
The magnetic-nanoparticle-assisted sensitive detection of nitrated α-syn in blood based on a sensitizing electrochemical layer†
Zhao-huan
Zhang‡
a,
Junjie
Hu‡
b,
Qiang
Chen
c,
Jie
Chen
bc,
Xiaojun
Hu
b,
Kwangnak
Koh
d,
Hongxia
Chen
 *b and
Xiao-hui
Xu
*ef
*b and
Xiao-hui
Xu
*ef
aDepartment of Laboratory Medicine, Changzheng Hospital, Naval Medical University, Shanghai, 200003, PR China
bCenter for Molecular Recognition and Biosensing, School of Life Sciences, Shanghai University, Shanghai 200444, PR China. E-mail: hxchen@shu.edu.cn
cSchool of Medicine, Shanghai University, Shanghai 200444, PR China
dInstitute of General Education, Pusan National University, Busan, 609-735, Republic of Korea
eSchool of Preclinical Medicine, Wannan Medical College, Wuhu 241001, PR China. E-mail: xxhxxh@shu.edu.cn
fSchool of Life Sciences, Shanghai University, Shanghai 200444, PR China
First published on 9th April 2021
Abstract
Parkinson's disease (PD) is the second most prevalent neurodegenerative disease. Nitrated α-synuclein (α-syn) in the blood is a potentially efficient biomarker for PD in its early stages. In this work, an ultrasensitive electrochemical immunosensor was developed for the specific detection of nitrated α-syn. Supramolecule-mediated AuNP composites (GNCs) were modified on the gold electrode as a sensing film to capture anti-nitrated α-syn. Basic characterization studies revealed that GNCs were composed of abundant binding sites and had high conductivity with a large surface area, biocompatibility, and remarkable electrochemical activity. Anti-α-syn-modified magnetic nanoparticles (MNPs) were used as signal amplification tags to construct a sensitive sandwich assay. With a high specific surface area, strong conductivity, and abundant active sites, GNCs as an amplifying matrix can enhance the performance of the immunoassay and obtain preliminary signal amplification. MNPs showed excellent stability and led to a net decrease in the charge-transfer resistance due to their unique spherical structure and high conductivity, resulting in a sensitive electrochemical signal change according to the nitrated α-syn concentration in the sample. Therefore, this simple nitrated α-syn immunoassay with sensitivity and selectivity has potential for practical clinical applications.
1. Introduction
Parkinson's disease (PD) is one of the more common neurodegenerative diseases, with a prevalence of 2–3% in the population over 65 years old.1 To date, the diagnosis of PD relies mostly on clinical features, but by that time, PD patients have lost many dopaminergic neurons and show functional system disability. The time frame between the first events initiating neuron loss and clinical manifestation may be decades.2 There is an increased need to diagnose PD as early as possible, as early diagnosis and treatment may be a key step in ensuring the survival of dopaminergic neurons and promoting improvements in the prognosis of Parkinson's disease. α-Synuclein (α-syn) represents the main component of amyloid aggregates formed during Parkinson's disease. Nitrated α-syn is an undesirable modification associated with oxidative and nitrative damage, and it has been found extensively in Lewy bodies in patients with neurodegenerative diseases such as PD, dementia with Lewy bodies, the Lewy body variant of Alzheimer's disease, and multiple system atrophy.3,4 The nitro group (–NO2) is added to replace a hydrogen atom in the 3′ position of the tyrosine phenolic ring, forming 3-nitrotyrosine. The four Y39, Y125, Y133, and Y136 residues of α-syn are all susceptible to nitration. Our group has demonstrated that nitrated α-syn is neurotoxic through the injection of nitrated α-syn into the substantia nigra pars compacta (SNpc) of rats, which recapitulated many of the pathological features of PD.5 Nitrated α-syn exists in the early stage and is probably a promising biomarker of PD.6 Also, nitrated α-syn is a highly transferable protein, causing an enlarged spreading of α-syn from the affected neuron toward more rostral brain regions.7 Recently, reports showed that the α-syn levels in monocytes from PD patients were similar to those in normal controls, whereas the α-syn nitration levels were significantly higher and correlated with the levels of reactive oxygen species.8 Another report also identified that the levels of Y39-nitrated α-syn in a PD group were significantly elevated compared to those in a control group via examining the nitrated α-syn levels in erythrocytes from 58 PD patients and 30 age-matched healthy individuals.9 As the above-mentioned studies strongly suggest that nitrated α-syn in the blood may be a potential early-stage biomarker for PD, we want to develop a more sensitive and convenient method to detect nitrated α-syn in the blood.Electrochemical sensing is an analytical assay approach for determining target concentrations based on a bio-specific recognition reaction, which converts a biological interaction into an electrical signal, such as current, voltage, or impedance.10 An insulating layer can be formed when target proteins bind to a probe on a modified electrode, which hinders electron transfer and results in the impedance increasing. Based on the impedance variations, a linear equation relating to the target concentration can be drawn for the impedance sensor. Though many impedance biosensors have been fabricated, the sensitive detection of trace targets in the blood still presents a huge challenge. To overcome these limits, two methods can be applied to enhance the impedance sensor signal. One is to enhance the substrate conductivity before target capture.11 The other is to design sandwich signal amplification strategies to increase the impedance.12
Currently, various kinds of substrates, such as polyamide film,13 graphene,14 molybdenum disulfide, and metal organic frameworks (MOFs), are used to make sensitive impedance sensors.15,16 Due to the complexity of fabrication, the need for multi-step production, excessive background signals, and the easily destroyed activities of immune molecules (aptamers, peptides, and antibodies), the above-mentioned sensing substrates suffer from lower repeatability and sensitivity. Given the high conductivity, large specific surface area, and excellent biocompatibility of gold nanoparticles (AuNPs), they have great potential for use in the construction of electrochemical impedance sensors.17–19 Recently, supramolecule-complex-modified nanomaterials have combined the properties of nanomaterials and molecular recognition, and they have been widely used in drug delivery, imaging, and sensing.20 In addition, the mild nature of the supramolecules on the surface of AuNPs will allow the native activity of the immobilized immune molecules to be maintained. In our previous study, supramolecule-mediated AuNPs can self-assemble to form AuNP composites (GNCs) after the addition of diamine guests, with a high specific surface area, strong conductivity, and various active sites.21 The one-step modification process not only reduces the time cost but also prevents poor repeatability caused by tedious operations. Therefore, GNCs as an amplifying matrix can enhance the performance of the immunoassay and obtain preliminary signal amplification. Further signal amplification strategies, such as sandwich assays, play a vital role in the fabrication of precise impedance sensors. Magnetic nanoparticles (MNPs) exhibit interesting properties, such as a high specific surface area, high biocompatibility, excellent dispersibility, and efficient separation abilities, which have significant applications in the field of sensing.22,23 Zheng's group reported that Fe3O4/polyaniline nanoparticles show increased electrical conductivity, which is caused by a circuit formed as a result of contact between some ions.24 The formation of the corresponding circuit enhanced the electronic hopping process between the Fe2+ and Fe3+ ions of MNPs; this can increase the electrical conductivity when utilized on the electrode interface, which paves the way to applying MNPs in electrochemical sensing.25
In short, an efficient sandwich assay has been successfully designed for the sensitive measurement of nitrated α-syn. This is, to the best of our knowledge, the first time that a nitrated α-syn immunosensor utilizing GNCs has been developed as a sensing platform, fixing anti-nitro-α-syn and anti-α-syn-modified MNPs as signal amplifiers (Fig. 1). GNCs provide a fast electron transfer rate and large specific surface area, and the incorporation of supramolecules offers abundant binding sites for the immobilized antibodies through host–guest interactions, ensuring the activity of anti-nitro-α-syn via noncovalent interaction forces.26 Anti-α-syn-modified MNPs have been used as signal amplification tags, and their unique spherical structure and high conductivity resulted in excellent stability and a net change of the charge-transfer resistance (Rct). Moreover, the MNPs in the sandwich assay not only provide hydrophilic abilities, which can reduce the nonspecific binding, but also the alignment of MNPs in a specific direction induced by the electric field promotes the flow of electrons, resulting in a significant decrease in Rct.27 This allows the constructed electrochemical biosensor to output enlarged impedance signal changes during target detection. Antibody-modified MNPs integrate the specificity of target recognition and the ability to promote electron transfer, which can effectively improve the detection sensitivity of the immunosensor to meet clinical application requirements. Finally, the specially developed impedance-based nitrated α-syn detection platform has been applied to practical clinical sample measurements. The results showed that this proposed nitro-α-syn sensor possesses tremendous potential for use in the clinical diagnosis and personalized treatment of PD, and related scientific research.
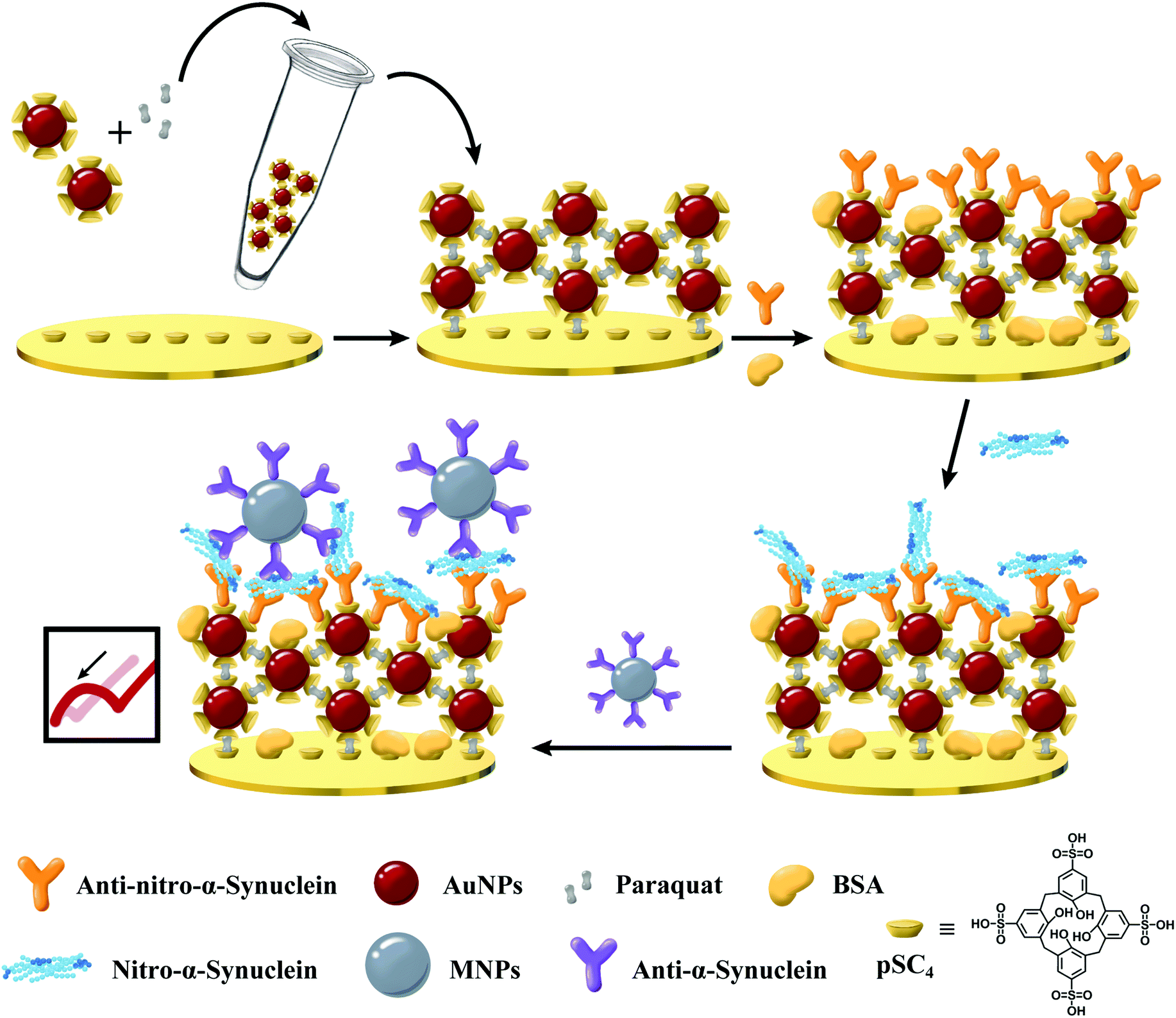 | ||
| Fig. 1 A schematic view of the sensitizing layer and MNP-based dual-signal amplification electrochemical immunosensor for detecting nitrated α-syn. | ||
2. Experimental
2.1. Reagents and apparatus
Paraquat (PQ), tetraethyl orthosilicate (TEOS), aminopropyltriethoxysilane (APTES), glutaric anhydride, 1-ethyl-3-(3-dimethylaminopropyl)ethylcarbodiimide hydrochloride (EDC), N-hydroxysuccinimide (NHS), hydrogen tetrachloroaurate trihydrate (HAuCl4·3H2O), and sodium borohydride (NaBH4) were purchased from Sigma–Aldrich, Inc. (Shanghai, China). para-Sulfonatocalix[4]arene (pSC4) was purchased from TCI Development Co., Ltd (Shanghai, China). Trisodium citrate dihydrate, potassium ferricyanide, 3-morpholinosydnonimine (SIN-1), and bovine serum albumin (BSA) were acquired from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Anti-α-syn antibodies, anti-nitro-α-syn (Tyr39) antibodies, and α-syn were purchased from Abcam (Shanghai, China). All solutions were treated with deionized water purified using a Milli-Q distillation system to a determined electrical resistance of 18.2 MΩ cm (Barnstead, USA).Transmission electron microscopy (TEM) images were recorded using a JEM-2010F microscope (JEOL, Japan). The modification of materials was studied using atomic force microscopy (Agilent Technologies Co Ltd, USA) in tapping mode (scan rate: 325 kHz). UV-vis spectra were recorded using a Shimadzu UV-2450 PC UV-vis spectrophotometer (Shimadzu, Japan) with a quartz cuvette, having a volume of 50 μL and a path length of 10 mm. The potential change and dynamic diameter were observed using Malvern Nano Zetasizer apparatus (Malvern Panalytical, England). Fourier-transform infrared (FT-IR) spectra were obtained using Bruker 70 apparatus (Bruker, Germany). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) measurements were obtained using an Autolab PGSTAT128N system (Metrohm Autolab, Switzerland) with a conventional three-electrode set-up.
2.2. Functionalization of magnetic nanoparticles (MNPs)
The synthesis of MNPs is based on a previous work.28 Briefly, 3 mL of 25 mg mL−1 Fe3O4 NPs and 1 mL of 28% ammonia water were injected into 20 mL of ethanol and sonicated for 10 min. 125 μL of TEOS was added and then the mixture was mechanically stirred at room temperature for 4 h. 750 μL of APTES was added in drop-wise, and stirring was continued for another 4 h. Following reflux at 90 °C for 1 h, the product was washed with ethanol and dried at 60 °C under vacuum for 6 h to gain amino-functionalized MNPs. 10 mL of 2 mg mL−1 amino-functionalized MNPs in ethanol was mixed with 10 mL of 0.2 M glutaric anhydride and stirred at 200 rpm for 24 h to synthesize carboxyl-functionalized MNPs.1 mL of 10 mg mL−1 carboxyl-functionalized MNPs was mixed with 1 mL of 20 mg mL−1 EDC and 1 mL of 10 mg mL−1 NHS thoroughly, and this was reacted for 1 h. After applying an external magnet for magnetic separation, the product was washed 3 times with PBS. The above solution was injected into 714 μL of 14 μg mL−1 anti-α-syn and reacted at room temperature. Afterwards, magnetic separation was performed, and the product was washed with PBS to obtain MNPs–Ab.
2.3. Synthesis of nitrated α-syn
1 mL of 50 μM α-syn solution was mixed with 1 mL of 100 μM SIN-1 at 37 °C for 30 min. After that, it was dialyzed in PBS for 24–48 h. It is worth noting that PBS should be replaced several times during this process, and unreacted SIN-1 was removed using an ultrafiltration membrane which can retain 5 kDa proteins.292.4. Preparation of GNCs
AuNPs were synthesized via the classic method of the citrate reduction of HAuCl4·3H2O. Herein, all glassware used for synthesis was thoroughly soaked in aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1, v/v) for 30 min and rinsed with deionized water (DW). 1 mL of 25.39 mM HAuCl4·3H2O was pipetted into 94.5 mL of boiling DW for 5 min, and then 3.5 mL of 10 mg mL−1 sodium citrate was quickly added to the boiling solution, which was stirred for 20 min. Heating was stopped when the color changed from light yellow to wine red, and the reaction was continued for another 40 min. After cooling the solution to room temperature, AuNPs were obtained and stored at 4 °C for subsequent experiments.
:HNO3 = 3:1, v/v) for 30 min and rinsed with deionized water (DW). 1 mL of 25.39 mM HAuCl4·3H2O was pipetted into 94.5 mL of boiling DW for 5 min, and then 3.5 mL of 10 mg mL−1 sodium citrate was quickly added to the boiling solution, which was stirred for 20 min. Heating was stopped when the color changed from light yellow to wine red, and the reaction was continued for another 40 min. After cooling the solution to room temperature, AuNPs were obtained and stored at 4 °C for subsequent experiments.
GNCs should be prepared for immediate use in each experiment, and the synthesis method is as follows. After mixing freshly prepared AuNPs and DW at a ratio of 1:1, 0.5 mM PQ and diluted AuNPs were mixed at a ratio of 1:4 and reacted for 30 min for subsequent experiments. The GNC structure formed from PQ-mediated AuNPs was studied based on TEM images.
2.5. Modification of gold electrodes
50 μL of 10 mM pSC4 was dropped onto the surface of gold electrodes and left overnight. After rinsing with DW, GNCs were added to form a sensitized layer. 50 μL of 10 μg mL−1 anti-nitro-α-syn (Tyr39) antibodies were added and modified was carried out for 1.5 h. 0.1 mg mL−1 BSA was utilized to avoid non-specific binding. Thereafter, 50 μL of 280 ng mL−1 nitro-α-syn was modified on the surface of the electrode for 1.5 hours, and then 50 μL of 50 μg mL−1 MNPs–Ab was reacted on the surface of the electrode for 1.5 h. The gold electrode modification process was studied using atomic force microscopy in tapping mode, and the scan rate was set at 325 kHz.In order to study the influence of the anti-nitro-α-syn concentration and modification time on the EIS changes of the electrochemical surface, after modifying with GNCs, different concentrations of anti-nitro-α-syn (2 μg mL−1, 4 μg mL−1, 6 μg mL−1, 8 μg mL−1, and 10 μg mL−1) were used to modify the surface of the gold electrode. Electrochemical measurements were performed every 0.5 h for a total of six measurements.
2.6. Sensor measurements
Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) measurements were performed using a typical three-electrode cell (WE, CE, and RE). The CV and EIS experiments using this electrochemical sensor were carried out in 6 mL of [Fe(CN)6]3−/4− solution. The CV scanning potential was from −0.8 V to 0.8 V, and the scanning rate was 50 mV s−1. The frequency range of EIS measurements was set from 0.1 Hz to 100 kHz. The working electrode was rinsed carefully with DW after each modification, and then electrochemical detection was performed.In order to conduct research into the sensor selectivity, 2.8 μg mL−1 dopamine (DA), histidine (His), Siglec 15, programmed death ligand 1 (PD-L1), and α-synuclein were modified on the surface of the gold electrode. The EIS changes were compared with 280 ng mL−1 nitro-α-synuclein.
2.7. Clinical serum sample analysis
The serum samples of patients at different stages of disease and healthy donors were gathered from Changzheng Hospital in Shanghai, China. All experiments were performed in accordance with WHO guidelines (clinical use of blood) and approved by the Ethics Committee of Shanghai Changzheng Hospital, affiliated to Naval Medical University. Informed consent was obtained from all human subjects prior to the collection of blood samples. The collected serum samples were centrifuged at 1500 rpm for 20 min to obtain the crude supernatant. The crude supernatant was further centrifuged at 1200 rpm for 20 min to obtain the supernatant with the centrifuged product, and this was stored at −20 °C for subsequent use. After being diluted ten-fold with PBS, 50 μL of the sample was dropped onto the surface of the gold electrode and it was modified for 1.5 h at room temperature for clinical sample testing.3. Results and discussion
3.1. Characterization of the nanomaterials
The characterization results relating to AuNPs and MNPs are illustrated in Fig. 2. The diameter of AuNPs, with good uniformity and dispersion, was approximately 8 ± 1.5 nm (Fig. 2A). Based on the host–guest recognition interactions between pSC4 and PQ, the TEM image of GNCs after AuNP self-assembly showed a spider-web-like network structure (Fig. 2B), which was continuous, tight, and staggered. The spider-web-like network structure could provide a larger specific surface area for the loading of immune probe molecules, which is beneficial for improving the sensitivity of the sensor. To further prove the successful synthesis of GNCs, the host–guest recognition interactions between PQ and pSC4–AuNPs were also measured via UV-vis spectroscopy.26 As shown in Fig. 2C, the maximum absorption of the synthesized pSC4–AuNPs appeared at 525 nm. After mixing PQ with pSC4–AuNPs, the absorption peak at 525 nm decreased, and a typical aggregation peak corresponding to interparticle-coupled plasmon excitons appeared at 650 nm. As shown in Fig. S1,† upon the addition of PQ, DLS studies showed a change in the particle size from 33.83 nm to 498.2 nm, and the ζ potential also changed from −31.1 mV to −14.9 mV. These results revealed that interactions occur between PQ and pSC4–AuNPs, and the successful synthesis of GNCs is achieved for subsequent sensing interface modification.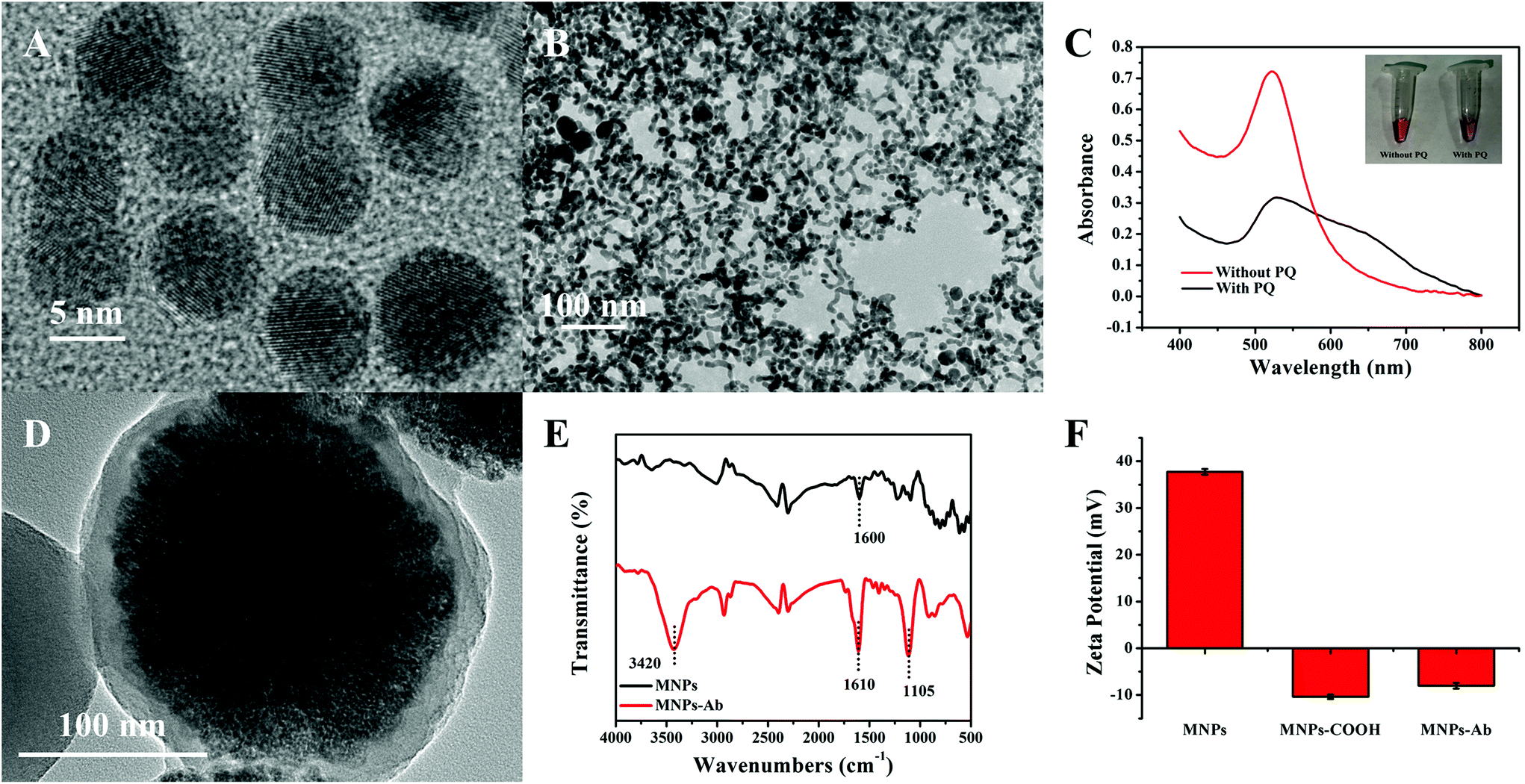 | ||
| Fig. 2 TEM images of (A) pSC4–AuNPs and (B) GNCs. (C) UV–vis spectra of pSC4–AuNPs with (red trace) and without (black trace) PQ. (D) A TEM image showing MNPs encapsulated in a SiO2 shell ranging between approximately 200 and 220 nm in diameter. (E) FT-IR spectra before (black) and after (red) the anti-α-syn modification of MNPs. (F) The Z-potential values of MNPs, MNPs–COOH, and MNPs–Ab. | ||
The prepared MNPs were characterized via TEM, as shown in Fig. 2D. The overall shape of the particle was a uniformly dispersed spherical structure. The particle size was 210 ± 10 nm, including a magnetic core and an encapsulating SiO2 shell (about 10 nm), facilitating chemical modification on the surface and imparting specific functionality. After the carboxyl functionalization of MNPs, specific antibodies were chemically linked to MNPs through EDC/NHS activation. The FT-IR spectrum of the modified MNPs in Fig. 2E shows peaks related to the N–H bond at 3420 cm−1, and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond peak shifted from 1600 cm−1 to 1610 cm−1, which confirmed the formation of peptide bonds. The C–N bond peak at 1105 cm−1 also indicated the successful combination of anti-α-syn and MNPs.30 Finally, the ζ potential of MNPs in water was measured to be 38.5 mV, while that of carboxyl-functionalized MNPs was −10.3 mV. Coupling MNPs–COOH with positively charged antibodies led to a change in the potential from −10.3 mV to −7.8 mV (Fig. 2F). These results confirmed the successful functionalization and modification of MNPs to facilitate nitrated α-syn signal amplification.
O bond peak shifted from 1600 cm−1 to 1610 cm−1, which confirmed the formation of peptide bonds. The C–N bond peak at 1105 cm−1 also indicated the successful combination of anti-α-syn and MNPs.30 Finally, the ζ potential of MNPs in water was measured to be 38.5 mV, while that of carboxyl-functionalized MNPs was −10.3 mV. Coupling MNPs–COOH with positively charged antibodies led to a change in the potential from −10.3 mV to −7.8 mV (Fig. 2F). These results confirmed the successful functionalization and modification of MNPs to facilitate nitrated α-syn signal amplification.
3.2. Feasibility of the biosensor
In order to investigate the response process of the strategy more intuitively, real-time dynamic monitoring was performed using SPR (Fig. 3A). The modification of pSC4 and GNCs on the gold chip resulted in increases in the SPR angles of 89 m° and 142 m°, respectively. These results indicated that the GNCs effectively modified the surface of the gold chip as a sensing substrate. The binding of anti-nitro-α-syn to pSC4 of GNCs on the gold-chip surface resulted in a significant increase (194 m°) in the SPR angle. BSA was used for blocking non-specific adsorption and it resulted in a change similar to the SPR angle shift caused by antibody modification. During this process, MNPs functionalized with anti-α-syn were bound with analytes in complex matrices and then efficiently separated using an external magnet. Finally, the MNP complexes were injected onto the gold chip surface. This coupling resulted in a considerable 78 m° SPR angle change, which demonstrated the successful binding of analytes and MNPs–Ab. Hence, these results illustrate that this detection strategy has good feasibility.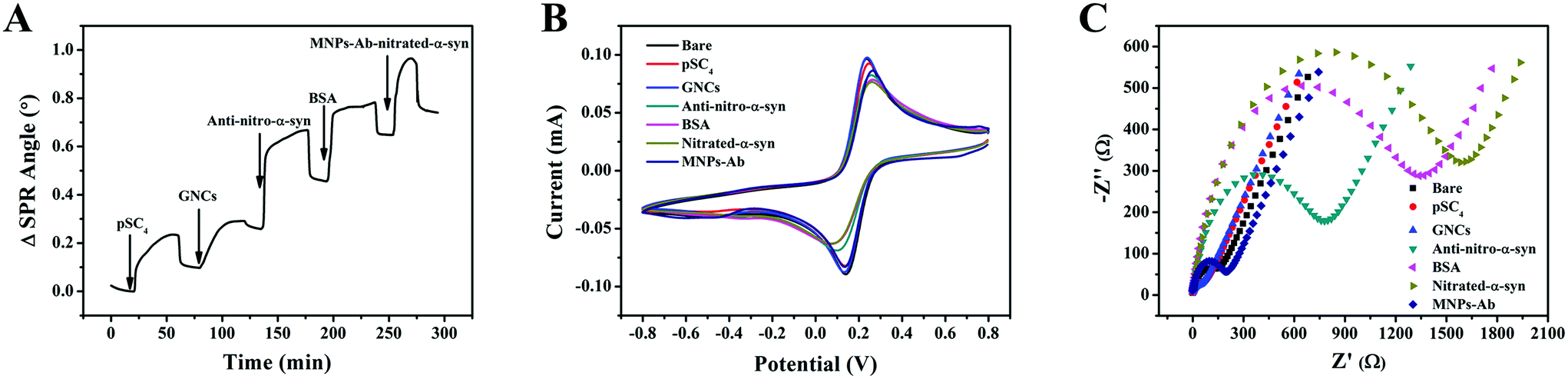 | ||
| Fig. 3 (A) A real-time SPR sensorgram of the different modification steps of the gold chip. (B) CV and (C) EIS data from the stepwise modification of a gold electrode in PBS (pH 7.4) containing 5.0 mM [Fe(CN)6]3−/4−. | ||
CV and EIS studies were performed to characterize the step-by-step modification process of the constructed sensor, as shown in Fig. 3B and C. Due to the great electron transfer capabilities of GNCs, they exhibited better sensing performance than the bare gold electrode, which includes a higher CV peak current and lower interfacial electronic resistance.31 Given the poor conductivity of antibodies and proteins, the CV peak current decreased when the value of EIS impedance increased after anti-nitro-α-syn, BSA and nitrated α-syn modification. Due to better electronic conductivity and the direction effect in the electric field, MNPs promoted ion exchange at the interface and resulted in a significant decrease in the impedance value.32 In summary, these results demonstrated the stepwise modification of the electrode interface.
Afterwards, AFM was used to characterize the successful modification of nanomaterials onto the surface of the gold chip. Fig. 4A and B depict 2D images and typical cross-sectional line drawings before and after modification with GNCs, respectively. The bare gold chip presented good uniformity, and the relative surface roughness was less than 3 nm. However, as seen in Fig. 4B, after the incorporation of GNCs and proteins, the height of the surface increased to an average of 40 nm, which corresponded to the size of three layers of AuNPs. After further interaction with MNPs–Ab, MNPs were modified onto the chip, as shown in Fig. 4C, and the surface exhibited higher roughness, with a height of around 110 nm. Therefore, AFM image results confirmed the feasibility of the amplified signal and sensing platform based on MNPs.
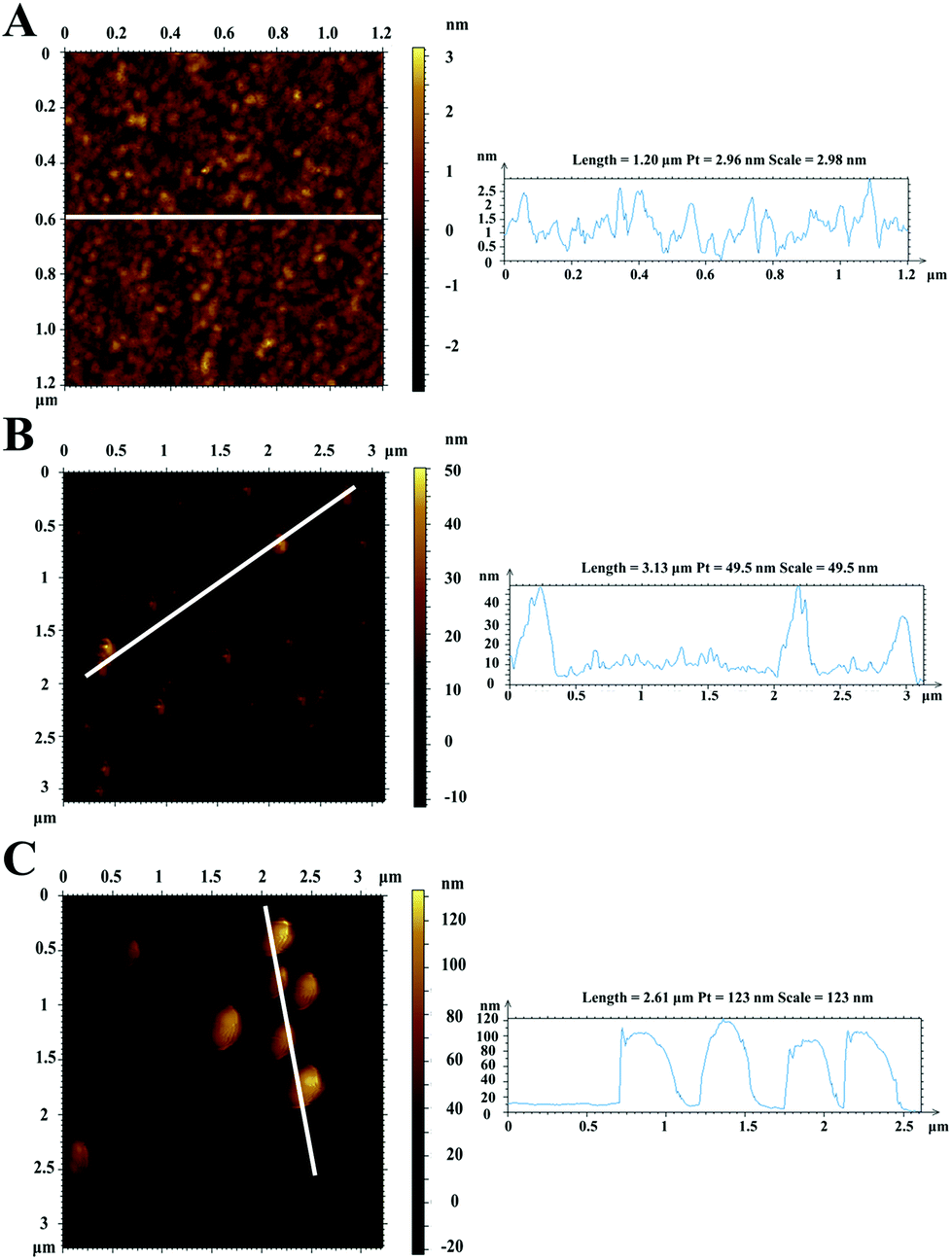 | ||
| Fig. 4 2D AFM images and line profiles of the bare gold chip (A), GNC- and protein-functionalized gold film (B), and the MNPs–Ab modified gold surface (C). | ||
3.3. Immunosensor amplifying matrix analysis
EIS was used to monitor the supramolecular assembly of GNCs mediated by different concentrations of PQ on the gold surface (Fig. 5). One of the most commonly used electrochemical sensitization strategies is to modify gold electrodes with materials with a high specific surface area, such as MOFs.33,34 MOFs have been used as a basal layer to increase the binding of immune molecules, given their porous crystal structures, high specific surface areas, and rich compositions. However, the improvement of sensitivity for electrochemistry is limited due to the inherent deficiencies of MOFs, including poor electron transfer abilities, significantly increased impedance values, and instability in aqueous environments. The gold electrode modified with GNCs exhibited a higher CV peak current, and the interfacial electronic resistance also decreased. It was confirmed that GNCs not only increased the specific surface area and created more active sites, but they also obviously facilitated nanoparticle-mediated electron transfer.35 As shown in Fig. 5A, the electron-transfer resistance decreased as the concentration of PQ increased from 0.1 mM to 10 mM because of the host–guest recognition resolved gold network. A gradual increase in PQ concentration may lead to an increase in the content of pSC4–AuNPs in the GNCs, and the interfacial electronic resistance significantly decreased. Excessive PQ may cause some of the calixarene cavities on the surface of the GNCs to be occupied by PQ, and the negative interface correspondingly inhibited the redox reaction of [Fe(CN)6]3−/4−, thus increasing the Rct value (Fig. 5B). Therefore, GNCs mediated using 1 mM PQ may form the most conductive amplifying matrix and are used in the following experiments.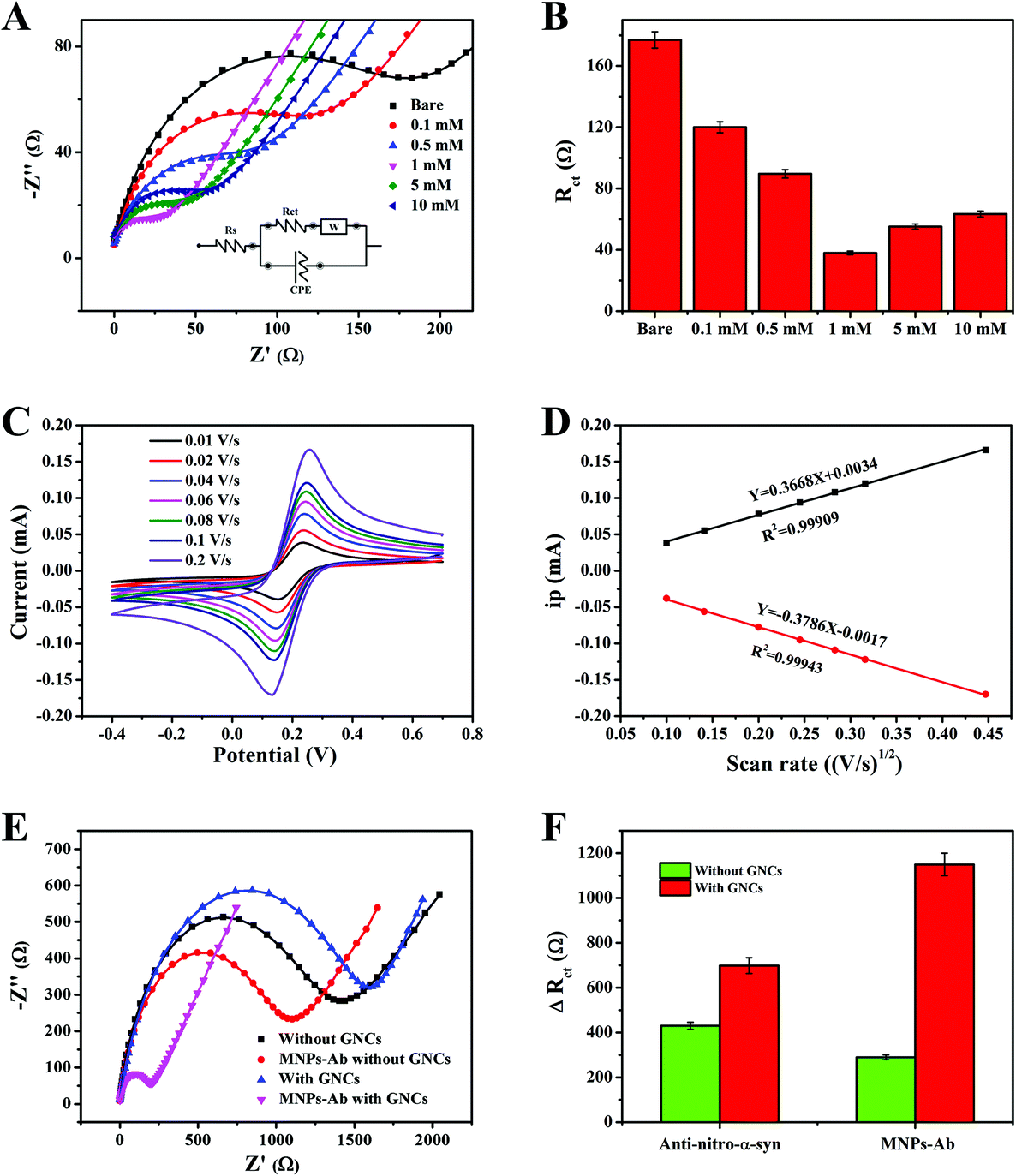 | ||
| Fig. 5 (A) EIS measurements for GNCs on gold electrodes formed via PQ-mediated self-assembly from pSC4–AuNPs with different concentrations of PQ. (B) Histograms of the EIS values obtained from (A). (C) The diffusion control process for GNC-modified electrodes formed from 1 mM PQ and pSC4–AuNPs self-assembly, and (D) the corresponding calibration curves. (E) EIS spectra for MNPs–Ab on electrodes constructed with and without GNC modification. (F) A histogram of EIS changes caused by anti-α-syn and MNP-Ab on electrodes with and without GNC modification. | ||
Fig. 5C shows CV studies of GNCs carried out at different scan rates (0.01–0.2 V s−1) in ferricyanide solution, which were recorded to calculate the effective electrochemical active surface area of the modified electrode. Evidently, the peak current value increases upon elevating the scan rate. Most significantly, a linear relationship was established between the peak current values of the cathode and anode versus the square root of the scan rate; the R2 values were 0.99943 and 0.99909 for the cathode and anode, respectively. The above results demonstrated that the redox reaction of the amplifying matrix involved a diffusion-controlled process.36 The corresponding data from the bare electrode are illustrated in Fig. S2.† The fitting curves of the anode peak and the cathode peak (Fig. 5D) have the forms: ipa = 0.3668V1/2 + 0.0034; and ipc = −0.3786V1/2 − 0.0017, respectively. The electrochemical surface areas were calculated via the Randles–Sevcik equation:37
| ip = 2.69 × 105D1/2V1/2n3/2AC | (1) |
The number of immune probes can affect the subsequent capture of target and signal amplification molecules.39 The number of immuno-probes on the substrate surface should be considered in order to make an effective biosensor. Anti-nitrated-α-syn was utilized as a model probe to study immune molecule adsorption on the GNC-matrix-modified interface. Fig. 5E showed the sensor signal changes compared between electrodes constructed with and without GNC modification. The signal after anti-nitrated-α-syn immobilization on the GNC-modified immuno-probe was 1.6 times that on a bare gold electrode, which may be due to the larger specific surface area and more abundant calixarene cavities of the GNCs (Fig. 5F). In addition, the signal relating to MNPs–Ab showed a further enhancement in difference when in the presence and absence of GNCs, and the signal change was 5 times larger than the signal change caused by nitrated α-syn on a bare electrode. The signal difference in the presence of GNCs was increased 3.8-fold compared to in the absence of GNCs. The extremely large signal changes caused by MNPs may be related to their alignment under an electric field and the electronic hopping process between Fe2+ and Fe3+ ions.27,40 Hence, with two-step signal amplification, the fabricated EIS immunosensor provided a significant gain in sensor sensitivity.
3.4. Optimization of the experimental conditions
Several experimental conditions of the bioassay were optimized to achieve maximum sensing sensitivity. The following parameters were optimized: (a) the reaction time during GNC formation, (b) the concentration of anti-nitro-α-syn, and (c) the anti-nitro-α-syn incubation time. We investigated the effects of the reaction time between AuNPs and PQ using UV-vis absorption spectroscopy. The aggregated state was represented by A600/A520; the larger the value of A600/A520, the higher the level of aggregation.41 As shown in Fig. 6A, in the first 22 min the value of A600/A520 rose rapidly, and then it reached a plateau, indicating that AuNPs and PQ completely reacted after 22 min. Therefore, we chose 22 min as the reaction time for subsequent experiments. The concentration and incubation time of anti-nitro-α-syn were optimized based on impedance spectroscopy. Rct gradually increased with increasing anti-nitro-α-syn concentration and remained stable after a concentration of 8 μg mL−1 was reached (Fig. 6B). Hence, the best concentration of anti-nitro-α-syn in this experiment was 8 μg mL−1. Rct after 2.5 h was more than 90% of that after 3 h, so the amount of surface anti-nitro-α-syn binding was not very different, as shown in Fig. 6C. In order to cut the time required for a single test, 2.5 h was selected as the anti-nitro-α-syn reaction time for subsequent experiments.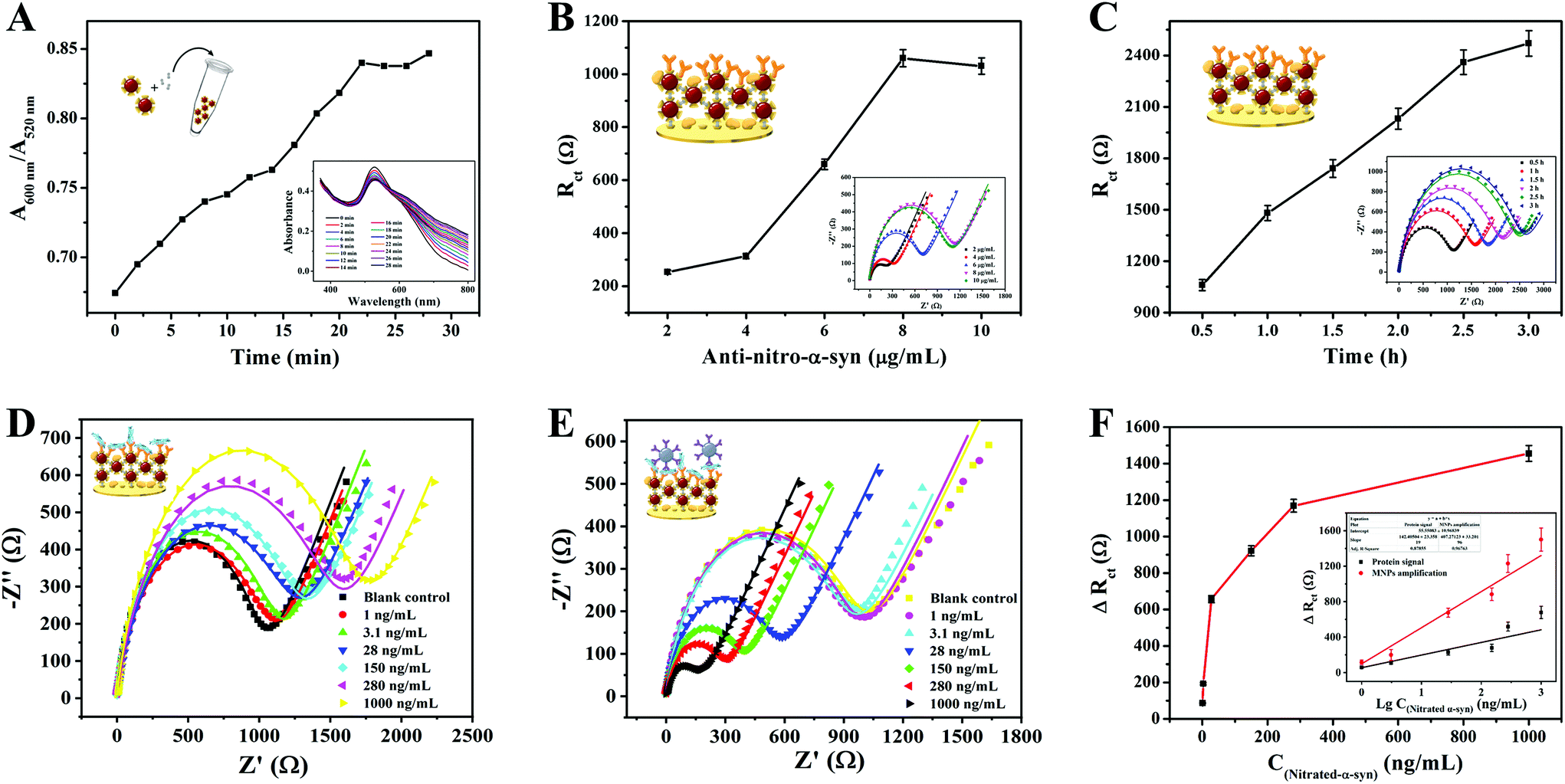 | ||
| Fig. 6 Optimization of the experimental parameters: (A) the reaction time of GNC formation, (B) the concentration of anti-nitro-α-syn, and (C) the anti-nitro-α-syn incubation time; the inset of (A) shows the UV-vis absorption spectra for GNCs after different reaction times, and the insets of (B) and (C) show EIS diagrams for anti-nitro-α-syn at different concentrations and for different incubation times (8 μg mL−1). (D) EIS diagrams for different nitrated α-syn concentrations. (E) EIS diagrams of MNPs–Ab bound with the electrodes from (D). (F) The resistance response vs. the concentration of nitrated α-syn; inset: the linear relationship between the resistance response and the logarithm of the nitrated α-syn concentration. | ||
3.5. Sensor sensitivity for nitrated α-syn detection
Under the abovementioned optimized experimental conditions, the analysis performance of the constructed biosensor was tested via measuring the EIS responses to α-syn at different concentrations (1 ng mL−1–1000 ng mL−1) in 5 mM [Fe(CN)6]3−/4−, as shown in Fig. 6D–E. As the second step of the signal amplification strategy, MNPs–Ab had a more significant effect on ΔRct than nitrated α-syn. The ΔRct values induced by MNPs–Ab bound to different concentrations of nitrated α-syn are shown in Fig. 6F. Furthermore, the relationships between the ΔRct responses before and after MNPs–Ab modification and the logarithmic values of the nitrated α-syn concentrations (lgC nitrated α-syn) are displayed in the inset of Fig. 6F. The signal was amplified about 2.5-fold after combining with MNPs–Ab compared with the signal change from the protein alone, achieving higher detection sensitivity and a lower detection limit. The linear equation of MNPs–Ab was ΔRct (Ω) = 459.54lgC + 14.99 (R2 = 0.9676) in the linear range of 1 ng mL−1–1000 ng mL−1. The calculated limit of detection (LOD) was 310 pg mL−1 (S/N = 3) based on three standard deviations. Relative to other earlier reported biosensors for neurodegenerative disease detection (Table S1†), these results demonstrate the lower LOD and superior analytical performance of the designed sensing platform. Based on the two-step signal amplification strategy utilizing GNCs and MNPs, the sensing platform can achieve more sensitive quantification than immunohistochemistry, which is the gold standard for nitrated α-syn detection.3,4,6 This sensing strategy based on dual-recognition also showed greater clinical application value than most sensors developed for neurodegenerative diseases. The signals obtained using the GNC-modified electrode were amplified about 5-fold compared with the bare electrode, which was attributed to the larger specific surface area of GNC, which could capture more immuno-probes, and the higher conductivity of the spider-web-like structure (Fig. S3†).42
3.6. Selectivity and stability of the constructed sensor
In order to verify the selectivity of our method, five possible interfering substances were tested, including PD-L1, Siglec 15, DA, His, and α-syn (multiple interferents widely present in serum), as shown in Fig. S4.† Compared with the high ΔRct obtained in response to nitrated α-syn, all five interferents displayed negligible and similar signal changes (Fig. 7A). It is notable that the concentrations of interferents were far greater than that of nitrated α-syn. The favorable selectivity arises from utilizing anti-nitrated-α-syn as the Tyr39 specific recognition element toward nitrated α-syn, which enhanced the specificity of recognition.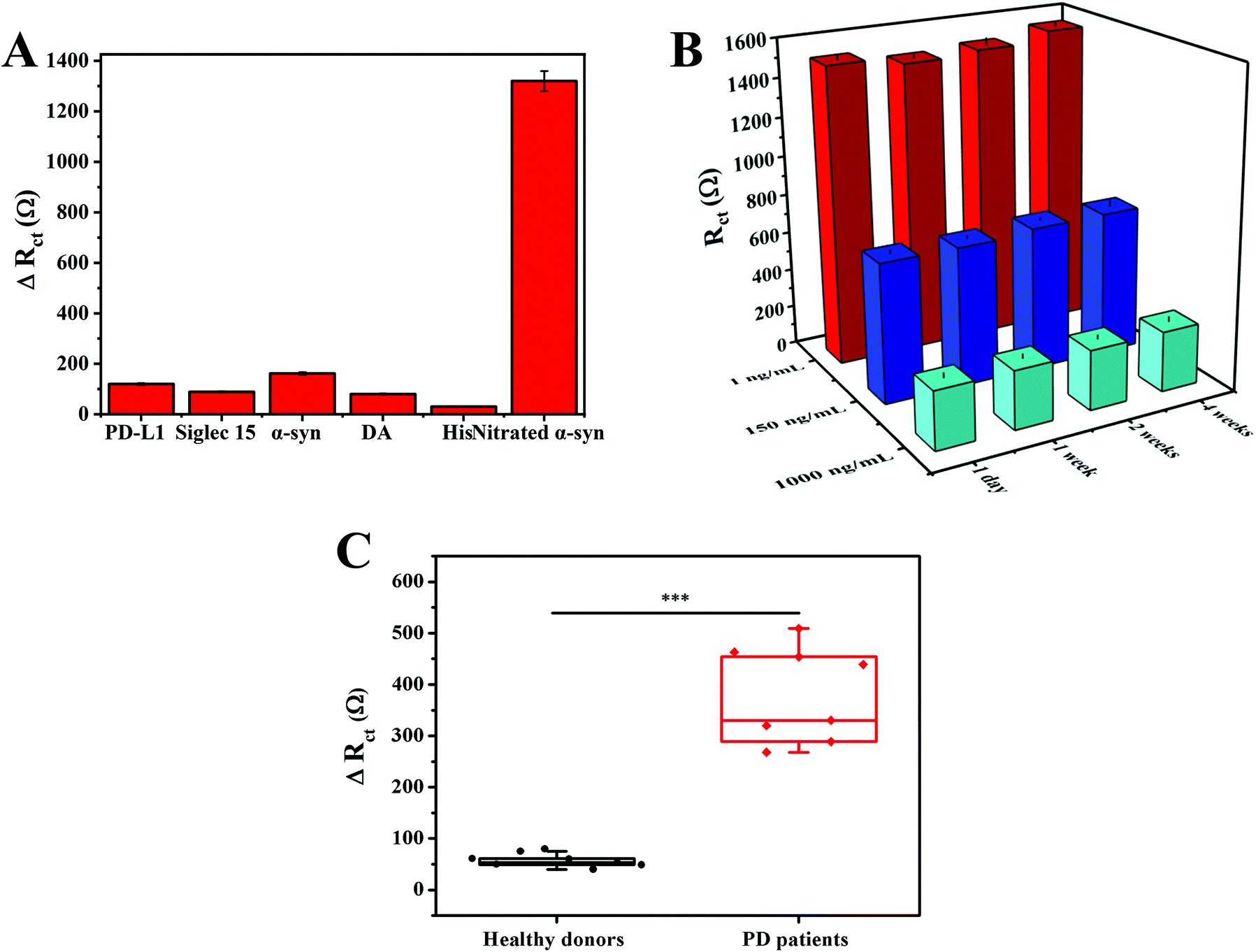 | ||
| Fig. 7 (A) ΔRct responses of the assay toward different interferents: PD-L1, Siglec 15, α-syn, DA, and His, all at concentrations of 2 μg mL−1 (nitrated α-syn was 200 ng mL−1). (B) The long-term stability of the biosensor for measuring different concentrations of nitrated α-syn. (C) A comparison of the ΔRct responses caused by serum nitrated α-syn detected via the electrochemical sensor between healthy donors (n = 8) and PD patients (n = 8). Statistical comparisons between the two groups were performed via t-tests. | ||
To evaluate the stability of the immunoassay, different sensors were stored in a dry and dark environment at 4 °C for a certain period of time, and then the corresponding impedance signals were detected in potassium ferricyanide solution (Fig. S5†). The impedance value was 103.3% of the original impedance value after one month of storage (Fig. 7B). These data indicate that the sensor has good stability.
3.7. Nitrated α-syn detection in real samples
First, the potential utility of the immunoassay was assessed utilizing the standard addition method.43 Three concentrations of nitrated α-syn were added to 50% real serum samples for detection. The results illustrated that the average recovery rate and RSD values of the biosensor were 90.40%–94.70%, and 2.41%–6.31%, respectively (Table S2†). The good practicability of the sensing strategy was proved.We then tested the applicability of the biosensor via measuring the levels of nitrated α-syn in diluted serum samples from healthy donors and PD patients (all donor information is shown in Table S3†). As shown in Fig. S6,† the ΔRct values of different clinical samples were compared. Due to the significantly improved sensitivity of the immunoassay caused by the GNCs and MNPs, we found that the ΔRct responses due to the serum nitrated α-syn concentrations of healthy blood donors and PD patients were significantly different (t-test, P < 0.001), as shown in Fig. 7C. These results confirm that this electrochemical immunoassay has potential clinical application value, and it could serve to assist existing detection methods and guide precision medicine.
4. Conclusions
In summary, a sandwich-type electrochemical immunoassay for the detection of nitrated α-syn was developed using GNCs as a sensing platform and MNPs–Ab as a signal amplification source. The high conductivity, large specific surface area, good stability, and supermolecule surface of GNCs provided suitable binding sites for immobilized antibodies, consequently resulting in higher specificity for nitrated α-syn detection. The addition of MNPs–Ab further enhanced the sensing advantages of the amplifying matrix. Under the best experimental conditions, the sensing platform showed an excellent and wide linear detection range (1–1000 ng mL−1), a low detection limit (310 pg mL−1), and good selectivity, stability, and feasibility. Based on the experimental results from the analysis of clinical samples, the developed immunosensor has great potential for clinical applications relating to nitrated α-syn. However, there are still some issues that need to be optimized, such as the simplification of the sensor preparation method and the portability of the device, which are critical for advancing the pace of clinical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 61875114 and 62005156).References
- W. Poewe, K. Seppi, C. M. Tanner, G. M. Halliday, P. Brundin, J. Volkmann, A.-E. Schrag and A. E. Lang, Nat. Rev. Dis. Primers, 2017, 3, 17013 CrossRef PubMed.
- M. J. Armstrong and M. S. Okun, JAMA, J. Am. Med. Assoc., 2020, 323, 548–560 CrossRef PubMed.
- B. I. Giasson, J. E. Duda, I. V. J. Murray, Q. Chen, J. M. Souza, H. I. Hurtig, H. Ischiropoulos, J. Q. Trojanowski and V. M.-Y. Lee, Science, 2000, 290, 985–989 CrossRef CAS PubMed.
- G. G. Kovacs, G. Botond and H. Budka, Acta Neuropathol., 2010, 119, 389–408 CrossRef CAS PubMed.
- Z. Yu, X. Xu, Z. Xiang, J. Zhou, Z. Zhang, C. Hu and C. He, PLoS One, 2010, 5, e9956 CrossRef PubMed.
- L. Y. Ma, L. Y. Gao, X. Li, H. Z. Ma and T. Feng, Neurosci. Lett., 2019, 704, 45–49 CrossRef CAS PubMed.
- R. E. Musgrove, M. Helwig, E.-J. Bae, H. Aboutalebi, S.-J. Lee, A. Ulusoy and D. A. D. Monte, J. Clin. Invest., 2019, 129, 3738–3753 CrossRef PubMed.
- A. Prigione, F. Piazza, L. Brighina, B. Begni, A. Galbussera, J. C. DiFrancesco, S. Andreoni, R. Piolti and C. Ferrarese, Neurosci. Lett., 2010, 477, 6–10 CrossRef CAS PubMed.
- H. V. Miranda, R. Cássio, L. Correia-Guedes, M. A. Gomes, A. Chegão, E. Miranda, T. Soares, M. Coelho, M. M. Rosa, J. J. Ferreira and T. F. Outeiro, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- U. Guth, W. Vonau and J. Zosel, Meas. Sci. Technol., 2009, 20, 042002 CrossRef.
- S. C. Barman, M. A. Zahed, M. Sharifuzzaman, S. G. Ko, H. Yoon, J. S. Nah, X. Xuan and J. Y. Park, Adv. Funct. Mater., 2020, 30, 1907297 CrossRef CAS.
- Y. Jia, Y. Li, S. Zhang, P. Wang, Q. Liu and Y. Dong, Biosens. Bioelectron., 2020, 149, 111842 CrossRef CAS PubMed.
- Y. Lin, M. Bariya, H. Y. Y. Nyein, L. Kivimaki, S. Uusitalo, E. Jonsson, W. Ji, Z. Yuan, T. Happonen, C. Liedert, J. Hiltunen, Z. Fan and A. Javey, Adv. Funct. Mater., 2019, 29, 1902521 CrossRef.
- Y. Shao, J. Wang, H. Wu, J. Liu, I. A. Aksay and Y. Lin, Electroanalysis, 2010, 22, 1027–1036 CrossRef CAS.
- M. Ghanei-Motlagh and M. A. Taher, Biosens. Bioelectron., 2018, 109, 279–285 CrossRef CAS PubMed.
- M. Wang, M. Hu, Z. Li, L. He, Y. Song, Q. Jia, Z. Zhang and M. Du, Biosens. Bioelectron., 2019, 142, 111536 CrossRef PubMed.
- P. A. Rasheed and N. Sandhyarani, Microchim. Acta, 2017, 184, 981–1000 CrossRef.
- T. Chen, J. Xu, P. Yang, Q. Sheng, J. Zheng, W. Cao, T. Yue, M. Zhou and C. Wang, Sens. Actuators B Chem., 2019, 298, 126724 CrossRef CAS.
- J. Xu, X. Qiao, Y. Wang, Q. Sheng, T. Yue, J. Zheng and M. Zhou, Mikrochim. Acta, 2019, 186, 1–8 CrossRef PubMed.
- G. A. Hudalla, T. Sun, J. Z. Gasiorowski, H. Han, Y. F. Tian, A. S. Chong and J. H. Collier, Nat. Mater., 2014, 13, 829–836 CrossRef CAS PubMed.
- H. Dong, F. Zou, X. Hu, H. Zhu, K. Kwangnak and H. Chen, Biosens. Bioelectron., 2018, 117, 605–612 CrossRef CAS PubMed.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2010, 46, 1222–1244 CrossRef PubMed.
- Z. Mei, A. Dhanale, A. Gangaharan, D. K. Sardar and L. Tang, Talanta, 2016, 151, 23–29 CrossRef CAS PubMed.
- H. Bai, Y.-P. Zheng, T. Wang and N. Peng, J. Mater. Chem. A, 2016, 4, 14392–14399 RSC.
- J. M. George, A. Antony and B. Mathew, Microchim. Acta, 2018, 185, 358 CrossRef PubMed.
- Z. Zhu, H. Li, Y. Xiang, K. Koh and H. Chen, Microchim. Acta, 2020, 187, 1–9 CrossRef PubMed.
- A. Kaushik, P. R. Solanki, A. A. Ansari, G. Sumana, S. Ahmad and B. D. Malhotra, Sens. Actuators B Chem., 2009, 138, 572–580 CrossRef CAS.
- H. He, D. W. Sun, H. Pu and L. Huang, Food Chem., 2020, 324, 126832 CrossRef CAS PubMed.
- M. Bixby, L. Spieler, T. Menini and A. Gugliucci, Life Sci., 2005, 77, 345–358 CrossRef CAS PubMed.
- C. Berthomieu and R. Hienerwadel, Photosynth. Res., 2009, 101, 157–170 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- L. Gloag, M. Mehdipour, D. Chen, R. D. Tilley and J. J. Gooding, Adv. Mater., 2019, 31, 1904385 CrossRef CAS PubMed.
- Y. Xu, Q. Li, H. Xue and H. Pang, Coord. Chem. Rev., 2018, 376, 292–318 CrossRef CAS.
- L. He, Z. Li, C. Guo, B. Hu, M. Wang, Z. Zhang and M. Du, Sens. Actuators B Chem., 2019, 298, 126852 CrossRef CAS.
- S. L. Young, J. E. Kellon and J. E. Hutchison, J. Am. Chem. Soc., 2016, 138, 13975–13984 CrossRef CAS PubMed.
- C. J. Slevin, A. Malkia, P. Liljeroth, M. Toiminen and K. Kontturi, Langmuir, 2003, 19, 1287–1294 CrossRef CAS.
- N. G. Tsierkezos, J. Solution Chem., 2007, 36, 289–302 CrossRef CAS.
- S.-Y. Lu, M. Jin, Y. Zhang, Y.-B. Niu, J.-C. Gao and C. M. Li, Adv. Energy Mater., 2018, 8, 1702545 CrossRef.
- M. Lin, P. Song, G. Zhou, X. Zuo, A. Aldalbahi, X. Lou, J. Shi and C. Fan, Nat. Protoc., 2016, 11, 1244–1263 CrossRef CAS PubMed.
- R. Rajagopalan, Z. Wu, Y. Liu, S. Al-Rubaye, E. Wang, C. Wu, W. Xiang, B. Zhong, X. Guo, S. X. Dou and H. K. Liu, J. Power Sources, 2018, 398, 175–182 CrossRef CAS.
- H. Li, X. Hu, J. Zhao, K. Koh and H. Chen, Electrochem. Commun., 2019, 100, 126–133 CrossRef CAS.
- J. Li, H. Liu, J. Guo, Z. Hu, Z. Wang, B. Wang, L. Liu, Y. Huang and Z. Guo, J. Mater. Chem. C, 2016, 5, 1095–1105 RSC.
- X. Huang, X. Hu, S. Song, D. Mao, J. Lee, K. Koh, Z. Zhu and H. Chen, Sens. Actuators B Chem., 2020, 305, 127543 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr01415c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |