
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetabolic labeling probes for interrogation of the host–pathogen interaction
Bob J.
Ignacio†
a,
Thomas
Bakkum†
 b,
Kimberly M.
Bonger
a,
Nathaniel I.
Martin
bc and
Sander I.
van Kasteren
*b
b,
Kimberly M.
Bonger
a,
Nathaniel I.
Martin
bc and
Sander I.
van Kasteren
*b
aInstitute for Molecules and Materials, Radbout Universiteit, Nijmegen, Gelderland, Netherlands
bLeiden Institute of Chemistry, Leiden University, Einsteinweg 55, Leiden, Zuid-Holland, Netherlands. E-mail: s.i.van.kasteren@chem.leidenuniv.nl
cInstitute of Biology, Leiden University, Sylviusweg 72, Leiden, Zuid-Holland, Netherlands
First published on 16th March 2021
Abstract
Bacterial infections are still one of the leading causes of death worldwide; despite the near-ubiquitous availability of antibiotics. With antibiotic resistance on the rise, there is an urgent need for novel classes of antibiotic drugs. One particularly troublesome class of bacteria are those that have evolved highly efficacious mechanisms for surviving inside the host. These contribute to their virulence by immune evasion, and make them harder to treat with antibiotics due to their residence inside intracellular membrane-limited compartments. This has sparked the development of new chemical reporter molecules and bioorthogonal probes that can be metabolically incorporated into bacteria to provide insights into their activity status. In this review, we provide an overview of several classes of metabolic labeling probes capable of targeting either the peptidoglycan cell wall, the mycomembrane of mycobacteria and corynebacteria, or specific bacterial proteins. In addition, we highlight several important insights that have been made using these metabolic labeling probes.
Introduction
Despite the near-ubiquity of antibiotics in modern times, bacterial infections are still among the leading causes of death globally.1 Overuse of antibiotics in high-income countries has contributed to an evolutionary selection pressure that is driving antibiotic resistance.2 New antibiotic resistance mechanisms are rapidly emerging,3 resulting in a staggering increase in multi-drug resistant (MDR), extensive drug resistant (XDR) and pan-drug resistant (PDR) strains.4 At the same time, the development of novel classes of antibiotics has been extremely slow, with only two new classes of broad-spectrum antibiotics introduced to the clinic since 1962.5,6 Development of antibiotics for a certain class of pathogenic bacteria has been particularly difficult: the intracellular bacteria. Intracellular bacteria reside inside host cells, where they can evade immune detection and persist for months or even years in a dormant state.7,8 In order for an antibiotic to be active against infections caused by intracellular bacteria, the drug not only has to cross the bacterial cell envelope but also the host cell membrane. The study of (obligate) intracellular pathogens is further complicated by the fact that they are often difficult to culture in vitro.9,10 In light of these challenges, novel antibiotics against intracellular bacteria are much needed, as the disease burden from these species is very high.The deadliest bacterial species is arguably Mycobacterium tuberculosis (Mtb), a facultative intracellular bacterium that is the major cause of tuberculosis (TB). This pathogen is responsible for approximately 2 million deaths annually11 and antibiotic-resistant strains are continually emerging. Treating infections due to Mtb is made difficult by the heterogeneity in its intracellular life cycle: after infecting macrophages it diverges into fast and very slow-growing forms.12,13 The slow growing form is generally hard to treat with conventional antibiotics such as those that target the nascent cell wall of the dividing bacteria.14–20 As a result, TB is typically treated with a complex cocktails of antibiotics that need to be taken for long periods of time. Addressing infections due to MDR Mtb is even more challenging requiring antibiotic treatment courses of up to 2 years (often with severe side-effects).21 One new therapy – the first in many years – was recently approved for drug-resistant forms of Mtb: pretomanid in combination with bedaquiline and linezolid. Whilst this is laudable – as is the clinical development of other Mtb active agents – the demand for new treatments for Mtb and other intracellular pathogens currently far outstrips supply.22Mtb is not the only intracellular pathogen causative of high-mortality/morbidity disease. Other intracellular pathogens of clinical concern include Salmonella enterica, Yersinia pestis, Chlamydia trachomatis, Listeria monocytogenes and Coxiella burnetii to name but a few.23 Whilst not displaying quite the same nefariousness as Mtb, these organisms are associated with a host of pathologies ranging from infertility to increased risk of bowel cancer.24,25 While these pathogens can largely still be controlled with available antibiotics, the increased emergence of resistance threatens our continued ability to do so and paints a bleak future.26
Bacterial pathogens have acquired highly effective mechanisms for infecting- and surviving inside hosts. These mechanisms lead to increased virulence and persistence within the host environment, along with suppression of the host immune system.27 Due to their importance for bacterial cell viability and infectivity inside the host, bacterial defense mechanisms at the host–pathogen interface are a promising target for novel antibiotics.28,29 Thus, effective chemical tools to study the host–pathogen interface at the molecular level are of paramount importance in deepening our understanding of the relationship between the fundamental biochemistry of the host–pathogen and their virulence. This in turn can provide new information in the fight against antibiotic resistance. To provide clinically significant data, these chemical tools must be useable in cell culture and in vivo in pathogenic bacteria. Moreover, the cellular processes they report on must be relevant to bacterial cell survival and/or virulence.
Recently, novel chemical probes and approaches have been developed that allow for the profiling of various metabolic processes as well as in cell cell wall and protein labeling. In metabolic profiling a chemical probe is exogenously supplied to an organism, which subsequently incorporates the chemical probe into its cellular architecture by means of endogenous enzymes. Novel chemical probes have been developed that carry fluorescent dyes allowing for facile, one-step metabolic labeling of bacterial structures of interest such as the peptidoglycan (PG) cell wall,30,31 or the mycomembrane of corynebacteria.32,33 Moreover, the introduction of bioorthogonal chemistry has furthered the field by allowing for chemical reactions to be performed inside the crowded environment of the bacterial cell.34 Bioorthogonal groups are by definition non-reactive, or orthogonal, to their biological environment, but highly reactive towards their bioorthogonal reaction partner.35 Moreover, bioorthogonal groups such azides or alkynes can be readily introduced into common bacterial metabolites with minimal perturbation of biological function. After metabolic incorporation into bacteria, bioorthogonal probes can be reacted with reporter molecules carrying a complimentary bioorthogonal group. Such reporters include fluorophores that allow for the visualization of bacterial biomolecules of interest or affinity tags to facilitate their enrichment. As such, bioorthogonal chemistry has driven advancements in two-step metabolic labeling approaches for host–pathogen interaction studies. As a whole, metabolic labeling in bacteria, in one step or two steps, has shed a light on previously uncharacterized bacterial cell processes, making it a valuable tool in the search for novel targets for antibiotic drug development.
Here, we review recent developments in the field of metabolic labeling in the context of the host–pathogen interface. We cover the initial discoveries of several chemical probes and labeling strategies and the subsequent improvements made to these probes and labeling strategies while also highlighting several examples of important findings achieved using these methods. Specifically, we highlight the use of D-amino acids (DAAs) for metabolic labeling of PG in the bacterial cell wall and the labeling of the mycomembrane of corynebacteria using trehalose-analogues. Also covered is the use of bioorthogonal non-canonical amino acid tagging for the labeling of new protein synthesis in bacteria and the application of so-called activity based probes that target enzyme activities specific to the bacterial pathogen of interest.
Metabolic labeling of peptidoglycan
Peptidoglycan (PG) is the major constituent of the bacterial cell wall. PG consists of linear glycan strands that are cross-linked through short peptides, creating a dense macromolecular network that is vital to bacterial survival. The bacterial cell wall provides structural integrity for the cell and protects it against the osmotic pressure established by high intracellular concentrations of proteins and other biomolecules.36 The PG cell wall also functions as an anchor for important macromolecules of the cell envelope, such as teichoic acids,37 and Braun's lipoprotein.38 Due to its vital role in bacterial survival, relative accessibility at the outside of the cell, and absence in eukaryotic cells, PG biosynthesis is a prime target for antibiotics.39 Many widely used antibiotics including the β-lactams exemplified by penicillin,40 glycopeptides such as vancomycin,41 and the lipopeptide bacitracin42 function by inhibiting various aspects of bacterial cell wall biosynthesis. However, due to misuse and overuse, bacteria are acquiring resistance against these antibiotics at an alarming rate.5,43 Nonetheless, the bacterial cell wall remains a very promising target for the development of novel antibiotics.PG metabolism has been studied using various methods, such as metabolic labeling with radioprobes.44 Other methods include labeling of the bacterial cell wall using fluorescently tagged antibiotics45,46 as well as by the incorporation of a fluorescently labeled tripeptide into the cell wall by exploiting the PG recycling machinery of Escherichia coli (E. coli).47 These techniques have multiple limitations: (1) labeling techniques that are dependent on the PG recycling pathway of E. coli can only be used to study this specific organism; (2) radiolabeling of PG is known to be a very laborious and technically complex technique; (3) labeling the bacterial cell wall using fluorescently tagged antibiotics is limited to bacterial species with an accessible cell wall and enzyme machinery tolerant to these large pendant fluorophores; and (4) the use of fluorescently tagged antibiotics has an inhibitory effect on bacterial cell growth, thereby limiting their use.
Recently, metabolic labeling of PG using new D-amino acid (DAA) analogues has led to significant advancements in the understanding of cell wall biosynthesis and recycling of PG. Whereas nearly all life forms use exclusively L-amino acids, most bacteria also use DAAs, which are incorporated into the PG cell wall.48D-Alanine and D-glutamic acid are the DAAs that are most often incorporated into PG and can thus be found in the cell wall of nearly all bacteria.48 Interestingly, studies have shown that the biosynthetic machinery involved in PG synthesis is highly tolerant to a variety of different DAAs.49,50 This phenomenon has been exploited to incorporate DAAs and analogues thereof, to act as reporter molecules (one-step metabolic PG labeling) or as chemical handles for the conjugation of reporter molecules (two-step metabolic PG labeling).
Kuru et al.30 were the first to employ fluorescent D-amino acids (FDAAs) for one-step PG labeling and imaging of bacterial cell walls in live cells. Starting from the D-amino acids 3-amino-D-alanine and D-lysine, they reacted reporter molecules 7-hydroxycoumarin-3-carboxylic acid (HCC), 4-chloro-7-nitrobenzofurazan (NBD), fluorescein and carboxytetramethylrhodamine (TAMRA) to synthesize a variety of fluorescent amino acids, such as HADA, NADA, FDL and TDL (Fig. 1).30 Subsequently, they succeeded in labeling a wide range of Gram-negative and Gram-positive bacteria, including, but not limited to, E. coli, Staphylococcus aureus and Bacillus subtilis, and showed that these FDAAs were selectively incorporated into the bacterial cell wall. In B. subtilis, they showed that these FDAAs were incorporated exclusively into PG and not into teichoic acids; which also contain D-alanine residues.30 Because of their broad species applicability and excellent selectivity for PG, FDAAs have been used to study cell wall synthesis and its role in pathogenesis,51–59 and to establish the presence of PG in many bacterial species.60–62 An interesting application of FDAAs is the construction of so-called ‘virtual time-lapse’ images of PG synthesis in individual organisms. By sequentially adding FDAAs with different spectral properties, the growing PG layer can be sequentially labeled with multiple, differently colored FDAAs. This creates a multi-colored image where each color represents the location and amount of PG that is synthesized during the time in which FDAA was added.30,63
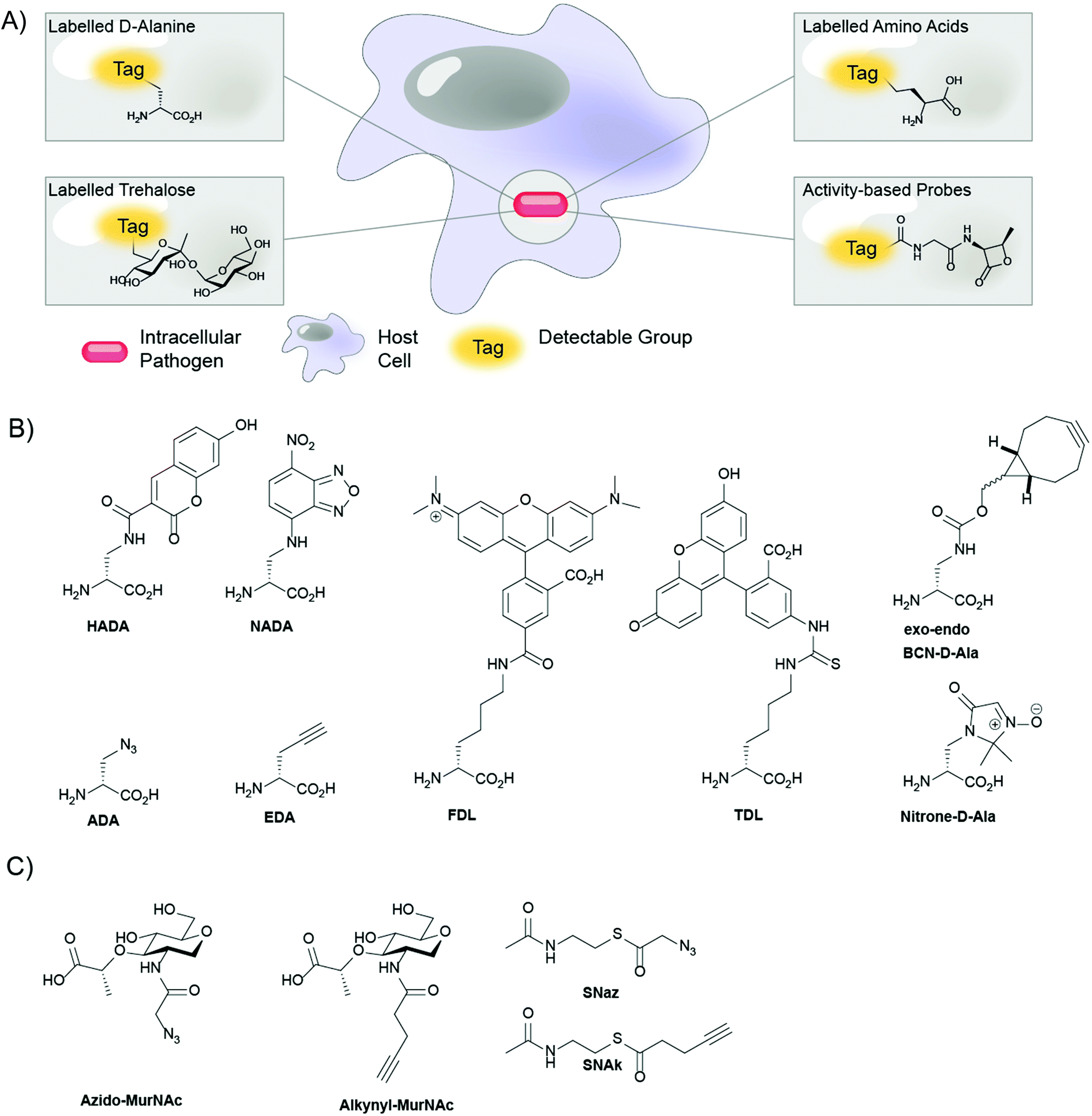 | ||
| Fig. 1 (A) Schematic overview of the labeling approaches; (B) D-amino acid probes for metabolic labeling of peptidoglycan (PG); (C) click-reactive muramic acid and acetyl-CoA analogues. | ||
FDAAs owe their broad applicability partially to their low molecular weight; labeling of the Gram-negative bacterial cell wall requires passage through the outer membrane, which is markedly less permeable to molecules over a molecular weight of ∼600 Da, such as antibiotics.64 Indeed, when Hsu et al.65 compared the labeling of E. coli with FDAAs with molecular weights between 300–700 Da, they found that FDAAs over a molecular weight of ∼500 Da have reduced access to the E. coli periplasm and cytoplasm and label the cell wall less efficiently. Today, the available toolkit of FDAAs covers the whole visible spectrum, from violet to far-red, providing many options for PG imaging in real time. Amongst these are FDAAs that are compatible with stochastic optical reconstruction microscopy (STORM), such as Cy3B and Atto 488, allowing imaging of PG nanostructures with far better spatial resolution.65 Improvements to other FDAAs include increased water solubility, increased photostability, and the possibility to excite with far-red light, reducing the phototoxic burden on the bacteria that is being probed. In conclusion, FDAA labeling is an excellent analysis technique to study the bacterial cell wall because of its selectivity for bacterial PG, it's relative experimental simplicity, and the ubiquity of DAAs in the cell walls of all bacterial taxa. Moreover, FDAAs only label nascent PG and are therefore an ideal tool to study spatial and temporal aspects of PG synthesis. The expansion of the FDAA toolkit into probes that are suitable for STORM has improved the spatial resolution that can be attained with FDAA labeling and cements the relevance of this technique for the study of the bacterial cell wall.65 Notably, however, FDAAs have not yet been applied to the imaging of intracellular pathogenic bacteria.
One downside of the FDAA-approach is that these probes are only incorporated into PG via the extracellular pathways reliant on the D,D- and L,D-transpeptidases, and are therefore not tolerated by the D-alanine racemase (Alr) and the UDP-N-acetylmuramoyl-tripeptide-D-alanyl-D-alanine ligase (MurF).62,66 These steps of the pathway in most species can, however, be labeled using a two-step labeling approach. The approach is based on metabolic incorporation of a D-amino acid equipped with a specific chemical handle, which, once incorporated into the PG network, can be labeled using a reporter molecule that is reactive to the handle. Two-step PG labeling has a lower risk of causing biological perturbation than one-step PG labeling, as the chemical handle is usually much smaller than the probes used in one-step PG labeling. Presently, bioorthogonal groups are commonly used as chemical handles in two-step PG labeling, because of their high reactivity towards their bioorthogonal reaction partners and their inertness towards biomolecules.34 PG labeling has been performed using D-cysteine as a chemical handle, effectively incorporating thiol groups into the cell wall for coupling to thiol-reactive reporter molecules.67 But, reporter molecules that target D-cysteine do not selectively label PG, as they are also reactive to L-cysteine residues in cellular proteins. To overcome such limitations, the introduction of D-amino acids containing bioorthogonal groups such as azides, alkynes, and nitrones allows for highly selective two-step metabolic labeling of PG.68
Kuru et al.30 and Siegrist et al.31 independently reported the use of DAAs containing bioorthogonal groups for PG labeling: R-propargylglycine (also known as ethynyl-D-alanine, EDA) and 2-amino-3-azidopropanoic acid (commonly known as azido-D-alanine, ADA) (Fig. 1). Kuru et al. validated the concept of bioorthogonal two-step metabolic PG labeling by labeling both B. subtilis and E. coli cells in vitro with EDA and ADA.30 Expanding on this work, Siegrist et al. showed that Listeria monocytogenes and Mtb could be efficiently labeled by EDA and ADA, in vitro and inside infected J774 macrophages. Notably, this work also demonstrated that macrophages take up sufficient EDA for effective labeling of intracellular bacteria without any apparent toxicity to either the macrophages or bacteria, highlighting the potential of two-step metabolic DAA labeling for cell culture-based applications.17 Combining the promiscuity of EDA labeling with the selectivity of RADA labeling, García-Heredia et al. showed that PG repair also occurs away from the sites of de novo PG synthesis in various mycobacterial species.66 Since then, Pidgeon et al. also reported probes specific for the L,D-transpeptidase to provide further tools to study bacterial cell wall synthesis. The possibility of applying these probes to the study of the intracellular lifecycle of pathogens offers a tantalizing opportunity as a tool in the development of drugs against such pathogens.69
The bioorthogonal toolkit for metabolic labeling of PG was further expanded by Shieh et al.70 They synthesized D-amino acids equipped with the strained cyclooctyne and bicyclononyne (BCN) moieties (Fig. 1), for copper-free, strain-promoted alkyne–azide “click” (SPAAC) reactions. Furthermore, they synthesized near-infrared (NIR) azide-functionalized fluorogenic Si-rhodamine dyes whose fluorescence quantum yield greatly increases upon reaction with alkynes. These NIR fluorogenic dyes are very useful for in vivo imaging as near infrared light penetrates deeper through tissue than higher wavelength light and no washing of excess dye is required as the dye becomes vastly more fluorescent upon reaction. The Pezacki group added an additional bioorthogonal reaction to the labeling repertoire by designing nitrone D-amino acids that readily undergo reactions with strained alkynes in a strain-promoted alkyne-nitrone cycloaddition (SPANC) reaction (Fig. 1).68,71
Metabolic incorporation of D-amino acid probes is well tolerated in most bacteria, with notable exceptions. For instance, the intracellular bacterium Chlamydia trachomatis required additional reagents to enable PG labeling. While circumstantial evidence for the existence of a PG containing cell wall in C. trachomatis had existed for decades,72,73 until recently no one had conclusively isolated or characterized PG from this bacterium.74,75 This resulted in a discrepancy called the ‘Chlamydial Anomaly’, where no PG was observed, yet the pathogen was sensitive to PG-targeting antibiotics.76,77 To address the matter Liechti et al. set out to conclusively establish the presence of PG in C. trachomatis by labeling with the D-amino acid probe EDA, but found that EDA failed to label the bacteria.62 They hypothesized that this was because chlamydial PG synthesis enzymes could not accommodate EDA, but that they might accommodate modified D-alanine dimers wherein one of the two amino acids was EDA (EDA-DA). Indeed, after incubating the bacteria with the alkyne containing EDA-DA dipeptide, they were subsequently able to label C. trachomatis with Alexa Fluor 488 azide. Interestingly, no labeling was detected when using the dipeptide analogue bearing the bioorthogonal modification on the C-terminal D-alanine residue (i.e. DA-EDA). The authors reasoned that the lack of labeling was due to removal of the C-terminal D-alanine residue in either transpeptidation or carboxypeptidation reactions. The work of Liechti et al. shows that labeling of PG in some bacteria may require modified D-amino acid probes, but also highlights the versatility of D-amino acid labeling for the detection and study of bacterial PG.
In addition to the use of unnatural D-amino acids, strategies based on other building blocks have been developed to probe PG assembly in bacteria. In this regard, Grimes and coworkers designed metabolic probes based on N-acetyl muramic acid (MurNAc) that allowed for labeling of the carbohydrate moiety of PG.78,79 Although labeling of PG with D-amino acids analogues is a powerful and versatile method, there are several benefits to labeling this carbohydrate moiety: firstly, D-alanine residues of the PG stem peptide can be removed by transpeptidation and carboxypeptidation reactions, which may decrease the probe's lifetime, whereas the carbohydrate backbone of PG is not subject to remodeling. Secondly, MurNAc is a superior probe for de novo PG synthesis, as it is only incorporated in newly synthesized PG, while D-alanine probes can also be incorporated into mature PG by exchange reactions.50,80,81 Finally, MurNAc binding to Nod-like receptors (NLRs) is involved in innate immune activation,82 while D-alanine residues are not involved in immune activation, making MurNAc analogues more suitable for probing immune activation by PG. Metabolic labeling of the carbohydrate moiety in PG was first achieved by Liang et al. using MurNAc analogues with azido- and alkyne modifications at the 2-N acetyl position (Fig. 1).78 In some Gram-negative bacteria, including, amongst others, Pseudomonas putida, MurNAc can be recycled to uridine diphosphate-MurNAc (UDP-MurNAc) by enzymes AmgK and MurU and subsequently incorporated into newly synthesized PG.83 Interestingly, Liang et al. showed that both E. coli and B. subtilis could incorporate MurNAc analogues into de novo synthesized PG after transfection with a vector for P. putida AmgK and MurU.78 Their results suggest that metabolic MurNAc labeling could be applied to a wide range of bacteria, both Gram-negative and Gram-positive, if transfected with amgK and murU. Metabolic MurNAc labeling of most bacteria does, however, require genetic modification of the bacteria and is limited to bacteria in which amgK and murU can be introduced, limiting the practical use of this particular metabolic labeling technique.
Building on these approaches, Wang et al. recently reported an elegant method for post-synthetic, metabolic labeling of the PG carbohydrate backbone.79 Their technique exploits the function of the bacterial enzyme peptidoglycan O-acetyltransferase B (PatB), which uses acetyl-CoA as an acetyl donor to O-acetylate the 6′ hydroxyl group of MurNAc residues in the carbohydrate moiety of PG.84 Post-synthetic O-acetylation of PG occurs in many intracellular pathogenic bacteria, such as Neisseria meningitidis, Neisseria gonorrheae and Staphylococcus aureus,85–87 where it contributes to bacterial virulence by providing resistance against PG degradation by the antimicrobial enzyme lysozyme.88 Wang et al. hypothesized that bioorthogonal groups could be introduced into PG post-synthetically by feeding azide- (SNAz) or alkyne- (SNAk) functionalized acetyl-CoA analogues (Fig. 1). They found that PatB efficiently O-acetylates PG in B. subtilis when supplied with SNAz or SNAk and in doing so enabled visualization of the incorporated probes by reaction with fluorescent dyes carrying complementary bioorthogonal groups.79 Interestingly, they also showed that the artificially O-acylated B. subtilis was, at least partially, protected from degradation by lysozyme.
Metabolic labeling of the mycomembrane using trehalose analogues
The mycobacterial cell wall contains an outer membrane – the mycomembrane (MM) – that lends itself well to selective labeling. It consists of long chain (C60–C90) mycolic acids that are covalently linked to the cell wall's PG layer through highly branched polysaccharides called arabinogalactan (AG).89,90 Components of the MM are important for mycobacterial cell survival; for example, blocking the biosynthesis of the nonmammalian disaccharide trehalose, a key component of the MM glycolipids trehalose dimycolate (TDM) and trehalose monomycolate (TMM), reduces cell viability and induces growth defects.91,92 TMM is a modulator of MM biosynthesis84 and a precursor to TDM, which promotes virulence and survival in host macrophages.93 Since trehalose metabolism is vital for mycobacterial cell viability and is not found in mammalian cells, its biosynthetic pathway makes an excellent target for the development of novel antibiotics against Mtb. Moreover, since trehalose is exclusively found in members of the suborder of Corynebacterineae, which does not include canonical Gram-negative or Gram-positive bacteria, it opens up the possibility to use metabolic labeling with trehalose-probes for clinical diagnosis of Mtb.Clinical diagnosis of Mtb in high-burden, developing countries is currently performed using either the Ziehl–Neelsen test or the Auramine Truant stain, both of which are only moderately reliable.86 Furthermore, these diagnostic tests also do not discriminate between live or dead Mtb cells and thus cannot be used by physicians to determine treatment efficacy.94 Thus, novel, Mtb-specific diagnostic tools are required to improve TB diagnosis efficacy. Metabolic labeling with trehalose analogues has shown promise as a novel diagnostic approach for the rapid detection of Mtb infection. Trehalose, in the form of TMM and TDM, is anchored non-covalently to the mycobacterial cell wall. Trehalose is synthesized and esterified to form TMM in the cytosol, after which TMM is transported to the periplasm.95 Once in the periplasm, the mycolyltransferase complex antigen 85 (Ag85) transfers mycolate from TMM to AG, to form a network of covalent mycolates that form the foundation of the MM, or to another molecule of TMM, forming TDM.96–98 In both cases, a molecule of trehalose is regained, which is recycled by active transport into the cytosol by the trehalose-specific transporter SugABCLpqY.99
Based on analysis of the trehalose/Ag85 co-crystal structure, Backus et al.32 hypothesized that trehalose could be modified with functional groups without affecting substrate activity for Ag85, exploiting a promiscuity that is similar to that of D-alanine metabolism. They synthesized various trehalose-analogues, including fluorescein isothiocyanate (FITC)-trehalose (FITC-Tre, Fig. 2), and tested their ability to label pathogenic Mtb using a one-step labeling method. FITC-trehalose was efficiently incorporated into growing Mtb mycomembranes by Ag85 in vitro and exclusively labeled live Mtb cells; heat-killed Mtb cells were not labeled, nor were cells of the Gram-negative pathogens Pseudomonas aeruginosa and Haemophilus influenza or Gram-positive Staphylococcus aureus.32 FITC-Tre could even be incorporated into Mtb inside macrophages; macrophages were infected with Mtb and successfully labeled with FITC-Tre.
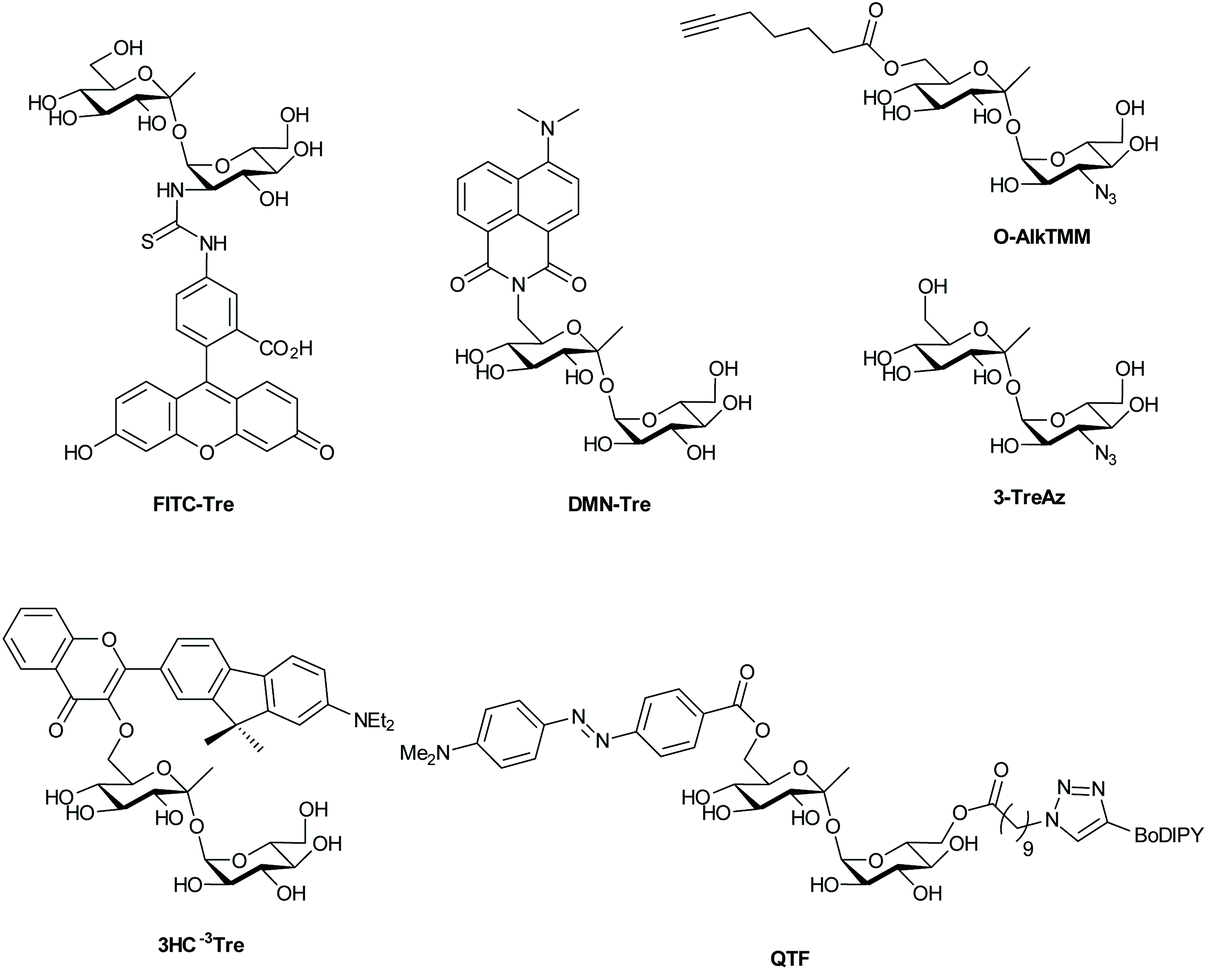 | ||
| Fig. 2 Fluorescent and fluorogenic trehalose analogues for labeling of the mycomembrane. | ||
Backus et al. also demonstrated that Ag85 enzymes tolerate a surprisingly wide range of substrate modifications. Moreover, their work demonstrates that the Ag85 pathway can be hijacked to incorporate exogenous trehalose into TMM and, subsequently, into the MM. They also speculate that, given the conserved nature of Ag85 amongst mycobacteria, the FITC-trehalose probe could presumably label other mycobacterial species equally as well as Mtb. However, Rodriguez-Rivera et al. found that FITC-trehalose labels other mycobacterial with poor efficiency and does not label corynebacteria at a detectable level at all.100 SAR studies showed that removal of an anomeric methyl group significantly increased labeling efficiency amongst myco- and corynebacteria, as did repositioning of the FITC functionality on the trehalose scaffold. Rodriguez-Rivera et al. applied metabolic trehalose labeling to assess the influence of ethambutol treatment on mycomembrane fluidity using fluorescence recovery after photobleaching (FRAP) experiments. They showed that ethambutol can increase MM fluidity at sub-inhibitory concentrations, thereby enhancing antibiotic drug accessibility.100
One-step metabolic labeling is an excellent method for metabolic incorporation of trehalose analogues into the mycomembrane via extracellular incorporation by the Ag85 complex. However, an important aspect of trehalose metabolism occurs intracellularly, where trehalose is formed and subsequently incorporated into various metabolites, which together constitute the ‘trehalome’.33 The importance of trehalose for bacterial survival calls for chemical probes that can provide insight into intracellular trehalose metabolism. Swarts et al.33 hypothesized that the trehalose-recycling pathway could be hijacked to actively transport azido-trehalose analogues into the mycobacterial cytosol through the trehalose-specific transporter SugABCLpqY, after which the trehalose-analogue would be incorporated into the ‘trehalome’ and subsequently labeled with azide-reactive reporter molecules. Trehalose-analogues with azido-groups (TreAz analogues) at the 2-, 3-, 4- or 6-position (Fig. 2) were added to a culture of Mycobacterium smegmatis, a non-pathogenic Mycobacterium that is frequently used as a model organism for Mtb, and then exposed to a fluorescent dye coupled to a cyclooctyne group leading to labeling via a strain-promoted [3 + 2] cycloaddition. They found that the TreAz analogues labeled M. smegmatis efficiently and that the route of trehalose incorporation was dependent on the structure of the TreAz analogue; 2-, 4- and 6-TreAz were incorporated via the recycling pathway, while 3-TreAz was incorporated only extracellularly by Ag85. This incorporation was also possible for pathogenic Mtb and Mycobacterium bovis. The recent development of chemoenzymatic routes towards unnatural trehalose analogues is expected to expand the application of this method by facilitating access to the new analogues.101
Two-step metabolic labeling with trehalose analogues has also been applied to study mycolation of AG and O-mycolated proteins that are found in the MM. The Swarts group hypothesized103 that the substrate specificity of Ag85 as a mycolyltransferase would extend to a 6-heptynoyl trehalose analogue (O-AlkTMM, Fig. 2), based on previously reported Ag85 activity studies.32,102 They hypothesized that O-AlkTMM could be taken up into the periplasm, where Ag85 would transfer the 6-heptynoyl group to mycolyl acceptors such as AG and O-mycolated proteins.103,104 Subsequent reaction with alkyne-reactive reporter molecules would allow for enrichment or visualization of mycolated targets. Foley et al.103 reported on the synthesis of O-AlkTMM and showed that O-AlkTMM effectively labels mycobacterial AG. Similar to TreAz analogues, O-AlkTMM selectively labels cell walls of mycobacteria and corynebacteria without labeling canonical Gram-negative and Gram-positive bacteria. In contrast to TreAz analogues, metabolic labeling with O-AlkTMM also provides the means to directly incorporate labels during the synthesis of AG mycolate (AGM), which is an essential component of the mycobacterial cell wall. O-Mycolation of proteins has only recently been discovered as a post-translational protein modification in corynebacteria.105 Notably, O-acylation of bacterial proteins was unheard of prior to the discovery of O-mycolated proteins, making it an intriguing phenomenon and worthy of further study. Using the O-AlkTMM probe, Kavunja et al.104 were able to label multiple O-mycolation target proteins in Corynebacterium glutamicum. Next, they visualized them with an in-gel fluorescence assay, by means of the well-established copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) click reaction with Alexa Fluor 488 azide. Their study uncovered multiple novel O-mycolated proteins and revealed that proteins can carry more than one O-mycolate group, highlighting the utility of the O-AlkTMM probe for studies into the ‘O-mycoloylome’. Much remains to be discovered about bacterial protein O-mycolation and the O-AlkTMM probe has proven to be a highly valuable tool for such O-mycolation studies. Taken together, the aforementioned investigations show the potential of trehalose analogues for studies into trehalose- and mycoloyl-metabolism. The same group recently expanded the bioorthogonal TMM library to include trans-cyclooctene TMM analogues reactive in an inverse electron-demand Diels–Alder ligation reaction.106
Recently, various trehalose analogues have emerged as novel potential point-of-care diagnostic tools for Mtb. This is a large unmet clinical need, as the current diagnostic methods are notoriously unreliable.107 Rundell et al.108 took the first steps towards synthesis of an 18F-labeled trehalose analogue that would facilitate targeted positron emission tomography (PET) imaging of patients lungs for quick diagnosis of TB infections. However, this approach requires advanced equipment for diagnosis – infrastructure often absent at remote diagnostic sites. To provide a facile low-tech Mtb-detection test, Kamariza et al.109 developed a solvatochromic trehalose probe (DMN-Tre), consisting of a trehalose-conjugated fluorescent dye, 4-N,N-dimethylamino-1,8-naphthalimide (DMN) (Fig. 2), that dramatically increases in fluorescence upon transition from a hydrophilic to a hydrophobic environment. The authors showed that the solvatochromic nature of DMN allowed for no-wash labeling of Mtb, since DMN-Tre should only become fluorescent once incorporated into the hydrophobic environment of the MM. DMN-Tre could indeed be used for efficient no-wash labeling of Mtb cells, even in sputum samples from TB-positive patients. This probe was further used for high-content image-based screening of the effect of various drugs on intracellular Mtb.110 Recently, new solvochromatic trehalose probes based on 3-hydroxychromone fluorophores (3HC, Fig. 2) were reported that show a 10-fold increase in fluorescence enhancement compared to DMN. This allowed the detection of Mtb cells within 10 minutes of treatment and may well represent a major step towards on-site testing for Mtb infection.111
Hodges et al. recently reported the use of a different strategy for fluorogenic Mtb cell wall detection. Their approach employs a trehalose variant dubbed “quencher-trehalose-fluorophore” (QTF, Fig. 2) consisting of a lipid-BODIPY construct and a DABCYL quencher attached to a trehalose scaffold.112 Esterase Ag85 cleaves the lipid-fluorophore from the quencher-trehalose, leading to enhancement (turn-on) of fluorescence (Fig. 2). Finally, the Swarts-group recently reported a similar approach, in which cleavage of a quencher group from a construct containing fluorescein and a DABCYL quencher by the mycomembrane remodeling hydrolase Tdmh, led to a the enhancement of green fluorescence.113 One downside to all these probes is that they exclusively label metabolically active cells. As such, they can only be used to diagnose dividing Mtb and monitor treatment efficacy. The metabolically dormant forms of Mtb, however, remain undetected by these technologies. There remains an urgent and unmet need for the development of probes for this subclass of Mtb.
Proteome labeling with BIOORTHOGONAL amino ACIDs
The methionine tRNA/tRNA synthetase pair of many species are rather promiscuous. At the turn of the millennium, the Tirrell and Bertozzi groups were the first to exploit this promiscuity to introduce bioorthogonal groups into bacterially expressed proteins and label these using click reactions.114 This technique was later applied by the Tirrell-lab to the pan-proteome Met-labeling of whole cells. The approach, latter dubbed bioorthogonal non-canonical amino acid tagging (BONCAT),115 has subsequently been used for the enrichment, identification, and visualization of exclusively nascent proteins, thus reducing the complexity of proteomic analysis of a given sample.116 Moreover, pulse-labeling with BONCAT probes allows for analysis of changes in protein production in response to internal and external cues.115 With regards to the study of intracellular pathogens, two BONCAT-based approaches have been taken. Those reliant on the hijacking of existing tRNA/synthetase pairs, and those that genetically incorporate new tRNA/synthetase pairs. Both approaches have been used extensively to interrogate pathogen's protein expression at the host–pathogen interface.The first approach, which relies on the promiscuity of the methionine tRNA and its synthetase, have shown that methionine analogues containing alkene-,117–120 alkyne-117,121 and azido-114 functionalities can be accepted substrates by the methionyl-tRNA synthetase (MetRS) in a variety of pro- and eukaryotes.122 Care has to be taken when incorporating these amino acids into new species, as the speed of protein synthesis and cell division can be altered by the presence of such unnatural amino acids, as demonstrated by homopropargylglycine (Hpg, Fig. 3) which shows toxicity towards certain bacteria at higher incubation rates, whereas azidohomoalanine does not (Aha, Fig. 3).123 The advantage of this approach is that it provides a facile way to image only the proteome of an intracellular pathogen after uptake by a host cell, without any need for further genetic modification of the species. Reacting these proteins with a clickable biotin- or FLAG-tag can also enable the enrichment of these target proteins for mass spectrometry.116 We ourselves have used this strategy to incorporate Aha or Hpg into E. coli ex vivo and then co-incubated these bacteria with mouse bone marrow-derived cells (BMDCs) to visualize bacterial cell degradation in situ.124 For this we used correlative light-electron microscopy (CLEM) imaging, to provide ultrastructural content of the degrading E. coli cells within BMDC phagosomes. We have also translated the technique to allow super-resolution correlative light-electron microscopy (STORM-CLEM) of pathogens in BMDCs to show the durability of Salmonella bacteria inside the host cell vacuole.123 Recently, we also combined the BONCAT labeling of the Mtb proteome with the labeling of its PG layer through the use of EDA-labeling with correlative imaging, showing that multiple click reactions can be performed on pathogens simultaneously. We used this approach to assess the heterogeneous effect of various antibiotics on intracellular Mtb (see Fig. 4 for an example image).125
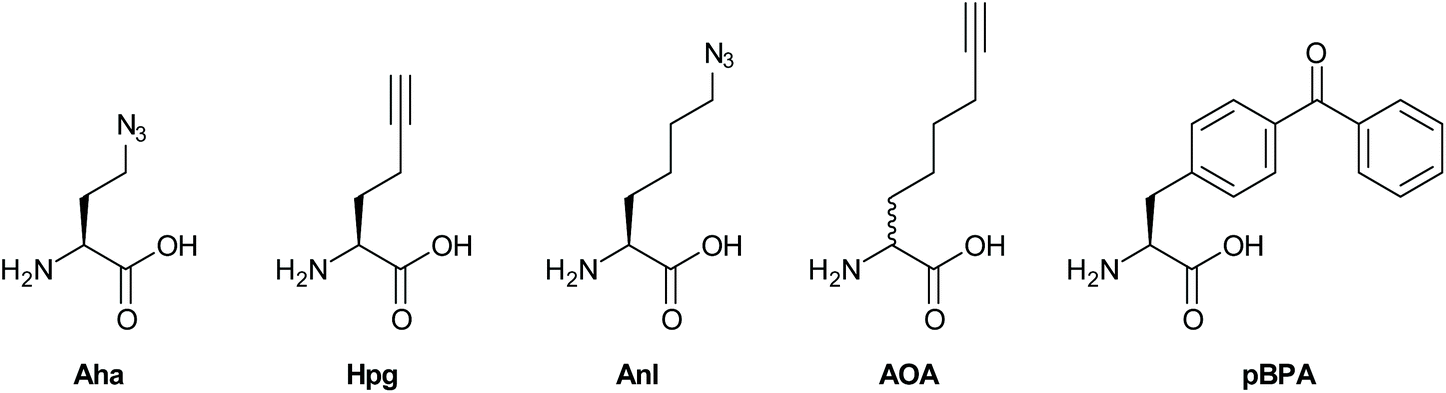 | ||
| Fig. 3 BONCAT probes for metabolic labeling of proteins. | ||
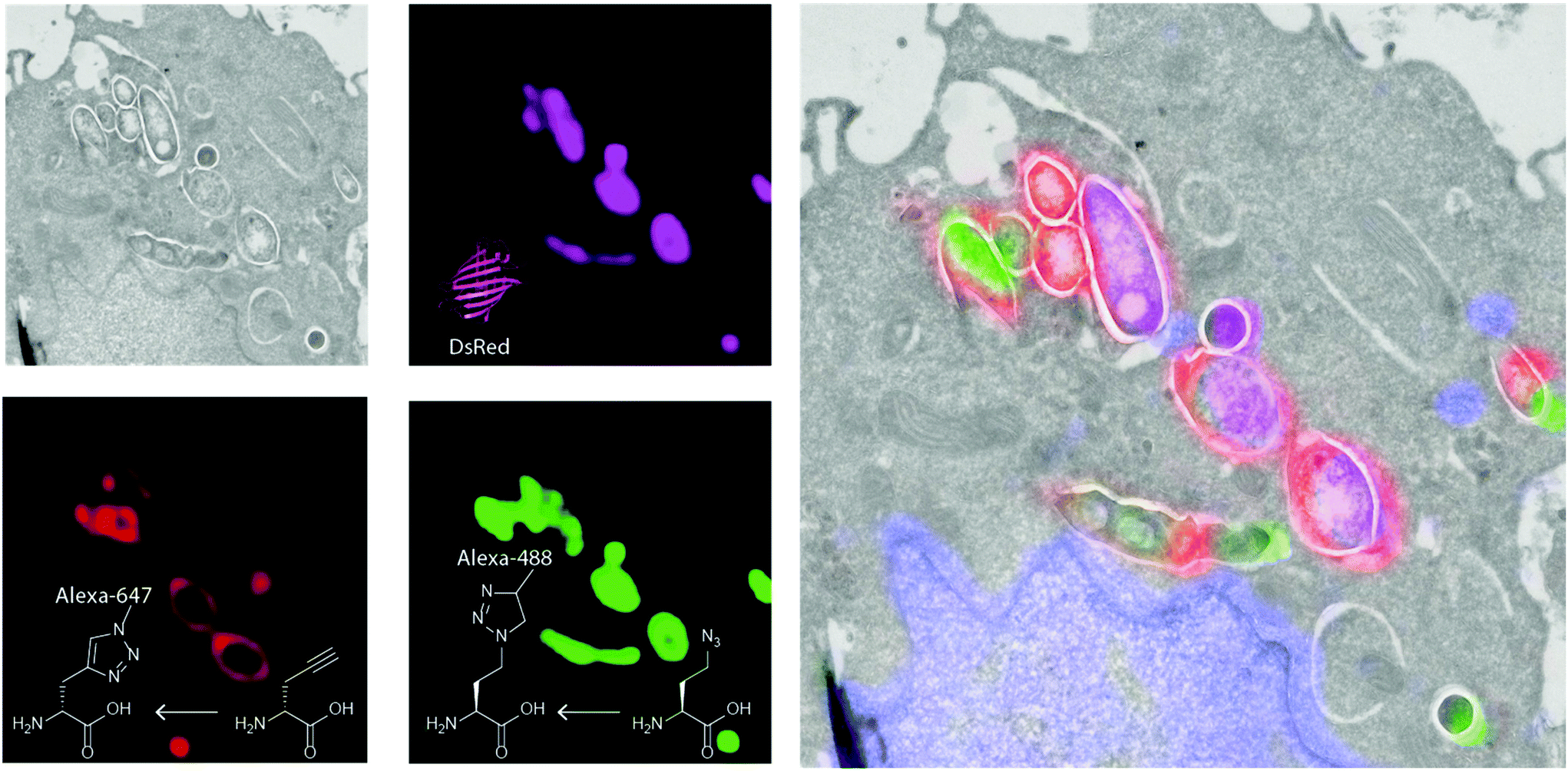 | ||
| Fig. 4 Correlative light-electron microscopy of triple labelled Mtb. Green: Alexa Fluor 488 alkyne reacted to Aha in the proteome; Red: Alexa Fluor 647 azide reacted to EDA; Pink: expressed DsRed signal. Grey: electron micrograph indicating the subcellular environment in which the bacteria reside. | ||
Approaches that allow expansion of the genetic code of pathogens have also become powerful tools to study host–pathogen interactions. For example, Ngo et al.126 devised a new BONCAT-like strategy that involved genetically modifying the cell type of interest to enable incorporation of NCAA for this cell type only. For this strategy, a mutant MetRS (NLL-MetRS) was developed that can append the NCAA azidonorleucine (ANL; Fig. 3) to endogenous tRNA.127 To achieve cell type selective protein labeling with bioorthogonal handles, the cell type of interest is transfected with the NLL-MetRS gene, enabling this cell type to incorporate ANL into newly synthesized proteins. ANL is not compatible with endogenous MetRS, leading the NCAA to be exclusively incorporated into proteins from the pathogen and not the host. Because of these features, BONCAT using NLL-MetRS is highly suited for analyzing pathogen's nascent protein expression at the host–pathogen interface. The potential of this approach was further demonstrated in the interrogation of host–pathogen interactions by infecting murine macrophages with E. coli that constitutively expressed NLL-MetRS and visualizing the bacteria selectively within the macrophage.
Using this cell selective BONCAT method, Mahdavi et al. were able to enrich and identify several key secreted Yersinia outer proteins (Yops) and other secreted factors in an infection model.128 To enrich Yops that were secreted into the host cytosol, HeLa cells were infected with NLL-MetRS-expressing Yersinia cells and subsequently lysed using digitonin, preserving the integrity of the Yersinia cells. Subsequent reaction to TAMRA-alkyne allowed for facile identification of fluorescent Yops in host cytosol fractions. Moreover, by applying BONCAT in a pulse-labeling manner at specific times after infections, the authors were able to determine the order of Yop secretion, demonstrating the potential of BONCAT for elucidating the temporal aspects of virulence factor secretion. The Hang group also reported a similar strategy for the labeling of intracellular salmonella with 2-aminooctynoic acid (AOA, Fig. 3) and showed that the bacterial proteome of Salmonella enterica could be labeled and imaged with a high degree of selectivity inside RAW-macrophages.129 In addition, this approach also enabled for the enrichment and proteomic analysis of endogenously expressed Salmonella proteins from infected mammalian cells.
Amber codon suppression – the approach whereby a tRNA/tRNA synthetase pair reactive to the amber stop codon are introduced in a species have also been applied to the study of pathogenic species. Wang et al. reported the application of this strategy to incorporate para-iodophenylalanine into GFP expressed in the Mtb-strain H37Ra inside RAW-macrophages.130 The same approach was later exploited by Touchette et al. for the incorporation of the photo-crosslinkable amino acid p-benzoyl-L-phenylalanine (pBPA, Fig. 3) into the mycobacterial outer cell wall of M. smegmatis to identify the interaction partners of this protein.131 It is likely that the application of such techniques to study Mtb interactions with the host proteome will deliver new insights. The expanded genetic code variant of Salmonella was also recently reported and used to incorporate p-azidophenylalanine (Fig. 3) into the bacterial proteome. However, no intracellular labeling was attempted yet by this approach.132 Intracellular labeling was achieved for the facultative intracellular pathogen Neisseria meningitides. Takahashi et al. also incorporated pBPA into the bacterium and used it to elucidate the role of NMB1345 in the infection pathway.133
ABPP CLEM of bacterial enzymes
A final area which is providing tantalizing new insights into the intracellular biology of the host–pathogen interaction is the use of probes to visualize the activity of specific pathogenic enzymes inside the host, so-called activity-based protein profiling (ABPP).134 This approach has been used to image host cell alterations upon bacterial infection,135,136 but the imaging of pathogenic enzymes – particularly those involved in virulence – is an exciting new area.134 An early example involved the use of the broad spectrum serine hydrolase probe FP-TAMRA (Fig. 5) to identify the secreted serine hydrolases of Mtb137,138 and V. cholera.139 In a later study, all four V. cholera serine hydrolases were identified using a proteomic approach in an infection model of this pathogen. FP-TAMRA was also used to identify 8 new serine hydrolases in the facultative140 intracellular pathogen Staphylococcus aureus.141 Lentz et al. used FP-TAMRA to develop activity-based probes [ABPs] for two of these hydrolases FphB and FphF. Using these inhibitors/probes the authors could delineate a role for FphB in the infection of liver and heart cells, although an exact mechanism for this role remains elusive. The recently resolved crystal structure of FphF in complex with the inhibitor may help in elucidating these roles.142 Perhaps this relates to the potential for intracellular survival of this pathogen under certain conditions. Recently, the group of Bogyo also used this approach to developed a covalent probe that allowed visualization of an Mtb hydrolase, namely the hydrolase important for pathogenicity-1 (HIP-1). Using probe CSL173 (Fig. 5), they could observe its activity when spiked into the host proteome to identify HIP-activity.143 They have recently described the further development of these types of probes to synthesize a chemiluminescent point-of-care test for Mtb that allowed the selective identification of live Mtb, providing a useful tool for the study of drug efficacy.144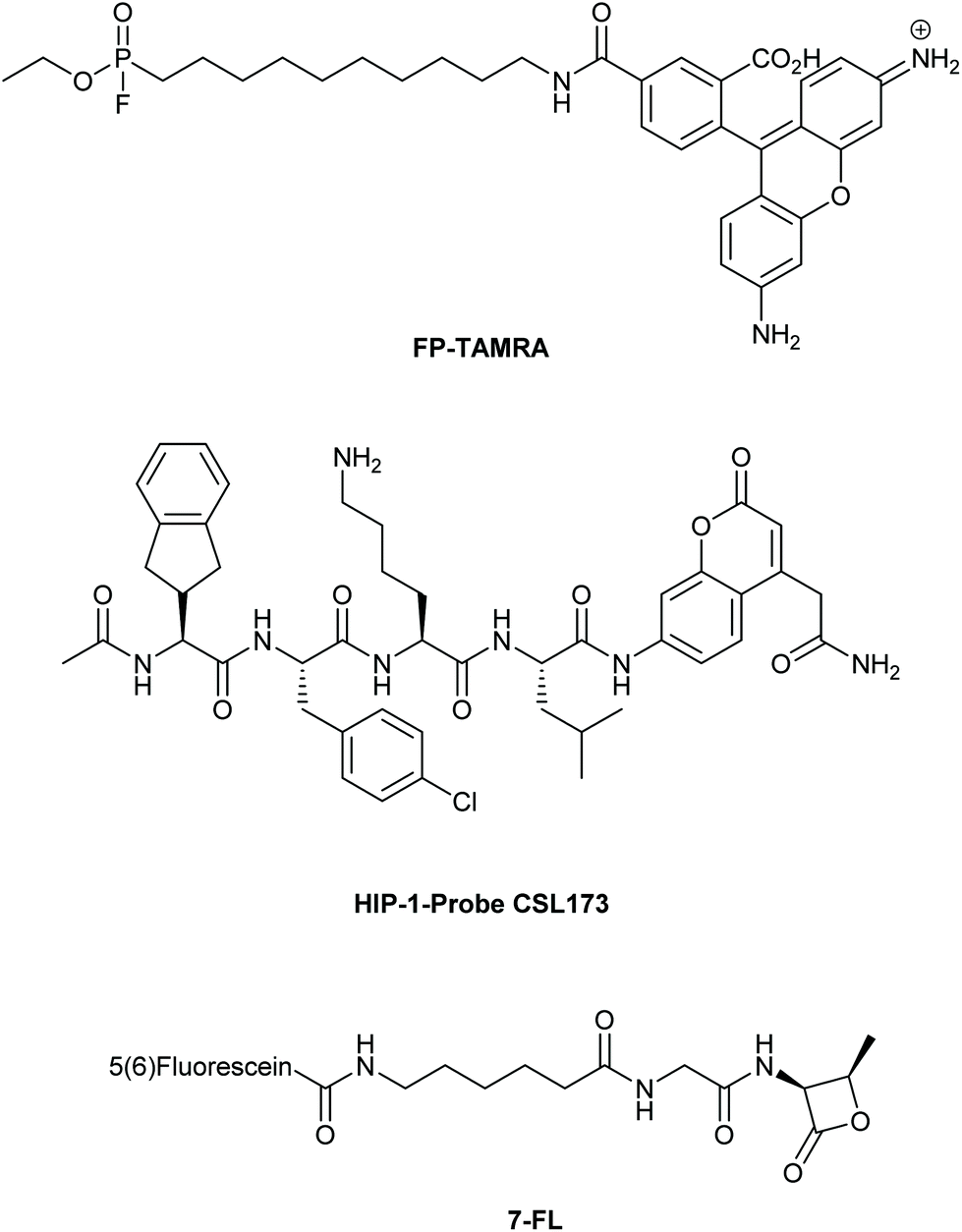 | ||
| Fig. 5 Covalent active-based probes used for imaging (active) bacterial enzymes. | ||
The electrophilic nature of the beta-lactam scaffold has also been used to image penicillin binding protein (PBPs) activities in pathogens.145 Initial approaches were marred by a poor substrate specificity of these probes for the various PBPs. This was recently solved for another facultative intracellular pathogen, Streptococcus pneumoniae.146 By modifying the beta-lactam core and attaching fluorescein (compound 7FL, Fig. 5) or TAMRA to these modified lactam-cores, the PBPs 1b and 2× could be selectively labeled and imaged in the dividing bacteria.147 With these probes it was found that these PBPs were restricted to a ring surrounding the bacterial division site. What these observations mean in the context of the recently discovered intracellular niche of the pathogen remains to be elucidated.
Conclusion
As these above examples clearly demonstrate, the application of novel chemical approaches to imaging is leading to rapid advances in the study of intracellular pathogens. The labeling of cellular components and individual enzyme activities is leading us to improved point of care tests.There are, however, areas in which we foresee major developments taking place. An area, which will likely move the field forward is the continuing development of better live-cell compatible chemistries. These will hopefully allow the development of detailed dynamic pictures of the host–pathogen interaction. Reactions, such as the Inverse Electron-Demand Diels–Alder ligation (IEDDA),148 can serve as excellent starting points for this development. For the near future, we envisage that the currently existing metabolic cell wall probes, D-alanine- and trehalose analogues, will maintain their relevance, as there are still many lingering questions concerning bacterial cell wall metabolism and growth modes. Here we foresee that the use of these methods, combined with advanced imaging and quantification techniques, will be increasingly deployed to study the efficacy of antibiotics in these host–pathogen interactions. Information on the precise effects on the intracellular live cycle of pathogens will increase understanding of the method of actions of new and existing antibiotics.
Moreover, secreted bacterial proteins, many of which are associated with virulence and persistence in the host environment, remain uncharacterized and their mechanisms of action poorly understood; we envisage that BONCAT-probes can contribute significantly to further the understanding of these secreted factors through the use of new retrieval-mass spectrometry approaches. Together, these findings may lead to novel vaccines, antibiotics and other therapies against bacterial infection.
Conflicts of interest
There are no conflicts to declare.References
- C. Dye, After 2015: infectious diseases in a new era of health and development, Philos. Trans. R. Soc., B, 2014, 369(1645), 20130426 CrossRef PubMed.
- R. Laxminarayan and D. L. Heymann, Challenges of drug resistance in the developing world, Br. Med. J., 2012, 344, e1567 CrossRef PubMed.
- P. Nordmann, et al., Does broad-spectrum β-lactam resistance due to NDM-1 herald the end of the antibiotic era for treatment of infections caused by Gram-negative bacteria?, J. Antimicrob. Chemother., 2011, 66(4), 689–692 CrossRef CAS PubMed.
- R. Laxminarayan, et al., Antibiotic resistance - the need for global solutions, Lancet Infect. Dis., 2018, 13(12), 1057–1098 CrossRef.
- A. R. M. Coates, G. Halls and Y. Hu, Novel classes of antibiotics or more of the same?, Br. J. Pharmacol., 2011, 163(1), 184–194 CrossRef CAS PubMed.
- C. Walsh, Where will new antibiotics come from?, Nat. Rev. Microbiol., 2003, 1, 65–65 CrossRef CAS PubMed.
- M. X. Byndloss and R. M. Tsolis, Chronic Bacterial Pathogens: Mechanisms of Persistence, Microbiol. Spectrum, 2016, 4(2), 513–528 Search PubMed.
- J. E. Gomez and J. D. McKinney, M. tuberculosis persistence, latency, and drug tolerance, Tuberculosis, 2004, 84(1), 29–44 CrossRef PubMed.
- A. Omsland, et al., Host cell-free growth of the Q fever bacterium Coxiella burnetii, Proc. Natl. Acad. Sci. U. S. A., 2009, 106(11), 4430–4434 CrossRef CAS PubMed.
- S. Singh, et al., Axenic culture of fastidious and intracellular bacteria, Trends Microbiol., 2013, 21(2), 92–99 CrossRef CAS PubMed.
- C. Dye, Global epidemiology of tuberculosis, Lancet, 2006, 367(9514), 938–940 CrossRef.
- B. Mahboub and M. Vats, Tuberculosis: Current Issues in Diagnosis and Management, BoD–Books on Demand, 2013 Search PubMed.
- D. Bald, et al., Targeting Energy Metabolism in Mycobacterium tuberculosis, a New Paradigm in Antimycobacterial Drug Discovery, mBio, 2017, 8(2), e00272-17 CrossRef PubMed.
- J. P. Sarathy, et al., Extreme drug tolerance of Mycobacterium tuberculosis in caseum, Antimicrob. Agents Chemother., 2018, 62(2), e02266-17 CrossRef PubMed.
- D. Evangelopoulos, J. D. da Fonseca and S. J. Waddell, Understanding anti-tuberculosis drug efficacy: rethinking bacterial populations and how we model them, Int. J. Infect. Dis., 2015, 32, 76–80 CrossRef PubMed.
- S. T. Pullan, et al., The effect of growth rate on pyrazinamide activity in Mycobacterium tuberculosis-insights for early bactericidal activity?, BMC Infect. Dis., 2016, 16(1), 205 CrossRef PubMed.
- J. Daniel, et al., Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages, PLoS Pathog., 2011, 7(6), e1002093 CrossRef CAS PubMed.
- J. J. Baker and R. B. Abramovitch, Genetic and metabolic regulation of Mycobacterium tuberculosis acid growth arrest, Sci. Rep., 2018, 8(1), 4168 CrossRef PubMed.
- N. K. Dutta and P. C. Karakousis, Latent tuberculosis infection: myths, models, and molecular mechanisms, Microbiol. Mol. Biol. Rev., 2014, 78(3), 343–371 CrossRef PubMed.
- C. Deb, et al., A novel in vitro multiple-stress dormancy model for Mycobacterium tuberculosis generates a lipid-loaded, drug-tolerant, dormant pathogen, PLoS One, 2009, 4(6), e6077 CrossRef PubMed.
- S. Sarkar, A. Ganguly and H. Sunwoo, Current overview of anti-tuberculosis Drugs: metabolism and toxicities, Mycobact. Dis., 2016, 6(209), 1000209 Search PubMed.
- D. T. Hoagland, et al., New agents for the treatment of drug-resistant Mycobacterium tuberculosis, Adv. Drug Delivery Rev., 2016, 102, 55–72 CrossRef CAS PubMed.
- K. N. Fadhilah, K. Sharon and G. Liam, Targeting the hard to reach: challenges and novel strategies in the treatment of intracellular bacterial infections, Br. J. Pharmacol., 2016, 174(14), 2225–2236 Search PubMed.
- L. Mughini-Gras, et al., Increased colon cancer risk after severe Salmonella infection, PLoS One, 2018, 13(1), e0189721 CrossRef PubMed.
- M. H. Ahmadi, A. Mirsalehian and A. Bahador, Association of Chlamydia trachomatis with infertility and clinical manifestations: a systematic review and meta-analysis of case-control studies, Infect. Dis., 2016, 48(7), 517–523 CrossRef PubMed.
- J.-L. Ditchburn and R. Hodgkins, Yersinia pestis, a problem of the past and a re-emerging threat, Biosaf. Health, 2019, 1(2), 65–70 CrossRef.
- P. Escoll, et al., Targeting of host organelles by pathogenic bacteria: a sophisticated subversion strategy, Nat. Rev. Microbiol., 2015, 14, 5–5 CrossRef PubMed.
- A. M. Krachler and K. Orth, Targeting the bacteria–host interface, Virulence, 2013, 4(4), 284–294 CrossRef PubMed.
- C. J. Cambier, S. Falkow and L. Ramakrishnan, Host Evasion and Exploitation Schemes of Mycobacterium tuberculosis, Cell, 2014, 159(7), 1497–1509 CrossRef CAS PubMed.
- E. Kuru, et al., In Situ Probing of Newly Synthesized Peptidoglycan in Live Bacteria with Fluorescent D–Amino Acids, Angew. Chem., Int. Ed., 2012, 51(50), 12519–12523 CrossRef CAS PubMed.
- M. S. Siegrist, et al., d-Amino Acid Chemical Reporters Reveal Peptidoglycan Dynamics of an Intracellular Pathogen, ACS Chem. Biol., 2013, 8(3), 500–505 CrossRef CAS PubMed.
- K. M. Backus, et al., Uptake of unnatural trehalose analogs as a reporter for Mycobacterium tuberculosis, Nat. Chem. Biol., 2011, 7, 228–228 CrossRef CAS PubMed.
- B. M. Swarts, et al., Probing the Mycobacterial Trehalome with Bioorthogonal Chemistry, J. Am. Chem. Soc., 2012, 134(39), 16123–16126 CrossRef CAS PubMed.
- E. M. Sletten and C. R. Bertozzi, Bioorthogonal chemistry: fishing for selectivity in a sea of functionality, Angew. Chem., Int. Ed., 2009, 48(38), 6974 CrossRef CAS PubMed.
- S. S. Nguyen and J. A. Prescher, Developing bioorthogonal probes to span a spectrum of reactivities, Nat. Rev. Chem., 2020, 4(9), 476–489 CrossRef CAS.
- A. Typas, et al., From the regulation of peptidoglycan synthesis to bacterial growth and morphology, Nat. Rev. Microbiol., 2011, 10, 123–123 CrossRef PubMed.
- T. J. Silhavy, D. Kahne and S. Walker, The Bacterial Cell Envelope, Cold Spring Harbor Perspect. Biol., 2010, 2(5), a000414 Search PubMed.
- A. Kovacs-Simon, R. W. Titball and S. L. Michell, Lipoproteins of Bacterial Pathogens, Infect. Immun., 2011, 79(2), 548–561 CrossRef CAS PubMed.
- T. Schneider and H.-G. Sahl, An oldie but a goodie – cell wall biosynthesis as antibiotic target pathway, Int. J. Med. Microbiol., 2010, 300(2), 161–169 CrossRef CAS PubMed.
- E. M. Wise and J. T. Park, Penicillin: its basic site of action as an inhibitor of a peptide cross-linking reaction in cell wall mucopeptide synthesis, Proc. Natl. Acad. Sci. U. S. A., 1965, 54(1), 75–81 CrossRef CAS PubMed.
- J. C. J. Barna and D. H. Williams, The Structure and Mode of Action of Glycopeptide Antibiotics of the Vancomycin Group, Annu. Rev. Microbiol., 1984, 38(1), 339–357 CrossRef CAS PubMed.
- G. Siewert and J. L. Strominger, Bacitracin: An Inhibitor Of The Dephosphorylation Of Lipid Pyrophosphate, An Intermediate In The Biosynthesis Of The Peptidoglycan Of Bacterial Cell Walls, Proc. Natl. Acad. Sci. U. S. A., 1967, 57(3), 767–773 CrossRef CAS PubMed.
- R. Draenert, et al., Novel antibiotics: Are we still in the pre–post-antibiotic era?, Infection, 2015, 43(2), 145–151 CrossRef CAS PubMed.
- J. Viala, et al., Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island, Nat. Immunol., 2004, 5, 1166–1166 CrossRef CAS PubMed.
- K. Tiyanont, et al., Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(29), 11033–11038 CrossRef CAS PubMed.
- R. A. Daniel and J. Errington, Control of Cell Morphogenesis in Bacteria: Two Distinct Ways to Make a Rod-Shaped Cell, Cell, 2003, 113(6), 767–776 CrossRef CAS PubMed.
- N. K. Olrichs, et al., A Novel in vivo Cell-Wall Labeling Approach Sheds New Light on Peptidoglycan Synthesis in Escherichia coli, ChemBioChem, 2011, 12(7), 1124–1133 CrossRef CAS PubMed.
- H. Lam, et al., D-Amino Acids Govern Stationary Phase Cell Wall Remodeling in Bacteria, Science, 2009, 325(5947), 1552–1555 CrossRef CAS PubMed.
- F. Cava, et al., Distinct pathways for modification of the bacterial cell wall by non–canonical D-amino acids, EMBO J., 2011, 30(16), 3442–3453 CrossRef CAS PubMed.
- T. J. Lupoli, et al., Transpeptidase-Mediated Incorporation of d-Amino Acids into Bacterial Peptidoglycan, J. Am. Chem. Soc., 2011, 133(28), 10748–10751 CrossRef CAS PubMed.
- T. M. Bartlett, et al., A Periplasmic Polymer Curves Vibrio cholerae and Promotes Pathogenesis, Cell, 2017, 168(1), 172–185 CrossRef CAS PubMed.
- A. W. Bisson-Filho, et al., Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division, Science, 2017, 355(6326), 739–743 CrossRef CAS PubMed.
- M. J. Boersma, et al., Minimal Peptidoglycan (PG) Turnover in Wild-Type and PG Hydrolase and Cell Division Mutants of Streptococcus pneumoniae D39 Growing Planktonically and in Host-Relevant Biofilms, J. Bacteriol., 2015, 197(21), 3472–3485 CrossRef CAS PubMed.
- M. A. Danae, et al., Pentapeptide–rich peptidoglycan at the Bacillus subtilis cell–division site, Mol. Microbiol., 2017, 104(2), 319–333 CrossRef PubMed.
- C. Emöke, et al., Dynamics of the peptidoglycan biosynthetic machinery in the stalked budding bacterium Hyphomonas neptunium, Mol. Microbiol., 2017, 103(5), 875–895 CrossRef PubMed.
- A. K. Fenton, et al., CozE is a member of the MreCD complex that directs cell elongation in Streptococcus pneumoniae, Nat. Microbiol., 2016, 2, 16237–16237 CrossRef PubMed.
- A. Fleurie, et al., MapZ beacons the division sites and positions FtsZ-rings in Streptococcus pneumoniae, Nature, 2014, 516(7530), 259–262 CrossRef CAS PubMed.
- J. M. Monteiro, et al., Cell shape dynamics during the staphylococcal cell cycle, Nat. Commun., 2015, 6, 8055–8055 CrossRef CAS PubMed.
- X. Yang, et al., GTPase activity–coupled treadmilling of the bacterial tubulin FtsZ organizes septal cell wall synthesis, Science, 2017, 355(6326), 744–747 CrossRef CAS PubMed.
- M. Pilhofer, et al., Discovery of chlamydial peptidoglycan reveals bacteria with murein sacculi but without FtsZ, Nat. Commun., 2013, 4, 2856–2856 CrossRef PubMed.
- M. C. F. van Teeseling, et al., Anammox Planctomycetes have a peptidoglycan cell wall, Nat. Commun., 2015, 6, 6878–6878 CrossRef CAS PubMed.
- G. W. Liechti, et al., A new metabolic cell-wall labelling method reveals peptidoglycan in Chlamydia trachomatis, Nature, 2013, 506, 507–507 CrossRef PubMed.
- E. Kuru, et al., Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ, Nat. Protoc., 2014, 10, 33–33 CrossRef PubMed.
- F. Yoshimura and H. Nikaido, Diffusion of beta-lactam antibiotics through the porin channels of Escherichia coli K-12, Antimicrob. Agents Chemother., 1985, 27(1), 84–92 CrossRef CAS PubMed.
- Y.-P. Hsu, et al., Full color palette of fluorescent d-amino acids for in situ labeling of bacterial cell walls, Chem. Sci., 2017, 8(9), 6313–6321 RSC.
- A. García-Heredia, et al., Peptidoglycan precursor synthesis along the sidewall of pole-growing mycobacteria, eLife, 2018, 7, e37243 CrossRef PubMed.
- M. A. de Pedro, et al., Murein segregation in Escherichia coli, J. Bacteriol., 1997, 179(9), 2823–2834 CrossRef CAS PubMed.
- A. R. Sherratt, et al., Dual Strain-Promoted Alkyne–Nitrone Cycloadditions for Simultaneous Labeling of Bacterial Peptidoglycans, Bioconjugate Chem., 2016, 27(5), 1222–1226 CrossRef CAS PubMed.
- S. E. Pidgeon, et al., L,D-Transpeptidase Specific Probe Reveals Spatial Activity of Peptidoglycan Cross-Linking, ACS Chem. Biol., 2019, 14(10), 2185–2196 CAS.
- P. Shieh, et al., Imaging bacterial peptidoglycan with near-infrared fluorogenic azide probes, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(15), 5456–5461 CrossRef CAS PubMed.
- D. A. MacKenzie, et al., Bioorthogonal labelling of living bacteria using unnatural amino acids containing nitrones and a nitrone derivative of vancomycin, Chem. Commun., 2015, 51(62), 12501–12504 RSC.
- R. S. Stephens, et al., Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis, Science, 1998, 282(5389), 754–759 CrossRef CAS PubMed.
- A. Tamura and G. P. Manire, Effect of penicillin on the multiplication of meningopneumonitis organisms (Chlamydia psittaci), J. Bacteriol., 1968, 96(4), 875–880 CrossRef CAS PubMed.
- A. G. Barbour, et al., Chlamydia trachomatis has penicillin-binding proteins but not detectable muramic acid, J. Bacteriol., 1982, 151(1), 420–428 CrossRef CAS PubMed.
- A. Fox, et al., Muramic acid is not detectable in Chlamydia psittaci or Chlamydia trachomatis by gas chromatography-mass spectrometry, Infect. Immun., 1990, 58(3), 835–837 CrossRef CAS PubMed.
- J. W. Moulder, Why is Chlamydia sensitive to penicillin in the absence of peptidoglycan?, Infect. Agents Dis., 1993, 2(2), 87–99 CAS.
- I. Chopra, et al., Antibiotics, peptidoglycan synthesis and genomics: the chlamydial anomaly revisited, Microbiology, 1998, 144(10), 2673–2678 CrossRef CAS PubMed.
- H. Liang, et al., Metabolic labelling of the carbohydrate core in bacterial peptidoglycan and its applications, Nat. Commun., 2017, 8, 15015–15015 CrossRef CAS.
- Y. Wang, et al., Postsynthetic Modification of Bacterial Peptidoglycan Using Bioorthogonal N-Acetylcysteamine Analogs and Peptidoglycan O-Acetyltransferase B, J. Am. Chem. Soc., 2017, 139(39), 13596–13599 CrossRef CAS PubMed.
- M. D. Lebar, et al., Reconstitution of Peptidoglycan Cross-Linking Leads to Improved Fluorescent Probes of Cell Wall Synthesis, J. Am. Chem. Soc., 2014, 136(31), 10874–10877 CrossRef CAS PubMed.
- T. J. Lupoli, et al., Lipoprotein Activators Stimulate Escherichia coli Penicillin-Binding Proteins by Different Mechanisms, J. Am. Chem. Soc., 2014, 136(1), 52–55 CrossRef CAS PubMed.
- N. Inohara, et al., Host Recognition of Bacterial Muramyl Dipeptide Mediated through nod2: implications for crohn's disease, J. Biol. Chem., 2003, 278(8), 5509–5512 CrossRef CAS PubMed.
- M. Borisova, J. Gisin and C. Mayer, Blocking peptidoglycan recycling in Pseudomonas aeruginosa attenuates intrinsic resistance to fosfomycin, Microb. Drug Resist., 2014, 20(3), 231–237 CrossRef CAS.
- P. J. Moynihan and A. J. Clarke, O-Acetylated peptidoglycan: Controlling the activity of bacterial autolysins and lytic enzymes of innate immune systems, Int. J. Biochem. Cell Biol., 2011, 43(12), 1655–1659 CrossRef CAS PubMed.
- J. K. Blundell and H. R. Perkins, Effects of beta-lactam antibiotics on peptidoglycan synthesis in growing Neisseria gonorrhoeae, including changes in the degree of O-acetylation, J. Bacteriol., 1981, 147(2), 633–641 CrossRef CAS PubMed.
- J.-M. Ghuysen and J. L. Strominger, Structure of the Cell Wall of Staphylococcus aureus, Strain Copenhagen. II. Separation and Structure of Disaccharides, Biochemistry, 1963, 2(5), 1119–1125 CrossRef CAS PubMed.
- A. Antignac, et al., Detailed Structural Analysis of the Peptidoglycan of the Human Pathogen Neisseria meningitidis, J. Biol. Chem., 2003, 278(34), 31521–31528 CrossRef CAS PubMed.
- W. Brumfitt, A. C. Wardlaw and J. T. Park, Development of Lysozyme-Resistance in Micrococcus Lysodiekticus and its Association With an Increased O-Acetyl Content of the Cell Wall, Nature, 1958, 181, 1783–1783 CrossRef CAS PubMed.
- L. J. Alderwick, et al., The Mycobacterial Cell Wall—Peptidoglycan and Arabinogalactan, Cold Spring Harbor Perspect. Med., 2015, 5(8), a021113 CrossRef PubMed.
- P. J. B. Crick and C. Dean, The Cell-Wall Core of Mycobacterium tuberculosis in the Context of Drug Discovery, Curr. Top. Med. Chem., 2007, 7(5), 475–488 CrossRef PubMed.
- P. J. Woodruff, et al., Trehalose Is Required for Growth of Mycobacterium smegmatis, J. Biol. Chem., 2004, 279(28), 28835–28843 CrossRef CAS PubMed.
- H. N. Murphy, et al., The OtsAB Pathway Is Essential for Trehalose Biosynthesis in Mycobacterium tuberculosis, J. Biol. Chem., 2005, 280(15), 14524–14529 CrossRef CAS PubMed.
- J. Indrigo, R. L. Hunter and J. K. Actor, Cord factor trehalose 6,6′-dimycolate (TDM) mediates trafficking events during mycobacterial infection of murine macrophages, Microbiology, 2003, 149(8), 2049–2059 CrossRef CAS.
- S. Datta, et al., Clinical Evaluation of Tuberculosis Viability Microscopy for Assessing Treatment Response, Clin. Infect. Dis., 2015, 60(8), 1186–1195 CrossRef PubMed.
- K. Tahlan, et al., SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2012, 56(4), 1797–1809 CrossRef CAS PubMed.
- J. T. Belisle, et al., Role of the Major Antigen of Mycobacterium tuberculosis in Cell Wall Biogenesis, Science, 1997, 276(5317), 1420–1422 CrossRef CAS PubMed.
- N. Sathyamoorthy and K. Takayama, Purification and characterization of a novel mycolic acid exchange enzyme from Mycobacterium smegmatis, J. Biol. Chem., 1987, 262(28), 13417–13423 CrossRef CAS.
- J. O. Kilburn, K. Takayama and E. L. Armstrong, Synthesis of trehalose dimycolate (cord factor) by a cell-free system of Mycobacteriumsmegmatis, Biochem. Biophys. Res. Commun., 1982, 108(1), 132–139 CrossRef CAS PubMed.
- R. Kalscheuer, et al., Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(50), 21761–21766 CrossRef CAS PubMed.
- F. P. Rodriguez-Rivera, et al., Visualization of mycobacterial membrane dynamics in live cells, J. Am. Chem. Soc., 2017, 139(9), 3488–3495 CrossRef CAS PubMed.
- B. L. Urbanek, et al., Chemoenzymatic Synthesis of Trehalose Analogues: Rapid Access to Chemical Probes for Investigating Mycobacteria, ChemBioChem, 2014, 15(14), 2066–2070 CrossRef CAS PubMed.
- L. Favrot, et al., Mechanism of inhibition of Mycobacterium tuberculosis antigen 85 by ebselen, Nat. Commun., 2013, 4, 2748–2748 CrossRef PubMed.
- H. N. Foley, et al., Bioorthogonal Chemical Reporters for Selective In Situ Probing of Mycomembrane Components in Mycobacteria, Angew. Chem., Int. Ed., 2016, 55(6), 2053–2057 CrossRef CAS PubMed.
- H. W. Kavunja, et al., A chemical reporter strategy for detecting and identifying O-mycoloylated proteins in Corynebacterium, Chem. Commun., 2016, 52(95), 13795–13798 RSC.
- E. Huc, et al., O-Mycoloylated Proteins from Corynebacterium: an unprecedented post-translational modification in bacteria, J. Biol. Chem., 2010, 285(29), 21908–21912 CrossRef CAS PubMed.
- T. J. Fiolek, et al., Engineering the Mycomembrane of Live Mycobacteria with an Expanded Set of Trehalose Monomycolate Analogues, ChemBioChem, 2019, 20(10), 1282–1291 CrossRef CAS PubMed.
- J. Dinnes, et al., A systematic review of rapid diagnostic tests for the detection of tuberculosis infection, Health Technol. Assess., 2007, 11(3), 1–196 CAS.
- S. R. Rundell, et al., Deoxyfluoro-d-trehalose (FDTre) analogues as potential PET probes for imaging mycobacterial infection, Org. Biomol. Chem., 2016, 14(36), 8598–8609 RSC.
- M. Kamariza, et al., Rapid detection of Mycobacterium tuberculosis in sputum with a solvatochromic trehalose probe, Sci. Transl. Med., 2018, 10(430), eaam6310 CrossRef PubMed.
- H. A. Sahile, et al., DMN-Tre Labeling for Detection and High-Content Screening of Compounds against Intracellular Mycobacteria, ACS Omega, 2020, 5(7), 3661–3669 CrossRef CAS PubMed.
- M. Kamariza, et al., Towards Mycobacterium tuberculosis detection at the point-of-care: a brighter solvatochromic probe permits the detection of mycobacteria within minutes, bioRxiv, 2020 DOI:10.1101/2020.05.29.124008.
- H. L. Hodges, et al., Imaging mycobacterial growth and division with a fluorogenic probe, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(20), 5271–5276 CrossRef CAS PubMed.
- N. J. Holmes, et al., A FRET-Based Fluorogenic Trehalose Dimycolate Analogue for Probing Mycomembrane-Remodeling Enzymes of Mycobacteria, ACS Omega, 2019, 4(2), 4348–4359 CrossRef CAS PubMed.
- K. L. Kiick, et al., Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(1), 19–24 CrossRef CAS PubMed.
- D. C. Dieterich, et al., Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT), Proc. Natl. Acad. Sci. U. S. A., 2006, 103(25), 9482–9487 CrossRef CAS PubMed.
- D. C. Dieterich, et al., Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging, Nat. Protoc., 2007, 2, 532–532 CrossRef PubMed.
- J. C. M. van Hest, K. L. Kiick and D. A. Tirrell, Efficient Incorporation of Unsaturated Methionine Analogues into Proteins in Vivo, J. Am. Chem. Soc., 2000, 122(7), 1282–1288 CrossRef CAS.
- J. C. M. van Hest and D. A. Tirrell, Efficient introduction of alkene functionality into proteins in vivo, FEBS Lett., 1998, 428(1), 68–70 CrossRef CAS PubMed.
- A. M. Saleh, et al., Non-canonical amino acid labeling in proteomics and biotechnology, J. Biol. Eng., 2019, 13(1), 43 CrossRef PubMed.
- W. Song, et al., A Metabolic Alkene Reporter for Spatiotemporally Controlled Imaging of Newly Synthesized Proteins in Mammalian Cells, ACS Chem. Biol., 2010, 5(9), 875–885 CrossRef CAS PubMed.
- K. L. Kiick, et al., Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(1), 19–24 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins, Chem. Rev., 2014, 114(9), 4764–4806 CrossRef CAS.
- D. M. van Elsland, et al., Ultrastructural Imaging of Salmonella-Host Interactions Using Super-resolution Correlative Light-Electron Microscopy of Bioorthogonal Pathogens, ChemBioChem, 2018, 19, 1766–1770 CrossRef CAS PubMed.
- D. M. van Elsland, et al., Detection of bioorthogonal groups by correlative light and electron microscopy allows imaging of degraded bacteria in phagocytes, Chem. Sci., 2016, 7(1), 752–758 RSC.
- T. Bakkum, et al., Bioorthogonal Correlative Light-Electron Microscopy of Mycobacterium tuberculosis in Macrophages Reveals the Effect of Antituberculosis Drugs on Subcellular Bacterial Distribution, ACS Cent. Sci., 2020, 6(11), 1997–2007 CrossRef CAS PubMed.
- J. T. Ngo, et al., Cell-Selective Metabolic Labeling of Proteins, Nat. Chem. Biol., 2009, 5(10), 715–717 CrossRef CAS PubMed.
- A. J. Link, et al., Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids, Proc. Natl. Acad. Sci. U. S. A., 2006, 103(27), 10180–10185 CrossRef CAS PubMed.
- A. Mahdavi, et al., Identification of secreted bacterial proteins by noncanonical amino acid tagging, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(1), 433–438 CrossRef CAS PubMed.
- M. Grammel, et al., Orthogonal Alkynyl Amino Acid Reporter for Selective Labeling of Bacterial Proteomes during Infection, Angew. Chem., Int. Ed., 2010, 49(34), 5970–5974 CrossRef CAS PubMed.
- F. Wang, et al., Genetic Incorporation of Unnatural Amino Acids into Proteins in Mycobacterium tuberculosis, PLoS One, 2010, 5(2), e9354 CrossRef PubMed.
- M. H. Touchette, et al., A Screen for Protein–Protein Interactions in Live Mycobacteria Reveals a Functional Link between the Virulence-Associated Lipid Transporter LprG and the Mycolyltransferase Antigen 85A, ACS Infect. Dis., 2017, 3(5), 336–348 CrossRef CAS PubMed.
- Q. Gan, et al., Expanding the genetic code of Salmonella with non-canonical amino acids, Sci. Rep., 2016, 6(1), 39920 CrossRef CAS PubMed.
- H. Takahashi, et al., Genetic incorporation of non-canonical amino acid photocrosslinkers in Neisseria meningitidis: New method provides insights into the physiological function of the function-unknown NMB1345 protein, PLoS One, 2020, 15(8), e0237883 CrossRef CAS PubMed.
- M. H. Wright, Chemical Proteomics of Host–Microbe Interactions, Proteomics, 2018, 18(18), 1700333 CrossRef PubMed.
- F. van Dalen, T. Bakkum, S. I. van Kasteren, M. Verdoes, et al., in press.
- D. M. Van Elsland, et al., Correlative light and electron microscopy reveals discrepancy between gold and fluorescence labelling, J. Microsc., 2017, 267(3), 309–317 CrossRef CAS PubMed.
- C. Ortega, et al., Systematic Survey of Serine Hydrolase Activity in Mycobacterium tuberculosis Defines Changes Associated with Persistence, Cell Chem. Biol., 2016, 23(2), 290–298 CrossRef CAS PubMed.
- K. R. Tallman, S. R. Levine and K. E. Beatty, Small-Molecule Probes Reveal Esterases with Persistent Activity in Dormant and Reactivating Mycobacterium tuberculosis, ACS Infect. Dis., 2016, 2(12), 936–944 CrossRef CAS PubMed.
- S. K. Hatzios, et al., Chemoproteomic profiling of host and pathogen enzymes active in cholera, Nat. Chem. Biol., 2016, 12(4), 268–274 CrossRef CAS PubMed.
- G. Rollin, et al., Intracellular Survival of Staphylococcus aureus in Endothelial Cells: A Matter of Growth or Persistence, Front. Microbiol., 2017, 8, 1354 CrossRef PubMed.
- C. S. Lentz, et al., Identification of a S. aureus virulence factor by activity-based protein profiling (ABPP), Nat. Chem. Biol., 2018, 14(6), 609–617 CrossRef CAS PubMed.
- M. Fellner, et al., Structural basis for active-site probes targeting Staphylococcus aureus serine hydrolase virulence factors, bioRxiv, 2020 DOI:10.1101/2020.04.21.054437.
- C. S. Lentz, et al., Design of Selective Substrates and Activity-Based Probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis, ACS Infect. Dis., 2016, 2(11), 807–815 CrossRef CAS PubMed.
- B. M. Babin, et al., A chemiluminescent protease probe for rapid, sensitive, and inexpensive detection of live Mycobacterium tuberculosis, bioRxiv, 2020 DOI:10.1101/2020.09.14.296772.
- A. P. Marshall, J. D. Shirley and E. E. Carlson, Enzyme-targeted fluorescent small-molecule probes for bacterial imaging, Curr. Opin. Chem. Biol., 2020, 57, 155–165 CrossRef CAS PubMed.
- K. Subramanian, B. Henriques-Normark and S. Normark, Emerging concepts in the pathogenesis of the Streptococcus pneumoniae: From nasopharyngeal colonizer to intracellular pathogen, Cell. Microbiol., 2019, 21(11), e13077 CrossRef CAS PubMed.
- S. Sharifzadeh, et al., Novel Electrophilic Scaffold for Imaging of Essential Penicillin-Binding Proteins in Streptococcus pneumoniae, ACS Chem. Biol., 2017, 12(11), 2849–2857 CrossRef CAS PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels-Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46(17), 4895–4950 RSC.
Footnote |
| † Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |