
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFolding and self-assembly of short intrinsically disordered peptides and protein regions
Pablo G.
Argudo
 *a and
Juan J.
Giner-Casares
b
*a and
Juan J.
Giner-Casares
b
aUniversité de Bordeaux, CNRS, Bordeaux INP, LCPO, 16 Avenue Pey-Berland, 33600 Pessac, France. E-mail: Pablo.Gomezargudo@enscbp.fr
bDepartamento de Química Física y T. Aplicada, Instituto Universitario de Nanoquímica IUNAN, Facultad de Ciencias, Universidad de Córdoba (UCO), Campus de Rabanales, Ed. Marie Curie, E-14071 Córdoba, Spain
First published on 18th January 2021
Abstract
Proteins and peptide fragments are highly relevant building blocks in self-assembly for nanostructures with plenty of applications. Intrinsically disordered proteins (IDPs) and protein regions (IDRs) are defined by the absence of a well-defined secondary structure, yet IDPs/IDRs show a significant biological activity. Experimental techniques and computational modelling procedures for the characterization of IDPs/IDRs are discussed. Directed self-assembly of IDPs/IDRs allows reaching a large variety of nanostructures. Hybrid materials based on the derivatives of IDPs/IDRs show a promising performance as alternative biocides and nanodrugs. Cell mimicking, in vivo compartmentalization, and bone regeneration are demonstrated for IDPs/IDRs in biotechnological applications. The exciting possibilities of IDPs/IDRs in nanotechnology with relevant biological applications are shown.
Introduction
Proteins and their fragments as short peptides are being revealed as relevant players in nanoscience and self-assembly.1 Peptide derivatives can be designed to reach any nanostructure, from nanotubes2 to vesicles.3 Given the excellent biocompatibility and the biological origin of peptides, the forefront field of application for self-assembled nanostructures based on peptides is biomedicine and controlled delivery.4,5 Other fields such as optical materials also benefit from protein-based nanostructures.6 The directed design of peptides can greatly enhance intrinsic properties and biological activity against bacteria.7,8 Hybrid derivatives including peptides for self-assembly allow the solubilization of hydrophobic molecules relevant in the field of biological applications as antimicrobial peptides.9 Purposefully designed peptides can show self-assembly upon being triggered by certain stimuli such as fluid forces.10 Stimuli can be used to promote the self-assembly of peptide-based nanostructures when required, e.g., under a pH variation in tumor cells.11 Peptide derivatives are also relevant when considering biomineralization at interfaces.12,13 Peptides can form hybrids with relevant nanostructures based on carbon for subsequent self-assembly into intriguing materials.14,15The structural features of proteins and peptide fragments are undoubtedly the focus of a large body of research. The traditional view assumes that proteins were flexible and reach a final shape depending on the targeting molecule.16 A more recent view accepts that proteins and peptides appear in an ensemble of conformations with certain regions showing a higher degree of disorder. Thus, intrinsically disordered proteins (IDPs) can be described as the maximum degree of disorganization presented in a protein. IDPs are isolated polypeptide chains with no stable tertiary structure while still being functional. Note that not the whole protein necessarily lacks a fixated structure. The unstructured segments are the so-called intrinsically disordered regions (IDRs).17–19 IDRs were present in at least 2.0% of archaeal, 4.2% of eubacterial and 33.0% of eukaryotic proteins.20 IDPs/IDRs are generally found in nature. The analysis of ca. 3500 protein species from three different domains of life (archaea, bacteria, and eukaryotes) and viruses allowed an estimation of the prevalence of IDPs/IDRs. Viruses had the widest spread of the proteome disorder content (7.3 to 77.3%). Eukaryotic cells present a higher ratio of IDPs/IDRs to the total content of proteins than prokaryotic cells. A higher eukaryotic proteome disorder might be used by nature to deal with the increased cell complexity due to the appearance of various cellular compartments.21
The state of the field and the current trends in the study of intrinsically disordered proteins and regions are summarized herein. This review is focused on short IDRs or completely disordered peptides. After a brief introduction of the structural basics of proteins and peptides, several examples of IDPs/IDRs are introduced. Relevant experimental and computational methods for characterization are discussed. An excellent book by Tanford and Reynolds is strongly recommended for a longer analysis of how protein structure characterization was approached at early stages.22
Structures
Proteins are ‘naturally occurring and synthetic polypeptides having molecular weights greater than about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da’.23 From this IUPAC official definition, it can be clearly inferred that peptides are small chains of amino acids linked by peptide bonds.24 Peptides can be folded into several structures due to the interaction between the atoms of the backbone, described as a secondary structure in proteins. The most common structures are α-helix25 and β-sheet26 structures, which also can appear along β-turns27 and omega loops.28 Other structures are also rarely found, e.g., 310-helix29 or π-helix.30 Proteins with a longer peptide chain allow subsequent folding leading to ternary and quaternary structures. No further folding but self-assembly processes appear in short peptides. Self-assembly is defined as the autonomous organization of components into ordered patterns or structures.31 The short peptide chains act as building blocks, adding value to the final structure. Several final structures can be obtained after a first folding and a second self-assembly organization. Examples of these highly ordered nanostructures can be nanoparticles, nanotubes, nanofibers, vesicles, gels or films.32 However, this behaviour cannot be directly extrapolated to IDPs/IDRs. Due to their completely disordered random coil structure, these peptides miss their folding process.33 Thus, their final self-assembled conformation is going to be related not only to their chemical inner composition but also to the media that surround them and the interactions that take place.
000 Da’.23 From this IUPAC official definition, it can be clearly inferred that peptides are small chains of amino acids linked by peptide bonds.24 Peptides can be folded into several structures due to the interaction between the atoms of the backbone, described as a secondary structure in proteins. The most common structures are α-helix25 and β-sheet26 structures, which also can appear along β-turns27 and omega loops.28 Other structures are also rarely found, e.g., 310-helix29 or π-helix.30 Proteins with a longer peptide chain allow subsequent folding leading to ternary and quaternary structures. No further folding but self-assembly processes appear in short peptides. Self-assembly is defined as the autonomous organization of components into ordered patterns or structures.31 The short peptide chains act as building blocks, adding value to the final structure. Several final structures can be obtained after a first folding and a second self-assembly organization. Examples of these highly ordered nanostructures can be nanoparticles, nanotubes, nanofibers, vesicles, gels or films.32 However, this behaviour cannot be directly extrapolated to IDPs/IDRs. Due to their completely disordered random coil structure, these peptides miss their folding process.33 Thus, their final self-assembled conformation is going to be related not only to their chemical inner composition but also to the media that surround them and the interactions that take place.
In this section, we give a brief overview of the most extended folding and self-assembly structures that can be obtained for IDRs or peptides and the bindings or recognition processes responsible for the final conformation. Additional examples can be found in the following chapters of the review.
Helices
As the α-helix is the most extended structure, this peptide bonding organization is a mainly right-handed helical structure. The N–H group of each amino acid forms a hydrogen bond with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the amino acid placed in the previous fourth position. This Pauling–Corey standard model can be expressed as i + 4 → i. Specifically, the α-helix measures about 5.4 Å in width, has 3.6 amino acid residues per turn and each two continuous residues adopt a total dihedral angle (φ, ψ) of around −105°. Similar structures 310-helix (i + 3 → i)29 and π-helix (i + 5 → i)34 can be found in the −75° and −130° region, respectively. For a deep helix structure analysis, see Barlow et al.35
O group of the amino acid placed in the previous fourth position. This Pauling–Corey standard model can be expressed as i + 4 → i. Specifically, the α-helix measures about 5.4 Å in width, has 3.6 amino acid residues per turn and each two continuous residues adopt a total dihedral angle (φ, ψ) of around −105°. Similar structures 310-helix (i + 3 → i)29 and π-helix (i + 5 → i)34 can be found in the −75° and −130° region, respectively. For a deep helix structure analysis, see Barlow et al.35
Zsila et al. observed unstructured peptide LL-37 folding into an α-helix structure by non-covalent association of anti-inflammatory drugs, pigments, bile salts and food dyes. By hydrogen bonding and salt bridges, Lys and Arg amino acids were able to interact with a wide range of small molecules, resulting in multimeric complexes (Fig. 1).36 Moreover, hemin and bile pigments were able to force the 26 amino acid IDP melittin, the major bee venom component, to fold into parallel β-sheet structures. In contrast, an α-helix promoting effect was observed with the also disordered but more cationic hybrid derivative 15 amino acid long CM15. The Trp and Phe residues induced π–π stacking interactions with the porphyrin dye.37 León et al. studied the 11-mer repeat disordered unit P1LEA-22 behaviour at different temperatures and salt concentrations. Compared to similar 11-mer peptides, the presence of several Ala amino acids enabled the addition of FeCl3 to enhance a PPII helical structure. The same behaviour was observed after choline chloride addition. A higher percentage of the PPII structure at a low temperature was found.38 Fealey et al. investigated the structural dependence of synaptotagmin 1's IDR (Syn1) after dielectric constant and phosphorylation changes. A reduced dielectric constant promoted helix formation in neutrally charged core region residues. Lys–Asp acid salt bridges contributed to the stabilization of a transient secondary structure. However, phosphorylation in this region resulted in the formation of salt bridges, unsuitable for helix formation.39 On a higher scale, Johnson et al. observed how IDP 4E-BP1 folded into an α-helix upon binding to its protein ligand, eIF4E. H-bond thiol–aromatic interaction between Phe58 and Cys62 at 4E-BP1 stabilized the helix.40 Saglam et al. also observed the disorder-to-helix transition of the disordered peptide p53 in the presence of its protein receptor MDM2. A single α-helix was formed by induced fit if the unfolded state of the peptide was more stable than its folded state or at elevated MDM2 concentrations. The folding process was otherwise dominated by conformational selection.41 Jephthah et al. studied the N-terminal MgtA IDR, referred to as KEIF. While disordered in aqueous solution, the helical content of this peptide increased if added to an organic solvent, similar to an aqueous solution containing anionic vesicles,42 leading to similar results to histatin 5. For this peptide, the presence of the His–Ser–His residue sequence was directly related to the α-helical structure formation.43
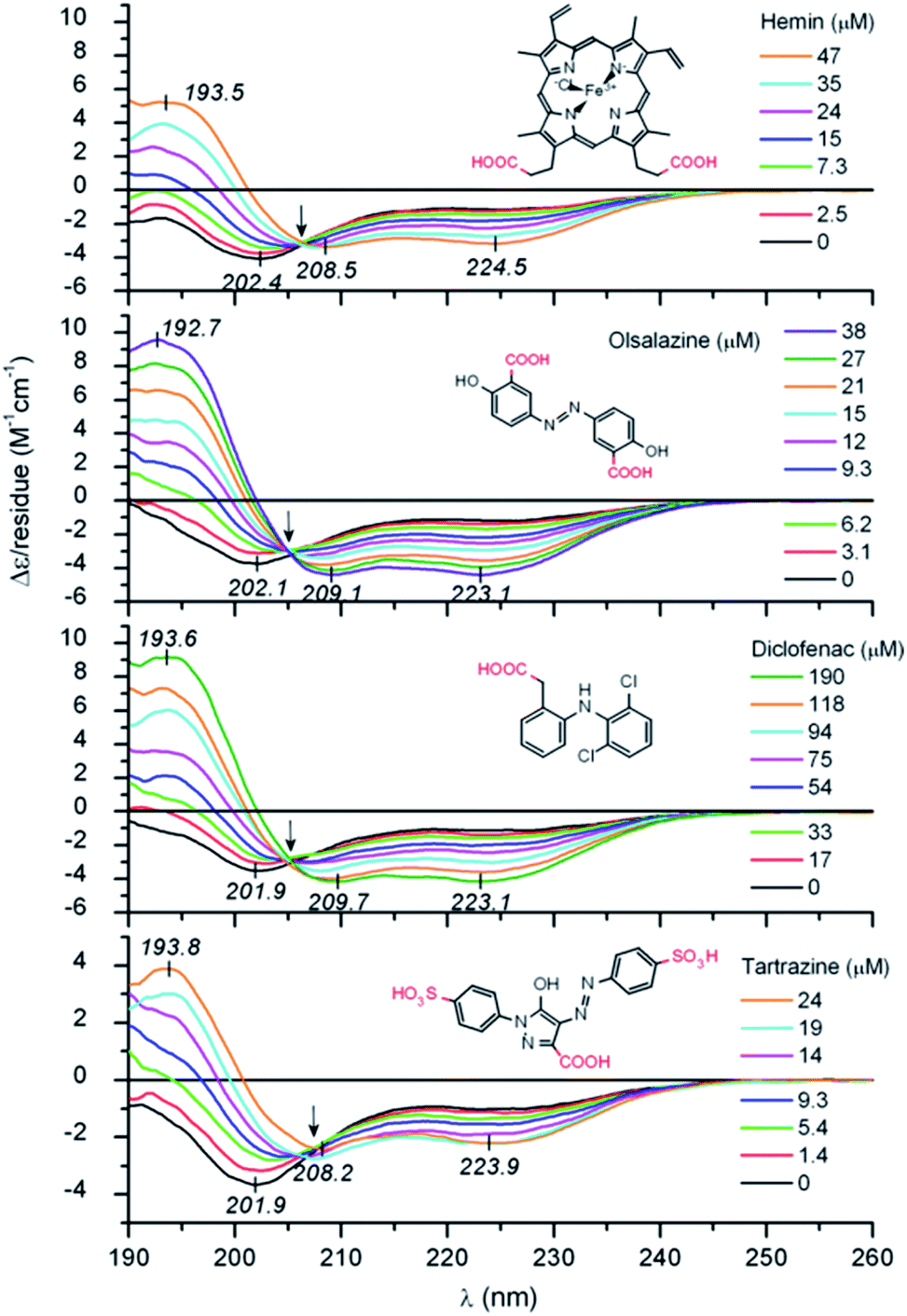 | ||
| Fig. 1 UV-Vis IDP LL-37 disorder-to-helix transitions under the addition of various organic compounds. Reprinted with permission from Zsila et al.36 Copyright 2019 Elsevier B.V. | ||
Sheets
Peptides in a β-sheet conformation zig-zag in a more extended conformation with φ and ψ angles in the −140° and 130° range, compared to the −60° and −45° of α-helix ones. The β-sheet axial distance between adjacent residues is 3.5 Å, while in the α-helix it is 1.5 Å. In a β-sheet, two or more polypeptide chains run alongside each other and are linked in a regular manner by hydrogen bonds between the main chain CO and N–H groups, while the side R chains point outward. The variations of the structure can be described depending on the strand orientation. All strands run towards the same direction in a parallel β-sheet conformation, while the strands are all alternated in an antiparallel β-sheet conformation. Mixed β-sheets show parallel and antiparallel strands. For extended structural characterization, we recommend the Salemme review.44
Coskuner et al. studied the relevance of the Tyr residue at the IDP Aβ42. After Tyr10–Ala mutations, the formation of β-sheet structures greatly diminished in the presence of adenosine triphosphate. They concluded that, after the Tyr10–Ala mutation, a decreased in the reactivity of Aβ42 toward various ligands and self-oligomerization in aqueous environments denoted the high structural control of the Tyr amino acid.45 Takekiyo et al. studied the β-sheet folding of the Aβ fragment Aβ1–11 in the presence of the ionic liquid (IL) 1-butyl-3-methylimidazolium thiocyanate. IDR Asp and Glu residues interacted with the IL imidazolium region, which led to their oligomerization. Moreover, ILs with lower denaturing ability could not promote the aggregation.46 Boopathi et al. showed how disordered Aβ42 was affected by Zn2+ and Cu2+. Zn2+ had a higher hydrophobic behaviour compared to Cu2+, directly related to the fastest self-assembly of Aβ42–Zn2+. Zn2+ increased the solvation free energy due to a higher tendency of forming the β-sheet structure at the Leu17–Ala42 residues.47
Micelles and vesicles
Micelles and vesicles are colloidal dispersions formed by the supramolecular assembly of amphiphilic molecules in a liquid media. These colloids usually show a spherical shape, with different molecule organizations. In aqueous solution, micelles are surfactants that have their hydrophilic part in the outer region of the sphere-like structure, in direct contact with the solvent. The hydrophobic section is placed at the core, surrounded by the hydrophilic one. In the case of an organic solvent, the orientation of the molecules will shift, leading to a reverse micellar structure. Vesicles, on the other hand, are spherical capsules formed by one or more bilayers entrapping an aqueous medium while being surrounded by an aqueous solution. In comparison, the number of molecules required to form a micelle and the size are mostly lower compared to a vesicle. Micelles are in the tens of nanometres diameter range, and vesicles are in the hundreds or even thousands. For an in depth understanding of the formation and design of both, we recommend the latest Lu et al.48 and Has et al.49 reviews, respectively.Ivanović et al. analysed the micellar behaviour of n-dodecyl-β-D-maltoside (DDM). Due to this peptide intrinsically disorder behaviour, a moderate shape fluctuation was observed in its self-assembly, leading to final DDM ellipsoidal, oblate and prolate, conformations.50 Accardo et al. synthetized two disordered peptide amphiphiles (PAs). PAs were characterized by two alkyl chains connected directly or by a linker to the R11 IDP, denoted as (C18)2–R11 and (C18)2–L1–R11, respectively. Presenting an ordered core and a ‘disordered’ surface, (C18)2–L1–R11 self-assembled into micelles (∼16 nm diameter) and small unilamellar vesicles (∼200 nm diameter), while the (C18)2–R11 PA was able to form only vesicles. With a mainly β-strand conformation of both PAs, the addition of the linker L1 gave a closer-to-liquid behaviour to the structure.51 Klass et al. imitated the process using diblock polymers that contained a hydrophobic and an intrinsically disordered hydrophilic domain (Fig. 2). These low polydispersity 27 nm diameter micelles were found to be formed across a broad range of pH (3.7–9.7), ionic strength (0–200 mM), and temperature conditions (25–70 °C). The authors concluded that at pH 7.9 or higher, significant heterogeneity and polydispersity in the micellar diameters were observed, due to the most collapsed state of the hydrophilic IDP portion. The micellar volume decreased reversibly with increasing temperature according to the interplay of intermolecular interactions of the hydrophobic tails and water with the hydrophilic headgroups. Finally, no obvious trends were observed after changing different salt concentrations.52 Acosta et al. proposed the use of antimicrobial peptides (AMPs) as self-assembling domains to drive hierarchical organization of intrinsically disordered protein polymers (IDPPs) based on an elastin-like recombinamer (ELR) (Val–Pro–Gly–Ser–Gly)50–(Ile–Pro–Gly–Val–Gly)60. At 5 °C, the ELR alone did not form any nanostructure. However, the AMP-ELR polymers formed nanofibers. Differences in the size and shape of the nanostructures as a function of the AMP sequence used were also observed. At a physiological temperature of 37 °C, both ELR and AMP-ELR self-assembled into micellar structures. In AMP-ELR samples, a small portion of nanofibers were present due to early AMP assemble processes. Moreover, after the incubation of the AMP-ELR samples, the presence of the AMP drove a second self-assembly in the form of aggregates with globular or amorphous shapes depending on the AMP structure.53 Rao et al. used several low complexity IDPs (LC-IDPs) to form vesicles. LC-IDRs of SM50, LSM34, MSP130, and Prisilkin-39, in the presence of Ca2+, self-associated by ionic interactions leading to 100–300 nm diameter vesicles. Moreover, THF was applied as an orthogonal solvent instead of the mineral precursor, forming the same structures.54 Going one step further, Costa et al. designed an IDP based on hydrophilic Val–Pro–Gly–Val–Gly and hydrophobic Val–Pro–Gly–Ser–Gly motifs. Self-assembly into spherical micelles was triggered by increasing the temperature above its critical micelle temperature in bulk solution. This reversible process could be combined with a UV irradiation process, while peptides were in their micellar form for the formation of nanogels, after the addition of para-azidophenylalanine groups.55
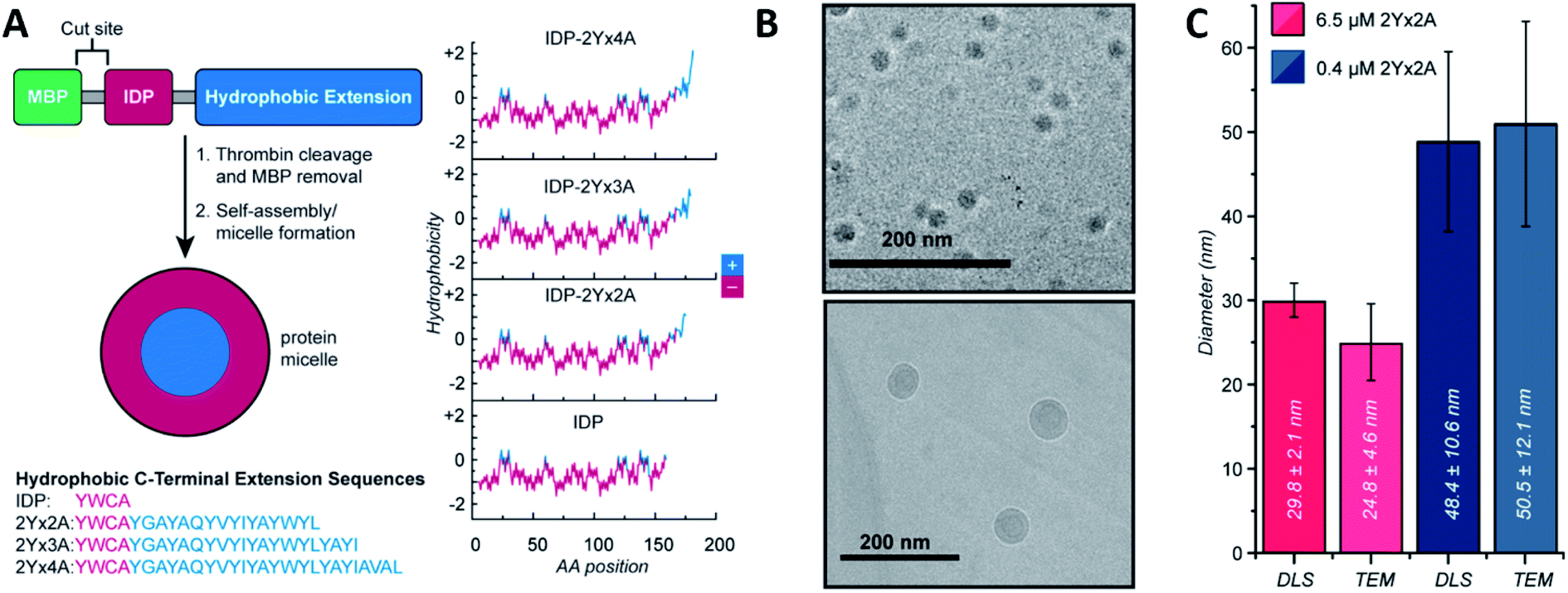 | ||
| Fig. 2 IDP-based self-assembly behaviour. (A) Structure. IDP segment fused to different hydrophobic sequences and hydrophobicity plots of each final amphiphilic protein. (B) Cryo-TEM images of 6.5 μM (top) and 0.4 μM (bottom) IDP-2Yx2A micelles in PBS, pH 5.7. (C) comparison of DLS and cryo-TEM diameters obtained at different concentrations. Reprinted with permission from Klass et al.52 Copyright 2019 American Chemical Society. | ||
Fibrils
Fibrils are linear 10–100 nm diameter chains differentiated from filaments (their precursor) and fibers (their product). Fibrils are a well-known structure in the biological field, quite commonly found in the form of amyloid fibrils. The peptide is folded in a β-sheet parallel or antiparallel structure with an inter-strand distance of ca. 4.8 Å (N–H⋯OC hydrogen bonds between two consecutive peptide backbones). Different β-sheets stack in parallel. In function of the residues and packaging, its inter-sheet distance varies from 8.8 to 14.6 Å. This structure is elongated to protofilaments and subsequently twisted into multistrand helical mature amyloid fibrils. Depending on their torsion, fibrils can lead to crystal or nanotube structures. For a wider view of protein fibrils and amyloid fibrils specifically, we recommend the Fändrich review.56
Humenik et al. observed the assembly of recombinant spider silk variants, denoted as eADF4(Cn), ‘n’ being the number of C-modules. This C-module was a 35 amino acid segment rich in Gly and Pro residues and one poly-Ala stretch. While monomers showed an intrinsically disordered behaviour by themselves, they could self-assemble into cross β-sheet fibrils. For C ≥ 2, the peptide folded towards antiparallel β-sheets followed by the formation of a nucleus via hydrophobic interactions of poly-Ala β-sheets. Finally, monomer addition occurred by the dock-lock mechanism forming the final fibril structure.57 Hernik-Magon et al. studied the influence of the poly-L-glutamic acid (PLGA) length in the self-assembly process. Long chained (Glu)200 molecules fibrillated more readily than short IDP (Glu)5 fragments. While both started with an alpha structure, only β2-(Glu)200 amyloid tended to form well-defined twisted superstructures. Moreover, their mixture accelerated the process. The intrinsically disordered pentapeptide, merged with structured (Glu)200 chains, followed the PLGA's fibrillation pattern. At different mixture ratios, (Glu)5 adopted a self-assembly β2-fibril pattern normally accessible only to long-chained PLGA.58 Similar results were reported by Zhang et al., who theoretically explained how Glu/Asp-rich peptides aggregated in β-sheet structures and self-assembled into highly ordered amyloid fibrils.59 Pan et al. showed the formation of amyloid-like fibrils from intrinsically disordered α-, β-, and k-caseins during heating (90 °C) at an acidic pH (2.0). The fibrillated caseins had increased contents of β-sheet organized structures with different nanomechanical properties and bulk viscosity.60 Bakou et al. described the self-assembly of an intrinsically disordered polypeptide islet amyloid polypeptide (IAPP) fibril. Phe, Leu, and Ile were the residues directly related to the fibrillar structure formation.61 Larini et al. analysed a construct that included the PHF6* region of the neuronal-related IDP Tau. Specifically, Tau273–284 self-assembled into full-fledged fibrils.62 Adamcik et al. studied the third repeat fragment (R3) of this protein and obtained similar results. The 26-amino acid Tau-derived peptide could self-assemble into amyloid fibrils with a β-sheet-based structure without any external induction. Complete ordered 2D laminated flat ribbons with on average 18.7 protofilaments were observed with a lateral size of 149.7 nm and 3.8 nm thickness (Fig. 3). Moreover, ribbons of >350 nm lateral size and 45 protofilaments could be observed, the biggest reported to date.63 The self-assembly of the complete microtubule-associated protein Tau into neurotoxic oligomers, fibrils, and paired helical filaments took place after the addition of polyanions, such as heparin, as described by Despres et al.64 Dec et al. analysed the H-fragment, disordered in aqueous solutions, of predominantly ordered α-helical insulin. Thin and structurally homogenous fibrils with a typical parallel β-sheet conformation appeared upon lowering of the pH value. It was concluded that, due to its acidity, the Ala-rich chain portion played a crucial role in the aggregation of the whole H-fragment.65 Kuhn et al. observed the p3 peptide behaviour and the IDP formed through alternative processing of Amyloid-β (Aβ).66 The self-assembly of this peptide proved to form oligomers and fibrils at higher aggregation rates than Aβ. In addition, p3 fibrils exhibited cross-β-sheet amyloid structures. A final hydrophobic steric zipper was a convincing organization given that the two amyloidogenic hydrophobic patches of Aβ are also found in p3.67 Focused on the Aβ IDP, Jana et al. showed how the addition of glycated Lys residues forced Aβ to self-assemble at early stages into protofibrillar conformations after folding of β-sheets. New and stronger inter-monomer salt bridging bindings took place in the glycated form with dispersion interactions playing no significant roles.68
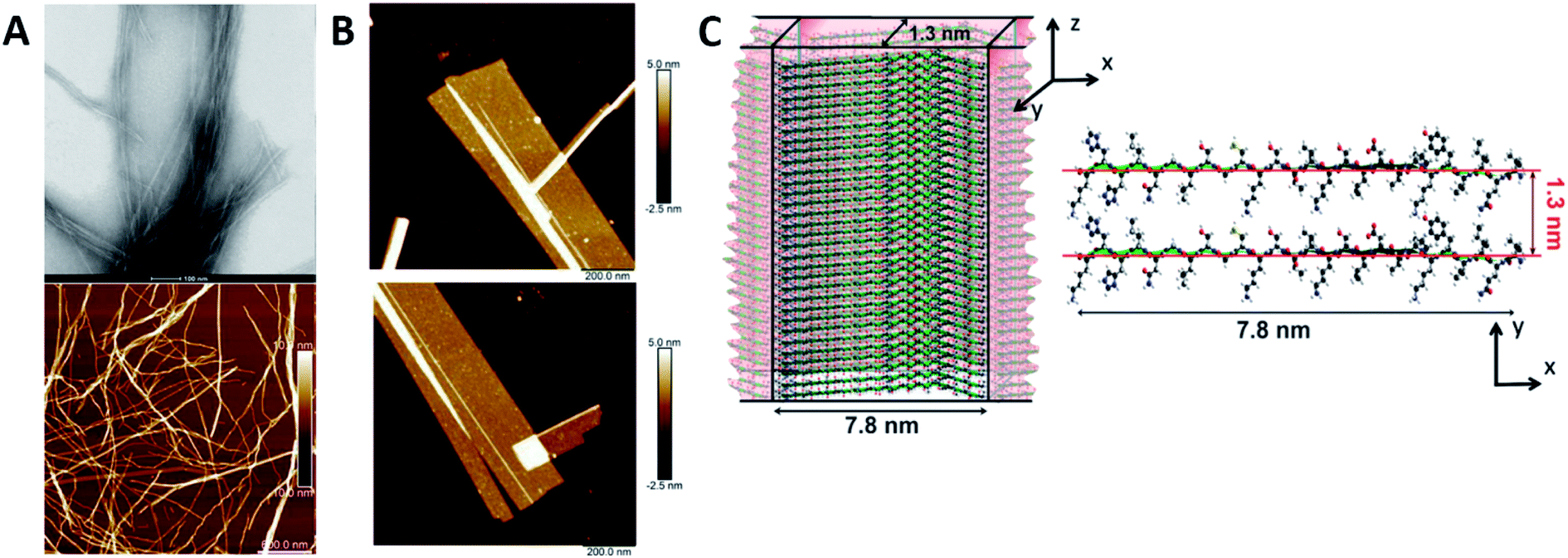 | ||
| Fig. 3 R3 peptide (A) TEM (up) and AFM (low) images of self-assembled fibril structures in the presence of heparin. (B) High-resolution AFM images of flat multistranded ribbons in the absence of heparin. (C) Structural illustration of protofilaments. The distance between β-sheets is 1.3 nm with an off-set of ca. 0.4–0.6 nm corresponding to the peptide residues on both sides of the β-sheet. Reprinted with permission from Adamcik et al.63 Copyright 2016 Wiley-VCH Verlag GmbH & Co. | ||
Other structures
Although most natural disordered peptides and proteins may have α-helix and β-sheet structures after folding and fibrils after self-assembling, some exceptions could be found. Mostly artificially synthesized, several IDPs/IDRs can be organized from more classical to complex structures, from rods to fractals, respectively.Khatun et al. analysed 37-residue IDP amylin. While several typical structures were already reported for this peptide, such as fibrils69 or micelles,70 they discovered that also fractal self-assembly processes could occur (Fig. 4). Affected by the solvent and the media, results indicated the main role of the hydrophobic interactions in the fractal self-assembly and aggregation of amylin. Relevant interactions between the anisotropically distributed hydrophobic residues and polar/ionic residues on the solvent-accessible surface of the protein drove the process.71 Quiroz et al. synthesized two IDPPs that exhibited lower and/or upper critical solution temperature phase behaviour. The IDPPs were composed of the same corona-forming ELP block and different hydrophobic IDPP core-forming blocks with distinct hysteretic phase behaviours. While the ELP formed 20 to 30 nm diameter micelles, IDPPs self-assembled into nanoparticles with a rod-like morphology, all thermodynamically controlled.72 Stehli et al. reported the formation of highly ordered spherulite structures after the self-assembly of intrinsically disordered PLGA. PLGA monomers at early stages could exist in either a collapsed globular state or an extended random coil conformation. An α-helical conformation promoted the spherulite formation, while a random coil promoted the formation of an amyloid fibril.73 Bishof et al. stated how the disordered low-complexity domain 1 (LC1) of the U1 small nuclear ribonucleoprotein (U1-70K) self-assembled into oligomers. The LC1 domain contains highly repetitive basic (Arg/Lys) and acidic (Asp/Glu) residues and behaves like a Gln/Asn-rich LC domain. These domains could form “glue” that drove the granule assembly processes.74 Dooley et al. designed an IDP based on a repeats-in-toxin (RTX) domain that allowed it to gain a β-roll secondary structure in a calcium rich environment. Based on the adenylate cyclase region obtained from Bordetella pertussis bacteria, the repeating polypeptide consisted of two parallel β-sheet faces separated by flexible turn regions. Aspartic acid residues were included in the turn regions to promote the coordination of the calcium ions by the carboxylic groups. Moreover, after engineering, the addition of L-Leu and D-Leu residues to the structure made this IDP self-assemble into hydrogels with minimal impact on its calcium affinity.75
 | ||
| Fig. 4 Optical microscopic images of an amylin fractal observed in PBS buffer at pH 6.5 ± 0.1 (A) at ∼10 μM concentration, (B) at ∼0.1 μM concentration, and (B*) inset showing the presence of different morphologies (C) at ∼1 μM concentration; in PBS buffer at ∼1 μM concentration (D) at pH 11.5 ± 0.1 and (E) at pH 2.5 ± 0.1; and (F) amylin fractal observed in DI water at ∼1 μM concentration at pH 6.5 ± 0.1. The table in the figure contains the df for the morphologies obtained with an optical microscope shown in (c)–(f). Reprinted with permission from Khatun et al.71 Copyright 2020 The Royal Society of Chemistry. | ||
Experimental techniques
While having biological activity, IDPs/IDRs lack a well-defined structure. A classical approach based on basic structural experimental techniques makes IDPs/IDRs impossible to characterize due to their highly heterogeneous conformational behaviour. Over the last few years, a large variety of physical techniques have been optimized and applied successfully to unveil the fundamental rules that make proteins fold into different structures and elucidate the conformational transition from their native disordered to a more structured state. Even more, the understanding of these transitions from a disordered to different secondary or tertiary structures can lead to remarkable changes in their implementation and future applicability. However, the mechanisms of these structure transitions are not fully understood, and a fundamental description of the kinetic and thermodynamic variables will undoubtedly help to recognize these transition processes and their relevance. The effect of the AA sequence, the length of the AA chain, and the biological media are the main parameters to be studied. Furthermore, the existence of domains showing this behaviour or IDRs is an intriguing question in the field.In this section, we provide examples of key types of systems and insights that can be addressed using the most relevant and widely used methods, summarized in Table 2.
Nuclear magnetic resonance (NMR)
NMR spectroscopy is undoubtedly one of the most preeminent methods for determining the molecular behaviour of IDPs/IDRs at the atomic resolution. The use of NMR for protein structural characterization was developed in the 1970s and 80s by scientists such as Ernst,76 Wüthrich,77 Clore78 or Gronenborn.79 Nowadays, after its optimization, NMR is applied to the biochemical field, specifically in the binding and self-assembly of peptides in aqueous media. In NMR spectroscopy, radiofrequency (RF) waves in the MHz range are applied to samples subjected to a strong and homogeneous magnetic field (B0). Their active nucleus (such as 1H, 13C or 15N) absorbs the electromagnetic radiation at a specific frequency characteristic of the isotope. To know more about the analysis of NMR spectra applied to peptides, how to assign the chemical shifts and how structural changes are reflected in the NMR signal, we recommend the Brutscher et al. chapter.80 Briefly, the Hamiltonian 〈H〉 describes the energy of interaction of the nuclear spins with internal and external electric and magnetic fields. In isotropic systems, like IDPs, the fast-molecular reorientation will average out any orientation dependence of the interactions, and only the isotropic parts of this Hamiltonian solution will remain. Therefore, despite the complexity of these molecules, a well resolved spectra can be obtained. It can be easily explained by the absence of structure. Similar shifts will be obtained due to protons in the same chemical group having the same chemical environment.Chaves-Arquero et al., using 1Hα and 13Cα conformational shifts in solution, could observe the phosphorylation effect on the structural behaviour of two derived IDPs.81 While both had a random coil structure in water (|ΔδHα| ≤ 0.05 ppm and |ΔδCα| ≤ 0.4 ppm, being ΔδHα = δHα,observed − δHα,random coil, ppm; and ΔδCα = δCα,observed − δCα,random coil, ppm), the IDP T118-H1.0 and its phosphorylated derivative, pT118-H1.0, consisted of two helical regions in the presence of trifluoroethanol (TFE). Clear changes in |ΔδHα| as well as |ΔδCα| denoted it (T118-H1.0: |ΔδHα| = −0.29 ppm and |ΔδCα| = 3.35 ppm; pT118-H1.0: |ΔδHα| = 0.27 ppm and |ΔδCα| = 2.92 ppm, (Table 1)). Even more, the helix populations could be estimated from ΔδHα and ΔδCα averaged for the helical residues as well as the orientation between the helices. Both systems contained a >80% helical structure, with a perpendicular arrangement exclusively for the non-phosphorylated IDP. This structural difference is directly related to the different biological roles. Kosol et al. elucidated the structural basis of the Bamb_5917 protein. They could observe low heteronuclear Nuclear Overhauser Effects (NOEs) in the Bamb_5917 PCP domain, typical behaviour of an IDR with high picosecond-nanosecond flexibility ({1H}–15N NOE close to 0).82 Based on 2D CONPro experiments, Murrali et al. showed how to obtain the fingerprint of the Pro residue in IDPs,83 a largely exploited AA used to prevent the formation of stable secondary structures. The lack of the peptide HN atom implied that this amino acid was not directly detectable in the commonly used 2D 1H, 15N NMR spectroscopy. 2D CON was the chosen experimental technique to determine the correlations between the backbone carbonyl carbon and nitrogen of neighbouring residues. Fast NMR assignments of IDPs can be done by combining 2D hNCA and 2D hNcoCA spectra as Sukumaran et al. presented for the human α-synuclein protein during its self-aggregation process.84 By solid-state NMR measurements, Reichheld et al. were able to observe the conformational transition of elastin cross-linking domains during their self-assembly. The Cα and Cβ chemical shift values of Ala and Lys AAs were used due to their particular sensitivity to backbone torsion angles. Lyophilized EP20-24 IDP Ala residues showed Cα–Cβ cross-peaks with chemical shift values indicative of a larger α-helix population combined with a smaller random coiled one. On the other hand, the hydrated EP20-24 coacervate displayed a conspicuous Cα–Cβ cross-peak with shifts indicative of a β-strand backbone conformation. If cross-linked with genipin, the polypeptide showed a very prominent cross-peak with chemical shift values characteristic exclusively of Ala α-helical conformations.85 Using 1H–15N HSQC and 3D HNCACB, Garry et al. studied the self-assembly of the Leucine-Rich Amelogenin Protein (LRAP) into a unique quaternary structure referred to as a ‘nanosphere’ in the presence of NaCl. They identified the specific residues involved in the early stages of the nanosphere assembly in this IDP by following its amide chemical shift perturbations as a function of salt concentration. The disappearance of amide cross peaks in the 1H–15N HSQC spectrum at high NaCl concentrations likely reflected a restricted motion at the protein–protein interface.86 With these techniques, Beck Erlach et al. also studied the pressure and temperature effects on the self-assembly of intrinsically disordered human IAPP (hIAPP) and Alzheimer peptide Aβ1–40. The hIAPP N-terminal region displayed large differences in pressure sensitivity compared to Aβ, pinpointing to a different structural ensemble in this sequence element. A helical origin was related to hIAPP and an amyloid deposit to Aβ1–40.87 Based on 2D [1H 1H] NOESY and TOCSY, Accardo et al. characterized the self-assembly and final organization of different IDP-amphiphilic molecules. In the case of (C18)2–L1–R11, the spectra revealed NOEs between the CH2 groups from the C18 alkyl chains and peptidic protons, which resonated at 3.55, 3.51, 2.55 and 2.42 ppm.51 Data indicated that the peptidic region did indeed interact with the C18 chains, endowing (C18)2–L1–R11 with a certain degree of flexibility in solution. In the case of the (C18)2–R11 peptide, NOEs between the CH2 protons related to the C18 alkyl chains and either peptidic HN or aromatic protons appeared rather clear with a line broadening effect. This confirmed the high tendency of this PA to self-associate in larger aggregates than (C18)2–L1–R11 due to the lack of the connector L1. Hou et al. could characterize the self-assembly mechanism of Aβ IDPs in a global β structure and how the oxidation of some regions prevented this structure from being promoted to a random coil instead. The relevance of His residues in the self-assembly process was studied by oxidation processes and pH variations, key for a final β-structure.68,88
| Peptide | Conditions | [θ]222nm (deg cm−2 dmol−1) | % helixa from [θ]222nm | Helix lengtha | ΔδHα [ppm] | % α helix from ΔδHα | ΔδCα [ppm] | % α helix from ΔδCα | Avgd% α helixa,c |
|---|---|---|---|---|---|---|---|---|---|
| a Note that the CD-estimated helix percentages correspond to an average for all the peptide residues, whereas the NMR-estimated helix percentages relate to the residues within the helix. b Values measured at 5 °C. c Reported errors are standard deviations for the mean of the percentages obtained from the ΔδHα and ΔδCα values. | |||||||||
| T118-H1.0 | H2O | −69.9 | 8 | 105–115 | −0.05b | 13b | +0.12b | 4b | 9 ± 5b |
| 90% TFE | −11229.6 |
37 | −0.29 | 75 | +3.35 | 100 | 87 ± 13 | ||
| pT118-H1.0 | H2O | 86.4 | 7 | 105–115 | −0.05b | 13b | +0.15b | 5b | 9 ± 4b |
| 90% TFE | −10988.9 |
36 | 0.27 | 66 | +2.92 | 95 | 81 ± 14 | ||
| T140-H1.0 | H2O | −915.8 | 10 | 141–147 | −0.06b | 16b | +0.21b | 7b | 12 ± 5b |
| 90% TFE | −7137.4 | 26 | −0.15 | 39 | +1.58 | 51 | 45 ± 6 | ||
| pT140-H1.0 | H2O | 1175.9 | 5 | 141–147 | −0.06b | 16b | +0.28b | 9b | 13 ± 4b |
| 90% TFE | −8168.4 | 29 | −0.16 | 40 | +1.80 | 58 | 49 ± 9 | ||
| Technique | Structural observation | References |
|---|---|---|
| Nuclear magnetic resonance (NMR) | 1Hα and 13Cα signal shifts (ΔδHα and ΔδCα) | 53, 66, 72, 73, 83–90, 94, 99, 109, 115, 116 and 165 |
| Circular dichroism (CD) | Maximums and minimums in the 190–250 nm CD region | 38–42, 44, 45, 48, 53, 55, 59, 60, 62, 65, 67, 70, 71, 75, 77, 83, 87, 93, 94, 97, 99, 102, 105, 109, 112, 117, 119 and 120 |
| Electron paramagnetic resonance (EPR) | Substituted Cys or coordinated Cu2+ tracking | 96–99 |
| Fluorescence spectroscopy | Tryptophan (Trp, 300–450 nm), tyrosine (Tyr, 250–370 nm) and phenylalanine (Phe, 250–350 nm) shifts | 39, 42, 53, 62, 64, 66, 69, 70, 71, 73, 75, 77, 87, 94, 101 and 102 |
| Raman spectroscopy | Amide I (1630–1700 cm−1), amide III (1230–1310 cm−1) and backbone skeletal stretch (870–1150 cm−1) regions | 104–107 |
| Fourier transform infrared spectroscopy (FT-IR) | Amide I (1700–1600 cm−1) and amide II (1600–1500 cm−1) regions | 48, 59, 60, 67, 75, 93, 106 and 109–112 |
| Small-angle X-ray scattering (SAXS) | Form factor, Kratky plot and pair distance-distribution function (PDDF) shape | 44, 45, 52, 65, 111, 115, 116, 117 and 120 |
| Static and dynamic light scattering (SLS & DLS) | Gyration (Rg) and hydrodynamic radius (RS) | 44, 53, 54, 55, 56, 57, 73, 75, 88, 99, 119 and 120 |
Circular dichroism (CD)
IDPs, due to the lack of any significant organized secondary structure, show the characteristic CD spectrum of an unordered polypeptide, with a strong negative band near 200 nm and either a weak negative shoulder or a weak positive maximum near 220 nm. IDRs are also recognized by CD, even though they show ordered and unordered regions, making the IDR difficult to diagnose. For these regions, a limited proteolysis approach followed by CD is commonly used.89 As IDPs/IDRs present dynamic conformations, measurements should be carried out not at a single and unique set-up but instead changing the chemical conditions and observing the peptide behaviour, as part of the determination of the intrinsically disordered nature. While we recommend Chemes et al. work for a complete understanding of the methodology to achieve the maximum the CD can contribute,90 we will try to show the relevance of this powerful tool. This essential technique is often used as a preliminary to more complex methodologies such as NMR, for a deep secondary structure characterization. Without going further, some studies cited in the NMR section also carried out CD measurements as a starting point.Chaves-Arquero et al. work is an example. Four IDPs derived from the C-terminal domain of Histone H1.0 showed a typical random coil strong minimum at ca. 195 nm and no other secondary structure features, confirming that they were predominantly disordered in water (Table 1 and Fig. 5). The addition of TFE caused an increase of structural organization on all the peptides with a progressive conversion of the 195 nm region into a maximum at ca. 197 nm, related to helix populations.81 Reichheld et al. used mainly CD to support the structural conformations observed in elastin. IDP EP20-24 and tropoelastin (its monomer), at 5 °C, had a similar behaviour with a strong minimum at 202 nm and another at 222 nm, characterized as the disordered and α-helical signals, respectively. At 37 °C or higher temperature values under physiological conditions, the absence of the 222 nm band denoted the significant loss of the α-helical structure. EP20-24 showed a totally different structure at temperatures above 40 °C after the mutation of the Lys monomers to Ala. The 202 nm signal was lost above 40 °C and a shift to a strong minimum at 217 nm was indicative of β-strand formation. Furthermore, a Lys-to-Tyr mutation made the IDP show at 5 °C a strong minimum at 217 nm with a very weak minimum at 202 nm, demonstrating that this mutant polypeptide was predominantly a β-strand even at low temperatures. Increasing the temperature to 37 °C resulted in the loss of the minimum at 202 nm, indicating that EP:Lys-to-Tyr had a greater propensity for β-strand formation than the EP:Lys-to-Ala mutant.85 Sun et al., while analysing the amyloid self-assembly fibrillation process of hIAPP8–20, revealed a negative peak at 200 nm at 0 h and a negative peak at 218 nm at 48 h, denoting a structural transition from random coil to β-strands.91 Rivera-Najera et al. tested the disordered self-association behaviour predicted for PvLEA6 protein in its secondary structure. Temperature changes could also promote modifications in the protein conformation, as demonstrated by CD in far-UV light. A negative band at 197–200 nm related to a random coil unordered structure described this IDP at low temperatures, which was altered to a β-like one for higher temperatures, denoted by an increase in the CD negative signal between 215 and 220 nm.92 Dooley et al. used an intrinsically disordered peptide isolated from the repeats-in-toxin (RTX) domain to show how the addition of Ca2+ folded the system into a β-roll secondary structure based on two parallel β-sheet faces. In calcium-free environments, the spectra exhibited large negative peaks at 198 nm, indicative of a randomly coiled peptide. A random coil to β-sheet transition was evidenced by the emergence of a negative peak at 218 nm after Ca2+ titration, indicative of this disordered-to-folded transition.75 Bakou et al. observed the key effects of Ala mutations on the conformation of IAPP. Mixtures of random coil and β-sheet/β-turn structural elements were observed for freshly dissolved IAPP and mutant samples. CD spectral deconvolutions suggested IAPP 30–40% β-turn/β-sheet, 50–60% random coil and 10% α-helix contents. In the case of most mutants, 30–50% β-turn/β-sheet, 40–60% random coil and 5–15% α-helical distributions were determined. Together, CD studies provided evidence that (a) mutations are not related to changes in the structure and (b) β-strand–loop–β-strand conformers can be observed in major populations for IAPP compared to its mutants.61
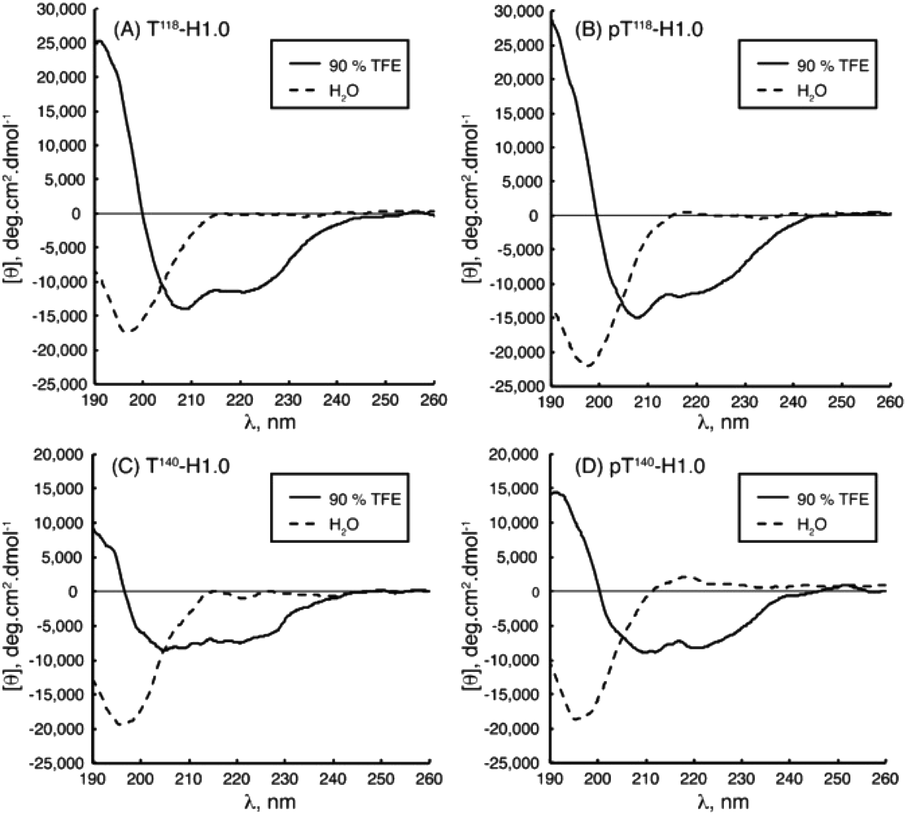 | ||
| Fig. 5 CD spectra of peptides (A) T118-H1.0, (B) pT118-H1.0, (C) T140-H1.0 and (D) pT140-H1.0 in aqueous solution (dotted line) and in 90% TFE (black line) at pH 5.5 and 25 °C. Reprinted with permission from Chaves-Arquero et al.81 Copyright 2020 Wiley-VCH Verlag GmbH &Co. | ||
Electron paramagnetic resonance spectroscopy (EPR)
EPR spectroscopy specifically detects unpaired electrons. The introduction of a site-directed spin labelling (SDSL) model at the protein of study is the common procedure when applied in the biological field. It is usually accomplished by Cys substitution mutagenesis followed by covalent modification of the unique sulfhydryl group with a selective nitroxide reagent. For an in-depth analysis of all EPR variety approaches depending on the system, see Weickert et al.93Pirman et al. characterized the disordered to α-helical transition of IA3 upon TFE addition. In this case, they modified the Cys AA side chain with methanethiosulfonate (MTSL), 4-maleimido-TEMPO (MSL) and 3-(2-iodoacetamido)-proxyl (IAP). They compared the peak-to-peak intensities of the low-field, h(+1), center-field, h(0), and high-field, h(−1), resonances. It was possible to conclude that (a) the line shapes obtained showed the following expected mobility trend: IAP > MTSL > MSL and (b) the addition of TFE produced a conformational change. The first conclusion was reached because the overall intensity of the signal is proportional to the mobility. The spin probe increases the motional average with the increase of intensity. The overall intensity of all the spectra decreased and was broader after the TFE addition. This decrease was directly related to the lower overall mobility of the spin label, arising from the conformational changes of the protein backbone.94 Bund et al. used this technique to observe how copper induced a self-assembly process in the 18.5 kDa intrinsically disordered Myelin Basic Protein (MBP). Because Cu2+ is an EPR-active ion (with S = 1/2, 63/65Cu:I = 3/2), direct investigations of the interaction between MBP and Cu2+ are feasible. Comparing the continuous wave EPR spectra of Cu2+/MBP with and without PBS, clear shifts of the peak positions as well as significant changes in the overall signal width were denoted. Multiple nitrogen coordinations of Cu2+ were indicated by the copper g- and hyperfine coupling values obtained in phosphate buffer. These results were in line with the values of Cu2+ coordinated to nitrogen atoms of imidazole rings of several His AAs and significantly differ from values for non-coordinated Cu2+. To summarize, at a MBP:Cu2+ ratio > 1:2, a coordination process occurred with significant aggregation of MBP into larger particles of 100–200 nm diameter in a PBS media.95 Kaminker et al. directly probed the backbone changes between IDPs that allowed the control over their preferred conformation (Fig. 6). For oligomer structures, the use of peptide-linkers had a high tendency to form trans configurations around the amide bonds due to their more extended conformations (distance distribution of 8.3 to 20.5 Å). In contrast, the peptoid octamer adopted a more compact conformation, likely due to a relative increase in cis configurations around the amide bonds (11 to 23 Å). The DS-peptoid octamer showed an intermediate behaviour, as both cis/trans options were viable given its alternating sequence (9 to 22 Å).96 Chinak et al. tracked the structural and aggregation features of the Human k-Casein fragment called lactaptin. The pH variation from 3.9 to 7.5 led to significant changes in the EPR spectrum of the main fraction and the appearance of a very wide line with a very short electron spin relaxation time. These results were in good agreement with the fact that most of the proteins form aggregates under physiological pH conditions, which greatly broadened the EPR spectra because (a) larger-size aggregates correspond to slow rotation and long correlation times and (b) modulation of the dipole–dipole interaction and the exchange interaction between spin labels in aggregates lead to short electron spin relaxation times.97
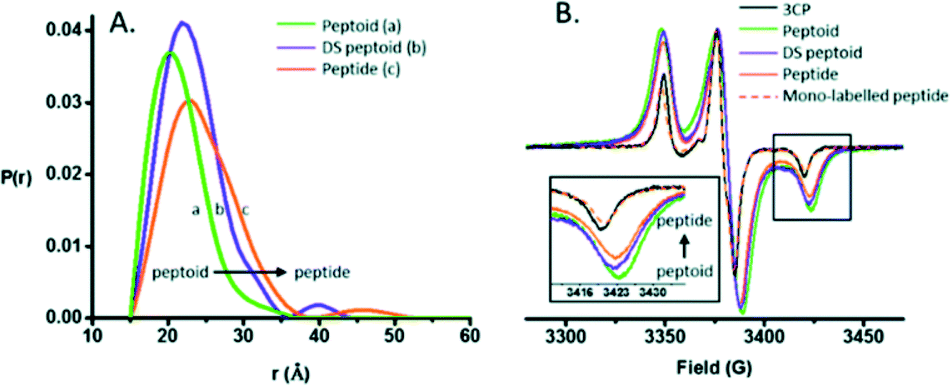 | ||
| Fig. 6 (A) DEER distance distributions obtained for the peptide, peptoid, and DS-peptoid octamers. (B) Low temperature CW EPR of the trimer series overlaid with the mono-labeled peptide and 3CP free radical. Insets: zoom in of the high-field region. Reprinted with permission from Kaminker et al.96 Copyright 2018 The Royal Society of Chemistry. | ||
Fluorescence spectroscopy
IDPs can be characterized by tracking the Trp residues using fluorescence spectroscopy. Trp is accessible to external fluorescence quenchers and has a redshifted fluorescence spectrum with a maximum at 340–353 nm. Moreover, interactions with other residues lead to a more rigid or more hydrophobic system, resulting in a displacement of the fluorescence maximum position to the blue region. This spectral effect can be used to evaluate not only self-assembly but also interaction processes. For a more comprehensive analysis, see Permyakov & Uversky.98Bakou et al. used fluorescence to analyse the binding between IDPs Aβ40, IAPP and several alanine-altered IAPPs. Using N-terminal fluorescein-labelled IAPPs (Fluos-IAPP), the alanine-altered IAPP affinity with IAPP and Aβ40 was quantified by fluorescence spectroscopic titrations. The results denoted that Fluos-IAPP, in the presence of Aβ40 or IAPP, led to a significant enhancement of its fluorescence emission, while the alanine-altered IAPP showed no fluorescence changes after the addition of Aβ40 or IAPP at the same low concentrations. The interactions were weaker when the number of Ala groups was increased, directly related to a decrease in the affinity. By mutating different AAs and looking at its effect on the fluorescence signal, they could conclude which residues were the ones taking part in the IAPP–IAPP self-assembly and IAPP–Aβ40 binding.61 Rivera-Najera et al. also used fluorescence spectroscopy to track the local environment around the aromatic residues of the PvLEA6 IDP. They observed a decrease in the fluorescence intensity of the Tyr residue, indicative of a structure reorganization, going from a mostly rigid and hydrophobic environment to a more relaxed structure with possible tertiary interactions. This explained a transition from random coil and PPII-like extended helices to β-like structures.92 Acharya et al. modified the α-synuclein IDP at positions 94 and 69 to promote Trp–Cys quenching. The binding was monitored using the fluorescence properties of the molecular tweezers CLR01. The incubation of α-synuclein alone over 6 h denoted a red shift in its fluorescence spectra, meaning that Trp was more solvent-exposed in oligomers than in the monomer form. Also, the increase in the sample turbidity denoted the formation of aggregates. When incubated with CLR01, increased values in the fluorescence of Trp94 were observed, related to a high-affinity binding between them. No red shift was found, denoting CLR01 changed the oligomer structure such that the solvent exposure of Trp did not increase during oligomerization.99 Zsila et al. studied Melittin binding with several bile pigments, in this case using the existence of a Trp residue in the molecule. At low pigment concentrations, the emission declined more sharply than at higher loadings, suggesting the formation of an initial complex serving as a scaffold for the binding of additional molecules and denoting the Trp stabilizing behaviour.100
Raman spectroscopy
Conventional polarized visible Raman is generally considered a low-resolution technique, which can solely be used to discriminate between helices, β-sheets, turns, and random coils. Raman spectroscopy mostly exploits the structural sensitivity of the amide I mode to obtain dihedral angles adopted by folded and unfolded peptides. The amide I band region is located in the Raman shift range of ca. 1500 to 1750 cm−1. Specifically, the ∼1650 cm−1 region is typical of an α-helix, ∼1670 cm−1 for β-sheet and ∼1680 cm−1 for random coil structures. Recent advances in the field have substantially improved its usability. Chiral techniques like Raman optical activity (ROA) or UV resonance Raman spectroscopy (UVRR) have been developed to probe the ordered and unordered structures of peptides and proteins. Insights in the study of IDPs by Raman spectroscopy can be found in the Zhu et al. review.101Signorelli et al. used Raman spectroscopy to carry out the structural characterization of the p53 IDP with potential therapeutic applications. A careful analysis of the amide I Raman band revealed the presence of extended random coils and predominant β-sheet regions in its DNA binding domain (DBD). A wide curve at 1675 cm−1 for p53 and a maximum peak at 1669 cm−1 for the DBD were observed by peak deconvolution, explaining their predominant random coil and β-sheet organization. Even more, they observed how the addition of MeOH to the PBS aqueous media affected the amide I structure, with both p53 and DBD peptides showing a major contribution of the β-sheet structure. In contrast, if TFE was added, a final α-helix could be observed.102 McCaslin et al. used UVRR to obtain new information concerning the structure and function of histatin 5 and its interactions with Zn2+ and Cu2+ (Fig. 7). Bands at 1315 cm−1, 1334 cm−1, 1371 cm−1, and 1565 cm−1 were assigned to the His side chain and the bands at 1176 cm−1, 1210 cm−1, and 1612 cm−1 were related to Tyr due to the phenol/phenolate and imidazole side chain contributions. While adding Cu2+ did not change the histatin 5 self-assembled structure, Zn2+ binding altered it. The 1315 cm−1 band underwent an upshift to 1334 cm−1, and the 1371 cm−1 band was decreased in intensity. In addition, the 1565 cm−1 band was reduced in intensity, and a zinc-induced shoulder was present at 1583 cm−1. Still, it was stated that more analysis were needed to finally elucidate the complete 3D structure of the IDP.103 Rawat et al. observed possible different IAPP structures after its aggregation. The Raman spectra of oligomers suggested the presence of mostly an α-helix due to a clear peak at 1656 cm−1 and a weak peak at 1261 cm−1, assignable to amide-I and amide-III, respectively. In the fibril state, there were strong peaks at 1668 cm−1 (in the amide-I region) and 1235 cm−1 (in the amide-III region), which confirmed their well-known β-sheet conformation. These results led to the understanding of why oligomers interact more with membranes, which prefer an α-helix structure.104 Again, for IAPPs, La Rosa et al. used Surface Enhanced Raman Scattering (SERS) to validate their hIAPP self-aggregation simulations after the addition of silver nanoparticles to the system. The secondary structure of the amyloidogenic proteins was revealed. It showed how proteins self-aggregate from monomers to oligomers, and eventually into proto-fibrils and fibrils. Three signals were used for the identification of different protein backbone confirmations: amide I (stretching vibration of CO ranging from 1600 to 1690 cm−1), amide II (1480–1580 cm−1) and amide III (1230–1300 cm−1) both associated with the coupled C–N stretching and N–H bending vibrations of the peptide group.105
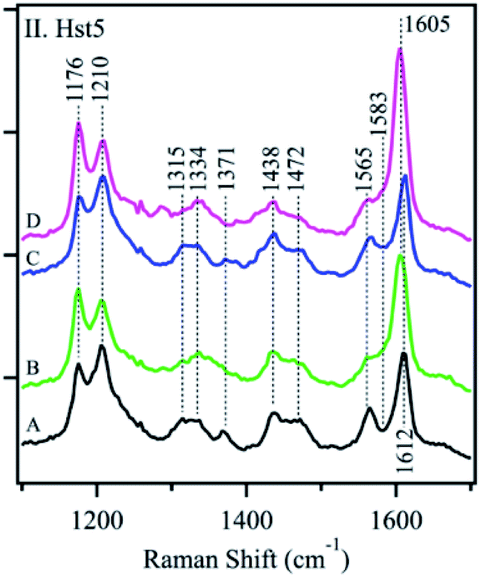 | ||
| Fig. 7 UVRR spectra of Hst-5 in the absence of added metals (A, black) and after the addition of Zn2+ (B, green), Cu2+ (C, blue), or a mixture of Zn2+ and Cu2+ (D, pink). Reprinted with permission from McCastlin et al.103 Copyright 2019 Springer Nature. | ||
Fourier transform infrared spectroscopy (FT-IR)
Infrared spectroscopy is a reliable tool to obtain information on the protein secondary structure and aggregation. Similarly to Raman, it mostly requires to identify and analyse the protein absorption components in the amide I (CO stretching vibrations, 1700–1600 cm−1) and amide II (C–N stretching vibrations in combination with N–H bending, 1600–1500 cm−1) regions, leading to the final structure characterization. Interestingly, this spectroscopy allows the examination of proteins under different environmental conditions such as in solution or in the form of solid films. For a complete overview of this procedure and its spectra analysis, see Natalello et al.106
Koubaa et al. investigated the structure of the 11-mer repeat motif TdLEA3, an IDR of the Late Embryogenesis Abundant (LEA) protein. The results showed that TdLEA3 was mostly disordered under aqueous conditions and acquired an α-helical structure in a dry medium (Fig. 8). Because H2O overlaps in the amide I region, measurements in D2O were carried out. Bands in the region between 1660 and 1650 cm−1 were assigned to the α-helix, between 1640 cm−1 and 1650 cm−1 to unordered regions and at around 1620 cm−1 to intermolecular β-sheet aggregates. The hydrated IDR was centred at 1648 cm−1, indicating a mainly unstructured protein. Upon drying, this maximum shifted to 1657 cm−1, indicating a more α-helix oriented structure.107 Mohammad et al. utilized Attenuated Total Reflection Fourier Transform Infrared (ATR-FTIR) spectroscopy to study two intrinsically disordered protein α-synuclein variants, the IDP wildtype αS (αS-wt) and the naturally occurring splicing variant (αS-Δexon3). A disordered state in the amide I spectra for both compounds as the initial state was observed. A slow aggregation process was observed over time, but with striking dissimilarities: αS-wt revealed two bands at 1665 cm−1 and 1618 cm−1, whereas αS-Δexon3 exhibited a broad band with a maximum at 1630 cm−1. In the long term, both variants showed a conformational heterogeneity of secondary structures and aggregates but with some differences. The fibrillar aggregates dominated in αS-wt and the oligomers prevailed in αS-Δexon3. αS-wt showed a very low frequency that indicated a well-ordered extended β-sheet and fibrils with strong hydrogen bonds formed between the backbone amide carbonyls along the fibril axis. For αS-Δexon3, an absorption below 1630 cm−1 denoted the formation of typical β-structured aggregates.108 Villarreal-Ramirez et al. used FT-IR to demonstrate how one IDR of dentin phosphoprotein (DPP), named P5, assumed different conformations when associated with Ca2+ or hydroxyapatite (HA). Furthermore, they showed that after P5 phosphorylation (P5P), DPP also adopted distinct conformations. In solution, P5 was disordered, while P5P displayed a more compact globular structure. P5 had a higher amide I intensity with a narrow band, whereas P5P had a broadened amide I signal. Also, the P5 amide II band corresponding to the COO− of the Asp side chain was less intense, whereas the absorbance for P5P increased due to the substitution of phosphoserine residues. In the presence of Ca2+ or HA, P5 adopted a random coil structure, whereas its phosphorylated counterpart had a more compact arrangement associated with conformations that showed β-sheet and α-helix motifs. P5, in the presence of Ca2+ or HA crystals, showed a slight decrease in the amide I region, whereas the amide II band intensity was increased. These changes were associated with a modest decrease in random coil and an increase in a beta turn structure. In P5P, after adding Ca2+ or HA, the amide I band was broadened due to the formation of β-sheet structures.109 Vitali et al. analysed the structures of three IDPs (α-casein, Sic1 and α-synuclein) after interacting with silica NPs. α-Casein did not show conformational changes and continued being dominated by a peak at 1644 cm−1, assigned to disordered structures. In the case of Sic1, the appearance of amide I shoulders at ∼1638 cm−1 and 1689 cm−1 denoted a clear random coil to β-sheet transition. Finally, α-synuclein displayed a similar behaviour, the silica induced intermolecular interactions with a final β-sheet morphology (1627 cm−1 and 1696 cm−1).110
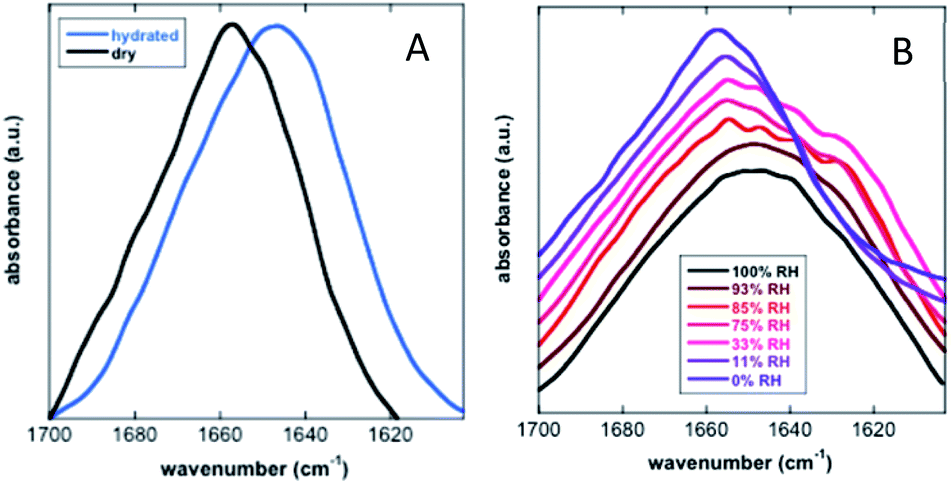 | ||
| Fig. 8 FT-IR analysis of the secondary structure of TdLEA3. (A) Amide I region in the hydrated (D2O, blue) and in the dry (black) state. (B) Amide I region at different relative humidities (RH). Reprinted with permission from Koubaa et al.107 Copyright 2019 Springer Nature. | ||
Small-angle X-ray scattering (SAXS)
SAXS was initially used exclusively to qualitatively monitor folding/unfolding processes. Currently, unlike most other structural methods, this technique is applied to equilibrium and non-equilibrium mixtures to monitor kinetic processes extracting the quantitative information of IDPs. For the reminder of the main theoretical and experimental aspects of X-ray scattering applied to IDPs, see Bernadó and Svergun.111Jephthah et al. studied the N-terminal IDR of the magnesium-transporter-A protein (KEIF). By the analysis of the form factor, Kratky plot and Pair Distance-Distribution Function (PDDF), they could observe the natively unfolded behaviour of this region. The absence of a minimum in the first one and a maximum in the second graph were typical curve shapes of a fully flexible and extended peptide.42 Lenton et al. studied the phosphorylation effect on the recombinant human-like osteopontin (rOPN). A plateau at high q values in the Kratky plot and the asymmetrical shape of the PDDF confirmed its highly unfolded and flexible behaviour. Also, phosphorylation appeared to have minimal effect on the solution scattering of rOPN, reflected in an overall unchanged conformation at the SAXS resolution.112 Didry et al. observed how a few amino acids in the IDR β-thymosin control the actin peptide self-assembly process. In a 1:1 stoichiometric ratio with actin, the β-thymosin inhibits its assembly by sequestering its monomers like thymosin-β4. In other words, an exchange in the β-thymosin linker –Phe–Asn–Gln–Asp–Lys– with a –Phe–Asp–Lys–Ser–Lys– one decreased the β-thymosin:actin binding affinity, showing the last linker mentioned a different structure in the SAXS spectra.113 Cragnell et al. proposed a molecular mechanism of oligomerization directed by divalent cations. After adding Zn2+ to histatin 5, a clear relationship between the cation concentration and the IDP was observed (Fig. 9). The addition of Zn2+ resulted in an increase of I(0) corresponded to an increase in the measured molecular mass. Furthermore, a less linear plateau in the Kratky plot also concluded that the cation led to a compaction of the overall protein. To conclude, this compaction was also supported by a redistribution of the PDDF towards shorter distances in the protein, moving to a more Gaussian-like structure in the presence of zinc.114 Hardouin et al. observed the behaviour of the RNaseY N-terminal IDR (BsRNaseY). The resulting SAXS curve averaged on the plateau gave values for Rg and the maximal extension, Dmax, significantly higher than those expected for a 176-residues compact protein. Therefore, the Dmax and Rg values indicated a highly elongated shape. Moreover, PDDF and Kratky curves were significantly different from that of a fully unstructured protein. Coupling this qualitative information with the strong propensity of BsRNaseY to form coiled-coil structures, they could finally fit the SAXS model factor to a representative central coiled-coil conformation appended with flexible ends.115
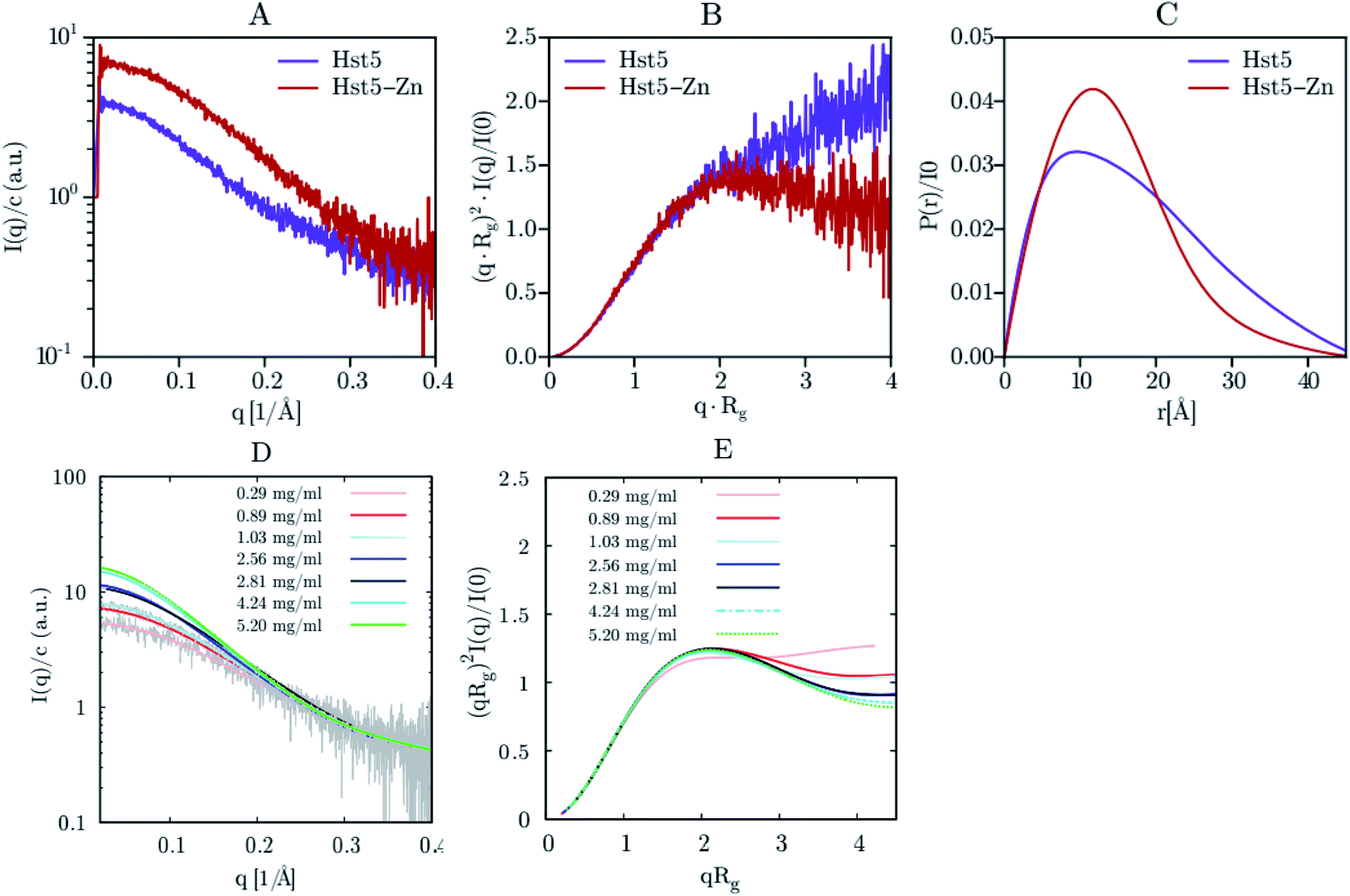 | ||
| Fig. 9 SAXS analysis of Hst5 in the absence and presence of ZnCl2. (A) Comparison of the intensity function normalised by concentration for 0.9 mg mL−1 Hst5, in 20 mM MES-buffer, pH 6.7, 150 mM NaCl and 4 mM ZnCl2. (B) SAXS data shown as a dimensionless Kratky plot. (C) Plot of the intra-peptide distance distribution determined by indirect Fourier transform, for Hst5, with either NaCl (purple curves) or ZnCl2 (red curves). (D and E) Concentration dependent SAXS-measurements of Hst5 in the presence of ZnCl2, showing the intensity curve normalised with protein concentration and the corresponding Kratky plot. Reprinted with permission from Cragnell et al.114 Copyright 2019 MDPI. | ||
Static and dynamic light scattering (SLS & DLS)
SLS and DLS can identify several different physical macromolecule parameters. SLS can measure molar masses within the 103–108 g mol−1 range, directly related to the state of association of IDPs in solution. On the other hand, DLS is an appropriate technique to monitor the expansion or compaction of protein molecules and their Stokes radius, RS. The radius of gyration, Rg, that could be obtained by SLS is not easily elucidated due to the small size of this systems. For a long and detailed explanation of the DLS and SLS basics applied to IDPs and IDRs, see Gast and Fiedler.116Chinak et al. used DLS to elucidate the structure–activity relationship of an analogue of lactaptin, RL2. They studied the structural and aggregation features of this fairly large intrinsically disordered fragment of human milk κ-casein. This IDR, due to its Pro and Gln-enriched AA sequences, self-assembled into micellar formation or amyloid fibrils, preventing casein precipitation in milk. Changes in pH from 5.5 to 8.0 and the addition of NaCl led to a dramatic increase in the diameter distributions, related to its oligomerization ratio. Under extracellular environmental conditions, RL2 led to large 700 nm diameter oligomers, while at pH 5.5 (corresponding to early endosomes), RL2 was predominantly in monomeric/dimeric forms, and its oligomers had a size of ca. 200 nm. Also, the presence of physiological ionic strength caused RL2 to oligomerize at lower pH (ca. 430 nm and ca. 290 nm diameter with and without NaCl, respectively).97 Zsila et al. observed how the addition of a drug or dye induced a self-organization on the cationic IDP CM15. After the addition of these ligands to the CM15 system in a ratio 1:1 or 1:2, an increase in the hydrodynamic radius of ca. 1000 nm was obtained, after the formation of large aggregates. The mutual charge neutralization within the complexes composed of cationic CM15 and its anionic partners could be reached. As a consequence, the resulting adducts became less hydrophilic and were prone to aqueous aggregation. Further increase of the ligand concentration (2:1 ratio) decreased the broadness of their size distribution.117 Khatun et al. used both techniques, DLS and SLS, to observe the possible structures of the IDP human amylin. The peptide self-assembled in aqueous media, as demonstrated by the presence of a small percentage of protofilaments with diameter values from 200 to 400 nm along with matured fibrils (>1000 nm). After a short sonication step, a reduction in the average size of the protofilaments (to 100–200 nm) as well as the fibril maturation (ca. 1000 nm) were observed. If extended to 30 min, even smaller protofilaments (50–100 nm) and slightly smaller fibrils (1000 nm) were detected.71 To conclude, Shou et al. also analysed the conformation selection, in this case, of the IDP COR15A. SLS was used to obtain a hydrodynamic radius of 2.5 nm, which corresponded to a typical IDP. At glycerol concentrations above 5.47 osM, an increase in RS up to 3.4 nm was observed, which is far above the scaling behaviour of IDPs and denoted the COR15A oligomer formation. After obtaining the Rg by DLS, the Rg/RS ratio concluded that the IDP had a slightly oblate shape in the absence of glycerol (ratios between 0.875 and 0.987) and adopted a more elongated conformation at values of osmotic concentrations between 1 and 3 osM (Rg/RS > 1). The aggregate structural change also could reverted back to an oblate ellipsoid at higher glycerol concentrations.118
Force fields and simulations
Experimental techniques for the characterization of IDPs/IDRs offer information on an average conformation upon binding or recognition process. The conformational ensembles of IDPs/IDRs still far exceed the number of available experimental observables. Thus, theoretical models are a suitable alternative for extracting detailed structural information at the atomic level, which experimental techniques cannot provide. Molecular Dynamics (MD) and Monte Carlo (MC) simulations are powerful tools to fill this gap. They produce a time sequence of atomic-level configurations and offer a potentially powerful complement to elucidate the key conformational characteristics of IDPs/IDRs. Even more, the atomistic details obtained from force field-based simulations can be used to help interpret experimental results. MD, while being the most commonly applied to IDPs/IDRs, rely on the accuracy of the underlying potential energy functions or force fields, so performing accurate structural characterization is a challenging task.Various force fields have been developed to describe biomolecular structures in aqueous environments. In this section, we will show some of the currently relevant force fields used in recent studies focused on the IDP/IDR field, which are also summarized in Table 3.
| Force fields | Parameter sets | Changes | References |
|---|---|---|---|
| AMBER | ff99 | First AMBER parameter set | 150 and 166 |
| ff99SB | Improved backbone torsional term | 74, 129, 143, 148, 150 and 166 | |
| ff99SB* | Corrected backbone energy term | 129, 143 and 146 | |
| ff99SB-ILDN | Improved side-chain torsion term | 44, 45, 49, 125, 129, 131, 132, 134, 135, 136, 150–152, 166 and 167 | |
| ff99SB*-ILDN | Improved side-chain torsion term | 128, 132, 137, 139, 147, 148 and 151 | |
| ff99SB-DISP | Corrected protein and water vdW terms | 127, 128 and 136 | |
| ff14SB | Improved backbone and side chain | 63, 126, 127, 130, 134, 137, 138, 149, 150 and 167 | |
| ff14IDPSFF | Corrected backbone torsional term | 126, 127 and 138 | |
| ff03 | Second AMBER parameter set | 135 and 166 | |
| ff03w | Corrected backbone torsional term | 132, 146 and 147 | |
| ff03ws | Modified protein–water interaction term | 127, 128 and 139 | |
| CHARMM | CHARMM22 | CHARMM parameter set | 47, 70, 129, 143, 158 and 166 |
| CHARMM22* | Corrected backbone energy term | 128, 136, 139, 146, 147, 148, 150–152 and 167 | |
| CHARMM36 | Modified backbone and side-chain torsional term | 41, 67, 129, 143, 146, 147, 149 and 150 | |
| CHARMM36m | Corrected backbone conformational term | 127–129, 132, 135–137, 144, 149 and 150 | |
| CHARMM36IDPSFF | Corrected backbone torsional term | 145 | |
| GROMOS | GROMOS96 43a1 | GROMOS parameter set | 111, 150 and 152 |
| GROMOS96 53a6 | Improved hydration thermodynamics reproduction | 107, 131, 150, 152, 158 and 166 | |
| GROMOS96 54a7 | Improved torsional term and hydration energy | 107, 131, 139, 150, 152, 158 and 159 | |
| OPLS | OPLS-AA | OPLS parameter set | 64, 139, 150, 158, 166 and 167 |
| OPLS-AA/L | Refitted Fourier torsional term | 135, 147 and 161 | |
| OPLS-AA/M | Refitted Fourier torsional term | 164 | |
| OPLSIDPSFF | Corrected backbone torsional term | 165 |
Assisted model building with energy refinement (AMBER)
AMBER is a suite of biomolecular simulation programs which started to be designed in the late 1970s by Peter Kollman.119 The energy function form of this force field used in protein, nucleic acid and organic molecule simulations is described as: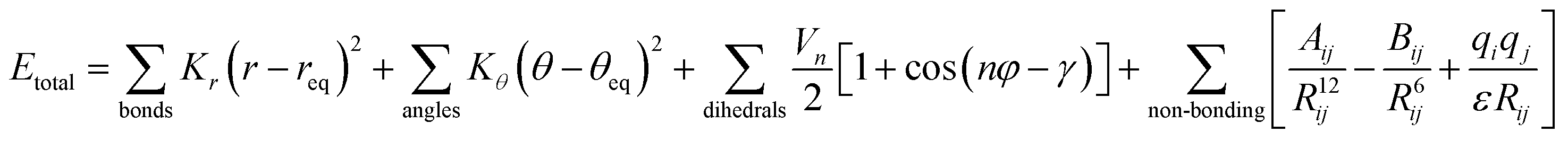 | (1) |
In brief, the model represents the bonds and angles by a simple diagonal harmonic expression, the dihedral energies by a simple set of parameters (often only specified by the two central atoms) and the non-bonding energies, electrostatic and van der Waals (vdW) interactions, are only calculated between atoms in different molecules or for atoms in the same molecule separated by at least a three bond distance. The first non-bonding energy value is modelled by a coulombic interaction of atom-centred point charges while vdW is represented by a 6–12 potential.120,121 To use the AMBER force field, it is necessary to have the preliminary parameter values of the force field (e.g. force constants, charges, equilibrium bond lengths and angles). Several authors during the last few decades have applied different base parameter sets, improving the original force field and finally leading to several parameter sets optimized for each analysed system. Here, we show the strength and weaknesses of the AMBER force field using some examples of its application to IDPs/IDRs. To know more about this package of computer programs, see Case et al.122
Based on relevant recent modifications of the force field for IDPs/IDRs, Chen group, based on AMBER ff99SB-ILDN, developed the AMBER ff99IDPs. They refined the IDPs sampling by transplanting residue-specific grid-based energy correction maps (CMAPs) corrections of eight disordered promoting residues (Ala, Arg, Gln, Glu, Gly, Lys, Pro, and Ser), improving the φ/ψ dihedral terms.123 This approach was followed by Song et al., who extended these CMAPs to all 20 amino acids, and proposed AMBER ff14IDPSFF, that raised its quality in the reproducibility of secondary chemical shifts of multiple short disordered proteins. 14 unstructured short peptides showed similar results between the simulated Cα chemical shifts obtained by NMR and the ff14IDPSFF force field. As an example, ff14IDPSFF produced diverse β-sheet conformers for the Tau protein, consistent with previous experimental observations.124 Continuing with this tendency, Song et al. also applied the CMAP approach and presented an environmental specific precise force field (ESFF1) to improve the accuracy and efficiency of MD simulations for both, IDPs and folded proteins.125 Meanwhile, Robustelli et al. proposed another force field, AMBER ff99SB-DISP, that could describe ordered, disordered, and transitional regions. They were able to achieve this goal by modifying the water model and iteratively testing small changes in backbone torsion corrections and the strength of the backbone O–H Lennard-Jones (LJ) pair.126 Best et al. started from AMBER ff03 and proposed AMBER ff03ws, strengthening the LJ potential for protein–water interactions and applying a scaling factor for protein–water interactions.127 Lately, Yu et al. introduced a residue-specific protein force field, ff99SBnmr2, derived from ff99SBnmr1. A different balance at the backbone dihedral angle potentials quantitatively better reproduced the dihedral angle distributions from a set of experimental coil systems.128
Focusing on their applicability, Henriques et al. applied AMBER ff99SB-ILDN and AMBER ff03ws to the IDP histatin 5, achieving a reasonable balance between protein–protein and protein–water dispersion interactions using a TIP4P-D and TIPAP/5 water model, respectively.129 Pietrek et al. observed the local and overall dimensions of the IDP α-synuclein. By using AMBER ff99SB-ILDN, they could match the modelling structure to the NMR and SAXS experimental data, complemented with AMBER ff03ws simulations for the possible force field issues of the full-length coiled structure.130 Rieloff et al. also compared this force field with CHARMM36m but in the 15-residue-long N-terminal fragment of the IDP fragment (SN15n) before and after phosphorylation (SN15p). While both force fields agreed regarding the size and shape of SN15n, for SN15p the CHARMM36m force field denoted strong interactions in the form of hydrogen bonding between the phosphorylated amino acids and the Arg residues. AMBER ff99SB-ILDN showed a less compacted structure higher in helical content, closer to what the CD experimental data suggested.131 Joseph et al. compared AMBER ff99SB-ILDN with older force fields such as AMBER ff14ipq or AMBER ff14SB in the human CD4 receptor, an IDR. Overall, ff99SB-ILDN performed better than its predecessors in terms of reproducing the HN-NMR shifts and J coupling constants. In the N-terminal peptidic region, ff14SB predicted more helical structures and ff14ipq more disordered ones than those observed experimentally.132 Ouyang et al. analysed the conformational features of the p53 distinct activation domain 2 (TAD2) IDR with different force fields. They concluded that AMBER ff99SB-ILDN showed a structural dimension closer to that theoretically predicted, with a more heterogeneous conformation. This force field provided correct results for p53 TAD2, whereas other force fields led to a collapse of the system. Force fields like CHARMM27 tended to over-stabilize a helical structure, CHARMM36m produced a most expended coil ensemble and OPLS-AA/L exhibited a strong preference on a β-sheet structure, far from experimental results.133 In a later study, Lui et al. demonstrated that the AMBER ff99SB-DISP force field had the best agreement with the experimental data obtained for, in this case, the p53 61-residue N-terminal TAD (Fig. 10). The AMBER ff99SB-DISP force field seemed capable of faithfully recapitulating virtually all experimental characterization results, including the overall chain dimensions, residual secondary structures, and transient long-range ordering. CHARMM36m and CHARMM36mw (CHARMM36m with a new water force field) failed to generate converged ensembles despite using multiple microsecond simulation time scales. CHARMM22* generated overly compact structural ensembles and an overestimation of the residual helicity, like AMBER ff99SB-ILDN.134 Kuzumanic et al. supported the use of AMBER ff99SB-DISP for the Von Willebrand Factor (VWF) study, being the force field that overall agreed best with the NMR data, followed by RSFF2+ and CHARMM36m ones. While all the force fields kept the β-sheets of the rigid VWF E′ domain in place, the TIL′ domain showed differences. By NMR, it could be inferred that this domain, as an IDR, lacked a secondary structure except for one 310-helical and three β-sheet regions. The AMBER ff99SB-DISP force field agreement with NMR came from the comparison of NOE distance restraints, chemical shifts, and backbone dihedral angles.135 Duong et al. were successful in simulating short peptides with a Glu–Gly–Ala–Ala–X–Ala–Ala–Ser–Ser structure (X = Asp, Gln, Glu, His, Leu, Lys, Pro, Trp, Tyr). Two force fields were tested and, while AMBER ff14SB denoted an increased helical content, a coiled content was obtained for ff14IDPSFF, with the latter in higher agreement with NMR and CD experiments.136 Henriques et al. also reported the improvement of AMBER ff03w compared to old AMBER and GROMOS force fields for the histatin 5 model. Previous models exhibited considerable bias towards overly compact conformational ensembles (force field independent) and certain secondary structure motifs (force field dependent), over-stabilizing the structure.129 Carballo-Pacheco et al. defended the use of AMBER ff03ws in the study of the aggregation and non-aggregation of the Alzheimer related Aβ16–22 IDP. GROMOS 54a7 and OPLS-AA strongly over-stabilized protein–protein interactions. AMBER99SB*ILDN and CHARMM22* were also considered, even though they were not that accurate.137
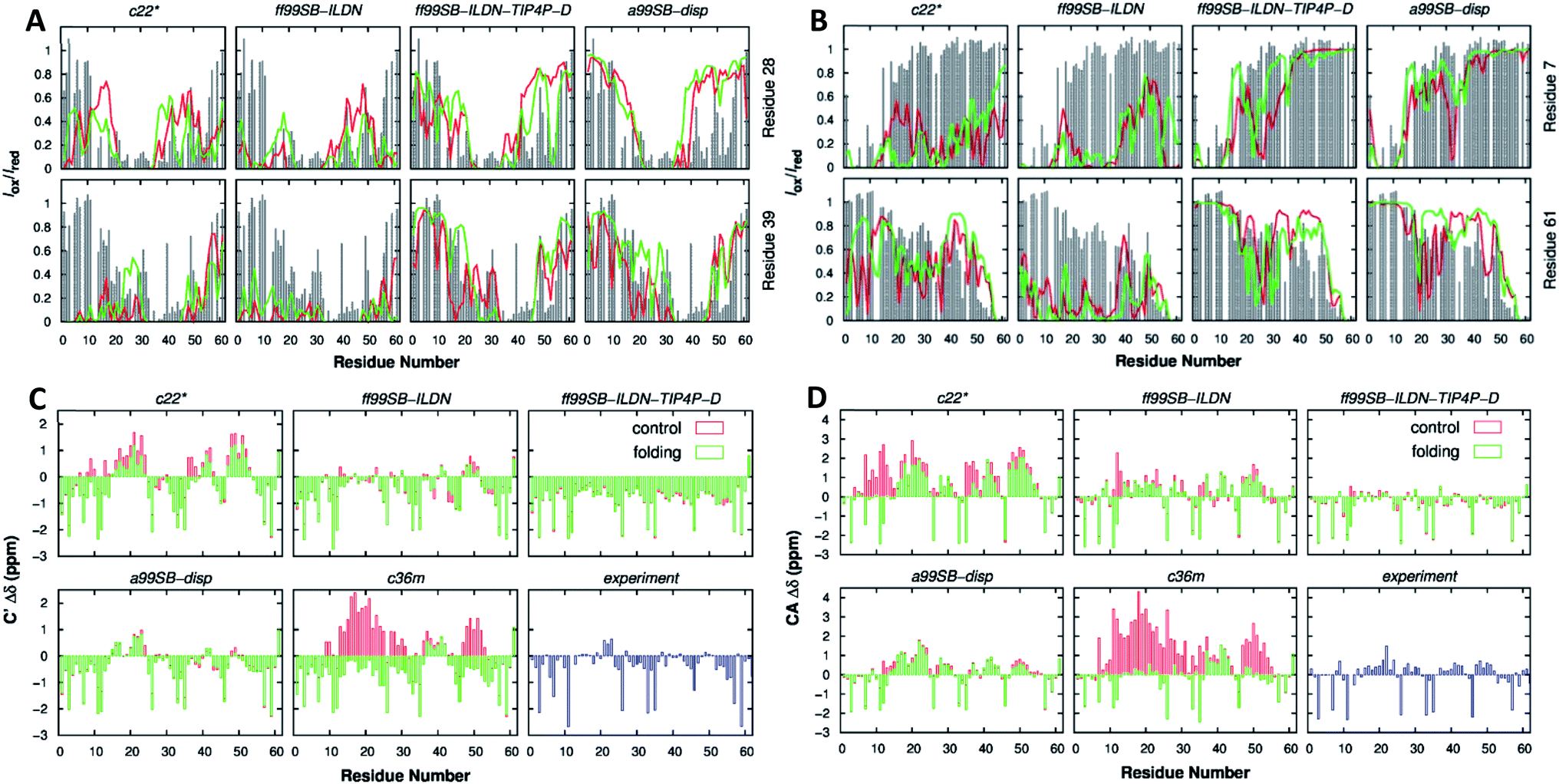 | ||
| Fig. 10 p53 61-residue N-terminal TAD (A) calculated (lines) and experimental (gray bars) paramagnetic relaxation enhancement effects induced by paramagnetic spin labeling at residues 28 (top row) and 39 (bottom row) and (B) residues 7 (top row) and 61 (bottom row). (C) Secondary chemical shift analysis for Cα atoms and (D) C′ atoms. Calculations were performed using independent control (red) and folding (green) simulations. Reprinted with permission from Lui et al.134 Copyright 2019 American Chemical Society. | ||
Chemistry at Harvard macromolecular mechanics (CHARMM)
CHARMM is a well-known and widely used set of force fields for molecular dynamics developed by Martin Karplus that can be used for DNA, RNA, lipids, drug-like molecules and especially proteins.138 The general form of the potential energy function most commonly used in CHARMM for self-assembling amphiphilic peptide simulations is based on fixed point charges and described as: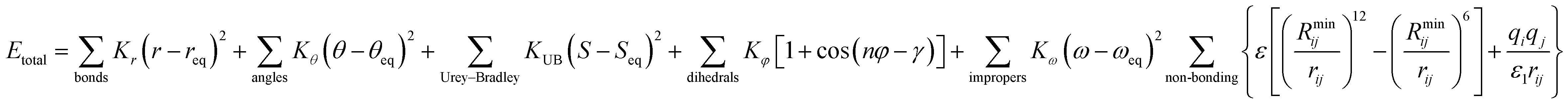 | (2) |
While terms like bond stretches, angles, dihedral force, or non-bonded forces appear as in the basic AMBER force field, two more are added in CHARMM. These terms account for the out of plane bending and the Urey–Bradley component, a cross-term accounting for the angle bending using 1,3 non-bonded interactions in the harmonic potential.139 A significant number of groups around the world are working on the development of the CHARMM package. Among them, the Charles L. Brooks III group deserves a special remark for their multiple improvements. Thus, we recommend their deep analysis of the program, from the basics to its implementation in different systems.140
After the improvement of the CHARMM22 force field by MacKerell et al. in the form of CHARMM22*,139 Best et al. proposed the CHARMM36 force field. They validated it by the comparison of: (i) simulations of eight proteins; (ii) backbone scalar couplings for each IDP/IDR; (iii) NMR residual dipolar couplings and scalar couplings for both the backbone and side-chains in folded proteins; (iv) folding equilibrium of peptides.141 Huang et al. presented the refinement of the CHARMM36 protein force field, the known CHARMM36m, improving the accuracy in generating polypeptide backbone conformational ensembles for intrinsically disordered peptides and proteins. The field was validated using a comprehensive set of 15 peptides and 20 proteins. In general, the sampling of αL-helical conformations in IDP ensembles generated with the CHARMM36m force field was significantly lower than in ensembles generated with CHARMM36, in agreement with experimental data. Examples of it were the arginine–serine peptide, the FG–nucleoporin peptide, a hen egg white lysozyme N-terminal fragment, and the N-terminal domain of HIV-1 integrase.142 On the other side, Liu et al. developed the CHARMM36IDPSFF, which showed an improvement over the CHARMM36 force field in 18 IDPs, even though some limitations were found in the radius of gyration of large disordered proteins and the stability of fast-folding ones.143
Lazar et al. carried out 24-residue Ser/Arg-rich (SR22-45) MD simulations using CHARMM22* and CHARMM36. The histogram of Rg distributions, compared to experimental data, showed a higher than real compactness in the CHARMM36 model, making the CHARMM22* force field the way to go. Results that also supported the use of CHARMM22* for this IDR were also reported by Rauscher et al.144,145 Carballo-Pacheco et al. tested the ability of five force fields to model the IDP Aβ42. Comparing their results to NMR experimental data, they observed how CHARMM22* was the best force field for reproducing Cα and HN chemical shifts associated with a β-hairpin structure. Particularly, CHARMM22* generated fewer compact conformations without the recalibration of protein–water interactions, as AMBER ff99SB*ILDN or AMBER ff03w. Older force fields like OPLS, GROMOS or CHARMM22 showed irreal structures.146 These results were updated by Krupa et al., who applied the main force fields currently used. They concluded that CHARMM36m > CHARMM36 > CHARMM22* force field for the IDP Aβ42. In CHARMM36m, the monomeric Aβ42 structure was less stable and more hydrophilic compared to AMBER. That could be explained by water interactions, which played a much more important role in CHARMM compared to AMBER.147 Man et al. compared 17 different force fields: 7 from the AMBER and GROMOS families, 3 from the CHARMM and one from the OPLS (Fig. 11). Applied to the seven-residue IDR fragment Aβ16–22, just 5 force fields were able to denote the real amyloid peptide assembly by providing good balances in terms of structures and kinetics. Among them, all the CHARMM force fields included (CHARMM22*, CHARMM36 and CHARMM36m) reported great results. While the old AMBER force fields predicted α-helices, far from real, and the GROMOS-family formed β-sheets too rapidly, CHARMM force fields matched the CD and NMR experimental data.148 Watts et al. compared the conformational space of the Aβ1–40 dimers using several force fields. They concluded that CHARMM22* and CHARMM36 were the chosen ones for explaining the collapse of the central and C-terminal hydrophobic cores from residues 17–21 and 30–36 and reproduced a theoretically expected β-sheet-turn-β-sheet conformational motif.149 These results were in agreement with Somavarapu et al., who defended the use of CHARMM22* over every AMBER, GROMOS or OPLS force field.150
 | ||
| Fig. 11 Aβ16–22 Dimer normalized distributions of the radius of gyration (Rg), the end-to-end distance (dee), the order parameter (P2), the intermolecular backbone H-bonds (NhbondC), the intermolecular side chain−side chain contacts (NscC), and the solvent accessible surface area (SASA). Reprinted with permission from Man et al.148 Copyright 2019 American Chemical Society. | ||
Groningen molecular simulation (GROMOS)
The GROMOS force fields are united atom force fields, i.e. without explicit aliphatic (non-polar) hydrogens. Developed in 1978 for the dynamic modelling of biomolecules, it was a simulation computer program package released by the research group of Wilfred van Gunsteren, who also realized a substantial rewrite of it in 1996.151,152 Known for having two different versions, GROMOS software can be applied to aqueous or non-polar solutions of proteins, nucleotides, and sugars (GROMOS force field A-version) or to simulate gas phase isolated molecules (B-version). For an understanding of how GROMOS, in general, and GROMOS05, specifically, work, see Christen et al.153The force field was updated twice during the last decade, leading to GROMOS 53a6 and GROMOS 54a7. The first one was done by Oostenbrink et al., introducing a new set of charges into the system to reproduce more accurately the hydration free enthalpies in water but with a drawback, an underestimation of the helical behaviour of peptides and proteins.154 The second one was developed by Schmid et al. Several corrections were applied, being the most relevant one the adjustment of the torsional angle terms to correct the helical inaccuracy mentioned before.155
While it is not the best force field for IDPs/IDRs compared to the AMBER and CHARMM families, authors such as Gerben et al. concluded that GROMOS96 54a7 and GROMOS 53a6, along with OPLS-AA, were the best force fields to explain the β-strand content in the intrinsically disordered amyloid β-peptide (Aβ). AMBER ff03 and CHARMM22 over-stabilized a helical structure and also could produce elongated Aβ structures, as for the last force field, far from what NMR shifts and Rg experimental data showed.156 Also, GROMOS96 54a7 was used by Bandyopadhyay et al. to study two IDRs (the scaffolding protein GPB from Escherichia virus phix174, 1CD3, and the human coagulation factor Xa, 1F0R) as well as two IDPs (α-synuclein, α-syn, and amyloid beta, Aβ42). 1CD3 showed three different structural conformations, α-syn had five and the last two peptides six. 1CD3 proposed structures were relatively more self-similar to each other by having the highest secondary structural as well as helical content, which made 1CD3 the closest peptide to the globular class out of all of them. α-Syn had more structural diversity yet a continuous transition behaviour. 1F0R and Aβ42 both had appropriate diverse structural phases with a substantial self-similarity among the conformational phases.157
Optimized potentials for liquid simulations (OPLS)
The OPLS force field was developed by William L. Jorgensen and started with a functional form very similar to the one used by AMBER. Currently used IDP-specific force fields have been mostly derived from the force fields of AMBER and CHARMM, while little attention has been paid to the widely used OPLS family. OPLS potential energy is expressed in a summary of 4 terms as:| Etotal = Ebonds + Eangles + Edihedrals + Enon-bonding | (3) |
One of the most relevant differences that can be observed is at the non-bonded interactions; while the charges used in the OPLS force fields are empirical, in AMBER they are obtained on a case-by-case basis from fitting to electrostatic potential surfaces from ab initio 6-31G* calculations. For a complete understanding and a comparison between OPLS and other force fields, such as AMBER and CHARMM, see Jorgensen et al.158
Focused just on the improvement of the force field and after the RSFF1 modifications of OPLS-AA/L by Jiang et al. and Xun et al.,159,160 Robertson et al. presented OPLS-AA/M. This force field demonstrated a significant improvement over previous OPLS-AA force fields. This model can be applied to normal peptides and IDPs out-performing previous OPLS-AA158 and OPLS AA/L161 dihedral parameters. Their ability to reproduce both gas phase conformer energies for longer peptides and aqueous phase experimental properties in molecular dynamics simulations was improved.162 The residue-specific force field OPLSIDPSFF, based on OPLS-AA/L, corrected the backbone dihedral term for all 20 residues by two-dimensional CMAPs (Fig. 12). IDPs and two short peptides were tested showing an agreement with NMR experimental results. The force field could obtain the β-sheet structures of GB1, while not stabilizing helix structures for the proteins AAQAA3 and GB1. In addition, the remaining disability of helical structures could be addressed by: (i) a novel CMAP refinement schedule, (ii) a more precise water model, or (iii) incorporating electronic polarization in a next step.163
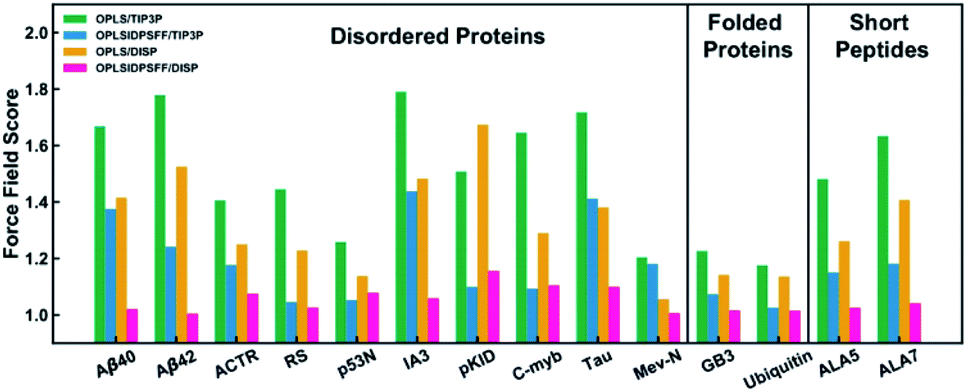 | ||
| Fig. 12 Normalized force field scores (lower the better) for short peptides, folded proteins, and disordered proteins. OPLS and OPLSIDPSFF represent the original OPLS-AA/L and the new force field, respectively. DISP means the disp-TIP4PD solvent model. Reprinted with permission from Yang et al.163 Copyright 2019 American Chemical Society. | ||
Smith et al. examined the dynamic behaviour of the IDR Aβ21–30 under seven force fields. Analysing the secondary structure, AMBER-family force fields, CHARMM27-CMAP, and GROMOS 53a6 were hindered in finding the local minima due to their enhancement of helical structures. OPLS-AA showed a substantially greater overall number of intrapeptide hydrogen bonds and suggested a metastable β-hairpin motif, associated with previous experimental results. Even more, OPLS was preferred as its sampling of Ramachandra space was more attuned to steric restrictions.164 Man et al. also supported the application of the OPLS-AA force field, but in this case, for the Aβ1–42 IDR. While AMBER SB14 and CHARMM22* ensembles significantly overestimated the CD-derived helix content, OPLS-AA, followed by AMBER ff99SB-ILDN, denoted a more accurate β-hairpin secondary structure. In the 17–21 and 30–36 regions, 8% and 13% β-hairpin were observed by both force fields, respectively, while AMBER SB14 showed only 1.5% and CHARMM22* 5%.165 Fluitt et al. observed that OPLS-AA/L, along with AMBER ff99SB and AMBER ff99SB*, was the most suitable for studies of polyglutamine (polyQ) folding and aggregation when comparing 12 force fields. OPLS-AA/L denoted predominantly disordered and collapsed conformations in water. CHARMM22*, and CHARMM36 exhibited no obvious biases in secondary structures but do exhibited larger persistence lengths, leading to more extended, aspherical, and diffuse conformations in water. CHARMM27 predicted a large fraction of helical secondary structures. GROMOS96 54a7 appeared to under-stabilize α-helices and over-stabilize β-sheets while GROMOS96 53a6 also failed in predicting a large fraction of β-strand content.166
Applications
Given all the possibilities that intrinsically disordered proteins or regions bring about, their applicability is also wide. Their multiple conformations, such as their core unique property position, make these biological structures highly relevant in the biomedical field. Already extended in several fields, IDP/IDR implementations are mostly in biology or biomedicine fields. Moreover, artificial disordered peptides are engineered to improve and tune up even more their performance and adapt their behaviour to specific functionalities. Due to the extension and all the possibilities that these short peptides structures can support, and not being the focus of this review, only a representative small sample of bio-related recent studies are summarized below.Focused on the biology field, cells present compartments called organelles to carry out their inner functions. However, membrane-less organelles formed via active liquid–liquid phase separation (LLPS) have garnered interest during the last few years. Proteins, peptides, and AAs can condense while being surrounded by a light phase, leading to a two-phase regime. Thermodynamically controlled, this process is based on intermolecular as well as water–water interactions, mostly by hydrogen bonding. However, this stimulus-responsive process is directed by external stimuli and environmental changes such as salt or molecule concentration, pH and temperature. Dzuricky et al. analysed a total of 63 IDPs that formed these membrane-less organelles in order to determine common structural features to exploit in future artificial IDPs. The octapeptide Gly–Arg–Gly–Asp–Ser–Pro–Tyr–Ser was the key to control LLPS processes by temperature and pH transitions. The formation and dynamics of their phase separation into coacervate droplets were controlled by two simple design parameters using in vitro and in vivo conditions: the molecular weight of the final octapeptide-based IDP and the aromatic:aliphatic ratio of residues in the octapeptide repeat.167 Savastano et al. used the IDP Tau at the AT180 epitope to regulate the cell compartmentation and form liquid-like droplets. While Tau assembled into microtubules, AT180 underwent LLPS in solution and on the surface of the microtubules. From these results, phosphorylation processes were suggested as a mechanism to modulate the LLPS of IDPs in a condensate-mediated cytoskeletal assembly.168 Metrick et al. reported the LLPS behaviour of the IDP UL11, from herpes simplex virus 1 (HSV-1). This tegument protein, while the process remains unclear, assembled as a biomolecular condensate in a complex network. Its disordered properties would form this membrane-less conformation, helping future biological processes such as membrane deformation during endocytosis.169 Dogra et al. also formed membrane-less organelles. In this case, they were controlled by using a pH-responsive IDR comprising 10 imperfect repeats rich in hydrophobic, polar, and acidic residues. Based on Ala, Gly, Thr, Pro, Ser and Val residues, this Pmel17 protein disordered domain promoted the formation of liquid droplets at neutral cytosolic pH that formed solid aggregates. At a mildly acidic melanosomal pH, the monomers self-assembled into amyloid fibrils in a reversible way.170 To study IDP LLPS relevance, Dignon et al. developed a model to predict temperature-dependent solvent-mediated interactions of each type of amino acid for further LLPS design. Sequences with an hourglass-shaped phase diagram or upper critical solution temperature behaviour generally were obtained for IDPs with more polar or charged residues than a typical IDP sequence.171 Using artificial simplified IDP models, Zhao et al. used elastin-like polypeptides (ELPs) in two compartmentalization strategies, namely bulk phase emulsion and cell-like compartment. ELP thermo-responsive phase transition properties allowed them to form membrane-less organelles via LLPS in the cellular milieu. This study is considered a significant step in the building of cell-mimicking systems with a higher degree of hierarchical complexity.172 Faltova et al. conjugated soluble globular domains to low complexity domains (LCDs) of a few disordered amino acids. In this way, they developed molecular adhesives that enabled sensitive and controlled self-assembly processes into final supramolecular architectures. LCD regions, which contained a high fraction of charged and polar amino acids, led to liquid–liquid phase separation processes due to their colocalization behaviour while the globular domain maintained its functionality. These chimera proteins reversibly self-assembled into liquid droplets which evolved into irreversible protein aggregates and finally solid particles over time. Finally, they applied active porous solid particles as microreactors, releasing soluble proteins over time.173 With a different application in mind, Urosev et al. used specific ELPs (hELPs) to restore the mechanical strength of fibrin networks, improve their clot development rate, reduce the plasmin degradation rate, and reduce the fibrin network pore size. IDPs mainly based on Val–Pro–Gly–X–Gly pentapeptides (with Ala, Glu and Val residues in guest X positions at a ratio of 2:8:1) coacervated at physiological temperature in β-spirals. The addition of a Gln residue to the N-terminal region, in the presence of the protein FXIIIa, covalently cross-linked the IDP by Lys–Gln interactions. After interacting with fibrinogen, thrombin and FXIII, hELP coacervates could be integrated into fibrin networks. These interactions took place through Gln- and Lys-residues on Fb γ-chains and α-chains, and AA cross-linked with hELP through its Gln- and Lys-blocks.174 Hossain et al. used intrinsically disordered peptide-polymers (IDPPs) for post-translational modifications (PTMs) adding a lipid chain to encode non-equilibrium phase behaviour transitions, an emergent frontier in biomacromolecular engineering. The IDR was based on a tropoelastin (Gly–X–Gly–Val–Pro)80 domain (containing a mixture of Ala:Val 2:8 in the X position), while the lipids tested were a canonical PTM (M-IDPP) and an azide (–N3) non-canonical PTM (ADA-IDPP). Both IDPPs self-assembled into spherical micelles at room temperature. When heated above the lower critical solubility temperature (LCST) around 31 °C and then cooled again, the azide based-IDPP behaviour was totally different. Unlike myristic acid, the ADA chain could not efficiently pack inside the hydrophobic core due to the forced linear arrangement of the terminal azide group. With heat, an increase in the mobility could facilitate the rearrangement of ADA-IDPP, leading to the shifting of the spherical micelles into rod-like aggregates.175 Wonderly et al., based on a marine mussel IDP (Mfp), improved the adhesion and cohesion of peptidic structures by changing their backbone to a peptoidic one.176 Bulutoglu et al. designed a stimulus responsive peptide based on two domains. The first domain was an IDR that self-assembled into a β-roll conformation when binding to Ca2+ due to its Leu residues. The second one was also a repeats-in-toxin domain that could recognize the lysozyme protein in specific situations. Ca2+ ions were responsible of the β-roll formation and final gelation, while the protein binding helped to obtain even more robust hydrogel networks.177
As stated before, IDPs/IDRs can participate in conditioning soft and hard extracellular matrices, among other structural processes. One of the most relevant is based on biomineral-associated protein interactions with final biomedical applications. Rao et al. showed how IDRs appeared to not only regulate the finally formed biomineral structure, but also modulate the formation and stability of crystal precursors. Four unstructured peptides with a vesicular shape were able to control and inhibit crystallization processes via a confinement-based mechanism. High Ca2+ concentrations forced organic–inorganic interactions and disorder-to-order transitions in these Gln, Thr or Ser rich peptides at high pH values. IDRs were able to interact with discrete mineral species and present lower free energy values, stabilizing and stopping the biomineralization process at intermediate structures between the Ca2+ ions and the final crystal conformation.54 In contrast, biomineralization processes can be enhanced and be directly applied to bone formation. Zhu et al. presented two biomolecules inspired by IDPs, denoted as P2 and P6, that helped the bone regeneration in 2D and 3D systems by increased biomineralization rates, cell attachment and proliferation. These rich-proline peptides were based on hydrophobic residues Leu, Met, Pro, and Val and polar Gln, His and Ser amino acids. The results showed how these amelogenin and ameloblastin hard tissue extracellular matrix protein imitations were more efficient that actual drugs such as Emdogain®.178 Roberts et al. studied synthetic partially organized polymers (POPs) based on ELP IDRs (a Val–Pro–Gly–X–Gly pentapeptide) attached to helix polyalanine (Ala–Ala–Ala–Ala–Ala) regions for tissue recovery (Fig. 13). While ELPs alone formed micrometre-sized coalescing aggregates, leading to a colloidal suspension of liquid-like droplets, POPs underwent arrested phase separation into porous networks. Moreover, the lower size the disordered ELP region presented, the more fractal-like architecture they showed in PBS media. Depending on the helical percentage, the pore size could be tuned, going from ca. 30–50 μm pores (90% polyalanine) to ca. 3–5 μm pores (60%). In vivo mice studies showed how POPs rapidly and robustly were integrated into the sub-cutaneous space, creating mechanical connections with the surrounding tissues and finally promoting wound healing and tissue growth.179 Recent studies by Chilkoti's group concluded how these POP structures self-assembled into fractal conformations. While using Val as a guest residue formed the already reported conformations, the use of Ala formed coacervate droplets with a physically crosslinked interconnected porous shell. The adjustment of the ELP/polyalanine ratio allowed the tuning of the porosity.180
 | ||
| Fig. 13 In vivo stability and tissue incorporation of POPs: (a) 125I radiolabelled E1-H5-25% POP subcutaneous injections were significantly more stable than their E1 counterparts, with just 5% of the injected dose (ID) degraded at 120 h; 200 μl 250 μM injections; p < 0.05 for all data points after 0 h, determined by two-tailed t-tests (n = 6 mice); data represent mean ± s.e.m. (b) Whereas ELPs diffuse into the subcutaneous space, POP deposits were externally apparent, retaining the shape and volume of the initial injection up to dissection and ex vivo analysis. (c) Representative CT-SPECT images of the deposits confirm the increased diffusivity of ELPs and the increased stability of POPs. (d) POPs were injected into BL/6 mice and explanted for analysis over 21 days. Representative images are shown with arrows pointing at externally evident vascularization. Scale bars: 5 mm. (e) POPs rapidly integrated into the subcutaneous environment with sufficient strength to endure moderate extension less than 24 h after injection. (f) There is a high initial cell incorporation with some change over the observed time periods; for *, p < 0.05 determined by ANOVA with Tukey post-hoc (day 1 n = 3, days 3–21 n = 4); data presented as 10–90% box plots. (g) Flow cytometry for cells involved in innate immunity reveals subsequent spikes in neutrophils, inflammatory monocytes, and macrophages, with a loss in all haematopoietic cells (CD45+) by day 21; for *, p < 0.05 determined by ANOVA with Tukey post-hoc (day 1 n = 3, days 3–21 n = 4); data represent mean ± s.e.m. (h) Population of haematopoietic-derived cells (CD45+) in time. (i) The loss in inflammation corresponds to an increase in vascularization, quantified by the number of visible capillaries in histological sections; for *, p < 0.05 as determined by ANOVA with Tukey post-hoc (n = 3); data represent mean ± s.e.m. (j) An example tissue slice 10 days post injection shows an area of particularly high vascularization density (scale bar: 100 μm). Reprinted with permission from Roberts et al.179 Copyright 2018 Springer Nature. | ||
Conclusions
The self-assembly and functional features of IDPs/IDRs constitute a hot and exciting topic. The relationship between the protein structure and disorder with the biological function of IDPs/IDRs is also a rich research area. Eight amino acid residues have been identified as the main promoters for the disordered behaviour: Ala, Arg, Gln, Glu, Gly, Lys, Pro, and Ser. Advanced experimental characterization methods connecting the amino acid sequence with the resulting disordered structure are highly desirable. Computational models and force fields accounting for the unique properties of IDPs/IDRs are to be developed. Preliminary studies with IDPs/IDRs show their promising performance in different areas. Biological and biotechnological applications stand out as the forefront field. With detailed understanding of the nature of IDPs/IDRs, nanotechnology will be one step closer to replicate real complex biological media and apply self-assembled nanostructures in biology and biotechnology.Conflicts of interest
There are no conflicts to declare.Acknowledgements
P. G. A. acknowledges the Région Nouvelle Aquitaine, the Université de Bordeaux, Bordeaux INP and CNRS for financial support. Support from the Ministry of Science and Innovation of Spain through the MANA project CTQ2017-83961-R and JEANS project CTQ2017-92264-EXP is acknowledged. J. J. G.-C. acknowledges the Ministry of Science and Innovation of Spain for a “Ramon y Cajal” contract (RyC-2014-14956). The Andalusian Government (Consejeria de Economia, Conocimiento, Empresas y Universidades, Junta de Andalucia) of Spain is acknowledged for the financial support through the UCO-1263193 Project.Notes and references
- B. O. Okesola and A. Mata, Chem. Soc. Rev., 2018, 47, 3721–3736 RSC.
- A. Méndez-Ardoy, J. R. Granja and J. Montenegro, Nanoscale Horiz., 2018, 3, 391–396 RSC.
- Z. Gong, X. Liu, J. Dong, W. Zhang, Y. Jiang, J. Zhang, W. Feng, K. Chen and J. Bai, Nanoscale, 2019, 11, 15479–15486 RSC.
- S. Li, Q. Zou, Y. Li, C. Yuan, R. Xing and X. Yan, J. Am. Chem. Soc., 2018, 140, 10794–10802 CrossRef CAS.
- S. Li, R. Xing, R. Chang, Q. Zou and X. Yan, Curr. Opin. Colloid Interface Sci., 2018, 35, 17–25 CrossRef CAS.
- D. Sanchez-deAlcazar, D. Romera, J. Castro-Smirnov, A. Sousaraei, S. Casado, A. Espasa, M. C. Morant-Miñana, J. J. Hernandez, I. Rodríguez, R. D. Costa, J. Cabanillas-Gonzalez, R. V. Martinez and A. L. Cortajarena, Nanoscale Adv., 2019, 1, 3980–3991 RSC.
- Z. Ye, X. Zhu, S. Acosta, D. Kumar, T. Sang and C. Aparicio, Nanoscale, 2019, 11, 266–275 RSC.
- Z. Ye and C. Aparicio, Nanoscale Adv., 2019, 1, 4679–4682 RSC.
- M. Gontsarik, M. Mohammadtaheri, A. Yaghmur and S. Salentinig, Biomater. Sci., 2018, 6, 803–812 RSC.
- C. L. Hedegaard, E. C. Collin, C. Redondo-Gómez, L. T. H. Nguyen, K. W. Ng, A. A. Castrejón-Pita, J. R. Castrejón-Pita and A. Mata, Adv. Funct. Mater., 2018, 28, 1703716 CrossRef.
- Y. Cong, L. Ji, Y. Gao, F. Liu, D. Cheng, Z. Hu, Z. Qiao and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 4632–4637 CrossRef CAS.
- B. O. Okesola, S. Ni, B. Derkus, C. C. Galeano, A. Hasan, Y. Wu, J. Ramis, L. Buttery, J. I. Dawson, M. D'Este, R. O. C. Oreffo, D. Eglin, H. Sun and A. Mata, Adv. Funct. Mater., 2020, 30, 1906205 CrossRef CAS.
- M. Cano and J. J. Giner-Casares, Adv. Colloid Interface Sci., 2020, 286, 102313 CrossRef CAS.
- Y. Wu, B. O. Okesola, J. Xu, I. Korotkin, A. Berardo, I. Corridori, F. L. P. di Brocchetti, J. Kanczler, J. Feng, W. Li, Y. Shi, V. Farafonov, Y. Wang, R. F. Thompson, M.-M. Titirici, D. Nerukh, S. Karabasov, R. O. C. Oreffo, J. Carlos Rodriguez-Cabello, G. Vozzi, H. S. Azevedo, N. M. Pugno, W. Wang and A. Mata, Nat. Commun., 2020, 11, 1182 CrossRef CAS.
- M. Liutkus, A. López-Andarias, S. H. Mejías, J. López-Andarias, D. Gil-Carton, F. Feixas, S. Osuna, W. Matsuda, T. Sakurai, S. Seki, C. Atienza, N. Martín and A. L. Cortajarena, Nanoscale, 2020, 12, 3614–3622 RSC.
- B. A. Shoemaker, J. J. Portman and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS.
- C. González-Obeso, M. González-Pérez, J. F. Mano, M. Alonso and J. C. Rodríguez-Cabello, Small, 2020, 2005191 CrossRef.
- H. Ruan, Q. Sun, W. Zhang, Y. Liu and L. Lai, Drug Discovery Today, 2019, 24, 217–227 CrossRef CAS.
- I. Yruela and J. L. Neira, Arch. Biochem. Biophys., 2020, 684, 108328 CrossRef CAS.
- J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 635–645 CrossRef CAS.
- B. Xue, A. K. Dunker and V. N. Uversky, J. Biomol. Struct. Dyn., 2012, 30, 137–149 CrossRef CAS.
- C. Tanford and J. Reynolds, Nature's Robots: A History of Proteins, 2001 Search PubMed.
- International Union of Pure and Applied Chemistry, Glossary of Class Names of Organic Compounds and Reactivity Intermediates Based on Structure, IUPAC, 1995 Search PubMed.
- W. T. Miller, Q. Rev. Biol., 1996, 71, 563 CrossRef.
- L. Pauling, R. B. Corey and H. R. Branson, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 205–211 CrossRef CAS.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 729–740 CrossRef CAS.
- A. M. C. Marcelino and L. M. Gierasch, Biopolymers, 2008, 89, 380–391 CrossRef CAS.
- J. S. Fetrow, FASEB J., 1995, 9, 708–717 CrossRef CAS.
- C. Tonlolo and E. Benedetti, Trends Biochem. Sci., 1991, 16, 350–353 CrossRef.
- B. W. Low and H. J. Greenville-Wells, Proc. Natl. Acad. Sci., 1953, 39, 785–801 CrossRef CAS.
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS.
- J. J. Panda and V. S. Chauhan, Polym. Chem., 2014, 5, 4418–4436 RSC.
- L. J. Smith, K. M. Fiebig, H. Schwalbe and C. M. Dobson, Folding Des., 1996, 1, 95–106 CrossRef.
- P. Kumar and M. Bansal, FEBS J., 2015, 282, 4415–4432 CrossRef CAS.
- D. J. Barlow and J. M. Thornton, J. Mol. Biol., 1988, 201, 601–619 CrossRef CAS.
- F. Zsila, G. Kohut and T. Beke-Somfai, Int. J. Biol. Macromol., 2019, 129, 50–60 CrossRef CAS.
- F. Zsila, T. Juhász, S. Bősze, K. Horváti and T. Beke-Somfai, Chirality, 2018, 30, 195–205 CrossRef CAS.
- D. Léon, M. P. Vermeuel, P. Gupta and M. R. Bunagan, J. Pept. Sci., 2020, 26, 1–7 CrossRef.
- M. E. Fealey, B. P. Binder, V. N. Uversky, A. Hinderliter and D. D. Thomas, Biophys. J., 2018, 114, 550–561 CrossRef CAS.
- O. T. Johnson, T. Kaur and A. L. Garner, ChemBioChem, 2019, 20, 40–45 CrossRef CAS.
- A. S. Saglam, D. W. Wang, M. C. Zwier and L. T. Chong, J. Phys. Chem. B, 2017, 121, 10046–10054 CrossRef CAS.
- S. Jephthah, L. K. Månsson, D. Belić, J. P. Morth and M. Skepö, Biomolecules, 2020, 10, 1–22 CrossRef.
- S. Jephthah, J. Henriques, C. Cragnell, S. Puri, M. Edgerton and M. Skepö, J. Chem. Inf. Model., 2017, 57, 1330–1341 CrossRef CAS.
- F. R. Salemme, Prog. Biophys. Mol. Biol., 1983, 42, 95–133 CrossRef CAS.
- O. Coskuner and V. N. Uversky, J. Chem. Inf. Model., 2017, 57, 1342–1358 CrossRef CAS.
- T. Takekiyo, N. Yamada, T. Amo and Y. Yoshimura, Pept. Sci., 2020, 112, 1–8 Search PubMed.
- S. Boopathi, P. Dinh Quoc Huy, W. Gonzalez, P. E. Theodorakis and M. S. Li, Proteins: Struct., Funct., Bioinf., 2020, 88, 1285–1302 CrossRef CAS.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11, 4985–4998 CrossRef.
- C. Has and S. Pan, J. Liposome Res., 2020, 1–22 Search PubMed.
- M. T. Ivanović, M. R. Hermann, M. Wójcik, J. Pérez and J. S. Hub, J. Phys. Chem. Lett., 2020, 11, 945–951 CrossRef.
- A. Accardo, M. Leone, D. Tesauro, R. Aufiero, A. Bénarouche, J. F. Cavalier, S. Longhi, F. Carriere and F. Rossi, Mol. BioSyst., 2013, 9, 1401–1410 RSC.
- S. H. Klass, M. J. Smith, T. A. Fiala, J. P. Lee, A. O. Omole, B. G. Han, K. H. Downing, S. Kumar and M. B. Francis, J. Am. Chem. Soc., 2019, 141, 4291–4299 CrossRef CAS.
- S. Acosta, Z. Ye, C. Aparicio, M. Alonso and J. C. Rodríguez-Cabello, Biomacromolecules, 2020, 21, 4043–4052 CrossRef CAS.
- A. Rao, M. Drechsler, S. Schiller, M. Scheffner, D. Gebauer and H. Cölfen, Adv. Funct. Mater., 2018, 28, 1802063 CrossRef.
- S. A. Costa, J. R. Simon, M. Amiram, L. Tang, S. Zauscher, E. M. Brustad, F. J. Isaacs and A. Chilkoti, Adv. Mater., 2018, 30, 1–9 CrossRef.
- M. Fändrich, Cell. Mol. Life Sci., 2007, 64, 2066–2078 CrossRef.
- M. Humenik, M. Magdeburg and T. Scheibel, J. Struct. Biol., 2014, 186, 431–437 CrossRef CAS.
- A. Hernik-Magoń, W. Puławski, B. Fedorczyk, D. Tymecka, A. Misicka, P. Szymczak and W. Dzwolak, Biomacromolecules, 2016, 17, 1376–1382 CrossRef.
- Y. Zhang, V. H. Man, C. Roland and C. Sagui, ACS Chem. Neurosci., 2016, 7, 576–587 CrossRef CAS.
- K. Pan and Q. Zhong, Soft Matter, 2015, 11, 5898–5904 RSC.
- M. Bakou, K. Hille, M. Kracklauer, A. Spanopoulou, C. V. Frost, E. Malideli, L. M. Yan, A. Caporale, M. Zacharias and A. Kapurniotu, J. Biol. Chem., 2017, 292, 14587–14602 CrossRef CAS.
- L. Larini, M. M. Gessel, N. E. Lapointe, T. D. Do, M. T. Bowers, S. C. Feinstein and J. E. Shea, Phys. Chem. Chem. Phys., 2013, 15, 8916–8928 RSC.
- J. Adamcik, A. Sánchez-Ferrer, N. Ait-Bouziad, N. P. Reynolds, H. A. Lashuel and R. Mezzenga, Angew. Chem., Int. Ed., 2016, 55, 618–622 CrossRef CAS.
- C. Despres, J. Di, F. X. Cantrelle, Z. Li, I. Huvent, B. Chambraud, J. Zhao, J. Chen, S. Chen, G. Lippens, F. Zhang, R. Linhardt, C. Wang, F. G. Klärner, T. Schrader, I. Landrieu, G. Bitan and C. Smet-Nocca, ACS Chem. Biol., 2019, 14, 1363–1379 CrossRef CAS.
- R. Dec, M. Koliński, M. Kouza and W. Dzwolak, Int. J. Biol. Macromol., 2020, 150, 894–903 CrossRef CAS.
- A. J. Kuhn and J. Raskatov, J. Alzheimer's Dis., 2020, 74, 43–53 CAS.
- A. J. Kuhn, B. S. Abrams, S. Knowlton and J. A. Raskatov, ACS Chem. Neurosci., 2020, 11, 1539–1544 CrossRef CAS.
- A. K. Jana, K. B. Batkulwar, M. J. Kulkarni and N. Sengupta, Phys. Chem. Chem. Phys., 2016, 18, 31446–31458 RSC.
- B. Konarkowska, J. F. Aitken, J. Kistler, S. Zhang and G. J. S. Cooper, FEBS J., 2006, 273, 3614–3624 CrossRef CAS.
- R. P. R. Nanga, J. R. Brender, S. Vivekanandan and A. Ramamoorthy, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2337–2342 CrossRef CAS.
- S. Khatun, A. Singh, S. Maji, T. K. Maiti, N. Pawar and A. N. Gupta, Soft Matter, 2020, 16, 3143–3153 RSC.
- F. G. Quiroz, N. K. Li, S. Roberts, P. Weber, M. Dzuricky, I. Weitzhandler, Y. G. Yingling and A. Chilkoti, Sci. Adv., 2019, 5, 1–12 Search PubMed.
- D. Stehli, M. Mulaj, T. Miti, J. Traina, J. Foley and M. Muschol, Intrinsically Disord. Proteins, 2015, 3, 1–12 Search PubMed.
- I. Bishof, E. B. Dammer, D. M. Duong, S. R. Kundinger, M. Gearing, J. J. Lah, A. I. Levey and N. T. Seyfried, J. Biol. Chem., 2018, 293, 11047–11066 CrossRef CAS.
- K. Dooley, B. Bulutoglu and S. Banta, Biomacromolecules, 2014, 15, 3617–3624 CrossRef CAS.
- W. P. Aue, E. Bartholdi and R. R. Ernst, J. Chem. Phys., 1976, 64(5), 2229–2246 CrossRef CAS.
- K. Wüthrich, Nat. Struct. Biol., 2001, 8, 923–925 CrossRef.
- G. M. Clore, eMagRes, 1996, 1–7 Search PubMed.
- A. M. Gronenborn and G. M. Clore, Biochem. Pharmacol., 1990, 40, 115–119 CrossRef CAS.
- B. Brutscher, I. C. Felli, S. Gil-Caballero, T. Hošek, K. Rainer, A. Piai, R. Pierattelli and Z. Sólyom, Adv. Exp. Med. Biol., 2015, 49–122 CrossRef CAS.
- B. Chaves-Arquero, J. M. Pérez-Cañadillas and M. A. Jiménez, Chem. - Eur. J., 2020, 26, 5970–5981 CrossRef CAS.
- S. Kosol, A. Gallo, D. Griffiths, L. Valentic, T. R. Masschelein, J. Jenner, M. de los Santos, E. L. C. Manzi, P. K. Sydor, D. Rea, S. Zhou, V. Fülöp, N. J. Oldham, S.-C. Tsai, G. L. Challis and J. R. Lewandowski, Nat. Chem., 2019, 11, 913–923 CrossRef CAS.
- M. G. Murrali, A. Piai, W. Bermel, I. C. Felli and R. Pierattelli, ChemBioChem, 2018, 19, 1625–1629 CrossRef CAS.
- S. Sukumaran, S. A. Malik, S. Sharma, K. Chandra and H. S. Atreya, Chem. Commun., 2019, 55, 7820–7823 RSC.
- S. E. Reichheld, L. D. Muiznieks, R. Stahl, K. Simonetti, S. Sharpe and F. W. Keeley, J. Biol. Chem., 2014, 289, 10057–10068 CrossRef CAS.
- W. B. Garry, J. T. Barbara, J. Roberts, L. S. Malcolm and W. J. Shaw, Biochim. Biophys. Acta, 2010, 1804, 1768–1774 Search PubMed.
- M. Beck Erlach, H. R. Kalbitzer, R. Winter and W. Kremer, Biophys. Chem., 2019, 254, 106239 CrossRef CAS.
- L. Hou, H. Shao, Y. Zhang, H. Li, N. K. Menon, E. B. Neuhaus, J. M. Brewer, I.-J. L. Byeon, D. G. Ray, M. P. Vitek, T. Iwashita, R. A. Makula, A. B. Przybyla and M. G. Zagorski, J. Am. Chem. Soc., 2004, 126, 1992–2005 CrossRef CAS.
- A. Fontana, P. P. De Laureto, B. Spolaore and E. Frare, Identifying Disordered Regions in Proteins by Limited Proteolysis, 2012, vol. 896 Search PubMed.
- L. B. Chemes, L. G. Alonso, M. G. Noval and G. De Prat-Gay, Methods Mol. Biol., 2012, 895, 387–404 CrossRef CAS.
- Y. Sun, A. Kakinen, Y. Xing, P. Faridi, A. Nandakumar, A. W. Purcell, T. P. Davis, P. C. Ke and F. Ding, Small, 2019, 15, 1–10 Search PubMed.
- L. Y. Rivera-Najera, G. Saab-Rincón, M. Battaglia, C. Amero, N. O. Pulido, E. García-Hernández, R. M. Solórzano, J. L. Reyes and A. A. Covarrubias, J. Biol. Chem., 2014, 289, 31995–32009 CrossRef CAS.
- S. Weickert, J. Cattani and M. Drescher, Electron Paramagn. Reson., 2019, 26, 1–37 CAS.
- N. L. Pirman, E. Milshteyn, L. Galiano, J. C. Hewlett and G. E. Fanucci, Protein Sci., 2011, 20, 150–159 CrossRef CAS.
- T. Bund, J. M. Boggs, G. Harauz, N. Hellmann and D. Hinderberger, Biophys. J., 2010, 99, 3020–3028 CrossRef CAS.
- R. Kaminker, I. Kaminker, W. R. Gutekunst, Y. Luo, S. Lee, J. Niu, S. Han and C. J. Hawker, Chem. Commun., 2018, 54, 5237–5240 RSC.
- O. A. Chinak, A. V. Shernyukov, S. S. Ovcherenko, E. A. Sviridov, V. M. Golyshev, A. S. Fomin, I. A. Pyshnaya, E. V. Kuligina, V. A. Richter and E. G. Bagryanskaya, Molecules, 2019, 24, 2919 CrossRef CAS.
- E. A. Permyakov and V. N. Uversky, in Instrumental Analysis of Intrinsically Disordered Proteins: Assessing Structure and Conformation, 2010, pp. 323–344 Search PubMed.
- S. Acharya, B. M. Safaie, P. Wongkongkathep, M. I. Ivanova, A. Attar, F. G. Klärner, T. Schrader, J. A. Loo, G. Bitan and L. J. Lapidus, J. Biol. Chem., 2014, 289, 10727–10737 CrossRef CAS.
- F. Zsila, T. Juhász, S. Bősze, K. Horváti and T. Beke-Somfai, Chirality, 2018, 30, 195–205 CrossRef CAS.
- F. Zhu, N. W. Isaacs, L. Hecht and L. D. Barron, Structure, 2005, 13, 1409–1419 CrossRef CAS.
- S. Signorelli, S. Cannistraro and A. R. Bizzarri, Appl. Spectrosc., 2017, 71, 823–832 CrossRef CAS.
- T. G. McCaslin, C. V. Pagba, J. Yohannan and B. A. Barry, Sci. Rep., 2019, 9, 1–14 CrossRef CAS.
- A. Rawat, B. K. Maity, B. Chandra and S. Maiti, Biochim. Biophys. Acta, Biomembr., 2018, 1860, 1734–1740 CrossRef CAS.
- C. La Rosa, M. Condorelli, G. Compagnini, F. Lolicato, D. Milardi, T. N. Do, M. Karttunen, M. Pannuzzo, A. Ramamoorthy, F. Fraternali, F. Collu, H. Rezaei, B. Strodel and A. Raudino, Eur. Biophys. J., 2020, 49, 175–191 CrossRef CAS.
- A. Natalello, D. Ami and S. M. Doglia, Methods Mol. Biol., 2012, 895, 229–244 CrossRef CAS.
- S. Koubaa, A. Bremer, D. K. Hincha and F. Brini, Sci. Rep., 2019, 9, 1–11 CrossRef CAS.
- M. A. Fallah, H. R. Gerding, C. Scheibe, M. Drescher, C. Karreman, S. Schildknecht, M. Leist and K. Hauser, ChemBioChem, 2017, 18, 2312–2316 CrossRef CAS.
- E. Villarreal-Ramirez, D. Eliezer, R. Garduño-Juarez, A. Gericke, J. M. Perez-Aguilar and A. Boskey, Bone, 2017, 95, 65–75 CrossRef CAS.
- M. Vitali, V. Rigamonti, A. Natalello, B. Colzani, S. Avvakumova, S. Brocca, C. Santambrogio, J. Narkiewicz, G. Legname, M. Colombo, D. Prosperi and R. Grandori, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 1556–1564 CrossRef CAS.
- P. Bernadó and D. I. Svergun, Mol. BioSyst., 2012, 8, 151–167 RSC.
- S. Lenton, M. Grimaldo, F. Roosen-Runge, F. Schreiber, T. Nylander, R. Clegg, C. Holt, M. Härtlein, V. García Sakai, T. Seydel and S. C. Marujo Teixeira, Biophys. J., 2017, 112, 1586–1596 CrossRef CAS.
- D. Didry, F. X. Cantrelle, C. Husson, P. Roblin, A. M. E. Moorthy, J. Perez, C. Le Clainche, M. Hertzog, E. Guittet, M. F. Carlier, C. Van Heijenoort and L. Renault, EMBO J., 2012, 31, 1000–1013 CrossRef CAS.
- C. Cragnell, L. Staby, S. Lenton, B. B. Kragelund and M. Skepö, Biomolecules, 2019, 9, 168 CrossRef CAS.
- P. Hardouin, C. Velours, C. Bou-Nader, N. Assrir, S. Laalami, H. Putzer, D. Durand and B. Golinelli-Pimpaneau, Biophys. J., 2018, 115, 2102–2113 CrossRef CAS.
- K. Gast and C. Fiedler, Methods Mol. Biol., 2012, 896, 137–161 CAS.
- F. Zsila, S. Bösze, K. Horváti, I. C. Szigyártó and T. Beke-Somfai, RSC Adv., 2017, 7, 41091–41097 RSC.
- K. Shou, A. Bremer, T. Rindfleisch, P. Knox-Brown, M. Hirai, A. Rekas, C. J. Garvey, D. K. Hincha, A. M. Stadler and A. Thalhammer, Phys. Chem. Chem. Phys., 2019, 21, 18727–18740 RSC.
- M. Levitt and V. Daggett, Nat. Struct. Biol., 2001, 8, 662 CrossRef CAS.
- P. K. Weiner and P. A. Kollman, J. Comput. Chem., 1981, 2, 287–303 CrossRef CAS.
- C. I. Bayly, K. M. Merz, D. M. Ferguson, W. D. Cornell, T. Fox, J. W. Caldwell, P. A. Kollman, P. Cieplak, I. R. Gould and D. C. Spellmeyer, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef.
- D. A. Case, T. E. Cheatham, T. Darden, H. Gohlke, R. Luo, K. M. Merz, A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef CAS.
- W. Wang, W. Ye, C. Jiang, R. Luo and H. F. Chen, Chem. Biol. Drug Des., 2014, 84, 253–269 CrossRef CAS.
- D. Song, R. Luo and H. F. Chen, J. Chem. Inf. Model., 2017, 57, 1166–1178 CrossRef CAS.
- D. Song, H. Liu, R. Luo and H. F. Chen, J. Chem. Inf. Model., 2020, 60, 2257–2267 CrossRef CAS.
- P. Robustelli, S. Piana and D. E. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E4758–E4766 CrossRef CAS.
- R. B. Best, W. Zheng and J. Mittal, J. Chem. Theory Comput., 2014, 10, 5113–5124 CrossRef CAS.
- L. Yu, D. W. Li and R. Brüschweiler, J. Chem. Theory Comput., 2020, 16, 1311–1318 CrossRef CAS.
- J. Henriques and M. Skepö, J. Chem. Theory Comput., 2016, 12, 3407–3415 CrossRef CAS.
- L. M. Pietrek, L. S. Stelzl and G. Hummer, J. Chem. Theory Comput., 2020, 16, 725–737 CrossRef.
- E. Rieloff and M. Skepö, J. Chem. Theory Comput., 2020, 16, 1924–1935 CrossRef CAS.
- J. A. Joseph and D. J. Wales, J. Phys. Chem. B, 2018, 122, 11906–11921 CrossRef CAS.
- Y. Ouyang, L. Zhao and Z. Zhang, Phys. Chem. Chem. Phys., 2018, 20, 8676–8684 RSC.
- X. Liu and J. Chen, J. Chem. Theory Comput., 2019, 15, 4708–4720 CrossRef CAS.
- A. Kuzmanic, R. B. Pritchard, D. F. Hansen and F. L. Gervasio, J. Phys. Chem. Lett., 2019, 10, 1928–1934 CrossRef CAS.
- V. T. Duong, Z. Chen, M. T. Thapa and R. Luo, J. Phys. Chem. B, 2018, 122, 10455–10469 CrossRef CAS.
- M. Carballo-Pacheco, A. E. Ismail and B. Strodel, J. Chem. Theory Comput., 2018, 14, 6063–6075 CrossRef CAS.
- M. Karplus, Annu. Rev. Biophys. Biomol. Struct., 2006, 35, 1–47 CrossRef CAS.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- B. R. Brooks, C. L. Brooks, A. D. Mackerell, L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS.
- J. Huang, S. Rauscher, G. Nawrocki, T. Ran, M. Feig, B. L. De Groot, H. Grubmüller and A. D. MacKerell, Nat. Methods, 2016, 14, 71–73 CrossRef.
- H. Liu, D. Song, Y. Zhang, S. Yang, R. Luo and H. F. Chen, Phys. Chem. Chem. Phys., 2019, 21, 21918–21931 RSC.
- T. Lazar, M. Guharoy, W. Vranken, S. Rauscher, S. J. Wodak and P. Tompa, Biophys. J., 2020, 118, 2952–2965 CrossRef CAS.
- S. Rauscher, V. Gapsys, M. J. Gajda, M. Zweckstetter, B. L. De Groot and H. Grubmüller, J. Chem. Theory Comput., 2015, 11, 5513–5524 CrossRef CAS.
- M. Carballo-Pacheco and B. Strodel, Protein Sci., 2017, 26, 174–185 CrossRef CAS.
- P. Krupa, P. D. Quoc Huy and M. S. Li, J. Chem. Phys., 2019, 151, 055101 CrossRef.
- V. H. Man, X. He, P. Derreumaux, B. Ji, X. Q. Xie, P. H. Nguyen and J. Wang, J. Chem. Theory Comput., 2019, 15, 1440–1452 CrossRef.
- C. R. Watts, A. Gregory, C. Frisbie and S. Lovas, Proteins: Struct., Funct., Bioinf., 2018, 86, 279–300 CrossRef CAS.
- A. K. Somavarapu and K. P. Kepp, ChemPhysChem, 2015, 16, 3278–3289 CrossRef CAS.
- W. F. v. Gunsteren, Biomolecular Simulation: GROMOS 96 Manual and User Guide, 1996, vol. 44 Search PubMed.
- W. R. P. Scott, P. H. Hünenberger, I. G. Tironi, A. E. Mark, S. R. Billeter, J. Fennen, A. E. Torda, T. Huber, P. Krüger and W. F. Van Gunsteren, J. Phys. Chem. A, 1999, 103, 3596–3607 CrossRef CAS.
- M. Christen, P. H. Hünenberger, D. Bakowies, R. Baron, R. Bürgi, D. P. Geerke, T. N. Heinz, M. A. Kastenholz, V. Kräutler, C. Oostenbrink, C. Peter, D. Trzesniak and W. F. Van Gunsteren, J. Comput. Chem., 2005, 26, 1719–1751 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. Van Gunsteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS.
- N. Schmid, A. P. Eichenberger, A. Choutko, S. Riniker, M. Winger, A. E. Mark and W. F. Van Gunsteren, Eur. Biophys. J., 2011, 40, 843–856 CrossRef CAS.
- S. R. Gerben, J. A. Lemkul, A. M. Brown and D. R. Bevan, J. Biomol. Struct. Dyn., 2014, 32, 1817–1832 CrossRef CAS.
- A. Bandyopadhyay and S. Basu, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140474 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- F. Jiang, C. Y. Zhou and Y. D. Wu, J. Phys. Chem. B, 2014, 118, 6983–6998 CrossRef CAS.
- S. Xun, F. Jiang and Y. D. Wu, Bioorg. Med. Chem., 2016, 24, 4970–4977 CrossRef.
- G. A. Kaminski, R. A. Friesner, J. Tirado-Rives and W. L. Jorgensen, J. Phys. Chem. B, 2001, 105, 6474–6487 CrossRef CAS.
- M. J. Robertson, J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2015, 11, 3499–3509 CrossRef CAS.
- S. Yang, H. Liu, Y. Zhang, H. Lu and H. Chen, J. Chem. Inf. Model., 2019, 59, 4793–4805 CrossRef CAS.
- M. D. Smith, J. S. Rao, E. Segelken and L. Cruz, J. Chem. Inf. Model., 2015, 55, 2587–2595 CrossRef CAS.
- V. H. Man, P. H. Nguyen and P. Derreumaux, J. Phys. Chem. B, 2017, 121, 5977–5987 CrossRef CAS.
- S.-H. Chong, P. Chatterjee and S. Ham, Annu. Rev. Phys. Chem., 2017, 68, 117–134 CrossRef CAS.
- M. Dzuricky, B. A. Rogers, A. Shahid, P. S. Cremer and A. Chilkoti, Nat. Chem., 2020, 12, 814–825 CrossRef CAS.
- A. Savastano, D. Flores, H. Kadavath, J. Biernat, E. Mandelkow and M. Zweckstetter, Angew. Chem., Int. Ed., 2021, 60, 726–730 CrossRef CAS.
- C. M. Metrick, A. L. Koenigsberg and E. E. Heldwein, mBio, 2020, 11, 1–22 CrossRef.
- P. Dogra, A. Joshi, A. Majumdar and S. Mukhopadhyay, J. Am. Chem. Soc., 2019, 141, 20380–20389 CrossRef CAS.
- G. L. Dignon, W. Zheng, Y. C. Kim and J. Mittal, ACS Cent. Sci., 2019, 5, 821–830 CAS.
- H. Zhao, V. Ibrahimova, E. Garanger and S. Lecommandoux, Angew. Chem., Int. Ed., 2020, 59, 11028–11036 CrossRef CAS.
- L. Faltova, A. M. Küffner, M. Hondele, K. Weis and P. Arosio, ACS Nano, 2018, 12, 9991–9999 CrossRef CAS.
- I. Urosev, J. Lopez Morales and M. A. Nash, Adv. Funct. Mater., 2020, 30, 2005245 CrossRef CAS.
- M. S. Hossain, C. Maller, Y. Dai, S. Nangia and D. Mozhdehi, Chem. Commun., 2020, 56, 10281–10284 RSC.
- W. R. Wonderly, T. R. Cristiani, K. C. Cunha, G. D. Degen, J. E. Shea and J. H. Waite, Macromolecules, 2020, 53, 6767–6779 CrossRef CAS.
- B. Bulutoglu, S. J. Yang and S. Banta, Biomacromolecules, 2017, 18, 2139–2145 CrossRef CAS.
- H. Zhu, M. Gomez, J. Xiao, G. Perale, F. Betge, S. P. Lyngstadaas and H. J. Haugen, ACS Appl. Bio Mater., 2020, 3, 2263–2274 CrossRef CAS.
- S. Roberts, T. S. Harmon, J. L. Schaal, V. Miao, K. Li, A. Hunt, Y. Wen, T. G. Oas, J. H. Collier, R. V. Pappu and A. Chilkoti, Nat. Mater., 2018, 17, 1154–1163 CrossRef CAS.
- S. Roberts, V. Miao, S. Costa, J. Simon, G. Kelly, T. Shah, S. Zauscher and A. Chilkoti, Nat. Commun., 2020, 11, 1 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |