DOI:
10.1039/D1PY00028D
(Paper)
Polym. Chem., 2021,
12, 1581-1587
The contribution of intermolecular forces to phototropic actuation of liquid crystalline elastomers†
Received
7th January 2021
, Accepted 8th February 2021
First published on 22nd February 2021
Abstract
Photomechanical effects in liquid crystal elastomers (LCEs) functionalized with photochromic moieties, such as azobenzene, have been widely studied. This prior work has demonstrated that isothermal, photoinduced (e.g. phototropic) disruption of order via isomerization of azobenzene affects the anisotropic chain configurations of the polymer network. Here, we examine the contribution of the strength of intermolecular interactions between polymer chains in the LCE to both the thermotropic and phototropic response. By incorporating liquid crystalline monomers with reduced aromatic content, both the temperature and irradiation conditions to induce mechanical response are reduced.
Introduction
Liquid crystal elastomers (LCE) are a unique class of functional materials that undergo mechanical deformation when subjected to stimuli capable of disrupting the order of these anisotropic polymer networks. Large scale deformations have been triggered in response to heat (thermotropic actuation) or light (phototropic actuation).1,2 Using heat as a stimulus is well-established as an approach to induce shape change in purely thermotropic systems where order-disorder transitions rely on intrinsic liquid crystalline phase properties. Light, however, represents a compelling approach to induce mechanical response in LCE, as it is contactless, triggerable and patternable.3,4 Azobenzene, a photoisomerizable chromophore, has been commonly incorporated in LCE to facilitate photochemical transduction of light into mechanical work. As occurs in low molar mass liquid crystals, UV light irradiation of LCE induces trans–cis isomerization of azobenzene, which reduces the order of the polymer network.5–8
Historically, aligned (e.g. monodomain) azobenzene-functionalized LCE (azo-LCE) have been prepared through two-step polymerization reactions combining polysiloxane backbones with azobenzene moieties in crosslinking groups that are aligned through application of mechanical load.9 As first shown by Finkelmann and coworkers, strains comparable to that of thermally actuated LCE can be generated by these systems through isomerization-driven disruption of order.9 More recently, methods for preparing LCE with liquid crystalline acrylate monomers have utilized chain-extension reactions followed by subsequent acrylate crosslinking. Specifically, this work will utilize the aza-Michael addition reaction to prepare acrylate-functionalized oligomeric starting materials. Prior reports have prepared LCE by mixing primary amines, diacrylate liquid crystalline monomer(s), and a photoinitiator. The thermally-initiated aza-Michael oligomerization proceeds to completion and subsequently the acrylate end-capped oligomers are polymerized via photoinitiation. By using liquid crystalline precursors, this method is amenable to surface-enforced alignment and is subject to complex patterning to prepare voxelated LCE that are sensitive to heat or light.10,11
The photomechanical response of azo-LCE is typically either photochemical or photothermal. Photothermal responses mirror the thermotropic actuation of these materials. Here, we are focused on photochemical disruption of order (e.g. phototropic). It has long been established that azobenzene in its trans form packs well with LC mesogens both in conventional liquid crystals as well as LCE matrices.6 Upon irradiation with a wavelength of light near the trans azobenzene absorption peak at 365 nm, azobenzene absorbs a photon and converts to a bent cis isomer. The geometric change in the azobenzene causes the order of the polymer chains in the LCE to decrease by disturbing the intermolecular interactions of mesogens in the material.12,13 As a result, the LCE shortens along the aligned axis and expands in orthogonal directions to facilitate the volume and molecular length changes associated with isomerization and phototropic influence on order.14,15 While azobenzene isomerization independently occurs on a timescale of picoseconds, photochemical responses in azo-LCE deformation can take orders of magnitude longer as dictated by the disruption of the aligned LC mesogens incorporated in the polymer network.16 Therefore, modification of the LC components will heavily influence the nature of phototropic response in the azo-LCE.
Because intermolecular interactions are a primary factor in order disruption and actuation, the liquid crystal mesogen structure plays a critical role in tuning photo-induced deformation. Previous reports show that these interactions are controlled through lateral substitution of aromatic groups or altering aliphatic chain length.17–19 As recently explored by our group in thermotropic LCE, is the aromatic content in the mesogenic cores.20–22 Given that the π–π interactions between the aromatic groups in LC mesogens are a key contributor to ordering in LCE, this method of tuning phototropic response is the focus of this work.23
Here, we systematically vary the composition of the azo-LCE to isolate the contribution of mesogen–mesogen interactions to the photomechanical response. We prepare the azo-LCE with varying ratios of a two-ring and a three-ring liquid crystalline monomer and compare the phototropic properties to the thermotropic properties. We ultimately correlate photo-induced disruption of order to deformation and strain generation in the azo-LCE to demonstrate that through modification of aromatic content in the mesogen structure, more effective photomechanical actuation is achieved.
Experimental
Synthesis of monodomain azobenzene-functionalized liquid crystalline elastomers
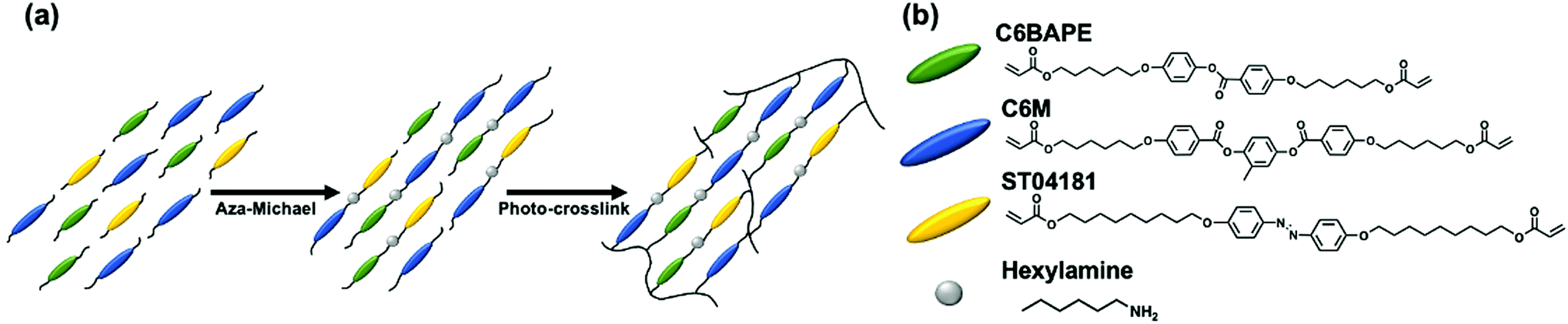
LCE containing azobenzene (azo-LCE) were synthesized by copolymerizing 4-(6-(acryloyloxy)hexyloxy)phenyl-4-(6-(acryloyloxy)hexyloxy)benzoate (C6BAPE) (Synthon Chemical), 1,4-bis-[4-(6-acryloyloxyhexyloxy)benzoyloxy]-2-methylbenzene (C6 M) (Wilshire Technologies), and 4,4′-bis(9-(acryloyloxy)nonyloxy) azobenzene (ST04181) (Synthon Chemical). These monomers were mixed in ratios detailed in Table 1 with 1-hexylamine in a functional group ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar (amine:acrylate). Omnirad 784 (2 wt%) and the co-initiator benzoyl peroxide were included for initiation of polymerization (2:1 molar ratio of co-initiator:initiator). The monomeric mixtures were heated to 150 °C and vortexed to ensure homogeneity. The amines and initiator were added at 90 °C and again the mixtures were vortexed. Melted mixtures were drawn by capillary action into 5 μm thick alignment cells at 95 °C (in the isotropic state) and slowly cooled to the nematic state. The alignment cells used in this study were fabricated with anti-parallel planar alignment enforced by Elvamide-coated glass slides that were rubbed with velvet. The coated glass substrates are adhered with a mixture of optical adhesive and 5 μm glass rod spacers. The compositions were held for 24 hours at elevated temperature within the nematic phase of the given mixture (47 °C, 52 °C, 57 °C, 65 °C for 1:1, 1:2, 1:3, and 0:1, respectively) to allow oligomerization of the acrylate monomers to proceed by aza-Michael addition. The 1:0 mixture of C6BAPE:C6 M retained a smectic phase and was not included in this study. After 24 hours, samples were photopolymerized within the alignment cells at the oligomerization temperature with 45 min of exposure to 50 mW cm−2 of 525 nm light. 525 nm light was used to photoinitiate polymerization so as to not disrupt the phototropic phase of the azo-LCE compositions. The polymerized azo-LCE films were removed from the alignment cells by soaking in water and gentle heating at 75 °C.
:2 molar (amine:acrylate). Omnirad 784 (2 wt%) and the co-initiator benzoyl peroxide were included for initiation of polymerization (2:1 molar ratio of co-initiator:initiator). The monomeric mixtures were heated to 150 °C and vortexed to ensure homogeneity. The amines and initiator were added at 90 °C and again the mixtures were vortexed. Melted mixtures were drawn by capillary action into 5 μm thick alignment cells at 95 °C (in the isotropic state) and slowly cooled to the nematic state. The alignment cells used in this study were fabricated with anti-parallel planar alignment enforced by Elvamide-coated glass slides that were rubbed with velvet. The coated glass substrates are adhered with a mixture of optical adhesive and 5 μm glass rod spacers. The compositions were held for 24 hours at elevated temperature within the nematic phase of the given mixture (47 °C, 52 °C, 57 °C, 65 °C for 1:1, 1:2, 1:3, and 0:1, respectively) to allow oligomerization of the acrylate monomers to proceed by aza-Michael addition. The 1:0 mixture of C6BAPE:C6 M retained a smectic phase and was not included in this study. After 24 hours, samples were photopolymerized within the alignment cells at the oligomerization temperature with 45 min of exposure to 50 mW cm−2 of 525 nm light. 525 nm light was used to photoinitiate polymerization so as to not disrupt the phototropic phase of the azo-LCE compositions. The polymerized azo-LCE films were removed from the alignment cells by soaking in water and gentle heating at 75 °C.
Table 1 Summary of monomer content, optical properties, order parameters (S) calculated by dichroic ratios, mechanical properties, and thermal properties for 5 μm azobenzene-functionalized LCE with varying C6BAPE:C6M ratios
| C6BAPE:C6M by weight |
wt% (mol%) C6BAPE |
wt% (mol%) C6M |
wt% (mol%) ST04181 |
T
g (°C) |
S
|
I/I0 |
|| Modulus (MPa) |
⊥ Modulus (MPa) |
| 0:1 |
0.0 (0.0) |
77.9 (54.9) |
15.0 (0.11) |
−4.1 |
0.13 |
0.015 |
180 ± 20 |
33 ± 4 |
| 1:3 |
19.4 (16.3) |
58.2 (39.2) |
15.0 (0.11) |
−4.2 |
0.17 |
0.015 |
160 ± 20 |
26 ± 7 |
| 1:2 |
25.8 (21.4) |
51.6 (34.2) |
15.0 (0.11) |
−2.3 |
0.18 |
0.014 |
170 ± 30 |
26 ± 7 |
| 1:1 |
38.6 (24.9) |
38.6 (31.1) |
15.0 (0.11) |
−0.3 |
0.16 |
0.016 |
140 ± 20 |
24 ± 5 |
UV-visible spectroscopy
The absorption of the azo-LCE was measured using UV/vis (Cary 7000, Agilent). Absorption spectra with unpolarized light were taken before and after irradiation of azo-LCE (20 min, 365 nm, 50 mW cm−2) and every 30 min for 24 h after irradiation to assess the kinetics of the cis–trans isomerization of azobenzene. Absorption spectra with polarized light were measured at 0° (parallel) and 90° (perpendicular) to the nematic director to determine the dichroic ratios of the materials. Given the concentration of azobenzene used in this examination, absorption measurements in the multi-path UV/vis were collected with an OD 3 neutral density filter placed in the reference beam. A control experiment is shown in Fig. S1† to confirm that absorption values of up to OD 5 are accurately quantifiable.
Tensile testing
Tensile experiments were conducted by dynamic mechanical analysis (RSA-G2, TA Instruments, DMA) on azo-LCE strips cut parallel and perpendicular to the nematic director. Measurements were taken at 5% strain per min. The modulus reported in Table 1 is taken from the linear stress/strain regime of the azo-LCE.
Differential scanning calorimetry
Thermal properties for monomer, oligomer, and polymer compositions were measured using differential scanning calorimetry (Discovery DSC 2500, TA Instruments). Second heating/cooling cycles (5 °C min−1) were used for analysis.
Photomechanical and thermomechanical actuation
Azo-LCE strips (∼2 mm × 12 mm × 0.005 mm) were subjected to isostress measurements at 0.001 N constant force (Discovery DMA 850, TA Instruments). Phototropic actuation of the azo-LCE was measured by the DMA as samples were exposed to UV light (365 nm, 50 mW cm−2) for 500 s, starting at 60 s. Strain recovery was monitored in the dark for 24 h with 0.001 N force maintained. For thermotropic actuation, again the azo-LCE were subject to a 0.001 N force, and the strain was measured as temperature in the DMA was increased from 20 °C to 220 °C at 3 °C min−1.
Photoinduced deformation
To visualize the photomechanical response of these materials, azo-LCE were cut into cantilevers (∼2 mm × 8 mm × 0.005 mm) and attached to a fixed surface, leaving the sample freely standing vertically. Cantilevers composed of azo-LCE were exposed to UV light (365 nm, 50 mW cm−2), and the deformation was recorded. Dimensional contraction of azo-LCE was observed due to prolonged UV exposure (365 nm light, 50 mW cm−2, 30 minutes) of an azo-LCE sample (5 mm × 10 mm × 0.05 mm). Images were taken before and immediately after the 30 minutes exposure to compare the length of the samples along the nematic director.
Results and discussion
Here, we are concerned with the contribution of mesogen–mesogen interactions (intermolecular forces) on the phototropic response of azo-LCE. Our approach to prepare monodomain azo-LCE via surface alignment is summarized in Fig. 1. Liquid crystalline monomers C6BAPE, C6 M, and ST04181 were mixed at various ratios and subjected to aza-Michael addition with hexylamine. After a 24 h oligomerization in the alignment cells, the compositions were irradiated with visible light to photoinitiate polymerization of the acrylate-terminated oligomers, resulting in monodomain azo-LCE.
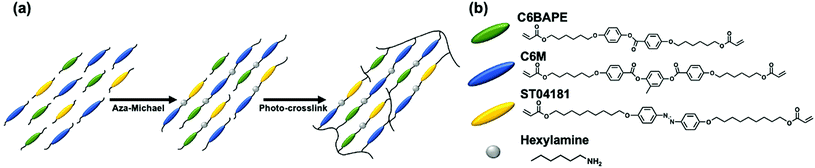 |
| | Fig. 1 (a) Generalized synthetic approach to prepare azobenzene-functionalized liquid crystalline elastomers (azo-LCE) by a one-pot, two-step polymerization method utilizing aza-Michael addition and subsequent photopolymerization. (b) The liquid crystalline monomers C6BAPE, C6M, and ST04181 were mixed with hexylamine to prepare monodomain azobenzene-functionalized LCE. | |
Thermal characterization of materials
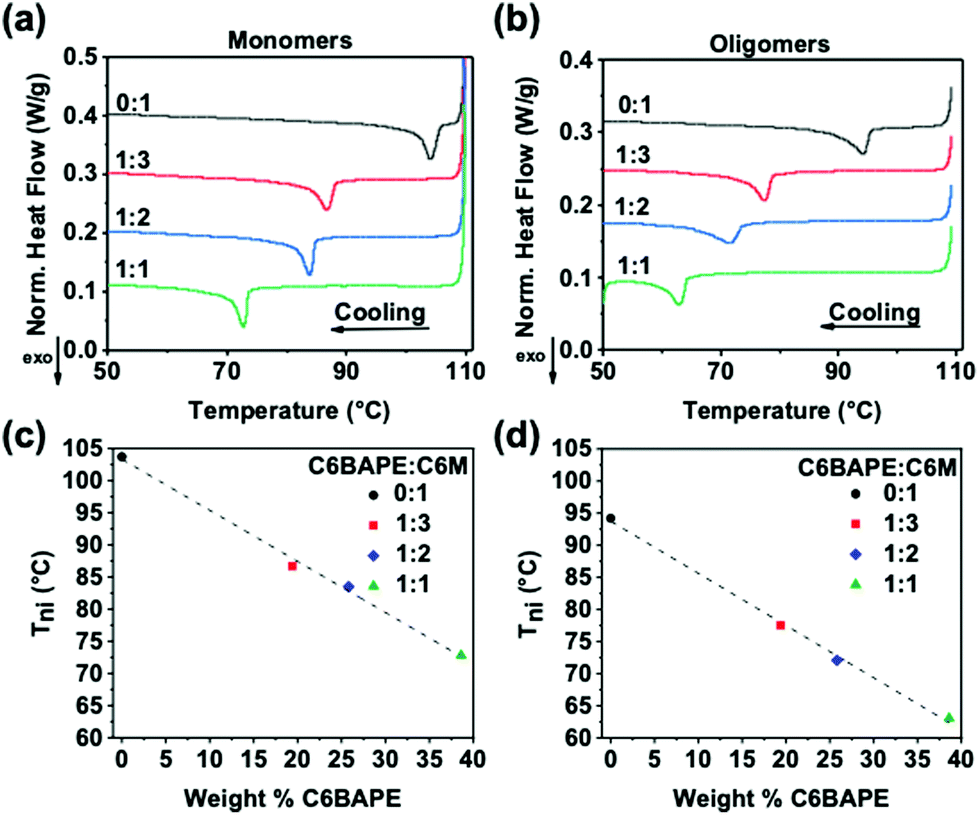
To inform subsequent study of phototropic properties, we first characterized the thermotropic phase behavior of these materials in the monomer state with DSC. As evident by the Tni (exothermic peaks) in Fig. 2a, incorporating C6BAPE at varying ratios with C6 M considerably affected the thermotropic properties of these compositions by shifting the Tni to lower temperatures as C6BAPE concentration was increased. Similarly, the variation in thermotropic phase behavior of the monomer mixtures was retained upon oligomerization via aza-Michael addition. The correlations between Tni of the monomers/oligomers and C6BAPE concentration are summarized in Fig. 2c and d. In both the monomer and oligomer mixtures, the Tni was reduced by nearly 30 °C when the C6BAPE concentration was increased from 0–40 wt%. As illustrated in Fig. 1b, C6BAPE is distinctive from C6 M in that it is based on a mesogenic core with two aromatic groups, rather than three. The reduction in aromatic content decreases the strength of the π–π coupling that strongly defines mesogen–mesogen interactions in the monomer melt and oligomers, leading to easier disruption of nematic ordering among mesogens and the resulting reduction in Tni.
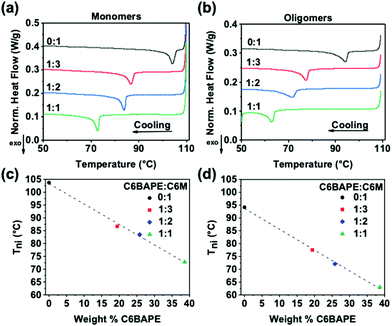 |
| | Fig. 2 DSC thermograms of (a) monomer mixtures and (b) oligomers used to prepare monodomain azobenzene-functionalized LCE. The Tni of these materials is plotted as a function of C6BAPE concentration for (c) monomer mixtures and (d) oligomers. | |
Thermomechanical response
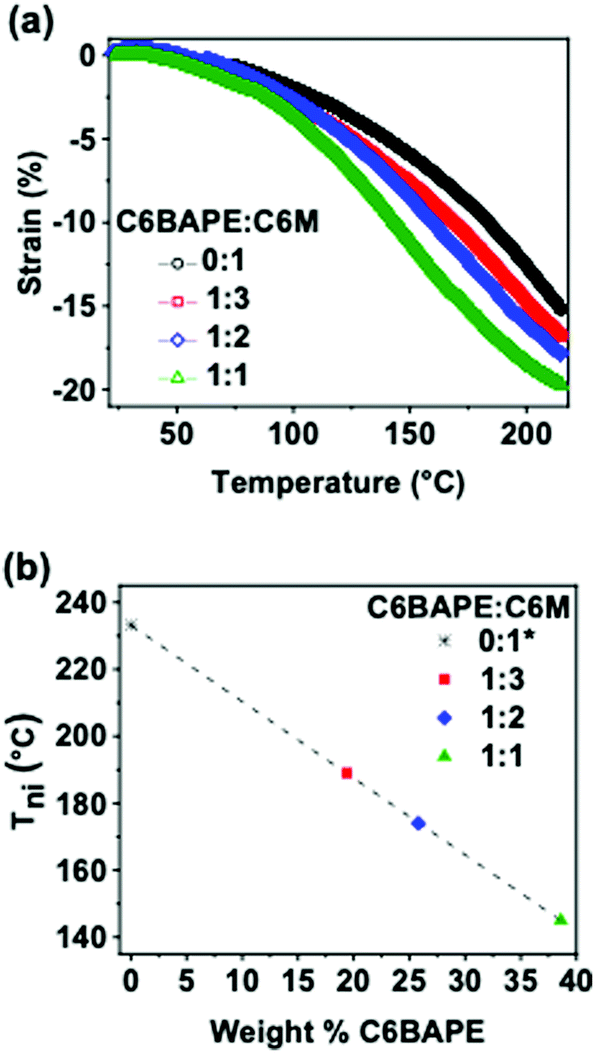
The thermomechanical responses of the azo-LCE materials were measured while held in tension (0.001 N). Strain was monitored as a function of temperature for the four azo-LCE compositions with 0:1, 1:3, 1:2, and 1:1 C6BAPE:C6 M. The results are summarized in Fig. 3a. The thermomechanical response of all four azo-LCE compositions is characterized by two strain generation regimes: (i) minimal strain generation at temperatures in the nematic state and (ii) rapid strain generation at temperatures which order disruption is occurring. As the C6BAPE concentration was increased in the azo-LCE, the temperature at which the increased strain rate is shifted to lower temperatures. Here, we use the maximum of the derivative of the strain-temperature response from each LCE as an indicator of the Tni of the materials.24Fig. 3b plots the Tni of the azo-LCE as a function of C6BAPE content and shows a decrease of nearly 90 °C as C6BAPE concentration increased from 0–40 wt%. Notably, the composition based on C6 M (0:1) had the highest Tni in the monomer/oligomer mixtures and did not reach a thermomechanical inflection point before thermal degradation. As evident in these data and detailed in a parallel report,20 the contribution of C6BAPE to mesogen–mesogen interactions seen in monomer and oligomer mixtures is retained in the polymer network and lowers the temperature of thermotropic response of the LCE.
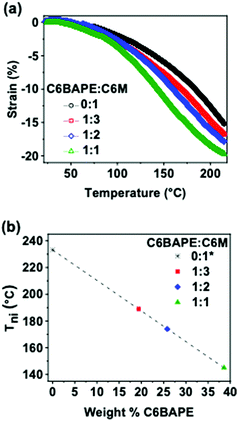 |
| | Fig. 3 (a) Thermomechanical strain generated by azobenzene-functionalized LCE (0.001 N constant force) as temperature was ramped at 3 °C min−1 (b) Tni calculated by inflection point of thermomechanical strain curves and plotted as a function of C6BAPE concentration. Composition 0:1 did not reach an inflection point before degradation, so Tni was extrapolated by linear fit of Tni plotted against C6BAPE concentration for the other three compositions and plotted as 0:1. | |
Optical and mechanical characterization
The photomechanical response of an azo-LCE is known to be dependent on the alignment, absorption, and thermal properties of the material.25 Accordingly, we characterized these properties of the azo-LCE to isolate their potential contribution to the stimuli-response. The monodomain alignment of the 5 μm films is evident in strong and uniform birefringence in polarized optical microscopy (Fig. S2†) and confirmed in order parameters for the azo-LCE calculated from the dichroic ratio26 measurements based on polarized UV-Visible spectroscopy (Fig. S3†) As summarized in Table 1, the Tg and moduli (parallel and perpendicular to nematic director) of the azo-LCE are similar (Table 1 and Fig. S4, S5†) despite increasing C6BAPE concentration reducing the aromatic content of the polymer network. Due to the incorporation of 15 wt% azobenzene in each of the azo-LCE materials, the absorption of these materials was also similar for all compositions (Fig. S6†) Since these materials have been shown to exhibit non-Beer behavior,27,28I/I0 values are reported in Table 1 rather than absorption coefficient. These characterizations validate that differences in photomechanical response is attributed to modifying mesogen–mesogen interactions in the azo-LCE.
Photomechanical response
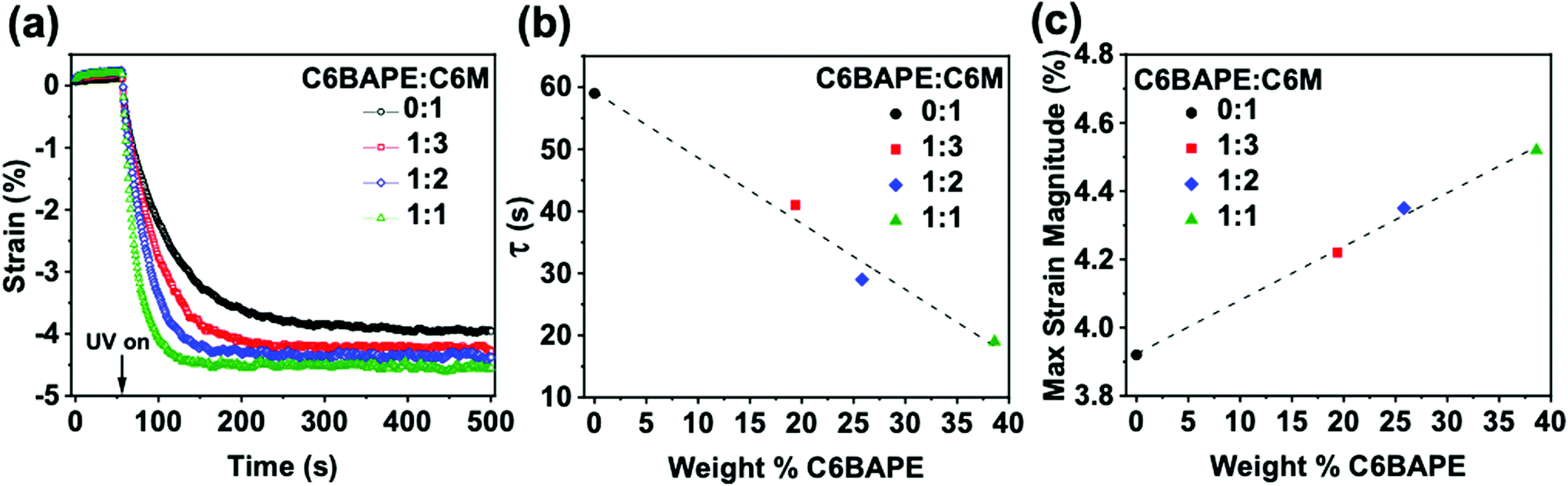
With other factors excluded, the effects of reduction in the strength of mesogen–mesogen interaction on the phototropic actuation of these materials are discussed in parallel with the thermotropic actuation of the azo-LCE. In an experiment analogous to thermomechanical characterization, azo-LCE samples composed of 0:1, 1:3, 1:2, and 1:1 C6BAPE:C6 M were held in tension (0.001 N) and irradiated with UV light. Fig. 4a presents the photogenerated strain over time in these materials (with error Fig. S7†). Upon UV light exposure after equilibrating for 60 s, all four azo-LCE generated strain. The rate and magnitude of strain generation in the azo-LCE increased as C6BAPE concentration increased from 0–40 wt%. The photomechanical response curves in Fig. 4a were fit with an exponential to determine the time constant (τ), which is plotted in Fig. 4b as a function of the C6BAPE concentration. Increasing the C6BAPE concentration from 0–40 wt% in the azo-LCE results in a nearly 40 s decrease in this value. The contribution of C6BAPE to the magnitude of strain generation is summarized in Fig. 4c. Photogenerated strain is attributed largely to photochemical mechanisms rather than photothermal, as supported by thermographic imaging that confirms only a small increase in the sample temperature that remains well below the Tni of the materials upon exposure (Video S1 and Fig. S9†).29 Relatedly, as evident in previous reports,9 it would be expected that performing the experiment at temperatures closer to the materials’ Tni would result in further increase in photomechanically generated strain and decrease in time constant. These results affirm that reducing the strength of the mesogen–mesogen interactions within the azo-LCE results in a faster and larger magnitude of the phototropic response.
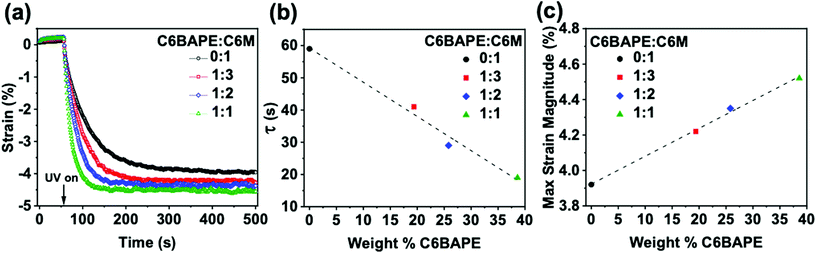 |
| | Fig. 4 (a) Photomechanical strain generated by azobenzene-functionalized LCE. (0.001 N constant force, 50 mW cm−2, 365 nm light exposure at 60 s) (b) Time constants as a function of C6BAPE concentration calculated from exponential fitting of photomechanical strain curve. (c) Maximum photomechanical strain as a function of C6BAPE concentration. | |
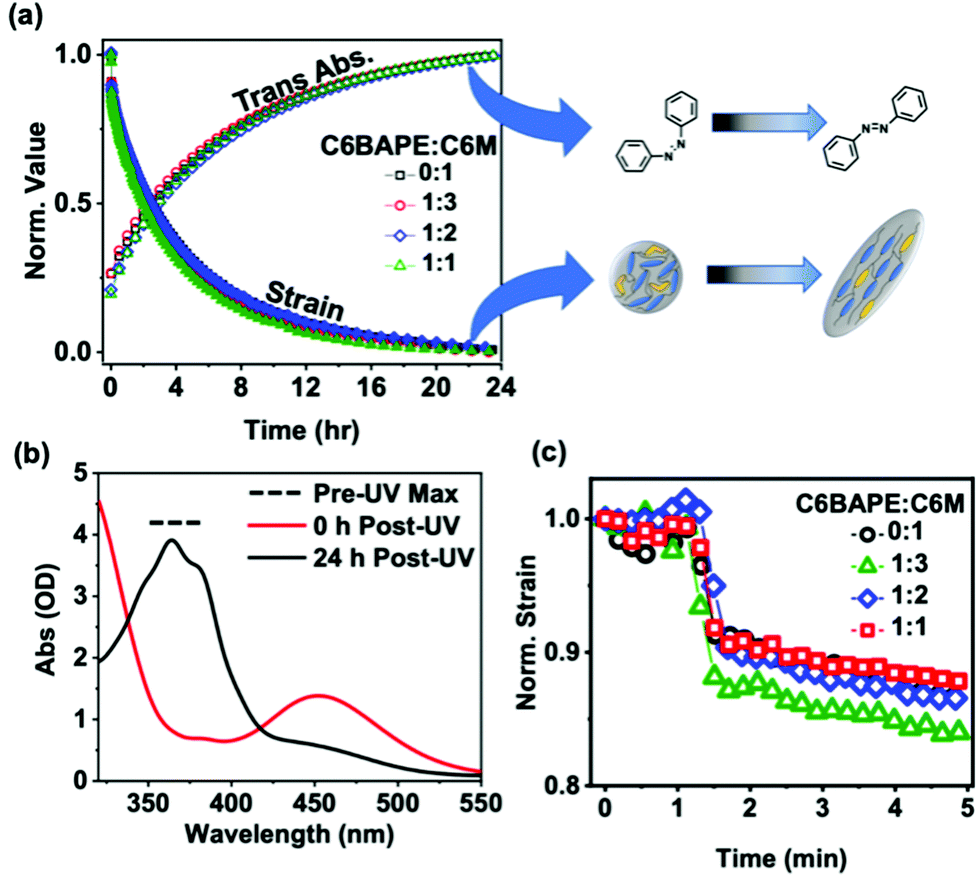
Relaxation studies
While decreasing mesogen–mesogen interactions resulted in more rapid actuation of the azo-LCE, we observe minimal contributions to the relaxation time. Fig. 5a illustrates that thermal back-isomerization of the azobenzene moieties to the trans state, indicated by increasing absorbance normalized to the initial pre-UV exposure value (open symbols), occurs over a timescale of approximately 24 hours for all compositions. This behavior is confirmed by the UV-Vis spectra shown in Fig. 5b, in which the trans isomer peak (absorbance at 365 nm) was not observable after UV exposure but was nearly fully restored after 24 h in the dark. Fig. 5a also shows a 24-hour timescale for relaxation of the polymer to its pre-UV exposure state, indicated by decreasing strain normalized to the initial pre-UV exposure value (closed symbols) for all compositions. Notably, the sharp recovery in strain evident in the first 5 minutes of relaxation (Fig. 5c, 1 min UV light exposure) is indicative of the small contribution from photothermal effects.30 Although the overall timescale of relaxation for both the azobenzene moieties and the azo-LCE was similar, a slight mismatch in the relaxation trends was observed, evident by the difference in half-lives occurring at 4.5 h for the azobenzene isomerization and 2.5 h for polymer relaxation. These observations indicate a mismatch in azobenzene and polymer relaxation, consistent with previous studies exploiting compositional decoupling of isomerization and polymer relaxation kinetics in azo-LCE.11 However, the mismatch is consistent in all ratios of C6BAPE:C6 M, indicating that thermal cis–trans isomerization and strain recovery may not be prominently impacted by mesogen–mesogen interactions. This is yet another example in which the relaxation of azo-LCE after irradiation exhibits a decoupling in cis–trans isomerization kinetics and mechanical recovery.11,31
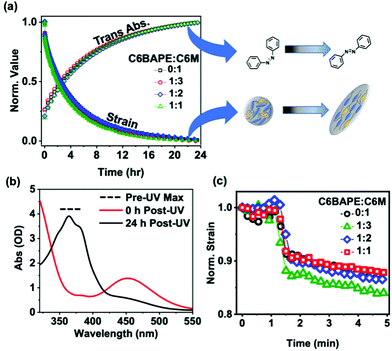 |
| | Fig. 5 (a) Normalized photoinduced strain relaxations (solid symbols) for monodomain azobenzene-functionalized LCE films monitored for 24 h in the dark following 365 nm light exposure (20 min, 50 mW cm−2) and normalized trans absorbance (open symbols) of azobenzene-functionalized films monitored for 24 h in the dark following 365 nm light exposure (20 min, 50 mW cm−2) to show cis–trans thermal relaxation. (b) Representative (1:3 composition) UV-Vis spectra collected at 0 h and 24 h after removal of UV light. (c) First five minutes of photoinduced strain relaxation experiments with removal of UV light occurring at 1 min. | |
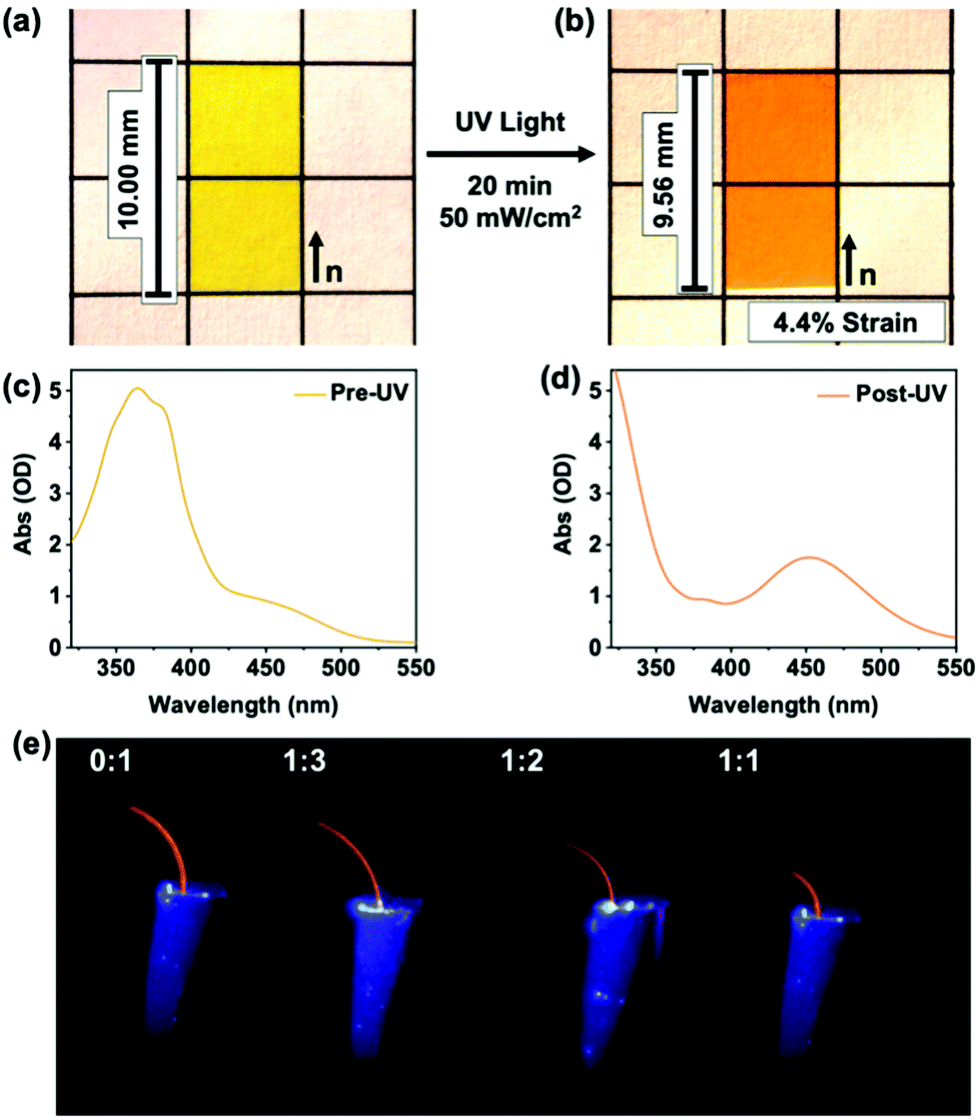
Visualization of stimuli-response
We conclude this study by visually demonstrating the contribution of mesogen–mesogen interactions on the photomechanical response of azo-LCE. The photogenerated strain is evident in comparing the images in Fig. 6a (pre-UV exposure) and 6b (post-UV exposure). Measurements confirm that the azo-LCE of 1:2 C6BAPE:C6 M composition contracts by 4.4%, matching the photomechanical experiments presented in Fig. 4.
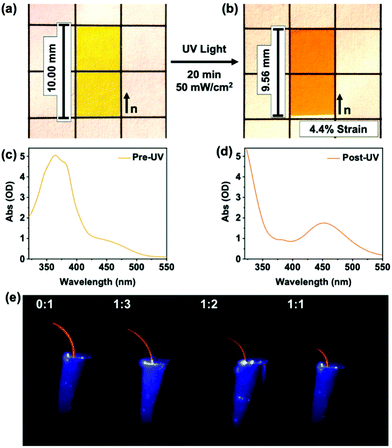 |
| | Fig. 6 Images of azobenzene-functionalized LCE (1:2 composition) (a) before and (b) after UV light exposure (365 nm, 20 min, 50 mW cm−2). UV-Vis spectra for film (c) before and (d) after UV exposure. (e) Monodomain azobenzene-functionalized LCE cantilevers (0.005 mm × 1 mm × 6 mm, increasing C6BAPE concentration from left to right) after 2 seconds of 365 nm light exposure (50 mW cm−2). | |
As demonstrated in Fig. 6c, the incorporation of C6BAPE affects the rate that light exposure produces strain. This outcome is visualized in experiments in which the azo-LCE were cut into cantilevers with the nematic director aligned parallel to the long axis of the cantilever and exposed to UV light (50 mW cm−2). The temporal response of photomechanical deformation is evident in images taken 2 seconds after exposure. Increasing the C6BAPE concentration in the azo-LCE increases the rate of photomechanical strain generation, evident here as bending. The time-dependent deflection of the cantilevers prepared from the azo-LCE is fully presented in Video S2† and the deformation of the azo-LCE is plotted against irradiation time in Fig. S10.† After 30 min of irradiation, the azo-LCE take on a flat state, indicative of near-complete conversion of trans isomers into cis isomers, evident in the photography in Fig. 6b.
Conclusions
The photomechanical response of azobenzene-functionalized liquid crystalline elastomers (azo-LCE) is shown to be strongly dependent on the strength of mesogen–mesogen interactions in the polymer network. Upon irradiation, azo-LCE compositions prepared with increasing concentrations of a non-traditional liquid crystalline monomer, C6BAPE, exhibit more rapid photomechanical responses and greater magnitudes of strain generation. The dependence of phototropic actuation of the azo-LCE with C6BAPE concentration is also evident in the thermotropic deformation of these materials, confirming the association of stimuli, order, and response.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
T.S.H. acknowledges Graduate Research Fellowship support from the National Science Foundation.
References
- P. M. Hogan, A. R. Tajbakhsh and E. M. Terentjev, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 2002, 65, 10 CrossRef.
- T. J. White and D. J. Broer, Nat. Mater., 2015, 14, 1087–1098 CrossRef CAS.
- T. J. White, J. Polym. Sci., Part B: Polym. Phys., 2018, 56, 695–705 CrossRef CAS.
- H. Koerner, T. J. White, N. V. Tabiryan, T. J. Bunning and R. A. Vaia, Mater. Today, 2008, 11, 34–42 CrossRef CAS.
- T. J. White, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 877–880 CrossRef CAS.
- J. Cviklinski, A. R. Tajbakhsh and E. M. Terentjev, Eur. Phys. J. E, 2002, 9, 427–434 CrossRef CAS.
- T. Ikeda and O. Tsutsumi, Science, 1995, 268, 1873–1875 CrossRef CAS.
- W. C. Xu, S. Sun and S. Wu, Angew. Chem., Int. Ed., 2019, 58, 9712–9740 CrossRef CAS.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- T. H. Ware, Z. P. Perry, C. M. Middleton, S. T. Iacono and T. J. White, ACS Macro Lett., 2015, 4, 942–946 CrossRef CAS.
- S. Ahn, T. H. Ware, K. M. Lee, V. P. Tondiglia and T. J. White, Adv. Funct. Mater., 2016, 26, 5819–5826 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201–205 CrossRef CAS.
- M. Warner and L. Mahadevan, Phys. Rev. Lett., 2004, 92, 134302 CrossRef CAS.
-
Y. Zhao and T. Ikeda, Smart Light-Responsive Materials, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2009 Search PubMed.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037–2057 RSC.
- M. A. Osman, Mol. Cryst. Liq. Cryst., 1985, 128, 45–63 CrossRef CAS.
- D. J. Broer, G. N. Mol and G. Challa, Makromol. Chem, 1991, 192, 59–74 CrossRef CAS.
- Z. Zhang, M. Chen, I. Schneider, Y. Liu, S. Liang, S. Sun, K. Koynov, H. J. Butt and S. Wu, Macromolecules, 2020, 53, 8562–8569 CrossRef CAS.
- J. M. McCracken, B. R. Donovan and T. J. White, Adv. Funct. Mater. DOI:10.1002/adfm.202100564.
- D. J. Broer, G. N. Mol and G. Challa, Die Makromol. Chem., 1991, 192, 59–74 CrossRef CAS.
- J. Lub, D. J. Broer and N. Van Den Brock, Liebigs Ann., 1997, 2281–2288 CrossRef CAS.
-
J. W. Goodby, E. J. Davis, R. J. Mandle and S. J. Cowling, in Handbook of Liquid Crystals, 2014 Search PubMed.
-
W. H. de Jeu, Liquid Crystal Elastomers: Materials and Applications, 2012 Search PubMed.
-
T. J. White, in Photomechanical Materials, Composites, and Systems: Wireless Transduction of Light into Work, John Wiley & Sons, Ltd, 2017, pp. 393–403 Search PubMed.
- L. Corvazier and Y. Zhao, Macromolecules, 1999, 32, 3195–3200 CrossRef CAS.
- F. Serra and E. M. Terentjev, J. Chem. Phys., 2008, 128, 1–6 CrossRef.
- D. Corbett and M. Warner, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2008, 77, 1–11 CrossRef.
- K. M. Lee and T. J. White, Macromolecules, 2012, 45, 7163–7170 CrossRef CAS.
- M. Pilz Da Cunha, E. A. J. Van Thoor, M. G. Debije, D. J. Broer and A. P. H. J. Schenning, J. Mater. Chem. C, 2019, 7, 13502–13509 RSC.
- D. Liu and D. J. Broer, Nat. Commun., 2015, 6, 1–7 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py00028d |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Christopher N.
Bowman
a,
Christopher N.
Bowman