
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The tridecaptins: non-ribosomal peptides that selectively target Gram-negative bacteria†
Samantha J.
Bann
 ,
Ross D.
Ballantine
and
Stephen A.
Cochrane
*
,
Ross D.
Ballantine
and
Stephen A.
Cochrane
*
School of Chemistry and Chemical Engineering, Queens University Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, UK. E-mail: s.cochrane@qub.ac.uk
First published on 22nd January 2021
Abstract
Tridecaptins are a re-emerging class of non-ribosomal antibacterial peptides (NRAPs) with potent activity against highly problematic strains of Gram-negative bacteria. An intricate mode of action has been reported to explain the bactericidal activity of these NRAPs, wherein they bind selectivity to the Gram-negative version of the peptidoglycan precursor lipid II on the outer leaflet of the inner membrane and disrupt the proton-motive force. Tridecaptins are highly amenable to synthetic modification owing to their linear structure, therefore, various synthetic analogues have been reported, several of which have enhanced antimicrobial activity, reduced cost of synthesis and/or improved stability towards D-peptidase mediated hydrolysis. It has also been demonstrated that unacylated tridecaptins can act synergistically with clinically relevant antibiotics by sensitizing the outer membrane. This review will summarize past literature on the development/discovery of novel tridecaptin analogues (up until the end of 2020), some of which may be useful therapeutic agents to treat insidious Gram-negative bacterial infections.
1. Introduction
Antimicrobial resistance (AMR) poses a major threat to healthcare systems and global economies and if overlooked, could have disastrous implications for modern medicine. Current projections paint an unsavoury picture; it is predicted that by 2050, 10 million people could die annually due to AMR.1 Tuberculosis (TB) claims 1.8 million lives each year2 and despite available therapies, approximately half a million cases of TB were reported in 2018 in which the infection was resistant to the front-line antibiotic, rifampicin.3 Although the imminent threat of AMR is clear, persistent overuse of antibiotics4,5 and a discovery void, which has seen many pharmaceutical companies withdraw from antimicrobial research and development,6,7 have perpetuated this issue.The need for novel antimicrobial agents is clear. While there are many clinically approved antibiotics with selectivity for Gram-positive bacteria,8 there remains an urgent demand for those with broader activity against the more robust Gram-negative bacteria. The latter are inherently more difficult to kill than Gram-positives, owing to the presence of an outer membrane (OM) in their structure. This extra layer of protection obstructs membrane crossing for larger antibiotics,9 thereby making discovery of Gram-negative-targeting agents more challenging. The ever-increasing prevalence of multidrug-resistant (MDR) bacteria is recognized by the World Health Organisation (WHO) as an imminent threat to human life, particularly the critical priority pathogens Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacteriaceae,10 all of which are Gram-negative pathogens. Fortunately, the current preclinical pipeline of antibacterial research is working towards addressing this issue, as a significant proportion of current research is towards treatments for Gram-negative bacterial infections.7
Peptides are experiencing ongoing interest as potential antibacterial candidates; as of 2019, 27 antimicrobial peptides (AMPs) were undergoing clinical trials, while 34 were under preclinical investigation.11 AMPs also offer the unique advantage that their often non-specific mechanism of action means resistance is less likely to arise11–13 compared to other classes of antibacterial agents, while their relative ease of synthesis makes them favourable antibacterial drug candidates. Non-ribosomal antibacterial peptides (NRAPs) are a promising class of antibiotic drug candidate owing to the nature of their synthesis. Produced by non-ribosomal peptide synthetases (NRPSs), the ability to incorporate both proteinogenic and non-proteinogenic amino acids into the peptide sequence, and the occurrence of enzyme-mediated modifications, allows for diverse structural scaffolds14,15 that can facilitate chemical modification to improve efficacy.
The tridecaptins are a linear class of NRAP that display selective activity against Gram-negative bacteria. They were initially discovered in 197816 but after this early work by Kato et al., there was a significant innovation gap until 2014, when research into this class of NRAPs by the Vederas group began.17 This review will summarize research relating to the tridecaptins up until November 2020. We have provided the structures of all known natural and synthetic tridecaptin derivatives, as well as any available antimicrobial activity and toxicity data in the supporting information. We recommend that readers refer to this document whilst reading our review.
In early work by Kato et al., three separate antimicrobial tridecapeptides were identified as tridecaptin A, B and C, with differences in their amino acid compositions and lipid tail structures postulated (Table 1). Edman degradation and acid hydrolysis were used to distinguish the structural components of each novel peptide. Tridecaptin A1 (TriA1) was reported to bear a 3-hydroxy-6-methyloctanoyl (aiC9h3) tail,20 while subsequent HPLC analysis identified a major and minor component, differing in amino acid composition. The major component of the native peptide, A1 (formerly Aα) (1), contains allo-Ile at position 12, while in the minor component, A2 (formerly Aβ) (2), this residue is replaced with Val12.18 Tridecaptins B and C were also reported to possess differing elemental compositions; both have L-Ser13 in place of L-Ala and the former contains a 6-methyloctanoyl (aiC9) tail while the latter was reported to contain two fatty acyl derivatives, methyl β-hydroxy anteiso undecanoate (aiC11h3) and methyl β-hydroxy isodecanoate (iC10h3).21 HPLC analysis also identified differing moieties within TriB and C. Tridecaptin B is comprised of Bα (5), Bβ (6), Bγ (7), and Bδ (8) components, differing only in the presence of Val, Ile or aIle residues (Table 1) whilst tridecaptin C is made up of Cα1 (9), Cα2 (10), and Cβ1 (11) distinguishable in both their lipid tails and Val/aIle ratios.18
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound number | Tridecaptin variant | Variable residues | Lipid tail (R)a | ||||
| 1 | 5 | 9 | 12 | 13 | |||
| a Only stereochemistry of aiC9h3 and aiC9 have been reported. b The lipid tail assignment of TriA1 (1) will henceforth be taken as aiC9h3 in line with elucidations by Lohans et al.17 Lipid tail nomenclature; ai = anteiso, i = iso. Uncommon amino acids native to tridecaptin; D-Dab (D-diaminobutyric acid), D-aIle (D-allo-isoleucine). | |||||||
| 1 17,18 | A1 (Aα) | D-Val | D-Trp | L-Phe | D-aIle | L-Ala | aiC9h3, aiC11h3, iC11h3b |
| 2 18 | A2 (Aβ) | D-Val | D-Trp | L-Phe | D-Val | L-Ala | aiC9h3 |
| 3 19 | A3 | D-Val | D-Trp | L-Phe | D-aIle | L-Ala | aiC11h3, iC11h3 |
| 4 19 | A4 | D-Val | D-Phe | L-Phe | D-aIle | L-Ala | aiC11h3, iC11h3 |
| 5 18 | Bα | Gly | D-Trp | L-Ile | D-aIle | L-Ser | aiC9 |
| 6 18 | Bβ | Gly | D-Trp | L-Ile | DVal | L-Ser | aiC9 |
| 7 18 | Bγ | Gly | D-Trp | L-Val | D-aIle | L-Ser | aiC9 |
| 8 18 | Bδ | Gly | D-Trp | L-Val | D-Val | L-Ser | aiC9 |
| 9 18 | Cα1 | D-Val | D-Trp | L-Phe | D-Val | L-Ser | aiC11h3 |
| 10 18 | Cα2 | D-Val | D-Trp | L-Phe | D-Val | L-Ser | iC10h3 |
| 11 18 | Cβ1 | D-Val | D-Trp | L-Phe | D-aIle | L-Ser | aiC11h3 |
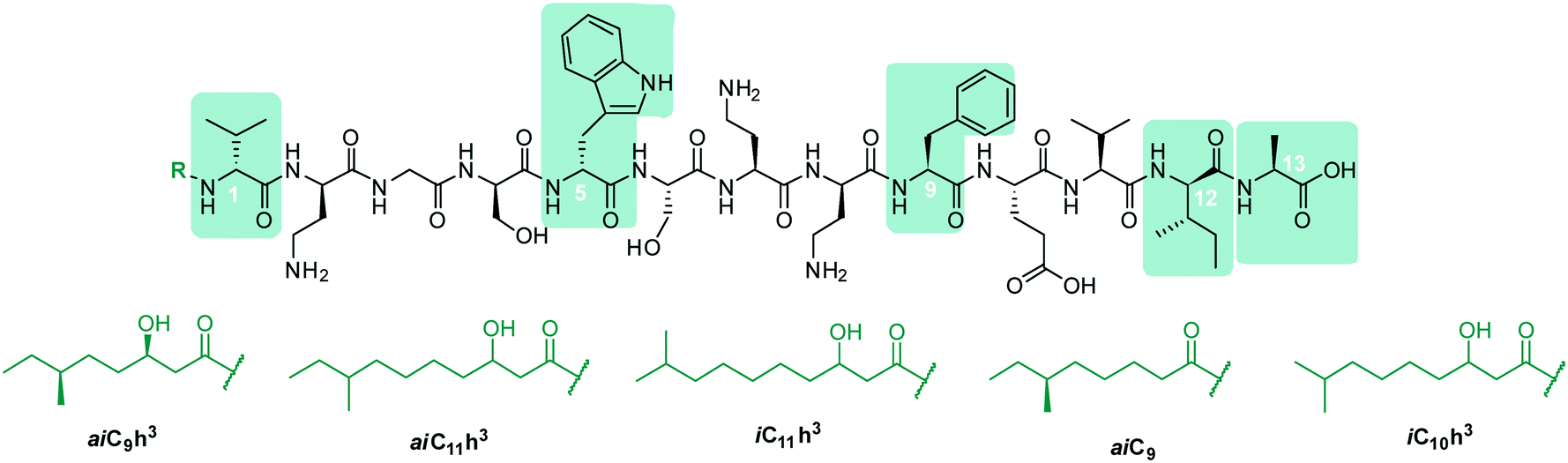
In recent years, the tridecaptin class became a focus of research for the Vederas group. In their first report, they showed that the antibacterial agent produced by Paenibacillus polymyxa NRRL B-30509 with activity towards Campylobacter jejuni was actually TriA1, rather than a previously reported bacteriocin (SRCAM 602).19 The two most prevalent variants were tridecaptin A3 (3) and A4 (4) for which two possible lipid tails were identified as 3-hydroxy-8-methyldecanoyl (aiC11h3) and 3-hydroxy-9-methyldecanoyl (iC11h3) (Table 1), the former being the major lipid component. Having elucidated amino acid and lipid tail discrepancies within the TriA class of AMP, Lohans et al. subsequently recommended that the original naming system be replaced with a more rudimentary numbering system (Aα was henceforth to be known as A1, Aβ as A2, etc.) to now encompass the tridecaptin A class.
After the initial resurgence of the tridecaptins, many new studies have been performed, including on their structural elucidation and/or biosynthesis,17,22–24 structure–activity relationships (SAR),25–29 mechanism of action,30 synergy with and conjugation to other antibiotics31,32 and as selective Gram-negative-staining agents.33
2. Resurgence of the tridecaptins: establishing a structure–activity relationship
TriA1, (1) is a NRAP with robust bactericidal activity against Gram-negative bacteria.26 The tridecaptins belong to the linear cationic family of antimicrobial peptides; they possess 13 amino acids, including non-proteinogenic residues, and have a chiral lipid tail. Unfortunately, the need to incorporate a chiral moiety during peptide synthesis significantly increases their cost of synthesis (as well as adding several solution-phase synthesis steps to obtain the chiral lipid). In the first complete structural elucidation of a tridecaptin analogue, the Vederas group showed that TriA1 contained a (3R,6S)-3-hydroxy-6-methyloctanoyl lipid tail.17 This was achieved by chemically synthesizing the four possible lipid variants of TriA1 ((3R,6S) (1); (3R,6R) (12), (3S,6S) (13), and (3S,6R) (14)) (Fig. 1) and comparing these to the natural peptide using high-performance liquid chromatography (HPLC) and nuclear magnetic resonance (NMR) experiments. Interestingly, the chirality of the lipid tail did not have a significant impact on antimicrobial activity.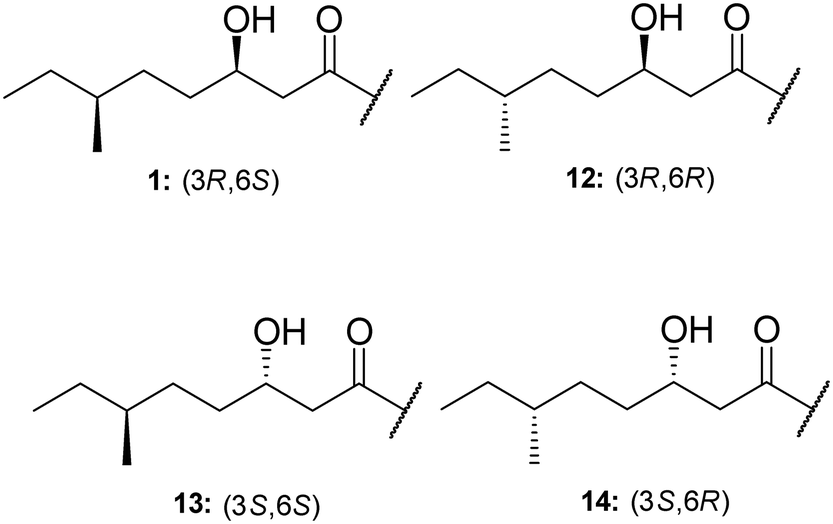 | ||
| Fig. 1 Chiral lipid tail variants of TriA1. A methodical approach was employed to assist in the complete stereochemical assignment of TriA1. Both HPLC and NMR were used to definitively ascertain the lipid tail stereochemistry as 3R,6S. | ||
In the same study, the Vederas group also sequenced the producer strain (Paenibacillus terrae NRRL B-30644) and identified the biosynthetic gene cluster (BGC) responsible for the production of the tridecaptins (Fig. 2), which contains a predicted thioesterase (triA), two putative ABC (ATP-binding cassette) transporter proteins (triB and triC) and two non-ribosomal peptide synthetases (triD and triE).
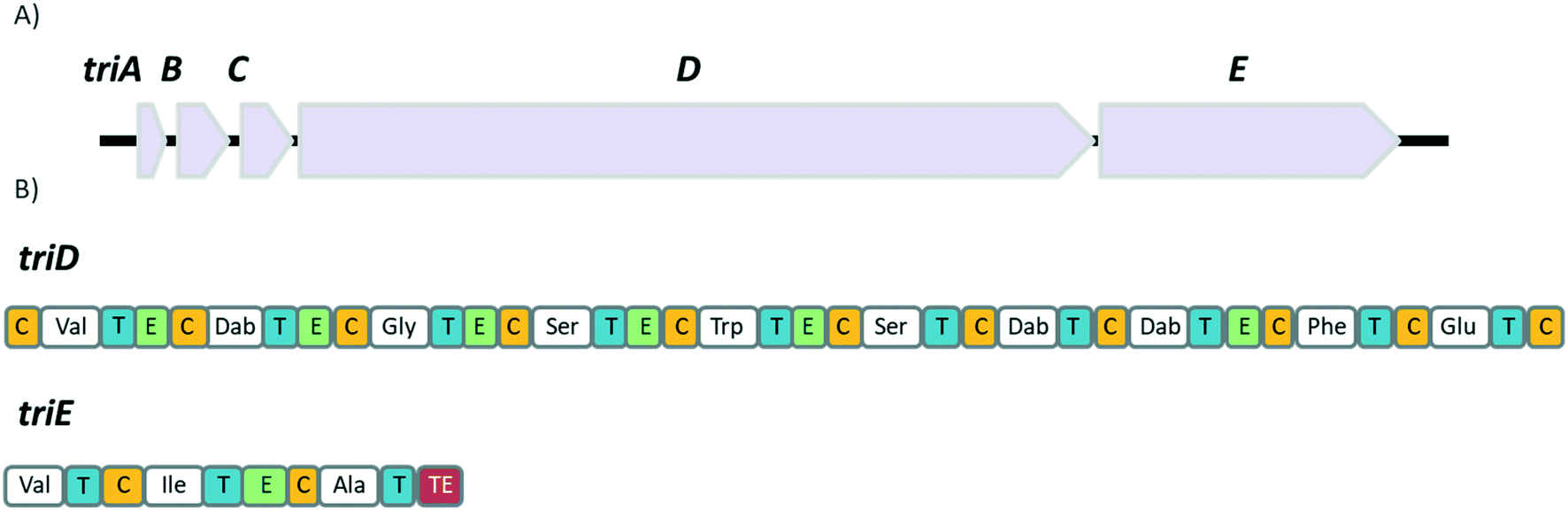 | ||
| Fig. 2 A) Biosynthetic gene cluster of TriA1 (from P. terrae NRRL B-30644) containing a predicted thioesterase (triA), two putative ABC transporter proteins (triB and triC) and NRPSs, triD and triE. B) Predicted domains of the NRPSs, triD and triE. Condensation domains (C) are depicted in gold, adenylation domains in white, thiolation domains (T) in blue, epimerase domains (E) in mint and the thioesterase domain (TE) in red. | ||
In a follow-up study, Cochrane et al. synthesized a library of TriA1 lipid-chain analogues (16–32).26 The major finding from this work was that that chirality of the lipid chain was not essential for activity (although it was noted that inversion of both stereochemical centres along the lipid tail gave a four-fold decrease in activity). The lipid tail on the tridecaptins is essential for activity, as truncated analogue H-TriA1 (15) (Fig. 3) displayed a significant reduction in antimicrobial activity against Escherichia coli (MIC = 100 μg mL−1) compared to the native peptide (1) (MIC = 3.13 μg mL−1). From the range of analogues tested, Oct-TriA1 (16) consistently gave the most potent activity against all strains tested. Various lipid tail lengths were reasonably well tolerated (C4–12, 18–24) and only showed two- to four-fold decreases in activity against the model Gram-negative strain, E. coli, when compared to the natural peptide (1).26 However, when longer lipid chains were used (C14/16, 25–26), activity was abolished across most strains (MIC >100 μg mL−1), likely due to the poor solubility of analogues. The necessity of a lipid tail for antimicrobial activity, and indeed the correlation to chain length, aligns with the behaviour of lipopeptides such as TriA1 to act as cell membrane disrupters. Indeed, it was found that the truncated derivative, H-TriA1 (15), exhibits reduced cytotoxic effects when compared to the native peptide, owing to its ability to sensitize the bacterial membrane, rather than disrupt it.26,31 The discovery that the synthetic analogue, Oct-TriA1 (16), retained all properties of the natural peptide, provided a significantly cheaper and more efficient method to access tridecaptin analogues. Oct-TriA1 was also found to exhibit strong activity against multidrug resistant Klebsiella and Enterobacter strains.26
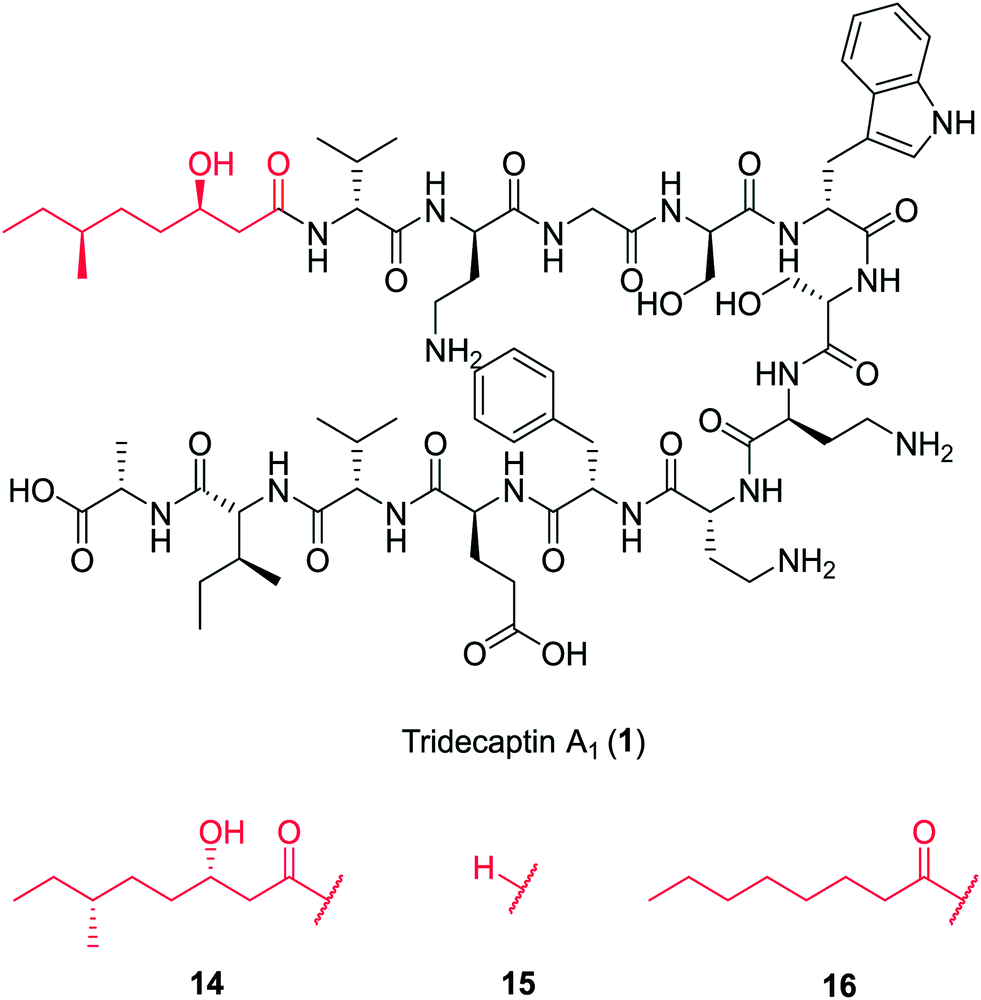 | ||
| Fig. 3 Variants of tridecaptin A1. The structures of natural tridecaptin A1 (1) and synthetic lipid tail analogues (14–16) are shown. | ||
Despite its lack of antimicrobial activity, the unacylated analogue H-TriA1 (15) has been shown to work synergistically with a range of antibiotics, where it acts as a strong outer-membrane sensitizer of Gram-negative bacteria.31 In the first study on the synergistic effects of H-TriA1 with other antibiotics, it was found that sub-MIC concentrations of H-TriA1 reduced the MIC of clinically relevant antibiotics rifampicin and vancomycin against E. coli by up to 512- and 64-fold, respectively. H-TriA1 (15) can also act as a sensitizer of MDR Klebsiella pneumoniae, improving susceptibility to antibiotic treatments by up to 512-fold. Recently, a collaborative effort between the Cochrane, Payne, Martin and Vederas groups, and the Ferring Research Institute, showed that H-TriA1 significantly improves the antimicrobial activity of teixobactin analogues to include Gram-negative strains.34 Perhaps the most significant observation was a 125-fold decrease in MIC of native teixobactin against Salmonella enterica when co-administered with H-TriA1 at 12.5 μg mL−1.
While partial membrane lysis was ruled out as a potential explanation for the synergistic effects of H-TriA1 (15),31 the similarity of its circular dichroism (CD) spectra in the presence of large unilamellar vesicles (LUVs) as model membranes, to that of the synthetic analogue, Oct-TriA1 (16),25 suggests that formation of a stable secondary structure on the OM may facilitate sensitization of Gram-negative bacteria. In a later paper describing the mechanism of action of the tridecaptins, it was shown by isothermal titration calorimetry (ITC) that these peptides bind to lipopolysaccharide (LPS), a glycopolymer found on the surface of the outer membrane.30 Therefore, it is likely that H-TriA1 binding to LPS is the mechanism of sensitization, which is how the related polymyxin B nonapeptide (PMBN) sensitizes Gram-negative bacteria.35
In another study on the synergistic effects of the tridecaptins with existing antibiotics, H-TriA1 and Oct-TriA1 were covalently linked to the antibiotics rifampicin, vancomycin and erythromycin.32 Vancomycin and erythromycin are inactive against Gram-negative bacteria due to their inability to cross the outer membrane, so it was hoped that conjugation to the tridecaptins could allow their passage through this barrier. Copper-assisted azide-alkyne cycloaddition (CuAAC) was the method used for conjugation and Glu10 was identified as an ideal site to install an azide group on H-TriA1 and Oct-TriA1. Azidopeptides H-TriA1(Glu10-PEG3N3) (33) and Oct-TriA1(Glu10-PEG3N3) (34) (Fig. 4) both retained their respective antimicrobial and synergistic activities. Conjugation to an erythromycin alkyne, vancomycin alkyne or rifampicin alkyne yielded tridecaptin antibiotic conjugates 35–40 in moderate – good yields (Fig. 4). When screened for in vitro activity against E. coli, K. pneumoniae and A. baumannii strains, the tridecaptin-antibiotic conjugates were much less effective than a synergistic mixture of the individual components. However, when tested in a mouse-infection model, it was found that the erythromycin–H-TriA1 conjugate (35) was more effective at promoting mouse survival than the synergistic combination. In this in vivo model, it was found that the synergistic combinations were not particularly effective, although this was just a single study with a small number of animals.
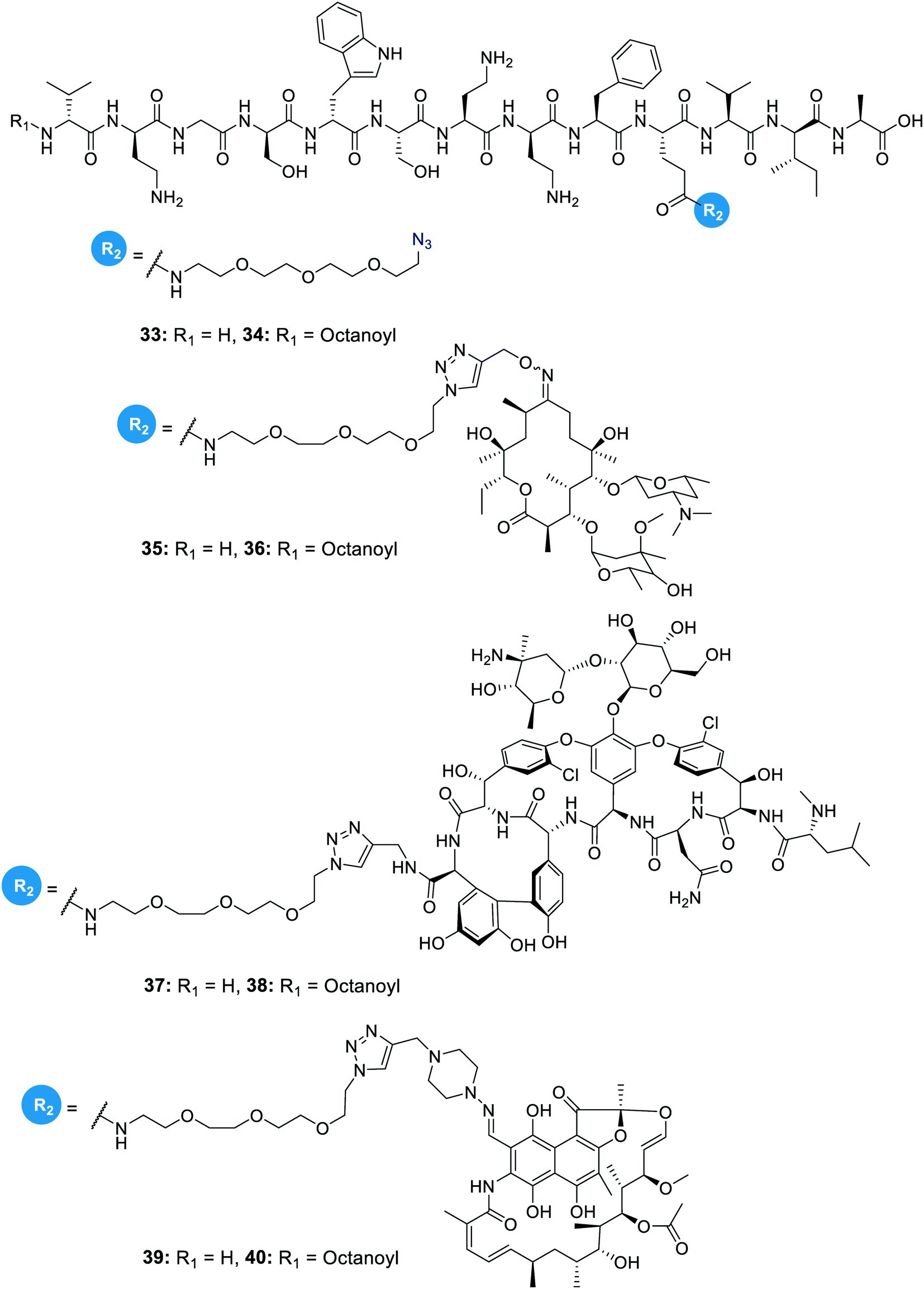 | ||
| Fig. 4 Azidopeptides and covalently linked tridecaptin–antibiotic conjugates. The structures of azidopeptides, H-TriA1(Glu10-PEG3N3) (33) and Oct-TriA1(Glu10-PEG3N3) (34), are shown alongside tridecaptin–antibiotic conjugates, H-TriA1-Eryc (35), Oct-TriA1-Eryc (36), H-TriA1-Van (37), Oct-TriA1-Van (38), H-TriA1-Rif (39) and Oct-TriA1-Rif (40). | ||
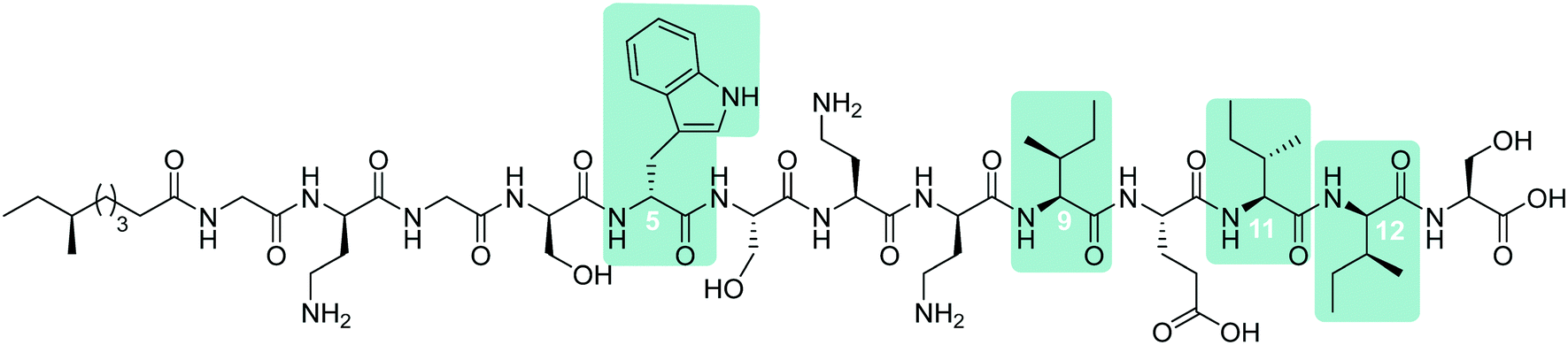
To probe the mechanism of action of the tridecaptins, the Vederas lab performed an alanine scan on Oct-TriA1 (16).25 The antimicrobial activity of all alanine scan analogues (41–52) was determined and their secondary structure in the presence of model membranes was analyzed using CD spectroscopy. Residue substitutions that proved most detrimental to antimicrobial activity were D-Dab8, D-allo-Ile12 and D-Trp5 (Fig. 5). In particular, substitution of the cationic residue D-Dab8 completely abolished antimicrobial activity, suggesting this site plays an important role in the mechanism of action of the peptide.25 Given that both D-allo-Ile12 and D-Trp5 are hydrophobic residues, it was speculated that their substitution causes a significant reduction in the hydrophobicity of Oct-TriA1 and impacts formation of a stable secondary structure. CD spectroscopy was employed to assess how alterations to the peptide sequence altered its secondary folding. 50 nm phospholipid LUVs were used to mimic the bacterial membrane environment.25
 | ||
| Fig. 5 Synthetic Oct-TriA1 analogue (16) with key residues highlighted. Substitution of these D-amino acid residues, Trp5, Dab8 and aIle12, resulted in the observed reductions in activity (relative to Oct-TriA1) against E. coli ATCC 25922. The most significant reduction in activity was associated with replacement of the D-Dab8 residue, suggesting it plays an intrinsic role in antimicrobial activity. | ||
The CD spectra of each alanine scan analogue was then recorded to assess the effects of amino acid substitution on formation of a stable secondary structure. It was found that reduction of the intensity of the negative band at 194 nm correlated with a loss in antimicrobial activity. Substitution of essential residues D-Dab8, D-allo-Ile12 and D-Trp5 significantly reduced activity and disrupted native peptide folding, verifying that a sequence-activity relationship exists, that when altered, impedes efficient peptide folding.25 In this study the authors also showed that the enantiomeric form of Oct-TriA1, Ent-Oct-TriA1 (53) was ∼four-fold less active, suggesting a chiral receptor may be involved in the mechanism of action.
In 2016, Cochrane, Findlay and Vederas finally identified a plausible mechanism of action for how the tridecaptins kill Gram-negative bacteria. Bactericidal growth kinetics and membrane lysis experiments showed that TriA1 rapidly kills bacteria, consistent with a membrane-targeting antimicrobial. However, follow-up membrane depolarization and membrane lysis assays showed that TriA1 does not significantly disrupt the cell membrane. This result was corroborated in a subsequent study by Sean Brady and co-workers as part of their work on synthetic-NRPs.36 Subsequent experiments suggested that TriA1 disrupts the proton-motive force via the formation of proton-specific pores within the membrane of Gram-negative bacteria, altering cytoplasmic pH and thus leading to bacterial cell death, likely due to prevention of ATP synthesis.30 With a mode of action identified, Cochrane et al. then looked for possible receptors to which TriA1 binds to to cross the outer membrane and interact with the inner membrane.
Isothermal titration calorimetry showed that TriA1 (1), Oct-TriA1 (16), the enantiomeric form of TriA1 (Ent-TriA1 (53)) and H-TriA1 (15) all bind to LPS with similar affinities. This suggested that TriA1 crosses the outer membrane by first binding to LPS. Further ITC experiments showed a remarkable selectivity of TriA1 for the Gram-negative version (contains diaminopimelic acid (DAP) rather than lysine) of the peptidoglycan precursor lipid II. Incubating TriA1 with Gram-negative lipid II before antimicrobial testing sequestered activity through complex formation and an intricate in vitro assay showed that LUVs doped with Gram-negative lipid II resulted in much faster proton leakage than undoped or Gram-positive lipid II doped vesicles. A solution NMR structure of Oct-TriA1 in the presence of Gram-negative lipid II and dodecylphosphocholine (DPC) micelles was determined and docking experiments used to produce a model of this interaction. The resulting model (Fig. 6) showed a key interaction between TriA1D-Dab8, the only truly essential residue, and lipid II DAP, the major structural difference between Gram-negative and Gram-positive lipid II.30 This may be why the tridecaptins specifically kill Gram-negative-bacteria. Taken together, this study provided a plausible mechanism of action for TriA1, by which it binds to LPS on the outer membrane, enters the periplasm, binds to lipid II on the outer leaflet of the inner membrane and disrupts the proton-motive force.
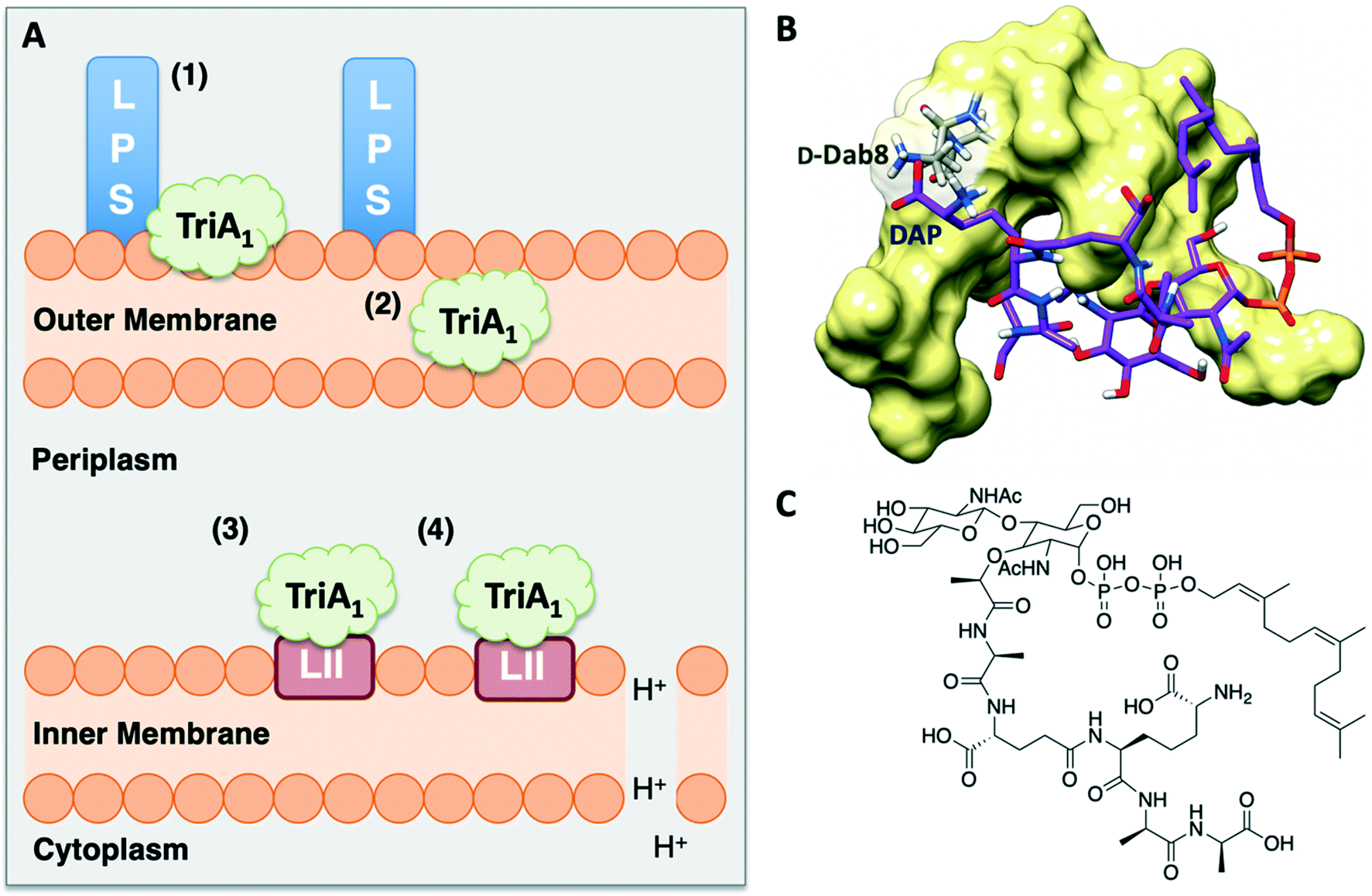 | ||
| Fig. 6 A) Summary of TriA1 mechanism of action.30 TriA1 interacts with LPS on the surface of the outer membrane (1). It then crosses the outer membrane (2) and enters the periplasm. TriA1 then binds to Gram-negative lipid II on the outer leaflet of the inner membrane (3) and disrupts the proton motive force (4). B) Model of TriA1-lipid II complex. Figure reproduced from deposited solution NMR structure of Oct-TriA1 in DPC micelles containing Gram-negative lipid II structure (PDB: 2N5Y) (obtained from Dr Stephen Cochrane). Lipid II is shown as a stick figure, with backbone in purple, and Oct-TriA1 shown as a surface representation. The interaction between D-Dab8 and DAP is highlighted, with the peptide visible as a stick figure with backbone in grey. C) Structure of synthetic Gram-negative lipid II analogue. In this study a synthetic Gram-negative lipid II analogue containing a shorter prenyl chain was used as this modification improved solubility for NMR studies. | ||
3. How robust are tridecaptins towards antimicrobial resistance?
Initial assessment of bacterial resistance towards the tridecaptins yielded positive results; a resistance development study in which E. coli cells were exposed to sub-MIC concentrations of Oct-TriA1 (16) for one month saw no resistance development but an eightfold reduction in activity was observed against the ciprofloxacin control.30 The lack of resistance development against the tridecaptins may be associated with the importance of their lipid II target in peptidoglycan biosynthesis. As this precursor is essential for formation of the bacterial cell wall, modification of this intermediate is unlikely due to potential deleterious effects downstream. Therefore, development of a ‘self-destructive’ resistance mechanism is unlikely. Unfortunately, a self-protection mechanism in tridecaptin-producing strains was recently identified that could have implications for their use as therapeutic agents.37D-Stereospecific resistance peptidase (DRP) enzymes, BogQ and TriF (from Brevibacillus laterosporus and P. polymyxa strains respectively), were heterologously expressed in E. coli and it was shown that the conserved catalytic region in each (Ser-XX-Lys and Tyr-XX) was the basis of peptidase activity.37 Both DRPs hydrolyse Oct-TriA1 (16) at the C-terminus of D-amino acid residues in the peptide chain, with BogQ showing specificity for D-cationic amino acids D-Dab2 and D-Dab8, and TriF showing selectivity for D-aromatic amino acid D-Trp5 (Fig. 7). Hydrolysis by either enzyme renders the peptide inactive. It is speculated that bacteria produce both BogQ and TriF D-stereospecific resistance peptidases as a self-protection strategy to evade cell death by their own antimicrobial metabolites. Worryingly, this level of inherent resistance has the potential to mobilize into pathogenic bacteria via horizontal gene transfer, potentially hindering the development of tridecaptins as novel therapeutic agents.37
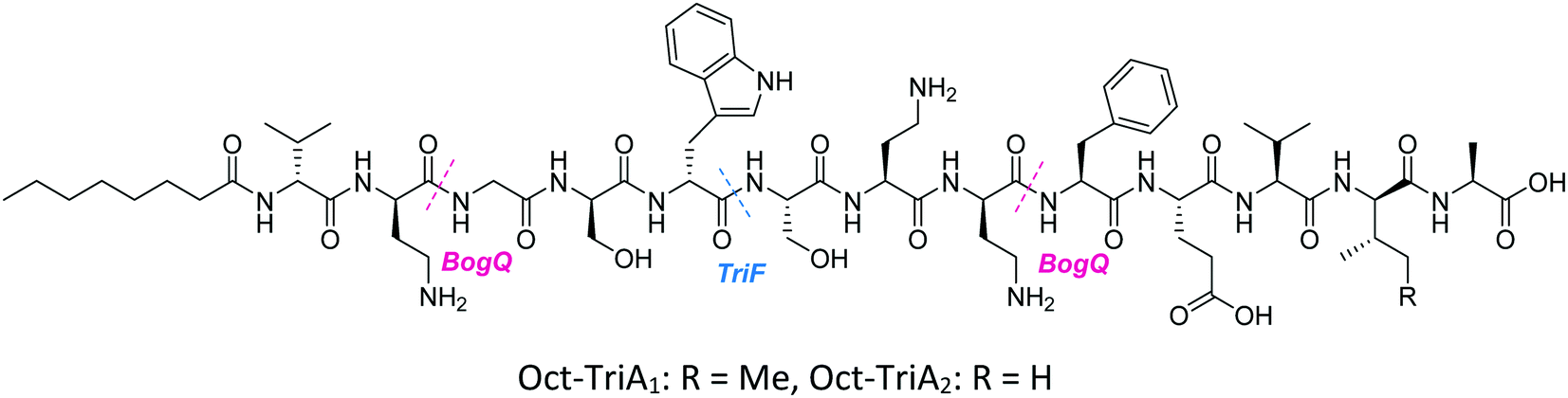 | ||
| Fig. 7 Cleavage sites on Oct-TriA variants of BogQ and TriF D-peptidase enzymes. BogQ hydrolyses the amide bond at the C-terminus of cationic D-Dab residues whilst TriF voids the peptide of antimicrobial activity by cleaving at the C-terminus of D-Trp5. | ||
The Cochrane lab recently identified modifications that improve the stability of the tridecaptins to D-peptidase hydrolysis.27–29 Analysis of the NMR solution structure of Oct-TriA1 in DPC micelles containing Gram-negative lipid II suggests that a π-stacking interaction between D-Trp5 and L-Phe9 may be important to maintain the looped structure of the peptide.30 TriF cleaves tridecaptins at D-Trp5, so the authors postulated that replacement of this π-stacking interaction with a covalent linkage to form a cyclic peptide could prevent TriF hydrolysis.27 A small library of peptides were synthesized in this study (54–61), including macrocyclic peptides wherein a D-Cys5 and L-Cys9 residue are crosslinked with various linkers. Macrocyclic peptides 58–60 (Fig. 8) retained strong activity against E. coli and even displayed comparable potency against the highly problematic A. baumannii strain,27 which the WHO currently rank as the no. one pathogen necessitating imminent novel therapeutic treatment.10 As anticipated, Xyl-linked analogues 58–60 were resistant to hydrolysis by the DRP enzyme, TriF. After a 12 h incubation period with the soluble periplasmic peptidase domain of TriF, TriFpep, UPLC-MS analysis revealed that while Oct-TriA1 (16) was hydrolysed, peptides 57–60 were stable to enzymatic degradation. The strategic design of these novel covalently linked macrocycles presents a new class of cyclic tridecaptin analogue with strong antimicrobial activity and an acquired ability to withstand D-peptidase mediated hydrolysis by TriF.
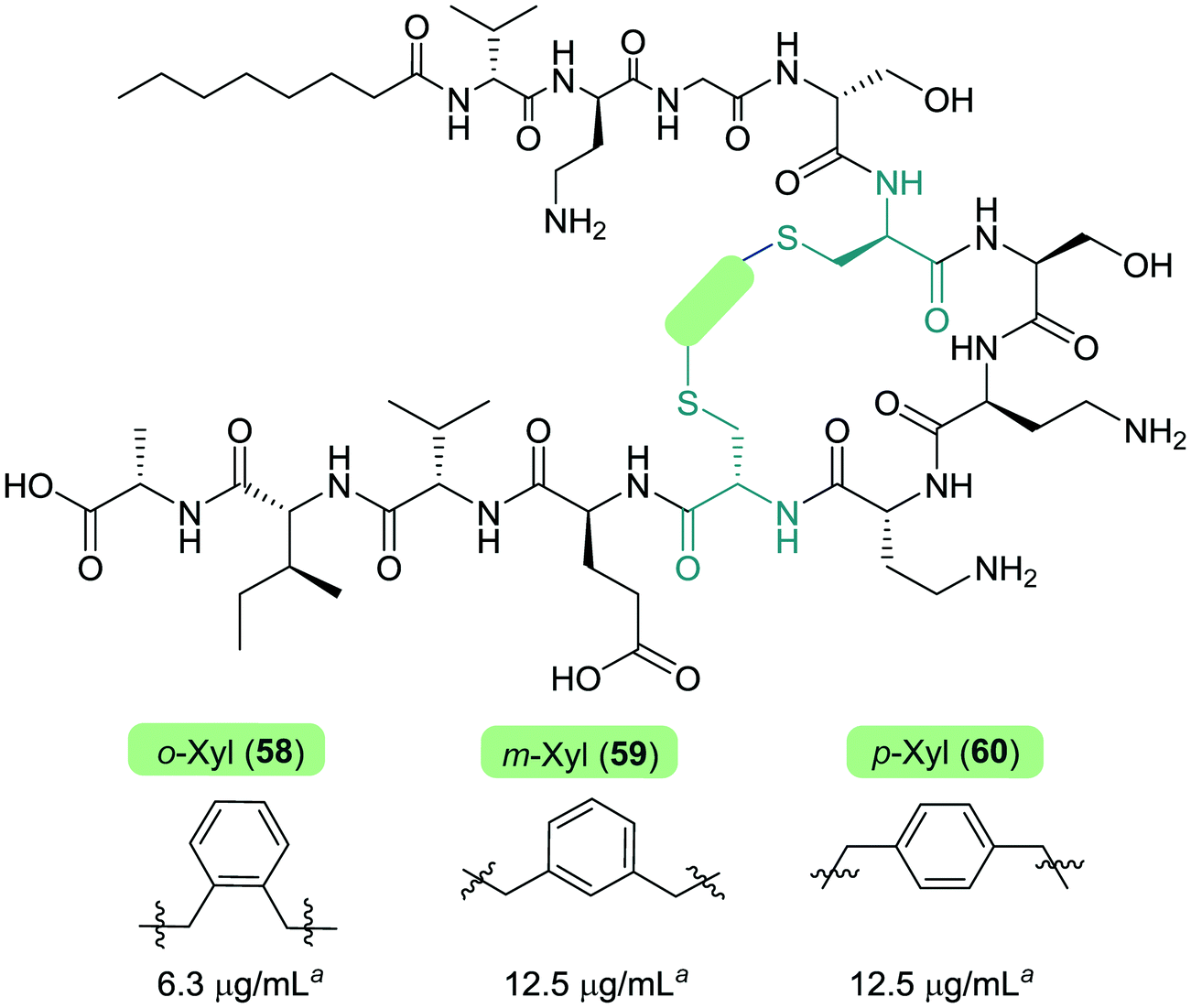 | ||
| Fig. 8 Cyclized Oct-TriA1 analogues (58–60), in which the characteristic π-stacking interaction has been replaced by regiospecific xylyl linkers. a MIC values against A. baumannii NCTC 13304. | ||
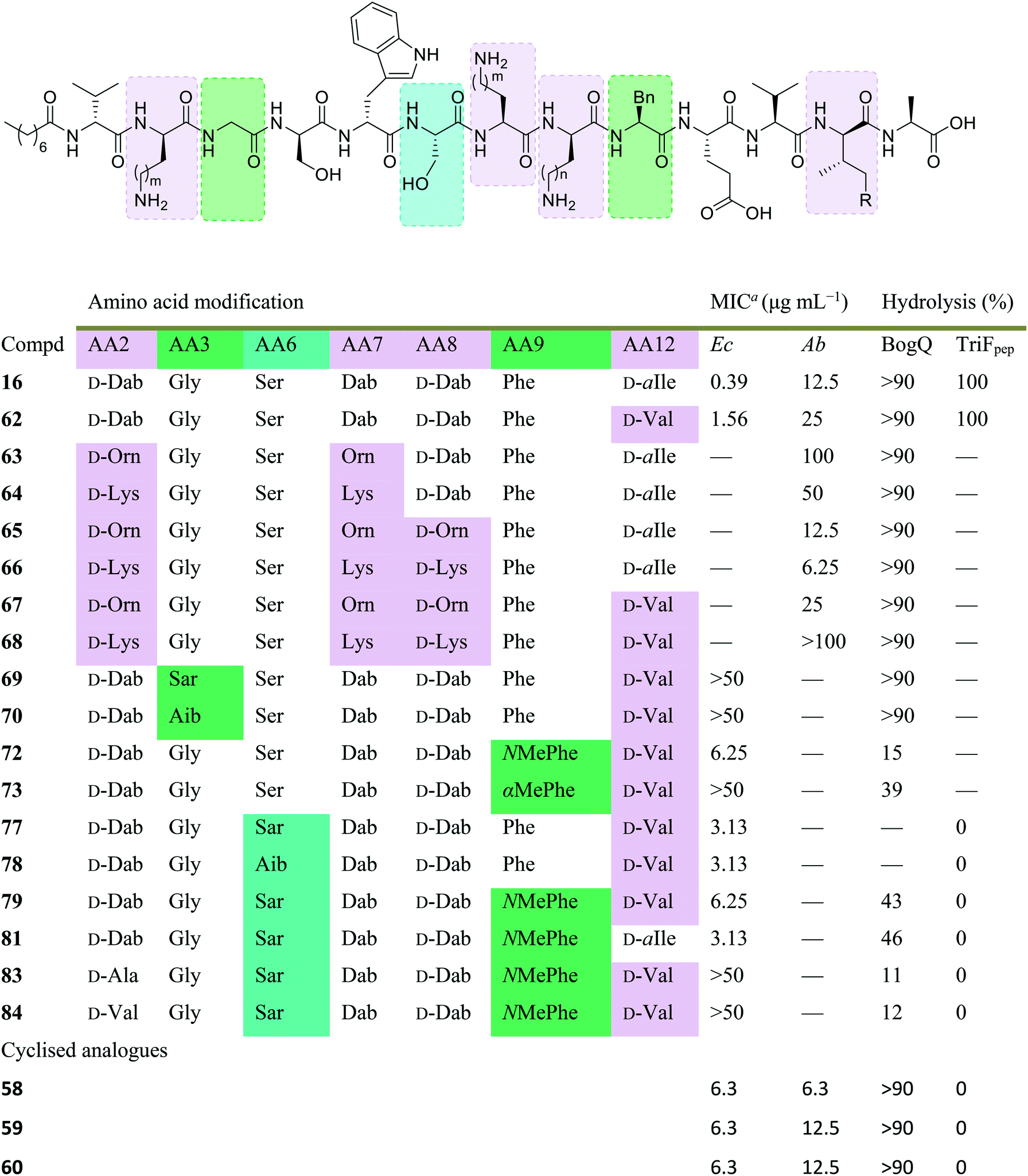
Conservative amino acid substitutions have also been applied to both Oct-TriA1 (16) and Oct-TriA2 (62) variants to mitigate hydrolysis by BogQ and TriF enzymes and reduce cost of synthesis. Initially, Dab at position 7 and/or D-Dab at positions 2 and 8 were replaced with the longer chain cationic amino acids (with the same L/D-configuration), lysine (Lys) or ornithine (Orn), yielding peptide library 63–68.28 It was postulated that these moderate alterations would not be deleterious to activity while producing analogues that could be synthesized at a considerably reduced cost. Oct-TriA1 (16) was found to be two- to four-fold more active than its variant Oct-TriA2 (62), which has D-Val12 in place of D-allo-Ile12. However, synthesis of the latter is much more cost-effective, as Fmoc-D-Val is much cheaper than Fmoc-D-allo-Ile.‡ Synthetic analogues substituted at positions 2 and 7 with Lys or Orn (of the correct L/D-configuration) showed comparable activity to one another, but significantly reduced activity against most strains when compared to control peptide Oct-TriA1 (16). Disubstituted analogues Oct-TriA1 (D-Orn2, Orn7) (63) and Oct-TriA1 (D-Lys2, Lys7) and (64) were found to be eight-fold less active against the carbapenemase-producing strain, Enterobacter cloacae, yet showed enhanced activity against Pseudomonas pseudoalcaligenes.28 Although switching native Dab residues for Lys/Orn seemed to hinder activity, preservation of antimicrobial activity led the authors to focus on triply substituted analogues. Analogues 65–68 would incorporate substitutions at positions 2, 7 and 8 to assess the impact, if any, on switching out the essential D-Dab8 residue, that is key to lipid II binding,30 for longer chain cationic amino acids. The most promising analogues to note were Oct-TriA1 (D-Orn2,8, Orn7) (65), Oct-TriA1 (D-Lys2,8, Lys7) (66) and Oct-TriA2 (D-Orn2,8, Orn7) (67); each is cheaper to synthesize than Oct-TriA1 (16) and retained antimicrobial activity relative to its unsubstituted precursor. Peptides 66 and 67 even showed enhanced activity against strains of the critical priority pathogen, A. baumannii, when compared to their relevant unsubstituted predecessor. It was found that replacing D-Dab8 with D-Lys was deleterious to activity, perhaps explained by the two-methylene increase in chain length when the native Dab residue is replaced with Lys rather than Orn. As the site in question is involved in lipid II binding, it would be reasonable to suggest that a considerable lengthening of the side chain would impede its bactericidal action, however binding studies are necessary to verify this assumption.
Despite the encouraging discovery of Oct-TriA2 (D-Orn2,8, Orn7) (67), with its strong antimicrobial activity and reduced synthetic cost, this peptide (and other analogues 16 & 62–68) were susceptible to hydrolytic cleavage by the D-peptidase BogQ (Table 2). When incubated with BogQ for 1 h, peptide 67 suffered >90% hydrolysis.29 The same pattern of degradation was also observed for xylyl-linked cyclic TriA1 analogues (58–60), further emphasising the ever-growing threat bacteria pose to the development of new antibiotic candidates.
| Dab: m/n = 1, Orn: m/n = 2, Lys: m/n = 3, D-allo-Ile: R = Me, D-Val: R = H. Boxes are colour-coded to reflect basis of change. Green and cyan denote chemical modifications to overcome BogQ and TriF hydrolysis, respectively. Substitutions in lilac boxes reduce synthetic cost.a MIC: minimum inhibitory concentration. Strains referenced are E. coli NCTC 12241 (Ec) and A. baumannii (Ab) (peptides 58–60: NCTC 13304, 16 & 62–68: environmental strain). |
|---|
|
In a follow-up study, the Cochrane lab looked for alternative modifications that could be made to a linear tridecaptin scaffold that improved their resistance to TriF and/or BogQ hydrolysis.29 To this end, a series of common modifications that improve peptide stability to peptidases were explored, including N-methylation, α-methylation and inversion of stereochemistry at hydrolysis sites. Residues 3, 6 and 9, residing at the C-termini of BogQ and TriF cleavage sites (Fig. 7), were chosen for modification, with the ultimate goal to sterically obstruct all cleavage sites by developing a triply substituted peptide that was completely stable to BogQ and TriF-mediated hydrolysis. As such, a library of new Oct-TriA2 analogues (69–84) were synthesized and tested. Replacement of Gly3 with bulkier N- and α-methylated residues, sarcosine (Sar, 69) or 2-aminoisobutyric acid (Aib, 70) was detrimental to antimicrobial activity at the upper limits tested (MIC >50 μg mL−1). Adding a methyl group at position 3 significantly reduces the rotational freedom of the archetypal peptide backbone29 and so was inferred to disturb the looped structure that Oct-TriA1 (16) forms when in contact with the lipid II target.30 Monosubstituted analogues bearing these alterations (69, 70) were also rapidly hydrolysed by BogQ (>90%) (Table 2). Perturbation of BogQ hydrolysis at D-Dab8 was achieved via replacement of Phe9 with its N-methylated variant, NMePhe (72). Enzymatic cleavage fell considerably to 15% and antimicrobial activity was retained (MIC = 6.25 μg mL−1). Despite boosting stability against BogQ-mediated hydrolysis (39%), substitution with the α-methylated residue, αMePhe (73), abolished activity at the limits tested. Knowing how such sequence alterations at positions 3 and 9 impacted antimicrobial activity, the focus was then directed to position 6 and how similar changes could prevent TriF hydrolysis. TriF cleaves Oct-TriA1 (16) at D-Trp5, therefore, Sar or Aib was placed at position 6 to increase steric bulk of the native L-Ser6 residue. Both analogues (77, 78) retained strong antimicrobial activity (MIC = 3.13 μg mL−1) relative to the parent peptide, Oct-TriA2 (62) and no TriF-mediated hydrolysis was observed. Analogues 77 and 78 are more active than the novel cyclic Xyl-linked peptides (58–60),27 showing that a simple amino acid substitution on the linear scaffold is a more efficient strategy to yield highly active peptides that are completely stable towards TriF-mediated hydrolysis.29
After establishing which single amino acid changes improved BogQ and TriF stability, multi-substituted analogues were then synthesized in an attempt overcome D-peptidase hydrolysis at all sites. Combining the most active single mutants produced disubstituted analogue Oct-TriA2 (Sar6, NMePhe9) (79), which retained strong antimicrobial activity (MIC = 6.25 μg mL−1), noticeably reduced BogQ-mediated hydrolysis (43%) and was completely resistant to TriF cleavage.29 Similar findings were obtained when D-Val12 was replaced with D-allo-Ile, yielding the more expensive Oct-TriA1 (Sar6, NMePhe9) (81). It too was completely stable to TriF and experienced comparative BogQ hydrolysis (46%) but was two-fold more active (MIC = 3.13 μg mL−1) than the cheaper counterpart (79). The authors also explored replacing the cationic D-Dab2 residue with hydrophobic amino acids D-Ala (83) or D-Val (84), and although the resulting analogues displayed high levels of resistance to both BogQ and TriF, they lacked antimicrobial activity at the upper limits tested.
Haemolytic assays were conducted on the propitious analogues Oct-TriA2 (NMePhe9) (72), Oct-TriA2 (Sar6) (77) and Oct-TriA2 (Sar6, NMePhe9) (79). Each exhibited low levels of haemolytic activity (30–60%) when compared to the parent peptide Oct-TriA2 (62, 53%).29 Additional spot-on-lawn assays with E. coli NCTC 12241, in the presence of lipid II, confirmed retention of their receptor-mediated mechanism of action. Discovery of these tridecaptin-inspired analogues demonstrates an ability to overcome D-peptidase resistance with cost-effective, yet highly active linear tridecaptin peptides, that retain their intricate mechanism of action and do not show signs of haemolytic toxicity.
4. Extended applications of tridecaptin and discovery of other native variants
The scope of the tridecaptins has gone beyond their use as antimicrobial peptides, as they have recently found application as a Gram-negative-specific fluorescent probe.33 Synthesis of a single-structure probe that selectively identifies Gram-negative bacteria helps overcome discrepancies inherent to the traditional Gram-staining technique,38 and indeed other chemical probes. The fluorophore tag, tetramethylrhodamine (TAMRA) was covalently linked to Oct-TriA1 (16) via the side chain of D-Dab2 to yield Oct-TriA1(D-Dab2-TAMRA) (85) (Fig. 9). This tridecaptin-conjugated probe (85) not only showed superior labelling of fixed bacteria compared to a polymyxin B based probe, which lends itself to practical applications, but also has capacity to selectively label Gram-negative bacteria in the midst of complex samples.33 Coupled with ease of synthesis, it is suggested that such a probe has the capability to differentially stain Gram-negative bacteria with a significant degree of accuracy that circumvents the intrinsic flaws of traditional Gram-staining.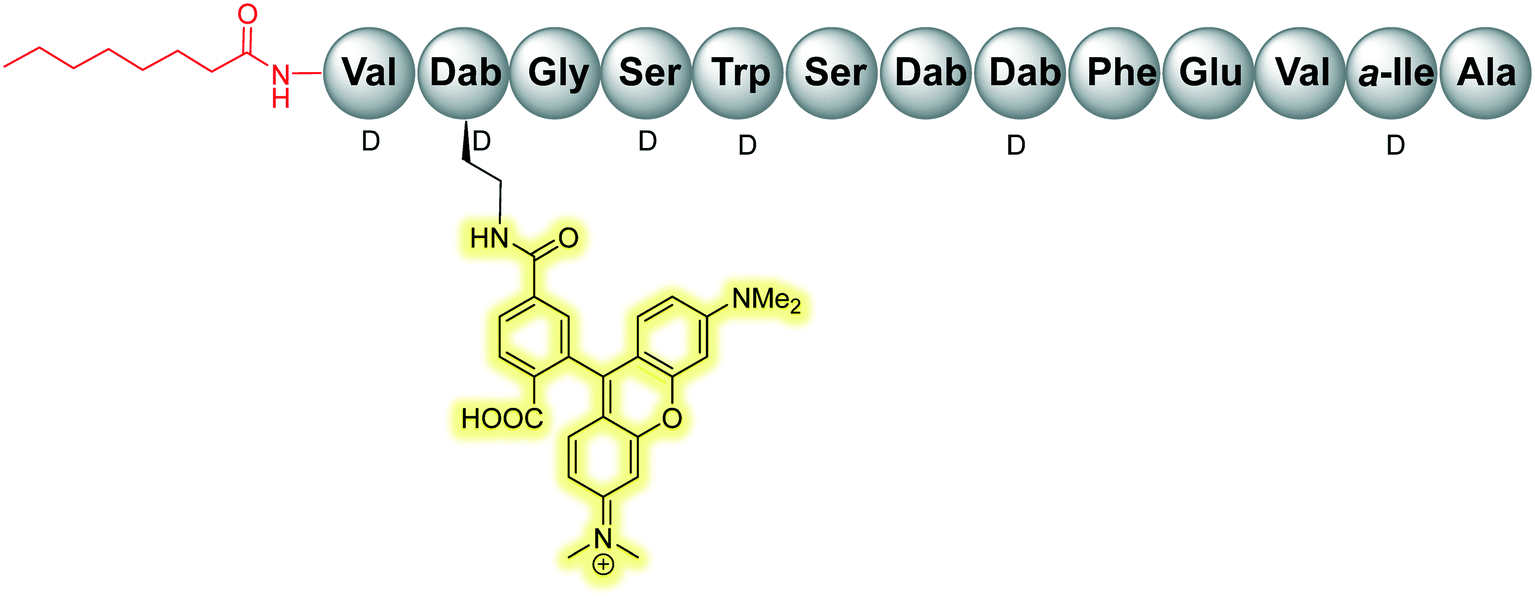 | ||
| Fig. 9 TriA-TAMRA probe (85) used for differential staining of Gram-negative bacteria. | ||
Other natural tridecaptin variants have also been reported, namely TriB1 (86) and TriM1 (90). TriB1 (86), isolated from a mixture of antimicrobials purified from an overnight culture of P. polymyxa NRRL B-30507, was fully characterized by Cochrane et al.22 C18-HPLC purification of this culture extract yielded three active fractions, two of which were determined to be polymyxin B1 and B2. However, the low level of anti-Campylobacter jejuni activity of the polymyxins39 suggested another metabolite must be responsible for this antimicrobial activity. Based on its analytical similarity to TriA1 (1) the third antimicrobial was identified as TriB1 (86) which differs slightly to TriA1, in that it contains a (6S)-methyloctanoic acid tail, glycine in place of D-Val1 and isoleucine replaces L-Phe9 (Fig. 10).22
 | ||
| Fig. 10 Structures of tridecaptin B variants: (6S)-TriB1 (86), (6R)-TriB1 (87), Oct-TriB1 (88) and H-TriB1 (89). | ||
Of particular note in this study is how the authors identified the stereochemical configuration of the TriB1 lipid tail. Unlike TriA1, it was not possible to distinguish between (6S)-methyloctanoyl-TriB1 (86) and (6R)-methyloctanoyl-TriB1 (87) (which were obtained by chemical synthesis) using HPLC or NMR. To identify the stereochemistry, the authors utilized the Ohrui–Akasaka method,40 where enantiomerically pure chiral lipids were converted into anthracenyl derivatives which, due to the different ring current experienced by an R- or S-methyl group, can be distinguished using 1H-NMR. The lipid tail was hydrolyzed from natural TriB1 and derivatized in the same manner, and comparison with the synthetic standards unambiguously showed that the 6-methyl group had the S-configuration (Fig. 11).
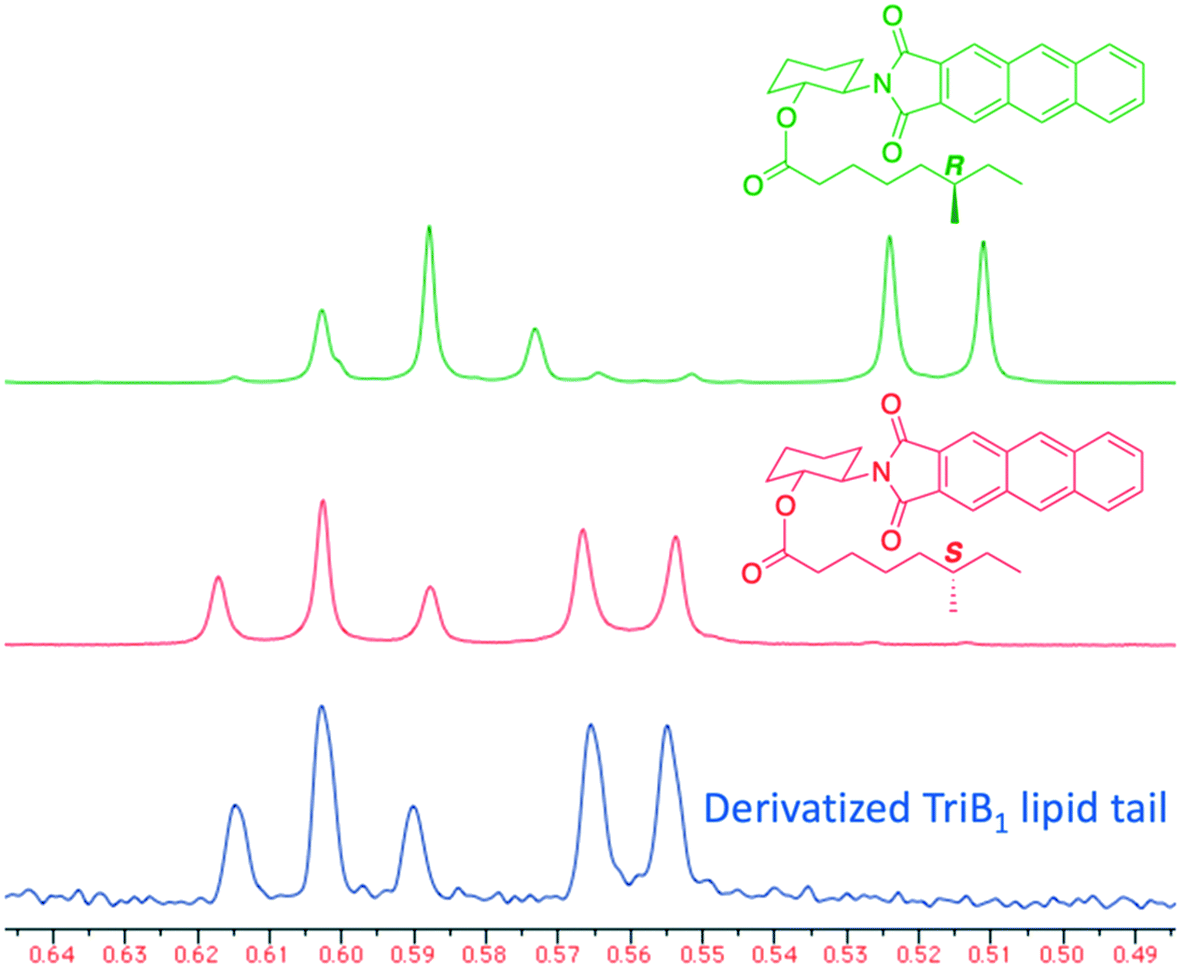 | ||
| Fig. 11 Elucidation of TriB1 lipid chain stereochemistry. (6R)-Methyl derivative (90, green) and (6S)-methyl derivative (91, red) were prepared by chemical synthesis. The lipid tail from TriB1 was obtained by hydrolysis and derivatized with the same anthracenyl unit (blue). 1H-NMR analysis allowed the chirality of the lipid tail to be determined as S. X-Axis units are parts per million (ppm). | ||
Much like TriA1 (1), TriB1 (86) exhibits strong activity against various Gram-negative strains (MIC = 3.13–6.25 μg mL−1), including a strain of MDR K. pneumoniae, but shows negligible activity against Gram-positive pathogens (MIC >100 μg mL−1). Although more cost-effective, synthetic variant Oct-TriB1 (88) experienced a two- to four-fold reduction in antimicrobial activity. Further similarities were found between TriA1 and B1; much like H-TriA1 (15), H-TriB1 (89) is devoid of antimicrobial activity but acts as an effective sensitiser of the front-line antibiotic, rifampicin, increasing its efficacy against E. coli 64×.22 The biosynthetic origins of TriB1 (86) were investigated via elucidation of its associated BGC. A BGC analogous to that of TriA1 (1) was found that differed only in the selectivity of adenylation domains 1 and 9. Although these differences could not be attributed to the flexibility of amino acids 1 and 9 found in tridecaptins A and B, identification of diversifying NRPSs emphasises the ability of these modular enzymes to produce heterogenous antimicrobial peptides that could be utilised as potent antibiotic drug candidates.
Recently, TriM1 (92) (Table 3) was isolated from a mud bacterium culture, Paenibacillus sp. M-152, and is reported to display activity against colistin-resistant and MDR Enterobacteriaceae strains, and K. pneumoniae.23 Similar to the classification of TriB1 (86), TriM1 was identified and its structure assigned via extensive analytical and NMR work and its initial presence postulated due to observed activity against resistant strains that could not be credited to existing antibiotics. TriM1 (92) has a similar structure to TriB1, with the exception of two amino acid positions; L-Val11 is replaced with L-Ile and L-Ala13 is instead L-Ser. Although two- to four-fold less active against polymyxin-sensitive strains of K. pneumoniae, TriM1 displayed superior activity against extensively drug-resistant (XDR) strains (MIC = 2 μg mL−1).23 Much like the tridecaptin A and B variants, analogue TriM1 (92) showed no activity against Gram-positive bacteria at the upper limits tested (MIC >128 μg mL−1) and its biosynthetic origins were attributed to a BGC comparable to that obtained for TriA and B.22
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Compound no. | Analogue | Variable residues | MIC (μg mL−1) | ||||
| 5 | 9 | 11 | 12 | K. pneumoniae AH-3 (Col-R) | S. marcescens MTCC 97 | ||
| a Residues 1–7 could not be conclusively determined. | |||||||
| 92 | M1 | D-Trp | L-Ile | L-Ile | D-aIle | 4 | 8 |
| 93 | M2 | D-Trp | L-Ile | L-Val | D-Val | 8 | >16 |
| 94 | M5 | D-Phe | L-Ile | L-Ile | D-Val | 16 | 8 |
| 95 | M6 | D-Trp | L-Ile | L-Ile | D-Val | 4 | 16 |
| 96 | M7 | D-Trp | L-Val | L-Ile | D-aIle | 8 | 8 |
| 97 | M8 | D-Phe | L-Ile | L-Ile | D-aIle | 8 | 16 |
| 98 | M11a | ND | L-Phe | L-Ile | D-aIle | 1 | 4 |
Time-kill assays of TriM1 at different concentrations confirmed it to be bactericidal in nature and exposure of K. pneumoniae to sublethal concentrations of TriM1 showed low-levels of resistance development. Mode-of-action studies aligned with data found for TriA1 (1)30 as it was found that TriM1 (92) disrupts the proton-motive force, likely blocking ATP synthesis.23 The susceptibility of polymyxin-resistant K. pneumoniae to TriM1 suggests that if the tridecaptins do indeed interact with LPS, as previously reported,30 it must be by a different mechanism than the polymyxins. Co-administration of TriM1 with subinhibitory concentrations of the OM-targeting antibiotic, colistin, reduced the MIC of TriM1 16-fold, further emphasising the role tridecaptins can play in forming synergistic relationships with clinically relevant antibiotics. TriM1 (92) has also been shown to act synergistically against the highly problematic bacterium A. baumannii in combination therapy with the clinically relevant antibiotic, rifampicin.41 The natural peptide (92) was proven to act as a outer membrane potentiator, reducing the MIC of rifampicin against the A. baumannii GMCH05 strain by 256×, when combined with TriM1 (92) at a concentration of 8 μg mL−1. This example of combination therapy was also demonstrated to tolerate a lag administration of each drug, as well as significantly reducing the presence of persister cells post treatment, a timely find in the continued fight to develop effective treatments for problematic bacterial infections.
Finally, in vitro experiments of novel TriM1 (92) found a favourable toxicology profile; the peptide exhibited low haemolysis and cytotoxicity, and no nephrotoxicity was observed at the same dose at which colistin was lethal.23 These observations are consistent with all available toxicity data for natural and synthetic TriA and TriB analogues. Such findings support the potential for the future development of tridecaptins as antibiotic agents, owing to their potency for highly problematic Gram-negative bacteria and their favourable in vivo activity.
Following on from the discovery of a new class of tridecaptin peptides, a further ten M variants were isolated from the same M-152 strain, however only six were completely characterised (92–97; M1–2, M5–8) with M11 (98) partially assigned (Table 3).24 These novel variants are attributed to the same BGC as the forerunner, TriM1 (92), highlighting the diversity afforded by non-ribosomally synthesized peptides. Of the six variants characterised, amino acids at positions 5, 9, 11 and/or 12 varied, while the lipid tail for each was assigned as (6S)-methyloctanoyl, as found in TriB1 (86). No clear structure–activity relationship could be drawn from the range of analogues isolated, except that there may exist a synergistic relationship between certain amino acids, such that substitutions may impede activity.24
The antimicrobial activity across the TriM family (92–98) is comparable to tridecaptin A and its variants such that they are active against Gram-negative E. coli, A. baumannii and even colistin-resistant strains of K. pneumoniae. TriM11 (98) displayed the most potent antimicrobial activity and was even found to be highly active (MIC = 4 μg mL−1) against the polymyxin-resistant bacteria, Proteus mirabilis and Serratia marcescens. Unfortunately, this superior activity is deterred by the high levels of haemolysis found for TriM11 (98) (∼50%), which is the highest reported across the analogues tested.
Membrane interaction and depolarisation assays were also conducted to ascertain the mode of action of this novel tridecaptin family. Uptake of N-(1-naphthyl)aniline (NPN) dye by various Gram-negative strains confirmed OM binding by both TriM1 and M11, while DiSC3(5) membrane depolarization assays are said to align with previous findings of disruption of the proton motive force.24,30 Although further mechanism of action studies are required to confirm the cellular interactions responsible for the antimicrobial activity of the TriM variants, this novel class of NRAP- further highlights the potential of the tridecaptins as novel therapeutics for the treatment of stubborn bacterial infections.
5. Conclusions and future outlooks
The current output of tridecaptin research has included isolation from nature as well as rational design of synthetic analogues in order to tackle the ever-growing threat of antimicrobial resistance. The nature of biosynthesis of tridecaptins favours their development as antimicrobial peptides as the class have a diverse structural capacity and display potent activity against many strains of multidrug-resistant Gram-negative bacteria. Tridecaptin variants studied to date are also reported to exhibit low levels of resistance development when compared to other clinically relevant antibiotics and have an innate biological profile that poses the potential for safe human administration. Moreover, unacetylated variants H-TriA1 and B1 have been proven to be effective sensitizers of problematic MDR bacteria when co-administered or covalently linked to other clinically relevant antibiotics, improving their efficacy. They have even found broader application in accurate differential staining of Gram-negative bacteria.Extensive development of novel synthetic tridecaptin analogues has presented cost-effective antimicrobial peptides with resistance to emerging D-peptidase enzymes that are intrinsically straightforward to synthesize. A complex mechanism of action has been identified for the tridecaptin class, in which lipid II has been confirmed as the chiral inner membrane receptor. The importance of this precursor in peptidoglycan biosynthesis suggests the chance for resistance development is low compared to other antibiotic candidates. While bacteria are usually a step ahead in the continued arms race to tackle antimicrobial resistance, continued research and development into the tridecaptin class and their subsequent implementation into the clinical setting could go a long way to overcome the increasing threat of Gram-negative bacterial infections to human health and global economies.
Funding sources
We thank the Engineering and Physical Sciences Research Council (EPSRC) and the Department for the Economy (DfE) for their respective funding of Samantha Bann's and Ross Ballantine's PhD studentships.Conflicts of interest
There is no conflict of interest to declare.References
- J. O'Neill, Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations, 2014 Search PubMed.
- World Health Organization, Antibacterial Agents in Clinical Development – An analysis of the antibacterial clinical development pipeline, including tuberculosis, 2017 Search PubMed.
- World Health Organization, Global Tuberculosis Report, 2019 Search PubMed.
- A. Coates, Y. Hu, R. Bax and C. Page, Nat. Rev. Drug Discovery, 2002, 1(11), 895–910 CrossRef CAS.
- E. Y. Klein, T. P. Van Boeckel, E. M. Martinez, S. Pant, S. Gandra, S. A. Levin, H. Goossens and R. Laxminarayan, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(15), E3463–E3470 CrossRef CAS.
- H. W. Boucher, G. H. Talbot, D. K. Benjamin, J. Bradley, R. J. Guidos, R. N. Jones, B. E. Murray, R. A. Bonomo and D. Gilbert, Clin. Infect. Dis., 2013, 56(12), 1685–1694 CrossRef.
- U. Theuretzbacher, K. Outterson, A. Engel and A. Karlen, Nat. Rev. Microbiol., 2020, 18, 275–285 CrossRef.
- D. Y. Aksoy and S. Unal, Clin. Microbiol. Infect., 2008, 14(5), 411–420 CrossRef CAS.
- R. O'Shea and H. E. Moser, J. Med. Chem., 2008, 51(10), 2871–2878 CrossRef.
- World Health Organization, Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis, 2017 Search PubMed.
- H. B. Koo and J. Seo, Pept. Sci., 2019, 111 DOI:10.1002/pep2.24122.
- W. C. Wimley and K. Hristova, J. Membr. Biol., 2011, 239, 27–34 CrossRef CAS.
- J. Wang, X. Dou, J. Song, Y. Lyu, X. Zhu, L. Xu, W. Li and A. Shan, Med. Res. Rev., 2019, 39(3), 831–859 CrossRef CAS.
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56(14), 3770–3821 CrossRef.
- Y. Liu, S. Ding, J. Shen and K. Zhu, Nat. Prod. Rep., 2019, 36(4), 573–592 RSC.
- J. Shoji, H. Hinoo, R. Sakazaki, T. Kato, Y. Wakisaka, M. Mayama, S. Matsuura and H. Miwa, J. Antibiot., 1978, 31(7), 646–651 CrossRef CAS.
- C. T. Lohans, M. J. Van Belkum, S. A. Cochrane, Z. Huang, C. S. Sit, L. M. McMullen and J. C. Vederas, ChemBioChem, 2014, 15(2), 243–249 CrossRef CAS.
- J. Shoji, T. Kato, S. Terabe and R. Konaka, J. Antibiot., 1979, 32(4), 313–319 CrossRef CAS.
- C. T. Lohans, Z. Huang, M. J. Van Belkum, M. Giroud, C. S. Sit, E. M. Steels, J. Zheng, R. M. Whittal, L. M. McMullen and J. C. Vederas, J. Am. Chem. Soc., 2012, 134(48), 19540–19543 CrossRef CAS.
- T. Kato, H. Hinoo and J. Shoji, J. Antibiot., 1978, 31, 652–661 CrossRef CAS.
- T. Kato, R. Sakazaki, H. Hinoo and J. Shoji, J. Antibiot., 1979, 32(4), 305–312 CrossRef CAS.
- S. A. Cochrane, C. T. Lohans, M. J. van Belkum, M. A. Bels and J. C. Vederas, Org. Biomol. Chem., 2015, 13(21), 6073–6081 RSC.
- M. Jangra, M. Kaur, R. Tambat, R. Rana, S. K. Maurya, N. Khatri, A. Ghafur and H. Nandanwar, Antimicrob. Agents Chemother., 2019, 63(6), e00338-19 CrossRef.
- M. Jangra, M. Kaur, M. Podia, R. Tambat, V. Singh, N. Chandal, N. Mahey, N. Maurya and H. Nandanwar, Sci. Rep., 2019, 9, 18870, DOI:10.1038/s41598-019-54716-8.
- S. A. Cochrane, B. Findlay, J. C. Vederas and E. S. Ratemi, ChemBioChem, 2014, 15(9), 1295–1299 CrossRef CAS.
- S. A. Cochrane, C. T. Lohans, J. R. Brandelli, G. Mulvey, G. D. Armstrong and J. C. Vederas, J. Med. Chem., 2014, 57, 1127–1131 CrossRef CAS.
- R. D. Ballantine, Y. X. Li, P. Y. Qian and S. A. Cochrane, Chem. Commun., 2018, 54, 10634–10637 RSC.
- R. D. Ballantine, C. E. McCallion, E. Nassour, S. Tokajian and S. A. Cochrane, MedChemComm, 2019, 10, 484–487 RSC.
- S. J. Bann, R. D. Ballantine, C. E. McCallion, P.-Y. Qian, Y.-X. Li and S. A. Cochrane, J. Med. Chem., 2019, 62, 10466–10472 CrossRef CAS.
- S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo and E. M. Rodriguez-lopez, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef CAS.
- S. A. Cochrane and J. C. Vederas, Int. J. Antimicrob. Agents, 2014, 44, 493–499 CrossRef CAS.
- S. A. Cochrane, X. Li, S. He, M. Yu, M. Wu and J. C. Vederas, J. Med. Chem., 2015, 58(24), 9779–9785 CrossRef CAS.
- W. Wang, Y. Wang, L. Lin, Y. Song and C. J. Yang, Anal. Bioanal. Chem., 2019, 411, 4017–4023 CrossRef CAS.
- S. Chiorean, I. Antwi, D. W. Carney, I. Kotsogianni, A. M. Giltrap, F. M. Alexander, S. A. Cochrane, R. J. Payne, N. I. Martin, A. Henninot and J. C. Vederas, ChemBioChem, 2020, 21(6), 789–792 CrossRef CAS.
- M. Vaara, FEMS Microbiol. Lett., 1983, 18, 117–121 CrossRef CAS.
- X. Vila-Farres, J. Chu, D. Inoyama, M. A. Ternei, C. Lemetre, L. J. Cohen, W. Cho, B. V. B. Reddy, H. A. Zebroski, J. S. Freundlich, D. S. Perlin and S. F. Brady, J. Am. Chem. Soc., 2017, 139(4), 1404–1407 CrossRef CAS.
- Y. X. Li, Z. Zhong, P. Hou, W. P. Zhang and P. Y. Qian, Nat. Chem. Biol., 2018, 14(4), 381–387 CrossRef CAS.
- L. P. Samuel, J. M. Balada-Llasat, A. Harrington and R. Cavagnolo, J. Clin. Microbiol., 2016, 54(6), 1442–1447 CrossRef CAS.
- J. Lin, Y. Wang and K. Van Hoang, Foodborne Pathog. Dis., 2009, 6(2), 173–185 CrossRef CAS.
- K. Akasaka and H. Ohrui, Biosci., Biotechnol., Biochem., 1999, 63(7), 1209–1215 CrossRef CAS.
- M. Jangra, V. Raka and H. Nandanwar, Molecules, 2020, 25(14), 3255, DOI:10.3390/molecules25143255.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0md00413h |
| ‡ Fmoc-D-allo-Ile-OH: £154 per g, Fmoc-D-Val-OH: £19.60 per g (prices from Alfa Aesar catalogue – accurate as of 27/11/20). |
| This journal is © The Royal Society of Chemistry 2021 |