
Copper catalysts for photo- and electro-catalytic hydrogen production†
Abdullah M.
Abudayyeh
a,
Olivier
Schott
b,
Humphrey L. C.
Feltham
a,
Garry S.
Hanan
 *b and
Sally
Brooker
*a
*b and
Sally
Brooker
*a
aDepartment of Chemistry and the MacDiarmid Institute for Advanced Materials and Nanotechnology, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: sbrooker@chemistry.otago.ac.nz; Fax: +64-3-4797906; Tel: +64-3-479 7912
bDépartment de Chimie, Université de Montréal, Montréal, Quebec H3T 1J4, Canada
First published on 11th December 2020
Abstract
Green production of hydrogen, a carbon-zero future fuel, requires long lived, high activity catalysts made from inexpensive, earth abundant metal ions. Only 15 molecular copper complexes catalyze the H2 evolving reaction (HER). Herein 3 such complexes are prepared and studied as catalysts for both photo- and electro-catalytic HER. Two new N5-donor analogues of the literature N4-donor Schiff base macrocycle HLEt (from [1 + 1] condensation of 2,2′-iminobisbenzaldehyde (dpa) and diethylenetriamine), macrocycle HLEt-MePy (2-bromomethylpyridine alkylation of HLEt) and non-cyclic HLEtPy2 (condensation of dpa and two 2-aminoethylpyridine), were prepared. Then literature [CuII(LEt)]BF4 (1), and new [CuII(LEt-MePy)]BF4 (2) and [CuII(LEtPy2)]BF4 (3), were prepared and structurally characterized, revealing square, square pyramidal and trigonal bipyramidal copper(II) geometries, respectively. Testing under photocatalytic conditions showed that 1–3 have modest turnover numbers (TON = 460–620), but the control, using Cu(BF4)2, had a higher TON (740), and the blank (no copper) also had significant activity (TONequiv = 290). So this is a cautionary tale: whilst 1–3 initially appeared to be promising catalysts for photocatalytic HER, running the control and blank – studies often not reported – shows otherwise. Hence the focus shifted to electrocatalytic HER testing. All three complexes show reversible redox events in MeCN vs. 0.01 M AgNO3/Ag: E1/2 = −1.39 V (1 and 2); −0.89 V (3). Unlike complexes 2 and 3 or the control, 1 is shown to be, or to form, an effective and stable electrocatalyst for HER in MeCN with acetic acid as the proton source (at 100 mV s−1, Ecat/2 = −1.64 V so overpotential necessary for catalysis = 0.23 V, and icat/ip = 34, where icat is peak catalytic current and ip is 1e− peak current for 1 in absence of acid): after 6 hours at −1.6 V, the TON for 1 is 12.5, despite the tiny glassy carbon working electrode used, and it retains good electrocatalytic activity. Results of both ‘rinse and repeat’ (for catalytically active deposit on working electrode) and drop of Hg (for formation of catalytically active nanoparticles) tests are consistent with homogeneous catalysis by 1, but a small copper stripping wave is seen after acetic acid is added, so it is probable that these initial test results are ‘false negatives’, and that there is a heterogenous catalytically active species present; so future studies will probe this point further.
Introduction
There are many acute global challenges which have the potential for unprecedented adverse impacts on human health and global ecosystems.1 These include climate change, exceptional wild fires, and ocean acidification – which are to a great extent due to the ongoing large scale combustion of fossil fuels, as this leads to ever increasing concentrations of greenhouse gases such as CO2 in the atmosphere.2 Therefore the development of carbon neutral (circular or closed carbon cycles), or better still carbon zero (H2) future fuels with production driven by green energy (renewables, such as solar, wind or wave generated energy) sources is an urgent necessity.3One of the most attractive strategies to achieve this aim is to mimic plants and store light energy in chemical bonds, forming so called ‘solar fuels’, such as hydrogen.4–6 Hydrogen is particularly attractive because it is a carbon zero fuel (produces only water and energy on combustion) and has a higher energy density than fossil fuels7 and batteries.8,9 Current commercial production of hydrogen uses a platinum catalyst, but for hydrogen to be widely adopted, it is essential that active long-lived new catalysts based on cheap, abundant metals be developed.10
Over the last few decades, various earth-abundant molecular cobalt,11–17 iron and nickel18,19 catalysts have exhibited activity for HER under photocatalytic conditions. Several very useful reviews of photochemical HER using earth-abundant molecular catalysts have been presented, and confirm that only 3 are based on copper.20–24 This is perhaps surprising, as copper complexes have well determined coordination chemistry, as well as rich redox and photo-chemistry, and are well known as catalysts in several other types of transformations,25–34 including CO2 reduction29 and water oxidation.30–34 But copper-based molecular catalysts have an enhanced propensity, under reducing conditions in the presence of water, to breakdown and form heterogeneous reduction products,35,36 such as metallic nanoparticles or deposits on the working electrodes, which can be active HER electrocatalysts in their own right.37–41 This fact may well have discouraged researchers from working with molecular copper catalysts for HER. But careful testing for the possible degradation of the molecular copper catalyst to form nanoparticles (mercury drop test or DLS) or deposits on the working electrode (rinse and repeat test, SEM/XPS) can, and should, be done, to help rule these out.42,43
To date, only 15 copper complexes (Fig. 1) have been reported to be active as molecular catalysts for HER:42,44–53 14 are active HER electrocatalysts,42,44–53 and 3 (Fig. 1, box) are active catalysts for light-driven HER.48,51
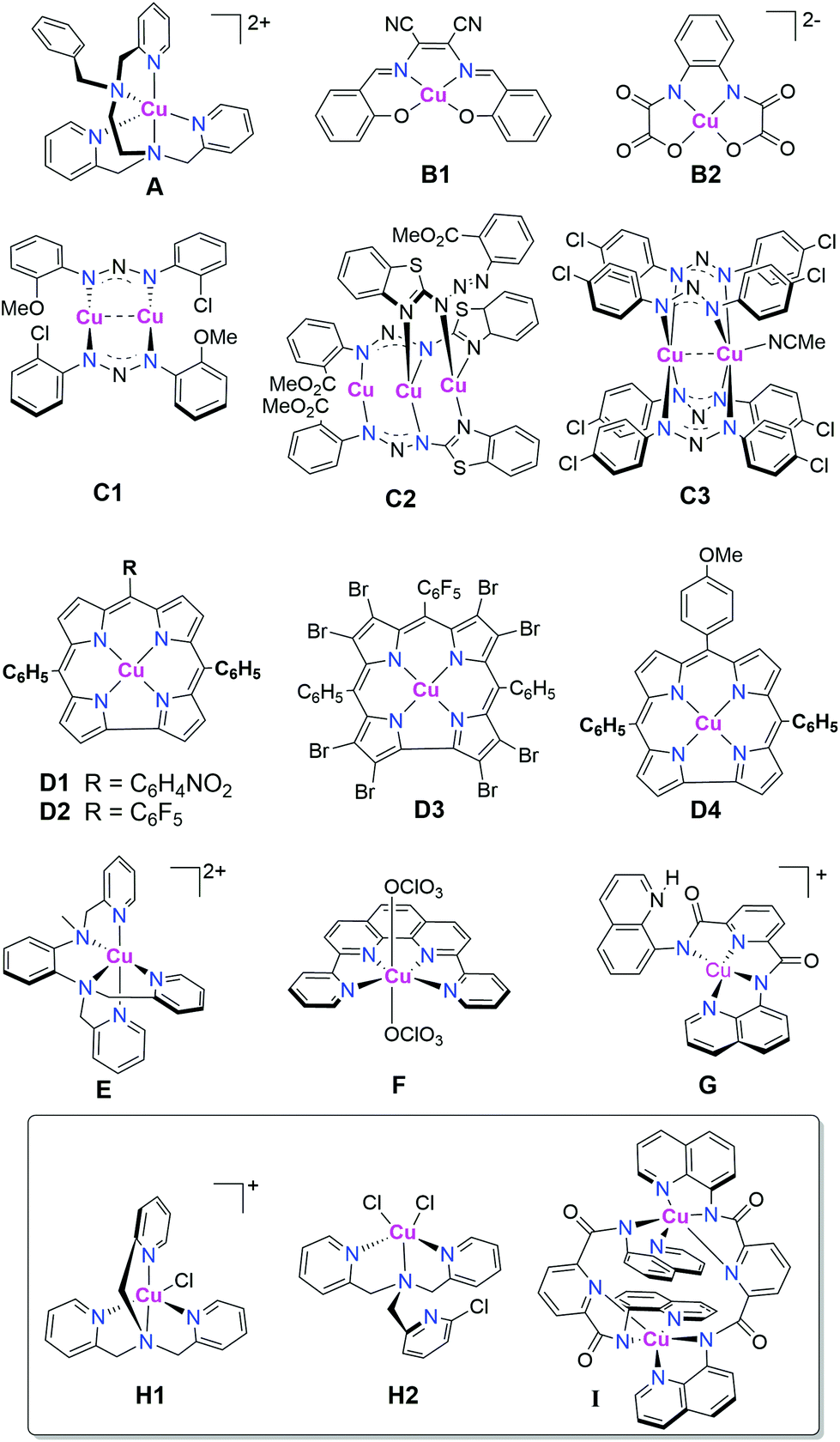 | ||
| Fig. 1 The 15 copper-based molecular catalysts reported to date to be active for HER;42,44–53 14 of which (all except I) are active electrocatalysts,42,44–53 whilst the box contains the 3 that are active under photocatalytic conditions.48,51 | ||
Considering electrocatalysis first: the first molecular copper catalyst for HER, A, was supported by an N5-donor extended tripodal ligand, and was reported in 2014 by Wang, Sun and co-workers.42 Complex A exhibited a turnover frequency (TOF) of 7000 h−1 cm−2 in water (i.e. TOF per electrode surface area). Then Zhan and co-workers reported copper(II) complexes a pair of mononuclear salen-like ligands (N2O2-donor), B1 & B2,45,46,54 as well as one tri- and two dinuclear copper complexes of bis-triazenido-type ligands (bridging N2 donor), C1–C3,47,52,53 all 5 of which were active HER electrocatalysts in water.45–47 Catalysts B1 & B2 had faradaic efficiencies of 91–96% and TOFs of up to 1331.7 h−1.45,46 Copper(I) complexes C1 & C2, and copper(II) complex C3, were active electrocatalysts in water, when acetic acid (AcOH) was used as the proton source, at overpotentials of 789–942 mV (applied potentials of −1.45 to −1.47 V vs. Ag/AgCl) with faradaic efficiencies of 94–97%.52,53 In the same year, Lai, Fu, Cao and co-workers reported that several copper(II) corroles (N4-donor macrocycle), D1–D4, were active electrocatalysts for HER, with the best of them, D1 & D2, featuring electron withdrawing groups on the meso positions of the corrole macrocycle, and an icat/ip of 303, (where icat is the maximum catalytic current and ip is the peak current in the absence of acid) in acetonitrile with trifluoroacetic acid (TFA) as the proton source.44
The pair of known copper complexes of a tripodal ligand, copper(II) chloride H1 & copper(II) dichloride H2,55,56 were subsequently examined, in 2016, by Hou, Wang and co-workers and found to exhibit electrocatalytic HER in acetonitrile, with acetic acid as the proton source, when −1.8 V vs. SCE was applied, yielding a faradaic efficiency of ∼95%.48 In 2017, probably inspired by A, the copper(II) complex of a slightly different extended tripodal N5-donor ligand, E, was reported by Mazumder, Verani and co-workers to exhibit electrocatalytic HER in water at pH 2.5, with a TON of 3900 mol H2 per mol of catalyst over 3 hours at −1.7 V vs. Ag/AgCl using a Hg pool working electrode, with no evidence of electrodeposition.49 Also in 2017, the water-soluble perchlorate salt of the copper(II) complex of an N4-donor pyridine-phen-pyridine ligand, F, prepared by Wang and co-workers, was reported to electrocatalyse HER in neutral water (phosphate buffer; SHE reference electrode) when held at an overpotential of 520 mV. A TON of 734 mol H2 per mol of catalyst was recorded over 2 hours.50 Finally, also in 2017, Padhi and co-workers reported that a mononuclear copper(II) complex of a dianionic N4-donor ligand, G, displayed HER electrocatalytic activity in 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 DMF/H2O (v/v) in the presence of acetic acid as the proton source, at −1.6 V vs. SCE (saturated calomel electrode) with icat/ip = 24 and TOF of ∼112 s−1.51
:5 DMF/H2O (v/v) in the presence of acetic acid as the proton source, at −1.6 V vs. SCE (saturated calomel electrode) with icat/ip = 24 and TOF of ∼112 s−1.51
Turning now to the 3 molecular copper catalysts shown to date to be active for HER under photocatalytic conditions: in 2016 Hou, Wang and co-workers showed that the two tripodal copper complexes H1 & H2 were active not only as HER electrocatalysts (see above), but that they were also the first examples of copper based molecular catalysts to be active under photocatalytic HER conditions.48 By using a relatively low concentration of catalyst (1 μM), high TONs, of 6108 and 10014 over 6 hours, were achieved for the copper(II) monochloride (H1) and copper(II) dichloride (H2), respectively, when driven by visible irradiation (λ = 400 nm; violet) with an Ir based photosensitizer (PS) (0.2 mM), in 9:1 MeCN/H2O (v/v), using triethylamine (TEA) as sacrificial reductant.48 Then in 2017, Padhi and co-workers demonstrated that visible light irradiation of the doubly μ-pyridine-bridged dicopper(II) complex I enabled HER in 80:20 DMF:H2O solution using fluorescein as PS, and TEA as sacrificial reductant, with a maximum TOF of 0.03 s−1. They also showed that, once it had plateaued, some of the activity of the system was restored on the addition of fresh catalyst, indicating that it is the lifetime of the catalyst, not the PS, that is the limiting factor.51
Of the above 15 molecular copper HER catalysts, testing to help rule out the presence of nanoparticles (either Hg drop or DLS), and/or the presence of a deposit on the working electrode (rinse and repeat, or SEM/XPS), was carried out in most, but not all, of the reported cases. These tests were used to try and help rule out heterogeneous catalysis in the case of the electrocatalysts A,42B2,46D,44E,49F,50 and G,51 although in the case of E some decomposition was observed after 8 hours of electrolysis.49 No nanoparticle or other heterogeneous deposit testing was done for the photocatalysts, H1 and H2,48 or I (despite evidence of degradation of I during photocatalysis).51 Clearly, the results of these tests should always be reported for molecular copper HER catalysts. But it is also critically important to point out that these tests are insufficient to rule out such heterogeneous species being the actual active catalytic species. Artero and co-workers reported a particularly comprehensive illustration of this in 2016, in which the active catalyst is not the molecular complex but is the heterogeneous deposit formed. But as the latter was meta-stable and readily redissolved before the rinse and repeat test, this led to a ‘false negative’ test result.57 Dempsey and co-workers have recently written a superb tutorial review on this issue.58
We recently reported16 that 17 cobalt complexes of a wide range of polydentate and macrocyclic ligands were effective catalysts for HER under photocatalytic conditions. The best of them, [CoLEt]+, was supported by an anionic N4-donor macrocycle (Fig. 2) and had a TON of 26, vs. TON(cobaloxime) of 9, both determined in DMF, with [Ru(bpy)3]2+ as the PS and triethanolamine (TEOA) as the proton source, under blue light irradiation.
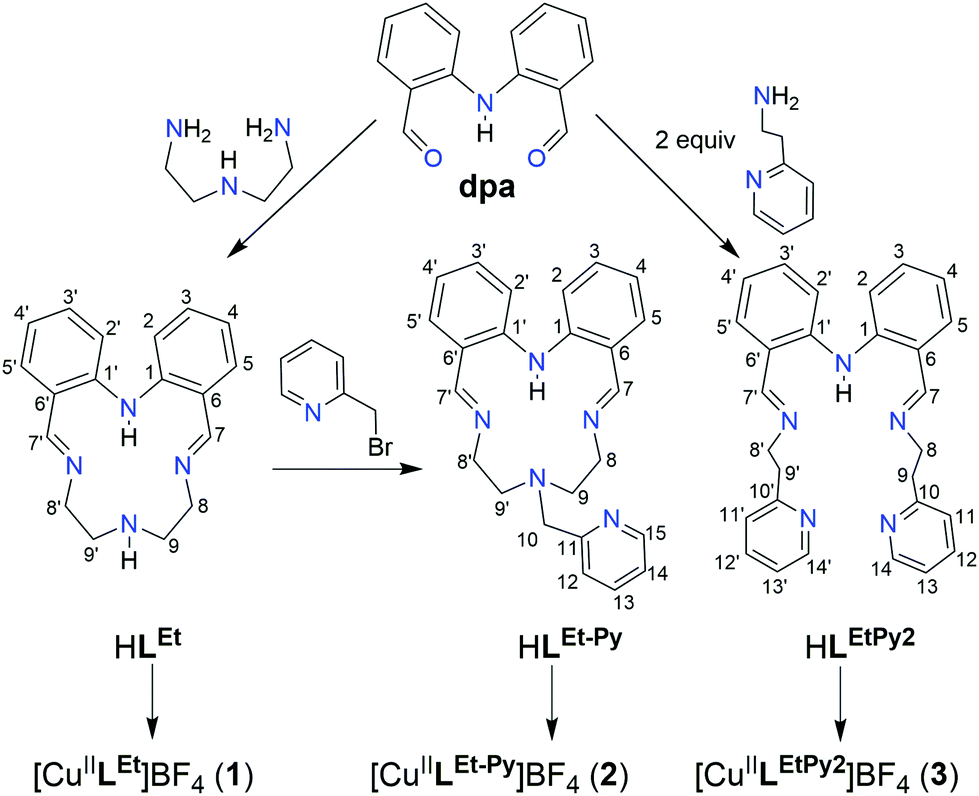 | ||
| Fig. 2 Synthesis of the three Schiff-base ligands, two macrocyclic (literature HLEt,59,60 and new HLEt-MePy) and one new acyclic (HLEtPy2) ligand, from which the three copper(II) complexes (1–3) were prepared by 1:1:1 reaction of ligand to TEA to CuII(BF4)2·H2O. | ||
Herein the analogous copper complex, [CuII(LEt)]BF4 (1, Fig. 2), of that N4-donor macrocycle, as well as two new copper complexes of two new N5-donor ligands that are prepared herein (Fig. 2), macrocyclic [CuII(LEt-MePy)]BF4 (2) and acyclic [CuII(LEtPy2)]BF4 (3), are tested for HER activity – as we look to grow the number of copper-based molecular HER catalysts. All 3 are shown to be active HER catalysts under photocatalytic conditions, despite presenting differing copper coordination geometries: square (1), square pyramidal (2) and trigonal bipyramidal (3). These findings double the number of known molecular copper catalysts for photocatalytic HER (only 3 prior examples; Fig. 1, box). But, disappointingly, on running the appropriate control and blank tests – not reported for the previous copper HER catalysts in the literature – these complexes are revealed to be no more active than the simple salt copper(II) tetrafluoroborate under photocatalytic conditions. This highlights the importance of always doing, and reporting, the results of blanks and controls, in order to put the observed activity of the complexes studied into proper context. In light of the disappointing photocatalysis results, our attention turned to the potential of 1–3 as electrocatalysts for HER. Pleasingly, all 3 complexes exhibit reversible redox processes in MeCN (E1/2 = −0.89 to −1.39 V vs. 0.01 M AgNO3/Ag). Even better is that in electrocatalysis tests on 1, in MeCN with acetic acid as the proton source, the electrocatalytic HER activity is retained for more than 6 hours.
Results and discussion
Ligand synthesis
The Schiff-base macrocycle HLEt was prepared, from 2,2′-iminobisbenzaldehyde (dpa) and diethylenetriamine, using the literature procedure (Fig. 1).59,60This N4-donor HLEt macrocycle was then converted into the N5-donor analogue, HLEt-MePy (Fig. 2), by adding a methylpyridyl ‘arm’; via N-alkylation with 2-(bromomethyl)pyridine in dry THF at room temperature in the presence of excess triethylamine (TEA). The desired ligand was obtained as HLEt-MePy·0.5DCM, according to both elemental analysis and the relative integration of the solvent peak in the 1H NMR spectrum of this red solid, in 63% yield. The third ligand, the N5-donor non-cyclic Schiff base ligand analogue, HLEtPy2 (Fig. 2), was prepared by condensation of dpa with 2 equivalents of 2-aminoethylpyridine in refluxing acetonitrile, in quantitative yield, as HLEtPy2·0.25(CH3)2CO, according to both elemental analysis and the relative integration of the solvent peak in the 1H NMR spectrum of this sticky red oil.
Synthesis of complexes
A metal templated cyclisation of dpa and diethylenetriamine was employed to prepare [CuII(LEt)]BF4 (1) in the literature,59 but unsurprisingly it is shown herein that it can also be prepared by metalation of the pre-formed macrocycle, as has been previously reported for other 3d metal complexes of this macrocycle.59All three complexes, 1–3, were prepared by 1:1:1 reaction of the appropriate ligand (HLEt, HLEt-MePy and HLEtPy2) with TEA and CuII(BF4)2·H2O. In all cases an instantaneous change in colour from yellow to dark red occurs on adding the copper salt. Dark orange crystals of complex 1 and dark red needles of complexes 2 and 3, suitable for single crystal X-ray structure determinations (see below), were grown by diethylether vapour diffusion into the reaction solutions. The complexes were pure by elemental analysis and were further characterized by electrospray mass spectrometry (ESI-MS), UV-vis spectroscopy and single crystal X-ray structure determinations. All three complexes, 1–3, are soluble in most common solvents: they are highly soluble in methanol, acetonitrile and DMF, and have moderate solubility in dichloromethane, chloroform and water.
The ESI-MS spectra of 1–3 all showed an intense peak attributed to the respective monocation: [Cu(LEt)]+ at m/z = 354.0902, [Cu(LEt-MePy)]+ at 445.1310 and [Cu(LEtPy2)]+ at 495.1451.
UV-vis spectra were obtained on 1–3 in DMF (300–1200 nm, Fig. 3). All feature two intense absorptions at low wavelengths, in the range 334–336 nm (ε = 3793–5733 M−1 cm−1) and 399–408 nm (ε = 3313–5371 M−1 cm−1), as well as a much more intense absorption in the visible, 466–491 nm (ε = 6216–13500 M−1 cm−1). Small red shifts of the most intense band, from 466 nm for the complex of the N4-donor macrocycle, 1, are seen for the complexes of the two N5-donor ligands, by +13 nm for 2 and by +23 nm for 3.
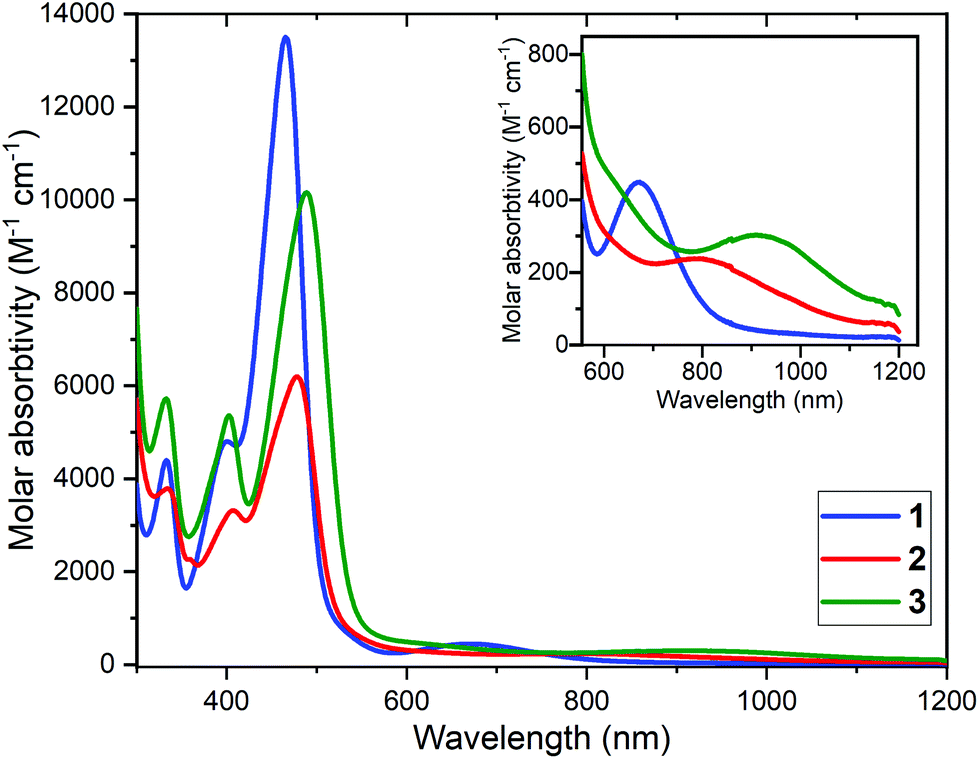 | ||
| Fig. 3 UV-vis spectra of 0.21 mM DMF solutions of: [CuIILEt](BF4) (1) blue line; [CuIILEt-MePy](BF4) (2) red line; [CuIILEtPy2](BF4)·0.5H2O (3·0.5H2O) green line. Inset: Expansion of 600–1200 cm−1 region. | ||
In all three cases a d–d band is also clearly seen: at 672 (453) for 1, 789 (342) for 2 and 917 nm (309 M−1 cm−1) for 3. Hathaway61 established that in general d–d transitions in copper(II) complexes that are square planar N4-coordinated occur at higher energy than for those that are square pyramidal N5-coordinated, which in turn occur at a higher energy than for trigonal bipyramidal analogues.62,63 This is in excellent agreement with the present findings: 14881 cm−1 (square planar 1) > 12674 cm−1 (square pyramidal 2) > 10905 cm−1 (trigonal bipyramidal 3).
Structures of complexes
As reported earlier,59 complex 1 features a square planar (τ4 = 0.01)64 copper(II) center with 4N donors, comprising the deprotonated diphenylamine, two imines and the tertiary amine (Fig. 4 top and Table 1).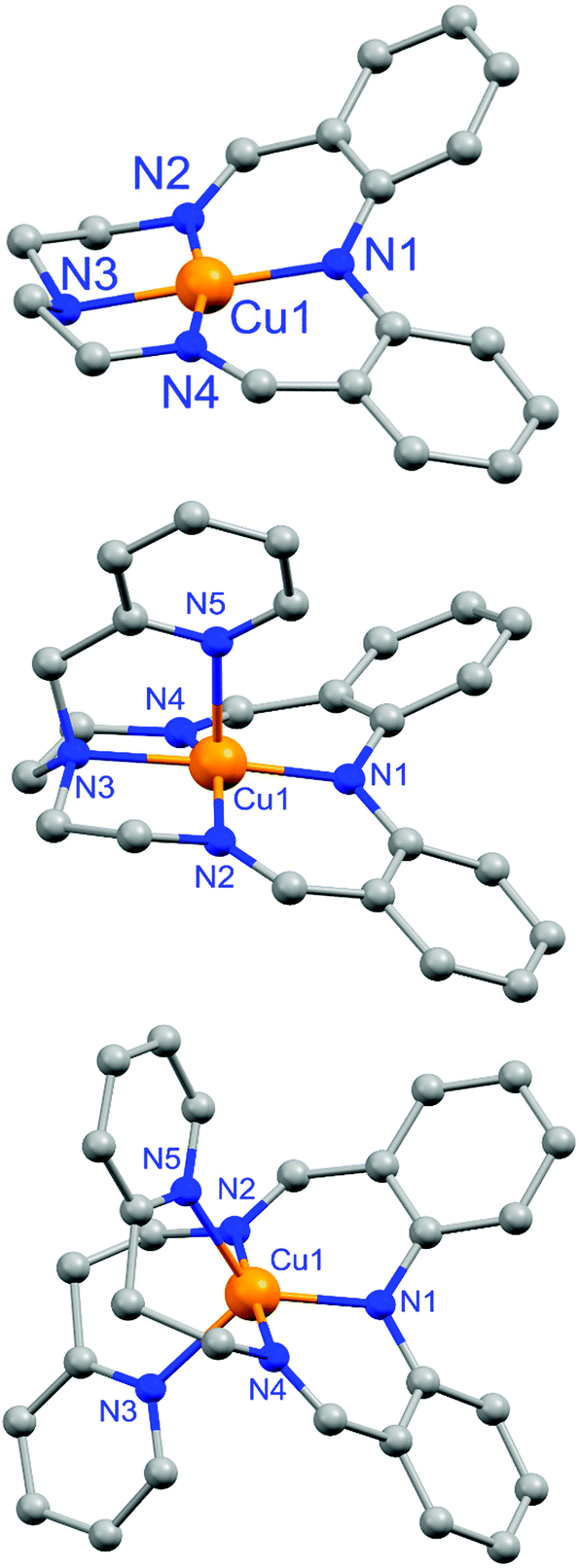 | ||
| Fig. 4 X-ray crystal structures (shown as ball and stick diagrams) of the monocations of (from top to bottom): macrocyclic complexes 1 (square planar CuII) and 2 (square pyramidal CuII), and non-cyclic complex 3 (trigonal bipyramidal CuII). | ||
| [CuIILEt](BF4) 159 | [CuIILEt-MePy](BF4) 2 | [CuIILEtPy2](BF4) 3 | |
|---|---|---|---|
| a 4-Coordinate distortion parameter τ4 (0 = square planar; 1 = tetrahedral).64 b 5-Coordinate distortion parameter τ5 (0 = square pyramidal; 1 = trigonal bipyramidal).65 c Only those within equatorial plane. d Only two trans angles (within square plane). e Only one trans angle (axial to trigonal plane). | |||
| Coordination | N4 – square planar | N5 – square pyramidal | N5 – trigonal bipyramidal |
| Distortion parameter | τ 4 = 0.01a | τ 5 = 0.27b | τ 5 = 0.83b |
| Cu(1)–N(1)dpa | 1.932(5) | 1.940(6) | 1.975(3) |
| Cu(1)–N(2)imine | 1.932(6) | 1.944(7) | 1.992(3) |
| Cu(1)–N(4)imine | 1.898(6) | 1.939(4) | 1.984(3) |
| Cu(1)–N(3)amine or pyridine | 2.036(6) amine | 2.106(4) amine | 2.206(3) pyridine |
| Cu(1)–N(5)pyridine | — | 2.247(4) | 2.173(3) |
| Average Cu–N | 1.950 | 2.035 | 2.066 |
| N(2)–Cu(1)–N(1) | 96.4(2) | 94.7(3) | 89.1(1) |
| N(2)–Cu(1)–N(4) | 166.2(2) | 157.9(3) | 178.4(2) |
| N(1)–Cu(1)–N(4) | 96.1(2) | 94.9(1) | 89.4(1) |
| N(1)–Cu(1)–N(3) | 179.3(3) | 174.4(3) | 123.9(1) |
| N(4)–Cu(1)–N(3) | 84.0(2) | 85.1(3) | 90.7(1) |
| N(2)–Cu(1)–N(3) | 83.6(2) | 83.5(2) | 90.3(1) |
| N(1)–Cu(1)–N(5) | — | 104.6(2) | 128.4(2) |
| Range cis Neq–Cu–Neqc |
83.6–96.4 | 83.5–94.9 | 107.7–128.4 |
| Range trans-N–Cu–N | 166.3, 179.3d | 157.9, 174.4d | 178.4e |
Herein, X-ray structure determinations are reported for complexes 2 and 3 (Fig. 4 and Table 1), for which the asymmetric unit comprised the entire cationic complex and a tetrafluoroborate anion. The Cu(II) center in the pyridyl-armed macrocycle, 2, is distorted square pyramidal (τ5 = 0.27; vs. 0 for a perfect square pyramid)65 through the addition of the axially coordinated pyridyl arm (Fig. 4, middle). In contrast, in the non-cyclic analogue complex 3 the Cu(II) center adopts a distorted trigonal bipyramidal geometry (τ5 = 0.83; vs. 1 for a perfect trigonal bipyramid)65 with the trigonal plane comprising the nitrogen donors from the deprotonated diphenylamine and two pyridine arms, and the two imine nitrogen atoms coordinating axially (Fig. 4, bottom).
The Cu–N bond lengths in 1–3 are interesting. In the square-based complexes, 1 and 2, the two imine nitrogen atoms are bound asymmetrically to the copper centre. As a result, one Cu–Nimine bond is the shortest Cu–N bond in the complex (1, 1.898(6); 2, 1.939(4) Å), followed by the Cu–Ndpa bond length (1, 1.932(6); 2, 1.940(6) Å), whilst the other Cu–Nimine bond is either the same length (1, 1.932(5) Å) or slightly longer again (2, 1.944(7) Å) than the Cu–Ndpa bond (Table 1). In both cases the longest bond is the Cu–Namine bond (1, 2.036(6); 2, 2.106(5) Å). The situation for the noncyclic complex, 3, is quite different from that in the cyclic complexes 1 and 2: the Cu–Nimine bonds are far closer to symmetrical, the Cu–Ndpa bond is the shortest, and the two axial Cu–Npyridine bonds are easily the longest (Table 1). As expected, the average Cu–N bond length increases on going from 4-coordinate (1.950 Å) to 5-coordinate (square pyramidal 2.035; trigonal bipyramidal 2.066 Å).
Photocatalytic hydrogen evolution
All experiments were conducted on a 5 mL DMF solution containing: 5 μM catalyst, 1 M triethanolamine (TEOA) as the sacrificial reductant, 0.2 mM [Ru(bpy)3](PF6)2 (bpy = 2,2′-bipyridyl) as the photosensitizer (PS), and 0.1 M (2.8 mL HBF4 (aq) 48% w/w dissolved in 100 mL DMF) as the proton donor, under irradiation by a blue LED (λ = 445 nm, 88 mW cm−2) at 20 °C (Fig. 5, 6 and Tables 2, 3). Blanks and controls were run under identical conditions but with the omission of either the catalyst or PS or LED irradiation, or replacement of the copper complex with the simple salt copper(II) tetrafluoroborate. No hydrogen evolution was seen in the absence of PS or of LED irradiation; the other results are discussed below (Fig. S2–S6†). Multiple tests were run for all of the complexes and average values of turnover number (TON), maximum turnover frequency (TOF) and volume of H2 produced are given in Table 2.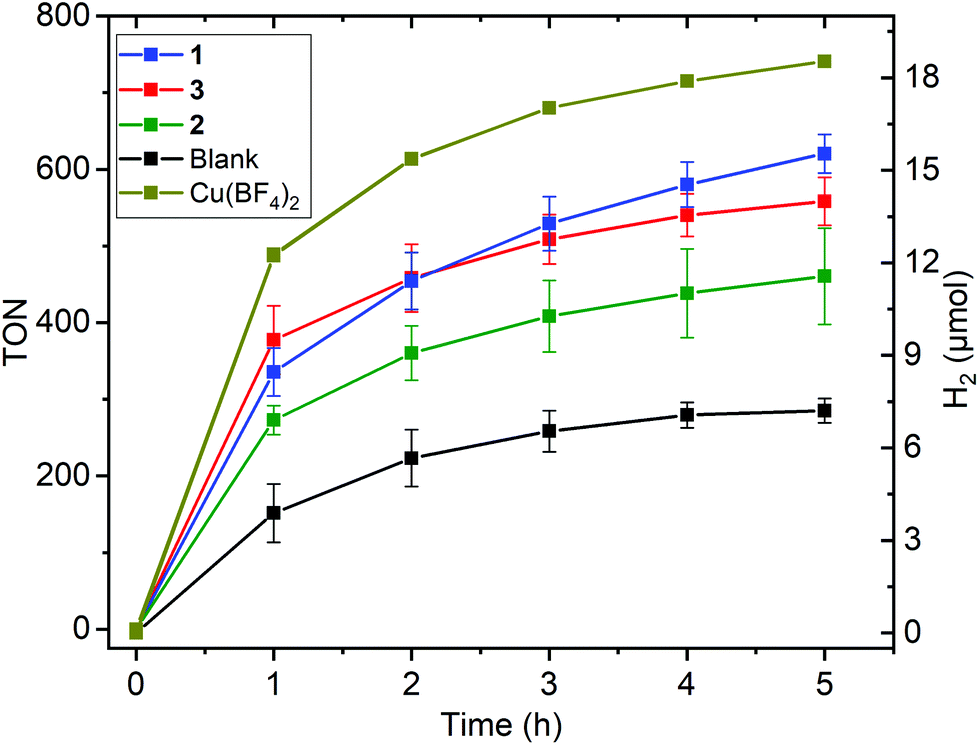 | ||
| Fig. 5 Hydrogen evolution (TON left, μmol right; see Fig. S12† for this plot in terms of hydrogen volume in mL) vs. time profile for copper complexes 1 [CuIILEt]BF4 (blue), 2 [CuIILEt-MePy]BF4 (green), and 3 [CuIILEtPy2]BF4 (red), as well as for the blank run with no copper catalyst (black) and the control experiment using the simple Cu(BF4)2 salt as the copper catalyst (olive), in DMF (Ccat = 5 μM) on irradiation with a blue LED (λ = 445 nm, 88 mW cm−2) at 20 °C, with 1.0 M TEOA, 0.2 mM [Ru(bpy)3](PF6)2 and 0.1 M HBF4/0.53 M H2O. Error bars show the standard deviation from the mean, calculated in Origin from multiple separate runs (Tables 2, S1 and Fig. S14–S17†). | ||
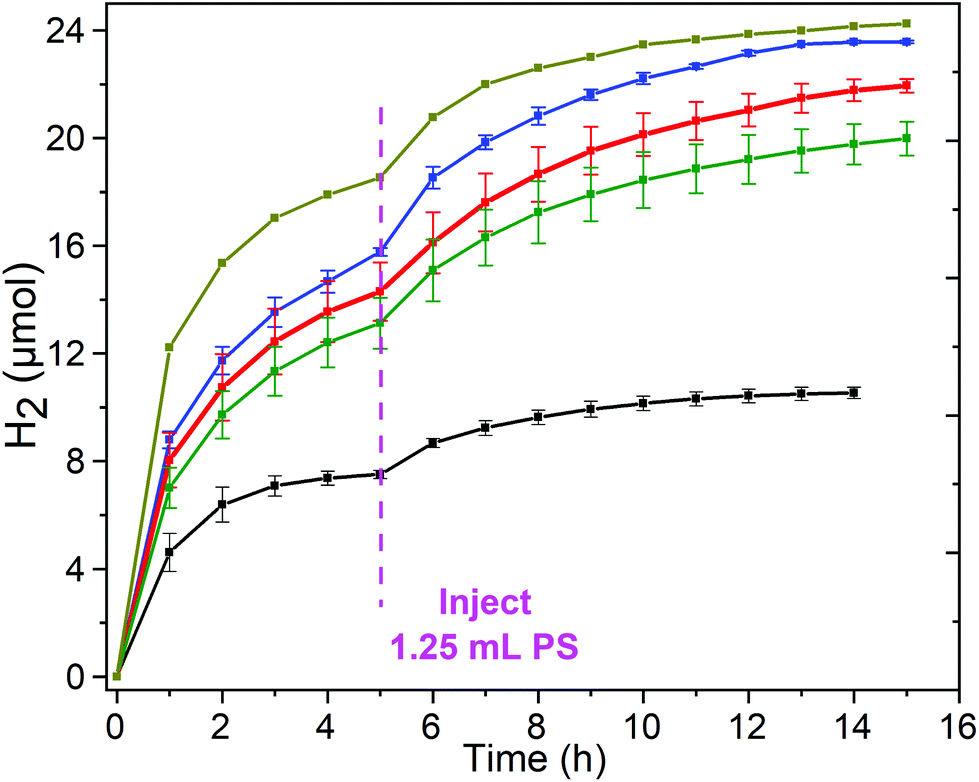 | ||
| Fig. 6 Hydrogen evolution-time profile for copper complexes 1–3 in DMF (Ccat = 5 μM), with a fresh 1.25 mL aliquot of PS solution (0.2 mM) added after 5 h, (blue) 1 [CuIILEt]BF4, (red) 3 [CuIILEtPy2]BF4 and (green) 2 [CuIILEt-MePy]BF4 as well as for the ‘blank’ run with 0.2 mM [Ru(bpy)3](PF6)2 (black) and the control experiment of Cu(BF4)2 salt (dark yellow). Conditions: Irradiation with a blue LED (λ = 445 nm, 88 mW cm−2) at 20 °C, with 1.0 M TEOA, 0.2 mM [Ru(bpy)3](PF6)2 and 0.1 M HBF4/0.53 M H2O. Error bars show the standard deviation from the mean, calculated from two separate runs (Table 3 and Fig. S18–S21†). | ||
| Catalyst | TON (molH2 molcat−1) | H2 (μmol) | TOFmax (min−1) |
|---|---|---|---|
| a Blanks run under the same conditions except: b cat = simple salt or c no cat or d no light. e Calculated assuming 5 μM catalyst was present (it's not) which enables comparison with the other TONs. NB. TON = 290 is TONPS = 7. | |||
| 1 | 620 ± 20 | 15.5 ± 0.6 | 16 ± 1 |
| 2 | 460 ± 60 | 11.4 ± 1.7 | 12 ± 3 |
| 3·0.5H2O | 560 ± 30 | 14.0 ± 0.7 | 16 ± 2 |
| Cu(BF4)2·xH2Oa,b | 740 | 18.5 | 26 |
| Cu(NO3)2·3H2Oa,b | 720 ± 20 | 17.9 ± 0.4 | 24 ± 3 |
| No coppera,c | 290 ± 10e | 7.2 ± 0.4 | 5 ± 1e |
| Darka,d | 0 | 0 | 0 |
| Complex | H2/μmol first cyclea | H2/μmol second cycleb | Restored activityc (%) |
|---|---|---|---|
| a 5 h irradiation with a blue LED (λ = 445 nm, 88 mW cm−2) at 20 °C, sacrificial reductant = 1 M TEOA, PS = 0.2 mM [Ru(bpy)3](PF6)2, proton source = 0.1 M HBF4/0.53 M H2O and 5 μM catalyst. b Reaction vessel was injected by 1.25 mL PS (same aliquot as used initially) after blue LED light irradiation for 10 hours. c Restored activity = H2 produced during the second cycle × (100)/H2 produced during the first cycle. | |||
| 1 | 15.8 | 7.8 | 49 |
| 2 | 13.1 | 6.7 | 51 |
| 3·0.5H2O | 14.3 | 7.6 | 53 |
| No copper | 7.5 | 2.5 | 33 |
| Cu(BF4)2 | 18.5 | 5.7 | 31 |
All three complexes, 1–3, are active for HER under photochemical conditions (Fig. 5 and Table 2), with approximately 11.4–15.5 μmol of hydrogen produced over 5 hours of blue light LED irradiation (Fig. 5). The corresponding TON values (mol of H2/mol of catalyst) increased from 460 for square pyramidal copper complex 2 to 560 for non-cyclic trigonal bipyramidal complex 3 to 620 for square planar 1 (Fig. 5 and Table 2). Comparison of these TON values to those of the only 3 molecular copper catalysts previously reported to be active for photocatalytic HER (Fig. 1), reveals that H1 and H2 had much higher TONs (6108 and 10014), albeit measured at 5 times lower catalyst concentration (1 μM) than herein, and using an Ir–PS.48 No TON was reported for the third such copper catalyst I.51
The blank test with no copper catalyst present produced an average of about half of that amount of hydrogen (7.2 μmol, TON = 290; Fig. 5, Table 2 and Fig. S16†). Disappointingly, control tests using a simple salt, CuII(BF4)2 or CuII(NO3)2, as the catalyst (Fig. 5, Table 2 and Fig. S7†) produced slightly more hydrogen (18.5 and 17.9 μmol, TON 740 and 720, respectively) than the copper complexes did. Eisenberg and co-workers have previously reported that the simple salt Ni(NO3)2, used as a control during photocatalytic HER testing in aqueous media, was highly active,66 and during the writing of this manuscript Wang, Fu and co-workers reported that CuSO4 in basic aqueous solution under photocatalytic conditions can form Cu2O and Cu nanoparticles that are active HER catalysts.41 But to the best of our knowledge no one has reported a simple copper salt control experiment in non-aqueous photocatalytic conditions prior to our report herein.
The trend in maximum turnover frequency (TOFmax/min−1) follows the same order at the TON values: 12 for square pyramidal 2 < 16 for non-cyclic trigonal bipyramidal 3 and square planar 1 < 24–26 for the simple copper salts (Table 2, see Fig. S8 of ESI† for TOF plot). In all cases this maximum occurred within 5–11 minutes of turning on the blue LED. A half-life, the time taken for this activity to drop by 50%, can be calculated (see ESI, Fig. S9–S12†) and is in the range 13–24 minutes for 1–3, and 17 minutes for the BF4 salt.
In an attempt to reactivate the system and thereby test whether the loss of activity was caused at least in part by decomposition of the PS, a fresh 1.25 mL aliquot of PS solution (same aliquot as added initially) was added to the test solutions of complexes 1, 2 and 3, and of the controls, after 5 hours of irradiation. In all cases this results in a partial recovery of the activity of the photocatalytic system (Fig. 6, Table 3 and Fig. S19–S21†). The total hydrogen produced over a further 10 hours, increased: 6.7 μmol for 2 to 7.6 μmol for 3 to 7.8 μmol for 1, which corresponds to restoration of 49–53% of the original activity. In the same way, restoration of the activity of the blank (no copper catalyst) and control (simple copper salt) was tested (Fig. 6, Table 3 and Fig. S18†), and found to restore 31–33% of the original activity, generating 2.5 and 5.7 μmol of hydrogen, respectively. These results are consistent with the deactivation of the system being caused in part by the decomposition of the PS,67 but also in part by decomposition of the catalyst and/or the sacrificial donor. Partial restoration of activity on addition of a second aliquot of [Ru(bpy)3]2+ has previously been reported for other photocatalytic HER systems: (a) a heptacoordinated CoII catalyst in aqueous media where 40% of the activity was restored,68,69 and (b) a CoII metallopeptide catalyst in neutral water where just 6% of activity was restored, and where they also noted that blue irradiation led to faster loss of activity than green light did.70
Clearly the fairly short time over which high photocatalytic HER activity is observed for these three copper complexes is a key limitation for the present system, so whilst these complexes double the number of molecular copper catalysts known to be active in photocatalytic HER (Fig. 1, box), the finding that the control is an even more active catalyst puts this into stark perspective. Hence our attention instead turned to the potential of 1–3 as electrocatalysts for HER.
Electrocatalytic hydrogen evolution
First the cyclic voltammetry data were obtained for 1–3 as well as for the control, Cu(BF4)2·xH2O, at 1 mM of copper(II) in dry MeCN with 0.1 M Bu4NPF6vs. 0.01 M AgNO3/Ag using a glassy carbon working electrode (Fig. 7 and S22†).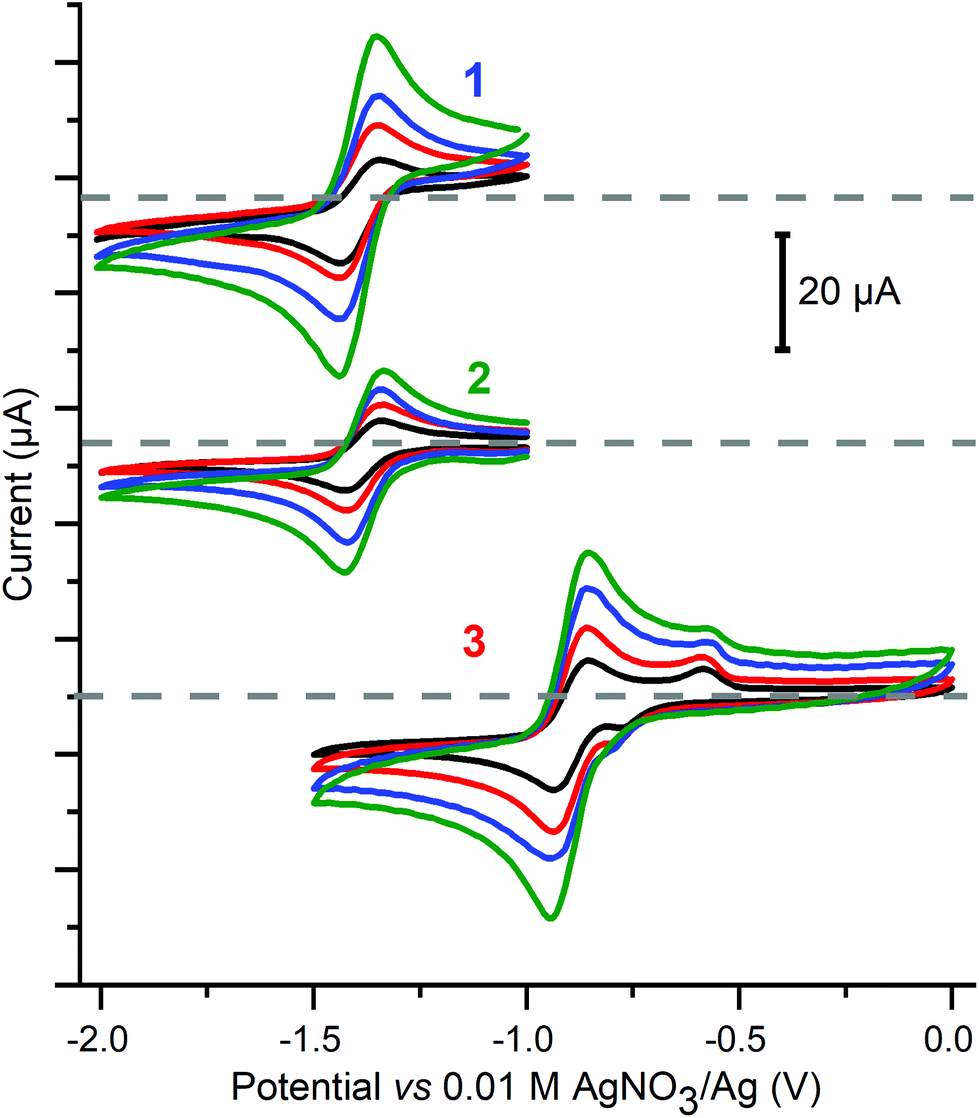 | ||
| Fig. 7 Cyclic voltammetry of the reversible redox processes seen at negative potentials for 1 mM MeCN solutions of 1 (top, E1/2 = −1.39 V, ΔE = 0.09 V, D = 7.7 × 10−6 cm2 s−1), 2 (middle, E1/2 = −1.39 V, ΔE = 0.09 V, D = 3.4 × 10−6 cm2 s−1), and 3 (bottom, E1/2 = −0.89 V, ΔE = 0.09 V, D = 5.4 × 10−6 cm2 s−1) Conditions: 0.1 M (Bu4N)PF6, glassy carbon working electrode (d = 3 mm, A = 0.071 cm2), 293 K, vs. 0.01 M AgNO3/Ag. In this system E1/2(Fc+/Fc) = 0.09 ± 0.01 V, with ΔE = 0.09 V, and this was unchanged even after 6 h of electrolysis (Fig. S31†). Scan direction: for 1 and 2 −1.0→−2.0→−1.0 V, for 3 0.0→−1.5→0.0 V; scan rate: 50 (black), 100 (red), 200 (blue) and 400 (green) mV s−1; grey dashed lines correspond to zero current for each set of CVs. | ||
All three copper complexes, 1–3, show a reversible redox process, at −1.39 V for square planar 1, −1.39 V for square pyramidal 2, and −0.89 V for trigonal bipyramidal 3 (Fig. 7), whereas the control experiment shows an irreversible process at approximately Epc = −1.0 V (Fig. S23†), all versus a 0.01 M AgNO3/Ag reference electrode. It is interesting to note that the pair of macrocyclic complexes (1 and 2), in which a square plane of donors is enforced upon the copper centre, have identical E1/2(Cu+/Cu2+) values (−1.39 V), whereas the non-cyclic ligand complex (3) allows the copper centre to adopt a trigonal bipyramidal geometry, and this complex is far easier to reduce (E1/2(Cu+/Cu2+) = −0.89 V) than the other two complexes. It is also important to note that (before adding acid) there is no evidence of a stripping wave in the CVs of 1–3 (Fig. S22, ESI†).
The linear current vs. square root of scan rate plots for 1 to 3 (Fig. S24†) confirm that these Cu2+ ↔ Cu+ redox events are reversible and diffusion controlled. Furthermore, this enables the use of the Randles–Sevcik equation71,72 to determine the diffusion coefficients (D, cm2 s−1) for each complex: 7.7 × 10−6 for 1, 3.4 × 10−6 for 2, and 5.4 × 10−6 for 3 (see the ESI† for more details). This is in the same ballpark as the diffusion coefficient of 1.24 × 10−5 cm2 s−1, determined in aqueous phosphate buffer (pH = 12), for a copper(II) complex of a tetradentate N4-donor ligand with a dangling OH head unit, used as a catalyst for the water oxidation reaction (WOR).73
Next, each of the three complexes 1–3, and Cu(BF4)2·xH2O as a control, were tested as HER electrocatalysts in the presence of acetic acid (Fig. 8–11 and S25†). Acetic acid was chosen as the acid source as in MeCN it has two key advantages, low homoconjugation,74,75 and being a weak acid (pKa = 23.5)76,77 which results in long catalyst lifetimes – but a key disadvantage is that it is challenging to reduce the protons to form H2, due to a relatively high thermodynamic potential (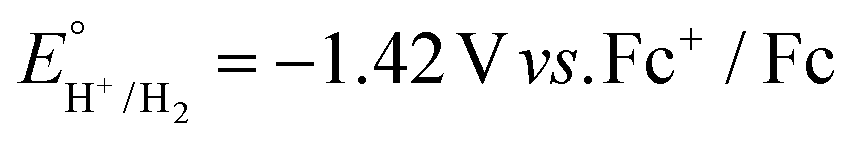 on glassy carbon in 0.1 M Bu4NPF6 in MeCN, which is approx. −1.41 V vs. 0.01 M AgNO3/Ag).74,77
on glassy carbon in 0.1 M Bu4NPF6 in MeCN, which is approx. −1.41 V vs. 0.01 M AgNO3/Ag).74,77
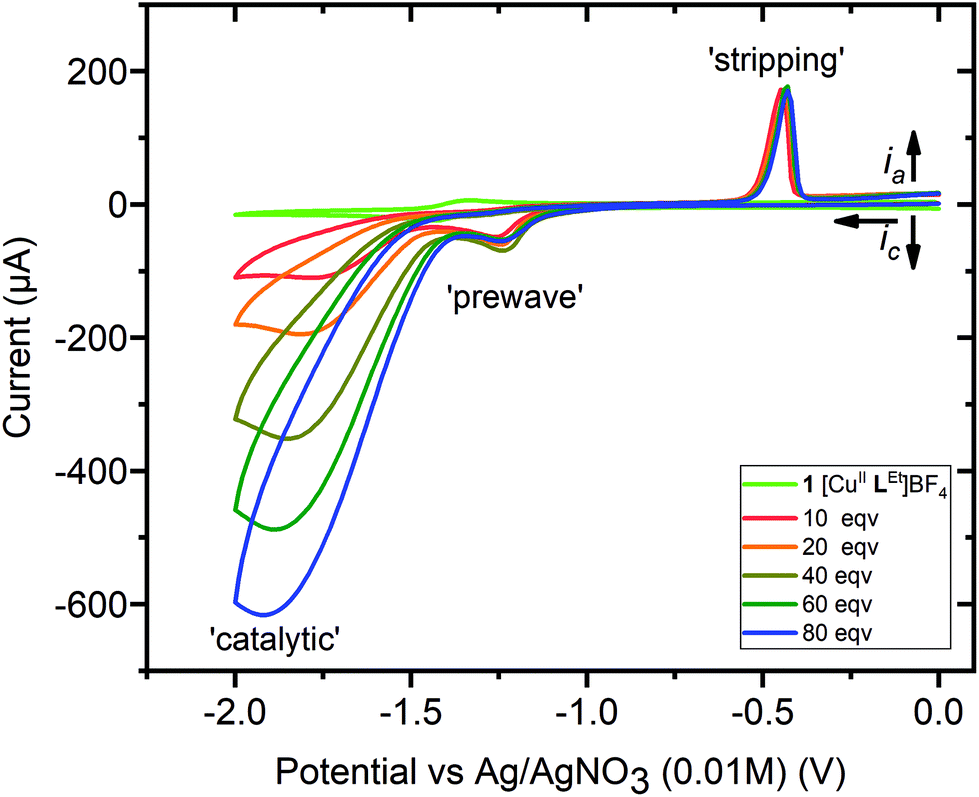 | ||
| Fig. 8 Cyclic voltammetry, 0→−2.0→0 V vs. 0.01 M AgNO3/Ag, for a 1 mM MeCN solution of 1 (light green, no acid), with successive additions of 10 or 20 equivalents of acetic acid (see key), up to a total of 80 equivalents (blue) = 80 mM, with increasing [acid] leading to increasing catalytic wave currents. Conditions: 0.1 M (NBu4)PF6, glassy carbon working electrode (d = 3 mm, A = 0.071 cm2), 293 K, scan rate 100 mV s−1. Before and after this study, E1/2(Fc+/Fc) = 0.09 ± 0.01 V, with ΔE = 0.09 ± 0.01 V (see Fig. S25†). | ||
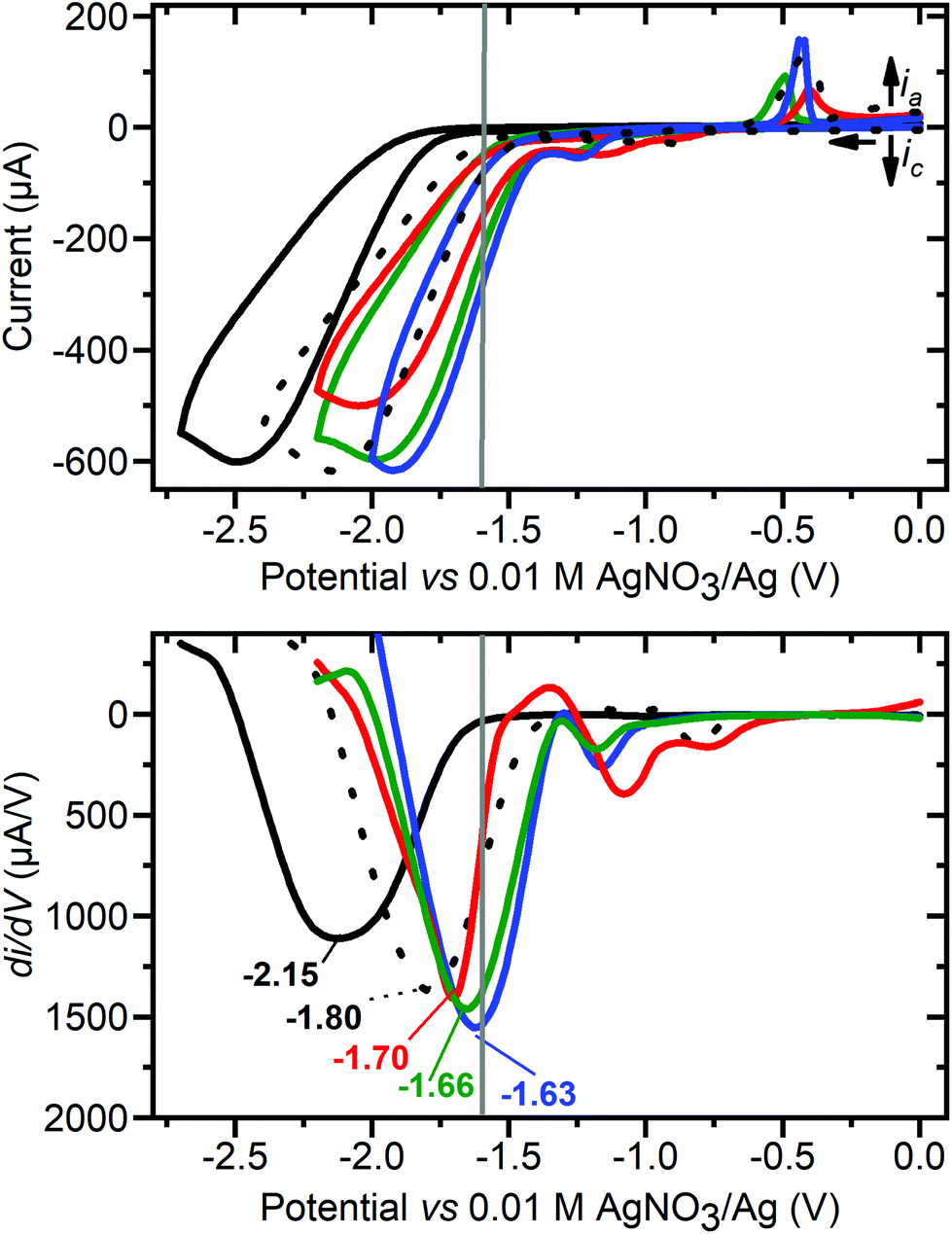 | ||
| Fig. 9 (Top) Cyclic voltammograms of 80 mM acetic acid in MeCN (control, black line) in the presence of 1 mM: CuII(BF4)2·xH2O (black dots), 2 (green), 3 (red) and 1 (blue). (Bottom) The first derivative of the forward scan of each CV, annotated with Einf (potential at inflection point in V vs. 0.01 M AgNO3/Ag), 80 mM acetic acid (−2.15, black line), in the presence of 1 mM: CuII(BF4)2·xH2O (−1.80, black dots), 3 (−1.70, red), 2 (−1.66, green) and 1 (−1.63, blue). Conditions: 100 mV s−1, 0.1 M (Bu4N)PF6, glassy carbon working electrode (d = 3 mm, A = 0.071 cm2), 293 K. The grey vertical line at −1.6 V is the Eapplied used in the controlled potential coulometry studies. Before and after each of these studies, E1/2(Fc+/Fc) = 0.09 ± 0.01 V, with ΔE = 0.09 ± 0.01 V (see Fig. S25†). | ||
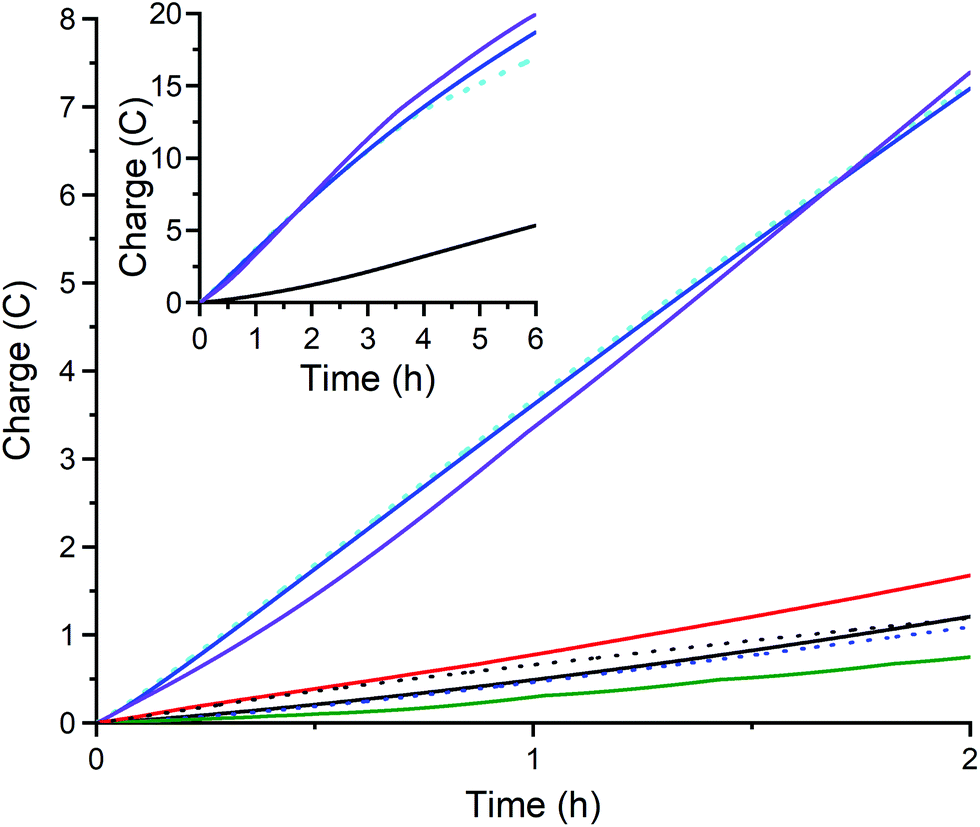 | ||
| Fig. 10 Charge transferred during controlled potential electrolysis at −1.60 V of an 8 mL solution of 80 mM acetic acid (black line, blank) in the presence of 1 mM: 1 (duplicate runs, blue and violet), 2 (green), 3 (red), CuII(BF4)2·6H2O (black dots, control). After electrolysis with 1 the glassy carbon working electrode (d = 3 mm, A = 0.071 cm2) was gently rinsed with acetonitrile and the electrolysis repeated in freshly made electrolyte with 80 mM acetic acid but without adding catalyst (blue dashes; rinse and repeat test; confirms there is no catalytically active deposit on the working electrode). Tests on 1 in the absence of mercury (duplicate runs, blue and violet) and in the presence of 1 mL of mercury drop (sky blue dots) confirm the absence of catalytically active nanoparticles. The inset shows extended electrolysis of 6 hours, carried out for the blank run and for the three runs of complex 1 (with and without mercury drop). Also see Table S3.† | ||
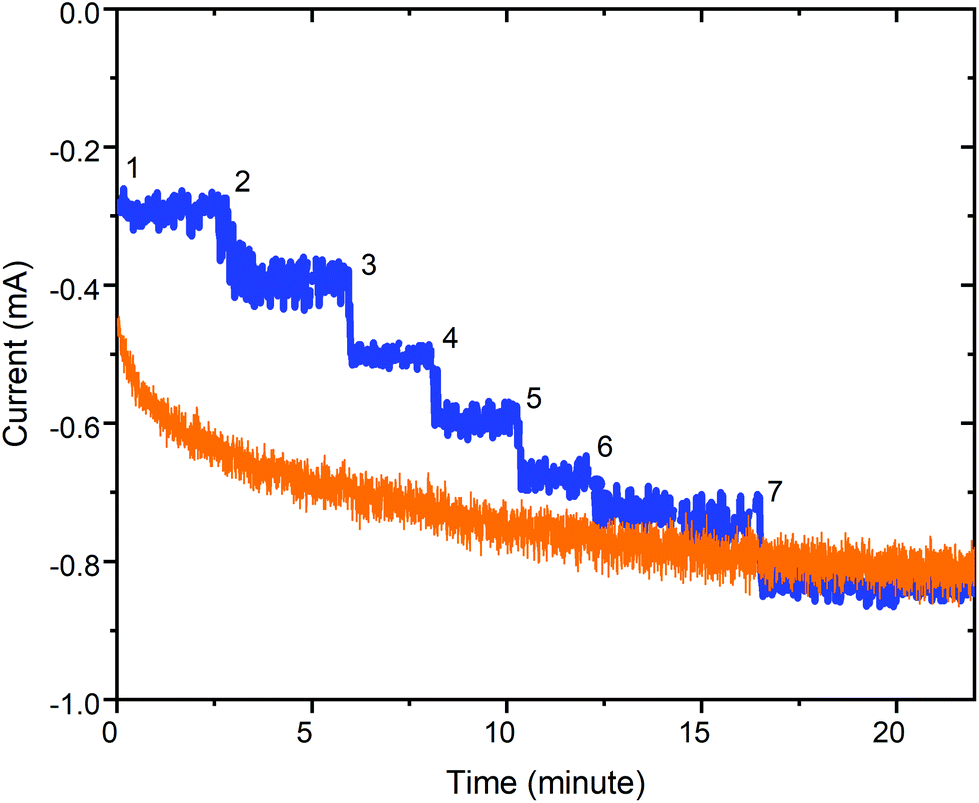 | ||
| Fig. 11 Plot of current response versus time at E = −1.6 V vs. Ag/AgNO3 (0.01 M) for a 0.33 M acetonitrile solution of 1 when the acetic acid is either (blue) added in 7 portions reaching 80 mM in H+ after the 7th addition or (orange) it is 80 mM from the start. Conditions: Glassy carbon working electrode (d = 3 mm, A = 0.071 cm2), 20 °C, and Pt counter electrode. Also see Table S3.† | ||
The CVs of each of the complexes, 1–3, were then obtained in the presence of increasing concentrations of acetic acid, and these showed catalytic waves, with the maximum current increasing with acid concentration (Fig. 8 and S25†). For complexes 1 and 2 a small prewave is observed (Fig. S32; Table S4†), at a more positive Ep value (−1.24 to −1.33 V) than the reversible process seen when no acid is present (−1.42 to −1.43 V), whilst for complex 3 the reversible process seen when no acid is present (−0.93 V) continues to be observed, and at a more positive Ep value than the prewave that is also seen after adding acid (−1.17 to −1.29 V).
In addition, whilst there is no evidence of a stripping wave in the CVs of these three complexes before adding acid (light green trace in Fig. 8; also Fig. S33, ESI†), after adding acetic acid a small stripping wave, consistent with the deposition of some Cu0 on the working electrode,35,36 is seen for all three complexes, 1–3, between −0.44 and −0.50 V (Fig. 8 and S25†). The results in the case of the simple salt are very different: the stripping peak is present before adding any acid, at −0.53 V, and this shifts to −0.33 V as acid is added (Fig. S25†).
Next the blank (Fig. 9, black, no added copper compound) for 80 mM acetic acid was run at 100 mV s−1, giving Epc = −2.45 V and Einf = −2.15 V vs. 0.01 M AgNO3/Ag (where Einf is the irreversible reduction potential inflection point74). These are similar to those reported by Dempsey and co-workers under similar conditions but with 25 mM acetic acid (Einf = −2.38 V vs. Fc+/Fc, which is approx. −2.30 V vs. 0.01 AgNO3/Ag).74 Then the various copper complexes were added to the 80 mM acetic acid: 1 resulted in the smallest Epc for proton reduction (Epc = −1.91 V, Einf = −1.63 V, and the potential corresponding to half the maximum catalytic current Ecat/2 = −1.60 V) with the onset potential of the catalytic wave at about −1.37 V, followed by 2 (Epc = −2.01, Einf = −1.66 and Ecat/2 = −1.66 V), 3 (Epc = −2.04, Einf = −1.70 and Ecat/2 = −1.66 V) and then Cu(BF4)2·xH2O (Epc = −2.16, Einf = −1.80 and Ecat/2 = −1.78 V).
The promising electrocatalytic HER activity seen in the case of 1 is also indicated by an icat/ip ratio of 34 at 100 mV s−1 with [cat] = 1 mM, [acetic acid] = 80 mM, and 20 °C, where icat is peak catalytic current and ip is peak current in absence of acid. This is comparable to that reported for copper catalyst G (icat/ipc = 24),51 and also to DuBois's nickel catalyst (icat/ipc = 38 and 74 in dry and wet MeCN, respectively).78 A much higher icat/ip of 303 was reported for copper corrole D1 using TFA (160 mM) and water (2.4 M) in acetonitrile.44
To further evaluate the catalytic activity of 1 for hydrogen evolution, controlled potential coulometry (see Fig. S26 for the electrochemical cell and Table S3† for key data) was carried out at Eapplied = −1.60 V for 2 hours (Fig. 9 and 10), using a small glassy carbon working electrode (diameter 3 mm, A = 0.071 cm2). As shown in Fig. 10, complexes 2 (green) and 3 (red) – as well as the control, CuII(BF4)2·xH2O (black dots) and the blank (black line) – all showed minimal activity (<1.7C, 2.2e per metal center, TON < 1.1) whereas complex 1 (blue and violet) stood out, transferring an average charge of 7.3C over 2 hours (9.5e per 1, TON = 4.7) with bubbles seen forming underneath the electrode (Fig. S26†).
The ‘overpotential necessary for catalysis’74,79 (Ecat/2 = −1.64 V) minus 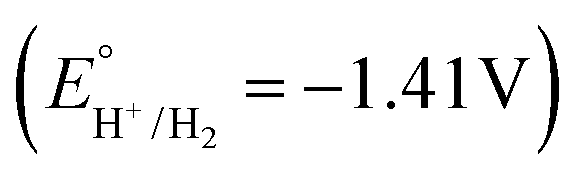 of about 0.23 V for complex 1 is at the low end of related values reported for the handful of molecular copper(II) HER electrocatalysts in the literature to date (Fig. 1): ‘onset overpotentials’ for A (0.420 V) in aqueous phosphate buffer solution (pH 2.5),42 for G (0.440 V) for acetic acid in DMF/H2O (95:5 v/v);51 ‘overpotentials’ for D (0.450 V) for trifluoroacetic acid (TFA) in acetonitrile (determined using Ecat/2),44 and in water at pH = 7 for B2 (0.636 V),46C3 (0.639 V)47 and B1 (0.817 V).45 But, as noted by Dempsey, such overpotentials “should not be considered a general parameter for direct, quantitative, catalyst comparison between independent reports because of the nonuniform use of the parameter Ecat”.80
of about 0.23 V for complex 1 is at the low end of related values reported for the handful of molecular copper(II) HER electrocatalysts in the literature to date (Fig. 1): ‘onset overpotentials’ for A (0.420 V) in aqueous phosphate buffer solution (pH 2.5),42 for G (0.440 V) for acetic acid in DMF/H2O (95:5 v/v);51 ‘overpotentials’ for D (0.450 V) for trifluoroacetic acid (TFA) in acetonitrile (determined using Ecat/2),44 and in water at pH = 7 for B2 (0.636 V),46C3 (0.639 V)47 and B1 (0.817 V).45 But, as noted by Dempsey, such overpotentials “should not be considered a general parameter for direct, quantitative, catalyst comparison between independent reports because of the nonuniform use of the parameter Ecat”.80
Given ongoing activity shown by 1 over the first 2 hours of controlled potential electrolysis at −1.6 V, this was continued for a total of 6 hours, resulting in an average total of 19.3C being transferred – with the catalyst retaining an almost constant level of activity throughout the 6 hours period (Fig. 10, inset, duplicate runs in blue and violet). i.e. after 2 hours 7.3C is transferred, so 3 times this gives an expected transfer of 21.9C after 6 hours, which is close to the 19.3C observed, consistent with 1 having a relatively long lifetime as an HER electrocatalyst. The 19.3C transferred equates to 25.0e per 1, so to a TON(H2) of 12.5 (assuming 100% FE). Clearly this TON could be further increased by use of a larger surface area electrode and/or running the electrolysis for longer as the catalyst clearly remains active after 6 hours. Nevertheless, the present TON(H2) of 12.5 compares favorably to the TON(H2) of 11 reported for DuBois's 0.90 M nickel catalyst in acetonitrile, using 0.43 M [(DMF)H]OTf, (OTf = triflate or trifluoromethanesulfonate) and 1.2 M of water, over 30 minutes, after which the catalyst has decomposed,78 and the TON of 23 for 0.30 M copper corrole D (Fig. 1) in acetonitrile, with TFA (180 mM), over 2 hours.44
Notably, further controlled potential electrolysis test runs at −1.6 V, with a third of the catalyst concentration (0.33 mM in 1), show that 1 remains an effective electrocatalyst for HER at this lower catalyst loading, regardless of whether the 80 mM acetic acid is present from the start (Fig. 11, orange trace), or is added in 7 aliquots (Fig. 11, blue trace). Immediate current growth is seen (steps) each time an aliquot of acetic acid is added (blue trace), and the overall result after 23 minutes is that it has approximately the same current flow i25 min = −0.85 mA (transferred a total of 0.82C; 3.2e− per 1) as that obtained after 23 minutes when it is 80 mM in acid from the start i25 min = −0.81 mA (total of 1.00C; 3.9e− per 1).
Given these promising findings, further checks were made on complex 1. Firstly, it was shown to be stable in 90 mM acetic acid for at least 5 hours, by monitoring the UV-vis spectrum (Fig. S28†). Secondly, before electrolysis the solution of 1 is dark golden yellow whereas during electrolysis it turns bright yellow (Fig. 12). After electrolysis, on exposure to air for 30 min (electrodes removed) it darkens again to gold yellow, albeit not returning fully to the original colour (Fig. 12 and S29†), even after longer air exposure. In part this is due to some diffusion of the catalyst into the central compartment of the “H” cell (Fig. S26†) during the 6 hours of electrolysis, reducing the concentration of the solution in the working electrode chamber: the pink trace in Fig. 12 shows an estimated correction to the UV-Vis spectrum (after exposure to air for 30 min) for this dilution, and is consistent with 76% of 1 being intact at this point.
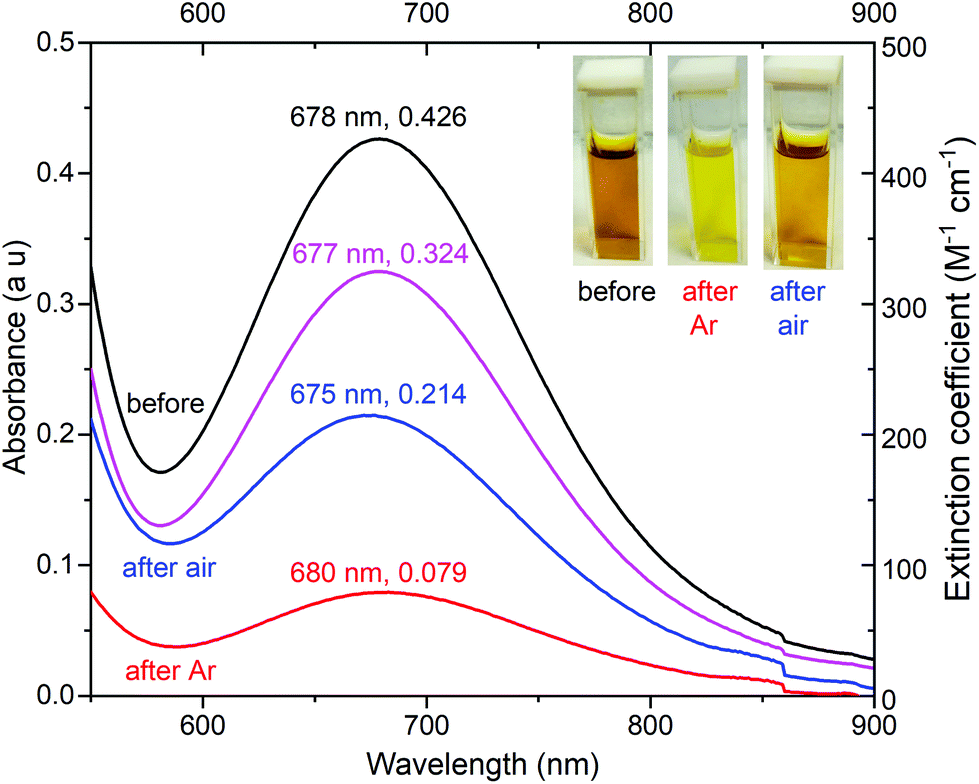 | ||
| Fig. 12 UV-vis spectra, focusing on the d–d band, of 1 mM [CuIILEt]BF4 (1), in the presence of 80 mM acetic acid in 0.1 M Bu4NPF6/MeCN: (black) before electrolysis commenced, (red) immediately after 6 hours electrolysis, (blue) after the post-electrolysis solution was exposed to air for 30 minutes, and (pink) is the blue curve recalculated in 10.5 not 8 mL (allowing for diffusion into the central compartment). Electrolysis conditions: Glassy carbon working electrode (d = 3 mm, A = 0.071 cm2), 20 °C, and Pt counter electrode. For the full range UV-vis spectra see Fig. S29.† | ||
Thirdly, the reference electrode was checked against Fc+/Fc before and after the 6 h electrolysis and was unshifted (Fig. S31†).
Fourthly, a rinse and repeat experiment – whereby the electrode was removed after 6 hours of controlled potential electrolysis at −1.60 V, gently rinsed with MeCN, and then placed into a fresh solution for electrolysis (without adding catalyst) showed minimal activity over the next 2 hours (blue dashed lines in Fig. 10), indicating that no catalytically active heterogeneous deposit was present on the electrode surface. Nevertheless, future studies will probe this point further, as this may well prove to be a case of a false negative (see introduction). Another key test performed was the mercury drop test: the 6 h electrolysis of 1 was repeated in same manner as before except that 1 mL of mercury was added (Fig. 10, sky blue dots). Pleasingly, a similar amount of charge (17.0C after 6 h; 7.2C after 2 h) was passed as when no mercury was present (average 19.3C after 6 h; 7.3 after 2 h; blue and violet), which indicates that the electrocatalytic activity is probably not due to nanoparticles or similar having formed. Whilst the results of both of these additional tests42,43 are consistent with the catalytically active species being homogeneous, not heterogeneous (see Fig. S29 and S30 in the ESI† for more details), future studies will probe this point further, as a small stripping wave is seen, and false negatives are not uncommon.
Conclusions
Inspired by the promising HER activity, under photocatalytic conditions, seen for (a) just three copper complexes to date24,48,51 and (b) the cobalt complex of the N4-donor [1 + 1] Schiff base macrocycle HLEt (made by the condensation of diphenylamine-2,2′-dicarboxaldehyde and diethylenetriamine) reported by some of us,16 herein the number of such copper catalysts is doubled.Firstly, two new N5-donor diphenylamine-based ligands have been prepared and characterised: an ‘armed’ macrocycle HLEt-MePy formed by alkylation of HLEt, and a non-cyclic analogue HLEtPy2. Secondly, 1:1 reactions of the respective ligand with copper(II) tetrafluoroborate in the presence of TEA, gave the literature complex [CuIILEt]BF4 (1)59 and the two new complexes [CuII(LEt-MePy)]BF4·0.5H2O (2·0.5H2O) and [CuII(LEtPy2)]BF4 (3). Single crystal structure determinations reveal contrasting copper(II) geometries: square planar in 1, square pyramidal in 2 and trigonal bipyramidal in 3.
Interestingly, despite the contrasting copper(II) geometries, all three of these readily prepared complexes, 1–3, have similar HER activities (TON 460–620) under the photocatalytic conditions employed (sacrificial reductant TEOA, PS [Ru(bpy)3]2+, proton source HBF4, irradiated by blue LED). But, disappointingly, on running the appropriate control and blank tests – not reported for the previous copper HER catalysts in the literature – complexes 1–3 are revealed to be no more active than the simple salts copper(II) tetrafluoroborate/nitrate (TON 720–740). This study therefore highlights the importance of always doing, and reporting, the results of blanks and controls, in order to put the observed activity of the complexes studied into proper context.
In all cases (copper complexes/salts), partial activity is restored on adding a second aliquot of PS after 5 hours, consistent with the drop in activity being due in part to PS decomposition, along with decomposition of the complex and sacrificial electron donor. This, taken with the fact that the blank run with no copper catalyst present shows about half the HER activity of complexes 1–3, indicates that a more stable PS (e.g. Ir–PS48), and/or irradiation wavelength (e.g. green not blue70), should be used in future tests.
But, given the disappointing photocatalytic HER results for 1–3, our attention instead turned to testing them for electrocatalytic HER. Pleasingly all three complexes, 1–3, show reversible redox processes at −0.89 (trigonal bipyramidal 3) and −1.39 (square planar 1 and square pyramidal 2) V vs. 0.01 M Ag/AgNO3. Furthermore, the square planar macrocyclic complex 1 shows good and ongoing electrocatalytic HER activity at −1.60 V for 6 h, with a TON(H2)6 h = 12.5, whereas both of the other complexes (2 and 3), and the control, showed similar, much more modest activity, with TON(H2) < 1.1 compared with 4.7 for 1, after 2 hours.
Hence the key finding of this study is that square planar copper complex 1 is, or forms, a promising and robust electrocatalyst for HER, showing (a) good and robust ongoing activity even after 6 hours, and (b) mercury drop, as well as ‘rinse and repeat’, test results that are consistent with homogeneous, not heterogeneous, electrocatalysis occurring. But false negatives for such tests can occur, and small stripping waves are seen, so the nature of the catalytically active species will be probed further in future studies. Given the identical redox potentials in MeCN (−1.39 V vs. 0.01 M Ag/AgNO3) for the pair of macrocyclic complexes, square planar 1 (NH in ligand backbone) and the closely related but square pyramidal 2 (no NH in ligand backbone), the difference in electrocatalytic HER activity is interesting and should also be probed further in the future. The importance of proton relays, facilitated by NH moieties, has been demonstrated by others,81–85 and may also be the key here. But if the electrocatalytically active species proves to be heterogeneous, then clearly the nature of that species is dependent on the choice of precursor.
In the longer term we aim to improve the activity and lifetime of these catalysts (a) for photocatalytic HER by further refining the ‘mix’ of components and the irradiation wavelength used, and (b) for electrocatalytic HER by employing other 3d metal ions and modifying the ligand skeleton further to develop new members of this promising new family, in particular aiming for new members that retain NH functionality. But our immediate priorities are more in depth electrocatalytic testing of the promising electrocatalyst 1. First we will carry out additional tests to identify whether or not a metastable heterogeneous deposit, not seen in the rinse and repeat test, is being formed, and if so what it is composed of. Other tests will include longer runs, runs with added water and in aqueous solution, probing the kinetics, and getting a local gc set up to quantify (H2), as well as testing these copper catalysts for activity in the photo- and electro-catalytic CO2 reduction reaction (CO2RR).
Experimental section
Elemental analyses (C, H, N) were determined at the Campbell Microanalytical Laboratory, University of Otago. 1H and 13C NMR spectra were recorded on a Varian 400 or 500 MHz NMR spectrometer at 298 K. Chemical shifts are reported in parts per million and referenced to the residual protonated solvent peak in the 1H NMR spectra and the residual solvent peak in the 13C NMR spectra [CDCl3: 7.26 ppm (1H) and 72.16 ppm (13C); d6-DMSO: 2.50 ppm (1H) and 39.52 ppm (13C)]. ESI MS spectra (Fig. S42–S45†) were collected on a Bruker MicrOTOFQ spectrometer. UV-vis spectra were obtained on a Varian 500 Scan UV-vis-NIR spectrophotometer. Infrared (IR) spectra were recorded on a Bruker ATR-IR spectrometer with a diamond anvil Alpha-P module, within the range of 400–4000 cm−1. Crystallographic data for the structures have been deposited with the Cambridge Crystallographic Data Centre, CCDC 1983967–1983968.† Photo- and electro-catalysis testing instrumentation and details are provided in the ESI.†General synthetic procedures
Methanol and acetonitrile were HPLC grade and other solvents were reagent grade. TEA (Fisher Scientific), 2-(bromomethyl)pyridine hydrobromide (AKSci) and 2-aminoethylpyridine (Aldrich) were bought and used as supplied. Diethylene triamine (Fisher) was purified by distillation under a reduced pressure of 200 mbar and temperature of 100 °C. Dpa and HLEt were prepared by the literature procedures.59,86 All reactions were carried out by AA (Otago) and were conducted in air unless otherwise stated.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–H), 2996 (C–H), 2911, 2843, 1630 (CN), 1589, 1514, 1433, 1316, 1144, 1045, 984, 741, 605, 511.
:1, v/v) was added TEA (0.070 mL, 0.54 mmol). The resulting bright yellow solution was stirred for 5 minutes at room temperature before CuII(BF4)2·H2O (0.138 g, 0.54 mmol) was added, causing a sudden color change to deep red. The solution was stirred at 60 °C for 3 hours, then stirred at room temperature overnight. The resulting brown solid was collected by filtration and washed with diethyl ether (2 × 15 mL) and dried in vacuo to give 1 (0.21 g, 88%). Microanalysis calcd for C18H19N4BF4Cu (%): C, 48.94; H, 4.34; N, 12.68. Found C, 48.88; H, 4.36; N, 12.70. ESI-MS m/z = 354.0902 (calcd for [C18H19CuN4]+ 354.0900). UV-Vis λmax/nm (DMF) (ε/dm3 mol−1 cm−1) = 334 (4334), 401 (3762), 467 (13500), 672 (453). Dark orange single crystals of 1 suitable for X-ray determination was grown by vapor diffusion of diethyl ether into a small sample of the reaction solution (1 mL).
:1, v/v) was added a blue methanol solution (2 mL) of CuII(BF4)2·H2O (0.040 g, 0.156 mmol). The resulting deep red solution was stirred at 60 °C for 3 h, then stirred at room temperature overnight, before being vapor diffused with diethylether. Dark red needle-like crystals of 2, suitable for a single crystal X-ray structure determination, formed over six days, and were collected by filtration, and washed with diethyl ether (2 × 15 mL) and dried in vacuo to give 2 (44 mg, 68%). Microanalysis calcd for C24H24N5BF4Cu (%): C 54.10, H 4.54, N 13.14. Found C 53.81, H 5.00, N 12.83. ESI-MS m/z = 445.1293 (calcd for [C24H24N5Cu]+ 445.1322). UV-Vis λmax/nm (DMF) (ε/dm3 mol−1 cm−1) = 336 (3793), 408 (3313), 479 (6216), 789 (342).
128), 917 (309). IR (ATR) (ν, cm−1): 3170, 3082, 1607, 1545, 1540, 1425, 1397, 1311, 1259, 1192, 1149, 1032, 879, 742, 633, 581, 513, 467.
C–H), 2996 (C–H), 2911, 2843, 1630 (CN), 1589, 1514, 1433, 1316, 1144, 1045, 984, 741, 605, 511.
:1, v/v) was added TEA (0.070 mL, 0.54 mmol). The resulting bright yellow solution was stirred for 5 minutes at room temperature before CuII(BF4)2·H2O (0.138 g, 0.54 mmol) was added, causing a sudden color change to deep red. The solution was stirred at 60 °C for 3 hours, then stirred at room temperature overnight. The resulting brown solid was collected by filtration and washed with diethyl ether (2 × 15 mL) and dried in vacuo to give 1 (0.21 g, 88%). Microanalysis calcd for C18H19N4BF4Cu (%): C, 48.94; H, 4.34; N, 12.68. Found C, 48.88; H, 4.36; N, 12.70. ESI-MS m/z = 354.0902 (calcd for [C18H19CuN4]+ 354.0900). UV-Vis λmax/nm (DMF) (ε/dm3 mol−1 cm−1) = 334 (4334), 401 (3762), 467 (13500), 672 (453). Dark orange single crystals of 1 suitable for X-ray determination was grown by vapor diffusion of diethyl ether into a small sample of the reaction solution (1 mL).
:1, v/v) was added a blue methanol solution (2 mL) of CuII(BF4)2·H2O (0.040 g, 0.156 mmol). The resulting deep red solution was stirred at 60 °C for 3 h, then stirred at room temperature overnight, before being vapor diffused with diethylether. Dark red needle-like crystals of 2, suitable for a single crystal X-ray structure determination, formed over six days, and were collected by filtration, and washed with diethyl ether (2 × 15 mL) and dried in vacuo to give 2 (44 mg, 68%). Microanalysis calcd for C24H24N5BF4Cu (%): C 54.10, H 4.54, N 13.14. Found C 53.81, H 5.00, N 12.83. ESI-MS m/z = 445.1293 (calcd for [C24H24N5Cu]+ 445.1322). UV-Vis λmax/nm (DMF) (ε/dm3 mol−1 cm−1) = 336 (3793), 408 (3313), 479 (6216), 789 (342).
128), 917 (309). IR (ATR) (ν, cm−1): 3170, 3082, 1607, 1545, 1540, 1425, 1397, 1311, 1259, 1192, 1149, 1032, 879, 742, 633, 581, 513, 467.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Otago for supporting this research (including a PhD scholarship AA) and the Catalyst Seed Fund (New Zealand) for supporting our collaborative studies (including exchange visits in both directions). GSH and OS also thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for funding. Finally, we would like to thank the referees very much for taking the time to provide such detailed and constructive feedback, which has enabled us to significantly improve this manuscript.Notes and references
- J. F. Johnstone, C. D. Allen, J. F. Franklin, L. E. Frelich, B. J. Harvey, P. E. Higuera, M. C. Mack, R. K. Meentemeyer, M. R. Metz, G. L. Perry, T. Schoennagel and M. G. Turner, Changing disturbance regimes, ecological memory, and forest resilience, Front. Ecol. Environ., 2016, 14, 369–378 CrossRef.
- T. P. Hughes, M. L. Barnes, D. R. Bellwood, J. E. Cinner, G. S. Cumming, J. B. C. Jackson, J. Kleypas, I. A. van de Leemput, J. M. Lough, T. H. Morrison, S. R. Palumbi, E. H. van Nes and M. Scheffer, Coral reefs in the Anthropocene, Nature, 2017, 546, 82 CrossRef CAS.
- A. L. Hammond, Solar energy: the largest resource, Science, 1972, 177, 1088–1090 CrossRef CAS.
- N. Armaroli and V. Balzani, The Future of Energy Supply: Challenges and Opportunities, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- J. Su and L. Vayssieres, A Place in the Sun for Artificial Photosynthesis?, ACS Energy Lett., 2016, 1, 121–135 CrossRef CAS.
- S. Berardi, S. Drouet, L. Francas, C. Gimbert-Surinach, M. Guttentag, C. Richmond, T. Stoll and A. Llobet, Molecular artificial photosynthesis, Chem. Soc. Rev., 2014, 43, 7501–7519 RSC.
- N. Armaroli and V. Balzani, The Hydrogen Issue, ChemSusChem, 2011, 4, 21–36 CrossRef CAS.
- J. A. Turner, Sustainable hydrogen production, Science, 2004, 305, 972–974 CrossRef CAS.
- Z. Han, F. Qiu, R. Eisenberg, P. L. Holland and T. D. Krauss, Robust photogeneration of H2 in water using semiconductor nanocrystals and a nickel catalyst, Science, 2012, 338, 1321–1324 CrossRef CAS.
- A. L. Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, From Hydrogenases to Noble Metal–Free Catalytic Nanomaterials for H2 Production and Uptake, Science, 2009, 326, 1384–1387 CrossRef.
- C. V. Krishnan, B. S. Brunschwig, C. Creutz and N. Sutin, Homogeneous catalysis of the photoreduction of water. 6. Mediation by polypyridine complexes of ruthenium(II) and cobalt(II) in alkaline media, J. Am. Chem. Soc., 1985, 107, 2005–2015 CrossRef CAS.
- P. Du, J. Schneider, G. Luo, W. W. Brennessel and R. Eisenberg, Visible Light-Driven Hydrogen Production from Aqueous Protons Catalyzed by Molecular Cobaloxime Catalysts, Inorg. Chem., 2009, 48, 4952–4962 CrossRef CAS.
- A. Fihri, V. Artero, A. Pereira and M. Fontecave, Efficient H2-producing photocatalytic systems based on cyclometalated iridium- and tricarbonylrhenium-diimine photosensitizers and cobaloxime catalysts, Dalton Trans., 2008, 5567–5569 RSC.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Cobaloxime-Based Photocatalytic Devices for Hydrogen Production, Angew. Chem., Int. Ed., 2008, 47, 564–567 CrossRef CAS.
- P. Zhang, M. Wang, C. Li, X. Li, J. Dong and L. Sun, Photochemical H2 production with noble-metal-free molecular devices comprising a porphyrin photosensitizer and a cobaloxime catalyst, Chem. Commun., 2010, 46, 8806–8808 RSC.
- R. W. Hogue, O. Schott, G. S. Hanan and S. Brooker, A smorgasboard of 17 cobalt complexes active for photocatalytic hydrogen evolution, Chem. – Eur. J., 2018, 24, 9820–9832 CrossRef CAS.
- W. T. Eckenhoff, Molecular catalysts of Co, Ni, Fe, and Mo for hydrogen generation in artificial photosynthetic systems, Coord. Chem. Rev., 2018, 373, 295–316 CrossRef CAS.
- M. Nippe, R. S. Khnayzer, J. A. Panetier, D. Z. Zee, B. S. Olaiya, M. Head-Gordon, C. J. Chang, F. N. Castellano and J. R. Long, Catalytic proton reduction with transition metal complexes of the redox-active ligand bpy2PYMe, Chem. Sci., 2013, 4, 3934–3945 RSC.
- Y. Huang and B. Zhang, Active Cocatalysts for Photocatalytic Hydrogen Evolution Derived from Nickel or Cobalt Amine Complexes, Angew. Chem., Int. Ed., 2017, 56, 14804–14806 CrossRef CAS.
- A. J. Esswein and D. G. Nocera, Hydrogen Production by Molecular Photocatalysis, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS.
- N. Queyriaux, R. T. Jane, J. Massin, V. Artero and M. Chavarot-Kerlidou, Recent developments in H2 evolving molecular cobalt(II)–polypyridyl catalysts, Coord. Chem. Rev., 2015, 304–305, 3–19 CrossRef CAS.
- T. S. Teets and D. G. Nocera, Photocatalytic hydrogen production, Chem. Commun., 2011, 47, 9268–9274 RSC.
- S. Losse, J. G. Vos and S. Rau, Catalytic hydrogen production at cobalt centres, Coord. Chem. Rev., 2010, 254, 2492–2504 CrossRef CAS.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Electro- and solar-driven fuel synthesis with first row transition metal complexes, Chem. Rev., 2019, 2752–2875 CrossRef CAS.
- J. A. Halfen, D. C. Fox, M. P. Mehn and L. Que Jr., Enhanced reactivity of copper catalysts for olefin aziridination by manipulation of ligand denticity, Inorg. Chem., 2001, 40, 5060–5061 CrossRef CAS.
- Y. Zhang, H.-C. Liang, L. N. Zakharov, S. K. Das, M. M. Hetu and A. L. Rheingold, Carboxyester hydrolysis promoted by Cu(II) complexes of pyridyl-amine carboxylate-pendant ligands, Inorg. Chim. Acta, 2007, 1691–1701 CrossRef CAS.
- L. Liang and D. Astruc, The copper(I)-catalyzed alkyne-azide cycloaddition (CUAAC) “click” reaction and its applications. An overview, Coord. Chem. Rev., 2011, 255, 2933–2945 CrossRef CAS.
- J. E. Hein and V. V. Fokin, Copper-catalyzed azide–alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides, Chem. Soc. Rev., 2010, 39, 1302–1315 RSC.
- R. Angamuthu, P. Byers, M. Lutz, A. L. Spek and E. Bouwman, Electrocatalytic CO2 Conversion to Oxalate by a Copper Complex, Science, 2010, 327, 313–315 CrossRef CAS.
- Z. Chen and T. J. Meyer, Copper(II) Catalysis of Water Oxidation, Angew. Chem., Int. Ed., 2013, 52, 700–703 CrossRef CAS.
- S. M. Barnett, K. I. Goldberg and J. M. Mayer, A soluble copper–bipyridine water-oxidation electrocatalyst, Nat. Chem., 2012, 4, 498 CrossRef CAS.
- J. Du, Z. Chen, S. Ye, B. J. Wiley and T. J. Meyer, Copper as a Robust and Transparent Electrocatalyst for Water Oxidation, Angew. Chem., Int. Ed., 2015, 54, 2073–2078 CrossRef CAS.
- M.-T. Zhang, Z. Chen, P. Kang and T. J. Meyer, Electrocatalytic Water Oxidation with a Copper(II) Polypeptide Complex, J. Am. Chem. Soc., 2013, 135, 2048–2051 CrossRef CAS.
- T. Zhang, C. Wang, S. Liu, J.-L. Wang and W. Lin, A Biomimetic Copper Water Oxidation Catalyst with Low Overpotential, J. Am. Chem. Soc., 2014, 136, 273–281 CrossRef CAS.
- D. J. Sconyers and J. D. Blakemore, Electrodeposition behavior of homoleptic transition metal acetonitrile complexes interrogated with piezoelectric gravimetry, Analyst, 2020, 145, 466–477 RSC.
- D. Grujicic and B. Pesic, Electrodeposition of copper: the nucleation mechanisms, Electrochim. Acta, 2002, 47, 2901–2912 CrossRef CAS.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS.
- J. Zhang, B. Xiao, X. Liu, P. Liu, P. Xi, W. Xiao, J. Ding, D. Gao and D. Xue, Copper dopants improved the hydrogen evolution activity of earth-abundant cobalt pyrite catalysts by activating the electrocatalytically inert sulfur sites, J. Mater. Chem. A, 2017, 5, 17601–17608 RSC.
- Q. Wu, M. Luo, J. Han, W. Peng, Y. Zhao, D. Chen, M. Peng, J. Liu, F. M. F. de Groot and Y. Tan, Identifying Electrocatalytic Sites of the Nanoporous Copper–Ruthenium Alloy for Hydrogen Evolution Reaction in Alkaline Electrolyte, ACS Energy Lett., 2020, 5, 192–199 CrossRef CAS.
- M. Kügler, J. Scholz, A. Kronz and I. Siewert, Copper complexes as catalyst precursors in the electrochemical hydrogen evolution reaction, Dalton Trans., 2016, 45, 6974–6982 RSC.
- S. Cao, C.-J. Wang, G.-Q. Wang, Y. Chen, X.-J. Lv and W.-F. Fu, Visible light driven photo-reduction of Cu2+ to Cu2O to Cu in water for photocatalytic hydrogen production, RSC Adv., 2020, 10, 5930–5937 RSC.
- P. Zhang, M. Wang, Y. Yang, T. Yao and L. Sun, A Molecular Copper Catalyst for Electrochemical Water Reduction with a Large Hydrogen-Generation Rate Constant in Aqueous Solution, Angew. Chem., Int. Ed., 2014, 53, 13803–13807 CrossRef CAS.
- H. I. Karunadasa, E. Montalvo, Y. Sun, M. Majda, J. R. Long and C. J. Chang, A Molecular MoS2 Edge Site Mimic for Catalytic Hydrogen Generation, Science, 2012, 335, 698–702 CrossRef CAS.
- H. Lei, H. Fang, Y. Han, W. Lai, X. Fu and R. Cao, Reactivity and Mechanism Studies of Hydrogen Evolution Catalyzed by Copper Corroles, ACS Catal., 2015, 5, 5145–5153 CrossRef CAS.
- J.-P. Cao, T. Fang, L.-Z. Fu, L.-L. Zhou and S.-Z. Zhan, First mononuclear copper(II) electro-catalyst for catalyzing hydrogen evolution from acetic acid and water, Int. J. Hydrogen Energy, 2014, 39, 13972–13978 CrossRef CAS.
- L.-Z. Fu, T. Fang, L.-L. Zhou and S.-Z. Zhan, A mononuclear copper electrocatalyst for both water reduction and oxidation, RSC Adv., 2014, 4, 53674–53680 RSC.
- J.-P. Cao, T. Fang, Z.-Q. Wang, Y.-W. Ren and S. Zhan, A dinuclear triazenido–copper complex: A new molecular electro-catalyst for generating hydrogen from acetic acid or water, J. Mol. Catal. A: Chem., 2014, 391, 191–197 CrossRef CAS.
- J. Wang, C. Li, Q. Zhou, W. Wang, Y. Hou, B. Zhang and X. Wang, Photocatalytic hydrogen evolution by Cu(ii) complexes, Dalton Trans., 2016, 45, 5439–5443 RSC.
- D. M. Ekanayake, K. M. Kulesa, J. Singh, K. K. Kpogo, S. Mazumder, H. Bernhard Schlegel and C. N. Verani, A pentadentate nitrogen-rich copper electrocatalyst for water reduction with pH-dependent molecular mechanisms, Dalton Trans., 2017, 46, 16812–16820 RSC.
- Z.-J. Xin, S. Liu, C.-B. Li, Y.-J. Lei, D.-X. Xue, X.-W. Gao and H.-Y. Wang, Hydrogen production in a neutral aqueous solution with a water-soluble copper complex, Int. J. Hydrogen Energy, 2017, 42, 4202–4207 CrossRef CAS.
- K. Majee, J. Patel, B. Das and S. K. Padhi, μ-Pyridine-bridged copper complex with robust proton-reducing ability, Dalton Trans., 2017, 46, 14869–14879 RSC.
- T. Fang, H.-X. Lu, J.-X. Zhao, S.-Z. Zhan and Q.-Y. Lv, A new copper(I)–triazenido electro-catalyst for catalyzing hydrogen evolution from acetic acid and water, J. Mol. Catal. A: Chem., 2015, 396, 304–309 CrossRef CAS.
- T. Fang, L.-L. Zhou, L.-Z. Fu, S.-Z. Zhan and Q.-Y. Lv, Synthesis and studies of a molecular copper(I)-triazenido electrocatalyst for catalyzing hydrogen evolution from acetic acid and water, Polyhedron, 2015, 85, 355–360 CrossRef CAS.
- M. J. Samide and D. G. Peters, Electrochemical reduction of copper(II) salen at carbon cathodes in dimethylformamide, J. Electroanal. Chem., 1998, 443, 95–102 CrossRef CAS.
- S. Kim, C. Saracini, M. A. Siegler, N. Drichko and K. D. Karlin, Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent, Inorg. Chem., 2012, 51, 12603–12605 CrossRef CAS.
- K. D. Karlin, J. C. Hayes, S. Juen, J. P. Hutchinson and J. Zubieta, Tetragonal vs. trigonal coordination in copper(II) complexes with tripod ligands: structures and properties of [Cu(C21H24N4)Cl]PF6 and [Cu(C18H18N4)Cl]PF6, Inorg. Chem., 1982, 21, 4106–4108 CrossRef CAS.
- N. Kaeffer, A. Morozan, J. Fize, E. Martinez, L. Guetaz and V. Artero, The Dark Side of Molecular Catalysis: Diimine–Dioxime Cobalt Complexes Are Not the Actual Hydrogen Evolution Electrocatalyst in Acidic Aqueous Solutions, ACS Catal., 2016, 6, 3727–3737 CrossRef CAS.
- K. J. Lee, B. D. McCarthy and J. L. Dempsey, On decomposition, degradation, and voltammetric deviation: the electrochemist's field guide to identifying precatalyst transformation, Chem. Soc. Rev., 2019, 48, 2927–2945 RSC.
- R. Sanyal, S. A. Cameron and S. Brooker, Synthesis and complexes of an N4 Schiff-base macrocycle derived from 2,2′-iminobisbenzaldehyde, Dalton Trans., 2011, 40, 12277–12287 RSC.
- R. K. Wilson and S. Brooker, Complexes of a porphyrin-like N4-donor Schiff-base macrocycle, Dalton Trans., 2013, 42, 7913–7923 RSC.
- I. M. Procter, B. J. Hathaway and P. Nicholls, The electronic properties and stereochemistry of the copper(II) ion. Part I. Bis(ethylenediamine)copper(II) complexes, J. Chem. Soc. A, 1968, 1678–1684 RSC.
- B. J. Hathaway and D. E. Billing, The electronic properties and stereochemistry of mono-nuclear complexes of the copper(II) ion, Coord. Chem. Rev., 1970, 5, 143–207 CrossRef CAS.
- I. M. Procter, B. J. Hathaway and P. G. Hodgson, The electronic properties and stereochemistry of the copper(II) ion—VIII: Mono(ethylenediamine)- and mono(2,2′-bipyridyl)-copper(II) complexes, J. Inorg. Nucl. Chem., 1972, 34, 3689–3697 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4, Dalton Trans., 2007, 955–964 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Vershoor, Synthesis, structure and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)2,6-dithiaheptane]copper(II) perchlorate, Dalton Trans., 1984, 1349–1356 RSC.
- A. Das, Z. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, Nickel Complexes for Robust Light-Driven and Electrocatalytic Hydrogen Production from Water, ACS Catal., 2015, 5, 1397–1406 CrossRef CAS.
- R. S. Khnayzer, V. S. Thoi, M. Nippe, A. E. King, J. W. Jurss, N. Roz, K. A. El, J. R. Long, C. J. Chang and F. N. Castellano, Towards a comprehensive understanding of visible-light photogeneration of hydrogen from water using cobalt(II) polypyridyl catalysts, Energy Environ. Sci., 2014, 7, 1477–1488 RSC.
- F. Lucarini, M. Pastore, S. Vasylevskyi, M. Varisco, E. Solari, A. Crochet, K. M. Fromm, F. Zobi and A. Ruggi, Heptacoordinate CoII complex: a new architecture for photochemical H2 production, Chem. – Eur. J., 2017, 23, 6768–6771 CrossRef CAS.
- Y. Kuramochi and O. Ishitani, Iridium(III) 1-Phenylisoquinoline Complexes as a Photosensitizer for Photocatalytic CO2 Reduction: A Mixed System with a Re(I) Catalyst and a Supramolecular Photocatalyst, Inorg. Chem., 2016, 55, 5702–5709 CrossRef CAS.
- S. Chakraborty, E. H. Edwards, B. Kandemir and K. L. Bren, Photochemical Hydrogen Evolution from Neutral Water with a Cobalt Metallopeptide Catalyst, Inorg. Chem., 2019, 58, 16402–16410 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- P. Zanello, Inorganic Electrochemistry: Theory, Practice and Application, Royal Society of Chemistry, Cambridge, UK, 2003 Search PubMed.
- S. Nestke, E. Ronge and I. Siewert, Electrochemical water oxidation using a copper complex, Dalton Trans., 2018, 47, 10737–10741 RSC.
- B. D. McCarthy, D. J. Martin, E. S. Rountree, A. C. Ullman and J. L. Dempsey, Electrochemical reduction of Brønsted acids by glassy carbon in acetonitrile—implications for electrocatalytic hydrogen evolution, Inorg. Chem., 2014, 53, 8350–8361 CrossRef CAS.
- V. Fourmond, P.-A. Jacques, M. Fontecave and V. Artero, H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation, Inorg. Chem., 2010, 49, 10338–10347 CrossRef CAS.
- K. Izutsu, Acid-Base Dissociation Constants in Dipolar Aprotic Solvents, 1990 Search PubMed.
- L. Tong, L. Duan, A. Zhou and R. P. Thummel, First-row transition metal polypyridine complexes that catalyze proton to hydrogen reduction, Coord. Chem. Rev., 2020, 402, 213079 CrossRef CAS.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s−1 for H2 Production, Science, 2011, 333, 863–866 CrossRef CAS.
- A. M. Appel and M. L. Helm, Determining the Overpotential for a Molecular Electrocatalyst, ACS Catal., 2014, 4, 630–633 CrossRef CAS.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Evaluation of Homogeneous Electrocatalysts by Cyclic Voltammetry, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS.
- M. Rakowski DuBois and D. L. DuBois, The roles of the first and second coordination spheres in the design of molecular catalysts for H2 production and oxidation, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- D. K. Bediako, B. H. Solis, D. K. Dogutan, M. M. Roubelakis, A. G. Maher, C. H. Lee, M. B. Chambers, S. Hammes-Schiffer and D. G. Nocera, Role of pendant proton relays and proton-coupled electron transfer on the hydrogen evolution reaction by nickel hangman porphyrins, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15001–15006 CrossRef CAS.
- R. M. Bullock and M. L. Helm, Molecular electrocatalysts for oxidation of hydrogen using earth-abundant metals: shoving protons around with proton relays, Acc. Chem. Res., 2015, 48, 2017–2026 CrossRef CAS.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, Hydrogen Oxidation and Production Using Nickel-Based Molecular Catalysts with Positioned Proton Relays, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS.
- X. L. Ho, H. Shao, Y. Y. Ng, R. Ganguly, Y. Lu and H. S. Soo, Visible Light Driven Hydrogen Evolution by Molecular Nickel Catalysts with Time-Resolved Spectroscopic and DFT Insights, Inorg. Chem., 2019, 58, 1469–1480 CrossRef CAS.
- R. K. Wilson and S. Brooker, Oxidative dehydrogenation of a new tetra-amine N4-donor macrocycle tunes the nickel(II) spin state from high spin to low spin, Dalton Trans., 2013, 42, 12075–12078 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthetic, crystallographic, photo- and electro-catalytic HER data and figures, as well as NMR and mass spectra. CCDC 19839671983968. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0qi01247e |
| This journal is © the Partner Organisations 2021 |