
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGalbofloxacin: a xenometal-antibiotic with potent in vitro and in vivo efficacy against S. aureus†
Apurva
Pandey
,
Dariusz
Śmiłowicz
and
Eszter
Boros
 *
*
Department of Chemistry, Stony Brook University, 100 Nicolls Road, Stony Brook, New York 11790, USA. E-mail: eszter.boros@stonybrook.edu
First published on 19th October 2021
Abstract
Siderophore-antibiotic drug conjugates are considered potent tools to deliver and potentiate the antibacterial activity of antibiotics, but only few have seen preclinical and clinical success. Here, we introduce the gallium(III) complex of a ciprofloxacin-functionalized linear desferrichrome, Galbofloxacin, with a cleavable serine linker as a potent therapeutic for S. aureus bacterial infections. We employed characterization using in vitro inhibitory assays, radiochemical, tracer-based uptake and pharmacokinetic assessment of our lead compound, culminating in in vivo efficacy studies in a soft tissue model of infection. Galbofloxacin exhibits a minimum inhibitory concentration of (MIC98) 93 nM in wt S. aureus, exceeding the potency of the parent antibiotic ciprofloxacin (0.9 μM). Galbofloxacin is a protease substrate that can release the antibiotic payload in the bacterial cytoplasm. Radiochemical experiments with wt bacterial strains reveal that 67Galbofloxacin is taken up efficiently using siderophore mediated, active uptake. Biodistribution of 67Galbofloxacin in a mouse model of intramuscular S. aureus infection revealed renal clearance and enhanced uptake in infected muscle when compared to 67Ga-citrate, which showed no selectivity. A subsequent in vivo drug therapy study reveals efficient reduction in S. aureus infection burden and sustained survival with Galbofloxacin for 7 days. Ciprofloxacin had no treatment efficacy at identical molecular dose (9.3 μmol kg−1) and resulted in death of all study animals in <24 hours. Taken together, the favorable bacterial growth inhibitory, pharmacokinetic and in vivo efficacy properties qualify Galbofloxacin as the first rationally designed Ga-coordination complex for the management of S. aureus bacterial infections.
Introduction
Infectious diseases are one of the leading causes of death worldwide.1 Among infective agents, the Gram-positive pathogen Staphylococcus aureus (S. aureus) can trigger a variety of pathologies, such as skin and soft tissue infections, endocarditis, osteomyelitis, lethal pneumonia, and causes the majority of hospital acquired bacterial infections and deaths in the United States.2,3 Depending on the susceptibility of pathogen towards currently used antibiotics, S. aureus can be divided into MSSA (methicillin susceptible S. aureus) and MRSA (methicillin resistant S. aureus).4 In recent decades, the opioid epidemic and the over-prescription of antibiotics, has resulted in rapid emergence of drug resistant S. aureus worldwide. The incidence of infection requiring hospital care has increased, yet clinical treatment of MRSA has become more challenging. The emergence of bacteria with resistance to currently used antibiotics, combined with the paucity of new compounds in the pipeline necessitates the development of targeted novel antibiotics with new mechanisms of action.5,6Bacterial virulence is closely associated with nutrient acquisition, which is essential for bacterial growth and proliferation.7 During an infection, iron – an indispensable micronutrient, essential for a plethora of cellular reactions in both humans and pathogens, is restricted for bacterial uptake as the available iron is tightly sequestered by host proteins such as – hemoglobin, transferrin and ferritin.7 Bacteria rely on scavenging essential transition metal ions from their host, specifically iron in the form of ferrous/ferric (Fe2+/Fe3+). The low solubility of Fe(OH)3 (Ksp = 6.3 × 10−38) at pH 7.4 results in an insufficient quantity of iron for bacteria to grow. Given the essential role of iron (Fe) in bacterial physiology and pathogenicity, and the ability of bacteria to actively transport essential nutrient suggests that the iron uptake and metabolism pathway can be highjacked for development of new theranostic agents that can circumvent the membrane permeability barrier of bacteria.8 Consequently, to maintain the Fe homeostasis and counteract iron limitation, bacteria have developed iron uptake strategies, by producing low molecular weight, water soluble iron chelators called siderophores.9
Siderophore-antibiotic conjugates are a compound class with a long history of efficacy against various bacterial strains. These conjugates act as Trojan horses for delivery of antibiotics across the bacterial membrane, taking advantage of the Fe assimilation pathway in bacteria.10–16 In 2019, the FDA (Food and Drug Administration) approved cefiderocol (Fig. 2), a siderophore based antibiotic conjugate for treatment of complicated urinary tract infections (cUTI).17 The conjugate consists of a bidentate catechol siderophore moiety that promotes the formation of chelated complexes with ferric iron and thereby facilitates siderophore-like transport across the outer bacterial membrane and a beta lactam-based cephalosporin drug counterpart.18 The conjugate is active against carbapenem-resistant Enterobacteriaceae (CRE) and extended-spectrum beta-lactamases (ESBLs) producing bacteria.19,20 However, the bidentate coordinating donor site limits recognition by a large fraction of Fe-siderophore transport proteins, especially those accessing the cytoplasm.21,22
While the coordinating moiety is not essential for periplasm targeting antibiotics such as cefiderocol, it could significantly limit siderophore-mediated delivery of antibiotics that interact with a cytoplasmic target. In this regard, a more complete, tetra- or even hexacoordinate, siderophore-like motifs are desired, as they are more efficiently recognized and able to be transported to the cytoplasm (Fig. 1). However, the successful demonstration of in vivo potency of other hexadentate siderophore antibiotic conjugates remains scarce.23,24 Two primary reasons emerge as the possible cause for this significant knowledge and data gap: (1) a common hypothesis for the lack of translatability of these constructs due to their need for an iron depleted environment for efficacy and (2) a sensitivity to premature proteolytic degradation of hexadentate siderophores in mammalian plasma.
 | ||
| Fig. 1 Proposed transport internalization pathway for ferrichrome based Galbofloxacin in wt S. aureus using active transmembrane transport. | ||
Previously, we introduced the Ga3+ complex of the hexadentate, ornithine-derived tripeptide siderophore linear desferrichrome (LDFC) linked to the FDA approved antibiotic, ciprofloxacin via a beta-alanine linker, Ga-D2.25 We showed that the complexation with the xenometal Ga(III) produces enhanced potency when compared with the apo-(metal free) or endogenous metal ion (Fe)-bound siderophore conjugate.25 Our work was inspired by the structure of the natural product and antibiotic albomycin (Fig. 2) and the recent clinical success of Ga(NO3)3 (ref. 26–29) in a phase 1 trial for treatment of Pseudomonas aeruginosa (P. aeruginosa) lung infections in adults with cystic fibrosis (CF).28 Ga(NO3)3 imparted significant therapeutic effects.30 However, the drug was dosed at a high concentration of 200 mg per m2 per day for 5 days, so we posited that delivery of the Ga cargo with a siderophore-conjugate could significantly reduce dosing needs. Indeed, our first-generation construct Ga-D2 (Fig. 2) demonstrated good, broad spectrum potency in E. coli, S. aureus, P. aeruginosa and Klebsiella pneumoniae (K. pneumoniae) with a minimum inhibitory concentration for 90% inhibition of growth (MIC90) of 0.2, 1.9, 3.8 μM and 12.5 μM, respectively but fell short of exceeding the parent antibiotic's potency. Using a medicinal inorganic chemistry approach, we sought to further potentiate the activity of the construct.
 | ||
| Fig. 2 Chemical structures of (top panel) structurally related hydroxamate siderophore-antibiotic conjugates synthesized and investigated previously, as well as subject of studies in this work: D2, D5, albomycin (Fe-D3), a “Trojan horse” conjugate produced by Streptomyces. (Bottom panel) Enterobactin–ciprofloxacin, mixed ligand biscatecholate–monohydroxamate siderophore–carbacephalosporin conjugate, 1b and cefiderocol, FDA approved siderophore based “Trojan horse” antibiotic. Black: metal ion, red: siderophore, green: linker, blue: antibiotic. | ||
Here, we introduce the second generation Trojan horse compound, Ga-D5 (Galbofloxacin) as a ciprofloxacin-bearing mimic of albomycin natural product with a serine linker and explore its potential for a concerted diagnosis and therapy approach (Fig. 2). We incorporated a protease-sensitive serine linker in close analogy to the parent natural product albomycin.31–34 We demonstrate that this structural modification is critical to further enhance the in vitro and in vivo antibiotic activity of Galbofloxacin against S. aureus beyond the potency of the parent antibiotic ciprofloxacin.
Experimental section
Materials and methods
All starting materials were purchased from Acros Organics, Alfa Aesar, Sigma Aldrich, or TCI America and used without further purification. NMR spectra (1H and 13C) were collected on a 700 MHz Advance III Bruker, 500 MHz or 400 MHz Bruker instrument at 25 °C and processed using TopSpin 3.5pl7. 19F NMR were collected on a 400 MHz Bruker instrument at 25 °C using TFA as an internal standard (δ: −76 ppm). Chemical shifts are reported as parts per million (ppm). Mass spectrometry: low-resolution electrospray ionization (ESI) mass spectrometry and high-resolution (ESI) mass spectrometry was carried out at the Stony Brook University Institute for Chemical Biology and Drug Discovery (ICB&DD) Mass Spectrometry Facility with an Agilent LC/MSD and Agilent LC-UV-TOF spectrometers, respectively. UV-VIS spectra were collected with the NanoDrop 1C instrument (AZY1706045). Spectra were recorded from 190 to 850 nm in a quartz cuvette with 1 cm path length. HPLC: preparative HPLC was carried out using a Shimadzu HPLC-20AR equipped with a Binary Gradient, pump, UV-Vis detector, manual injector on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed). Method A (preparative purification method): A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. Method B (preparative purification method): A = 10 mM sodium acetate (pH = 4.5) in water, B = 100% MeCN. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. Analytical HPLC analysis was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-Vis detector, autoinjector and Laura radio detector on a Gemini-NX C18 column (100 mm × 3 mm, 110 Å, AXIA packed). Method C: A = 0.1% TFA in water, B = 0.1% TFA in MeCN with a flow rate of 0.8 mL min−1, UV detection at 254 and 280 nm. Gradient: 0–2 min: 95% A. 2–14 min: 5–95% B gradient. 14–16 min: 95% B. 16–16.6 min: 95–5% B. 16.5–20 min: 5% B. Method D (analysis of 67Ga/Ga complexes): A = 10 mM sodium acetate (pH = 4.5) in water, B = 100% MeCN. Gradient: 0–2 min: 95% A. 2–14 min: 5–95% B gradient. 14–16 min: 95% B. 16–16.6 min: 95–5% B. 16.5–20 min: 5% B, UV detection at 254 and 280 nm. Purity of all intermediates and final products including radiochemical species was determined using analytical HPLC. All conjugates and complexes were ≥95% pure. ICP-OES was carried out using an Agilent 5110 ICP-OES. A 10-point standard with respect to gallium and iron was used and lines of best fit were found with R2 of 0.999. IVIS Lumina Series II from Caliper LifeSciences small animal imager was used to image mice and S. aureus Xen 29 MIC plates. All scans were collected with blocked excitation filter, open emission filter (from 515 nm to 840 nm) and collection time t = 5 minutes. Images were analyzed with Living Image software version 4.3.1. Regions of interest were determined in quintuplicate with the ROI tool for each concentration as well as a general background ROI for background correction. 5-(N-Acetyl-N-acetoxyamino)-2-{5-(N-acetyl-N-acetoxy-amino)-2-[5-(N-acetyl-N-acetoxyamino)-2-(benzyloxy-carbonylamino)valerylamino]valerylamino}valeric acid (P5) and 7-(4-β-alanyl-1-piperazinyl)-1-cyclopropyl-6-fluoro-4-oxo-1H-quinoline-3-carboxylic acid (fragment A) were synthesized according to previously published procedures.25 A detailed account of the chemical synthesis of D5, including NMR spectral assignments, HPLC traces, HRMS and corresponding data is provided in the ESI (Fig. S1–S17†).Synthesis of coordination complexes of D5
Ga and Fe complexes of D5 were synthesized using the following protocol. Galbofloxacin. D5 (0.010 g, 0.009 mmol, 1 eq.) was dissolved in DMF (1 mL) and Ga(NO3)3 (0.007 g, 0.029 mmol, 3 eq.) dissolved in 10 mM CH3COONa (pH = 5) was added. The pH of the solution was adjusted to 6 by adding 0.1 M NaOH. The reaction mixture was stirred for 1 h at 60 °C and overnight at room temperature. Solvent was removed in vacuo and product was purified by preparative HPLC (Method A, product elutes at 46% B) to afford Ga-D5/Galbofloxacin (0.006 g, 0.005 mmol, 62%) as a white solid. Calculated mass for Ga-D5/Galbofloxacin (C44H61FGaN11O15): 1071.3; found 1072.4 [M + H]+. Retention time (method D): 7.13 min (purity/peak area: >99%). Fe-D5. D5 (0.005 g, 0.004 mmol, 1 eq.) was dissolved in DMF (1 mL) and FeCl3 (0.002 g, 0.012 mmol, 3 eq) was added. The reaction mixture was stirred for 1 h at room temperature. Solvent was removed in vacuo and product was purified by preparative HPLC (method A, product elutes at 48% B) to afford Fe-D5 (0.006 g, 0.003 mmol, 87%) as a red solid. Calculated mass for Fe-D5 (C44H61FFeN11O15): 1058.3; found 1059.3 [M + H]+. Retention time (method D): 7.10 min (purity/peak area: >99%).Antimicrobial activity assay
Antibacterial activity of apo-D5, its corresponding Fe and Ga complex and fragment A was determined by measuring their minimum inhibitory concentrations (MIC) using the broth microdilution method according to the Clinical and Laboratory Standards Institute (CLSI) guidelines. All aqueous solutions and media were prepared using deionized water. All liquids and media were sterilized by autoclaving (220 °C, 1 h) before use. Iron deficient Mueller–Hinton broth (MHB) (cation adjusted) was used for these assays. 5.25 g of MHB was added to 250 mL of deionized water. Aqueous solutions of 1 M Ca2+ (0.418 mL) and 1 M Mg2+ (0.155 mL) to 250 mL of MHB was added to the broth. The broth was autoclaved for 1 h at 220 °C. To make the broth iron-deficient, 4.06 mL of 1 mg mL−1 sterile (autoclaved) aq. solution of 2,2′-bipyridine (DP) was added to 250 mL of cation adjusted MHB. MIC experiments were conducted in triplicate and carried out at 3 different instances, resulting in n = 9 for each reported MIC98, to ascertain a maximum error margin of <5% for each reported MIC value.In general, 0.3 mM stock solutions of testing compounds were prepared. 10 μL solution of testing compound (0.3 mM) was added to the first well of the 96-well plate and serial dilutions were made down each row of the plate. 40 μL of growth media and 50 μL of diluted bacterial inoculum was also added to each well, resulting in a total volume of 100 μL and a concentration gradient of 0.3 × 10−4 M to 0.92 × 10−12 M. The dilution rate was adjusted keeping in mind the MIC of ciprofloxacin (positive control). The plates were incubated at 37 °C for 18 h and each plate was examined for bacterial growth using a plate reader (EPOCH2NS, Biotek microplate spectrometer). The MIC was recorded as the lowest compound concentration (μM) required to inhibit >90% of bacterial growth as judged by the absorbance of the culture media relative to the negative control.
Cytotoxicity experiments
HEK-293 cells were grown in MEM with 1% sodium pyruvate, 1% L-glutamine, 100 units per mL Pen Strep and 10% Fetal Bovine Serum. The cells were maintained at 37 °C in a humidified incubator under an atmosphere containing 5% CO2. Modified Eagle's Medium (MEM), containing 10% fetal calf serum, 1% penicillin and streptomycin, was used as growth medium. HEK-293 cells were detached from the wells with trypsin and EDTA, harvested by centrifugation and re-suspended again in cell culture medium. The assays were carried out on 96 well plates with 6000 cells per well for normal human embryonic kidney cells (HEK-293). After 24 h of incubation at 37 °C and 5% CO2, the cells were treated with the compounds (with final DMSO concentrations of 0.5%) with a final volume of 200 mL per well. For a negative control, one series of cells was left untreated. The cells were incubated for 48 h followed by adding 50 mL MTT (2.5 mg mL−1). After an incubation time of 2 h, the medium was removed and 200 mL of DMSO were added. The formazan crystals were dissolved, and the absorption was measured at 550 nm, using a reference wavelength of 620 nm. Each test was repeated in quadruplicates in three independent experiments. Results were plotted as % cell viability at different concentrations of samples tested.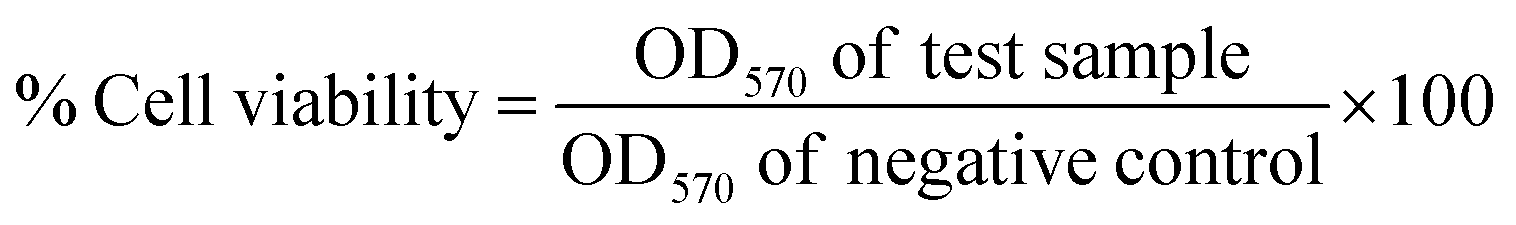
In vitro cleavability assessment
In vitro cleavage of D5 and Galbofloxacin was performed using a proteinase K enzyme assay. Conjugates (10 μM) were incubated with proteinase K (20×, 200 μM, dissolved in Tris buffer, pH = 7) at 37 °C. Aliquots were removed at select time points, and were monitored for cleavage on analytical HPLC (D5 – method C, Galbofloxacin – method D). Cleavage was confirmed by appearance of a second peak corresponding to the cleaved fragment A and was expressed as % cleavage over time.Radiolabeling with 67Ga and bacterial uptake of 67Ga in wt bacterial strains
67Ga-citrate was received from Jubilant Radiopharma at an average specific activity of 140.9 MBq mL−1. The 67Ga-citrate solution was converted to 67Ga-chloride using a previously described protocol.25 The average specific activity of the resultant 67Ga-chloride solution used for radiolabeling was 131.3 MBq mL−1. For radiolabeling of D5, an aliquot of (67GaCl3 3.7 MBq, 30 μL) was mixed with a solution of D5 (30 μL, 0.01 mM) in chelex resin treated water. The pH of the solution was adjusted with HEPES (100 mM, 100 μL) to 7.4. Complexation was monitored by radio-HPLC, method D. Radiolabeling was found to proceed after 5 minutes at room temperature. 67Ga–D5: tR = 7.13 min (purity/peak area: 99%).Wt E. coli (Mg 1655), wt S. aureus (RN4220) and wt P. aeruginosa (PAO1) were grown overnight in 5 mL LB (Fe-deficient) at 37 °C. The overnight cultures were inoculated in 10 mL iron deficient LB and incubated at 37 °C until the OD600 reached 0.4. Uptake was initiated by adding 67Ga complexes (10 μL, 0.18 MBq) to culture tubes containing 10 mL bacterial inoculum and incubating at 37 °C. Aliquots (1 mL) were removed after 10 min, 20 min, 30 min, 1 h and 2 h and centrifuged for 3 min (34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). The supernatant was removed, and the bacterial pellet was washed with DPBS (1 mL, 3×). The assay was performed in five replicates and in parallel with a 67Ga-citrate control. All tubes were counted using an automated gamma counter to quantify retained radioactivity in pellets in comparison to the 1 mL parent inoculum.
000 rpm). The supernatant was removed, and the bacterial pellet was washed with DPBS (1 mL, 3×). The assay was performed in five replicates and in parallel with a 67Ga-citrate control. All tubes were counted using an automated gamma counter to quantify retained radioactivity in pellets in comparison to the 1 mL parent inoculum.
To challenge active transporter-mediated uptake, the bacterial cultures were preincubated with 200× Fe-LDFC. 67Galbofloxacin (10 μL, 0.18 MBq) was added after 30 min. Aliquots (1 mL) were removed after 10 min, 20 min, 30 min, 1 h and 2 h and centrifuged for 3 min. Pellet were washed with DPBS (3×) and retained activity was quantified as described above.
Biodistribution of naïve and infected Balb/c mice
All animal experiments were performed using protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Stony Brook University accredited through Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International, Federal assurance #A3011-01). 0.3–0.6 MBq of 67Galbofloxacin and 67Ga-citrate (control) were intravenously injected via tail vein catheter in naïve mice. Mice were sacrificed 1 h p.i. and select organs were harvested. Radioactivity was counted by using a gamma counter, and the radioactivity associated with each organ was expressed as % ID g−1. For infected mice biodistribution, mice were given a S. aureus (105 CFU, 50 μL) in left triceps. After 1 h p.i. mice were sacrificed and select organs were harvested. Radioactivity was counted by using a gamma counter, and the radioactivity associated with each organ was expressed as % ID g−1.Metabolite analysis
100 μL of urine was directly injected on the radio HPLC. Eluate was collected in 30 s increments from 0–15 minutes. Activity in each tube was quantified using a gamma counter. The counts were used to reconstruct the metabolite trace which was then compared to the HPLC traces of the original 67Galbofloxacin complex.Galbofloxacin efficacy study
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) at Stony Brook Medicine. Bacterial stocks were freshly prepared before inoculation. In general, bioluminescent S. aureus Xen 29 strain was cultured in MHB at 37 °C to mid log phase (OD600 = 0.4, 2 × 108 cells per mL). Bacterial cells were harvested by centrifugation (34000 rpm, 3 min) and washed with sterilized MHB. Final inoculums were generated by diluting bacteria to the desired concentrations in MHB. To induce localized infection model, mice were injected with 2 × 107 bacterial cells (2 × 108 cells per mL inoculum, 50 μL) into the left forelimb muscle of mice. Test compounds were administered intravenously 1 h and 25 hours post bacterial inoculation. The study included five groups of mice: ciprofloxacin as positive control: 284.4 μmol kg−1 (4 mice) and 9.3 μmol kg−1 (4 mice); vehicle (saline) as a negative control (4 mice); and Galbofloxacin: 9.3 μmol kg−1 (4 mice) and 2.3 μmol kg−1 (4 mice). Muscle size and survival was monitored for 7 days post-infection. The mice were also imaged on a small animal imager every 12 hours for 7 days to monitor the infection burden and treatment efficacy. After 7 days, mice were euthanized, infected and control muscle from each cohort was extracted and plated for CFU determination.
Results and discussion
Synthesis of siderophore antibiotic conjugate D5 and its metal complexes: galbofloxacin and Fe-D5
The structure of D5 draws direct structural inspiration from the natural product albomycin; the presence of the serine linker is essential for the release of the t-seryl transferase inhibitor portion of the natural product inside the bacterial cytoplasm.31,35–37 Thus, we proposed that the introduction of serine to our ciprofloxacin-conjugates could result in enhanced efficacy by way of drug cargo release, a concept validated by Nolan and coworkers on enterobactin derived constructs.34,38 We implemented a conjugation strategy mirroring the connectivity of the natural product albomycin and our first-generation lead D2. The acetylated, amino protected linear desferrichrome precursor P5 was obtained by employing the synthetic strategy pioneered by Miller and coworkers.25,39–41 The serine linker was introduced to the building block P5 by functionalizing ciprofloxacin first with Fmoc-β-alanine,38 followed by N-terminal deprotection and subsequent reaction of the free amine of fragment A25 with Fmoc-Ser(OtBu)-OH with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, Hexafluorophosphate Benzotriazole Tetramethyl Uronium (HBTU) mediated amidation using a solution phase coupling approach. A second amidation step with the protected, linear desferrichrome-derived building block P5 following Fmoc-deprotection of the serine residue (Scheme S1†) was employed to link the proto-siderophore P5 with the drug cargo precursor (2). The final product D5 was attained in a <2% over-all yield after 16 steps following final purification using reverse phase chromatography (Fig. S1–S17†).Complexation of D5 with Ga(III) and Fe(III) salts formed Galbofloxacin and Fe-D5 in aqueous, buffered media at pH 6 and pH 7, respectively. The corresponding complexes were further purified using reverse phase chromatography and isolated as white and orange-red powders, respectively. Characterization via high performance liquid chromatography (HPLC), high resolution mass spectrometry (HRMS) and inductive coupled plasma-optical emission spectrometry (ICP-OES) was employed to affirm purity and produce consistent complex concentrations for subsequent in vitro and in vivo studies based on the quantitation of metal content (Fig. S5, S6, S16 and S17†).
Growth inhibition in wt bacterial strains
With apo- (metal-free), Ga and Fe complexes of D5 in hand, we next probed their ability to inhibit bacterial growth in a conventional minimum inhibitory concentration for 98% inhibited growth (MIC98) assay. We selected E. coli K12 and P. aeruginosa PA01 as Gram-negative model organisms, and S. aureus RN4220 as the Gram-positive model organism. Fe-deficient media was prepared by addition of 2,2′-bipyridine (DP) to mimic the tightly iron controlled in vivo environment.15,42,43 The parent drug ciprofloxacin was utilized as a control giving an MIC98 of 0.93 μM, which is consistent with literature values. apo-D5 and Galbofloxacin were determined to have MIC98 of 0.93 μM in wt E. coli, retaining the growth inhibitory action of ciprofloxacin and comparable to the antibiotic activity of our first-generation conjugate Ga-D2. In accordance with our previous findings, Fe(III) complexes remain significantly less active: Fe-D5 showed a 2-fold decrease in activity in E. coli in comparison to apo-D5 and Galbofloxacin. We hypothesize that the concerted delivery of Fe at elevated concentrations is growth promoting and efficiently offsets the inhibitory action of the siderophore-mediated delivery of the ciprofloxacin payload (Fig. S18,†Table 1). In P. aeruginosa, the activity of apo-D5 and Galbofloxacin activity decreased 10-fold with an MIC98 value of 30 μM and 15 μM respectively, while Fe-D5 produced growth promoting effects (Fig. S18,†Table 1). However, S. aureus exhibited exceptional sensitivity to Galbofloxacin with an MIC98 of 93 nM, an order of magnitude higher activity than parent drug ciprofloxacin (Fig. 3A, Table 1). This qualifies Galbofloxacin as one of the most active synthetic Trojan horse compounds against S. aureus measured to date. The apo-D5 compound showed and MIC90 of 0.93 μM, whereas Fe-D5 showed decreased activity (MIC90 = 7.5 μM) (Fig. 3A, Table 1), indicating that complexation with Ga(III) is essential to produce the synergistic, growth inhibitory effect.

|
 | ||
| Fig. 3 (A) MIC assay of Galbofloxacin in S. aureus shows that the Galbofloxacin complex exhibits an order of magnitude higher potency when compared to ciprofloxacin, while apo-D5 and Fe-D5 exhibit similar or attenuated growth inhibition respectively, when compared to ciprofloxacin (n = 3 × 3). (B) Schematic description of proteinase K assay with Galbofloxacin at 37 ∘C, pH 7. (C) MIC assay of cleaved fragment A in S. aureus shows no growth inhibitory activity for fragment A (n = 3 × 3). (D) In vitro assay using proteinase K shows time dependent cleavage of Galbofloxacin (tR = 7.13 min) by appearance of a second peak corresponding to the cleaved fragment A (tR = 7.03 min) (n = 2). | ||
D5 and its metal complexes were also accessed for its antimicrobial activity in the FhuA transporter deficient mutant E. coli AN193. Fig. S19† summarizes the corresponding results: the previously observed growth inhibitory activity in E. coli of apo-D5, Galbofloxacin and Fe-D5 is completely muted, while ciprofloxacin retains its growth inhibitory activity as found with the wt E. coli strain. This demonstrates that internalization and activity of Galbofloxacin remains FhuA-dependent, affirming that siderophore-mediated active transport of D5-constructs remains essential to produce growth inhibition.
In vitro cleavage of galbofloxacin and apo-D5
As growth inhibition experiments confirmed that Galbofloxacin exhibited increased potency, we assessed apo-D5 and Galbofloxacin as substrates for proteinase mediated cleavage adjacent to the serine residue. In vivo, the antibacterial active moiety is released by peptidase N.31,32 The same reaction can be mimicked in vitro by proteinase K,44,45 a broad-spectrum serine protease (Fig. 3B). We incubated 10 μM of apo-D5 (tR = 7.28 min), Fe-D5 (tR = 6.96 min) or Galbofloxacin (tR = 7.13 min) with proteinase K (20×, 200 μM, in tris buffer, pH = 7) at 37 °C and monitored cleavage by analytical HPLC, which reveals release of fragment A (tR = 7.03 min, Fig. 3B and D). Under these conditions, proteinase K cleaved apo-D5 completely in 15 min (Fig. S21, Table S1†), whereas the cleavage for Galbofloxacin is slower, with a t1/2 of 4 h, with complete cleavage to fragment A after 8 h (Fig. 3D, Table S1†). The cleavage of Fe-D5 was slowest with 34% conjugate still intact after 24 h incubation with proteinase K (Fig. S21,† Table S1†). The results for apo-D5, Fe-D5 and Galbofloxacin are consistent with prior enzymatic cleavage studies on albomycin and desferri-albomycin.44 The results also indicate higher dissociation constant for Fe-D5 as compared with Galbofloxacin.While apo-D5, Fe-D5 and Galbofloxacin are proteinase substrates, it is conceivable that dissociation of the metal ion in the bacterial cytoplasm results in accelerated cleavage of the conjugate to release the antibacterial payload. Importantly, cleavage of the construct does not occur outside the bacterial envelope, as fragment A is completely inactive in all tested wt bacterial strains (E. coli, S. aureus and P. aeruginosa, Fig. 3C and S20†). These results affirm the importance of siderophore mediated active uptake of Galbofloxacin prior to release of the antibiotic payload.
Cytotoxicity analysis
As we had identified Galbofloxacin as a potent antibiotic against S. aureus, and with potential for prospective in vivo applications, we explored its toxicity to mammalian cells next. Specifically, we determined the anti-proliferative activity of Galbofloxacin and apo-D5 against normal human embryonic kidney cells (HEK-293) using cisplatin, gallium(III) citrate and ciprofloxacin as controls. As indicated in Fig. 4, the Galbofloxacin conjugate displays no in vitro toxicity up to 80 μM, with a slightly reduced cell viability (85%), comparable to ciprofloxacin, at 100 μM (Fig. S22†). In comparison, the benchmark cytotoxin cisplatin produces reduced cell viability of 60% at 2 μM compound concentration (Fig. S22†). apo-D5 and Ga(III) citrate are non-toxic to HEK-293 human cells at all tested concentrations (Fig. 4). | ||
| Fig. 4 MTT assay of Galbofloxacin, apo-D5 and Ga-citrate in HEK-293 cell line shows low cytotoxicity for all tested conjugates (n = 3 × 5). | ||
Radiochemical labeling with 67Ga and radiochemical uptake experiments in wt bacterial strains
We next employed a radiochemical tracer approach to further elucidate and quantitate in vitro and in vivo properties of Galbofloxacin. To this end, we employed 67Ga (t1/2 = 79 hours, Eγ (avg) = 93.3 keV), an isotope with multi-day half-life that enables bacterial uptake and pharmacokinetic studies. The formation of the 67Galbofloxacin complex is feasible under mild radiolabeling conditions (pH 7, 100 mM HEPES, 5 minutes) (Fig. 5A) quantitatively with ligand concentrations of 10−6 M. The complexation was monitored using a radio-HPLC (Fig. 5B, method D).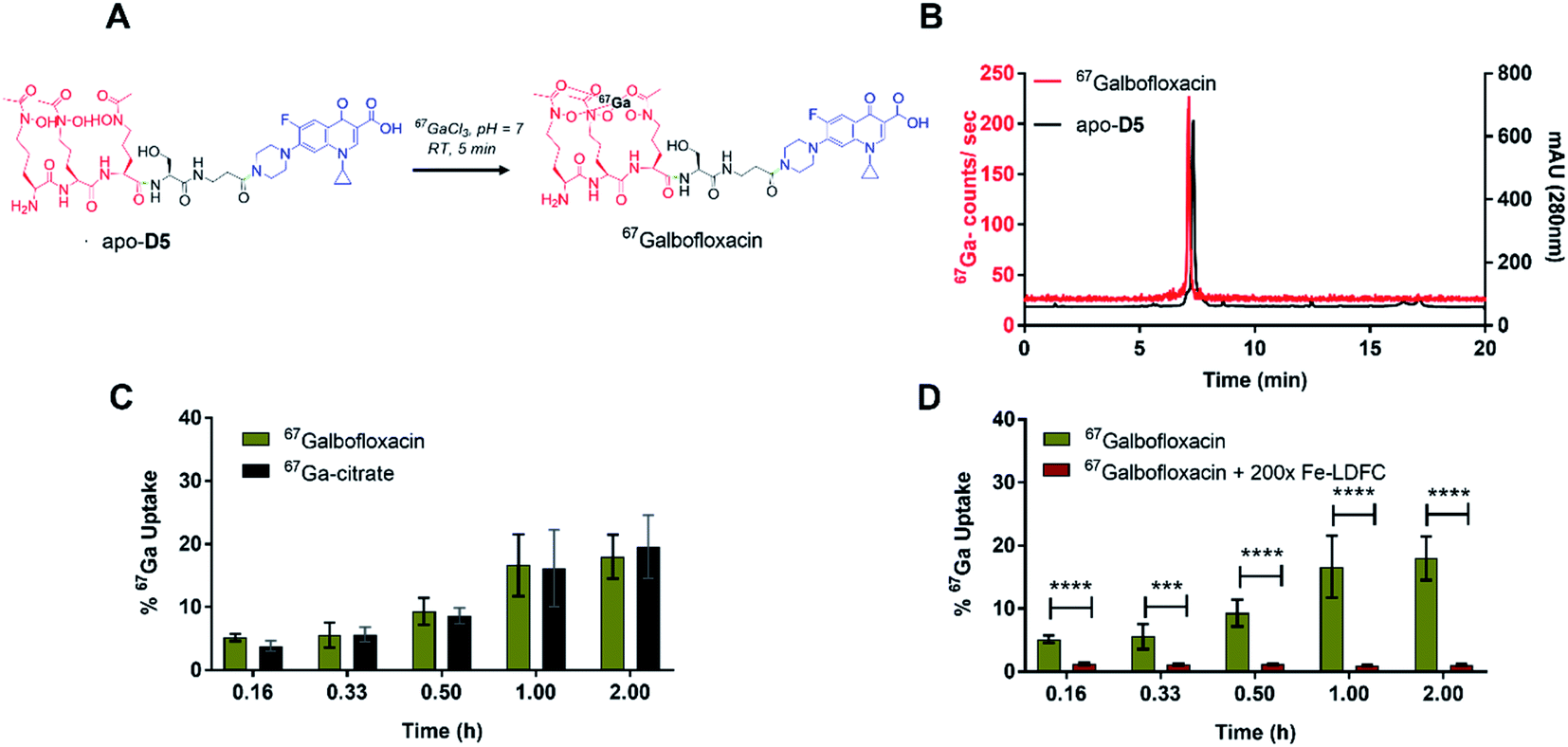 | ||
| Fig. 5 (A) Schematic description of the radiochemical complexation of apo-D5 with 67Ga. 67Ga-complexation proceeds in 5 min, 25 ∘C, pH 7. (B) A representative radiolableing HPLC trace for the characterization of 67Galbofloxacin (tR = 7.13 min, left axis, 67Ga counts per second), in comparison with HPLC characterization of the apo-D5 ligand (tR = 7.28 min, right axis, absorbance at 280 nm) is shown. Free 67Ga elutes at 0.7 minutes. (C) Time-dependent, radiochemical bacterial uptake studies in S. aureus of 67Galbofloxacin in iron deficient, DP-treated media show that uptake is comparable with 67Ga-citrate, which acts as a staphyloferrin (natural siderophore) mimic for S. aureus (n = 5). (D) On pre-incubation with 200× Fe-LDFC, the uptake of 67Galbofloxacin in S. aureus is attenuated significantly after 10 min (n = 5). | ||
The radiolabeled 67Galbofloxacin complex provides a convenient tool to probe time-dependent compound uptake in bacteria relative to weakly complexed 67Ga-citrate. 67Galbofloxacin was prepared as outlined previously, with an average specific activity of 1.9 MBq μmol−1. E. coli, S. aureus and P. aeruginosa (3.2 × 108 CFU) were incubated in Fe-deficient LB with 0.18 MBq of 67Galbofloxacin at 37 °C. Aliquots were removed at 10, 20, 30, 60 and 120 minutes followed by separation of pellets from supernatant and counting of retained radioactivity in each fraction. For all the tested strains, 67Galbofloxacin shows time dependent increase in uptake with a peak uptake at 120 minutes and statistically significant, enhanced uptake in comparison to 67Ga-citrate, for E. coli and P. aeruginosa (Fig. S23, S24, Tables S2 and S3†). Notably, for S. aureus,67Galbofloxacin uptake was comparable to 67Ga-citrate uptake (Fig. 5C, Table S4†). We hypothesize that 67Ga-citrate is efficiently taken up by S. aureus due to its similarity to staphyloferrin, a citric acid based siderophore naturally produced by S. aureus.46 To probe siderophore specific active transport to the bacterial cytoplasm, we also challenged 67Galbofloxacin uptake with the Fe complex of parent siderophore (Fe-LDFC). All three bacterial cultures were incubated for 30 min with 200× excess Fe-LDFC before addition of 67Galbofloxacin. We observed statistically significant depression of 67Galbofloxacin uptake within the bacterial pellet at all time points (Fig. 3D, S25, S26, Tables S5, S6 and S7†).
In vivo biodistribution and pharmacokinetics in naïve and infected mice models
With 67Galbofloxacin uptake validated in vitro using bacterial uptake assays, we investigated biodistribution 67Galbofloxacin in vivo next. Balb/c mice (female, 8 weeks) were injected with 67Galbofloxacin and 67Ga-citrate intravenously, followed by biodistribution and analysis of metabolites in the urine using radio-HPLC at 1 hour post injection. Naïve biodistribution revealed rapid, renal clearance of all both 67Galbofloxacin and 67Ga-citrate (Fig. 6, Table S8†). Metabolite study revealed 5% intact 67Galbofloxacin radiochemical complex in the urine (Fig. S27†). To quantitate the bacterial uptake of 67Galbofloxacin in vivo, we induced a localized, soft tissue infection in mice using 105 CFU S. aureus infection in the left triceps. 67Galbofloxacin showed a significant, enhanced uptake (P ≤ 0.05) in the infected muscle as compared to the control muscle (Fig. 6, Table S8†), in contrast to 67Ga-citrate, which did not distinguish infected and control muscle tissues. Overall, favorable pharmacokinetics, as indicated by rapid renal excretion, no significant off-target organ uptake, and increased uptake in infected muscle qualifies 67Galbofloxacin for further evaluation for therapy in an animal model of soft tissue infection. | ||
| Fig. 6 Comparative biodistribution of 67Galbofloxacin and 67Ga-citrate in naïve mice (n = 3) shows predominantly renal clearance. (Right to the dotted line) Comparative biodistribution of 67Galbofloxacin and 67Ga-citrate in S. aureus infected mice (n = 3), 67Galbofloxacin shows a significant higher uptake (P ≤ 0.05) in the infected muscle as compared to the control muscle (n = 3). | ||
Growth inhibition in bioluminescent S. aureus strain Xen 29 and in vivo therapy studies
The bioluminescent S. aureus strain Xen 29 has previously been used to monitor in vivo antibiotic efficacy using preclinical bioluminescence imaging non-invasively.47–50 Therefore, before using S. aureus Xen 29 for in vivo therapy studies, the in vitro antibacterial activity of Galbofloxacin was accessed in S. aureus Xen 29. MIC assays were performed in Fe deficient media as described for the wt strains. The parent drug ciprofloxacin was employed as a control with MIC98 of 3.8 μM. Galbofloxacin exhibits MIC98 of 0.93 μM, 4× more potent than ciprofloxacin. The 96 well plates for both ciprofloxacin and Galbofloxacin were imaged on small animal optical imager (Fig. 7A and C) to quantify the radiance value due to the bioluminescent bacteria for each well. The MIC results based on OD600 and ROI analysis coincided for both test compounds as shown in Fig. 7B and D. The 4-fold higher activity of Galbofloxacin as compared to ciprofloxacin in S. aureus Xen 29 affirmed this strain as an ideal model system for in vivo therapy studies. | ||
| Fig. 7 (A) Bioluminescence image of ciprofloxacin 96 well plate tested in S. aureus Xen 29 and imaged on an IVIS small animal scanner. (B) MIC assay results of ciprofloxacin in S. aureus Xen 29 (left axis, OD600), in comparison with bioluminescence characterization (right axis, radiance) is shown (n = 3 × 3). (C) Bioluminescence image of Galbofloxacin 96 well plate tested in S. aureus Xen 29 and imaged on an IVIS small animal scanner. (D) MIC assay results of Galbofloxacin in S. aureus Xen 29 (left axis, OD600), in comparison with bioluminescence characterization (right axis, radiance) is shown (n = 3 × 3). | ||
The in vivo therapeutic activity of Galbofloxacin was evaluated in S. aureus Xen 29 triceps infection model. 8 week-old female Balb/c mice were infected intramuscularly with 2 × 107 bacterial cells in the left forelimb and treatments were administered intravenously at 1 h and 25 h post infection. The treatments included 4 mice in each cohort of ciprofloxacin high dose (284.4 μmol kg−1), ciprofloxacin medium dose (9.3 μmol kg−1), Galbofloxacin medium dose (9.3 μmol kg−1), Galbofloxacin low dose (2.3 μmol kg−1) and vehicle (saline) (Table S9†). Mice were imaged on a small animal optical imager every 12 hours and monitored for change in muscle size and survival (Fig. 8A) for 7 days. The muscle size for the vehicle and ciprofloxacin (9.3 μmol kg−1) increased rapidly, for Galbofloxacin (2.3 μmol kg−1) it increased gradually, whereas for ciprofloxacin (284.4 μmol kg−1) and Galbofloxacin (9.3 μmol kg−1) cohort the muscle size decreased over the course of 7 days (Fig. S28 and S29†). Bioluminescence imaging and ROI analysis revealed increased and eventually fatal infection for ciprofloxacin (9.3 μmol kg−1) on day 1 (Fig. 8C, S31, S40, Table S10†), whereas the infection burden decreased for Galbofloxacin (9.3 μmol kg−1) over the course of 7 days (Fig. 8C, S47, S44, Table S10†) and efficiently reduced morbidity. Untreated mice had a survival of 0% after 12 h (Fig. 8B–D, S40,†Tables 2 and S10†). Infected muscle from each cohort was plated after euthanizing and infection burden was determined by counting the CFUs (Fig. S39,†Table 2) and imaging the agar plates on IVIS scanner (Fig. S30, S32, S34, S36 and S38†). Mice treated with medium dose ciprofloxacin (9.3 μmol kg−1) had a survival of 0% after day 1 (Fig. 8C, Table 2), whereas the survival was 100% for same dose Galbofloxacin mice at day 7 (Fig. 8D, Table 2).
 | ||
| Fig. 8 (A) In vivo therapy experimental timeline. (B) Survival plot showing 100% survival for ciprofloxacin (284.4 μmol kg−1) after 7 days, whereas mice dosed with 9.3 μmol kg−1 ciprofloxacin were euthanized on day 1. Saline mice were euthanized after 12 h. (C) Bioluminescence imaging of mice dosed with vehicle (saline), ciprofloxacin (9.3 μmol kg−1) and Galbofloxacin (9.3 μmol kg−1). (D) Survival plot showing 100% survival for Galbofloxacin (9.3 μmol kg−1) after 7 days, whereas % survival was 25% for mice dosed with 2.3 μmol kg−1 Galbofloxacin after 7 days. Saline mice were euthanized after 12 h due to meeting study endpoint criteria of muscle enlargement and distress. | ||

|
Mice receiving Galbofloxacin at 2.3 μmol kg−1 dose exhibited increased survival, albeit infection persisted. By day 4.5 the low dose Galbofloxacin cohort showed slowed progression of infection when compared to the vehicle treated cohort (Fig. S35†). The survival rate was 25% on day 7 for mice dosed with 2.3 μmol kg−1 Galbofloxacin (Fig. 8D, Table 2). Mice dosed with ciprofloxacin (284.4 μmol kg−1) survived for 7 days and showed efficient reduction of infection, indicating expected dose dependency (Fig. 8C, S33,†Table 2).
Conclusions
Here, we show for the first time that a Ga-siderophore antibiotic conjugate, Galbofloxacin exhibits both superior in vitro potency and in vivo efficacy when compared to its parent antibiotic ciprofloxacin. In our study, Galbofloxacin shows an order of magnitude higher potency in comparison to the parent drug ciprofloxacin against S. aureus with MIC98 of 93 nM. Moreover, Galbofloxacin shows higher potency than apo-D5 (MIC98 = 0.93 μM) indicating the synergistic effect of Ga complexation, siderophore-mediated transmembrane transport and cytoplasmic release of the antibiotic. MIC assay with FhuA deficient E. coli results in no growth inhibition, demonstrating siderophore mediated active uptake remains essential for Galbofloxacin. MTT assay in HEK-293 cells demonstrates no toxicity for Galbofloxacin up to 80 μM, comparable to ciprofloxacin at 100 μM. Proteinase K assay was carried out to demonstrate the time dependent, proteolytic cleavage of Galbofloxacin. The results indicate 50% cleavage of Galbofloxacin after 4 h in the presence of 20× proteinase K. We also quantified bacterial uptake for the conjugate and measured in vivo pharmacokinetics using 67Galbofloxacin. 67Galbofloxacin can be synthesized radiochemically under mild conditions and with high radiochemical yields. The complex shows time dependent siderophore mediated active uptake in wt bacterial strains. Biodistribution of 67Galbofloxacin in naïve mice and in a mouse model of intramuscular infection reveals renal clearance and significantly enhanced uptake in infected muscle when compared to 67Ga-citrate, which showed no difference. Finally, an in vivo therapy study with Galbofloxacin in a S. aureus Xen 29 murine infection model demonstrates that the high in vitro potency of Galbofloxacin is directly reflective of its in vivo efficacy. Mice dosed twice with 9.3 μmol kg−1 of Galbofloxacin survived for 7 days with the infection burden decreasing and eventually resolving completely over the course of study, whereas the infection burden was fatal on day 1 for mice dosed with same molar dose of ciprofloxacin.Galbofloxacin constitutes the first, rationally designed gallium coordination complex with superior potency to combat Gram-positive soft tissue infection when compared to its parent antibiotic. Galbofloxacin also demonstrates that Trojan horse antibiotics can successfully reprise their in vitro potency in vivo. The expansion of the scope of treatable infections by modification of the metal, antibiotic payload as well as mechanistic studies to further elucidate the potentiating, growth inhibitory effect of Ga-sideromycins on bacterial cells are underway.
Data availability
All experimental and image data is shown in the ESI,† and additional raw data files can be obtained from the authors upon request.Author contributions
Experiments were designed by AP and EB, experiments were conducted by contributions from authors. The manuscript was written and edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The National Institutes for General Medicine (NIGMS) are acknowledged for funding (R35GM142770). Yong Li is acknowledged for his valuable suggestions during the therapy study.References
- CDC, Biggest Threats and Data | Antibiotic/Antimicrobial Resistance, CDC, 2019 Search PubMed.
- Y. Guo, G. Song, M. Sun, J. Wang and Y. Wang, Front. Cell. Infect. Microbiol., 2020, 10, 107 CrossRef PubMed.
- S. Y. C. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- S. Y. C. Tong, J. S. Davis, E. Eichenberger, T. L. Holland and V. G. Fowler, Clin. Microbiol. Rev., 2015, 28, 603–661 CrossRef CAS PubMed.
- R. J. Turner, Microbiol. Biotechnol., 2017, 10, 1062–1065 CrossRef PubMed.
- A. Pandey and E. Boros, Chem.–Eur. J., 2021, 27, 7340–7350 CrossRef CAS PubMed.
- U. E. Schaible and S. H. E. Kaufmann, Nat. Rev. Microbiol., 2004, 2, 946–953 CrossRef CAS PubMed.
- T. A. Wencewicz and M. J. Miller, Top. Med. Chem., 2018, 26, 151–183 Search PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- G. L. A. Mislin and I. J. Schalk, Metallomics, 2014, 6, 408–420 CrossRef CAS PubMed.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297–313 CrossRef CAS PubMed.
- S. C. Andrews, A. K. Robinson and F. Rodríguez-Quiñones, FEMS Microbiol. Rev., 2003, 27, 215–237 CrossRef CAS PubMed.
- M. J. Miller, H. Zhu, Y. Xu, C. Wu, A. J. Walz, A. Vergne, J. M. Roosenberg, G. Moraski, A. A. Minnick, J. McKee-Dolence, J. Hu, K. Fennell, E. Kurt Dolence, L. Dong, S. Franzblau, F. Malouin and U. Möllmann, BioMetals, 2009, 22, 61–75 CrossRef CAS PubMed.
- T. A. Wencewicz and M. J. Miller, J. Med. Chem., 2013, 56, 4044–4052 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677–9691 CrossRef CAS PubMed.
- H. K. Zane, H. Naka, F. Rosconi, M. Sandy, M. G. Haygood and A. Butler, J. Am. Chem. Soc., 2014, 136, 5615–5618 CrossRef CAS PubMed.
- J. A. Karlowsky, M. A. Hackel, M. Tsuji, Y. Yamano, R. Echols and D. F. Sahm, Int. J. Antimicrob. Agents, 2019, 53, 456–466 CrossRef CAS PubMed.
- T. Aoki, H. Yoshizawa, K. Yamawaki, K. Yokoo, J. Sato, S. Hisakawa, Y. Hasegawa, H. Kusano, M. Sano, H. Sugimoto, Y. Nishitani, T. Sato, M. Tsuji, R. Nakamura, T. Nishikawa and Y. Yamano, Eur. J. Med. Chem., 2018, 155, 847–868 CrossRef CAS PubMed.
- G. G. Zhanel, A. R. Golden, S. Zelenitsky, K. Wiebe, C. K. Lawrence, H. J. Adam, T. Idowu, R. Domalaon, F. Schweizer, M. A. Zhanel, P. R. S. Lagacé-Wiens, A. J. Walkty, A. Noreddin, J. P. Lynch III and J. A. Karlowsky, Drugs, 2019, 79, 271–289 CrossRef CAS PubMed.
- J. Y. Wu, P. Srinivas and J. M. Pogue, Infect. Dis. Ther., 2020, 9, 17–40 CrossRef PubMed.
- W. Neumann, M. Sassone-Corsi, M. Raffatellu and E. M. Nolan, J. Am. Chem. Soc., 2018, 140, 5193–5201 CrossRef CAS PubMed.
- T. J. Sanderson, C. M. Black, J. W. Southwell, E. J. Wilde, A. Pandey, R. Herman, G. H. Thomas, E. Boros, A.-K. Duhme-Klair and A. Routledge, ACS Infect. Dis., 2020, 6, 2532–2541 CrossRef CAS PubMed.
- M. Ghosh, Y. M. Lin, P. A. Miller, U. Möllmann, W. C. Boggess and M. J. Miller, ACS Infect. Dis., 2018, 4, 1529–1535 CrossRef CAS PubMed.
- M. Ghosh, P. A. Miller, U. Möllmann, W. D. Claypool, V. A. Schroeder, W. R. Wolter, M. Suckow, H. Yu, S. Li, W. Huang, J. Zajicek and M. J. Miller, J. Med. Chem., 2017, 60, 4577–4583 CrossRef CAS PubMed.
- A. Pandey, C. Savino, S. H. Ahn, Z. Yang, S. G. Van Lanen and E. Boros, J. Med. Chem., 2019, 62, 9947–9960 CrossRef CAS PubMed.
- C. E. Arnold, A. Bordin, S. D. Lawhon, M. C. Libal, L. R. Bernstein and N. D. Cohen, Vet. Microbiol., 2012, 155, 389–394 CrossRef CAS PubMed.
- C. R. Chitambar, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2044–2053 CrossRef CAS PubMed.
- C. H. Goss, Y. Kaneko, L. Khuu, G. D. Anderson, S. Ravishankar, M. L. Aitken, N. Lechtzin, G. Zhou, D. M. Czyz, K. McLean, O. Olakanmi, H. A. Shuman, M. Teresi, E. Wilhelm, E. Caldwell, S. J. Salipante, D. B. Hornick, R. J. Siehnel, L. Becker, B. E. Britigan and P. K. Singh, Sci. Transl. Med., 2018, 10, eaat7520 CrossRef PubMed.
- C. Goss, D. Hornick and M. Aitken, Pediatr. Pulmonol., 2012, 47, 303 Search PubMed.
- Y. Wang, B. Han, Y. Xie, H. Wang, R. Wang, W. Xia, H. Li and H. Sun, Chem. Sci., 2019, 10, 6099–6106 RSC.
- Z. Lin, X. Xu, S. Zhao, X. Yang, J. Guo, Q. Zhang, C. Jing, S. Chen and Y. He, Nat. Commun., 2018, 9, 3445 CrossRef PubMed.
- G. F. Gause, Br. Med. J., 1955, 2, 1177–1179 CrossRef CAS PubMed.
- T. E. Clarke, V. Braun, G. Winkelmann, L. W. Tari and H. J. Vogel, J. Biol. Chem., 2002, 277, 13966–13972 CrossRef CAS PubMed.
- Z. L. Reitz, M. Sandy and A. Butler, Metallomics, 2017, 9, 824–839 CrossRef CAS PubMed.
- V. Braun, K. Gunthner, K. Hantke and L. Zimmermann, J. Bacteriol., 1983, 156, 308–315 CrossRef CAS PubMed.
- A. Pramanik, U. Stroeher, J. Krejci, A. Standish, E. Bohn, J. Paton, I. Autenrieth and V. Braun, Int. J. Med. Microbiol., 2007, 297, 459–469 CrossRef CAS PubMed.
- A. Hartmann, H. P. Fiedler and V. Braun, Eur. J. Biochem., 1979, 99, 517–524 CrossRef CAS PubMed.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 134, 18388–18400 CrossRef CAS PubMed.
- E. K. Dolence, A. A. Minnick, C. E. Lin, M. J. Miller and S. M. Payne, J. Med. Chem., 1991, 34, 968–978 CrossRef CAS PubMed.
- C. J. Adams, J. J. Wilson and E. Boros, Mol. Pharm., 2017, 14, 2831–2842 CrossRef CAS PubMed.
- C. J. Adams, J. J. Wilson and E. Boros, Mol. Pharm., 2017, 14, 2831–2842 CrossRef CAS PubMed.
- R. E. Juárez-Hernández, P. A. Miller and M. J. Miller, ACS Med. Chem. Lett., 2012, 3, 799–803 CrossRef PubMed.
- E. W. Hunsaker and K. J. Franz, Inorg. Chem., 2019, 58, 13528–13545 CrossRef CAS PubMed.
- A. Pramanik, U. Stroeher, J. Krejci, A. Standish, E. Bohn, J. Paton, I. Autenrieth and V. Braun, Int. J. Med. Microbiol., 2007, 297, 459–469 CrossRef CAS PubMed.
- W. Saenger, in Handbook of Proteolytic Enzymes, 2013, vol. 3, pp. 3240–3242 Search PubMed.
- S. Konetschny-Rapp, G. Jung, J. Meiwes and H. Zahner, Eur. J. Biochem., 1990, 191, 65–74 CrossRef CAS PubMed.
- M. van Oosten, T. Schäfer, J. A. C. Gazendam, K. Ohlsen, E. Tsompanidou, M. C. de Goffau, H. J. M. Harmsen, L. M. A. Crane, E. Lim, K. P. Francis, L. Cheung, M. Olive, V. Ntziachristos, J. M. van Dijl and G. M. van Dam, Nat. Commun., 2013, 4, 2584 CrossRef PubMed.
- T. Hertlein, V. Sturm, U. Lorenz, K. Sumathy, P. Jakob and K. Ohlsen, Antimicrob. Agents Chemother., 2014, 58, 1630–1638 CrossRef PubMed.
- T. Hertlein, V. Sturm, U. Lorenz, K. Sumathy, P. Jakob and K. Ohlsen, Antimicrob. Agents Chemother., 2014, 58, 1630–1638 CrossRef PubMed.
- Y. Li, F. Daryaee, G. E. Yoon, D. Noh, P. M. Smith-Jones, Y. Si, S. G. Walker, N. Turkman, L. Meimetis and P. J. Tonge, ACS Infect. Dis., 2020, 6, 2249–2259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of intermediates for the synthesis of D5, representative HPLC traces of ligands and their corresponding complexes, MIC experiments, radiochemical characterization, radiochemical bacterial uptake data and tabulated biodistribution data. See DOI: 10.1039/d1sc04283a |
| This journal is © The Royal Society of Chemistry 2021 |