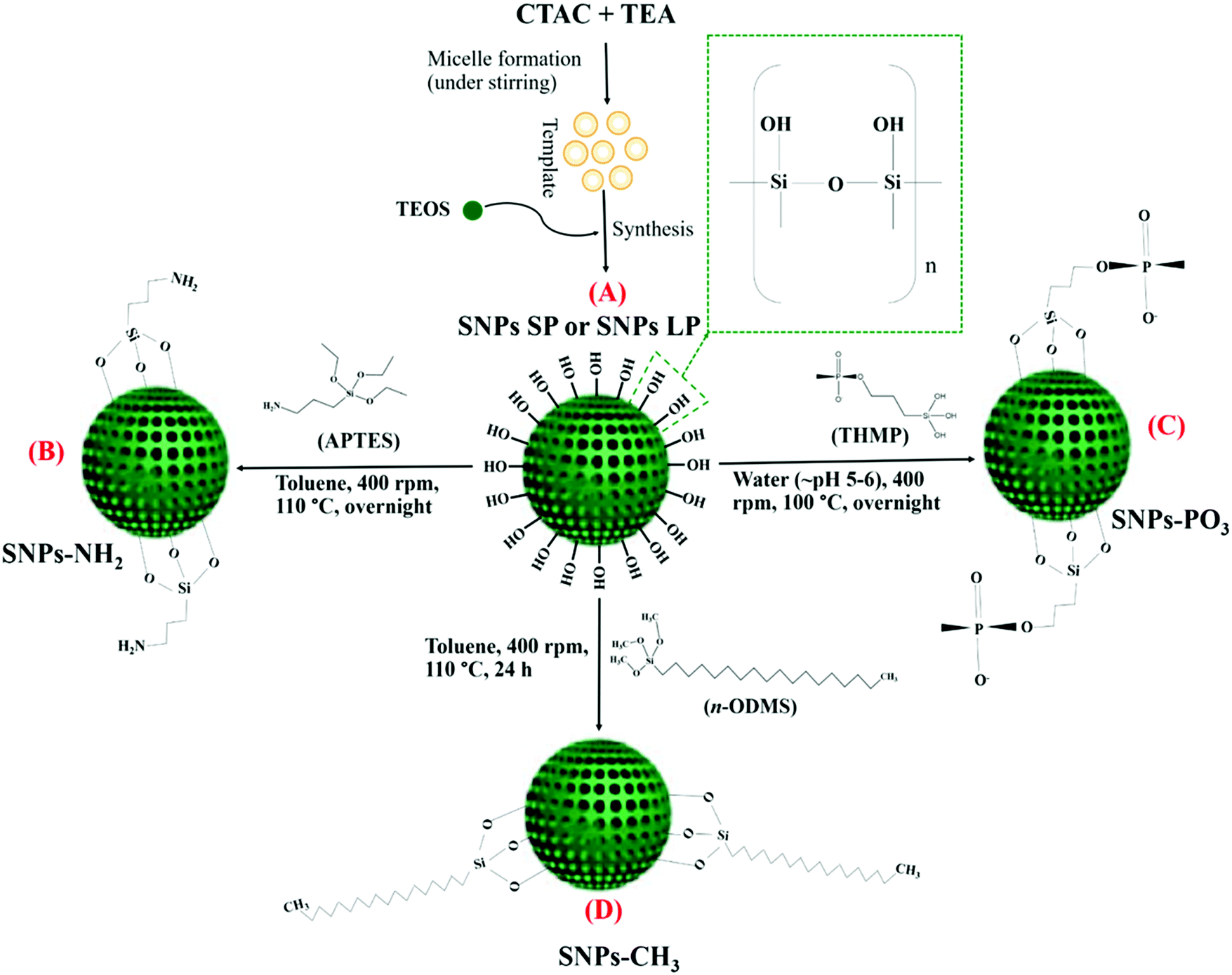 | ||
| Scheme 1 Schematic illustration of (A) SNPs synthesis, (B) functionalisation with aminopropyl groups, (C) functionalisation with propyl methyl-phosphonate groups, and (D) functionalisation with octadecyl methyl groups. | ||
2.3. Functionalisation of SNPs
SNPs functionalisation with aminopropyl groups (Scheme 1B) was done according to a previously reported method55 with minor modifications. Prior to functionalisation, calcined SNPs (300 mg) were vacuum-dried (overnight) to remove off any possible excess moisture in the SNPs pores. The vacuum-dried SNPs (300 mg) were then added into toluene (45 mL), followed by bath sonication (5 min). To this dispersion, under stirring (400 rpm, at 110 °C), 0.21 mL of APTES was added and the mixture was aged overnight under the same conditions. Functionalized SNPs (SNPs SP-NH2 or SNPs LP-NH2) were then centrifuged (10950 × g for 10 min), washed with acetone two times and with ethanol once, then oven-dried overnight at 60 °C.For functionalisation with propyl methyl-phosphonate groups55 (Scheme 1C), THMP (0.13 mL) was dissolved in 13 mL of Milli-Q water, and the pH of the solution, which was highly basic (pH 12.0), was adjusted to slight acidic pH (pH 5–6) using concentrated hydrochloric acid solution (1 M). This lowering of pH is done to prevent hydroxylation and dissolution of silica during functionalisation. The above-prepared solution was added to SNPs suspension (100 mg of SNPs in 13 mL of Milli-Q water), then the mixture was stirred (400 rpm, 100 °C) overnight. The functionalized SNPs were collected by centrifugation (10950×g for 10 min), washed thrice with water and with ethanol twice, then oven-dried at 60 °C. The resulting functionalised particles were denoted “SNPs SP-PO3 or SNPs LP-PO3”.
For hydrophobic (octadecyl methyl groups) functionalization (Scheme 1D), octadecyl methyl groups of ODMS were functionalised onto the SNPs surfaces. In this experiment, 200 mg of calcined SNPs were suspended in 30 mL of toluene containing 2% (v/v) of n-ODMS. After stirring (400 rpm) the mixture at 110 °C for 24 h, the particles were centrifuged (10950×g for 10 min), washed three times with toluene and two times with ethanol, and then oven-dried at 60 °C. The particles after functionalisation with octadecyl methyl groups were designated “SNPs SP-CH3 or SNPs LP-CH3”.43,57
2.4. Loading of Van in SNPs
Van was loaded in SNPs using a freeze-drying assisted method. In this method, Van loading in SNPs was done according to a previously reported procedure35,36 with some modifications. 4 mL of aqueous-ethanolic solution containing 70 mg SNPs (SNPs SP or SNPs LP, with/without functional groups) was prepared as a suspension. 2 mL of Van aqueous solution (15 mg mL−1) was added to each SNPs suspension to make a mixture of 70:30% w/w (SNPs:Van, respectively). The suspension mixture was then stirred (300 rpm, at 4 °C) overnight. Subsequently, the suspension mixture was frozen and then freeze-dried for 48 h using an Alpha Freeze Dryer (ALPHA 1–2 LD plus, Germany) and Van-loaded SNPs were obtained. The loading capacity of Van in SNPs were determined using a thermo-gravimetric analysis (detailed in Section 2.5.6).
2.5. Characterisation of SNPs
:30-line shapes, and the Kratos Relative Sensitivity Factor (RSFs) library.
cm−1 and the data were graphed as relative transmittance (%) vs. wave numbers (cm−1). TGA of the samples was done using Mettler Toledo DSC 2 STAR System and the sample weight was 5–7mg. The purge gas was nitrogen and heating was done under compressed air at the heating rate of 10°C min−1, over 25°C to 900°C. For every analysis, an empty crucible was used as a reference.
2.6. In vitro release studies
The method used for the release studies of Van-loaded SNPs was adapted from the method reported by Lopes et al.59 with minor modifications, using SnakeSkin® Dialysis Tubing (Thermo Scientific, IL, USA). Van-loaded SNPs (equivalent to 3 mg Van, based on TGA data) were dispersed in 3 mL of donor medium and the resulting suspension was loaded into the dialysis tubing (molecular weight cut off 10 kDa, tubing i.d of 22 mm) and knotted. The knotted dialysis tubing was placed in the reservoir containing the dialysate (PBS buffer pH 6.8, mimicking small intestinal pH) and the release was monitored for 10 h. The systems were maintained at 37 °C ± 1 under moderate stirring (200 rpm) throughout the whole experiment. At predetermined time intervals, 0.5 mL of the release medium was taken for Van analysis, and it was replaced by the same volume of fresh medium pre-conditioned at 37 °C. The released Van amount was quantified by HPLC. The HPLC system and conditions used are described below.2.7. High performance liquid chromatography method
In this study the HPLC method used for Van quantification was adapted from the methods reported earlier60,61 with modifications. An HPLC system (Agilent Technologies) equipped with a diode array detector was used. The mobile phase consisted of 88% (v/v) of 50 mM ammonium dihydrogen phosphate (adjusted to a pH of 4 using phosphoric acid) and 12% (v/v) of acetonitrile at a flow-rate of 0.15 mL min−1. Separation was carried out isocratically (at room temperature) on a GRACE Alltima HP C18 3 μm column (150 × 2.1 mm) at 220 nm. For Van analysis, 10 μL of an aliquot withdrawn was injected, and the run time was 12 min. Van eluted at about 7 min. Van quantification was done from the standard calibration curve of Van with a linearity range of 0.5 to 250 μg mL−1. The calibration curve was linear (Fig. S2, ESI†). Both Pearson coefficient (r) and correlation coefficient (r2) were 0.9999.2.8. In vitro cytotoxicity assays
Prior to permeability experiments, SNPs were investigated for their potential cytotoxicity to Caco-2 cell lines (Caco-2, ATCC HTB-37) using an MTT viability assay. Caco-2 cells (passage number: 40) were cultured in DMEM high glucose medium supplemented with 10% v/v of FBS and 1% v/v each of pen-strep, sodium pyruvate, MEM nonessential amino acids and L-glutamine and incubated at 37 °C in a humidity-controlled environment with 5% CO2 in a cell culture incubator. After 90% confluence, the cells were trypsinized, harvested and then seeded into 96-well plates (cell density: 105 cells per well), then incubated for 24 h under the same incubation conditions and subsequently treated with 100 μL (per well) of suspensions of SNPs (concentrations ranging from 25–500 μg SNPs mL−1 DMEM). This SNPs concentration range was chosen by considering the equivalent concentrations of SNPs used in transport experiments. Cells without treatment (DMEM only or DMEM + 1% DMSO) were used as controls. After a further 24 h incubation, treatments were aspirated, and 100 μL of MTT reagent (0.5 mg mL−1 in PBS) was added and then incubated at 37 °C for additional 4 h. After that, 100 μL DMSO per well was added to dissolve the crystals of formazan and the absorbance signal of formazan was measured at 570 nm using a microplate reader (SPECTROstar Nano, BMG LABTECH).2.9. Permeability experiments
| TEER (Ω cm2) = [TEER (Ω) − TEER background (Ω)] × A (cm2) | (1) |
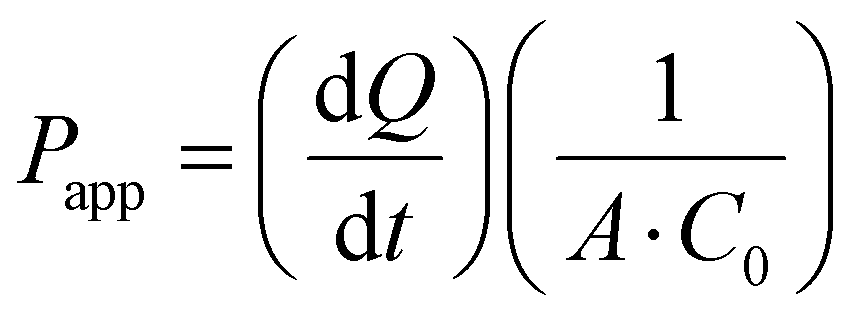 | (2) |
2.10. Data processing and statistical analysis
Descriptive statistics and analysis of variance were carried out with GraphPad Prism 9.0.0 and the results are presented as mean values ± SDs (n = 3, unless otherwise specified). One-way ANOVA (assuming Gaussian distribution of residuals) with Tukey's multiple comparisons test was carried out and the significant difference was considered when the P value was <0.05. Where other specific statistical tests were performed, they are given in the table or figure legends.3. Results and discussion
3.1. Particle size and morphology of SNPs
Particle size and morphology are key attributes when designing nanomaterials for drug delivery systems. Specifically for oral delivery, size and morphology of nanoparticles matter since the particles have to be of particular size and morphology to permeate the intestinal mucus layer and cross the intestinal epithelium.30 Specific size and morphology of SNPs can be accomplished by fine-tuning the amount of CTAC (an organic chloride salt of cetyltrimethylammonium, a cationic surfactant used during synthesis for micelle formation to make a template), TEA (a tertiary amine and a triol, used as a base catalyst to catalyse the hydrolysis and condensation reactions of silicate species),65 and TEOS (an ethyl ester of orthosilicic acid, which is used as a precursor to silica template in SNPs synthesis). TEA also plays an important role as surface capping agent to stabilize the particles against aggregation and thus producing monodispersed system.66,67 Other process conditions, including temperature at which the process is carried out and time of the process, play a major role in the size of the produced SNPs.68 After the preparation and surface functionalisation of SNPs, the SNPs were firstly characterized for their size characteristics by DLS, and the results are shown in Table 1 and Fig. S3 (ESI†). In general, un-functionalised SNPs SP showed slightly lower mean particle size and narrow distribution compared to un-functionalised SNPs LP. As shown in Table 1, the mean particle sizes were 128 ± 41 and 152 ± 46, respectively for SNPs SP and SNPs LP and their respective PDIs were 0.26 ± 0.02 and 0.32 ± 0.06, which underscores the effect of process conditions on the rate of precursor conversion, nucleation, and growth of SNPs (which affect the particle size characteristics). The results in Table 1 also illustrate the impact that the molar ratio variation of precursors had on particle size characteristics of synthesised SNPs.69,70 Specifically, to the amount of precursor (TEOS), the more TEOS is added, the more the particles could grow and vice versa,69,70 which concur with our experiments since the amount of TEOS used for the SNPs SP synthesis was 1.5 mL, with an increase to 8 mL for SNPs LP.| SNPs | Count ratea (kcps) | Mean particle size (nm) ± SD | Polydispersity index (PDI) ± SDd | |
|---|---|---|---|---|
| DLSb | TEMc | |||
|
a Count rates are for DLS measurements.
b Particle size were measured by DLS. The means are average of the means of three measurements. SDs are average of the SDs of three measurements.
c Mean particle size measured by SEM (count number: 100). The numbers are not the average of three measurements. Here the SDs are indicative of size distribution.
d PDIs are from DLS measurements. The numbers are average of PDIs of three measurements.
#Indicates that there was no significant difference in particle size between SNPs SP and SNPs LP. *Functionalised SNPs showed the significant difference in comparison to their un-functionalised SNPs. nsFunctionalised SNPs showed no significant difference in comparison to their un-functionalised SNPs. One-way ANOVA with Tukey's multiple comparisons test was performed. ns: no significance; significance level was *p < 0.033, **p < 0.002, ***p < 0.0002, ****p < 0.0001. |
||||
| SNPs SP | 266 | 128 ± 41# | 100 ± 7 | 0.26 ± 0.02# |
| SNPs SP-NH2 | 270 | 232 ± 81* | 110 ± 24 | 0.37 ± 0.07* |
| SNPs SP-PO3 | 360 | 292 ± 99** | 105 ± 10 | 0.42 ± 0.09** |
| SNPs SP-CH3 | 290 | 779 ± 125*** | 116 ± 54 | 0.89 ± 0.2**** |
| SNP LP | 324 | 152 ± 46# | 130 ± 12 | 0.32 ± 0.06# |
| SNPs LP-NH2 | 330 | 212 ± 87ns | 132 ± 15 | 0.39 ± 0.04ns |
| SNPs LP-PO3 | 380 | 158 ± 60ns | 130 ± 16 | 0.34 ± 0.04ns |
| SNPs LP-CH3 | 286 | 329 ± 51** | 140 ± 18 | 0.42 ± 0.08* |
Particle size characteristics of functionalised SNPs are shown in Table 1. In general, compared to un-functionalised SNPs, functionalised SNPs showed higher hydrodynamic diameters and broader particle size distribution in aqueous medium when measured by DLS. For example, when SNPs SP was functionalised with –NH2 and –PO3, the particle size and PDI values increased significantly to 232 ± 81 and 292 ± 99 and 0.47 ± 0.07 and 0.52 ± 0.09, respectively, which was almost a two-fold increase compared to un-functionalised ones. Similarly, –NH2 and –PO3 functionalised SNPs LP showed an increase in mean particle size and PDI compared to un-functionalised SNPs LP although it was not at the same magnitude as SNPs SP. This suggests that in aqueous medium the modified SNPs might have aggregated to some extent, due to the presence of functional groups which might have decreased the electrical double layer thickness under the condition of high ionic strength, even if their surface charges were higher compared to pure SNPs.57 Moreover, specifically to –NH2 functionalised SNPs, this might have originated from the electrostatic attraction between cationic amino groups and free (un-functionalised) silanol groups remaining at the surfaces of SNPs-NH2.71 Surface functionalisation with octadecyl methyl groups even further increased the mean particle size and PDI values. The mean particle size increased to 779 ± 125 for SNPs SP-CH3 and 329 ± 51 for SNPs LP-CH3 with similar increase in PDI (0.89 ± 0.2 and 0.42 ± 0.08, respectively). This large increase suggests that that severe aggregation among SNPs-CH3 might have occurred due to the hydrophobic nature of octadecyl chains of ODMS which might have hindered the solubilisation and promoted the aggregation even if 10% of ethanol was used in suspension for SNPs-CH3 in DLS. Here the SNPs functionalized with octadecyl methyl groups have a smaller shear or slippage plane, and the octadecyl groups extending from SNPs-CH3 surfaces are outside the shear plane, which leads to strong hydrophobic interactions between neighbouring particles, and this might lead to aggregation.72,73
TEM images were analysed for mean particle size using ImageJ and results are presented in Table 1. The mean particle sizes and the standard deviations for TEM mean particle sizes were lower than DLS. For instance, the TEM mean particle size for SNPs SP and SNPs LP were 100 ± 7 and 130 ± 12 down from 128 ± 41 and 152 ± 46 (DLS). Also, TEM mean particle sizes of functionalised SNPs were lower compared to DLS. This significant difference between TEM and DLS data in terms of mean particle size values might be explained by understanding the working principles of these two techniques. In TEM particles are directly observed under the microscope, while the DLS technique indirectly measures size using the principle of dynamic light scattering based on Brownian motion to detect diffusion coefficient of particles.74 Formation of a hydration layer around the particles (since particles are dispersed in DW in the case of DLS technique) will lead to larger apparent particle sizes (hydrodynamic diameters) compared to TEM.
The particle morphology and pore structures of all SNPs samples were assessed by TEM and SEM analysis. TEM and SEM images are displayed in Fig. 1 and Fig. S4, S5 (ESI†). The TEM images show particles with well-defined spherical shape, although SNPs SP-CH3 were somewhat aggregated (Fig. S4C, ESI†) compared to other functionalised SNPs, which concurs with their comparatively high mean particles size and PDI values. These SNPs were spherical with slightly rough outer surfaces (Fig. S6, ESI†) and they showed a highly ordered network of mesopores. Moreover, from TEM and SEM images (Fig. 1), there is clear difference in the pore structures between SNPs SP and SNPs LP. The highly ordered network of pores in SNPs SP are present as an array of small pores that are still readily observed from the images (Fig. 1 and Fig. S6, ESI†). In comparison, the large pores of the SNPs LP exhibit flower-like (or worm-like) structures (Fig. 1 and Fig. S6, ESI†). Furthermore, in the case of SNPs LP, two populations of pores can be differentiated: smaller pores in the centre and larger ones at the rim (Fig. S6, ESI†).66,75 After functionalization of the SNPs, TEM and SEM images showed that the original structure of the particles and pores were still preserved (Fig. S4 and S5, ESI†), although the surface of SNPs-CH3 was slightly smoother, most likely due the longer octadecyl chains on the SNPs surfaces after –CH3 functionalisation.
 | ||
| Fig. 1 Morphology of SNPs imaged by TEM (A) SNPs SP & (B) SNPs LP and SEM (C) SNPs SP & (D) SNPs LP. | ||
3.2. Surface area and porosity of SNPs
The specific surface area, pore volume and size, and pore size distribution of nanoparticles are important factors for drug encapsulation and/or adsorption and other characteristics of loaded drugs.76 They play important role in drug loading capacity, drug dissolution, kinetics of drug release and other characteristics.77 Although EM techniques give us information about the morphology and pore structures of SNPs, they still are qualitative techniques. Therefore, it was important to investigate the surface characteristics and porosity of SNPs with other techniques which are more quantitative. The nitrogen physisorption analysis is one technique which provides more detailed and quantitative analyses for surface area properties and porosity of SNPs. The technique is based on Brunauer–Emmett–Teller (BET) theory (an extension of the Langmuir theory, which is a theory of monolayer molecular adsorption), where it applies to systems of multilayer molecular adsorption and uses probing gases to quantify specific surface area and porosity of particles. Therefore, nitrogen adsorption/desorption experiments were used here to investigate the surface area and porosity nature of SNPs (functionalised and un-functionalised) and the results are shown in Fig, S7, S8 (ESI†) and Table 2.| SNPs | BET surface area [m2 g−1] | Pore volume [cm3 g−1] | Pore size [nm] |
|---|---|---|---|
| SNPs SP | 411.50 | 0.583 | 2 |
| SNPs SP-NH2 | 300.9 | 0.207 | 2 |
| SNPs SP-PO3 | 365.2 | 0.379 | 2 |
| SNPs SP-CH3 | 269.97 | 0.180 | 2 |
| SNPs LP | 623.87 | 1.343 | 9.2 |
| SNPs LP-NH2 | 395.82 | 0.760 | 8.2 |
| SNPs LP-PO3 | 457.18 | 0.938 | 9.2 |
| SNPs LP-CH3 | 486.07 | 0.989 | 8.2 |
The nitrogen adsorption/desorption isotherms for un-functionalised and functionalised SNPs are shown in Fig. S7 (ESI†). Fig. S7A (ESI†) shows the nitrogen adsorption/desorption isotherms for SNPs SP. It shows a type IV isotherm characteristic of pores of silica nanoparticles, indicating a mesoporous structure. The figure shows that the isotherm is composed of different regions depend on the adsorption process. Here at very low pressures, the pores are filled with adsorbate (nitrogen gas). At the knee (around P/P0 = 0.05), monolayer formation begins. The plateau region in the middle shows the formation of a monolayer, followed by multi-layer formation at medium pressures. At higher pressures, capillary condensation occurs. Here the adsorption/desorption patterns exhibited a major capillary condensation at a high relative pressure (P/P0) of around 0.9. Moreover, SNPs SP N2 sorption isotherms showed Type H1 hysteresis loops, which is characteristic of silica materials with narrow range of mesopores, as for example in templated silicas.78 Fig. S7B (ESI†) shows the nitrogen adsorption/desorption isotherms for SNPs LP. It was found that all SNPs LP exhibited type IV isotherms.78 Similar to SNPs SP, SNPs LP isotherms show that the adsorption process occurred via multilayer adsorption followed by capillary condensation.78 As can be seen on the Fig. S7B (ESI†), there is a slight increase in the initial part of the isotherm which shows the stage where the monolayer formation is completed, which is followed by multilayer formation, although the pattern and hysteresis of isotherms are slightly different from SNPs SP. Here, at low relative pressure range, the gas (N2 in this case) adsorbs and condense in the tiny capillary pores of the SNPs at pressures below the saturation pressure of the gas, due to the presence of smaller pores of about 2–4 nm, as can be seen in the profile of pore size distribution of SNPs LP (Fig. S8B and S6, ESI†). However, since SNPs LP has also large pores (Fig. S7B and S6, ESI†), after the initial region, there is a gradual increase of adsorption process observed at relative pressure range (P/P0) of 0.8–0.85, followed by the pore condensation step happening at higher relative pressure range, with a sharp hysteresis loop, characteristic of capillary condensation of the nitrogen occurring inside the mesopores, still with type IV isotherm nature.75 Moreover, SNPs LP N2 sorption isotherms showed Type H5 hysteresis loops, which is a distinctive form associated with certain pore structures containing mesopores with wider openings.78 Functionalising SNPs (both SNPs SP and SNPs LP) did not change the shape and type of the N2 adsorption/desorption isotherms although the adsorption plateau values and the quantity (cm3 g−1) of N2 adsorbed decreased, reflecting the decrease of total surface area of functionalised SNPs in comparison to un-functionalised ones.
Table 2 shows BET surface areas and porosity of functionalised and un-functionalised SNPs. SNPs SP showed BET surface area of 411.50 m2 g−1 while SNPs LP exhibited BET surface area of 623.87 m2 g−1, probably due to the difference in the amount of precursors and surfactants and process conditions used in synthesis of these two SNPs.79 BET surface area values decreased remarkably when SNPs surfaces were modified with different functional groups. For example, the BET surface area of SNPs SP was remarkably decreased to 300.9, 365.2, 269.97 m2 g−1 when SNPs SP was functionalised with –NH2, –PO3, and –CH3, respectively. Similarly, the functionalisation of SNPs LP induced a remarkable reduction of BET surface area to 395.82, 457.18, 486.07 m2 g−1, when functionalised with –NH2, –PO3, and –CH3, probably due to the presence of additional grafted groups on the internal and external pore surfaces of SNPs, which reduced surface areas.66 In addition, it is noteworthy to mention that different functional groups produced different reduction levels of BET surface areas. For instance, with SNPs SP, while –NH2 functionalised SNPs SP showed a 27% reduction in BET surface area compared to un-functionalised SNPs SP, –PO3 functionalised SNPs SP showed only a 11% reduction, while –CH3 functionalised SNPs SP showed a 34% reduction. This might be due to a different number of functional groups and the chemical structure of the anchored groups as octadecyl methyl chains are longer than the short propyl chains of APTES and THMP (Scheme 1Dvs. B and C).
The size distribution of pores is another important characteristic of porous nanomaterials. Pore size distribution curves were determined from the adsorption branch by the BJH method, and the results are shown in Fig. S8 (ESI†) and summarised in Table 2. SNPs SP showed narrow-distributed pores with a high population at 2 nm whereas SNPs LP were widely distributed with majority of pores in the range of 4–18 nm and a peak population at 9 nm. TEM and SEM images corroborate these above results as it can be seen in Fig. 1, where SNPs SP and SNPs LP show distinctive pores size variations roughly matching the BJH results. As shown in Table 2, SNPs LP, irrespective of whether functionalised or not, have relatively higher pore volumes ranging from 1.343 to 0.989 cm3 g−1 compared to SNPs SP (0.583–0.180 cm3 g−1). Functionalisation affected pore volume, decreasing from 0.583 to 0.207, 0.379, and 0.180 cm3 g-1 when SNPs SP were functionalised with –NH2, –PO3, and –CH3, respectively and from 1.343 to 0.760, 0.938, and 0.989 cm3 g−1 when SNPs LP were functionalised with –NH2, –PO3, and –CH3, respectively. The pore size of functionalised SNPs-SP did not change whereas pore size of functionalised SNPs LP were only slightly reduced to 8.2 nm (from 9.2 nm) in the case of –NH2 and –CH3 functionalised SNPs LP. In general though, regarding functionalisation of SNPs, as it can be seen in TEM and SEM images, neither the change of pore size distribution nor the reduction of BET surface areas have greatly affected the morphology of either SNPs SP or SNPs LP.
3.3. Anionic and cationic properties of SNPs
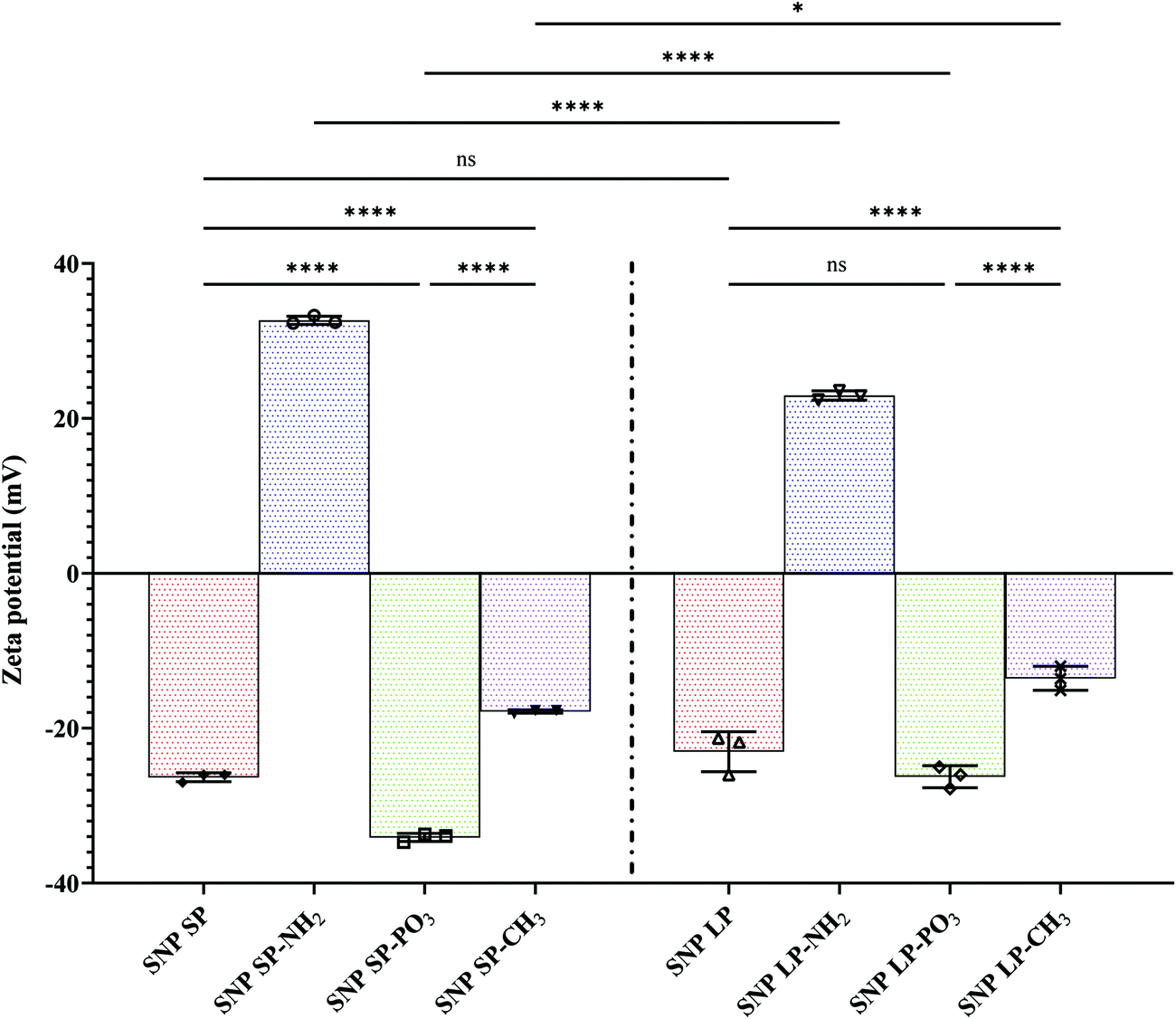 | ||
| Fig. 2 Surface charge of SNPs with/without functional groups. Values are presented as mean ± standard deviation (n = 3). One-way ANOVA with Tukey's multiple comparisons test was performed. ns: no significance; significance level was *p < 0.033, ****p < 0.0001. | ||
The FTIR spectra of functionalised SNPs are presented in Fig. S10 (ESI†) and chemical changes in surface structures consistent with the new chemical groups can be clearly seen. After functionalization with –NH2, the SNPs maintained its structure with no major changes in the Si–O–Si framework. However, the existence of aminopropyl functional groups was confirmed by the appearance of peaks at 2966 cm−1 and 2901 cm−1, which were stretching vibration peaks of methylene groups (–(CH2)–). Furthermore, a new absorption peaks appeared at 1491 cm−1 and 1409 cm−1 which correspond to the N–H stretching vibrations of the primary and secondary amines of APTES.66 For –PO3 functionalised SNPs, the existence of the propyl methyl-phosphonate groups was confirmed by the presence of the symmetric and asymmetric stretching vibration bands at 2966 cm−1 and 2901 cm−1 (methylene), which shows the presence of propyl groups from THMP. Also, weak additional peaks are observable at 1406 and 1392 cm−1 which might correspond to phosphonate P–O groups. The FTIR spectra also confirmed the grafting of octadecyl methyl groups to the SNPs due to the presence of characteristic vibration bands of the octadecyl methyl groups of ODMS. The band at 2897 cm−1 (symmetric C–H vibrations) accompanied by an intensive band at 2922 cm−1 (asymmetric C–H vibration of methyl group) in the spectra of SNPs-CH3 indicate a high concentration of grafted octadecyl methyl groups (alkyl chain of silane molecules on the SNPs surfaces) which also correlate with the data of elemental composition (discussed in detail in Section 3.4) and TGA showing high yield of octadecyl methyl groups grafted on the SNPs surfaces.72 Another important observation in all the functionalised SNPs was that the intensity of the peak 3440 cm−1, the broad peak corresponding to Si–OH groups, was reduced or in some cases disappeared from the spectra of modified SNPs, due to the silanol groups being modified by the functional groups.82 Moreover, we can see that the water physisorption band at 1620 cm−1 is seen in all functionalised SNPs, indicating the hygroscopic nature of SNPs,83 although this peak intensity slightly diminishes for SNPs functionalised by the hydrophobic octadecyl methyl groups.
3.4. Hydrophilicity and hydrophobicity of SNPs
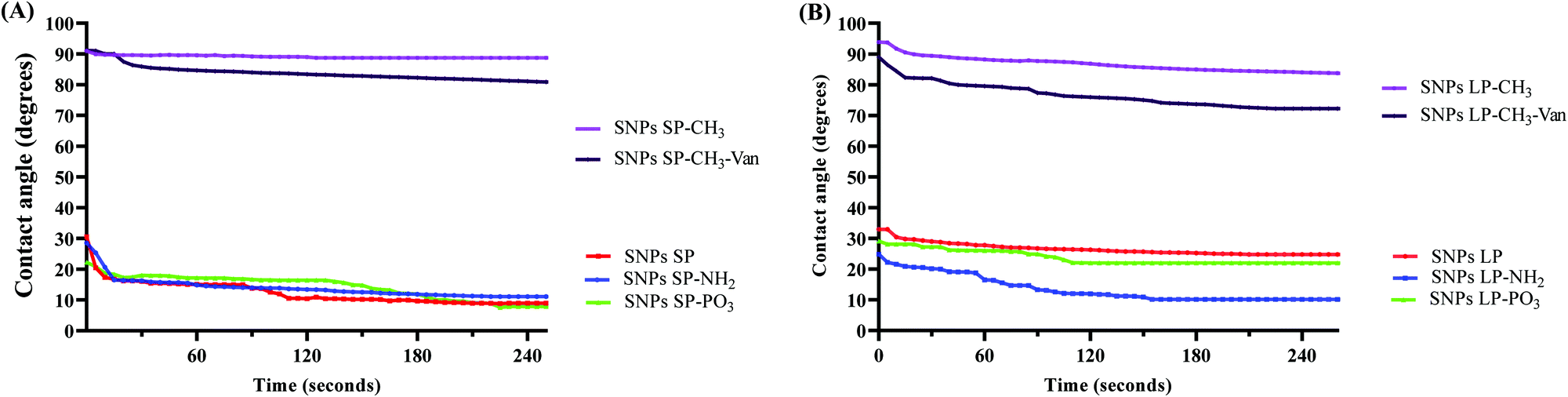 | ||
| Fig. 3 Functionalisation of SNPs with octadecyl methyl groups reduced their hydrophilicity. (A) The change in contact angle for SNPs SP with/without various functional groups at different time intervals, showing increase of hydrophobicity in SNPs SP functionalised with octadecyl methyl groups. (B) The change in contact angle for SNPs LP with/without various functional groups at different time intervals, showing increase of hydrophobicity in SNPs LP functionalised with octadecyl methyl groups. | ||
When the droplet (water droplet in this case) is sufficiently small, surface tensions dominate over gravity and the forces that acts on the droplet set the contact angle. If the droplet volume and surface tension of the system do not change, the contact angle remains constant.85 But, as time goes, the change in the volume of the droplet, shape, and surface tension may occur86 and different SNPs with different surfaces in their nature (in this case SNPs vs. –CH3 modified SNPs) may behave differently over time, and consequently a new equilibrium of forces which act on a droplet might produce a different contact angle. Therefore, the variation of contact angles over time were investigated. For un-functionalised SNPs, initially the contact angle slightly decreased then becomes almost constant for over 4 minutes. For SNPs functionalised with –NH2 and –PO3, there is a more noticeable change of contact angle values over time. The behaviour of SNPs functionalised with –CH3 differed between the two types of particles. While the contact angle of SNPs SP-CH3 was almost unchanged over 4 minutes, there was an initial slight decrease (from 94° to 90°) followed by a very small gradual change of contact angle values for SNPs LP-CH3, although their contact angle values were still high. This might have been due to the difference in the number of octadecyl methyl groups on surfaces of –CH3 functionalised SNPs, or the larger pore size of the SNPs LP.
When the surface modified SNPs are loaded with a drug (hydrophilic or hydrophobic), their surface polarity character (compared to un-loaded ones) in medium might change due to the nature of the drug and the effect caused by the encapsulated or free (un-encapsulated) drug on the surfaces,87 which might in turn have an effect on drug delivery. Therefore, depending on the drug encapsulated or adsorbed in SNPs, the degree of wetting ability of SNPs or the hydrophilic/hydrophobic interaction with the medium can change which would have an implication in the delivery of the drug. Although the in vitro release medium or a physiological medium in intestines is different from DW that we used in contact angle measurements, it was very important to investigate how the contact angles might change when the SNPs-CH3 were loaded with Van. Fig. 3 and Fig. S12, S13 (ESI†) shows the time-lapse contact angles measured for Van-loaded SNPs-CH3. As can be seen, SNPs-CH3 loaded with Van showed a slight reduction of contact angles compared to their corresponding un-loaded SNPs-CH3, possibly due to the influence that Van on the surfaces might have had (note: there was no change of contact angles for hydrophilic SNPs loaded with Van compared to their un-loaded counterparts, data not shown). Van-loaded SNPs LP-CH3 showed a more noticeable change of contact angle over time. In comparison to Van-loaded SNPs SP-CH3, although SNPs SP-CH3 loaded with Van showed a slight reduction of contact angles initially, the contact angle value became roughly constant over 4 minutes. This might indicate that there might have been Van on the SNPs surfaces which initially reduced slightly the contact angles (since Van is a hydrophilic molecule) once the water droplet fell on the surface but as time went the hydrophobic nature of octadecyl methyl groups contributed and overcame the initial slight decrease hence the contact angle values remained almost constant.
The wide scan XPS spectra of un-functionalised SNPs (Fig. 4) revealed three distinctive bands at a binding energy of 531.0, 151.0, and 102.0 eV, which corresponded to O 1s, Si 2s, and Si 2p, respectively, and there was no noticeable difference between SNPs SP and SNPs LP in terms of their elemental compositions on the surfaces (Table S14, ESI†). Even if the high-resolution XPS spectra of O 1s, Si 2s, and Si 2p were not carried out, those bands and binding energies can be associated with bonds of Si–O–Si and Si–OH, showing a silica network with silanol groups on the surface of SNPs. XPS spectra of functionalised SNPs are also presented in Fig. 4. From the figure, SNPs (SP and LP) functionalised with –NH2 showed the bands of O 1s, Si 2s, and Si 2p similar to un-functionalised ones but with additional bands at a binding energy of 282.0 and 397.0 eV, which were attributed to C 1s and N 1s, respectively, showing that aminopropyl groups from APTES were successfully functionalised on the SNPs surfaces. Looking at their elemental composition (%) (Table S14, ESI†), it can be seen that SNPs LP-NH2 has a slightly higher composition of C 1s and N 1s adsorbed on their surfaces compared to SNPs SP-NH2 (19.84 vs. 17.63% for C 1s and 3.69 vs. 2.39% for N 1s), which would be ascribed to higher surface area of SNPs LP (discussed in Section 3.2) where more groups might have been attached in comparison to SNPs SP. Although these surface elemental composition (%) results for SNPs-NH2 concur with TGA data, we do not see any positive correlation with SNPs-NH2 surface charge data and this indicates that other parameters like pore sizes of SNPs-NH2 might have involved.74 For SNPs-PO3 functionalisation, the surveyed XPS spectra exhibited four peaks at 531.0, 282.0, 151.0 and 102.0 eV representing the binding energies of O 1s, C 1s, Si 2s and Si 2p, respectively. The presence of C 1s in the spectrum suggested that propyl methyl-phosphonate groups were successfully functionalised on SNPs (since un-functionalised SNPs did not show C 1s in their XPS spectra and this was not due to contamination). The absence of phosphorus in the spectrum indicates that if P was present, it was at concentrations below the detection limit of the XPS (generally 0.1–1 atom %).90 Moreover, C 1s elemental composition was also slightly lower in SNPs SP-PO3 compared to SNPs LP-PO3 (7.49% vs. 8.74%), which might be due to surface area (as mentioned above). The XPS spectra scans of –CH3 functionalised SNPs also showed the peak of C 1s which is associated with octadecyl methyl groups anchored to the SNPs surfaces. Regarding the % composition of C 1s in SNPs-CH3 (Table S14, ESI†), SNPs SP-CH3 has almost four-fold the amount of C 1s compared to SNPs LP-CH3 (58.33% vs. 15.76%), which can also be correlated with their corresponding TGA and porosity data. This shows that unlike functionalisation with –NH2 and –PO3, the higher surface area of SNPs LP did not help increase the degree of grafting of octadecyl methyl silanes on SNPs. Also, since condensation may occur during synthesis, it is difficult to exclude the possibility that some of the C 1s might also arise from the condensed and/or un-adsorbed octadecyl silanes, although multiple washing steps were done to ensure the complete removal of un-grafted ODMS.
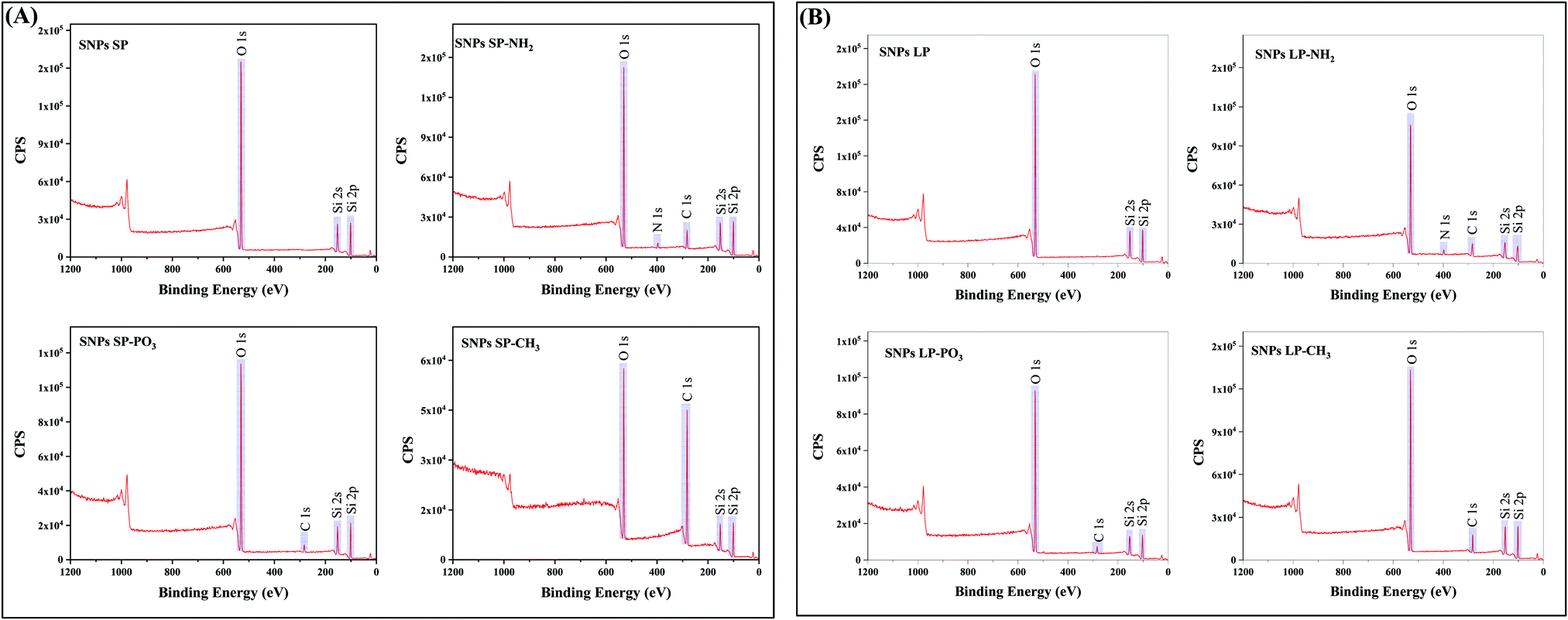 | ||
| Fig. 4 Wide scan XPS spectra of un-functionalised and functionalised SNPs. (A) SNPs SP with/without different functional groups and (B) SNPs LP with/without different functional groups. | ||
3.5. Loading of Van in SNPs
Van (30% w/w theoretical loading) was initially loaded into un-functionalised and functionalised SNPs and TGA was used to determine the loading capacity of Van. The loading capacity of Van into un-functionalised SNPs was ca. 25 wt% (Fig. 5A), with SNPs SP and SNPs LP showing no impact of pore size on the loading capacity. However, the loading capacity of functionalised SNPs did differ depending on the functional groups used (Fig. 6). For functionalised SNPs SP, the Van loading was slightly higher then un-functionalised SNPs SP, at about 29 wt% and 28 wt%, respectively for SNPs SP functionalised with –NH2 and –PO3. In contrast, Van loading decreased significantly to approximately 18 wt% for SNPs SP-CH3, showing that the surface area and hydrophobic modification impacted the Van loading. For functionalised SNPs LP, Van loading was around 29 wt% for all the samples, which demonstrates that despite functionalising the SNPs successfully, SNPs pores were not blocked, and thus Van loading was not affected. Moreover, in comparison to the Van loading capacity of un-functionalised SNPs these results suggest that the functionalization of the surface of SNPs might have reduced the pore size and increased the interactions forces between Van and matrix mesoporous surfaces and hence higher loading capacity.91 However since the physisorption of the drug on SNPs is a surface phenomenon and it would depend on the surface area of the SNPs where the drug can physically adsorb,91 the significant reduction of surface area in the case of SNPs SP-CH3 might have impacted negatively the adsorption of Van since there was no enough space on the SNPs SP-CH3 surfaces for Van to physically adsorb or the hydrophobic nature of SNPs SP-CH3 might contributed to repel Van. The loading of Van into the mesopores of SNPs was further confirmed by N2 adsorption/desorption measurement. The loading of Van in SNPs leads to a decreased BET surface area and pore volume (Table 3). For example, BET surface area decreased from 411.50 and 623.87 m2 g−1 (for SNPs SP and SNPs LP, respectively) to 118.26 m2 g−1 and 251.17 m2 g−1 after being loaded with Van. Similarly, the pore volume was markedly reduced from 0.583 cm3 g−1 and 1.343c m3 g−1 for SNPs SP and SNPs LP, respectively to 0.1 m3 g−1 and 0.26 m3 g−1 after being loaded with Van, which indicates the entrapment and presence of Van molecules in the pores.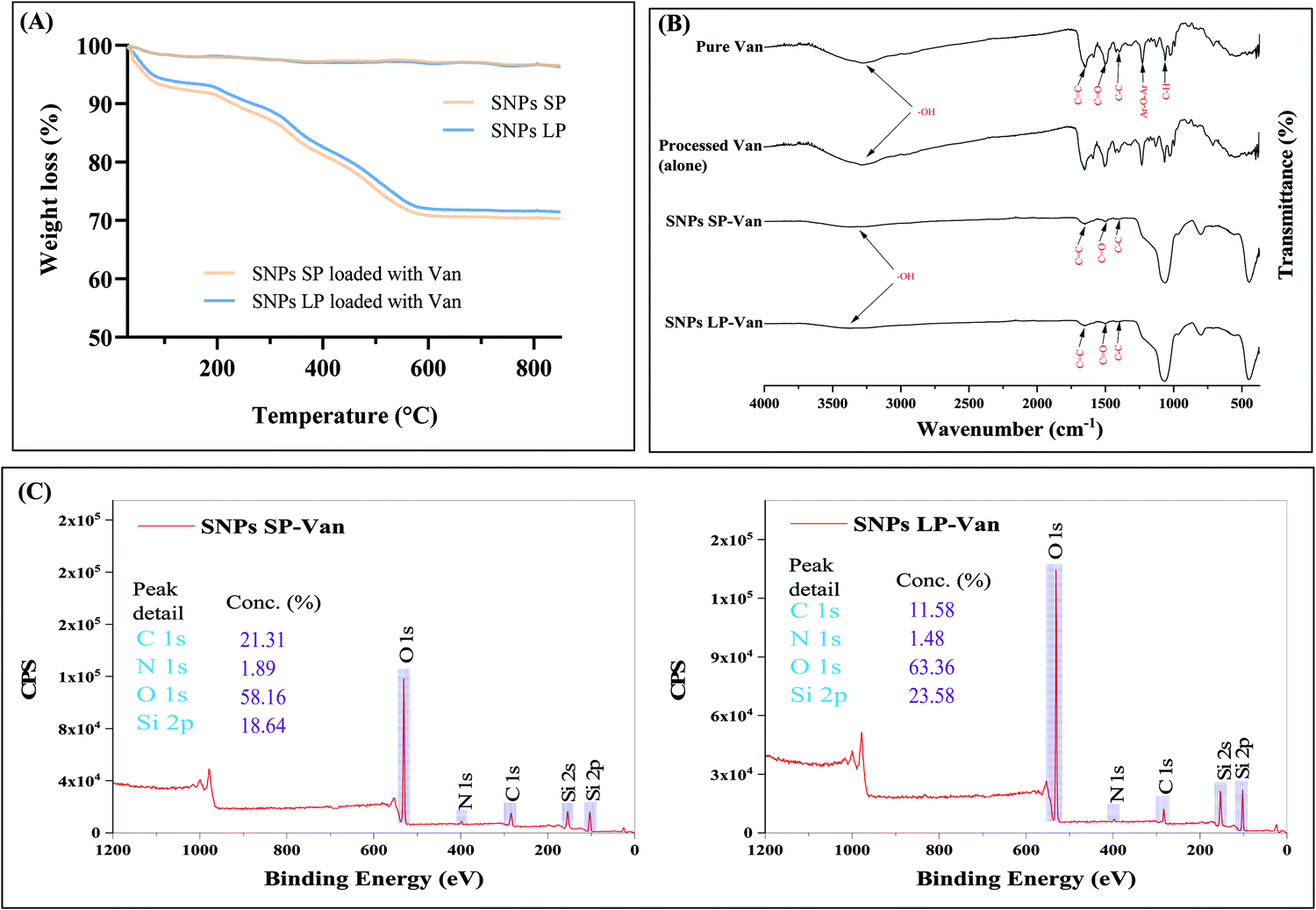 | ||
| Fig. 5 (A) TGA of SNPs SP-Van and SNPs LP-Van. (B) FT-IR spectra of pure Van, SNPs SP-Van, and SNPs LP-Van. (C) XPS survey spectra of SNPs SP-Van and SNPs LP-Van. | ||
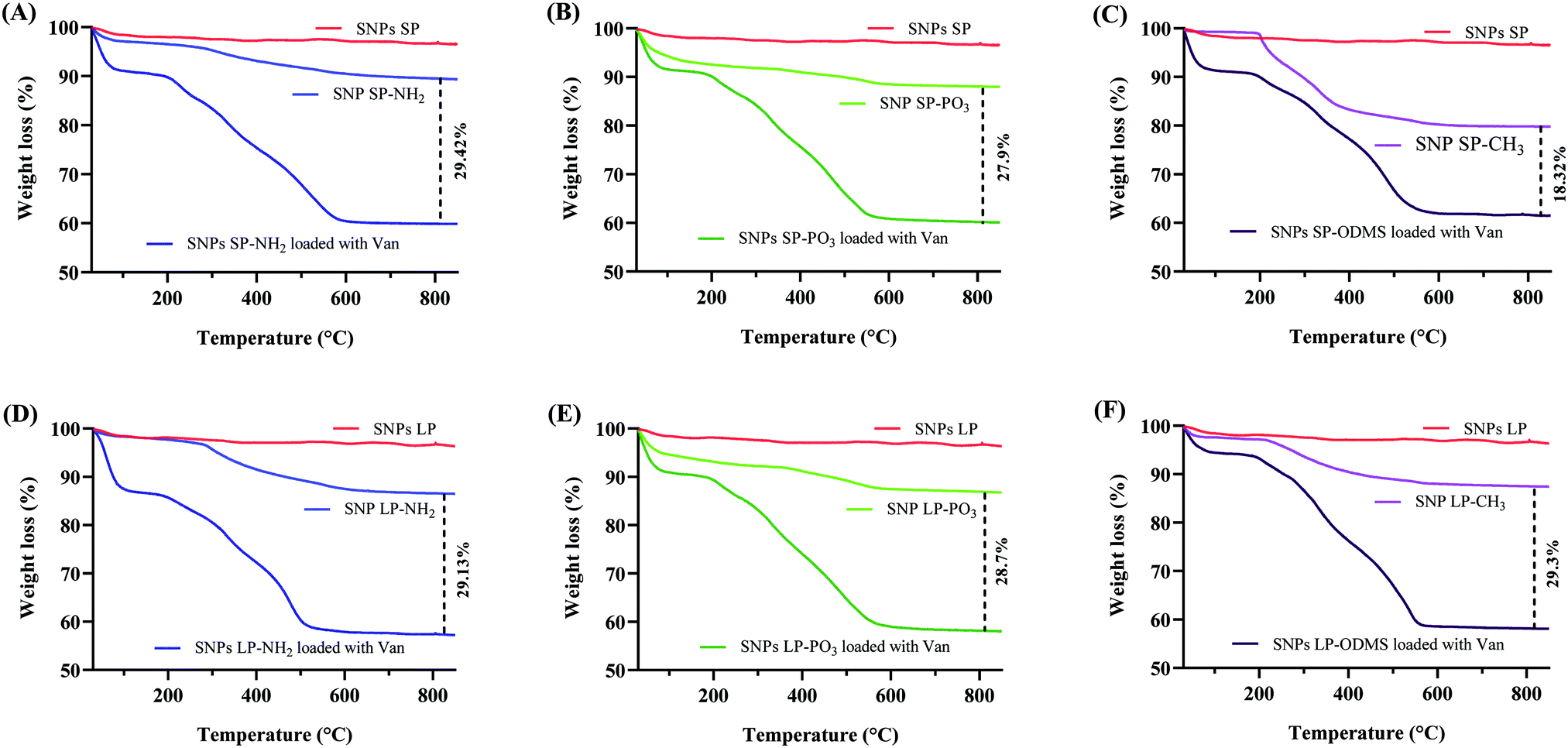 | ||
| Fig. 6 TGA of (A) SNPs SP-NH2 loaded with Van, (B) SNPs SP-PO3 loaded with Van, (C) SNPs SP-CH3 loaded with Van, (D) SNPs LP-NH2 loaded with Van, (E) SNPs LP-PO3 loaded with Van, and (F) SNPs LP-CH3 loaded with Van. | ||
| Van-loaded SNPs | Surface charge (mV) | Mean particle size (nm) ± SD | Polydispersity index (PDI) ± SD | BET surface area [m2 g−1] | Pore volume [cm3 g−1] | ||
|---|---|---|---|---|---|---|---|
| Van-loaded SNPsa | Un-loaded SNPsb | Van-loaded SNPs | Un-loaded SNPs | ||||
| a,bUnpaired two tailed t test was performed and there was a significant difference in particle size of samples in a in comparison to samples in b and the values with different letter in the same row are significantly different (p < 0.05). cIndicates the significant difference (p < 0.05) in particle size in comparison to un-functionalised SNPs presented in Table 1. dIndicates that there was no significant difference (p > 0.05) in particle size in comparison to un-functionalised SNPs presented in Table 1. eIndicates that there was no significant difference (p > 0.05) in surface charge in comparison to un-functionalised SNPs presented in Fig. 2. | |||||||
| SNPs SP-Van | −24.6 ± 1.08e | 230 ± 79a,c | 130 ± 40b,d | 0.33 ± 0.02 | 0.28 ± 0.03 | 118.26 | 0.1 |
| SNPs LP-Van | −21.53 ± 2.15e | 273 ± 49a,c | 153 ± 37b,d | 0.39 ± 0.03 | 0.37 ± 0.03 | 251.17 | 0.26 |
Furthermore, FTIR was performed for Van-loaded SNPs and the results are presented in Fig. 5B. The characteristics spectral bands of pure Van (Fig. 5B, 1st spectrum from up) were aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C at 1651 cm−1, CO stretching vibrations associated with amino groups at 1503 cm−1, C–C bending vibrations in aromatic rings at 1400 cm−1, Ar–O–Ar at 1227 cm−1, and C–H bending vibration at 1064 cm−1. FTIR spectra of SNPs loaded with Van showed both vibrational characteristics of Van and SNPs. The bands associated with chemical groups (at 1651, 1503, 1400 cm−1) of Van appeared in the spectra of Van-loaded SNPs, which demonstrated that Van has been loaded into SNPs, although the peaks associated with Van (Ar–O–Ar and C–H bending vibrations in the region of 1250–900 cm−1) are not observed due to the substantial peak from the big Si–O–Si asymmetric stretching band in that region which masks the eventual contribution of Van peaks in that same region. In addition, some changes in the shape and intensity of FTIR bands of SNPs in Van-loaded SNPs were noticed compared to un-loaded SNPs spectra (Fig. 5B, 1st and 2nd spectra from bottom). For instance, compared to un-loaded SNPs, the sharp peaks of the Si–O–Si asymmetric stretching band (at 1072 cm−1), symmetric stretching of Si–O–Si (at 798 cm−1), and Si–O–Si bending vibration (451 cm−1) became slightly wider when loaded with Van, suggesting that Van loading did affect those SNPs IR peaks, probably due to weak intermolecular interactions between Van molecules and SNPs. Van was also processed alone (without SNPs) to see whether the loading method used had an impact on the chemical structure of Van, and FTIR spectrum of processed Van (Fig. 5B, 2nd spectrum from up) showed similar spectrum as of pure Van indicating that the FD process did not affect the chemical structure of Van.
C at 1651 cm−1, CO stretching vibrations associated with amino groups at 1503 cm−1, C–C bending vibrations in aromatic rings at 1400 cm−1, Ar–O–Ar at 1227 cm−1, and C–H bending vibration at 1064 cm−1. FTIR spectra of SNPs loaded with Van showed both vibrational characteristics of Van and SNPs. The bands associated with chemical groups (at 1651, 1503, 1400 cm−1) of Van appeared in the spectra of Van-loaded SNPs, which demonstrated that Van has been loaded into SNPs, although the peaks associated with Van (Ar–O–Ar and C–H bending vibrations in the region of 1250–900 cm−1) are not observed due to the substantial peak from the big Si–O–Si asymmetric stretching band in that region which masks the eventual contribution of Van peaks in that same region. In addition, some changes in the shape and intensity of FTIR bands of SNPs in Van-loaded SNPs were noticed compared to un-loaded SNPs spectra (Fig. 5B, 1st and 2nd spectra from bottom). For instance, compared to un-loaded SNPs, the sharp peaks of the Si–O–Si asymmetric stretching band (at 1072 cm−1), symmetric stretching of Si–O–Si (at 798 cm−1), and Si–O–Si bending vibration (451 cm−1) became slightly wider when loaded with Van, suggesting that Van loading did affect those SNPs IR peaks, probably due to weak intermolecular interactions between Van molecules and SNPs. Van was also processed alone (without SNPs) to see whether the loading method used had an impact on the chemical structure of Van, and FTIR spectrum of processed Van (Fig. 5B, 2nd spectrum from up) showed similar spectrum as of pure Van indicating that the FD process did not affect the chemical structure of Van.
In addition, the presence of Van in SNPs was revealed from the comparison of chemical analysis through XPS measurements of Van-loaded SNPs, as shown in Fig. 5C. The binding energy peaks of N 1s (397 eV) were observed at the survey scan of Van-loaded SNPs. This shows the presence of nitrogen-containing groups in Van-loaded SNPs, which are the typical fingerprint groups of Van molecules and are not found in un-loaded SNPs. C 1s peaks (282 eV) were also present in Van-loaded SNPs confirming the presence of Van. Moreover, the elemental composition (%) was different for both SNPs SP-Van and SNPs LP-Van. For insistence, SNPs SP-Van showed higher composition (%) of C 1s and N 1s compared to SNPs LP-Van (21.31 vs. 11.58% for C 1s and 1.89 vs. 1.48 for N 1s), although they both had approximately the same loading capacity. This difference in XPS elemental composition can be explained by looking into the morphology and structure of their pores. While SNPs SP had smaller and superficial pores, SNPs LP had bigger, deep, and channel-like pores, which are symmetric around the core (as we discussed above). So, in the case of SNPs SP, more Van might have adsorbed on the surfaces and in the pores which might have been accessible to be detected by XPS (which is specific for surface analysis, around 5 nm of depth). On the other hand, with SNPs LP more Van might have penetrated deeply into the channel-like pores of SNPs LP (deeper than the detection depth of XPS) during the loading process and hence the amount of Van to be detected by XPS on the surface was lower compared to SNPs SP therefore lower % of C 1S and N 1s for SNPs LP-Van.
To investigate the particle size characteristics of SNPs-Van, SNPs after release studies were collected from the dialysis bag and analysed by DLS for their particle size characteristics. As shown in Table 3, after Van release studies in pH simulated intestinal medium, the mean measured particle sizes of SNPs SP and SNPs LP increased significantly to 230 ± 79 and 273 ± 49 nm, respectively, compared to un-loaded ones (un-functionalised SNPs in Table 1), suggesting that released Van—at least in this study—might have somewhat bound to SNPs after diffusion from the pores and increased the particle size (although Van is hydrophilic and we kept sink conditions during release studies). To investigate whether it was the influence of release medium (compared to DW used as dispersion medium in DLS), the un-functionalised and un-loaded SNPs were dispersed in release medium for 10 h and were subsequently analysed by DLS. As it can be seen in Table 3, the mean particle sizes of SNPs SP and SNPs LP were 130 ± 40 and 153 ± 37 nm, which were not significantly different from the ones in Table 1, showing that the particle size characteristics were affected by Van loading but not by the release medium. In addition, the surface charge values of SNPs after Van release (Table 3) were not significantly different from the values of un-loaded SNPs (in Fig. 2), showing that the Van loading did not significantly alter the surface charge of SNPs under the release conditions (pH 6.8) despite Van containing ionizable groups (one carboxyl, two amino, and three phenolic).92 Here in the solution with pH of 4 Van is slightly cationic (with net molecular charge of +2). However, when the pH of the medium increases the net molecular charge slightly decreases and it reaches net molecular charge of zero at around pH of 7.092 and that's probably the reason why in the release medium of 6.8, the surface charge of Van-loaded SNPs were not significantly altered.
3.6. In vitro release studies of Van-loaded SNPs
The release behaviour of the drug is a crucial parameter for the controlled and sustained delivery of therapeutics.93 For antibiotics, the effectiveness of different antibiotic is driven by different pharmacokinetic parameters, such as the maximum concentration, the time at which the concentration is above the MIC, or the area under the curve.94 The influence of the SNPs and their functionalisation on the release behaviour of Van in response to pH conditions similar to the small intestine was investigated and the release profiles are presented in Fig. 7.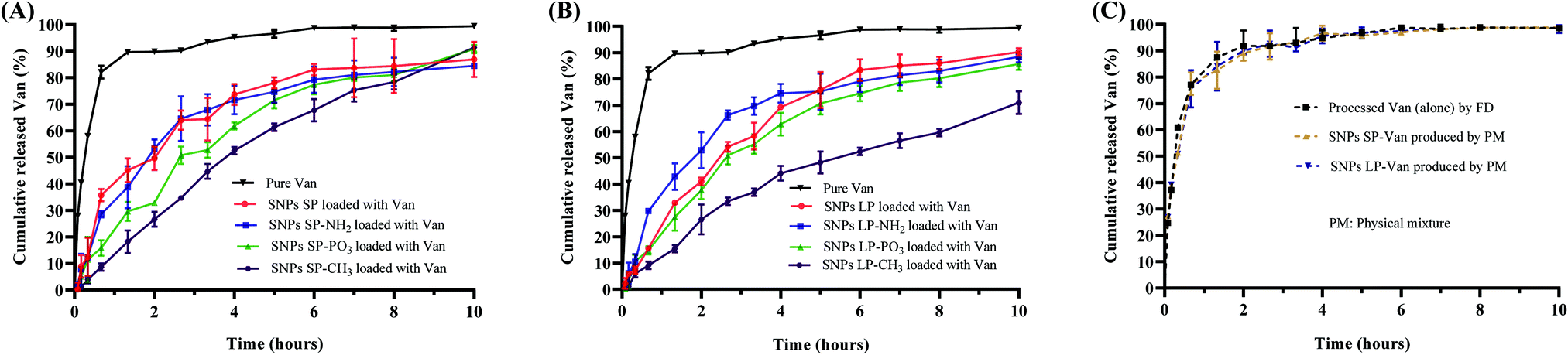 | ||
| Fig. 7 Release profiles of (A) Van-loaded SNPs SP, (B) Van-loaded SNPs LP, and (C) processed Van and Van SNPs produced by physically mixing them at a specified ratio. The error bars on the graphs represent the (n = 3 ± SD). | ||
Regardless of whether Van was loaded in un-functionalised or functionalised SNPs, Van-loaded SNPs showed a more prolonged release than pure (un-encapsulated Van) (Fig. 7A and B). For instance, the initial dissolution and release rate of pure Van was higher with 82 ± 2.4% of Van being released within first 40 min. In comparison, Van-loaded SNPs SP and Van-loaded SNPs LP showed a relatively slower release with a cumulative release of 36 ± 2.3% and 16 ± 1%, respectively, in 40 min (Fig. 7A and B, red lines) and this was 2.3 and 5.1 times less in comparison to percentage release of pure Van. This initial release pattern for Van-loaded SNPs indicates that although there might have been Van on the surfaces of SNPs, Van was either adsorbed (by physisorption) on the SNPs surfaces or in low amount since we do not see a burst release of Van (due to rapid dissolution of free Van on the surfaces) especially in the first 20 min. Moreover, looking at the release patterns it is evident that the release pattern of pure Van (as time progressed) was different from that of Van-loaded SNPs. For instance, after reaching a 90 ± 0.3% cumulative release within 80 min, as time progressed the release rate of pure Van slightly declined and almost plateaued. In contrast, the cumulative amount of released Van from SNPs over time kept increasing in a more linear and quadratic manner, exhibiting a steady-state flux with for example 50 ± 4.4% and 41 ± 1.5% cumulative release of Van from SNPs SP-Van and SNPs LP-Van, respectively, within 80 min. This sustained and more linear release pattern of Van from SNPs compared to pure (un-encapsulated) Van could be due to the entrapment of Van inside the pores and/or adsorption onto the SNPs surfaces, which results in a longer time for Van to be diffused from inside the pores to the release medium. Although we did not perform in vivo release studies and although there are many other factors which can impact the Van release behaviour in vivo,95 these results suggest a more controlled release of Van from SNPs compared to pure Van which could be beneficial in reducing the dosage frequency of Van in case of Van-loaded SNPs based formulations.96 Regarding the type of SNPs (SNPs SP or SNPs LP), there was a slight difference in their release profiles where release rate of Van from SNPs LP was slightly lower than that from SNPs SP (red line in Fig. 7Avs. red line in Fig. 7B). This might be attributed to the fact that Van in SNPs LP was loaded and trapped deeply in the channels of SNPs LP and thus it took longer to be accessed by release medium whereas Van in SNPs SP was adsorbed in the more superficial pores.
When the Van-loaded un-functionalised SNPs were compared to Van-loaded functionalised SNPs, there were slight differences in their release rates. For instance, compared to un-functionalised SNPs loaded with Van, within first 4 h the cumulative release of Van in SNPs LP-NH2 was slightly higher in comparison to SNPs LP-Van (Fig. 7B, blue line vs. red line) although the difference was not remarkable in SNPs SP. In SNPs-PO3, regardless of pore size, the cumulative release of Van was slightly lower throughout the time of release compared to SNPs LP-Van (Fig. 7A and B, green line vs. red line). In comparison to un-functionalised SNPs, SNPs-NH2 and SNPs-PO3, the SNPs-CH3 showed a substantially slower Van release throughout 10 h release (Fig. 7A and B, maroon line vs. other lines), suggesting that hydrophobic modification was more effective in retarding Van release compared to hydrophilic functional groups. Here when SNPs-CH3-Van were suspended in release medium they might self-aggregate and form a cluster-like structure due to their surface hydrophobicity, blocking the release of Van from the pores.
When drug is loaded in SNPs or in any carrier, the process used not only helps to load or adsorb the drug of interest, but it also impact the drug characteristics.11 Drug solid state, particle size and other characteristics may be altered, which can consequently have an impact on release behaviour.11,97 It is therefore important to understand that the way the drug dissolves and releases from carriers might be the result of (1) the drug being adsorbed and/or encapsulated in the carrier (SNPs in this case) and/or (2) the impact of process on the drug itself. Therefore, we investigated the processed Van (alone) to assess whether there was any possible impact of the process on Van. The FTIR data of pure Van and processed Van were similar, which shows that the process did not change the molecular structure of Van (Fig. 5B). Similarly, the release behaviour of processed Van was not different from that of pure Van (Fig. 7C), confirming that the release behaviours in Fig. 7A and B were due to the Van encapsulation and/or adsorption in mesopores of SNPs and not the impact of the FD process.
As discussed earlier, the slow-release behaviour of Van-loaded SNPs is proposed to be due to Van that is loaded and trapped and/or adsorbed into the pores is slowly released compared to free drug. To ensure that simple mixing of Van and SNPs without any processing did not lead to the same impact on the release behaviour of Van, we tested the release profiles of Van-loaded SNPs prepared by physical mixture. As shown in Fig. 7C they showed no difference in comparison to pure Van, confirming that the slow-release behaviour of Van-loaded SNPs was due to the entrapment and adsorption of Van molecules into the pores of the SNPs.
3.7. In vitro cytotoxicity assays
The cytotoxicity assays for SNPs in Caco-2 cells were performed by MTT assay. SNPs (functionalised/un-functionalised) in different concentrations ranging between 25–500 μg mL−1 were studied for their cytocompatibility in Caco-2 cells. The results are shown in Fig. S15 (ESI†). A slight decrease of cell viability is seen compared to controls, with a slight concentration-dependent cytotoxic effect evident for SNPs-CH3, but not other SNPs. Overall, more than 80% cell viability was observed for all the SNPs in the concentration range tested, suggesting a lack of cytotoxicity on Caco-2 cells. This concurs with previous studies about the in vitro cytotoxicity of mesoporous silica nanomaterials.55,983.8. Permeability studies of Van-loaded SNPs
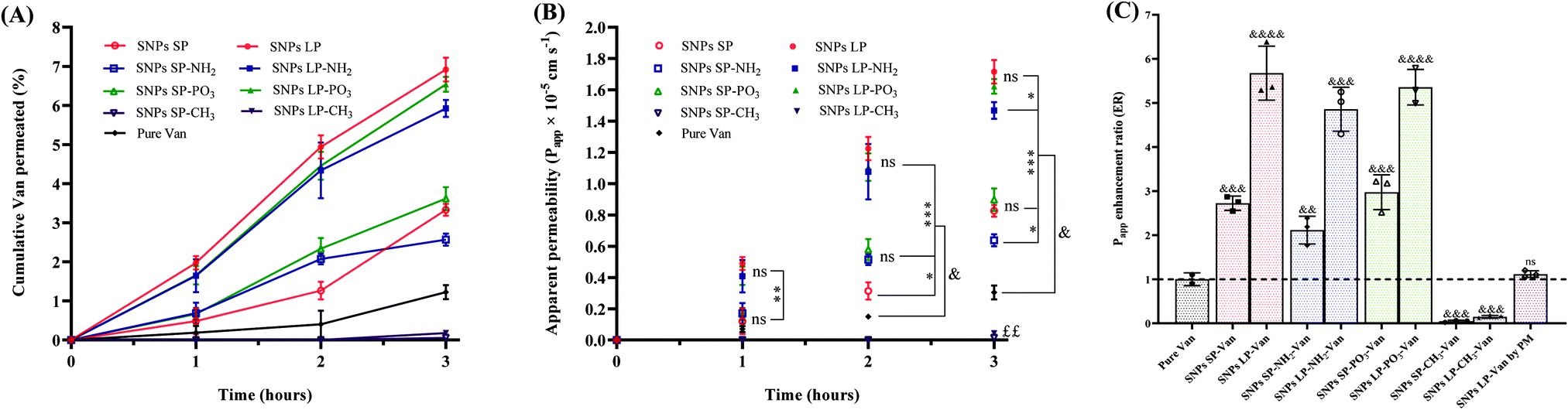 | ||
| Fig. 8 SNPs (functionalised or un-functionalised, except SNPs-CH3) significantly increased the permeability of Van across Caco-2 cells monolayers. All the values in the figure are presented as means ± SDs of n ≥ 3. (A) Cumulative permeability profiles of Van-loaded SNPs in Caco-2 cell monolayers for 3 h period. (B) Apparent permeability (Papp) of Van-loaded SNPs across Caco-2 cell monolayers; statistical variance among the samples was analysed by one-way ANOVA, followed by Tukey's test for multiple comparisons; ns: no significance; significance level was *p < 0.033, **p < 0.002 and ***p < 0.0002 with respect to pure Van; &indicates the significant increase of Papp for Van-loaded SNPs in comparison to pure Van; ££Van-loaded SNPs-CH3 showed no improvement of Papp, in fact SNPs-CH3 reduced significantly the Papp of Van in comparison to pure Van. (C) Enhancement ratio (ER) of the apparent permeability (Papp) of Van from different SNPs (at 3 h); ER was determined using the following equation: ER = Papp, Van-loaded SNPs/Papp, pure Van; also, un-paired two tailed t-test was performed to analyse the statistical variance for each sample vs. pure (un-encapsulated) Van; significance level was &&p < 0.002, &&&p < 0.0002 and &&&&p < 0.0001. Van-loaded SNPs LP produced by physical mixture was also studied and compared to pure Van. | ||
Moreover, pore size (SNPs SP or SNPs LP) and morphology had substantial effects on the permeability of Van (Fig. 8A–C). Van-SNPs SP and Van-SNPs LP showed different trends in terms of permeated amount of Van (Fig. 8B) and Papp profiles (Fig. 8B). For example, at 3 h (Fig. 8B), the Papp values increased significantly from 0.826 × 10−5 cm s−1, 0.637 × 10−5 cm s−1, and 0.898 × 10−5 cm s−1 for SNPs SP, SNPs SP-NH2, and SNPs SP-PO3, respectively, to 1.716 × 10−5 cm s−1, 1.468 × 10−5 cm s−1, and 1.622 × 10−5 cm s−1 when their SNPs LP counterparts were tested, and that was consistently more than a 2-fold increase in Papp for SNPs LP in comparison to SNPs SP (Fig. 8C). In addition to the fact that pore size and morphology might have played a crucial role,110 the degree of Van adsorption on the SNPs (SNPs LP vs. SNPs SP) might have played a significant role too. Our hypothesis here is that even if there was no significant difference in the loading capacity between SNPs SP and SNPs LP, a high amount of Van might have been encapsulated and adsorbed deep in the channel-like pores of SNPs LP, whereas in the case of SNPs SP a high amount of Van might have been in the superficial small pores of SNPs or even free on the surfaces (as discussed in detail in Section 3.6). Why does this matter? Well, as we discussed above, SNPs disrupt and open the tight junctions and then the drug is carried through the epithelium and reach the basolateral chamber. At this point we do not know which Van permeation mechanism is involved. Does the Van diffuse and release from SNPs first and then permeate (free without SNPS) via the already open tight junctions to reach the basolateral chamber? If this was the case, then Van-SNPs from PM (physical mixture) should have shown improved permeability compared to pure Van, which was not the case (Papp value of Van-SNPs LP was not significant different from pure Van, Fig. 8C). Based on the data, our hypothesis is that the SNPs open the tight junctions and then they act as delivery vehicles to carry Van across the epithelium and shuttle the Van payload to the receiving chamber. Correlating this with different ways Van might have been encapsulated and/or adsorbed in SNPs LP compared to SNPs SP, it might have happened that the high amount of free and superficially adsorbed Van in SNPs SP might have diffused, released, and deposited in the apical chamber and hence even if SNPs SP might have reached the basolateral chamber, the high amount of Van might had been already released and deposited in the apical chamber. On the other hand, Van in SNPs LP are entrapped and adsorbed in their deep channel-like pores which does not allow Van to quickly be diffused and deposited in the apical chamber. Therefore, in the latter, the SNPs LP are able to carry the Van payload via the epithelium and shuttle Van in the basolateral compartment hence the Van permeability in SNPs LP is higher than SNPs SP. This explanation is based on the permeability data but if we correlate these data with release studies, we see that although SNPs SP (un-functionalised/hydrophilically functionalised) showed slightly higher release at 3 h compared to SNPs LP counterparts, the difference was not significant in all the samples.
In addition to pore size, SNPs surface chemistry had a significant effect on Van permeability. Initially our hypothesis was that altering the SNPs surfaces charge and polarity by functionalising them with different groups would influence the permeability behaviours of Van. Irrespective of pore size, SNPs functionalised with –NH2 showed lower Papp values compared to SNPs-PO3 or un-functionalised SNPs. For example (at 3 h), the value of Papp was significantly (p > 0.05) reduced from 1.716 × 10−5 cm s−1 and 1.622 × 10−5 cm s−1, respectively, for SNPs LP (un-functionalised) and SNPs-PO3 to 1.468 × 10−5 cm s−1 for SNPs LP-NH2. This reduction in Papp value for –NH2 functionalised SNPs can be attributed to the fact that there might have a strong electrostatic interactions between the cationic SNPs-NH2 and the negatively charged cellular membranes111 and this did not permit the Van-loaded SNPs-NH2 to easily carry the Van across the epithelium to basolateral compartment. Moreover, the polarity of SNPs significantly affected the permeability of Van. For Van-loaded SNPs-CH3 (either SNPs SP or SNPs LP), the Van permeability was not only remarkably lower compared with other Van-loaded SNPs but also it was significantly low in comparison to pure Van, with only 0.015 × 10−5 cm s−1 and 0.044 × 10−5 cm s−1 for Van-loaded SNPs SP-CH3 and Van-loaded SNPs LP-CH3, respectively, at 3 h. This might have been due to the hydrophobic nature of SNPs-CH3, where the diffusion, release, and dissolution of Van in the incubation media might have hindered by the hydrophobic nature of SNPs-CH3, although we cannot entirely attribute this to their release behaviours. Furthermore, as we discussed above, SNPs disrupt and transiently open the tight junctions, which allow the drug to be carried to the basolateral chamber and this has been shown to correlate well with TEER values (the reduction of TEER values indicates the disruption, relaxation, and/or opening of tight junctions and vice versa) especially for molecules with size from 180 Da to at least 10 kDa.112,113 By correlating this with our results for example, as you can see from the Fig. 8, for the first 2 h of experiment, there was no amount (0%) of Van permeated from Van-SNPs-CH3. The question here is why SNPs-CH3 still showed significant reduction of TEER values in the first 2 h (Fig. S16, ESI†) despite 0% of Van permeation in the basolateral chamber? Our hypothesis here is (1) Van-loaded SNPs-CH3 were able to transiently open the tight junctions, and to traverse across the epithelium to reach the basolateral chamber, but as they are hydrophobic in nature, encapsulated and/or adsorbed Van was not able to be diffused and released (in 1 h or 2 h, even if 1% DMSO was used as a co-solvent and although their release profiles show that they can be in this time frame); (2) since SNPs-CH3 are hydrophobic, they were able to bind and fuse with cell membrane phospholipid bilayers (as we initially hypothesized), hence the disturbance of tight junctions and TEER values decrease but they stuck either in the phospholipid bilayers of cytoplasmic membrane or in the cytoplasm and were not able to escape and/or transcytose across the cytoplasm to reach the basolateral compartment. Whether we consider the first hypothesis or second the detailed investigation, elucidating which Caco-2 cells monolayers-SNPs-CH3 interaction mechanism is involved, is needed.
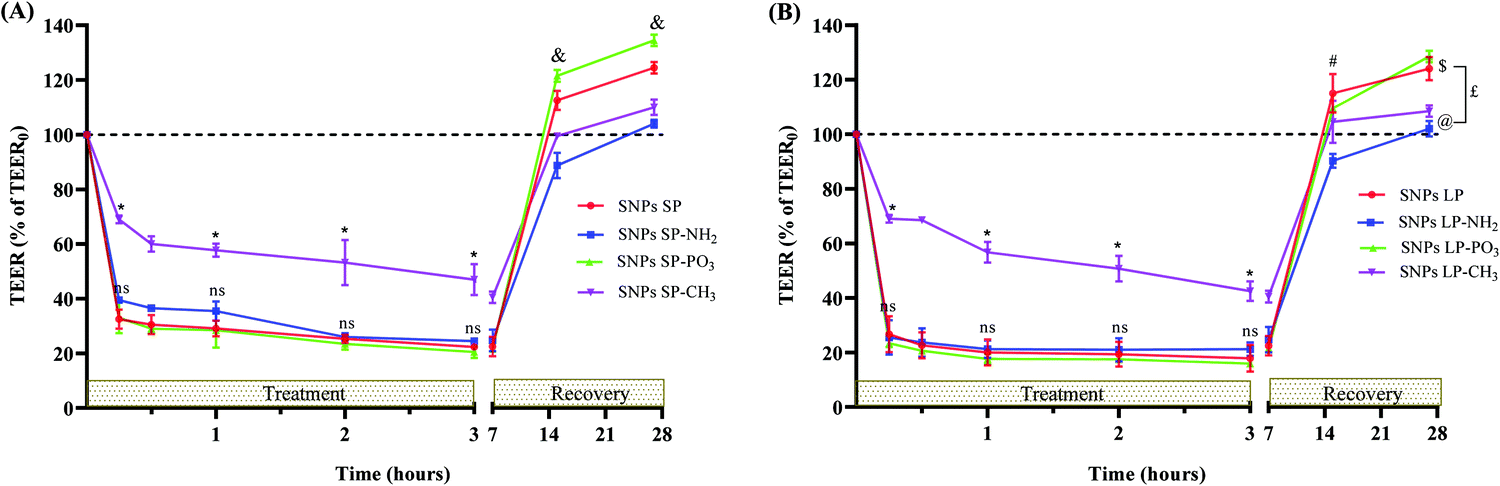 | ||
| Fig. 9 The ability of SNPs to disrupt the integrity of Caco-2 cell monolayers and whether the effect is reversible was investigated by treating the Caco-2 cell monolayers with Van-free (A) SNPs SP (functionalised/un-functionalised) and (B) SNPs LP (functionalised/un-functionalised) for 3 h. After 3 h of treatment, SNPs were removed, and the Caco-2 cell monolayers were rinsed with DMEM and equilibrated, and their recovery was studied within the next 24 h. TEER0 means TEER (Ω cm2) values at initial time point (t = 0). TEER values are presented as means ± SDs (n = 3) considering the TEER values of un-treated Caco-2 cells (Caco-2 cell monolayers with only HBSS or HBSS + 1% DMSO without SNPs, N.B.: we wanted to assume that DMSO might have an impact on the Caco-2 cell monolayers integrity, so we performed experiments by dosing the apical chamber with 0.5 mL of HBSS containing 1% DMSO and there was no significant difference in TEER values in comparison to DMSO-free HBSS). Statistical variance among the samples was analysed by one-way ANOVA, followed by Tukey's test for multiple comparisons. *indicates the significant difference (p < 0.05) in TEER values (throughout treatment experiment) between SNPs-CH3vs. other samples, &indicates the significant difference (p < 0.05) in all the samples, #indicates the significant difference (p < 0.05) between SNPs LP-NH2vs. other samples, nsindicates that there was no significant difference (p > 0.05) (throughout treatment experiment) between SNPs, SNPs-NH2, and SNPs-PO3, $indicates that there was no significant difference (p > 0.05) between SNPs LP and SNPs LP-PO3, @indicates the significant difference (p < 0.05) between SNPs LP-NH2 and SNPs-CH3, £indicates the significant difference (p < 0.05) (SNPs LP and SNPs LP-PO3vs. SNPs LP-NH2 and SNPs-CH3). | ||
4. Conclusion
The oral delivery of Van has the potential to improve disease outcomes and patient experience by alleviating the drawbacks associated with Van injections. Unfortunately, Van can only be administered intravenously for systemic infections since its physico-chemical properties limit its permeability through intestinal epithelium. In this work we designed and surface-engineered SNPs to encapsulate and improve the permeability of Van. We successfully synthesized SNPs of different pore sizes and morphology and surface-modified their charge and polarity, and tested their impact on the release and permeation behaviour of loaded Van. Van-loaded SNPs showed high loading and a controlled release behaviour of Van compared to pure Van. Van-loaded SNPs exhibited significant enhancement of Van permeability through an epithelial cell monolayer, with Van permeability enhancement dependent on the SNPs pore sizes and morphologies as well as their surface chemistry. Although the Van-loaded SNPs showed substantial improvement of Van permeation in the Caco-2 cell model, with high Papp values compared to pure Van, future work must conduct these permeability experiments in animal models to see whether this improvement is translatable in vivo. Furthermore, studies to decipher the exact mechanisms that SNPs use to increase the permeation of Van across Caco-2 cell monolayers are needed.Author contributions
John Ndayishimiye: conceptualization, methodology, visualization, data curation, software, investigation, writing, original draft, review & editing. Yuxue Cao: methodology, reviewing & editing. Tushar Kumeria: supervision, conceptualization, writing – review & editing. Mark A. T. Blaskovich: supervision, conceptualization, writing – review & editing. James Robert Falconer: supervision, conceptualization, writing – review & editing. Amirali Popat: supervision, conceptualization, writing – review & editing.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
J. N. holds an Australian Government Research Scholarship and research funding from The University of Queensland. Y. C. acknowledges support from a University of Queensland post-graduate scholarship. A. P. acknowledges financial support from National Health and Medical Research Council for the Early Career (GNT1071796) and Career Development Fellowship (GNT1146627). T. K. acknowledges financial support from National Health and Medical Research Council (NHMRC) for the Career Development Fellowship (GNT1143296) and Australian Research Council (ARC) for the Discovery Fellowship (DP200102723). The authors acknowledge the support from the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, the University of Queensland. Also, this work was performed in part at the Queensland node of the Australian National Fabrication Facility, a company established under the National Collaborative Research Infrastructure Strategy to provide nano- and micro-fabrication facilities for Australia's researchers.References
- J. O’Neill, Tackling drug-resistant infections globally: final report and recommendations (The Review on Antimicrobial Resistance), 2016 Search PubMed.
- S. Y. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- A. Simon, M. L. A. Moreira, I. F. d. J. B. Costa, V. P. de Sousa, C. R. Rodrigues, T. Sisnande, F. A. do Carmo, I. C. R. Leal, K. R. N. Dos Santos and L. C. R. P. da Silva, Nanotechnology, 2020, 31, 375101 CrossRef CAS PubMed.
- R. C. Moellering Jr, J. Antimicrob. Chemother., 2012, 67, 4–11 CrossRef PubMed.
- I. M. Gould, M. Z. David, S. Esposito, J. Garau, G. Lina, T. Mazzei and G. Peters, Int. J. Antimicrob. Agents, 2012, 39, 96–104 CrossRef CAS PubMed.
- A. Schilling, E. Neuner and S. J. Rehm, Clevel. Clin. J. Med., 2011, 78, 465–471 CrossRef PubMed.
- J. F. Mohr and B. E. Murray, Clin. Infect. Dis., 2007, 44, 1536–1542 CrossRef CAS PubMed.
- Y. Yang, M. V. McBride, K. A. Rodvold, F. Tverdek, A. M. Trese, J. Hennenfent, G. Schiff, B. L. Lambert and G. T. Schumock, Am. J. Health-Syst. Pharm., 2010, 67, 1017–1024 CrossRef PubMed.
- P. E. Reynolds, Eur. J. Clin. Microbiol. Infect. Dis., 1989, 8, 943–950 CrossRef CAS PubMed.
- S. Patel, C. V. Preuss and F. Bernice, 2020, https://pubmed.ncbi.nlm.nih.gov/29083794/.
- J. Ndayishimiye, A. Popat, M. Blaskovich and J. R. Falconer, J. Controlled Release, 2020, 324, 728–749 CrossRef CAS PubMed.
- R. Fekety, J. Silva, C. Kauffman, B. Buggy and H. G. Deery, Am. J. Med., 1989, 86, 15–19 CrossRef CAS PubMed.
- J. Silva, D. Batts, R. Fekety, J. Plouffe, G. Rifkin and I. Baird, Am. J. Med., 1981, 71, 815–822 CrossRef PubMed.
- Vancomycin hydrochloride, https://pubchem.ncbi.nlm.nih.gov/compound/Vancomycin-hydrochloride, accessed on December 3, 2020, 2020.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- Vancomycin hydrochloride https://www.chemicalbook.com/ChemicalProductProperty_EN_CB8117577.htm, accessed on December 3, 2020, 2020.
- J. M. Cyriac and E. James, J. Pharmacol. Pharmacother., 2014, 5, 83 CrossRef PubMed.
- A. J. Kaasch, G. Fätkenheuer, R. Prinz-Langenohl, U. Paulus, M. Hellmich, V. Weiß, N. Jung, S. Rieg, W. V. Kern and H. Seifert, Trials, 2015, 16, 450 CrossRef PubMed.
- J. McLenon and M. A. Rogers, J. Adv. Nurs., 2019, 75, 30–42 CrossRef PubMed.
- C. J. Sokolowski, J. A. Giovannitti and S. G. Boynes, Dent. Clin., 2010, 54, 731–744 Search PubMed.
- S. Sivagnanam and D. Deleu, Crit. Care, 2002, 7, 1–3 Search PubMed.
- M. P. Wilhelm, Mayo Clin. Proc., 1991, 66, 1165–1170 CrossRef CAS PubMed.
- C. Liu, Y. Kou, X. Zhang, H. Cheng, X. Chen and S. Mao, Expert Opin. Drug Delivery, 2018, 15, 223–233 CrossRef CAS PubMed.
- D. J. Drucker, Nat. Rev. Drug Discovery, 2020, 19, 277–289 CrossRef CAS PubMed.
- E. M. Pridgen, F. Alexis and O. C. Farokhzad, Expert Opin. Drug Delivery, 2015, 12, 1459–1473 CrossRef PubMed.
- A. J. Almeida and E. Souto, Adv. Drug Delivery Rev., 2007, 59, 478–490 CrossRef CAS PubMed.
- M. Liu, J. Zhang, W. Shan and Y. Huang, Asian J. Pharm. Sci., 2015, 10, 275–282 CrossRef.
- S. Maher, D. J. Brayden, L. Casettari and L. Illum, Pharmaceutics, 2019, 11, 41 CrossRef CAS PubMed.
- N. G. Lamson, A. Berger, K. C. Fein and K. A. Whitehead, Nat. Biomed. Eng., 2020, 4, 84–96 CrossRef CAS PubMed.
- L. Wright, P. Joyce, T. J. Barnes, R. Lundmark, C. A. Bergström, M. Hubert and C. A. Prestidge, J. Pharmaceut. Sci., 2020, 110(1), 217–227 CrossRef PubMed.
- M. M. Abeer, P. Rewatkar, Z. Qu, M. Talekar, F. Kleitz, R. Schmid, M. Lindén, T. Kumeria and A. Popat, J. Controlled Release, 2020, 326, 544–555 CrossRef CAS PubMed.
- M.-C. Desjonqueres and D. Spanjaard, Concepts in surface physics, Springer Science & Business Media, 2012 Search PubMed.
- J. Kurczewska, P. Sawicka, M. Ratajczak, M. Gajęcka and G. Schroeder, Int. J. Pharm., 2015, 486, 226–231 CrossRef CAS PubMed.
- Z. Gounani, M. A. Asadollahi, J. N. Pedersen, J. Lyngsø, J. S. Pedersen, A. Arpanaei and R. L. Meyer, Colloids Surf., B, 2019, 175, 498–508 CrossRef CAS PubMed.
- X. Zhou, W. Weng, B. Chen, W. Feng, W. Wang, W. Nie, L. Chen, X. Mo, J. Su and C. He, J. Mater. Chem. B, 2018, 6, 740–752 RSC.
- G. Qi, L. Li, F. Yu and H. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 10874–10881 CrossRef CAS PubMed.
- N. Hao, L. Li and F. Tang, Int. Mater. Rev., 2017, 62, 57–77 CrossRef CAS.
- J. Florek, R. Caillard and F. Kleitz, Nanoscale, 2017, 9, 15252–15277 RSC.
- C. Xu, C. Lei and C. Yu, Front. Chem., 2019, 7, 290 CrossRef CAS PubMed.
- M. Vallet-Regí, F. Balas, M. Colilla and M. Manzano, Prog. Solid State Chem., 2008, 36, 163–191 CrossRef.
- I. Izquierdo-Barba, E. Sousa, J. C. Doadrio, A. L. Doadrio, J. P. Pariente, A. Martínez, F. Babonneau and M. Vallet-Regí, J. Sol-Gel Sci. Technol., 2009, 50, 421–429 CrossRef CAS.
- J. Zhang, S. Karmakar, M. Yu, N. Mitter, J. Zou and C. Yu, Small, 2014, 10, 5068–5076 CAS.
- X. Hong, X. Zhong, G. Du, Y. Hou, Y. Zhang, Z. Zhang, T. Gong, L. Zhang and X. Sun, Sci. Adv., 2020, 6, eaaz4462 CrossRef CAS PubMed.
- C. G. Bavnhøj, M. M. Knopp, C. M. Madsen and K. Löbmann, Int. J. Pharm.: X, 2019, 1, 100008 Search PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Int. J. Pharm. Invest., 2015, 5, 124–133 CrossRef CAS PubMed.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- C. Dombu, R. Carpentier and D. Betbeder, Biomaterials, 2012, 33, 9117–9126 CrossRef CAS PubMed.
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941–951 CrossRef CAS PubMed.
- M. Barisik, S. Atalay, A. Beskok and S. Qian, J. Phys. Chem. C, 2014, 118, 1836–1842 CrossRef CAS.
- R. Cornu, C. Chrétien, Y. Pellequer, H. Martin and A. Béduneau, Arch. Toxicol., 2020, 94, 1191–1202 CrossRef CAS PubMed.
- H. Baghirov, D. Karaman, T. Viitala, A. Duchanoy, Y.-R. Lou, V. Mamaeva, E. Pryazhnikov, L. Khiroug, C. de Lange Davies, C. Sahlgren and J. M. Rosenholm, PLoS One, 2016, 11, e0160705 CrossRef PubMed.
- X. Tan, N. Yin, Z. Liu, R. Sun, J. Gou, T. Yin, Y. Zhang, H. He and X. Tang, Mol. Pharmaceutics, 2020, 17, 3177–3191 CrossRef CAS PubMed.
- J. Leal, H. D. Smyth and D. Ghosh, Int. J. Pharm., 2017, 532, 555–572 CrossRef CAS PubMed.
- Z. Chaudhary, G. M. Khan, M. M. Abeer, N. Pujara, B. W.-C. Tse, M. A. McGuckin, A. Popat and T. Kumeria, Biomater. Sci., 2019, 7, 5002–5015 RSC.
- E. Juère, G. Del Favero, F. Masse, D. Marko, A. Popat, J. Florek, R. Caillard and F. Kleitz, Eur. J. Pharm. Biopharm., 2020, 151, 171–180 CrossRef PubMed.
- Y. Niu, M. Yu, A. Meka, Y. Liu, J. Zhang, Y. Yang and C. Yu, J. Mater. Chem. B, 2016, 4, 212–219 RSC.
- D. Sriramulu, E. L. Reed, M. Annamalai, T. V. Venkatesan and S. Valiyaveettil, Sci. Rep., 2016, 6, 1–10 CrossRef PubMed.
- M. Lopes, N. Shrestha, A. Correia, M.-A. Shahbazi, B. Sarmento, J. Hirvonen, F. Veiga, R. Seiça, A. Ribeiro and H. A. Santos, J. Controlled Release, 2016, 232, 29–41 CrossRef CAS PubMed.
- M. J. de Jesús Valle, F. G. López and A. S. Navarro, J. Pharm. Biomed. Anal., 2008, 48, 835–839 CrossRef PubMed.
- M. Usman and G. Hempel, SpringerPlus, 2016, 5, 124 CrossRef PubMed.
- G. Kigen and G. Edwards, BMC Pharmacol. Toxicol., 2017, 18, 20 CrossRef PubMed.
- P. Artursson, J. Pharm. Sci., 1990, 79, 476–482 CrossRef CAS PubMed.
- I. Hubatsch, E. G. Ragnarsson and P. Artursson, Nat. Protoc., 2007, 2, 2111 CrossRef CAS PubMed.
- M. Bouchoucha, M.-F. Cote, R. C. Gaudreault, M. A. Fortin and F. Kleitz, Chem. Mater., 2016, 28, 4243–4258 CrossRef CAS.
- N. S. Zaharudin, E. D. Mohamed Isa, H. Ahmad, M. B. Abdul Rahman and K. Jumbri, J. Saudi Chem. Soc., 2020, 24, 289–302 CrossRef CAS.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605–612 CrossRef.
- E. Juère, J. Florek, M. Bouchoucha, S. Jambhrunkar, K. Y. Wong, A. Popat and F. Kleitz, Mol. Pharmacol., 2017, 14, 4431–4441 CrossRef PubMed.
- T. Prasad, S. Halder and S. S. Dhar, Mater. Today: Proc., 2020, 22, 1669–1675 CAS.
- X. Lv, L. Zhang, F. Xing and H. Lin, Microporous Mesoporous Mater., 2016, 225, 238–244 CrossRef CAS.
- E. Juère, R. Caillard and F. Kleitz, Microporous Mesoporous Mater., 2020, 306, 110482 CrossRef.
- T. Ji, C. Ma, L. Brisbin, L. Mu, C. G. Robertson, Y. Dong and J. Zhu, Appl. Surf. Sci., 2017, 399, 565–572 CrossRef CAS.
- R. P. Bagwe, L. R. Hilliard and W. Tan, Langmuir, 2006, 22, 4357–4362 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- E. Juère, G. Del Favero, F. Masse, D. Marko, A. Popat, J. Florek, R. Caillard and F. Kleitz, Eur. J. Pharm. Biopharm., 2020, 151, 171–180 CrossRef PubMed.
- M. Guo, Q. Liu, S. Lu, R. Han, K. Fu, C. Song, N. Ji, X. Lu, D. Ma and C. Liu, Langmuir, 2020, 36, 11528–11537 CrossRef CAS PubMed.
- M. Bouchoucha, M.-F. Côté, R. C.-Gaudreault, M.-A. Fortin and F. Kleitz, Chem. Mater., 2016, 28, 4243–4258 CrossRef CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- N. I. Vazquez, Z. Gonzalez, B. Ferrari and Y. Castro, Bol. Soc. Esp. Ceram. Vidrio, 2017, 56, 139–145 CrossRef CAS.
- A. Yildirim, E. Ozgur and M. Bayindir, J. Mater. Chem. B, 2013, 1, 1909–1920 RSC.
- M. Mousavi and E. Fini, ACS Sustainable Chem. Eng., 2020, 8, 3231–3240 CrossRef CAS.
- Y. Zhao, Y. Wang, F. Ran, Y. Cui, C. Liu, Q. Zhao, Y. Gao, D. Wang and S. Wang, Sci. Rep., 2017, 7, 4131 CrossRef PubMed.
- J. H. Park, S.-D. Baek, J. I. Cho, J. Y. Yoo, S.-Y. Yoon, S. Kim, S. Lee, Y. S. Kim and J.-M. Myoung, Composites, Part B, 2019, 175, 107188 CrossRef CAS.
- M. Castellano, E. Marsano, A. Turturro and L. Conzatti, Adsorption, 2012, 18, 307–320 CrossRef CAS.
- J. Seo and L. P. Lee, Sens. Actuators, B, 2006, 119, 192–198 CrossRef CAS.
- H. Y. Erbil, G. McHale and M. Newton, Langmuir, 2002, 18, 2636–2641 CrossRef CAS.
- E. Vargha-Butler, S. Sveinsson and Z. Policova, Colloids Surf., 1991, 58, 271–286 CrossRef CAS.
- M. H. Engelhard, T. C. Droubay and Y. Du, in Encyclopedia of Spectroscopy and Spectrometry, ed. J. C. Lindon, G. E. Tranter and D. W. Koppenaal, Academic Press, Oxford, 3rd edn, 2017, pp. 716–724 DOI:10.1016/B978-0-12-409547-2.12102-X.
- O. Sublemontier, C. Nicolas, D. Aureau, M. Patanen, H. Kintz, X. Liu, M.-A. Gaveau, J.-L. Le Garrec, E. Robert, F.-A. Barreda, A. Etcheberry, C. Reynaud, J. B. Mitchell and C. Miron, J. Phys. Chem. Lett., 2014, 5, 3399–3403 CrossRef CAS PubMed.
- A. G. Shard, Surf. Interface Anal., 2014, 46, 175–185 CrossRef CAS.
- J. C. Doadrio, E. M. Sousa, I. Izquierdo-Barba, A. L. Doadrio, J. Perez-Pariente and M. Vallet-Regí, J. Mater. Chem. B, 2006, 16, 462–466 RSC.
- R. P. Ralph, Rev. Infect. Dis., 1981, 3, S205–S209 CrossRef.
- Y. Perrie and T. Rades, FASTtrack Pharmaceutics: Drug Delivery and Targeting, Pharmaceutical Press, 2012 Search PubMed.
- E. I. Nielsen, O. Cars and L. E. Friberg, Antimicrob. Agents Chemother., 2011, 55, 4619–4630 CrossRef CAS PubMed.
- S. Hussain, J. Joo, J. Kang, B. Kim, G. B. Braun, Z.-G. She, D. Kim, A. P. Mann, T. Mölder and T. Teesalu, Nat. Biomed. Eng., 2018, 2, 95–103 CrossRef CAS PubMed.
- D. Hassan, C. A. Omolo, R. Gannimani, A. Y. Waddad, C. Mocktar, S. Rambharose, N. Agrawal and T. Govender, Int. J. Pharm., 2019, 558, 143–156 CrossRef CAS PubMed.
- J. Ndayishimiye, A. Popat, T. Kumeria, M. A. T. Blaskovich and J. Robert Falconer, Int. J. Pharm., 2021, 596, 120240 CrossRef CAS PubMed.
- A. Raza, F. B. Sime, P. J. Cabot, J. A. Roberts, J. R. Falconer, T. Kumeria and A. Popat, ACS Biomater. Sci. Eng., 2021, 7, 1836–1853 CrossRef CAS PubMed.
- P. Artursson and J. Karlsson, Biochem. Biophys. Res. Commun., 1991, 175, 880–885 CrossRef CAS PubMed.
- C. A. Larregieu and L. Z. Benet, AAPS J., 2013, 15, 483–497 CrossRef CAS PubMed.
- I. J. Hidalgo, T. J. Raub and R. T. Borchardt, Gastroenterology, 1989, 96, 736–749 CrossRef CAS.
- W. Du, Y. Fan, N. Zheng, B. He, L. Yuan, H. Zhang, X. Wang, J. Wang, X. Zhang and Q. Zhang, Biomaterials, 2013, 34, 794–806 CrossRef CAS PubMed.
- M. Pinto, Biol. Cell., 1983, 47, 323–330 Search PubMed.
- N. Shrestha, F. Araújo, M.-A. Shahbazi, E. Mäkilä, M. J. Gomes, B. Herranz-Blanco, R. Lindgren, S. Granroth, E. Kukk, J. Salonen, J. Hirvonen, B. Sarmento and H. A. Santos, Adv. Funct. Mater., 2016, 26, 3405–3416 CrossRef CAS.
- N. N. Salama, N. D. Eddington and A. Fasano, Tight Junctions, Springer US, Boston, MA, 2006, pp. 206–219 DOI:10.1007/0-387-36673-3_15.
- P. Artursson and S. D. Knight, Science, 2015, 347, 716–717 CrossRef CAS PubMed.
- L. L. Mitic, C. M. V. Itallie and J. M. Anderson, Am. J. Physiol.: Gastrointest. Liver Physiol., 2000, 279, G250–G254 CrossRef CAS PubMed.
- S. Mendiratta, M. Hussein, H. A. Nasser and A. A. A. Ali, Part. Part. Syst. Charact., 2019, 36, 1900195 CrossRef CAS.
- C. Saraiva, C. Praça, R. Ferreira, T. Santos, L. Ferreira and L. Bernardino, J. Controlled Release, 2016, 235, 34–47 CrossRef CAS PubMed.
- Y. Wang, C. Pi, X. Feng, Y. Hou, L. Zhao and Y. Wei, Int. J. Nanomed., 2020, 15, 6295 CrossRef CAS PubMed.
- V. Selvarajan, S. Obuobi and P. L. R. Ee, Front. Chem., 2020, 8, 602 CrossRef CAS PubMed.
- N. G. Lamson, G. Cusimano, K. Suri, A. Zhang and K. A. Whitehead, Mol. Pharmaceutics, 2016, 13, 578–585 CrossRef CAS PubMed.
- K. Whitehead, N. Karr and S. Mitragotri, Pharm. Res., 2008, 25, 1782–1788 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb01430g |
| ‡ In this paper Vancomycin Hydrochloride is simply referred to as Vancomycin (Van). |
| § The anatomy of GI epithelium and how it can be a physiological barrier for the permeation of macromolecules are detailed in ref. 11. |
| This journal is © The Royal Society of Chemistry 2021 |