
Ionic liquids: “normal” solvents or nanostructured fluids?
Salvatore
Marullo
 ,
Francesca
D'Anna
*,
Carla
Rizzo
and
Floriana
Billeci
,
Francesca
D'Anna
*,
Carla
Rizzo
and
Floriana
Billeci
Università degli Studi di Palermo, Dipartimento STEBICEF, Viale delle Scienze, Ed. 17, 90128 Palermo, Italy. E-mail: francesca.danna@unipa.it
First published on 4th February 2021
Abstract
Ionic liquids (ILs) are a class of non-conventional solvents, which, for almost two decades, have continued to generate burgeoning interest in different fields of present-day chemical research with few similar precedents. Among the various aspects related to ILs, a topic worthy of in-depth analysis is their influence on organic reactivity and reaction rates. In light of this, the present short review aims to provide an overview of the literature from 2010 to the present day that addresses this issue. In particular, we herein present two main different viewpoints by which the solvent effect of ILs is explained: the first is mainly based on considering the bulk polarity of ILs and linear solvation energy relationships, while the other treats ILs as nanostructured fluids. In both cases, studies dealing with IL mixtures are also covered. Finally, literature addressing the area of supramolecular catalysis “by” or “in” ILs is also reported. This is one of the few reviews covering these specific aspects, aiming to provide a useful framework to guide future research into the effects of ILs on organic reactivity.
1. Introduction
Ionic liquids (ILs) are organic salts with a melting point conventionally lower than 100 °C and for almost two decades have been the object of intense interest in chemical research.1 This interest is still high due to the wide range of fields in which they find application.The first examples of ILs can be traced back to Walden, who sought to obtain a molten salt, liquid at room temperature, and to the research of J. Atwood with his “red oil”.2,3 In a few decades, this topic witnessed a surge in interest, creating a real “trend”.2 Few classes of chemical compounds can boast as rapid an evolution as ILs. These organic salts have found application in different fields of chemistry4 as green solvents,5 electrolytes,6,7 lubricants,8,9 liquid crystals,5 heat storage fluids,10,11 separation and extractive phases,12,13 soft materials,14–16 catalysts17–19 and biocatalysis.20 Furthermore, interest in ILs has progressively also involved industry, and industrial processes utilizing ILs as solvents are now a reality.21
Initially, ILs emerged as eco-compatible alternatives to organic solvents, due to convenient properties like negligible vapour pressure and flammability, high thermal stability, good conductivity, wide liquid ranges and electrochemical windows, high tunability of structure and ability to dissolve organic and inorganic compounds. Moreover, many ILs have low miscibility in organic solvents, enabling their potential recovery and recycling. Some of the most frequently used ions constituting ILs are reported in Scheme 1.
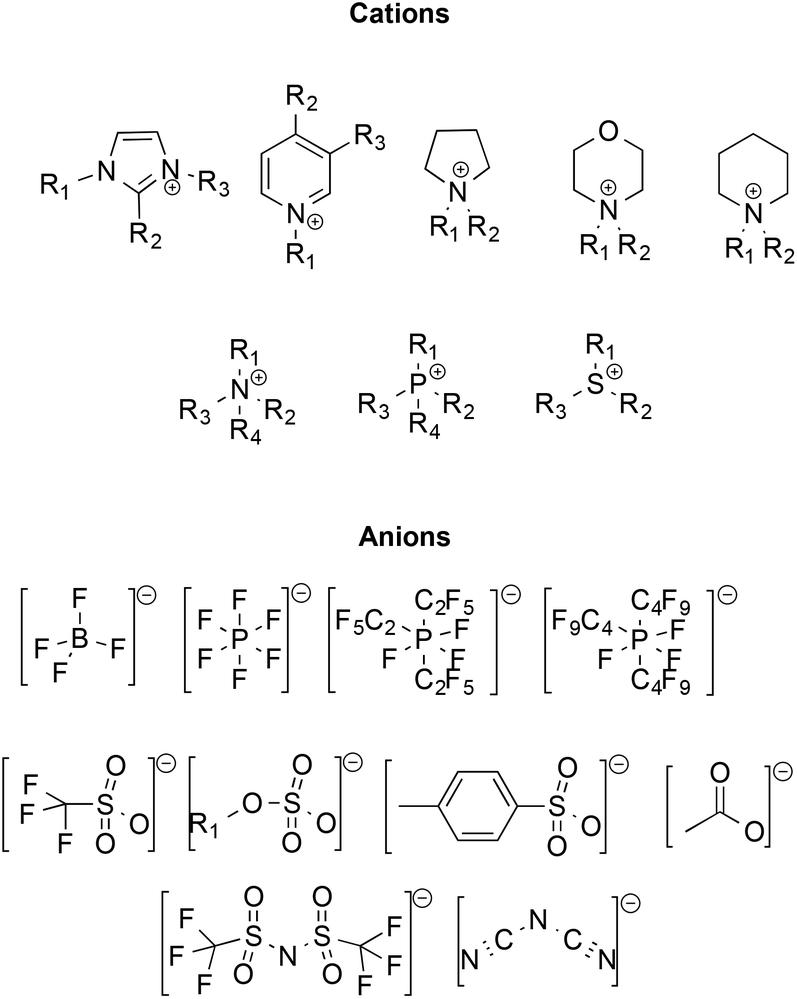 | ||
| Scheme 1 Structures of common cations and anions constituting ILs; cations (from left to right): imidazolium, pyridinium, pyrrolidinium, morpholinium, piperidinium, ammonium, phosphonium, and sulfonium; anions (from left to right): tetrafluoroborate, hexafluorophosphate, tris(perfluoroalkyl)triphosphates (FAP), trifluoromethanesulfonate, alkylsulfate, p-toluensulfonate, acetate, bis(trifluoromethanesulfonyl)imide, and dicyanamide. | ||
Despite these premises, recently the eco-sustainability of ILs has been subject to close scrutiny, as in a number of cases their synthesis fails to meet key sustainability criteria,22,23 and this has led to obtaining a new series of ILs derived from natural sources, paying attention also to the sustainability of the synthetic process.24,25
As previously mentioned, the distinctive characteristic of ILs is that they are composed entirely of ions, which provides a completely different solvent environment from conventional solvents. An important point is the relatively straightforward possibility to vary their properties to a great extent by simply changing the constituting ions. The tunability of the structure allows ILs to “fit” a particular application.
The ionic nature of ILs has fundamental consequences on their microscopic properties, arising from the occurrence of a range of interactions that are completely different to those present in molecular solvents. In particular, oppositely charged ions interact by means of strong coulombic interactions. However, since both cations and anions in ILs are generally characterized by a lower charge density and a higher delocalized character compared with ordinary salts, crystallization is hampered and hence the melting point is much lower than the latter. This allows their existence as liquids at room temperature. The stronger coulombic interactions are associated with other weaker but cooperative interactions that contribute to the liquid structure of ILs. More specifically, a thick network of hydrogen bonds is established between the anions and cations, coupled with van der Waals interactions involving alkyl chains borne on the cations or anions. A further pivotal set of interactions is represented by π–π interactions operating between aromatic ions. The sum of these intermolecular interactions results in significant ionic aggregation, which in turn endows ILs with a resilient and persistent nanostructure, retained to some extent even in the gas phase.26 ILs thus display a dynamic structural heterogeneity, with the presence of nano-segregated polar and non-polar domains.27,28 The polar domains are mainly composed of the cation core and the anion, while the non-polar ones consist mainly of the alkyl chains.29 Although some conventional solvents, like long-chain alcohols, can display nanostructure,30 this microphase separation between polar and non-polar domains is a prominent feature of ILs, affecting their ability to interact with solutes.31
Furthermore, especially for ILs constituted of aromatic ions, the concomitant occurrence of hydrogen bonds and π–π interactions results in a marked structural organization so that these ILs can be considered supramolecular fluids, i.e. liquids with a structural organization underpinned by non-covalent interactions.32,33
All these factors, in turn, affect the interactions of ILs with solutes, which fall under the broad concept of polarity i.e., the ability of the solvent to solvate dissolved charged or dipolar species.34 As will be discussed later, the nanostructure also exerts effects on reactivity. Indeed, any solute solubilised in the IL will interact differently depending on which domain is considered. Additional distinctions appear if solutes are charged or neutral.27
The topic of ILs has been the subject of recent reviews, to which the reader is directed for a more general overview.2,27,35–37
The aim of the present review is instead to critically examine how the solvent behaviour of ILs on organic reactions has been rationalized. In particular, we focused on studies published after 2010, in which the features of ILs affecting reactivity have been specifically examined. The reactivity in mixtures of ILs will also be covered. In contrast, we will not address issues concerning the sustainability or greenness of ILs. In addition, it is well known that IL reactivity can be drastically influenced by the presence of impurities such as halide residues or water content as, for example, halides and water can coordinate the transition metals used as catalysts, negatively affecting the reaction rates. Even if the presence of IL impurities could be crucial in the explanation of some unusual reactivity results, it is often a neglected issue, especially with regards to trace level contamination.38,39 For this reason, the discussion on reactivity results obtained in ILs will assume that there are no IL impurities in the articles reported.
In section 2, the different viewpoints used to this aim will be presented. In the subsequent section, we will review literature covering specific organic reactions and examples of supramolecular catalysis in/by ILs.
2. Different viewpoints on the effects of ionic liquids on reactivity
Organic reactivity in ILs has been rationalized by different viewpoints such as the polarity and entity of cation–anion interactions or the supramolecular nanostructure of these solvents. In the first viewpoint, ILs are considered like conventional solvents, so that reaction outcomes and rates are rationalized in terms of bulk polarity or solvation interactions occurring between solvent and reactants.402.1 Treatment as conventional solvents
An established way to rationalize solvent effects on reactivity takes into account linear solvation energy relationships (LSERs), in which a given property, such as the kinetic catalytic constant, is expressed quantitatively as a combination of suitable descriptors, accounting for different properties of the solvent. The most widely used is the LSER based on the Kamlet–Taft parameters, typically in the form reported in eqn (1).| (XYZ) = (XYZ)0 + aα + bβ + pπ* | (1) |
Welton et al. described the effect of ILs on the rate of the nucleophilic aliphatic substitution reactions of methyl-p-nitrobenzesulfonate (Scheme 2A).41 The reaction was studied in ILs and, for comparison, in some conventional solvents. They explained the reactivity observed on the grounds of LSER based on the Kamlet–Taft parameters. Notably, the same LSERs were significant for ILs and conventional solvents alike, suggesting that no special “ionic liquid effect” occurs. This revealed that reactivity is mainly correlated to the parameter α, which expresses the ability of the solvent to donate hydrogen bonds to the nucleophiles. The effect of the anion is secondary and acts by modulating the entity of the hydrogen bond between the IL cation and substrate: a stronger hydrogen bond accepting an anion interacts more strongly with the cation, thus reducing its availability to interact with the substrate.
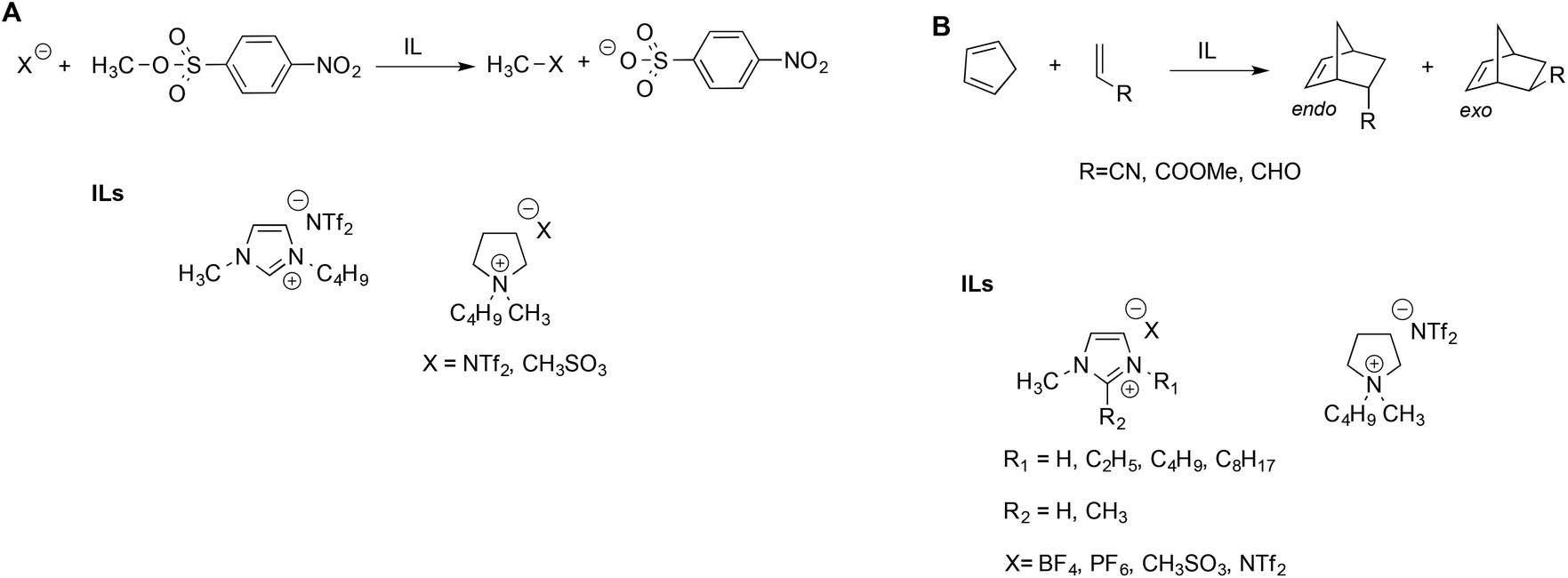 | ||
| Scheme 2 (A) Nucleophilic substitution and (B) the Diels–Alder reaction in ILs. | ||
Interestingly, the aforementioned favorable effect of hydrogen bonding by the cation was also evidenced by hybrid quantum mechanics/molecular mechanics (QM/MM) simulations.42 Similar considerations were used to explain the reactivity of Diels–Alder reactions of cyclopentadiene with different dienophiles in ILs (Scheme 2B).43 In general, the reaction occurred faster in ILs compared to conventional solvents, but the solvent effect on endo–exo selectivity and rate depended on both the IL and the dienophile. The endo–exo selectivity was rationalized in terms of Kamlet–Taft LSER with a prominent role of the α parameter, arising from the beneficial effect of the hydrogen bond between the IL cation and the carbonyl groups on acrolein and methyl acrylate. The effect of the IL anion is more important in the case of acrylonitrile, which has a similar hydrogen bond accepting ability compared to other dienophiles but is a poor hydrogen bond acceptor. The selectivity of the Diels Alder reaction has been studied in a broad range of ILs and has been reviewed elsewhere.44 While the LSER approach proved suitable to explain the trend of selectivity, Chiappe et al. remarked that LSERs account less efficiently for reactivity, which, in turn, appear to reflect a solvophobic effect and specific interactions between the reactants and IL components considered.44
The aforementioned Kamlet–Taft based LSERs were also shown to account for the reactivity of sulfonium salts with amines45 or ligand exchange reactions at a Pt(II) center.46 In the former case, (Scheme 3A), a dominant effect was exerted by the ability of the solvent to accept hydrogen bonds. Stronger hydrogen bond acceptor solvents cause a reduction in rate, due to the higher stabilization of the sulfonium substrate, which possesses more acidic protons compared to the transition state. A more modest influence of the hydrogen bond donating ability of the solvents was found, which, in turn, involves the amine and moderates its nucleophilicity.
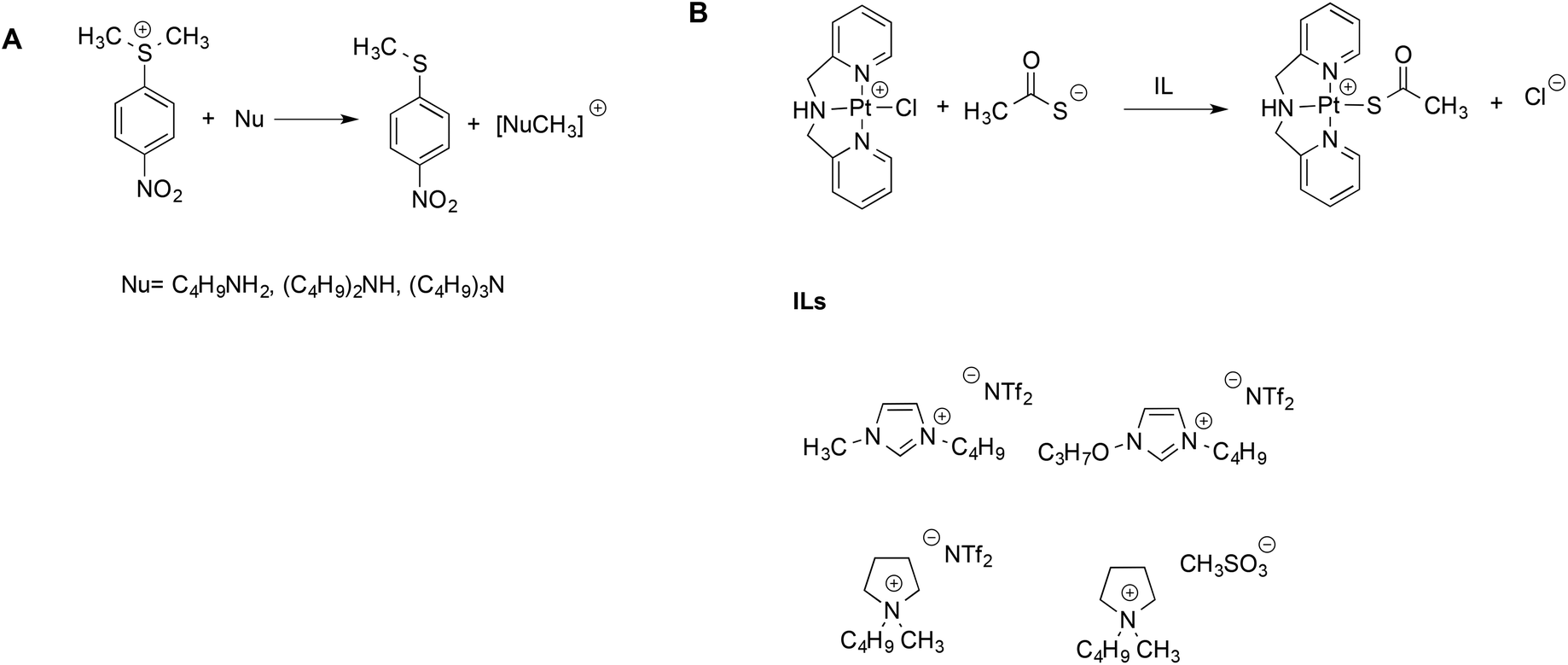 | ||
| Scheme 3 (A) The reaction between amines and sulfonium electrophiles and (B) the ligand exchange reaction at the Pt(II) center. | ||
On the other hand, for the latter reaction (Scheme 3B), the dominant and favorable contribution is brought about by the dipolarity/polarizability of the solvent, which is expected for a soft metal center like Pt(II). The mechanism was the same in ILs and conventional solvents alike, and rates measured in ILs were intermediate between the ones in methanol and water. Notably, once again, for both the reactions alike, the same LSERs were valid for ILs and conventional solvents, indicating the absence of any special IL-effect.
On a similar note, studying the proline-catalyzed asymmetric Michael reaction between β-nitrostyrene and n-pentanal in 10 ILs, Rahman et al. found that the hydrogen bond accepting ability of the IL anion is key in determining the reaction outcome.47 In particular, the reaction afforded higher yields in shorter times in ILs compared with conventional organic solvents like DMSO and short-chain alcohols, with the best results in terms of yield and stereoselectivity found in the presence of imidazolium-based ILs bearing weakly coordinating anions. This was explained considering that more basic and strongly coordinating anions, like carboxylates, interact more strongly with the catalyst, reducing its availability to the substrate.
Furthermore, Priede et al. applied the LSER approach to a Knoevenagel reaction between 4-(dimethylamino)-benzaldehyde and ethylcyanoacetate which, in contrast to what happens in conventional solvents, occurs without an external catalyst in ILs. In particular, they found a dominant correlation between the rate constants and the IL hydrogen bond basicity, as expressed by the Kamlet–Taft parameter β.48 This was ascribed to the interaction of the IL anion with the active methylene of ethylcyanoacetate, with stronger hydrogen bond accepting anions favouring the formation of the carbanion. The hydrogen bond acidity of the cation had a more marginal role, ascribed to an increased polarization of the carbonyl groups on both reactants, heightening the electrophilic character of the aldehyde and favouring enolization of ethylcyanoacetate, respectively.
The use of similar LSERs has been further extended to describe the interaction of ILs with solutes beyond organic reactivity like, for instance, the behavior of ILs in chromatography stationary phases.49
In other cases, reaction rates are mainly affected by electrostatic interactions due to the ionic nature of ILs. This is the case of the irreversible dimerization of acetophenone radical anions as monitored by cyclic voltammetry measurements.50 Conducting the reaction in dialkylimidazolium and trialkylimidazolium-based ILs sharing the same anion, the authors showed that the dimerization rate is only marginally affected by the apparent polarity or viscosity of the solvent, with a prominent influence of charge stabilization brought about by favourable coulombic interactions with the IL cations. This is possibly aided by hydrogen bonding with the protons at the C2-position in the imidazolium moieties. Furthermore, dimerization rates are higher in ILs than in the conventional ethanol-based electrolyte.
The stabilizing effect provided by the ionic nature of ILs has also been evidenced in radical reactions like the TiO2-promoted oxidative coupling of anilines.51 This reaction normally affords a mixture of diazo-, azoxy-compounds and nitroarenes (Scheme 4).
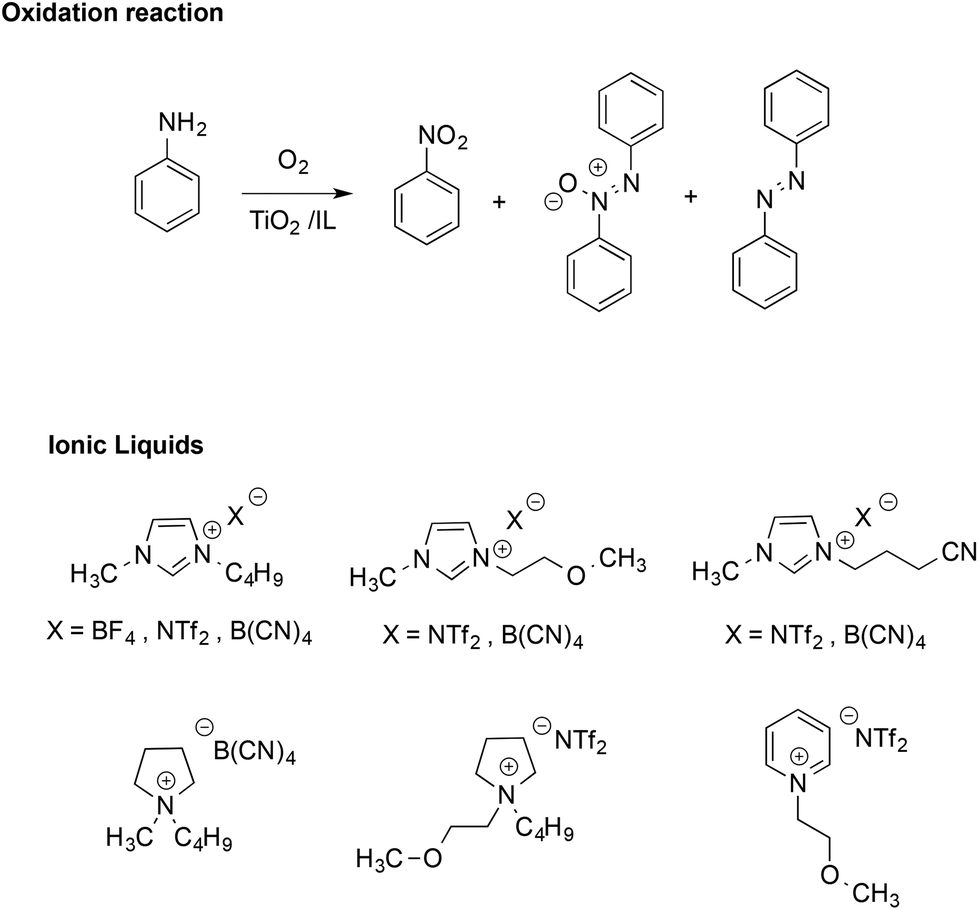 | ||
| Scheme 4 The oxidation of aniline and the ILs considered. | ||
Performing the reactions in functionalized ILs yielded a higher selectivity in the azoxy-compounds over the diazo-ones, whereas in most conventional solvents, the product was almost exclusively azobenzene. In ILs, azoxybenzene is the main product and selectivity is mainly affected by the structure of the IL cation, with the highest values found in methoxy-functionalized imidazolium and pyridinium-based ILs. Conversely, the effect of the IL anion on conversion and selectivity appeared less marked. The higher selectivity in ILs was attributed to the stabilizing effect of their ions, which enhances the cage lifetime for radical ion pairs over those of neutral radicals.
Another approach to the study of the effect of ILs on organic reactions is based on observing how reactivity changes as the composition of the solvent system shifts from conventional solvents to neat ILs, through the whole composition range.52 This allows us to get information complementary to that solely based on comparisons between neat-IL and conventional solvents. However, in our discussion, we will limit our attention to situations in which ILs make up the bulk of the solvent medium. In such studies, mostly focused on aliphatic nucleophilic substitution reactions, generally the trend of the observed kinetic constants as a function of solvent composition differs depending on the electrophile used and can be explained considering the extent of interaction between IL components and transition states. Such a rationale has been used to explain reactivity in bimolecular53 as well as unimolecular54 reactions (Scheme 5A and B).
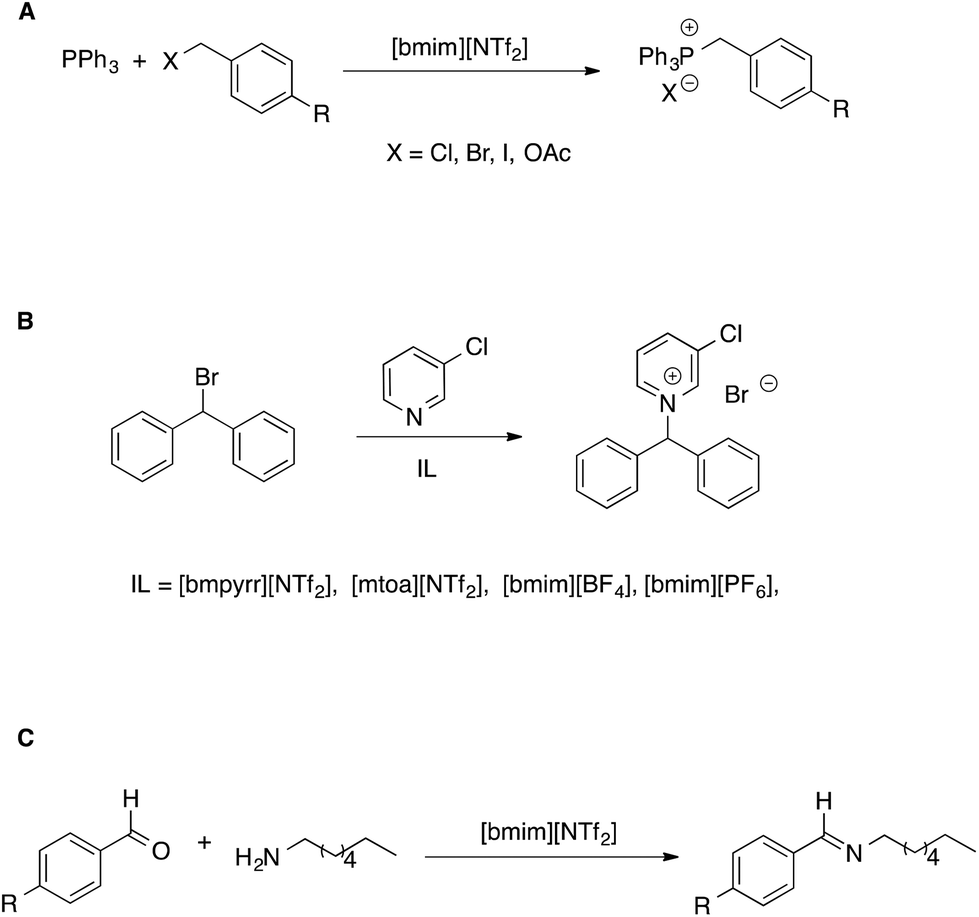 | ||
| Scheme 5 (A) Bimolecular and (B) unimolecular nucleophilic substitution reactions in ionic liquids. (C) A nucleophilic addition reaction. | ||
In the former case (Scheme 5A), the reaction was faster or slower in the IL, depending on what electrophile is considered. From a detailed kinetic analysis, it was found that interactions between the IL components and transition state dominate the changes in reactivity, more so in the presence of the I- and OAc-substituted electrophile which gave rise to a more charge-diffuse transition state. In the second case, the reaction between 3-chloropyridine and bromodiphenylmethane proceeds both by a SN1 and a SN2 pathway. Focusing on the unimolecular pathway, the authors found that the reaction is faster in all pure ILs than in acetonitrile, with the exception of [mtoa][NTf2], due to steric hindrance of the bulky ammonium cation. The effect of the IL anion was however dominant, with smaller and spherical IL anions leading to higher rates. A Kamlet–Taft based LSER also showed the prominent influence of solvent polarizability on the rate of the SN1.
A study of the kinetics of nucleophilic addition of 1-hexaneamine to several substituted benzaldehydes in mixtures of acetonitrile and [bmim][NTf2] shed light on the effect of ILs on a multistep process (Scheme 5C).55 In particular, once again the reaction occurred faster in the IL as compared with the conventional solvent, acetonitrile. Extensive kinetic analysis, including temperature-dependent kinetic investigations, revealed that in ILs the rates of each individual step increase and that ILs show a greater sensitivity than acetonitrile to the degree of charge development in each transition state.
Kinetic investigations on the reaction between 2,4-dinitrobenzenesulfonyl chloride and amines in 10 ILs showed that the reaction occurred faster in ILs with a stronger ability to interact through hydrogen bonding, which impacts on the nucleophilicity of the amine. Among the ILs, the effect exerted by the nature of the IL anion is more significant than that of the IL cation.56 Moreover, comparison with conventional solvents showed that reactivity followed the same pattern in both classes of solvents.
ILs are widely recognized as suitable reaction media for the valorization of lignocellulosic biomass. In this context, Hallett et al. have carried out a detailed kinetic investigation on the β-O-4 ether cleavage of lignin model compounds in protic and aprotic ILs bearing the [HSO4]− anion (Scheme 6A and B).57
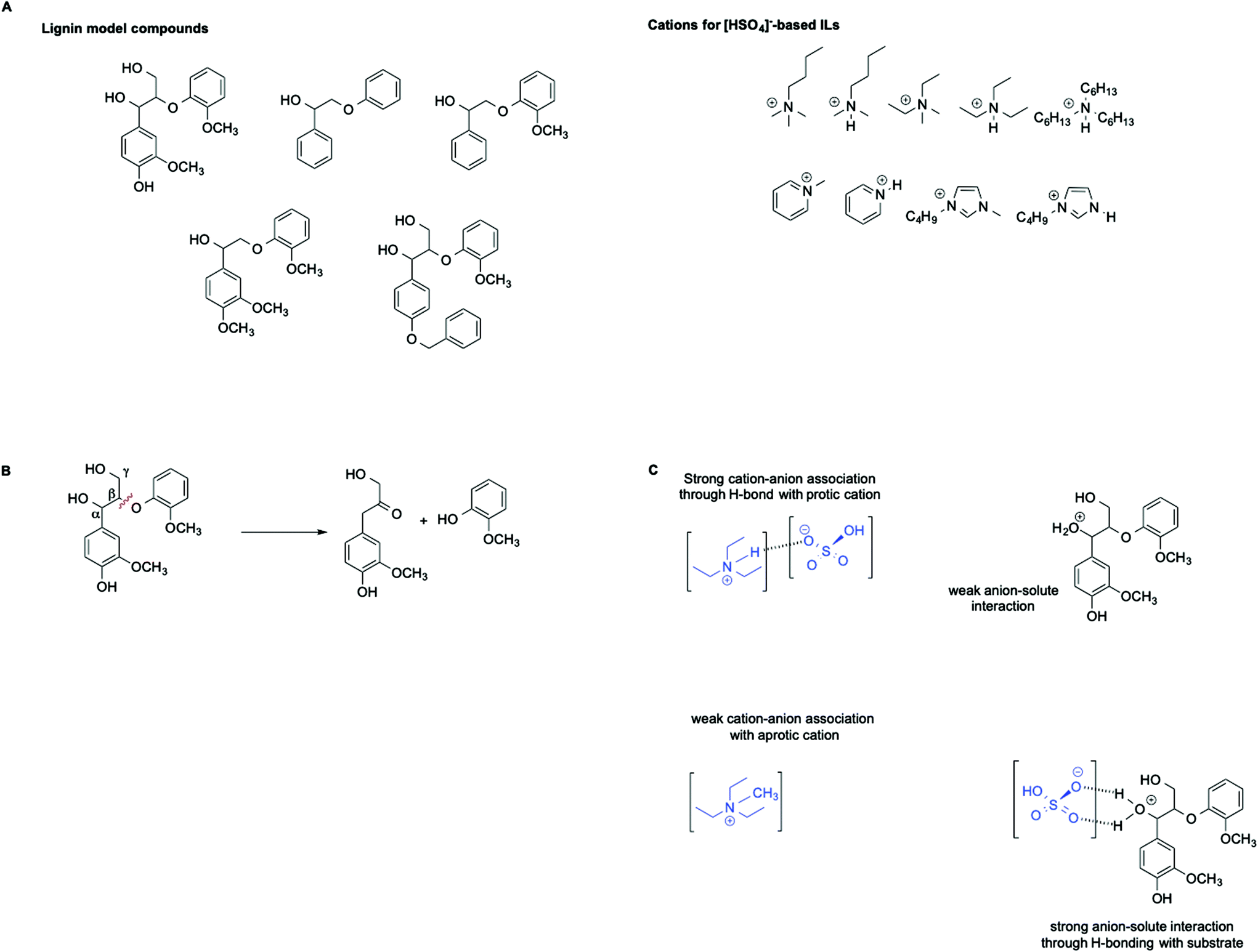 | ||
| Scheme 6 (A) Lignin model compounds and the [HSO4]−-based ILs used, (B) an example of the reaction, and (C) a schematic illustration of the repercussions of cation–anion interactions on the solvation of the protonated substrate. | ||
They found that the rate of the process is influenced not only by the acidity of the medium, as estimated by the Hammett acidity function H0, but also by a distinct effect of hydrogen bonding as well as the magnitude of the cation–anion interaction within each IL. In particular, an increase in rate was observed in ILs featuring a stronger cation anion association. This results in a less solvated and less stabilized protonated substrate compared with the transition state, which ultimately leads to an enhanced rate. This effect is schematically represented in Scheme 6C.
ILs endowed with suitable reactive functionalities, known as task specific ionic liquids (TSILs) can fulfil the dual role of solvent and catalyst. For example, Hardacre reported the use of ILs bearing the non-nucleophilic base functionality (iPr2N), which resembles the structure of Hünig's base, N,N-diisopropylethylamine (Scheme 7).58 These were used both as catalysts and solvent for the Knoevenagel reaction between benzaldehyde and ethyl cyanoethanoate.
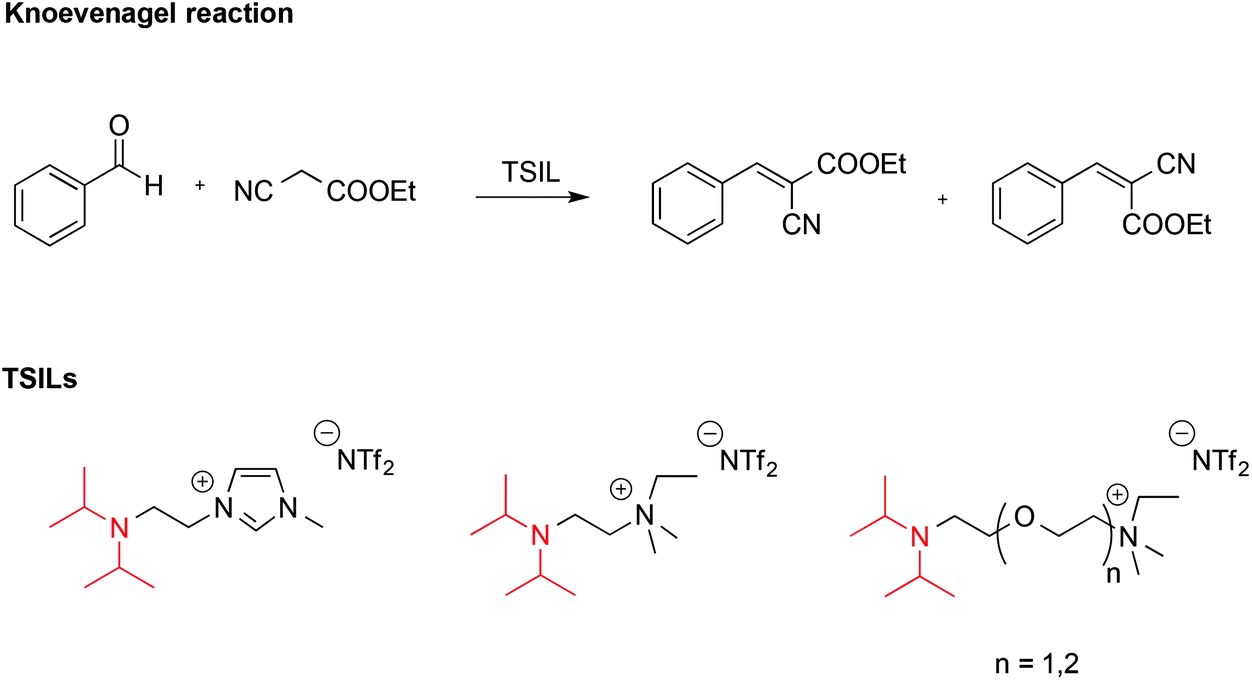 | ||
| Scheme 7 The Knoevenagel reaction and Hünig's base-appended ILs. | ||
It was found that the basicity of these TSILs was higher on increasing the length of the linker between the nitrogen atoms due to reduced electrostatic repulsion between the IL cation head and the protonated tertiary amine group. Basicity also increased on increasing the number of ether linkages due to intramolecular hydrogen bonding involving the protonated amino-group and the oxygen. The catalytic efficiency of these ILs was in agreement with their basicity, and in the best cases conversions of benzaldehyde were comparable with the one obtained using free Hünig's base in [bmim][NTf2] but with easier product separation.
2.2 Treatment as nanostructured fluids
A different point of view explains reactivity in ILs on the grounds of their peculiar nanostructure and microheterogeneity.5,36,59 Early suggestions on the occurrence of local organized structures in ILs came from the observation that the Raman spectrum of liquid [bmim]Cl indicated that the imidazolium cation had an arrangement similar to that of the single crystal.60 Further evidence for ionic aggregation of ILs was provided by the 1H NMR spectra of [emim][NTf2], which showed two distinct sets of resonance, hinting at the occurrence of ion-pair aggregates with remarkably long lifetimes.61 Aggregates formed by imidazolium-based ILs were also directly observed by ESI-MS spectrometry, further supported by concentration-dependent conductivity measurements.62 The latter, in particular, allowed determination of a critical aggregation concentration (CAC) for water-miscible and water-immiscible ILs.Delving into the microscopic structure and dynamics of ILs, Samanta et al. probed the microheterogeneity of ILs by fluorescence correlation spectroscopy and lifetime measurements.63 In particular, they studied the diffusion behavior of suitable fluorescent probes in different solvents. Unlike what happens in conventional solvents, in imidazolium-based ILs the probes reveal bimodal diffusion, which was suggestive of the occurrence of two different microenvironments within the ILs. Combining this evidence with lifetime measurements for ILs with increasing alkyl chain lengths, they were able to identify these microenvironments as polar and non-polar regions.
Raman spectroscopy also provided a powerful tool to investigate and prove the occurrence of local structure in ILs.64 For example, coherent anti-Stokes Raman scattering (CARS) measurements showed that ILs such as [bmim][PF6], [hmim][PF6] and [omim][PF6] are optically inhomogeneous at the microscopic level and more extensive local structures emerge with longer alkyl chains as a consequence of stronger interactions among alkyl chains.
However, the microheterogeneous nature of ILs is not limited only to ILs with aromatic cations. In this regard, 2D-NMR spectroscopy on the IL [bmpyrr][NTf2] revealed intermolecular interactions involving the fluorine atoms of the anion with the protons of the cation. The magnitude of the NOE effect showed that the approach of the anion is more oriented towards the protons on the nitrogen linked carbons, leading to a mesoscopic structure.
Finally, in a recent report, the microheterogeneity of chloride-based imidazolium ILs was probed by observing how the water-phenol phase diagram changes in their presence.65 In particular, it was found that above a critical aggregation concentration these ILs behaved like a well-known surfactant, TX-100. This suggested that the aggregates of these ILs could be similar, with an elliptical shape and length of ∼300 Å. For a more specific view of the nanostructure of ILs, the reader is directed to other reviews.36,66,67
All these examples are only a part of the extensive body of work showing that ILs, particularly ones composed of aromatic ions, feature polar and non-polar domains28 and can be considered as supramolecular fluids, i.e. liquids with a structural organization underpinned by non-covalent interactions such as hydrogen bond networks between anions and cations, π-stacking interactions between aromatic ions and van der Waals interactions involving alkyl chains (Fig. 1).68
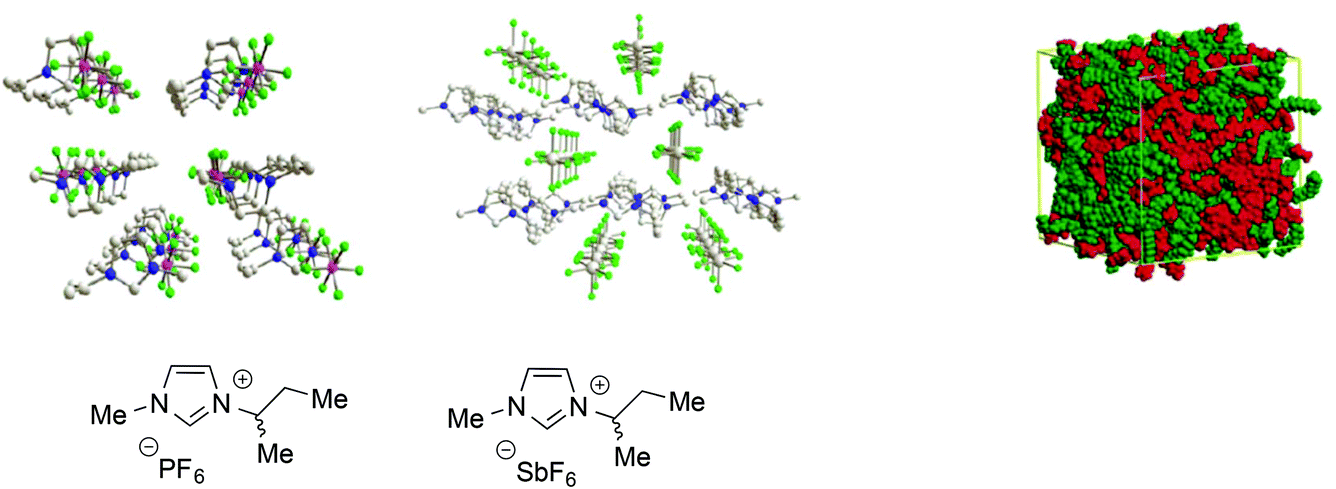 | ||
| Fig. 1 Left: Examples of the 3D-ionic network in aromatic ILs (adapted with permission from ref. 68. Copyright (2011) American Chemical Society). Right: A representation of polar and non-polar domains (adapted with permission from ref. 28. Copyright (2006) American Chemical Society). | ||
In this light, the structural organization of ILs is expected to affect the reaction rates and outcomes by inducing significant changes in the activation parameters. Consequently, bulk polarity-based parameters, in these cases, fail to account for the reactivity trends.
A direct consequence of this organized structure has been recognized, for instance, in the synthesis of inorganic materials with precisely controlled size and geometry,35 with ILs acting as “entropic drivers” for the obtainment of well-defined, ordered nanostructures.69,70 This also has important repercussions on organic synthesis and catalysis since the use of organized liquids as solvent media may induce enhanced reactivity due to these supramolecular effects.71
The effect of nanodomains in ILs on reactivity was studied by Maschmeyer et al., considering as a model reaction the bimolecular alcoholysis of N-(p-fluorophenyldiphenylmethyl)-4-picolinium chloride [Ar3Pic][Cl] in a solution of imidazolium ILs with increasing alkyl chain length (Scheme 8A).72
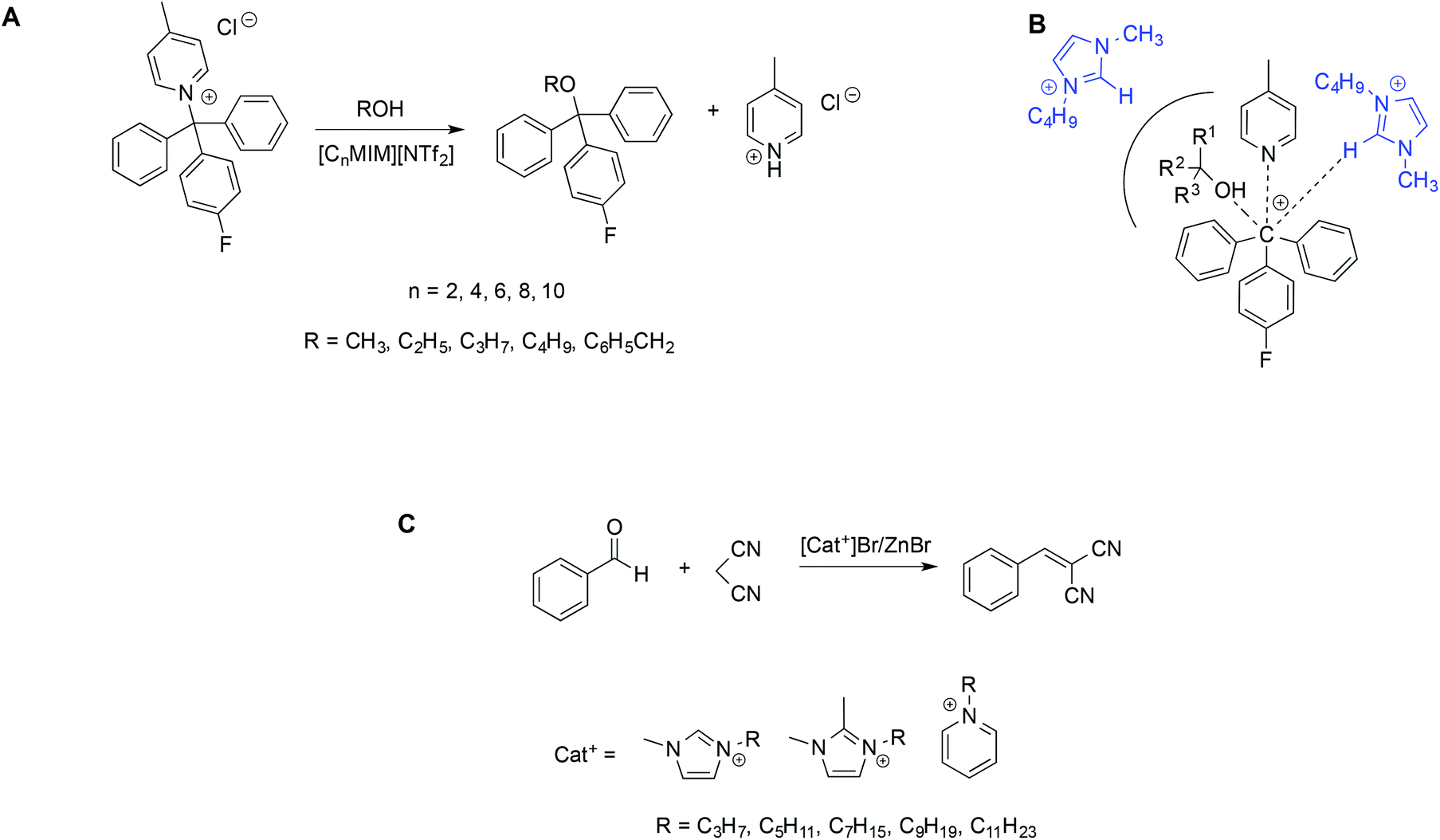 | ||
| Scheme 8 (A) Bimolecular alcoholysis in ILs and (B) a schematic representation of potential IL cation–nucleophile interactions in the transition state. (C) The Knoevenagel reaction in bromozincate ILs. | ||
The authors found a significant increase in the second-order rate constant upon increasing the IL alkyl chain length. This is due to a pseudo-encapsulation effect: the longer the alkyl chain, the more extended the non-polar domains within the IL. Given the ionic nature of the substrate, both the initial and transition states are expected to reside in the polar domains which, in turn, occupy a smaller volume on increasing the alkyl chain length, thus leading to a local concentration effect. A kinetic model based on this interpretation accurately represented the rates observed. Expanding the range of nucleophiles for the same reaction, they also observed significant steric effects on the reactivity, as well as disparities between the predicted and actual reaction rates, ascribed to the IL structural heterogeneity.73 A schematic depiction of the steric effects influencing cation-nucleophile interactions in the transition state is reported in Scheme 8B. To lend further support to the observation reported above, rates in water–IL mixtures instead follow a predictable trend, likely due to the partial disruption of the IL nanostructure. Accordingly, rates in IL–water mixtures, where the effects brought about by the IL nanostructure disappear, appear well-explained on the grounds of the bulk polarity of the solvent mixture.74
A similar effect of the nanodomain structure of ILs on reactivity has been observed in the Knoevenagel reaction of benzaldehyde with malononitrile in bromozincate-based aromatic ILs. In this case, ILs act both as the Lewis acid-catalyst and solvents (Scheme 8C).75 In particular, the ILs used differed in the cation and the alkyl chain length in order to modulate the relative size of the polar and non-polar domains. Kinetic experiments, carried out maintaining in all cases the same volume fraction of IL (99%), showed that conversions were affected by different factors including the Lewis acidity of the ILs and the domain nanostructure. In particular, higher conversions were found in ILs for which a smaller size of non-polar domains was calculated.
It is apparent how, in all cases, a suitable reaction has been used to probe the solvent behavior of ILs. Such reactions are ideally characterized by a fully established mechanism in conventional solvents, and should proceed through simple pathways. Following this “probe reaction” approach, D'Anna et al. obtained information on the influence of the IL structure on reactivity by studying a mononuclear rearrangement of heterocycles (MRH). In particular, the thermally induced MRH of the Z-phenylhydrazone of the 3-benzoyl-5-phenyl-1,2,4-oxadiazole into the relevant triazole was studied in neat ILs differing in terms of π-surface extension, alkyl chain length and charge (Scheme 9A).76 The reaction was carried out in the absence of any catalyst, and the trend of the obtained yield was compared with the polarity of the solvent media, estimated by Kamlet–Taft parameters and ET(30) and the extent of the structural organization of the ILs, as investigated by resonance light scattering (RLS) measurements, variable temperature NMR and UV-vis spectra of the solvatochromic probe Nile red.
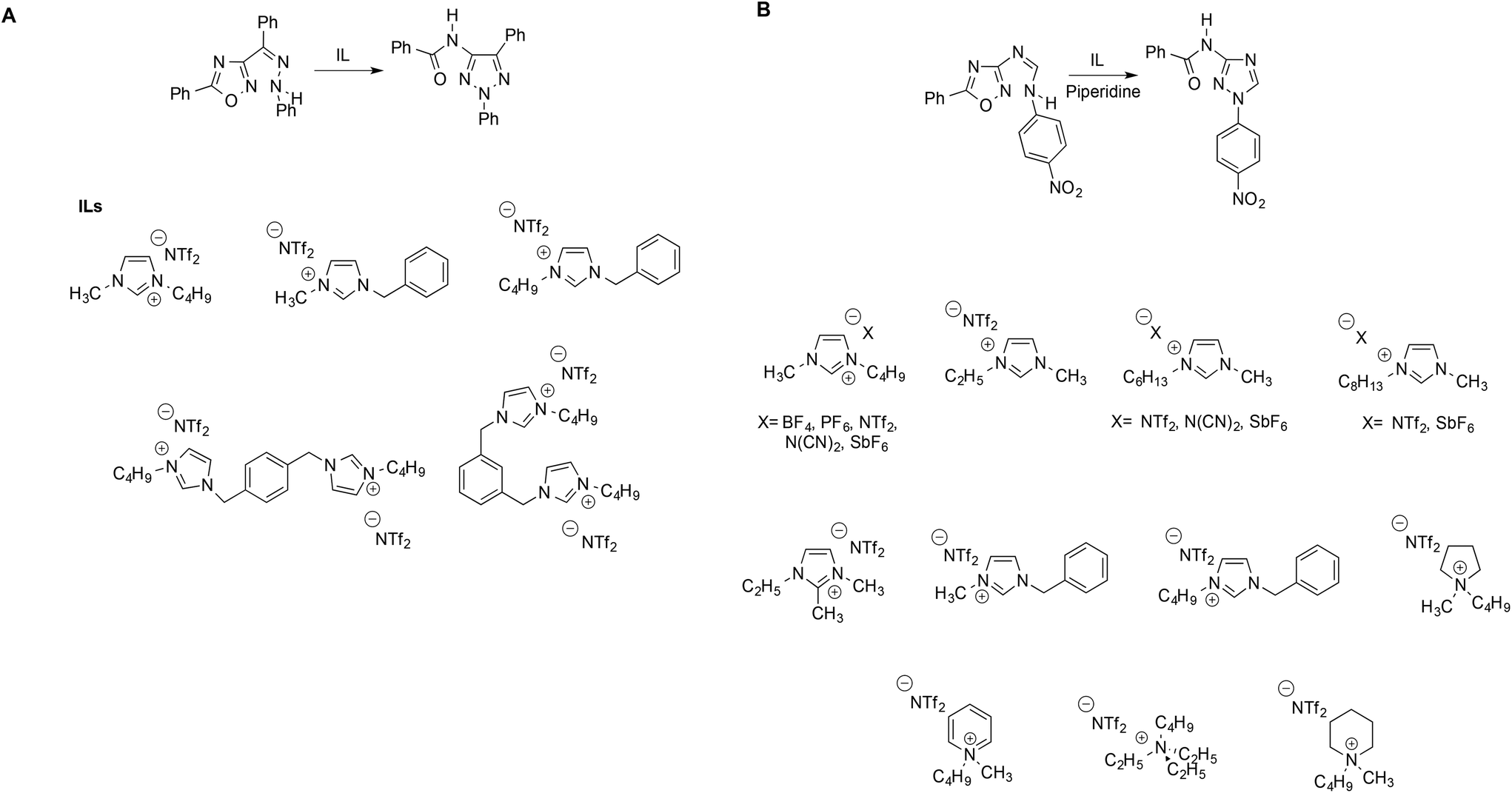 | ||
| Scheme 9 The MRH in (A) imidazolium-based ILs and (B) ILs bearing aliphatic and aromatic cations. | ||
The structural study conducted allowed establishment of an order of structural organization for the ILs used. Interestingly, polarity-based parameters failed to account for the reactivity observed, whereas the trend of the yields was explained considering the structural organization of the solvent with higher yields found in more extensively aggregated ILs. The rate-enhancing effect of the ordered structure of IL received further support from QM/MM calculations with free energy perturbation theory,77 where the formation of an IL clathrate, by means of π–π stacking interactions, enforces a coplanar arrangement of the phenyl rings. Notably, a similar favourable organizing effect was also revealed by the same calculation methods for a different reaction such as nucleophilic aromatic substitution.78
D'Anna et al. studied the piperidine-catalyzed MRH of N-(5-phenyl-1,2,4-oxadiazol-3-yl)-N′-(4-nitrophenyl)-formamidine into the relevant triazole in a solution of 19 ILs (Scheme 9B), covering a diverse range of anions, cations and alkyl chain lengths.79 Unlike what happens in conventional solvents, no satisfactory correlations with the trends of second order rate constants could be found using polarity based parameters like ENR and ET(30) or LSER based on Kamlet–Taft descriptors. Instead, the reactivity observed could be explained on the basis of the concomitant influence of solvent structural organization, again probed by RLS, and solvent viscosity, thus evidencing that even for the same reaction, the IL effect can be significantly affected by the nature of the substrate.
The stabilization of the transition state by means of π–π interactions with IL cations is shown to be effective not only in enhancing catalytic efficiency, but also in stereoselection, compared with conventional solvents, by means of a mechanism switch. In particular, for the thiol–ene reaction between alkenes and 4-methylthiophenol in the IL [hmim]Br (Scheme 10A),80 experimental and computational evidence showed that in the IL the reaction proceeds through a concerted mechanism, in which the imidazolium moiety interacts with the transition state by means of T-shaped π+–π interactions, forming only the anti-Markovnikov product (Scheme 10B). Conversely, in conventional solvents the reaction requires an external catalyst and proceeds through a stepwise mechanism, as exemplified in Scheme 10C for the thiol–ene reaction of related substrates in chloroform, catalyzed by an air stable trityl cation-based salt.81 The reaction is thus regiospecific, whereas in conventional solvents it is instead regioselective.
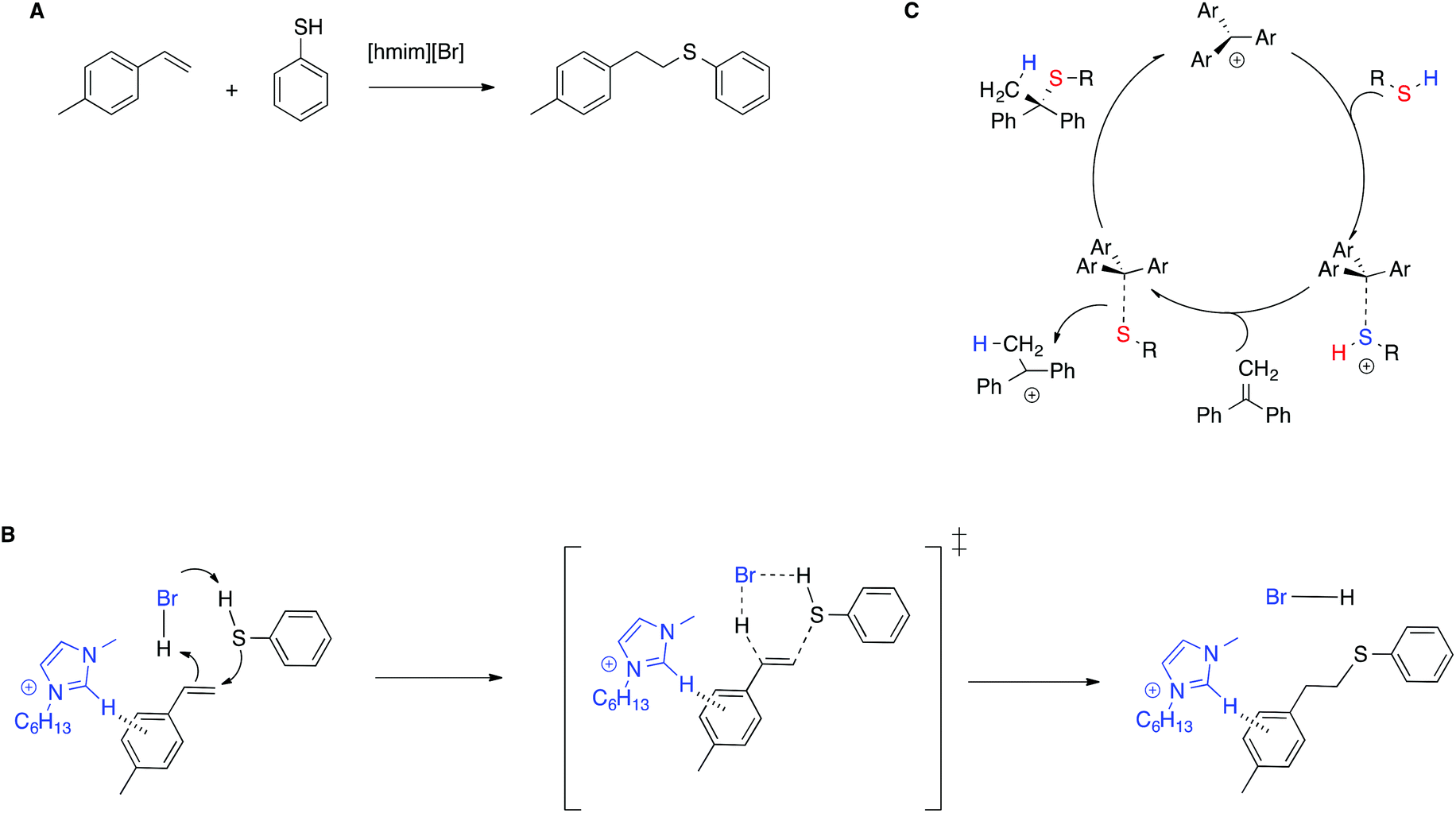 | ||
| Scheme 10 (A) The thiol–ene reaction, (B) the transition state proposed, and (C) an example of the stepwise mechanism in conventional solvents. | ||
Further experimental proof was given by the drop in reactivity upon methylation of the C2 in the imidazolium cation or in the presence of aliphatic alkenes for which CH–π interactions are not operative.
Nucleophilic aromatic substitutions are a class of reactions often employed to study the solvent effect of ILs. For instance, studying the rate of the reaction between amines and 4-chloroquinazoline, Campódonico et al. found an activating effect when the reaction is conducted in a solution of protic ILs due to the specific hydrogen bond interaction between solvent and substrate.82 It is worth noting that the reaction does not occur in conventional organic solvents nor in non-protic ILs.
Furthermore, ILs proved to be suitable solvents for the synthesis of aryl azides under mild conditions, employing imidazolium ILs bearing the azide anion as a nucleophile source. In particular, the azidation of three nitrothiophenes, in the presence of [bmim][N3] as a nucleophile was studied in a solution of several ILs, either aliphatic or aromatic, by means of kinetic experiments (Scheme 11A).83
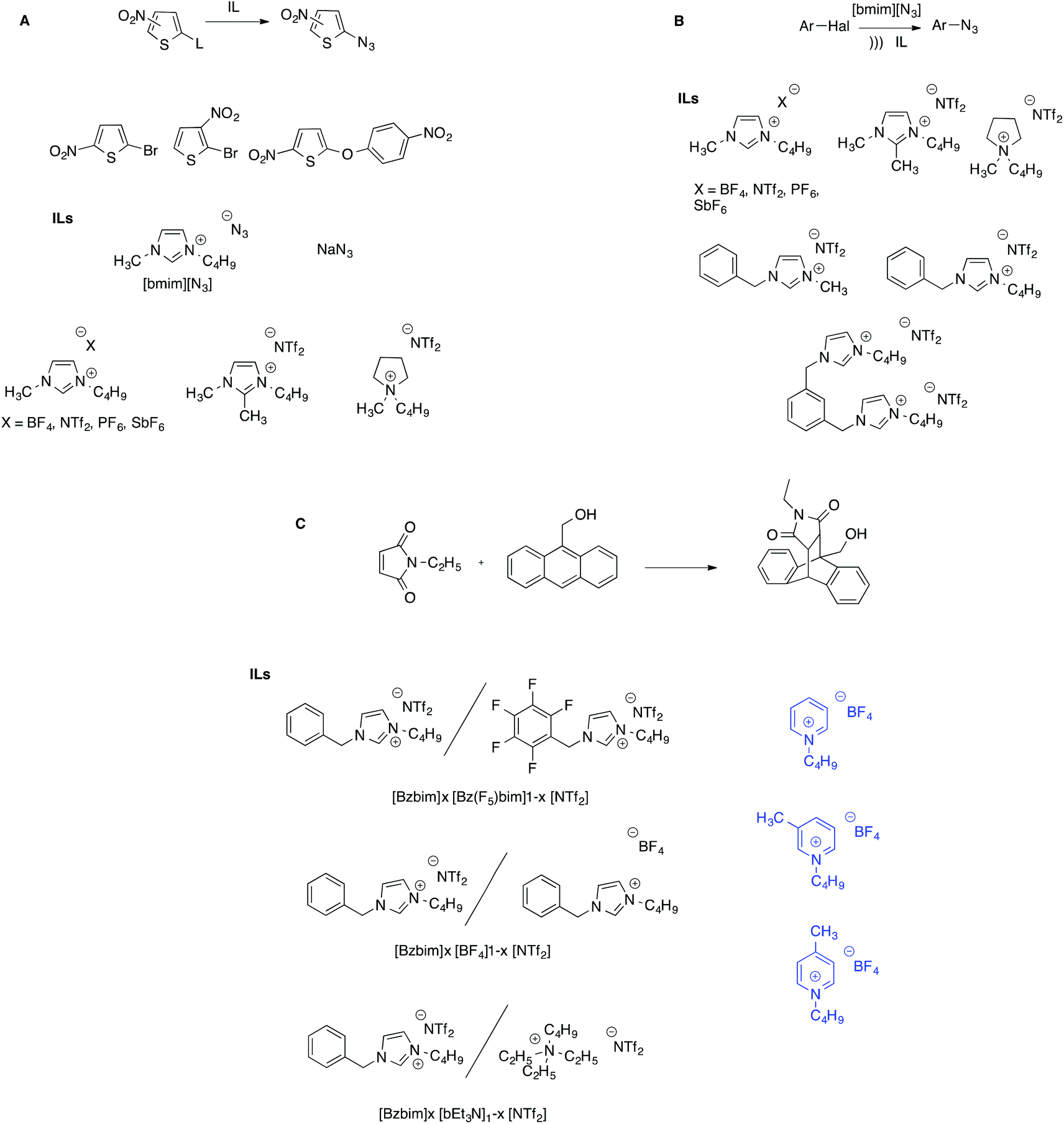 | ||
| Scheme 11 (A) Substrates and ILs for the study of the azidation of nitrothiophenes. (B) The synthesis of arylazides under ultrasonic activation. (C) The Diels–Alder reaction in (left) IL binary and (right) IL-conventional solvent mixtures. | ||
The trend of reactivity was dominated by the influence of the cation, which both stabilizes the transition state through π–π interactions and exerts an unfavorable effect by interacting through hydrogen bonding with the azide ion which, as a result, becomes less available to interact with the substrates. To explain the anomalous trend in rates as a function of the substrate nature, the organized structure of ILs was invoked, which was responsible for the larger reactivity ratios in ILs compared with a conventional solvent like DMF. Notably, a very different pattern arises when the same reaction is carried out in ILs under ultrasonic activation (Scheme 11B).84 In this case, the trend of reactivity as a function of the solvent is dominated by the IL anion, suggesting that, under sonochemical conditions other factors influence reactivity such as solvent viscosity. Further support for this hypothesis comes from the investigation of the Cu(I)-catalysed azide–alkyne addition between 4-chloroquinoline and phenylacetylene in a wide range of ILs.85 It is worth noting that the presence of inorganic salts does not significantly influence the cation–anion nanostructure of the ILs, which is notably resilient.86
This reaction was investigated both under silent and sonochemical conditions: in the former case, the changes in yield as a function of the IL cation could be satisfactorily explained on the grounds of the structural order of the solvent, as probed by RLS measurements, suggesting that the reaction was favored in more extensively aggregated solvent media. On the other hand, switching to sonochemical activation gave rise to a completely different trend and a more pronounced influence of the IL cation compared with what was observed under silent conditions.
The probe reaction approach was also used to investigate the structural features of IL mixtures.
In particular, the piperidine-catalysed MRH of the Z-phenylhydrazone of 3-benzoyl-5-phenyl-1,2,4-oxadiazole into the relevant triazole was studied in mixtures of ILs differing in terms of the cation and anion.87 The trend of observed kinetic constants, measured in the whole mole fraction range, revealed a monotonic trend in the case of the mixture with different cations, whereas a bell-shaped curve was found in solution of the mixture featuring different anions. Notably, such trends could not be rationalized on the basis of the variation of Kamlet–Taft and polarity based-parameters, although they agreed with the change in structural organization, as probed by RLS, NMR and UV-vis spectra of Nile red at variable compositions. The most significant changes in the nanostructural organization of the IL mixture occurred in the presence of anions with significantly different sizes, as previously put forward by Quitevis et al.88
The same mixtures were employed to study a different reaction, to assess whether the influence of the structural organization of the IL mixture on reactivity could be generalized. The model process employed was the Diels–Alder reaction between 9-anthracenemethanol and N-ethylmaleimide (Scheme 11C).89 In contrast to the previous case, the trend of observed kinetic constants gave a non-monotonic trend as a function of composition in all mixtures. Furthermore, the results showed that in the mixture with different anions, the observed kinetic constants gave a trend comparable to the one detected for the MRH, showing that the two reactions responded in a similar way to the same nanostructure changes. Conversely, for the mixtures with different cations, in which less pronounced structural organization variations were detected, the results obtained were explained considering the concomitant effect of solvent organization and viscosity. Interestingly, the difference in the importance of solvent viscosity on the rate of this reaction, depending on the nature of the solvent mixture, was also reported by Kumar et al. in mixtures of ILs with conventional organic solvents.90 In particular, they found that in IL–chloroform mixtures, the trend of reactivity as a function of composition closely followed that of solvent viscosity and thus was dominated by frictional forces. A more articulate picture arose in IL–water and IL–MeOH mixtures, in which additional solvent–solute interactions also became important, based on a pairwise model.
In an effort to single out the effect of pairwise interactions between ILs and reactants on the rate of the process,91 the same authors devised a model relating the ratios of reactivity measured in purely aqueous media and in dilute aqueous solutions of ILs, with the magnitude of these pairwise interactions. It is shown that in the presence of some ILs, reactivity is explained in terms of pairwise interactions, whereas others interact through bulk interactions. While providing a means to quantify the molecular interactions between ILs and reactants during C–C forming reactions, these results were however obtained in an extremely dilute regime and thus are not comparable with the reaction carried out in an ionic medium.
The sensitivity of Diels–Alder reactions towards the nanostructure of the solvent medium is evidenced by the intramolecular reaction of some ester-tethered 1,3,9-hexatrienes in diimidazolium ILs bearing a naphthalene core as solvents.92 These salts are indeed mesogens and form highly organized smectic T mesophases. It is observed that carrying out the reaction in these ILs results in a higher selectivity toward the intramolecular product compared with the results detected in the traditional IL [bmim][NTf2]. The highly organized structure of the smectic T phase enhances selectivity by preventing two substrate molecules from reacting with each other and affording the intermolecular product.
The organization of ILs and IL-mixtures has been shown to significantly affect the conversion of carbohydrates in 5-HMF. For instance, harnessing specific interactions between functionalized imidazolium cations led to a superior performance compared to commonly-used imidazolium chlorides in the conversion of glucose, cellobiose and maltotetraose in 5-HMF, catalyzed by CrCl2.93 In particular, the best results were obtained by performing the reaction in a mixture of imidazolium-based ILs functionalized with a hydroxyl or a phenyl ring in the side chains. 1H NMR mechanistic investigations, aided by a computational study, allowed the authors to identify a labile coordination complex between the hydroxy-functionalized IL and CrCl2 as the catalytically active species.
The hydroxyl group activates glucose through stronger hydrogen bonds compared with the one on the imidazolium ring protons. In turn, the phenyl ring in the side chain of the other IL can weakly interact with the Cr(II) complex, thus preventing its interaction with 5-HMF, which would result in formation of by-products like humins and lower selectivity.
Hydrogen bond interactions are key in determining the outcome of such processes, and using IL mixtures is an easy way to vary the hydrogen bond network as well as other properties of the IL solvent like structural organization or viscosity. In this regard, mixtures of ILs with aromatic or aliphatic cations proved suitable reaction media for the transformation of monosaccharides, like glucose and fructose, and disaccharides like sucrose into 5-HMF. In general, the best results were obtained by using equimolar mixtures, either in the presence of the ion exchange resin Amberlyst 1594 or HY zeolite95 as catalysts.
D'Anna et al. studied the same aforementioned MRH reaction in the presence of dicationic imidazolium salts bearing an imidazole functionality acting as a base catalyst. These salts, also acting as solvents, featured carboxylate anions with different charges, both aromatic and aliphatic (Scheme 12).96
 | ||
| Scheme 12 (A) Diimidazolium-based ILs used for the MHR and (B) a schematic depiction of the interaction between the ILs and transition state. | ||
Not surprisingly, the trend of yields as a function of the IL used was quite complex and was shown to arise from the interplay of different factors, such as salt basicity, flexibility of the anion and tightness of the cation–anion ion pair, as evaluated by VT-1H NMR measurements.97 Specifically, higher yields were obtained in the presence of salts bearing more flexible anions and a looser cation–anion ion pair. This was explained considering that such salts could better conformationally adapt to stabilize the transition state through π–π interactions. A proposed schematic interaction based on both a structural and reactivity investigation is shown in Scheme 12B.
Remarkably, a comparable effect was found by using similarly functionalized diimidazolium salts as catalysts and solvents for the Michael addition between t-chalcone and malononitrile.17
3. Supramolecular catalysis in/by ILs
This section will be focused on the supramolecular catalysis in/by ILs. Indeed, the previous paragraphs showed how the supramolecular structural organization of ILs affects reactivity. Furthermore, the same interactions determining the 3D-structure of ILs can enhance the catalytic efficiency of the catalyst in ILs, including catalytically active ILs.This section will be an excursus of the main classical reactions studied in organic chemistry, in which the supramolecular interactions involving ILs and reactants positively affect catalysis.
As previously mentioned, classical organic reactions can be used as suitable probes to investigate new reaction media, catalysts, and heterogeneous systems. Among these, C–C bond forming reactions like Diels–Alder are positively impacted by solvophobic interactions in ILs. We previously showed how the Diels–Alder reaction is affected by the hydrogen bonding ability of ILs, which makes it well-adapted to be effectively promoted by ILs as the catalyst.
Shen et al. synthesised imidazolium functionalized catalysts with different anions like I−, [BF4]− and [PF6]−, and employed them to promote the Diels–Alder reaction between trans-cinnamaldehyde and cyclopentadiene (Scheme 13).98
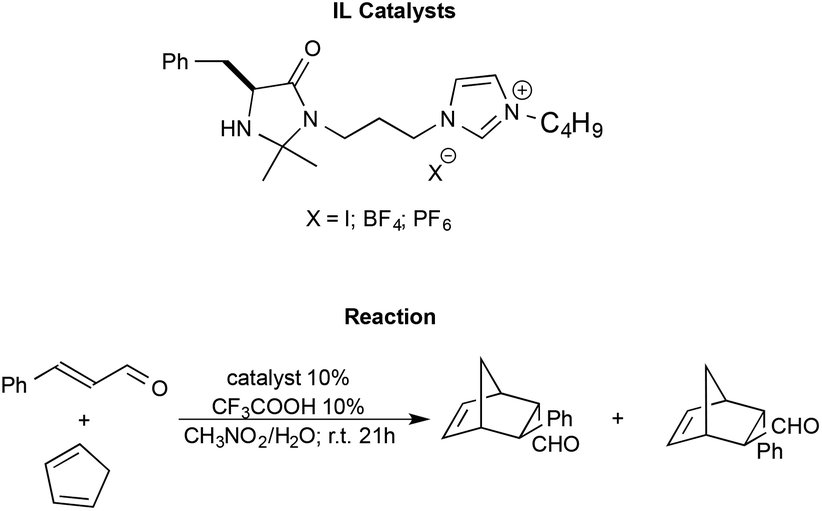 | ||
| Scheme 13 The Diels–Alder reaction between trans-cinnamaldehyde and cyclopentadiene with functionalized ILs as the catalyst synthesised by Shen et al. | ||
The reaction generally afforded almost quantitative yields and excellent enantioselectivities. The nature of the anion is shown to modulate the strength of the hydrogen bond between the N–H group of the catalyst and the substrate, affecting the outcome. In particular, the catalyst bearing the halide anion afforded the best results. Moreover, the catalysts were also recovered and recycled without a loss in performance.
The importance of hydrogen bonds is therefore apparent in the use of protic ILs (PILs) in catalysis, as previously highlighted by Chiappe et al.99 Indeed, the catalytic ability of this class of ILs is due to their Brønsted-acidic nature. In particular, studying the Diels–Alder reaction of cyclopentadiene with three dienophiles (Scheme 14A) by calculations at DFT level, they found that ILs bearing protic cations can exert a “clamp effect”, which favours the stacking and approach of the diene towards the dienophile, increasing reactivity.
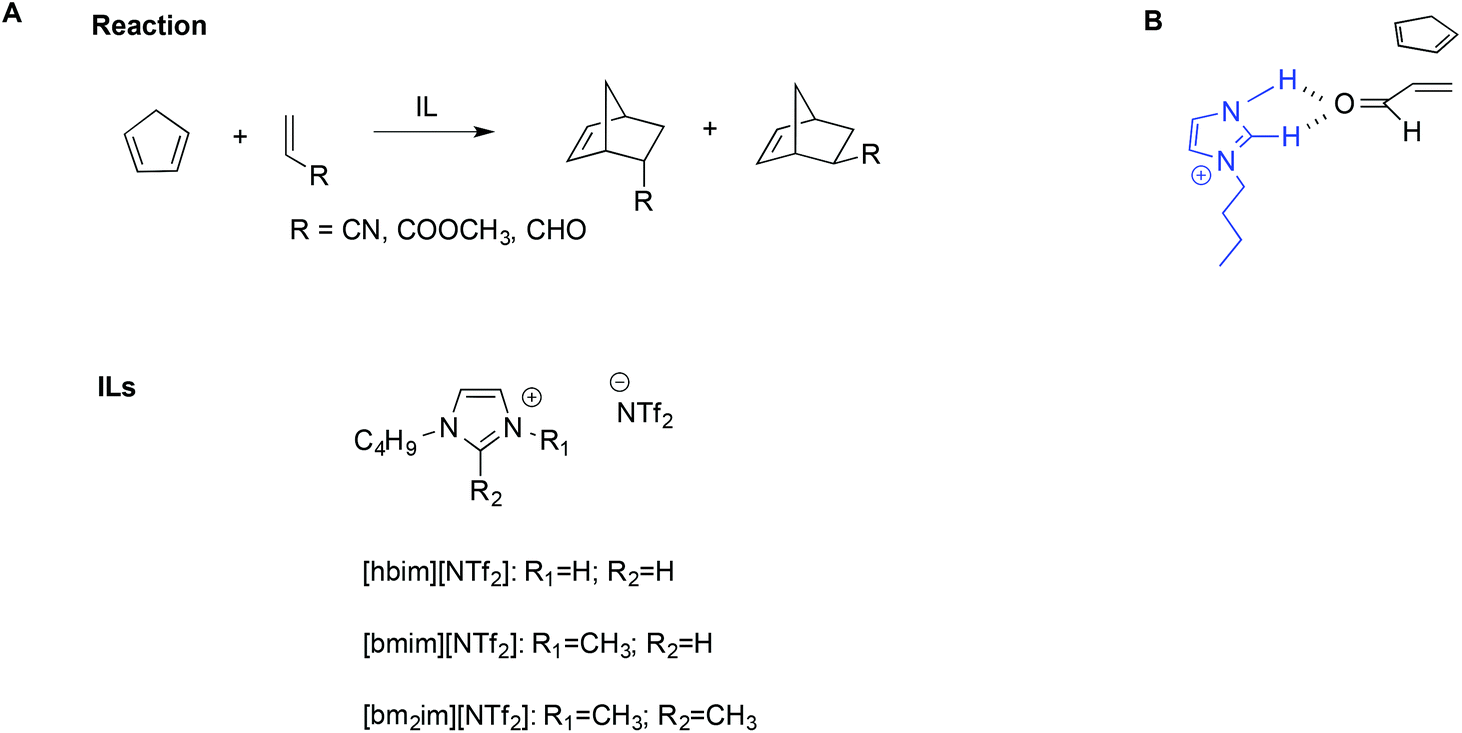 | ||
| Scheme 14 (A) The Diels–Alder reaction, the ILs considered, and (B) a simplified depiction of the clamp effect. | ||
Moreover, favourable polarization of the dienophile double bond occurs. A simplified schematic depiction is shown in Scheme 14B.
The same interactions involved in the Diels–Alder reaction proved beneficial for the aldol reaction, which takes advantage of the supramolecular organization of ILs on yields and selectivity. Moreover, control of the reaction conditions is fundamental to ensure obtainment of the aldol and avoid dehydration to α,β-unsaturated carbonyl compounds. The use of the appropriate catalyst is key to obtaining the desired product. The asymmetric aldol reaction usually employs organocatalysts to induce enantioselectivity and one of the most used for this purpose is L-proline.100–103 Thanks to its cyclic structure and secondary amino-group, it establishes strong hydrogen bonds and can form an iminium ion or enamine intermediates depending on the nature of the electrophile or nucleophile.104
In this regard, Porcar et al. investigated the effect of ILs on a widely used benchmark process, such as the L-proline-catalysed aldol reaction between acetone and p-nitrobenzaldehyde. The reaction was studied using CILs and, for comparison, an achiral IL such as [bmim]Cl (Scheme 15).105
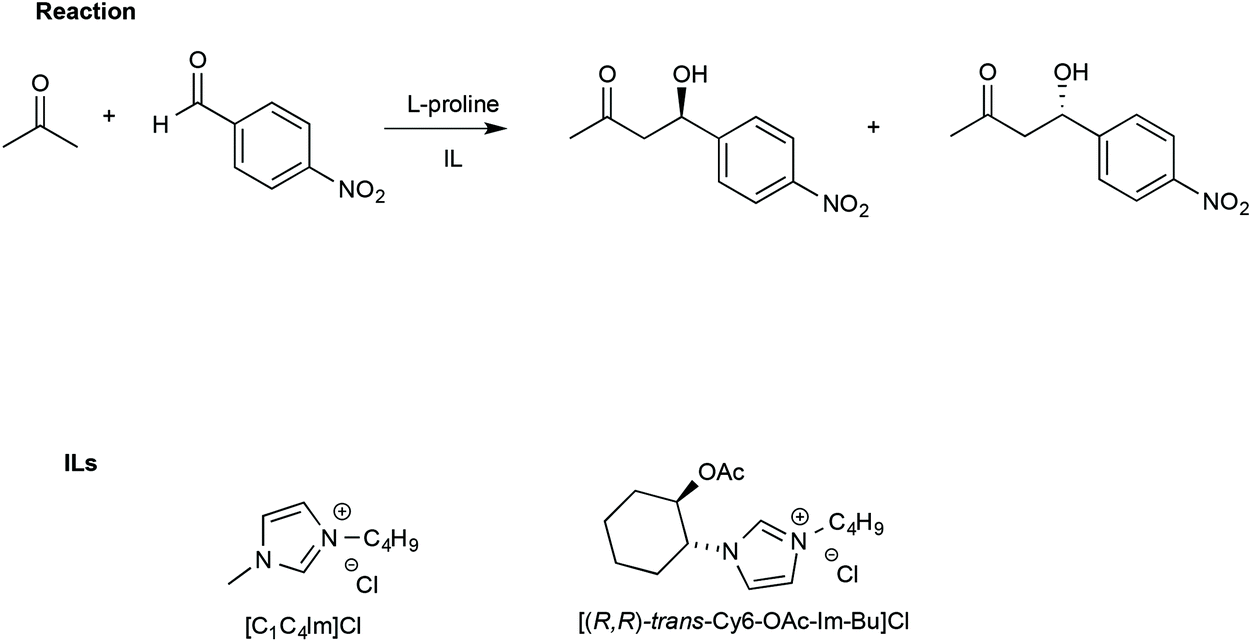 | ||
| Scheme 15 The aldol reaction between acetone and p-nitrobenzaldehyde and the structure of the ILs used with L-proline. | ||
The use of an IL such as [bmim]Cl led to higher yields (90%) and slightly lower ee (65%) compared with a conventional solvent like DMSO (yield = 68% and ee = 75%, respectively).
A detailed 1H NMR and FTIR study revealed that the presence of proline in the IL modifies its structural organization, inducing the formation of a sample-spanning hydrogen bonded network even leading to the formation of an organogel (Fig. 2).
 | ||
| Fig. 2 A schematic model of the supramolecular structure of an imidazolium IL network based on hydrogen bonding interactions induced by (L)-proline in [bmim]Cl (adapted with permission from ref. 105. Copyright (2016) American Chemical Society). | ||
It is worth noting that this occurs by the establishment of a network of hydrogen bonds and not by formation of an imidazolium prolinate salt. Moreover, the establishment of the hydrogen bond network was found to be essential for the chirality information transfer, since lowering the amount of IL, and therefore the presence of organized structures, led to a more modest asymmetric induction. In light of this, when the reaction was carried out in the CIL, [(R,R)-trans-Cy6-OAc-Im-Bu]Cl (Scheme 15), enhanced chirality transfer with respect to [bmim]Cl was observed (71% ee vs. 68% ee, CIL vs. [bmim]Cl respectively). The CIL is more effective in asymmetric induction since the enantiomeric excess remained constant even when lowering the catalyst loading from 40 mol% to 5 mol%, while a much lower ee was obtained in [bmim]Cl in the latter condition (ee < 48%).
Using the same aldol reaction between acetone and p-nitrobenzaldehyde (Scheme 16), González et al. studied how enantiomers of the proline, in combination with a CIL, affect catalytic activity. They synthesised imidazolium CILs derived from L-valine, L-phenylalanine, and L-leucine, studying the effect of the L-valine-based one (R = CH(CH3)2, Scheme 16) as reaction media in the presence of L- or D-proline, to promote the reaction.106
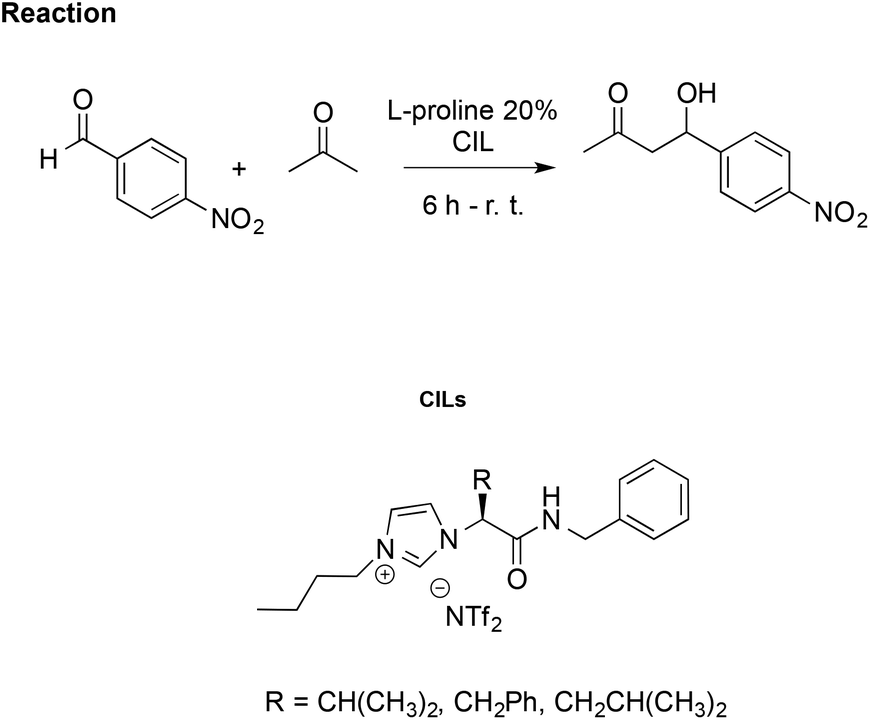 | ||
| Scheme 16 The aldol reaction between acetone and p-nitrobenzaldehyde catalysed by L-proline in CILs. | ||
Using CILs as reaction media, they achieved better enantioselection (ee = 77%) compared to either conventional solvents such as methanol, toluene and dichloromethane or to the non-chiral IL [bmim][NTf2] with ee values of 9%, 71%, 67% and 62%, respectively. However, lower selectivity and enantiomeric excess (70%) were obtained using D-proline as the catalyst, together with a drop-in reaction rate. This was explained by a “match/mismatch” effect due to the arrangement of the hydrogen bond network between the catalyst and CIL, which determines the formation of different diastereomeric supramolecular complexes dependent on the proline enantiomer. The one formed between CIL and D-proline is less active as the catalyst due to stronger hydrogen bond interactions, as probed by 1H NMR and FTIR.
The concomitant presence of ILs and L-proline can significantly improve the progress of the reaction both in terms of reaction rate and selectivity. In particular, incorporating proline derivatives in the structure of ILs could positively affect both the catalytic efficiency and recycling ability. On this topic, Schmitzer and Gauchot synthesised two different imidazolium catalysts bearing trans-L-hydroxyproline derived anions, characterized by an ester or an amide functionality (Scheme 17).107 Also in this case, the same model reaction was used and carried out in neat [bmim][NTf2] and, for comparison, in conventional solvents like methanol, dichloromethane, DMSO and DMF.
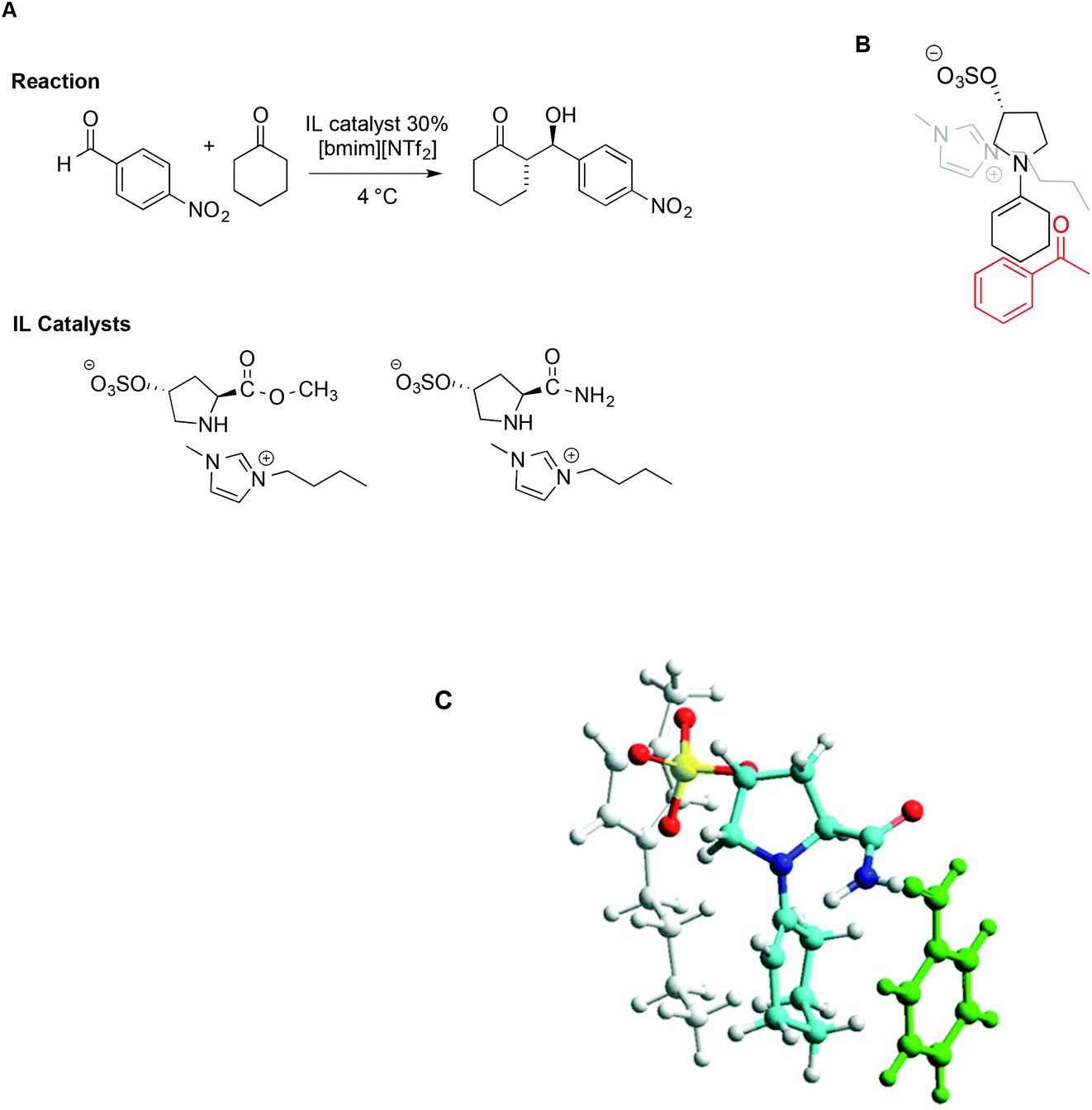 | ||
| Scheme 17 (A) The aldol reaction between cyclohexanone and p-nitrobenzaldehyde, catalysed by trans-L-proline derivatives, (B) a schematic depiction of the aldehyde approach to enamine, and (C) a representation based on molecular modelling with the imidazolium cation in grey, the anion in yellow, and benzaldehyde in green (adapted with permission from ref. 107. Copyright (2012) American Chemical Society). | ||
When the reaction was carried out in the presence of the ester-functionalized catalyst in conventional solvents, the yields ranged from 20% to 53%, while the ee obtained was between 22% in methanol and 79% in DMF. In the IL, under optimized conditions, both catalysts afforded good to excellent yields and selectivity, along with good enantiomeric excess (up to 85%). Due to lower steric hindrance, and the ability of the amide group to interact with the aldehyde by hydrogen bonding, the amide-functionalized catalyst leads to higher rates. Notably however, despite these differences, the major diastereoisomer is the same with both catalysts, suggesting a similar transition state. Following this reasoning, the source of selectivity was found in the proximity of the imidazolium cation to the negative charge in the sulfonate group, which prevents access of the aldehyde from that face, as schematically depicted in Scheme 17B. Molecular modelling based on PM6 methods lent further support to this hypothesis, as reported in Scheme 17C.
In a notable example, the use of ILs in the synthesis of 2,6-dibenzylidenecycloalkanone derivatives allowed circumvention of the low selectivity and difficult product isolation encountered in the presence of strong bases as catalysts.108 In particular, Kang et al. showed that using 2-hydroxyethylammonium acetate improved yields and selectivity. This was explained invoking a simultaneous activation of the carbonyl oxygen by hydrogen bonding with the [NH3]+ protons on the cation, coupled with the assistance of the hydroxyl group, favouring the abstraction of the α-H of the cyclohexanone (Scheme 18). The abstraction of the α-H of the cyclohexanone can also be favoured by the IL anion. A mechanism showing the catalytic action of the IL is shown in Scheme 18B.
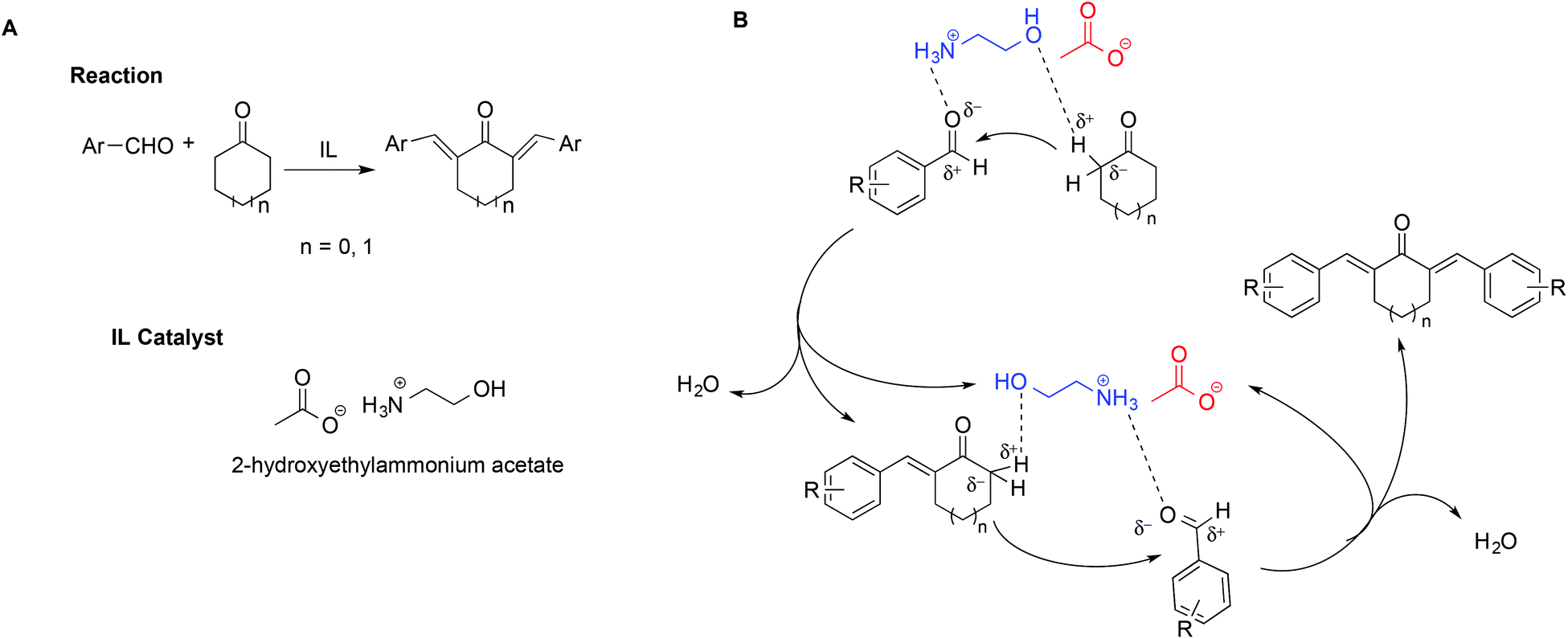 | ||
| Scheme 18 (A) The synthesis of 2,6-dibenzylidenecycloalkanones and (B) catalytic action of the IL. | ||
Similarly, α,α′-bis-(substituted benzylidene)cyclopentanones have been synthesised efficiently with the assistance of ILs. In particular, an amine-functionalized IL/H2O catalytic system has been utilized to facilitate the cross-aldol condensation between aromatic aldehydes with cyclopentanone (Scheme 19).109
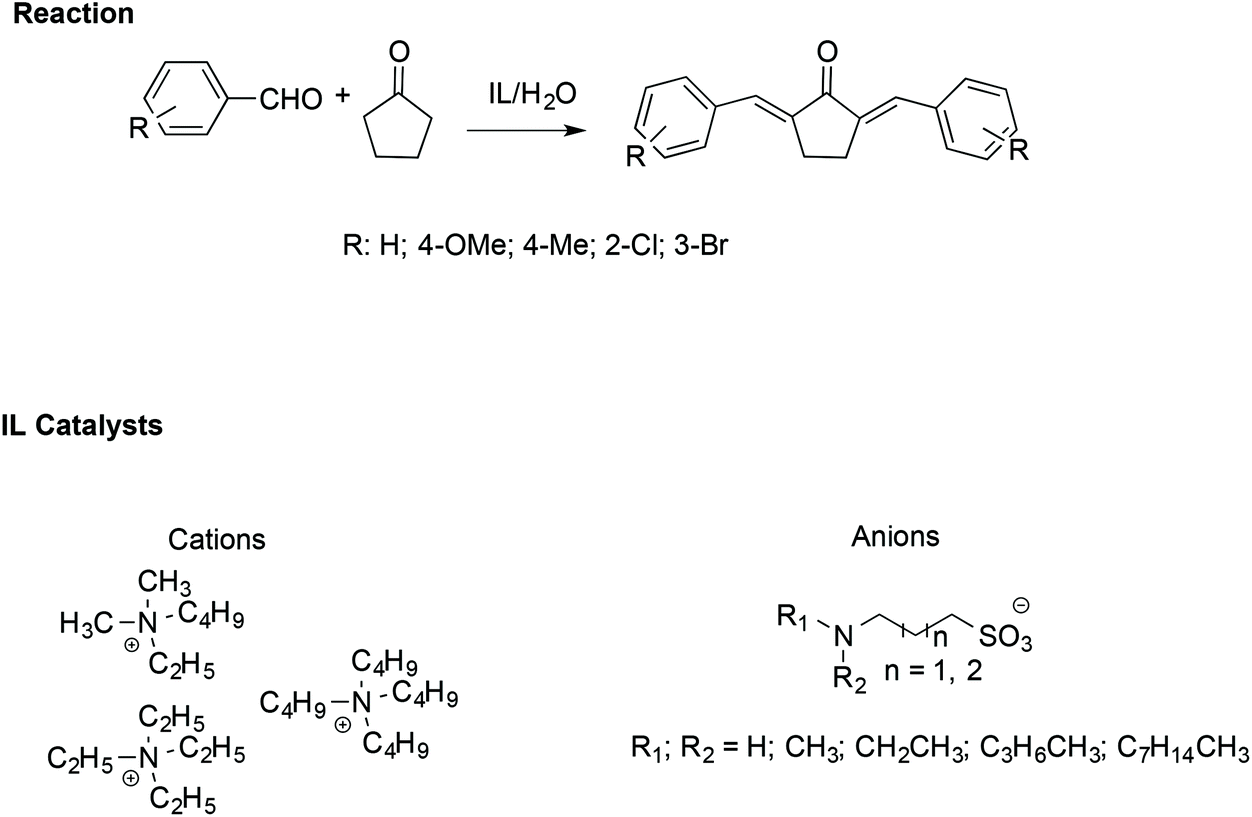 | ||
| Scheme 19 The synthesis of α,α′-bis(substituted benzylidene)cyclopentanones catalysed by amine-functionalised ILs. | ||
In this case, the reaction proceeds via an enolate pathway, in which the nucleophilic amine functionalised ILs act as a base and push enolate formation. Moreover, it has been found that the anion of the IL is the crucial catalytic portion; indeed, if a steric hindrance on the nitrogen of the anion occurs, deprotonation of cyclopentanone becomes difficult, slowing down the reaction.
The main advantage of this catalytic system over conventional organic solvents can be found in the easier separation of the product, at the end of the reaction as the product spontaneously separates due to the low solubility.
Together with the aldol and Diels–Alder reactions, the Michael reaction is one of the most widely used C–C forming reactions in synthesis. In this context, the involvement of IL-based supramolecular catalysis has been investigated by Chakraborti and Roy, considering the aza-Michael reaction between aniline and 1,3-diphenyl-2-propenone reported in Scheme 20.110
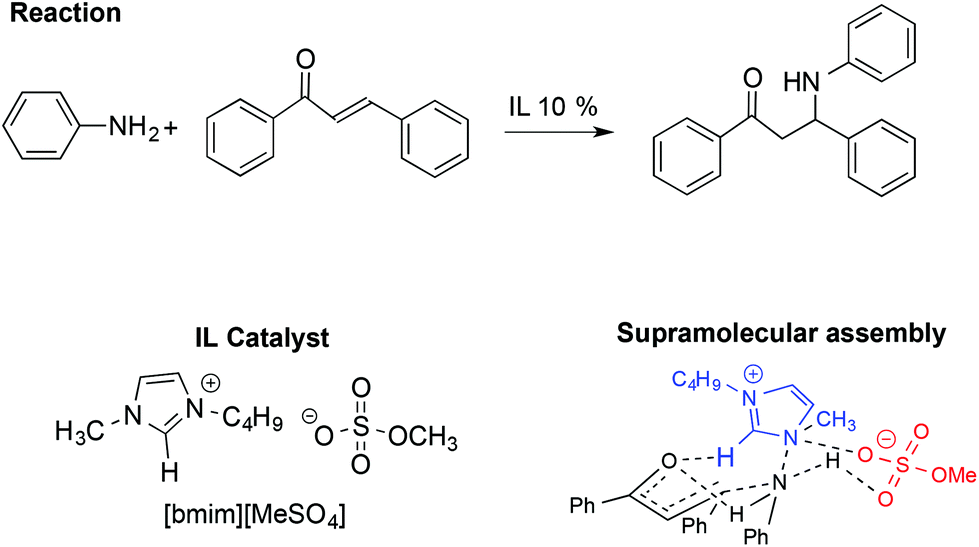 | ||
| Scheme 20 The IL-catalysed aza-Michael reaction and supramolecular assembly formed following the interaction between the IL and starting materials. Supramolecular interactions are represented by dashed lines. | ||
In particular, the enhanced reactivity observed using ILs and [bmim][MeSO4] is due to a supramolecular interaction in which the IL acts as an electrophile and nucleophile activator.
The supramolecular assembly reported in Scheme 20 is strongly related to the IL structure as the hydrogen bond between the C2–H on the [bmim]+ cation and the carbonyl oxygen of the ketone plays a fundamental role. Conversely, the role of the anion is recognizable through the formation of a six membered chair-like cyclic supramolecular assembly among the quaternary ammonium of the cation, the N–H hydrogen of the aniline and the O–S of the anion. A good correlation was also found between the abundance of the supramolecular assembly, detected by ESI-MS and catalytic efficiency, in the presence of different amounts of ILs.
A similar influence of supramolecular assemblies in an IL-promoted thia-Michael reaction was observed by the same authors (Scheme 21).111
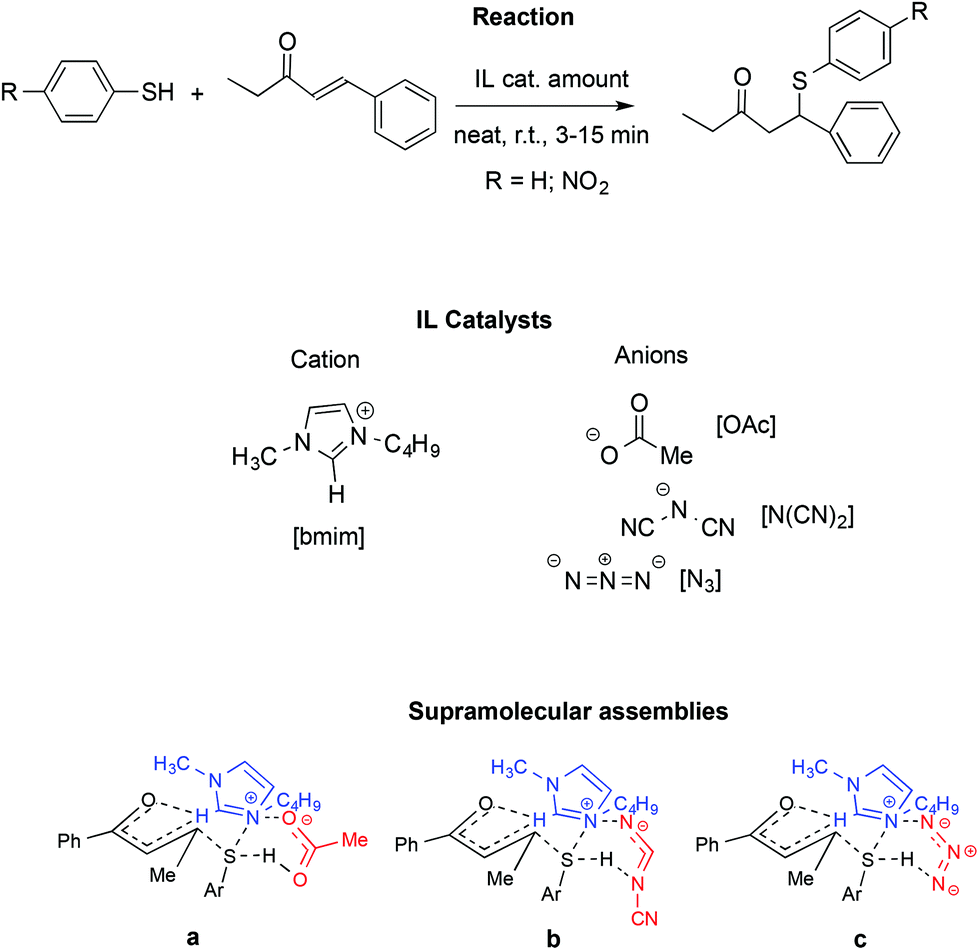 | ||
| Scheme 21 A thia-Michael reaction with ILs in catalytic amounts. a–c: Supramolecular assemblies formed following the interaction of the IL and starting materials. Supramolecular interactions are represented by dashed lines. | ||
In particular, they studied the reaction between 4-phenyl-3-buten-2-one and thiophenol or 4-nitrothiophenol in the presence of [bmim]-based ILs. Once again, activation of the carbonyl and S–H groups is achieved through a relay of hydrogen bonds involving the starting materials and both the IL cation and anion.
Similarly to the aza-Micheal reaction, the interaction between the quaternary ammonium of the [bmim]+ with the oxyanionic, amidic or imidic functionalities, in concomitance with the hydrogen bonding of the anion with the cation and the S–H group lead to cyclic supramolecular assembly. The fundamental role of the C2–H on the IL cation was confirmed by substitution of this hydrogen with a methyl group and the consequent drop in yield, thus ruling out the possibility that the reactivity observed comes only from the basicity of the IL anion. Accordingly, bases like sodium acetate fail to induce the reaction. Similarly, no reaction occurred in conventional solvents like DMSO or acetonitrile.
Further support for the involvement of supramolecular catalysts by ILs comes from the synthesis of (2E)-1-aryl-3-dimethylamino-2-propenones from aryl methyl ketone with DMF/DMA (Scheme 22).112
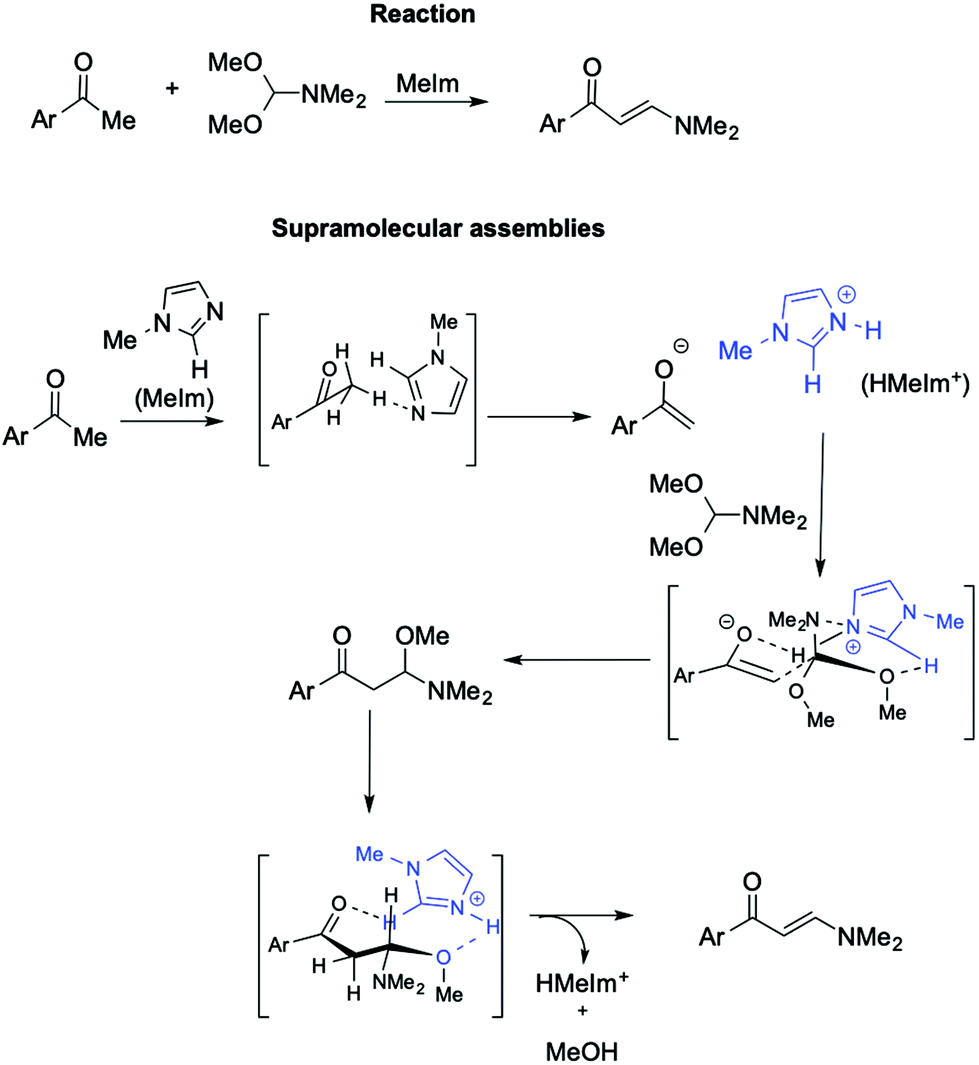 | ||
| Scheme 22 The synthesis of (2E)-1-aryl-3-dimethylamino-2-propenones from aryl methyl ketones with DMF-DMA catalyzed by an in situ generated imidazolium IL (in square brackets, supramolecular interactions are represented by dashed lines). | ||
In this base-promoted reaction, the best results were obtained in the presence of methylimidazole (MeIm), despite not being the strongest base used. This can be explained by considering the in situ formation of the [HMeIm]+ cation due to proton abstraction by MeIm from the 2,4-dimethoxyacetophenone. The ensuing enolate forms a hydrogen bond with the NH proton of [HMeIm]+ (Scheme 22). At the same time, hydrogen bonding is established between the C2–H of the imidazolium cation and the O of the OMe-groups of DMF-DMA. Moreover, the charge–charge interaction between the quaternary nitrogen of the cation and the lone pair of the NMe2 group of the DMF-DMA lends rigidity to the supramolecular structure obtained, driving the reaction to the nucleophilic displacement of the OMe-group. Once again, the yield drastically decreases when a cation devoid of C2–H hydrogen, such as 2,3 dimethylimidazolium IL, is used.
4. Conclusions
The aim of the present review was to provide a “bird's-eye view” of the different approaches used to rationalize the solvent effect and reactivity related to ILs. We showed that these can be essentially traced back to two viewpoints. According to the first one, the reactivity and reaction rates can be suitably explained on the basis of LSERs, mostly based on combinations of Kamlet–Taft polarity parameters. We discussed how, in this case, no special IL effect is often observed, and reactivity could be predicted in ILs and conventional solvents alike. By contrast, the other view takes into account the supramolecular nanostructures of ILs, with particular regard to ILs composed of aromatic ions. Under this viewpoint, the degree of structural organization appears to play a prominent role, along with specific weak interactions between IL ions and reactants and transition states. We discussed how these aspects have repercussions for different kinds of reactions. We then showed that these same approaches have been successfully extended in some cases to reactivity in IL mixtures, although these systems are obviously more complex. We then covered, in the closing section, examples of supramolecular catalysis “by” and “in” ILs.Describing the IL solvent effect on organic reactivity is a complex and multifaceted task, and this is not surprising if one looks at the fundamentally different nature of ILs compared with molecular solvents. A further source of complexity is the marked dependence of this behaviour on the nature of the reactants or reaction investigated, as originally pointed out by Armstrong.113,114 Therefore, future exploration of organic reactivity in ILs should increasingly consider aspects related to the influence of the IL structure, and, to this aim, not only will kinetic and mechanistic studies be beneficial but computational investigations can also be fruitful. Furthermore, future research in this field will be required to cover the effect of ILs in non-classical reaction environments, such as confined media, as well as in non-conventional methodologies, such as ultrasonic activation. We therefore envisage that this review could be helpful in that respect.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Palermo for financial support (FFR 2018).References
- R. D. Rogers, K. R. Seddon and S. Volkov, Green Industrial Applications of Ionic Liquids, Springer Netherlands, 2002 Search PubMed.
- T. Welton, Biophys. Rev., 2018, 10, 691–706 CrossRef CAS.
- J. S. Wilkes, Green Chem., 2002, 4, 73–80 RSC.
- A. M. Inamuddin, A. Asiri and K. Suvardhan, Green Sustainable Process for Chemical and Environmental Engineering and Science, Elsevier, 1st edn, 2019 Search PubMed.
- J. D. Scholten, B. C. Leal and J. Dupont, ACS Catal., 2012, 2, 184–200 CrossRef CAS.
- K. Matsumoto, J. Hwang, S. Kaushik, C.-Y. Chen and R. Hagiwara, Energy Environ. Sci., 2019, 12, 3247–3287 RSC.
- R. A. Perera Jayawickramage and J. P. Ferraris, Nanotechnology, 2019, 30, 155402 CrossRef.
- D. Blanco, R. González, J. L. Viesca, A. Fernández-González, M. Bartolomé and A. Hernández Battez, Tribol. Lett., 2017, 65, 66 CrossRef.
- H. Xiao, Tribol. Trans., 2017, 60, 20–30 CrossRef CAS.
- M. E. V. Valkenburg, R. L. Vaughn, M. Williams and J. S. Wilkes, Thermochim. Acta, 2005, 425, 181–188 CrossRef.
- F. Billeci, H. Q. N. Gunaratne, F. D'Anna, G. G. Morgan, K. R. Seddon and N. V. Plechkova, Green Chem., 2019, 21, 1412–1416 RSC.
- C. Rizzo, S. Marullo, N. T. Dintcheva and F. D'Anna, Molecules, 2019, 24, 2788 CrossRef.
- C. Rizzo, J. L. Andrews, J. W. Steed and F. D'Anna, J. Colloid Interface Sci., 2019, 548, 184–196 CrossRef CAS.
- F. Billeci, F. D'Anna, H. Q. N. Gunaratne, N. V. Plechkova and K. R. Seddon, Green Chem., 2018, 20, 4260–4276 RSC.
- C. Rizzo, S. Marullo, N. T. Dintcheva, C. Gambarotti, F. Billeci and F. D'Anna, J. Colloid Interface Sci., 2019, 556, 628–639 CrossRef CAS.
- P. C. Marr and A. C. Marr, Green Chem., 2016, 18, 105–128 RSC.
- C. Rizzo, F. D'Anna and R. Noto, RSC Adv., 2016, 6, 58477–58484 RSC.
- J. Zhao, Y. Yue, G. Sheng, B. Wang, H. Lai, S. Di, Y. Zhai, L. Guo and X. Li, Chem. Eng. J., 2019, 360, 38–46 CrossRef CAS.
- R. L. Vekariya, J. Mol. Liq., 2017, 227, 44–60 CrossRef CAS.
- T. Itoh, Chem. Rev., 2017, 117, 10567–10607 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123–150 RSC.
- F. Billeci, F. D'Anna, M. Feroci, P. Cancemi, S. Feo, A. Forlino, F. Tonnelli, K. R. Seddon, H. Q. N. Gunaratne and N. V. Plechkova, ACS Sustainable Chem. Eng., 2020, 8, 926–938 CrossRef CAS.
- C.-W. Cho, S. Stolte and Y.-S. Yun, Sci. Rep., 2016, 6, 33403 CrossRef CAS.
- T. Klejdysz, B. Łęgosz, D. Czuryszkiewicz, K. Czerniak and J. Pernak, ACS Sustainable Chem. Eng., 2016, 4, 6543–6550 CrossRef CAS.
- J. Hulsbosch, D. E. De Vos, K. Binnemans and R. Ameloot, ACS Sustainable Chem. Eng., 2016, 4, 2917–2931 CrossRef CAS.
- F. C. Gozzo, L. S. Santos, R. Augusti, C. S. Consorti, J. Dupont and M. N. Eberlin, Chem. – Eur. J., 2004, 10, 6187–6193 CrossRef CAS.
- J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508–3576 CrossRef CAS.
- J. N. A. Canongia Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 3330–3335 CrossRef CAS.
- B. Kirchner, F. Malberg, D. S. Firaha and O. Hollóczki, J. Phys.: Condens. Matter, 2015, 27, 463002 CrossRef.
- G. Cevc, I. Berts, S. F. Fischer, J. O. Rädler and B. Nickel, Langmuir, 2018, 34, 6285–6295 CrossRef CAS.
- J. N. Canongia Lopes, M. F. Costa Gomes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 16816–16818 CrossRef.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- K. Fumino, A. Wulf and R. Ludwig, Angew. Chem., Int. Ed., 2008, 47, 8731–8734 CrossRef CAS.
- A. R. Katritzky, D. C. Fara, H. Yang, K. Tämm, T. Tamm and M. Karelson, Chem. Rev., 2004, 104, 175–198 CrossRef CAS.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Green Chem., 2013, 15, 2655–2679 RSC.
- R. Hayes, G. G. Warr and R. Atkin, Chem. Rev., 2015, 115, 6357–6426 CrossRef CAS.
- A. K. Sinha, R. Singh and R. Kumar, Asian J. Org. Chem., 2020, 9, 706–720 CrossRef CAS.
- H. Olivier-Bourbigou and L. Magna, J. Mol. Catal. A: Chem., 2002, 182–183, 419–437 CrossRef CAS.
- K. R. Seddon, A. Stark and M.-J. Torres, Pure Appl. Chem., 2000, 72, 2275–2287 CAS.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- L. Crowhurst, R. Falcone, N. L. Lancaster, V. Llopis-Mestre and T. Welton, J. Org. Chem., 2006, 71, 8847–8853 CrossRef CAS.
- C. Velez, B. Doherty and O. Acevedo, Int. J. Mol. Sci., 2020, 21, 1190 CrossRef CAS.
- R. Bini, C. Chiappe, V. L. Mestre, C. S. Pomelli and T. Welton, Org. Biomol. Chem., 2008, 6, 2522–2529 RSC.
- C. Chiappe, M. Malvaldi and C. S. Pomelli, Green Chem., 2010, 12, 1330–1339 RSC.
- G. Ranieri, J. P. Hallett and T. Welton, Ind. Eng. Chem. Res., 2008, 47, 638–644 CrossRef CAS.
- I. Correia and T. Welton, Dalton Trans., 2009, 2009, 4115–4121 RSC.
- E. M. Omar, M. B. A. Rahman, B. Ni and A. D. Headley, Synth. Commun., 2019, 49, 1578–1591 CrossRef CAS.
- E. Priede, S. Brica, E. Bakis, N. Udris and A. Zicmanis, New J. Chem., 2015, 39, 9132–9142 RSC.
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS.
- F. André, P. Hapiot and C. Lagrost, Phys. Chem. Chem. Phys., 2010, 12, 7506–7512 RSC.
- M. I. Qadir, J. D. Scholten and J. Dupont, Catal. Sci. Technol., 2015, 5, 1459–1462 RSC.
- F. D'Anna, V. Frenna, S. La Marca, R. Noto, V. Pace and D. Spinelli, Tetrahedron, 2008, 64, 672–680 CrossRef.
- K. S. Schaffarczyk McHale, R. S. Haines and J. B. Harper, ChemPlusChem, 2018, 83, 1162–1168 CAS.
- A. Gilbert, R. S. Haines and J. B. Harper, Org. Biomol. Chem., 2019, 17, 675–682 RSC.
- S. T. Keaveney, R. S. Haines and J. B. Harper, Org. Biomol. Chem., 2015, 13, 8925–8936 RSC.
- M. Gazitúa, R. A. Tapia, R. Contreras and P. R. Campodónico, New J. Chem., 2018, 42, 260–264 RSC.
- G. F. De Gregorio, C. C. Weber, J. Gräsvik, T. Welton, A. Brandt and J. P. Hallett, Green Chem., 2016, 18, 5456–5465 RSC.
- S. A. Forsyth, U. Fröhlich, P. Goodrich, H. Q. N. Gunaratne, C. Hardacre, A. McKeown and K. R. Seddon, New J. Chem., 2010, 34, 723–731 RSC.
- A. A. H. Pádua, M. F. Costa Gomes and J. N. A. Canongia Lopes, Acc. Chem. Res., 2007, 40, 1087–1096 CrossRef.
- S. Saha, S. Hayashi, A. Kobayashi and H.-O. Hamaguchi, Chem. Lett., 2003, 32, 740–741 CrossRef CAS.
- J. D. Tubbs and M. M. Hoffmann, J. Solution Chem., 2004, 33, 381–394 CrossRef CAS.
- S. Dorbritz, W. Ruth and U. Kragl, Adv. Synth. Catal., 2005, 347, 1273–1279 CrossRef CAS.
- S. Patra and A. Samanta, J. Phys. Chem. B, 2012, 116, 12275–12283 CrossRef CAS.
- K. Iwata, H. Okajima, S. Saha and H.-O. Hamaguchi, Acc. Chem. Res., 2007, 40, 1174–1181 CrossRef CAS.
- A. Verma, N. E. Prasad, J. Srivastava and S. Saha, ChemistrySelect, 2019, 4, 49–58 CrossRef CAS.
- Y.-L. Wang, B. Li, S. Sarman, F. Mocci, Z.-Y. Lu, J. Yuan, A. Laaksonen and M. D. Fayer, Chem. Rev., 2020, 120, 5798–5877 CrossRef CAS.
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, J. Phys. Chem. Lett., 2012, 3, 27–33 CrossRef CAS.
- J. Dupont, Acc. Chem. Res., 2011, 44, 1223–1231 CrossRef CAS.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS.
- R. Elfgen, O. Hollóczki and B. Kirchner, Acc. Chem. Res., 2017, 50, 2949–2957 CrossRef CAS.
- L. Leclercq, G. Douyere and V. Nardello-Rataj, Catalysts, 2019, 9, 163 CrossRef.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Angew. Chem., Int. Ed., 2012, 51, 11483–11486 CrossRef CAS.
- C. C. Weber, A. F. Masters and T. Maschmeyer, Org. Biomol. Chem., 2013, 11, 2534–2542 RSC.
- C. C. Weber, A. F. Masters and T. Maschmeyer, J. Phys. Chem. B, 2012, 116, 1858–1864 CrossRef CAS.
- L. C. Player, B. Chan, P. Turner, A. F. Masters and T. Maschmeyer, Appl. Catal., B, 2018, 223, 228–233 CrossRef CAS.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, Eur. J. Org. Chem., 2011, 5681–5689 CrossRef.
- C. Allen, R. Ghebreab, B. Doherty, B. Li and O. Acevedo, J. Phys. Chem. B, 2016, 120, 10786–10796 CrossRef CAS.
- C. Allen, B. W. McCann and O. Acevedo, J. Phys. Chem. B, 2015, 119, 743–752 CrossRef CAS.
- F. D'Anna, D. Millan and R. Noto, Tetrahedron, 2015, 71, 7361–7366 CrossRef.
- L. Feng, R. Ye, T. Yuan, X. Zhang, G.-P. Lu and B. Zhou, New J. Chem., 2019, 43, 5752–5758 RSC.
- E. Mosaferi, D. Ripsman and D. W. Stephan, Chem. Commun., 2016, 52, 8291–8293 RSC.
- B. Sánchez, C. Calderón, R. A. Tapia, R. Contreras and P. R. Campodónico, Front. Chem., 2018, 6, 509 CrossRef.
- F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2010, 75, 767–771 CrossRef.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, Ultrason. Sonochem., 2012, 19, 136–142 CrossRef.
- S. Marullo, F. D'Anna, C. Rizzo and R. Noto, Ultrason. Sonochem., 2015, 23, 317–323 CrossRef CAS.
- L. M. Varela, T. Méndez-Morales, J. Carrete, V. Gómez-González, B. Docampo-Álvarez, L. J. Gallego, O. Cabeza and O. Russina, J. Mol. Liq., 2015, 210, 178–188 CrossRef CAS.
- F. D'Anna, S. Marullo, P. Vitale and R. Noto, ChemPhysChem, 2012, 13, 1877–1884 CrossRef.
- D. Xiao, J. R. Rajian, L. G. Hines, S. Li, R. A. Bartsch and E. L. Quitevis, J. Phys. Chem. B, 2008, 112, 13316–13325 CrossRef CAS.
- S. Marullo, F. D'Anna, P. R. Campodonico and R. Noto, RSC Adv., 2016, 6, 90165–90171 RSC.
- N. D. Khupse and A. Kumar, J. Phys. Chem. A, 2011, 115, 10211–10217 CrossRef CAS.
- A. Manna and A. Kumar, ChemPhysChem, 2014, 15, 3067–3077 CrossRef CAS.
- T. D. Do and A. R. Schmitzer, RSC Adv., 2015, 5, 635–639 RSC.
- S. Siankevich, Z. Fei, R. Scopelliti, G. Laurenczy, S. Katsyuba, N. Yan and P. J. Dyson, ChemSusChem, 2014, 7, 1647–1654 CrossRef CAS.
- F. D'Anna, S. Marullo, P. Vitale, C. Rizzo, P. Lo Meo and R. Noto, Appl. Catal., A, 2014, 482, 287–293 CrossRef.
- S. Marullo, C. Rizzo, A. Meli and F. D'Anna, ACS Sustainable Chem. Eng., 2019, 7, 5818–5826 CrossRef CAS.
- C. Rizzo, F. D'Anna, S. Marullo and R. Noto, J. Org. Chem., 2014, 79, 8678–8683 CrossRef CAS.
- F. D'Anna, H. Q. Nimal Gunaratne, G. Lazzara, R. Noto, C. Rizzo and K. R. Seddon, Org. Biomol. Chem., 2013, 11, 5836–5846 RSC.
- Z.-L. Shen, H.-L. Cheong, Y.-C. Lai, W.-Y. Loo and T.-P. Loh, Green Chem., 2012, 14, 2626–2630 RSC.
- R. Bini, C. Chiappe, V. L. Mestre, C. S. Pomelli and T. Welton, Theor. Chem. Acc., 2009, 123, 347–352 Search PubMed.
- B. List, P. Pojarliev and C. Castello, Org. Lett., 2001, 3, 573–575 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- A. B. Northrup and D. W. MacMillan, Science, 2004, 305, 1752–1755 CrossRef CAS.
- E. J. Sorensen and G. M. Sammis, Science, 2004, 305, 1725–1726 CrossRef CAS.
- J. F. Schneider, C. L. Ladd and S. Bräse, in Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 1, The Royal Society of Chemistry, 2016, pp. 79–119 Search PubMed.
- R. Porcar, M. I. Burguete, P. Lozano, E. Garcia-Verdugo and S. V. Luis, ACS Sustainable Chem. Eng., 2016, 4, 6062–6071 CrossRef CAS.
- L. González, J. Escorihuela, B. Altava, M. I. Burguete and S. V. Luis, Eur. J. Org. Chem., 2014, 5356–5363 CrossRef.
- V. Gauchot and A. R. Schmitzer, J. Org. Chem., 2012, 77, 4917–4923 CrossRef CAS.
- L. Q. Kang, Y. Q. Cai, H. Wang and L. H. Li, Monatsh. Chem., 2014, 145, 337–340 CrossRef CAS.
- C. Wang, X. Liu, M. Yang, H. Ma, P. Yan, J. M. Slattery and Y. Gao, RSC Adv., 2013, 3, 8796–8804 RSC.
- S. R. Roy and A. K. Chakraborti, Org. Lett., 2010, 12, 3866–3869 CrossRef CAS.
- A. Sarkar, S. R. Roy and A. K. Chakraborti, Chem. Commun., 2011, 47, 4538–4540 RSC.
- A. Sarkar, S. Raha Roy, D. Kumar, C. Madaan, S. Rudrawar and A. K. Chakraborti, Org. Biomol. Chem., 2012, 10, 281–286 RSC.
- D. W. Armstrong, L. He and Y.-S. Liu, Anal. Chem., 1999, 71, 3873–3876 CrossRef CAS.
- D. R. MacFarlane and S. A. Forsyth, in Ionic Liquids as Green Solvents, American Chemical Society, 2003, vol. 856, ch. 22, pp. 264–276 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |