
Microstructural origin of selective water oxidation to hydrogen peroxide at low overpotentials: a study on Mn-alloyed TiO2†
Jiahui
Li‡
abc,
Devan
Solanki‡
 ab,
Qianhong
Zhu
a,
Xin
Shen
ab,
Grace
Callander
ab,
Jaehong
Kim
a,
Yaogang
Li
c,
Hongzhi
Wang
*c and
Shu
Hu
*ab
ab,
Qianhong
Zhu
a,
Xin
Shen
ab,
Grace
Callander
ab,
Jaehong
Kim
a,
Yaogang
Li
c,
Hongzhi
Wang
*c and
Shu
Hu
*ab
aDepartment of Chemical and Environmental Engineering, Yale University, New Haven, Connecticut 06520, USA. E-mail: shu.hu@yale.edu
bEnergy Sciences Institute, Yale University, 810 West Campus Drive, West Haven, Connecticut 06516, USA
cState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, People's Republic of China. E-mail: wanghz@dhu.edu.cn
First published on 5th August 2021
Abstract
One key objective in electrocatalysis is to design selective catalysts, particularly in cases where the desired products require thermodynamically unfavorable pathways. Electrochemical synthesis of hydrogen peroxide (H2O2) via the two-electron water oxidation reaction (2e− WOR) requires a +0.54 V higher potential than four-electron O2 evolution. So far, best-performing electrocatalysts require considerable overpotentials before reaching peak faradaic efficiency. We present Mn-alloyed TiO2 coatings prepared by atomic layer deposition (ALD) and annealing as a stable and selective electrocatalyst for 2e− WOR. Faradaic efficiency of >90% at < 150 mV overpotentials was achieved for H2O2 production, accumulating 2.97 mM H2O2 after 8 hours. Nanoscale mixing of Mn2O3 and TiO2 resulted in a partially filled, highly conductive Mn3+ intermediate band (IB) within the TiO2 mid-gap to transport charge across the (Ti,Mn)Ox coating. This IB energetically matched that of H2O2-producing surface intermediates, turning a wide bandgap oxide into a selective electrocatalyst capable of operating in the dark. However, the high selectivity is limited to the low overpotential regime, which limits the system to low current densities and requires further research into increasing turn-over frequency per active site.
1. Introduction
Hydrogen peroxide (H2O2) is an important green oxidant1 routinely used for chemical and environmental applications2 such as pulp bleaching,3 chemical-mechanical actuation,4 water treatment, and disinfection.5 In addition, H2O2 is receiving growing interest as a potential energy carrier, capable of long-duration energy storage.6 H2O2 is primarily produced by the anthraquinone oxidation process.2,7 However, this process involves sequential hydrogenation and oxidation steps which require energy-intensive gas transport and product separation—processes only economical at plant-scale, hindering distributed use for water treatment or disinfection. Direct synthesis of H2O2 from H2 and O2 over heterogeneous catalysts is one route for distributed synthesis,8 but recently, (photo-)electrochemical synthesis of H2O2 which uses abundant water or air as feedstock and renewable electricity or sunlight as energy inputs has provided an attractive alternative to the industrial anthraquinone-based process.9H2O2 can be (photo-)electrochemically synthesized via a two-electron (2e−) water oxidation pathway, as shown in eqn (1):
| 2H2O → H2O2 + 2H+ + 2e−, E = +1.77 VRHE | (1) |
However, existing catalysts often exhibit low faradaic efficiency (FE) due to the competitive four-electron (4e−) pathway of O2 evolution reaction (OER; eqn (2)):
| 2H2O → O2 + 4H+ + 4e−, E = +1.23 VRHE | (2) |
The thermodynamic driving force for OER is always at least +0.54 V greater than that of the 2e− H2O2 pathway which can allow the undesired O2 evolution pathway to overcome any kinetic barriers and dominate. In addition, the produced H2O2 can be over-oxidized to O2 at an excess potential of at least +1.09 V (eqn (3)):
| H2O2 → O2 + 2H+ + 2e−, E = +0.68 VRHE | (3) |
Finally, catalytic surfaces should not disproportionate the adsorbed H2O2 molecules:
| 2H2O2 → 2H2O + O2 | (4) |
Thus, the selectivity of 2e− water oxidation reaction (WOR) represents a common conundrum in electrocatalysis and strategies to address this challenge can be broadly applicable.9–11
Various water-oxidation materials have been both experimentally and theoretically investigated, including aluminum porphyrins,12 carbon-based materials,13 BiVO4,14,15 WO3,16 CaSnO3,17 and various composite materials.18 Several electrocatalytic19 and photoelectrocatalytic15 systems have showed faradaic efficiency (FE) values as high as 86%. However, best performing 2e− WOR catalysts ZnO20 and CaSnO3 (ref. 17) reach their peak FE at +2.6 VRHE and +3.2 VRHE, respectively, requiring considerable overpotentials to reach the 81% and 76% FE for H2O2 selectivity, respectively. Thus, strategies to achieve high selectivity at low overpotentials should be a major milestone for the field.
MnOx is a conductive oxide that has been used for H2O2 production.21 However, MnOx has not been successfully utilized in aqueous environments as it is capable of 4e− O2-evolution and has been shown to disproportionate H2O2. TiO2-rich surfaces may be promising for H2O2 production, since TiO2 is ineffective for either O2 evolution or H2O2 decomposition. Typically, TiO2 undergoes a 1e− pathway to produce OH˙, which then produces H2O2 or O2.22
In this study, the authors synthesize Mn-alloyed TiO2via atomic layer deposition (ALD), denoted as (Ti,Mn)Ox. The introduction of Mn 3d electronic states leads to the formation of a highly conductive Mn IB inside the otherwise forbidden bandgap of an oxide coating. Based on materials characterizations, we confirmed the nanoscale mixing of conductive and catalytically active phases otherwise not possible without ALD and post-growth annealing. We then investigated the effects of the electronic tuning of TiO2 and found that the nanocrystalline microstructure resulted in a multifunctional electrocatalyst for selective water oxidation to hydrogen peroxide.
2. Experimental methods
2.1 Atomic layer deposition
The composite (Ti,Mn)Ox films were grown by using 2, 4, 8, or 16 ALD cycles of TiO2 per 1 cycle of MnOx, which are denoted as 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 4:1, 8:1, and 16:1 (TiO2:MnOx) films (ESI S1†). Tetrakis-dimethylamido-titanium (TDMAT, Sigma-Aldrich, 99.999%) and bis-(ethyl-cyclopentadienyl) manganese (Mn(EtCp)2, Strem Chemicals, 99.999%) precursors for ALD were used as the Ti and Mn sources, respectively. The growth rates of MnOx and TiO2 were 0.97 and 0.47 Å per cycle, respectively. The MnOx cycle was placed in the middle of the TiO2 cycles. For example, 4:1 cycle ratio should lead to TiO2-rich surfaces with subsurface Mn atoms. The sequences for each film composition were repeated until each film reached the desired thickness of ca. 50 nm. All (Ti,Mn)Ox films were annealed in air at 500 °C for 2 hours and oven cooled. For electrocatalysis, fluorine-doped tin oxide (FTO) substrates were used, while fused silica substrates were used for material characterizations. The Mn elemental composition was correlated with, but not equal to, the nominal cycle ratio.
:1, 4:1, 8:1, and 16:1 (TiO2:MnOx) films (ESI S1†). Tetrakis-dimethylamido-titanium (TDMAT, Sigma-Aldrich, 99.999%) and bis-(ethyl-cyclopentadienyl) manganese (Mn(EtCp)2, Strem Chemicals, 99.999%) precursors for ALD were used as the Ti and Mn sources, respectively. The growth rates of MnOx and TiO2 were 0.97 and 0.47 Å per cycle, respectively. The MnOx cycle was placed in the middle of the TiO2 cycles. For example, 4:1 cycle ratio should lead to TiO2-rich surfaces with subsurface Mn atoms. The sequences for each film composition were repeated until each film reached the desired thickness of ca. 50 nm. All (Ti,Mn)Ox films were annealed in air at 500 °C for 2 hours and oven cooled. For electrocatalysis, fluorine-doped tin oxide (FTO) substrates were used, while fused silica substrates were used for material characterizations. The Mn elemental composition was correlated with, but not equal to, the nominal cycle ratio.
2.2 Electrochemical measurements
The electrochemical behavior of (Ti,Mn)Ox electrodes was measured with a three-electrode setup. A Bio-Logic S200 potentiostat system connected the (Ti,Mn)Ox working electrode, saturated Ag/AgCl reference electrode, and Ti foil counter electrode. The Ti counter electrode was chosen to keep consistent with a previous study.14 A cation exchange membrane separates the (TiMn)Ox and Ti foil electrodes to confine the H2O2 production in the working electrode compartment. Cyclic voltammetry was acquired with a scan rate of 20 mV s−1.2.3 H2O2 quantification
FE for H2O2 production was quantified by measuring the molar quantities of accumulated H2O2 produced by 4:1 (Ti,Mn)Ox as a function of current densities (geometric electrode area), ranging from 0.1 to 5.0 mA cm−2 (Fig. 2c). H2O2 concentration was quantified and validated using multiple titration methods in an H-type membrane cell (S1.5).† High-performance liquid chromatography (HPLC, Fig. S15†) quantification was used only to validate product identity and titration precision. Extended discussion can be found in ESI (S1.5 H2O2 quantification).†
2.4 XPS characterization of intermediate-band electronic structures
Valence band XPS was used to characterize the electronic structures of catalytic surfaces. The valence XPS spectra of the (Ti,Mn)Ox films were compared with that of a pure TiO2 film and used to derive the IB energy levels in a band diagram.23 Peak deconvolution was conducted under the following assumptions: (1) bandgap measurements for MnOx-alloyed TiO2 by UV-Vis spectroscopy (Fig. S9†) showed that (Ti,Mn)Ox bandgaps remain the same as pure TiO2 (2) extended X-ray absorption fine structure (EXAFS) characterizations (Fig. S10†) show the presence of Mn–Mn feature as the second coordination shell, indicating that MnOx forms clusters or nanocrystals inside the host oxide of crystallized TiO2. Therefore, we consider the 4:1 (Ti,Mn)Ox, coating has nanoscale mixing of rutile TiO2 and bixbyite Mn2O3 crystallites, and therefore contains two types of oxygen, one bridging neighboring Ti atoms and the other connecting neighboring Mn atoms. Each type has a set of non-overlapping O 2p–π and O 2p–σ peaks. Therefore, the peak deconvolution involved six peaks for 4:1(Ti,Mn)Ox.
Valence XPS analysis of pure TiO2 resulted in two main peaks. This analysis is discussed later in the paper and presented in Fig. 4. One peak occurred at the binding energy (BE) of 7.10 eV and the other peak occurred at 5.03 eV.24 The valence XPS spectra of 4:1 (Ti,Mn)Ox has two extra oxygen peaks that are slightly (∼0.3 eV) more negative to these two peaks. In addition to the oxygen peaks, two additional peaks were assigned to the Mn 3d states. The Mn 3d-t2g and Mn 3d-eg bands corresponded to the Mn 3d3/2 and Mn 3d5/2 peaks, respectively.25,26 The two primary O 2p peaks were still assigned at 7.10 ± 0.10 eV and 4.90 ± 0.10 eV, while the two Mn 3d peaks were located at 3.03 ± 0.10 eV and 1.14 ± 0.10 eV. All these peaks are referring to Fermi level position (EF, designated at zero binding energy using Au calibration standards, as shown in Fig. S11†).
3. Results & discussion
As deposited (Ti,Mn)Ox contains atomically mixed Mn and Ti atoms in an amorphous phase due to the 150 °C growth temperature.27 After annealing, diffraction peaks of 4:1 (Ti,Mn)Ox (Fig. 1a) indicate mixed nanocrystalline phases of rutile TiO2 (PDF#01-078-1510) and bixbyite Mn2O3 (PDF#00-041-1442). Bixbyite Mn2O3 represented the dominant phase for annealed 2:1 and 4:1 (Ti,Mn)Ox while rutile represented the dominant phase for annealed 8:1 and 16:1 (Ti,Mn)Ox as no obvious XRD peaks for Mn2O3 were observed. However, extended X-ray absorption fine structure analysis at the Ti and Mn edges of the annealed 4:1 (Ti,Mn)Ox (Fig. S10†) revealed distinct second coordination shells for both Ti and Mn elements, consistent with the nanocrystallinity of both TiO2 and Mn2O3 in the annealed coating.
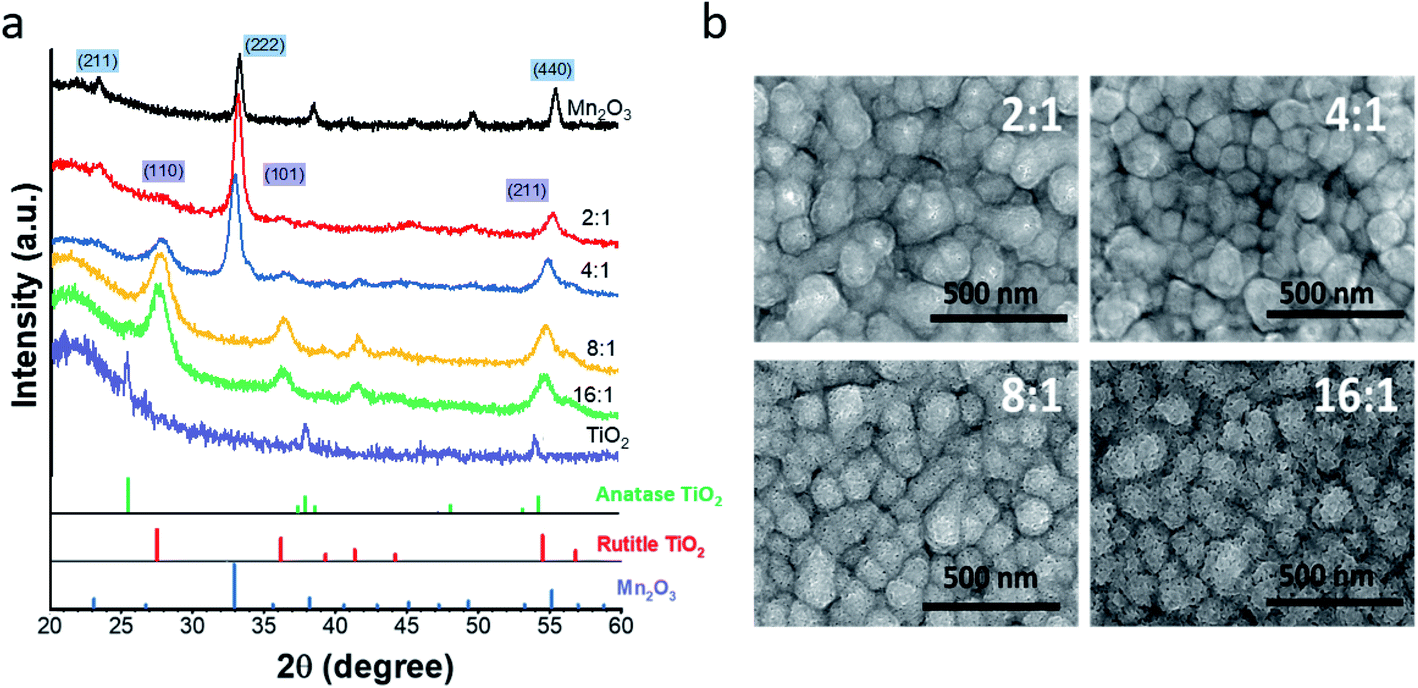 | ||
| Fig. 1 (a) Grazing incident X-ray diffraction (GI-XRD) spectra and (b) plan-view scanning electron microscope (SEM) images of annealed (Ti,Mn)Ox. Samples denoted Mn2O3 and TiO2 represent annealed ALD MnOx and TiOx, respectively. | ||
Thus, rutile TiO2 is present in 4:1 (Ti,Mn)Ox, and XRD analysis represents only a starting point for understanding the microstructure of (Ti,Mn)Ox, which is the topic of a further report. Diffraction peaks of annealed ALD TiOx without Mn alloying indicated anatase TiO2. Therefore, Mn-alloyed TiOx results in a tuned electronic structure due to a proportion of common anion mixed-metal bonds, which are distinctive from previously reported mixed Ti–Mn oxides.28,29
Plan-view (Fig. 1b) and cross-sectional SEM images of (Ti,Mn)Ox (Fig. S1†) indicate a conformal coating with a uniform thickness on the FTO substrate. TEM images (Fig. S4†) showed that Mn2O3 crystallites were uniformly distributed across the (Ti,Mn)Ox surface due to the diffusion of Mn atoms and Mn2O3 ripening during annealing. Image analysis indicated that the average sizes of Mn2O3 grains were 4.5 ± 0.5 and 7.7 ± 0.5 nm for 4:1 and 2:1 (Ti,Mn)Ox, respectively. The TEM images showed bending contours, suggesting residual strain in the annealed films. Williamson–Hall analysis of the GI-XRD data suggested a strained status of Mn2O3 crystallites with 0.5% lattice tension. The fitted crystallite sizes were consistent with the analysis of TEM bright-field images.
XPS core-level spectroscopy was used to quantify the surface composition of rutile TiO2 and bixbyite Mn2O3 (see Fig. S2†) and to verify the Mn3+ oxidation state (see Table S2 and ESI Results S2.1†). Angle-resolved X-ray photoelectron spectroscopy (XPS) indicated uniform compositional distribution of Ti and Mn from the surface to the bulk (Fig. S3†). The 4:1 (Ti,Mn)Ox has 36.09 at% of Mn within the 3 nm skin depth of the coating surface.
(J–E) behavior of (Ti,Mn)Ox in a phosphate-buffered (PB) solution of pH 7 (Fig. 2a), revealed that current density correlated with increasing Mn concentration. The films, except for 16:1 which was resistive due to sparse Mn concentration, share a similar on-set potential of approximately +1.8 VRHE. Annealed ALD MnOx showed no activity for H2O2 production and annealed TiO2 did not generate significant currents (Fig. S5†). Numerous O2 bubbles were observed on the annealed Mn2O3 control and at the cell sidewall. The redox feature of annealed MnOx cyclic voltammogram (Fig. S5b†) agrees with previous reports, which indicated the stable OER window at +1.6–1.8 VRHE and the formation of Mn(VII) species when the potential is above of +1.8 VRHE.30 These findings indicate that catalytic activity is dependent on alloying TiO2 with Mn2O3 nanocrystallites. Though 2:1 and 4:1 have similar J–E behavior, the crystal grain sizes for 2:1 (Ti,Mn)Ox of 7.7 ± 0.5 nm made it difficult to maintain a TiO2-rich surface. As surface MnOx compounds are known to disproportionate H2O2 in aqueous environments, 2:1 (Ti,Mn)Ox would pose challenges accumulating H2O2,31 and thus 4:1 (Ti,Mn)Ox was chosen as the focus of this study.
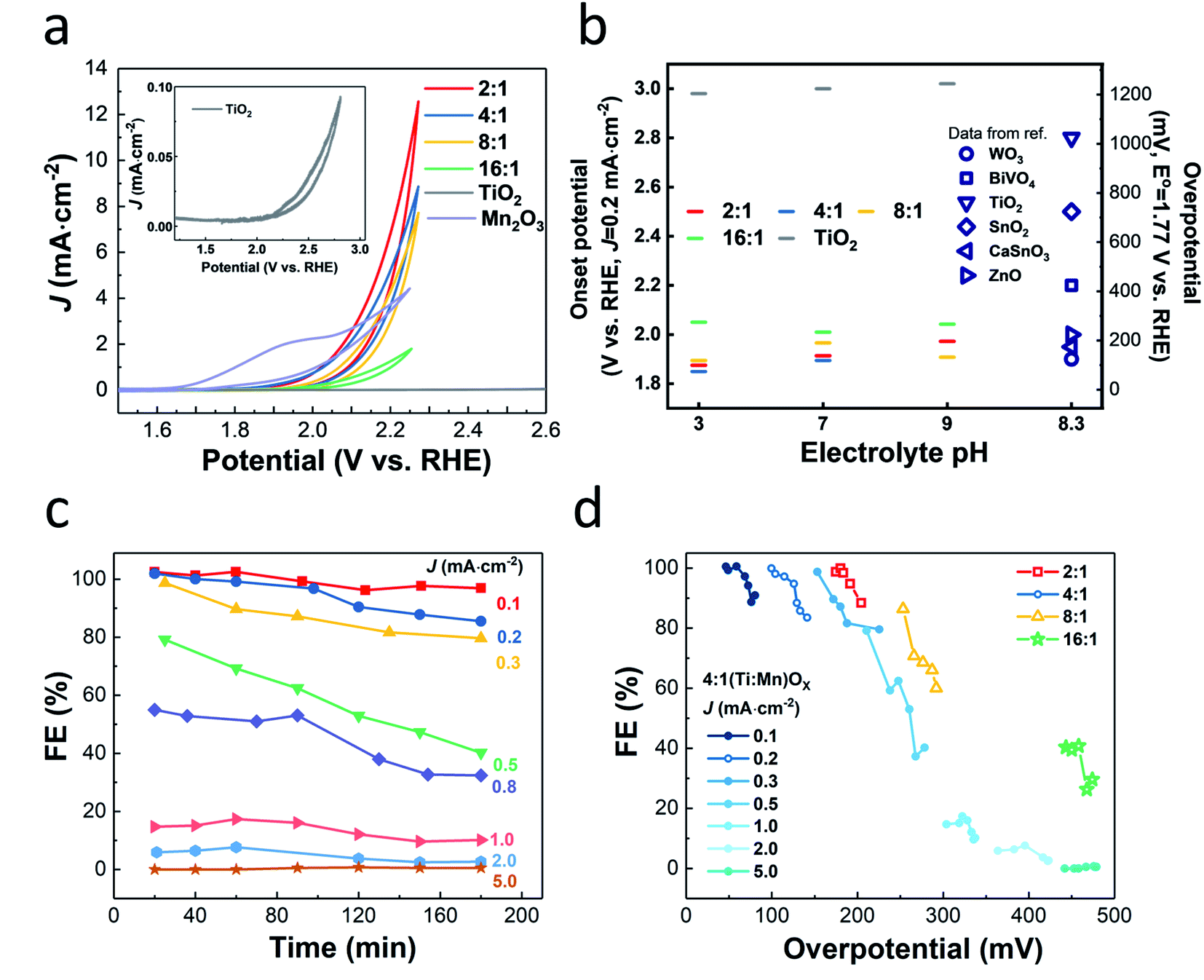 | ||
| Fig. 2 Electrochemical performance. (a) J–E behavior of (Ti,Mn)Ox in 0.5 M pH = 7 phosphate buffer at a scan rate of 20 mV s−1, (b) onset potentials at the respective J = 0.2 mA cm−2 (left y-axis) and overpotentials (right y-axis) derived from the (Ti,Mn)OxJ–E curves, plotted together with the data obtained from references of WO3, BiVO4, TiO2, and SnO2,10 ZnO,20 and CaSnO3.17 The measurement error for overpotentials is ± 0.02 V. (c) FE vs. electrolysis time using a 4:1 (Ti,Mn)Ox coating. (d) FE vs. overpotential at various current densities for the 4:1 (Ti,Mn)Ox coating in (c), overlaid with the FEs for 2:1, 8:1, and 16:1 (Ti,Mn)Ox films at J = 0.2 mA cm−2. | ||
J–E behavior measured in pH 3 and 9 PB solutions (Fig. S6†) showed similar trends, however, the pH 7 PB solution was chosen as the electrolyte for further testing to be compatible with reported membrane-free electrolyte-free H2O2 electrolysis designs that minimize transport loss.32
The onset potentials (at J = 0.2 mA cm−2) of 4:1 (Ti,Mn)Ox (Fig. 2b) was +1.89 VRHE, comparable to +1.90 and +1.96 VRHE for the 2:1 and 8:1 films, respectively. The onset potentials of the (Ti,Mn)Ox coatings are non-variant in changing pH (Fig. 2b) and follow the trend that 4:1 (Ti,Mn)Ox has the lowest onset potential. Onset potentials were lower than the many catalysts, including BiVO4 (+2.20 VRHE), TiO2 (+2.80 VRHE), SnO2 (+2.50 VRHE), and were comparable to WO3 (+1.90 VRHE).14 CaSnO3 reaches 0.2 mA cm−2 with an overpotential of 230 mV but requires +3.20 VRHE to reach peak Faraday efficiency of 76%.17 However, WO3 was reported to be unstable due to surface H2O2 absorption, which may lead to low efficiency in the long term.16
Computational work suggests that there are optimal potential windows for H2O2 production, due to the competition with one-electron and four-electron oxidation reactions.14 For 4:1 (Ti,Mn)Ox, FE values of over 95% were obtained at overpotentials less than 150 mV, i.e., at +1.9 VRHE, or at equivalent current densities of 0.1 mA cm−2. As FE begins near unity at just above +1.77 VRHE and decreases with increasing current density (Fig. 2c) or with increasing overpotentials, (Ti,Mn)Ox suffers from a small optimal potential window (Fig. 2d). However, this range of FE values was 20% higher than the highest reported FE of 66% for the hydrophobic carbon dark anode measured in NaHCO3(aq).14 The average FE of 98% at 0.1 mA cm−2 was comparable to reported photochemical results, such as a FE of 98% for BiVO4 photoanodes14 and a FE of 80% for BiVO4–Al2O3 photoanodes.33 However, when the current density was increased to 1.0 mA cm−2, corresponding to an overpotential of 250 mV, the FE dropped to less than 20%, and while this decline in FE is undesirable, it is consistent with other reports which show that selectivity can drop off with increased overpotentials.14 (Ti,Mn)Ox is unique as the measured FEs start >90% at near zero overpotentials and drop off while FE in other systems such as BiVO4 and ZnO ramp up, whereas the FEs in other systems such as BiVO4 and ZnO initially ramp up, reaching comparable selectivity only at >400 mV overpotentials.14,20
Electrochemical impedance spectroscopy (EIS) was used to correlate electrical conductivity with composition-dependent J–E behavior (Fig. S7 & Table S3†). Analyses showed that the charge-transport resistance through the coatings were 48.6, 47.2, 50.1, 78.0 Ω for the 2:1, 4:1, 8:1, and 16:1 films, respectively. The impedance data also provided an estimated roughness factor of 10.7, which is calculated by dividing the geometric area of the electrode by the electrochemical surface area (see ESI Results S2.2).†
Eight-hour electrolysis experiment using 4:1 (Ti,Mn)Ox in a batch reactor accumulated 2.97 mM of H2O2 in 20 mL, or 59.4 μmol (Table S4†). This accumulation was achieved in a static cell demonstrating that (Ti,Mn)Ox is capable of H2O2 desorption without overoxidation. As shown in Fig. 3, the cumulative FE begins near unity then gradually decreases to 85.8% after eight hours. However, this figure represents only the lower bound of FE as it does not account for product decomposition in the cell over the same period. As membranes have been reported to be highly effective at preventing H2O2 crossover between chambers, all of the H2O2 is accounted as being solely produced via WOR.34,35
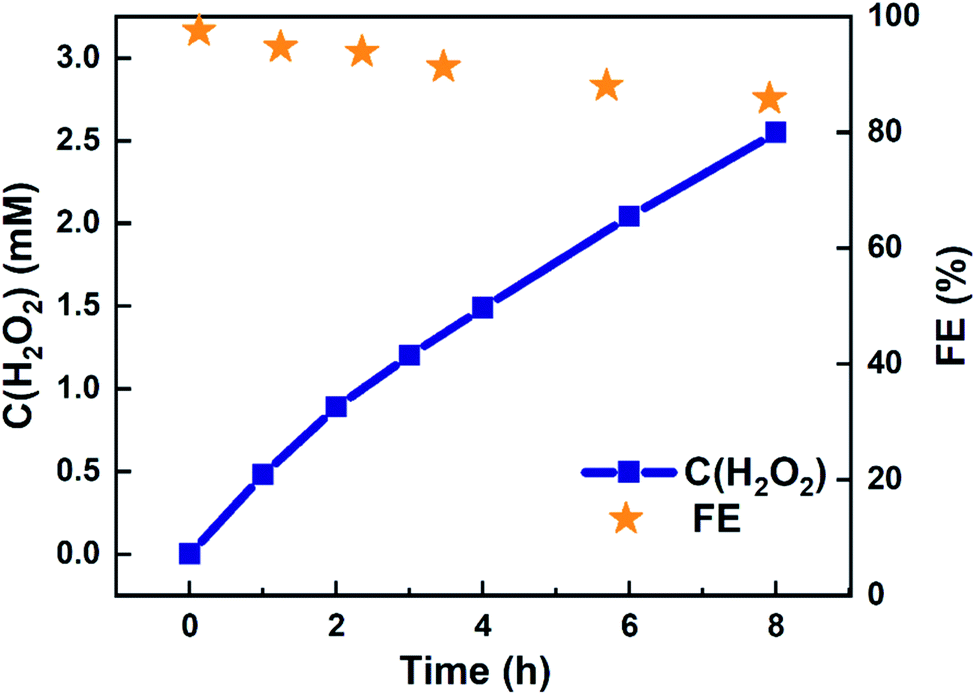 | ||
| Fig. 3 H2O2 accumulation using 4:1 (Ti,Mn)Ox-coated FTO electrode operated in the dark. Cumulative H2O2 concentration and the respective cumulative FE vs. time. The electrode area was 1.2 cm2, and the applied potential was +1.31 V vs. Ag/AgCl (equivalent to an overpotential of 150 mV) in a 20 mL 0.5 M pH = 7.0 PB solution. | ||
We further tested the long-term stability for 4:1 (Ti,Mn)Ox in PB solutions of pH 5, 7, and 9 to evaluate its potential as a protective layer. Chronopotentiometry studies at 0.2 mA cm−2 showed a moderate increase in overpotential, with the electrode potentials increasing from +1.8 VRHE to less than +2.1 VRHE after 200 hours (Fig. S8†). The potential increase was partially accelerated by the formation of O2 bubbles on the surface reducing the total surface area of the catalyst. As the chronopotentiometry experiment varies applied potential to sustain a current, this decrease in surface area led to an increase in required potentials during the constant current testing. As demonstrated in Fig. 2c, the selectivity towards H2O2 decreased at higher applied potentials, creating a feedback loop where higher applied potentials resulted in more oxygen generation. This process would slowly increase the required potential, until some breakdown voltage which would eventually lead to the failure of the coating. However, the coating was stable in all three pH conditions for over 200 hours, showing promise as protective coatings for semiconductors in corrosive environments.
The free energies of the relevant intermediates of the one, two, and four electron water oxidation reactions, i.e., OH* and O* free energy have been shown to be key in determining reaction pathways.14 While experimental evidence for the exact identity of the active site would allow for optimization and improved simulation, as recent literature has highlighted,36,37 it can be challenging identifying the nanocrystalline active sites, even with advanced microscopy and EXAFS analysis. TiO2 and MnOx are on opposite sides of this volcano plot of a OH*-overpotential scaling relationship, with TiO2 too large a OH* free energy and MnOx not large enough.38,39 Neither TiO2 nor Mn2O3 is active for hydrogen peroxide production (Fig. S5a and b†), demonstrating that Mn-alloying is the necessary for activity. TiO2 was chosen to be the surface termination layer as it was found not to disproportionate H2O2. Thus, (Ti,Mn)Ox promises favorable thermodynamics for selective peroxide production.
Valence XPS analyses indicated that a spatially continuous Mn-impurity IB was located within the TiO2 bandgap due to unfilled Mn 3d electronic states (see ESI Results S2.3†). The red arrows in Fig. 4b represent the relative difference in binding energy (BE) between the valence band maximum (VBM) and the Fermi level, namely EVBM − EF, while the blue arrows represent the difference of BE between the IB center and the Fermi level, EIB − EF. With the addition of Mn, the position of the EF shifted towards the VBM. For instance, EVBM − EF reduced from 3.20 V for pure TiO2 (i.e., EF at 0.14 VRHE) to 2.31 V for 4:1 (Ti,Mn)Ox films (i.e., EF at 1.09 VRHE). This EF lowering occurred because the Mn 3d states created inside the TiO2 bandgap hosted free carrier electrons.
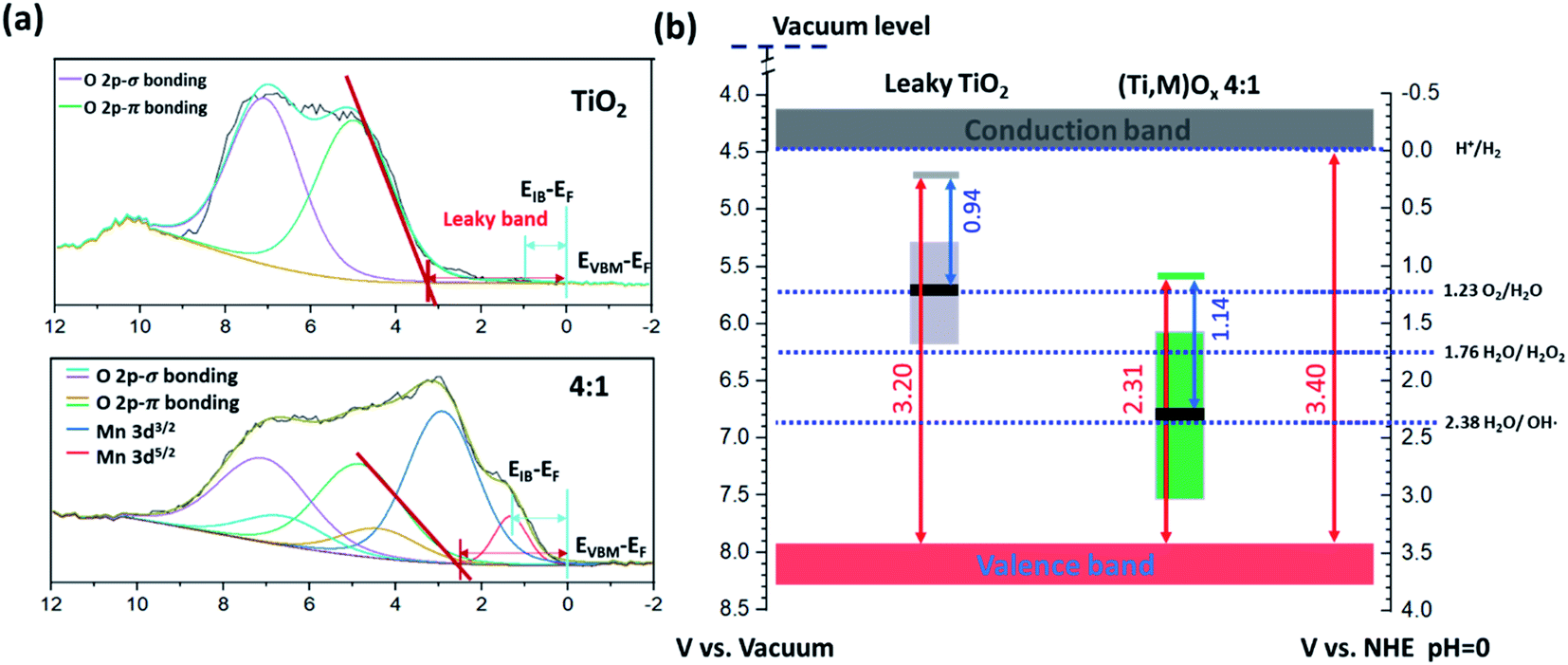 | ||
| Fig. 4 Electronic structure characterizations of (Ti,Mn)Ox catalytic coatings. (a) Valence XPS spectra and (b) respective band diagrams (in the potential scale versus Reversible Hydrogen Electrode, RHE) of 4:1 (Ti,Mn)Ox and “leaky” TiO2 surfaces. The positions of valence band maximum (EVBM), intermediate band center (EIB), and the Fermi level (EF) are indicated in (a), and their respective energetics are drawn in (b). EVBM − EF and EIB − EF are labelled and are listed in Table S1.† The height of colored boxes indicates the full width at half maximum (FWHM) of Ti3+-defect band (0.88 ± 0.10 eV) of “leaky” TiO2 quoted from ref. 36, and of the Mn intermediate bands (Mn 3d5/2) derived from the valence spectra shown in (a). The energetic error from XPS analyses is ±0.10 eV. | ||
Based on the valence XPS analyses, we assigned the potentials of band edges, Fermi levels, and IBs with respect to the RHE scale (right vertical axis), shown under flat-band conditions (Fig. 4b). At the liquid interface, the CB and VB edges of TiO2 were located at −0.05 and 3.29 VRHE, respectively.40 For (Ti,Mn)Ox the Mn IB center was 1.17 ± 0.11 eV above the VB edge or at 2.31 ± 0.11 VRHE. Under applied potentials, their relative positions were considered fixed.
Fig. 5a (“leaky TiO2”) was derived from a recent report (see ESI Discussions S3.1†), which was supported by direct in situ ambient-pressure XPS of the liquid junction interface of “leaky” TiO2 and KOH(aq).40 The band bending in the TiO2 is due to the positive and immobile space charge of their oxygen vacancies. The charge density of Mn2O3 crystallites, due to the point defects in Mn2O3 bixbyite, was at the degenerately p-type doping level due to the Mn vacancy defects in this class of p-type oxides. The Mn IB was always partially filled with electrons that were transferred from nearby rutile TiO2 crystallites. The high charge density of partially-filled Mn IB (>1021 cm−3) was supported by the 47–50 Ω resistance of (Ti,Mn)Ox for a range of Mn composition (26–51 at% Mn). The stoichiometric TiO2 achieved by ALD23 and the lower formation energy of Mn2O3 than TiO2 (ref. 41 and 42) ensured the metallic nature of the Mn IB states after annealing. The Mn2O3 nanocrystallites formed a conductive network throughout the (Ti,Mn)Ox coating.
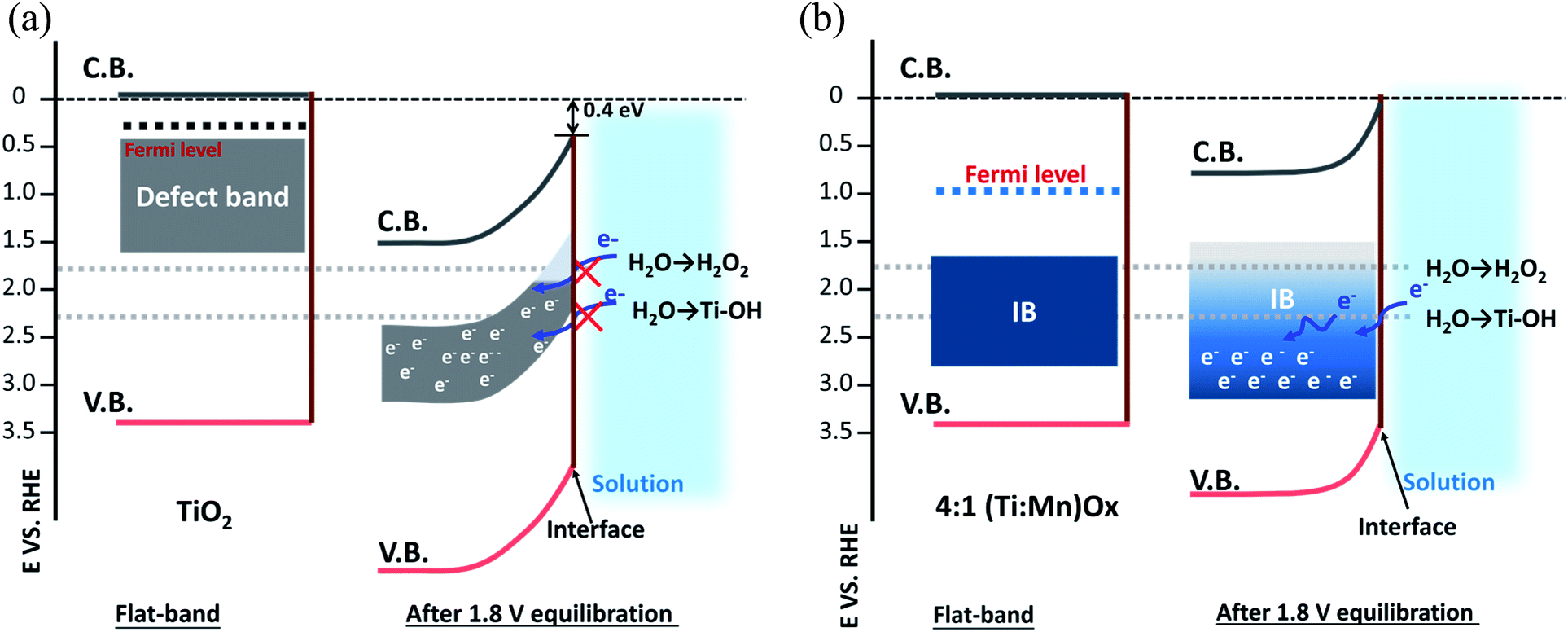 | ||
| Fig. 5 Interfacial band energetics of semiconductive electrocatalysts. (a) “Leaky” TiO2 and (b) 4:1 (Ti,Mn)Ox films. The left panel of each diagram was drawn under flat band conditions, and the right panel of each diagram was drawn where the electrode was poised at +1.8 VRHE during water oxidation. Box height was drawn based on the estimated top edge of the Ti3+-defect band or the Mn3+ intermediate band. | ||
Under bias, the band bending for TiO2 and Mn2O3 follow their respective properties of charge density and band positions at the liquid interface. As shown in Fig. 5a, applying +1.8 VRHE creates a band bending potential profile. This is because the ionic charge density of 1021 to 1023 cm−3 is typically observed at the TiO2-liquid interfaces.43 For the “leaky” TiO2 with a Ti3+-defect band, the positive space-charge density was calculated to be 2.1 × 1020 cm−3, but its free-electron concentration was less than 1017 cm−3 due to electron trapping by the Ti3+-defect band.44 In contrast, the metallic Mn2O3 should accumulate holes under the applied potential. Accordingly, the Mn IB shifted its potential energy downward to align with the applied potential. As shown in Fig. 5b, at +1.8 VRHE, the in-gap Mn states shifted to align with the EF of Mn2O3 crystallites of (Ti,Mn)Ox due to their metallic behavior. Higher overpotentials would lead to increased band bending for the TiO2 host oxide, but a shift in the position of the Mn intermediate band. This may enable other oxidation pathways, resulting in parasitic anodic current and an overall decrease in selectivity.
Under the applied potential of +1.8 VRHE there exists a small and positive overpotential to the 2e− water oxidation for H2O2 formation. Under such a small overpotential, the electron potential of the (Ti,Mn)Ox coatings lies more positive than the formal potential of the H2O2/H2O couple, i.e., 24 mV positive to E(H2O2/H2O). During operation, the energy levels of CB and VB of TiO2 are fixed, or “pinned”, at −0.05 and 3.29 VRHE, respectively, and due to the semiconductor-like band bending of the TiO2 bulk and the “unpinning” behavior of Mn-IB at the liquid-junction interface, the empty states of the Mn IB can precisely align with the energetics of H2O2 producing intermediates. Under a higher applied potential, i.e., +2.3 VRHE, there would exist a moderate overpotential for the 2e− water oxidation, but such a potential also enables the other pathways including 1e− water oxidation to produce radicals, leading to an overall reduction in H2O2 production rates due to the reduced selectivity. Thus, the strategy for tuning the intermediate band towards highly selective electrocatalysis is best suited for the desired low-overpotential electrolysis and other strategies are therefore necessary for increasing geometric current density and, eventually, turnover frequency per site.
4. Conclusion
This study offered a theory-supported design of electrocatalytic surfaces that favor a H2O2 pathway. The authors design a surface that does not exhibit favorable kinetics for O2 evolution, over-oxidize H2O2, nor disproportionate the as-produced H2O2. The mixed TiO2/Mn2O3 nanocrystallites are chemically stable and protected by the TiO2-rich surface. (Ti,Mn)Ox coatings have been shown to be stable over 200 hours in modestly acidic and modestly basic solutions suggesting that those (Mn,Ti) sites are not as labile and soluble as the ordinary MnOx sites. Their acid stability is comparable to the ordered ternary phases of recently reported Mn-based acid stable catalysts.45This study elucidates a new strategy of introducing charge-transport IBs and led to the design of H2O2-producing surfaces that oxidize water to H2O2 with high selectivity at very low overpotentials. The Mn3+-impurity IBs were created within the otherwise forbidden bandgap of TiO2 which enhanced the electrical conductivity across the coating and matched the electronic states for H2O2-producing surface intermediates. Given the growing literature related to H2O2 selective catalysis, this approach is expandable to a range of host oxides that prevent H2O2 disproportionation to favor 2e− H2O2 electrochemistry. However, this strategy is limited to the low overpotential regime, which limits the system to low current densities and requires further molecular or materials discovery for increasing turn-over frequency per active site.11
Author contributions
S. H. conceived the project. J. L., S. H., and Q. Z. designed the experiments. X. S. fitted and analyzed the valence XPS. J. L., G. C., and D. S., performed catalytic reactions and titration experiments. J. L., D. S., and Q. Z. performed XRD, XPS, and TEM characterizations and data analyses. J. L., S. H., X. S., Q. Z., and D. S. analyzed the data and co-wrote the manuscript. All co-authors discussed the results, finalized the conclusions, and edited the manuscript.Conflicts of interest
Yale University has filed a provisional U.S. patent application directly related to the work described in this paper (patent application no. 62/843856, filed on 6 May 2019).Acknowledgements
The authors would like to thank Dr Min Li at Yale's Materials Characterization Core (MCC) for his invaluable help with SEM, GIXRD, and XPS characterizations. The authors would like to acknowledge the start-up support from the Tomkat Foundation. J. L. would like to acknowledge the China Scholarship Council (CSC) for its financial support and the Donghua University Doctoral Innovation Fund Program (17D310606, 106-06-0019058).3References
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- R. Hage and A. Lienke, Angew. Chem., Int. Ed., 2006, 45, 206–222 CrossRef CAS PubMed.
- M. Wehner, R. L. Truby, D. J. Fitzgerald, B. Mosadegh, G. M. Whitesides, J. A. Lewis and R. J. Wood, Nature, 2016, 536, 451–455 CrossRef CAS PubMed.
- K. Kosaka, H. Yamada, K. Shishida, S. Echigo, R. A. Minear, H. Tsuno and S. Matsui, Water Res., 2001, 35, 3587–3594 CrossRef CAS.
- S. Fukuzumi, Y. Yamada and K. D. Karlin, Electrochim. Acta, 2012, 82, 493–511 CrossRef CAS PubMed.
- G. Goor, J. Glenneberg, S. Jacobi, J. Dadabhoy and E. Candido, Ullmann's Encycl. Ind. Chem., 2019, 1–40, DOI:10.1002/14356007.a13_443.pub3.
- D. W. Flaherty, ACS Catal., 2018, 8, 1520–1527 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal and T. W. Hansen, Nat. Mater., 2013, 12, 1137 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- J. Tang, T. Zhao, D. Solanki, X. Miao, W. Zhou and S. Hu, Joule, 2021, 5, 1432–1461 CrossRef CAS.
- S. Mathew, F. Kuttassery, S. N. Remello, A. Thomas, D. Yamamoto, S. Onuki, Y. Nabetani, H. Tachibana and H. Inoue, ChemPhotoChem, 2018, 2, 240–248 CrossRef CAS.
- Y. Ando and T. Tanaka, Int. J. Hydrogen Energy, 2004, 29, 1349–1354 CrossRef CAS.
- X. Shi, S. Siahrostami, G.-L. Li, Y. Zhang, P. Chakthranont, F. Studt, T. F. Jaramillo, X. Zheng and J. K. Nørskov, Nat. Commun., 2017, 8, 701 CrossRef PubMed.
- K. Zhang, J. Liu, L. Wang, B. Jin, X. Yang, S. Zhang and J. H. Park, J. Am. Chem. Soc., 2020, 142, 8641–8648 CrossRef CAS PubMed.
- J. C. Hill and K.-S. Choi, J. Phys. Chem. C, 2012, 116, 7612–7620 CrossRef CAS.
- S. Y. Park, H. Abroshan, X. Shi, H. S. Jung, S. Siahrostami and X. Zheng, ACS Energy Lett., 2019, 4, 352–357 CrossRef CAS.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 1801909 CrossRef.
- C. Xia, S. Back, S. Ringe, K. Jiang, F. Chen, X. Sun, S. Siahrostami, K. Chan and H. Wang, Nat. Catal., 2020, 3, 125–134 CrossRef CAS.
- S. R. Kelly, X. Shi, S. Back, L. Vallez, S. Y. Park, S. Siahrostami, X. Zheng and J. K. Nørskov, ACS Catal., 2019, 9, 4593–4599 CrossRef CAS.
- A. Izgorodin, E. Izgorodina and D. R. MacFarlane, Energy Environ. Sci., 2012, 5, 9496–9501 RSC.
- J. Zhang and Y. Nosaka, J. Phys. Chem. C, 2014, 118, 10824–10832 CrossRef CAS.
- P. Nunez, M. H. Richter, B. D. Piercy, C. W. Roske, M. Cabán-Acevedo, M. D. Losego, S. J. Konezny, D. J. Fermin, S. Hu, B. S. Brunschwig and N. S. Lewis, J. Phys. Chem. C, 2019, 123, 20116–20129 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, J. Phys. Chem. Solids, 2002, 63, 1909–1920 CrossRef CAS.
- V. E. Henrich and P. A. Cox, The Surface Science of Metal Oxides, Cambridge University Press, Cambridge, 1994 Search PubMed.
- R. J. Lad and V. E. Henrich, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 38, 10860–10869 CrossRef CAS PubMed.
- G. Siddiqi, Z. Luo, Y. Xie, Z. Pan, Q. Zhu, J. A. Röhr, J. J. Cha and S. Hu, ACS Appl. Mater. Interfaces, 2018, 10, 18805–18815 CrossRef CAS PubMed.
- K. L. Pickrahn, A. Garg and S. F. Bent, ACS Catal., 2015, 5, 1609–1616 CrossRef CAS.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 5054–5058 CrossRef CAS PubMed.
- A. Plauck, E. E. Stangland, J. A. Dumesic and M. Mavrikakis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E1973 CrossRef CAS PubMed.
- S. Hu, Sustainable Energy Fuels, 2019, 3, 101–114 RSC.
- X. Shi, Y. Zhang, S. Siahrostami and X. Zheng, Adv. Energy Mater., 2018, 1801158 CrossRef.
- E. Lobyntseva, T. Kallio, N. Alexeyeva, K. Tammeveski and K. Kontturi, Electrochim. Acta, 2007, 52, 7262–7269 CrossRef CAS.
- Q. Zhu, M. Hinkle, D. J. Kim and J.-H. Kim, ACS ES&T Engg, 2021, 1, 446–455 Search PubMed.
- K. Feng, H. Zhang, J. Gao, J. Xu, Y. Dong, Z. Kang and J. Zhong, Appl. Phys. Lett., 2020, 116, 191903 CrossRef CAS.
- L. Liu and A. Corma, Nat. Catal., 2021, 4, 453–456 CrossRef.
- S. Siahrostami, G.-L. Li, V. Viswanathan and J. K. Nørskov, J. Phys. Chem. Lett., 2017, 8, 1157–1160 CrossRef CAS.
- V. Viswanathan, H. A. Hansen and J. K. Nørskov, J. Phys. Chem. Lett., 2015, 6, 4224–4228 CrossRef CAS PubMed.
- M. F. Lichterman, S. Hu, M. H. Richter, E. J. Crumlin, S. Axnanda, M. Favaro, W. Drisdell, Z. Hussain, T. Mayer, B. S. Brunschwig, N. S. Lewis, Z. Liu and H.-J. Lewerenz, Energy Environ. Sci., 2015, 8, 2409–2416 RSC.
- N. Birkner and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E1046 CrossRef CAS PubMed.
- M. R. Ranade, A. Navrotsky, H. Z. Zhang, J. F. Banfield, S. H. Elder, A. Zaban, P. H. Borse, S. K. Kulkarni, G. S. Doran and H. J. Whitfield, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6476 CrossRef CAS PubMed.
- P. N. R. L. J. Lipkowski, The Electrochemistry Of Novel Materials (Frontiers In Electrochemistry, Wiley-VCH, 1993 Search PubMed.
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005 CrossRef CAS PubMed.
- L. Zhou, A. Shinde, J. H. Montoya, A. Singh, S. Gul, J. Yano, Y. Ye, E. J. Crumlin, M. H. Richter, J. K. Cooper, H. S. Stein, J. A. Haber, K. A. Persson and J. M. Gregoire, ACS Catal., 2018, 8, 10938–10948 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta05451a |
| ‡ Jiahui Li and Devan Solanki contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |