
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFuroic acid and derivatives as atypical dienes in Diels–Alder reactions†
Răzvan C.
Cioc
a,
Tom J.
Smak
a,
Marc
Crockatt
b,
Jan C.
van der Waal
 b and
Pieter C. A.
Bruijnincx
*a
b and
Pieter C. A.
Bruijnincx
*a
aOrganic Chemistry and Catalysis, Debye Institute for Nanomaterials Science, Faculty of Science, Utrecht University, Universiteitsweg 99, 3584 CG, Utrecht, The Netherlands. E-mail: p.c.a.bruijnincx@uu.nl
bDepartment of Sustainable Process and Energy Systems, TNO, Leeghwaterstraat 44, 2628 CA Delft, The Netherlands
First published on 23rd June 2021
Abstract
The furan Diels–Alder (DA) cycloaddition reaction has become an important tool in green chemistry, being central to the sustainable synthesis of many chemical building blocks. The restriction to electron-rich furans is a significant limitation of the scope of suitable dienes, in particular hampering the use of the furans most readily obtained from biomass, furfurals and their oxidized variants, furoic acids. Herein, it is shown that despite their electron-withdrawing substituents, 2-furoic acids and derivatives (esters, amides) are in fact reactive dienes in Diels–Alder couplings with maleimide dienophiles. The reactions benefit from a substantial rate-enhancement when water is used as solvent, and from activation of the 2-furoic acids by conversion to the corresponding carboxylate salts. This approach enables Diels–Alder reactions to be performed under very mild conditions, even with highly unreactive dienes such as 2,5-furandicarboxylic acid. The obtained DA adducts of furoic acids are shown to be versatile synthons in the conversion to various saturated and aromatic carbocyclic products.
Introduction
Since its discovery in 1929,1 the furan Diels–Alder (DA) reaction has been extensively applied in organic chemistry, with the resulting 7-oxabicyclo[2.2.1]hept-2-enes being exploited in natural product synthesis, drug discovery, bioconjugation, as well as in polymer and materials science applications.2–7 Indeed, furan DA reactions allow versatile synthons of considerable molecular complexity to be generated in an atom-economical fashion, making it a highly attractive strategy for green chemical synthesis of cyclic compounds.8 The reactivity of furan derivatives as dienes has been the subject of numerous theoretical and experimental studies. The general consensus is that good kinetics requires electron-rich furanic rings, as found in the parent furan and derivatives decorated with electron-donating substituents (e.g. alkyl-, alkoxyalkyl-, acetals etc., Scheme 1A)9–13 and accordingly most applications employ such dienes. | ||
| Scheme 1 Diels–Alder reactions with 2-furoic acid-derived dienes. | ||
Conversely, Diels–Alder reactions involving electron-poor derivatives such as furoic acids (furans substituted with a COOH group) are very much underexplored.14–21 That such highly oxygenated furanics cannot be readily used in DA-based synthesis strategies is a missed opportunity. Furoic acids are readily available via renewable platform molecules such as furfural and 5-hydroxymethyl furfural (5-HMF).22 Furoic acids also offer the advantage of being stable renewable platform molecules, this in contrast to, for instance, 5-HMF and its hydrogenated derivatives (2,5-dimethyl furan, 2,5-bishydroxymethyl furan) which can readily degrade and/or polymerize.23 Together, this makes them attractive building blocks for the atom- and redox-efficient synthesis of a large number of renewable value-added carboxylic acid- or ester-containing chemical products, including biobased aromatics.
Unfortunately, only a handful of recent reports describe attempts to capitalize on these advantages. Instead, the vast majority of chemistry developed in this area relies on the well-established use of electron-rich furan dienes. While this conventional approach may be efficient for the individual DA step, it is often redox uneconomical overall, as exemplified for instance by the cycloaddition between 2,5-dimethyl furan and ethylene targeting renewable terephthalic acid (Scheme 1A).24–27 This route is, in essence, the conversion of a highly oxygenated bio-derived resource (e.g. 5-HMF) to an oxygen-rich product (terephthalic acid) via a non-oxygenated hydrocarbon intermediate, p-xylene. The non-productive use of redox reactions featured in this approach (the so-called “redox-detour”) greatly reduces the incentive for scale-up of such routes.
In contrast, as solution to this problem, the groups of Davis,15–17 Clark18 and Jae19 and a patent by Avantium20 have disclosed new routes towards terephthalic acid starting from furan carboxylic (di)acids or their esters and ethylene, but these diene/dienophile combinations require very harsh conditions and the overall yields are low (Scheme 1B). Relatedly, Sibi et al. was successful in the valorisation of the dimethyl 2,5-furandicarboxylate with benzyne as highly reactive dienophile (Scheme 1C).21 Finally, the furoate ester/maleimide couple has on occasion been described in macromolecular applications,28–31 even though the molecular version was deemed unfeasible by Boutevin et al.10
To the best of our knowledge, there is a single study thoroughly investigating the reactivity of furoic acids in Diels–Alder chemistry. Bowman et al. showed that reactions of 3-furoic acid with maleimides are actually favoured both kinetically and thermodynamically;32 on the other hand, the more readily available 2-regioisomer was found to be much less reactive, with the coupling also being strongly entropically disfavoured. Interestingly, the authors note that reactivity could be tuned by solvent effects: rate and equilibrium conversions were significantly higher in water compared to reactions in dimethyl formamide.
Encouraged by this precedent and following our interest in the development of synthetic applications based on the Diels–Alder chemistry of readily available oxygenated bio-based furans,33,34 we decided to investigate 2-furoic acids as underexplored class of dienes.
Results and discussion
Reaction optimization
In line with the observations of Bowman et al., the reaction between furoic acid 1a and N-methyl maleimide 2a was slow in all common organic solvents. No obvious reactivity trend (Table 1) could be discerned, although polar solvents seemed more beneficial for the reaction (MeOH, EtOH, DMSO, AcOH). Hydrogen bonding interactions with the solvent might be important as only traces of product could be detected in apolar aprotic solvents (CH2Cl2, CHCl3; toluene being a curious exception with 5% yield). Undoubtedly, the electron-withdrawing effect of the COOH substituent on the furan ring translates into a lowered energy level for the HOMO of the diene and consequently a high activation barrier for the cycloaddition. We reasoned that the addition of a base would counteract this effect, as neutralization to the carboxylate diminishes the electron-withdrawing capability of the COOH substituent:35 indeed, in all solvents tested, the yields increased substantially in the presence of 1 equiv. of triethylamine. Adduct yields of nearly 50% (6 h reaction time, 50 °C) were now observed in CH2Cl2, CHCl3 and EtOH. Correlating reactivity with solvent properties is again not straightforward; plausibly, the extent of proton transfer and charge separation play a major role here.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Solvent | Yields in neutral conditions | Yields with NEt3 (1 equiv.) | ||||
| exo-3, % | endo-3, % | Total, % | exo-3, % | endo-3, % | Total, % | ||
| General procedure: 2-Furoic acid 1a (0.5 mmol), (NEt3, 1 equiv.) and N-methyl maleimide 2a (1.5 equiv.) stirred in the respective solvent (1 M) at 50 °C for 6 h; yields determined from the 1H-NMR ratios of product isomers and starting material; conversion of 1a was clean; solvolysis of 2a was detected as side reaction, in alcoholic solvents (in the presence of NEt3). | |||||||
| 1 | MeOH | 10 | 0 | 10 | 34 | 6 | 40 |
| 2 | EtOH | 8 | Trace | 8 | 49 | 1 | 50 |
| 3 | AcOH | 6 | 0 | 6 | n.a. | n.a. | n.a. |
| 4 | DMSO | 7 | Trace | 7 | 35 | 1 | 36 |
| 5 | DMF | 3 | 0 | 3 | 25 | 2 | 27 |
| 6 | MeCN | 3 | 0 | 3 | 28 | 5 | 33 |
| 7 | AcOEt | 4 | 0 | 4 | 28 | 3 | 31 |
| 8 | THF | 3 | Trace | 3 | 27 | 1 | 28 |
| 9 | Acetone | Trace | 0 | Trace | 27 | 5 | 32 |
| 10 | CHCl3 | 2 | 0 | 2 | 50 | 0 | 50 |
| 11 | CH2Cl2 | Trace | 0 | Trace | 41 | 6 | 47 |
| 12 | Toluene | 5 | 0 | 5 | 38 | 2 | 40 |
| 13 | H2O | 56 | 7 | 63 | 83 | 3 | 86 |
Next, we turned our attention to water as solvent. The rate-enhancement ability of water in Diels–Alder chemistry is well known and many arguments have been proposed to explain it.36,37 In our system, the rate of reaction between 1a and 2a (63% conversion in 6 h at 50 °C, Table 1, entry 13) is one order of magnitude higher than in all organic solvents tested; in fact, the reaction of the free acid in water even proved faster than the best result obtained in an organic solvent in the presence of base (Table 1, entry 2 or entry 10).
The reaction also clearly benefited from the synergy of using aqueous solvent and the base effect: nearly quantitative yield of 1a was obtained when the cycloaddition was performed in water in the presence of 1 equiv. of NaOH (Table 2, entry 4). The reaction efficiency correlates with the base strength (NaOH ≈ Na2HPO4 > NEt3 > NaH2PO4); clearly, the effect of the base is not catalytic, as lowering the NaOH loading (entries 7 and 8) produced results comparable to those obtained when using the weaker base NaH2PO4. Finally, the effect of temperature is typical for a reversible [4 + 2] cycloaddition: at lower temperatures (20 °C, entry 10), the reaction is under kinetic control, with the major product being the endo adduct, while at elevated temperatures (80 °C, entry 9), the formation of exo-3a is nearly exclusive (see also Fig. 1, bottom); noteworthy, the total yield diminishes with increasing temperature due to the unfavourable entropy contribution.
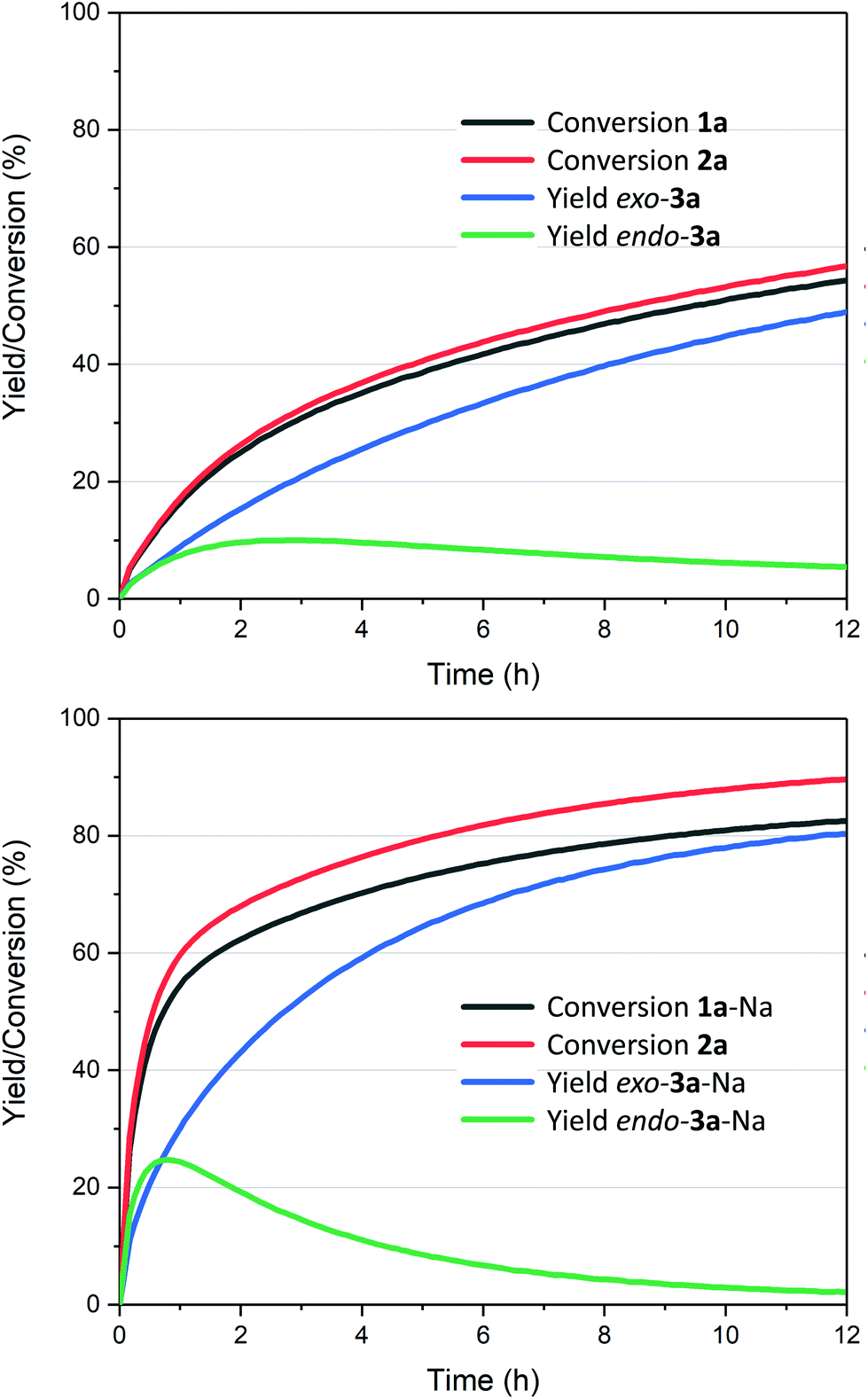 | ||
Fig. 1 Kinetic traces for the cycloadditions between 2-furoic acid 1a and maleimide 2a (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at 0.5 M) in water (top), with the addition of 1 equiv. NaOH (bottom); see ESI† for details; cf. Tables 2 and 3: reactions run at 1:1.5 ratio 1:2 and 1 M concentration. :1 ratio at 0.5 M) in water (top), with the addition of 1 equiv. NaOH (bottom); see ESI† for details; cf. Tables 2 and 3: reactions run at 1:1.5 ratio 1:2 and 1 M concentration. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Additive | Amount, equiv. | Temp., °C | Time, h | exo-3, % | endo-3, % | Total, % |
| General procedure: 2-Furoic acid 1a (0.5 mmol), (additive) and N-methyl maleimide 2a (1.5 equiv.) stirred in aqueous solution (1 M) at the indicated temperature for the indicated time; yields determined from the 1H-NMR ratios of product isomers and starting material. | |||||||
| 1 | None | n/a | 50 | 6 | 56 | 7 | 63 |
| 2 | None | n/a | 50 | 16 | 76 | 3 | 79 |
| 3 | NaOH | 1 | 50 | 6 | 90 | 5 | 95 |
| 4 | NaOH | 1 | 50 | 16 | 94 | 3 | 97 |
| 5 | Na2HPO4 | 1 | 50 | 6 | 89 | 6 | 95 |
| 6 | NaH2PO4 | 1 | 50 | 6 | 71 | 8 | 79 |
| 7 | NaOH | 0.5 | 50 | 6 | 75 | 6 | 81 |
| 8 | NaOH | 0.25 | 50 | 6 | 67 | 7 | 74 |
| 9 | NaOH | 1 | 80 | 6 | 81 | 3 | 84 |
| 10 | NaOH | 1 | 20 | 6 | 25 | 35 | 60 |
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | 3 | Conv. 1a | exo-3, % | endo-3, % | Isolatedb, % |
| General procedure: 2-Furoic acid, NaOH (1 equiv.) and maleimide (1.5 equiv.) stirred in water (1 M) at 50 °C for 16 h.a Conversion determined from the 1H-NMR ratios of product isomers and starting material in the crude mixture.b Isolated yield after acidification.c 40 mmol scale.d Poor dissolution of 2.e Methanol was used as cosolvent.f After hydrogenation on Pd/C.g Extensive hydrolysis of 2a to maleic acid.h With 2 equiv. of NaOH. | |||||||
| 1 | H | Me | 3a | 98 | 97 | 1 | 77(92)c |
| 2 | H | H | 3b | 95 | 95 | Trace | 68 |
| 3 | H | n Pr | 3c | 96 | 93 | 3 | 72 |
| 4d | H | Ph | 3d | 51 | 51 | Trace | 21 |
| 5d | H | Cy | 3e | 18 | 16 | 2 | n.d. |
| 6d,e | H | Cy | 3e | 56 | 53 | 3 | 31 |
| 7 | Me | Me | 3f | 93 | 88 | 5 | 75 |
| 8 | CH2OH | Me | 3g | 91 | 72 | 19 | 51f |
| 9d | CH2OH | Ph | 3h | 28 | 28 | Trace | 11 |
| 10g | CHO | Me | 3i | <10 | ∼5 | Trace | n.d. |
| 11g | COOH | Me | 3j | 20 | 20 | 0 | n.d. |
| 12g,h | COOH | Me | 3j | 56 | 56 | 0 | n.d. |
The reaction profiles depicted in Fig. 1 illustrate the significant enhancement of the rate of the cycloaddition in the presence of stoichiometric base: compared to the additive-free reaction (Fig. 1, top), the NaOH-mediated conversion is approx. 5 times faster, in the first 30 min. In addition, the system is nearly at equilibrium within 12 h, which is not the case for the base-free experiment. Importantly, the conversion of 1a was clean in both cases, with no side products originating from the diene detected. On the other hand, some hydrolysis of 2a (towards maleic acid) does occur, to a low extent, and if the NaOH stoichiometry is increased beyond the 1:1 ratio, the sodium salt of N-methyl maleamic acid is formed (see also note52).
Reaction scope
The choice of water as reaction solvent allows the convenient isolation of DA adducts 3 by precipitation. To maximize conversion and simplify purification, we performed the title reaction between 1a and 2a under more concentrated conditions (2 M) and with 1:1 stoichiometry; the precipitated product was isolated by filtration in a 63% yield. Unreacted starting materials were recovered from the filtrate and reused without loss of reaction efficiency (see ESI† for details). This example is an excellent showcase of the green chemistry principles: renewable raw materials use, 100% atom-economy, eco-friendly solvent, simple isolation, no additives and no waste.
In studying the reaction scope, we chose the conditions shown in entry 4, Table 2 (with 1 equiv. NaOH), with the aim of minimizing the amount of minor adduct endo-3 and simplifying the isolation of a pure product following a general protocol. Thus, after reaction, the excess maleimide can be washed away (and recovered) with an organic solvent, while acidification of the aqueous phase typically leads to the selective precipitation of exo-3. This procedure allowed for the isolation of the exo-adducts 3a–c in good yields (Table 3, entries 1–3); the performance of the reactions is only modestly influenced by the nature of the maleimide substituent in the series H/Me/nPr. Higher homologues (N-Ph, N-Cy) proved problematic, however, due to poor solubility of the dienophile in the aqueous medium (entries 4 and 5); the addition of a cosolvent (MeOH) was beneficial in this case (entry 6) and although the conversion levels were moderate, synthetically useful yields of pure adducts 3d and 3e could be obtained without excessive adjustment of the general procedure. Next, we proceeded with the investigation of the furan diene scope, with the focus on easily accessible biomass-derived 5-substituted 2-furoic acids. We found that both the kinetics and the thermodynamics of the reaction are greatly influenced by the nature of the 5-substituent. Expectedly, electron-donating groups (Me) increase the reaction rate (entry 7, see ESI†) while electron-withdrawing groups (CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, COOH) showed the opposite effect, in line with the generally-accepted Frontier Molecular Orbital theory-derived interpretation of kinetics in DA reactions. Substitution at the furan 5-position, regardless of the nature of the substituent, not only influences kinetics but also the equilibrium position and likely destabilizes the DA adduct with respect to its addends, plausibly due to increased steric encumbrance in 3.11 The implication is that the most reactive diene in a series does not necessarily lead to the most thermodynamically favourable cycloaddition and hence the highest adduct yield at equilibrium. In the series R1 = H/Me/CH2OH, the parent reaction (formation of 3a, R1 = H) was found to exhibit the highest equilibrium conversion (Table 3, entry 1 vs. 7 and 8; see also ESI† for further details) while the relative reactivity was Me > H > CH2OH. Despite the somewhat less favourable equilibrium, adduct 3f (R1 = Me) could nonetheless be isolated in a good yield (75%). Next, a high isolated yield was anticipated based on the analysis of the crude reaction mixture for adduct 3g (R1 = CH2OH) but unfortunately this very polar molecule is highly water soluble, which complicated its isolation. Nonetheless, a yield of 51% of pure exo product was obtained after the sequential, one-pot conversion of 3g to a more stable derivative, i.e. by hydrogenation over Pd/C, a method used before in furan DA chemistry.38–41 The less water-soluble Ph-analogue could be obtained utilizing our standard protocol, albeit in a low yield and purity (11%, entry 9). As expected, the presence of a second electron-withdrawing substituent is highly detrimental for the kinetics of the reaction (and likely also for the thermodynamics9,12,34): adducts 3i and 3j (R1 is CHO and COOH respectively) were formed in low amounts in the crude reaction mixtures. On the other hand, with 2 equiv. of NaOH, 2,5-furandicarboxylic acid (FDCA) gave a fast equilibration to the exo-bis-Na salt of its adduct with 2a. In this case, isolation after acidification was hampered by the highly polar nature, as noted previously, and facile cycloreversion of the (neutral) adduct back to the addends. Noteworthy, all these reactions feature high stereoselectivity for the exo adduct, typically above 15:1 (with the exception of adduct 3g); the isolated products were single diastereoisomers.
O, COOH) showed the opposite effect, in line with the generally-accepted Frontier Molecular Orbital theory-derived interpretation of kinetics in DA reactions. Substitution at the furan 5-position, regardless of the nature of the substituent, not only influences kinetics but also the equilibrium position and likely destabilizes the DA adduct with respect to its addends, plausibly due to increased steric encumbrance in 3.11 The implication is that the most reactive diene in a series does not necessarily lead to the most thermodynamically favourable cycloaddition and hence the highest adduct yield at equilibrium. In the series R1 = H/Me/CH2OH, the parent reaction (formation of 3a, R1 = H) was found to exhibit the highest equilibrium conversion (Table 3, entry 1 vs. 7 and 8; see also ESI† for further details) while the relative reactivity was Me > H > CH2OH. Despite the somewhat less favourable equilibrium, adduct 3f (R1 = Me) could nonetheless be isolated in a good yield (75%). Next, a high isolated yield was anticipated based on the analysis of the crude reaction mixture for adduct 3g (R1 = CH2OH) but unfortunately this very polar molecule is highly water soluble, which complicated its isolation. Nonetheless, a yield of 51% of pure exo product was obtained after the sequential, one-pot conversion of 3g to a more stable derivative, i.e. by hydrogenation over Pd/C, a method used before in furan DA chemistry.38–41 The less water-soluble Ph-analogue could be obtained utilizing our standard protocol, albeit in a low yield and purity (11%, entry 9). As expected, the presence of a second electron-withdrawing substituent is highly detrimental for the kinetics of the reaction (and likely also for the thermodynamics9,12,34): adducts 3i and 3j (R1 is CHO and COOH respectively) were formed in low amounts in the crude reaction mixtures. On the other hand, with 2 equiv. of NaOH, 2,5-furandicarboxylic acid (FDCA) gave a fast equilibration to the exo-bis-Na salt of its adduct with 2a. In this case, isolation after acidification was hampered by the highly polar nature, as noted previously, and facile cycloreversion of the (neutral) adduct back to the addends. Noteworthy, all these reactions feature high stereoselectivity for the exo adduct, typically above 15:1 (with the exception of adduct 3g); the isolated products were single diastereoisomers.
We then turned our attention to other furoic acid derivatives, such as esters and amides, anticipating that the electronic properties of the furan diene would not be significantly different for the substituent series COOH/COOR/CONR2. However, the physical properties, water miscibility in particular, are certainly strongly modulated by the nature of the substituent and this might impact the performance of the aqueous Diels–Alder cycloaddition. This proved indeed to be the case (Table 4). The 2-furoic acid esters tested (Me, Et, iPr, tBu) are liquids with poor water miscibility; however, reactions still proceeded smoothly with so-called ‘on-water’ activation.42,43
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | R2 | 3 | Conv. 1a | exo-3, % | endo-3, % | Isolatedb, % |
| General procedure: 2-Furoic acid derivative and maleimide (1.5 equiv.) stirred in/on water (1 M) at 50 °C for 16 h.a Conversion determined from the 1H-NMR ratios of product isomers and starting material.b Isolated yield after (chromatographic) workup (in brackets, yield corrected on reacted starting material).c 2 M initial concentration. | |||||||
| 1 | OMe | Me | 3k | 70 | 65 | 5 | 52(74) |
| 2 | OMe | H | 3l | 67 | 65 | 2 | 43(64)/82c |
| 3 | OMe | Et | 3m | 65 | 61 | 4 | 47(72) |
| 4 | OEt | Me | 3n | 63 | 59 | 4 | 29(46) |
| 5 | OiPr | Me | 3o | 54 | 50 | 4 | 26(49) |
| 6 | OtBu | Me | 3p | 54 | 51 | 3 | 25(46) |
| 7 | NH2 | Me | 3q | 94 | 91 | 3 | 77(83) |
| 8 | NMe2 | Me | 3r | 81 | 77 | 4 | 41(51) |
| 9 | NHOH | Me | 3s | 92 | 76 | 16 | 69(75) |
For ease of comparison, we employed reaction conditions similar to those found in the optimization of the cycloadditions with 1a (1 mL water per mmol, 50 °C, 16 h). The reactivity differences in the ester homologous series were marginal under these conditions. Product properties did vary, however, as the adducts of the methyl ester turned out to be much more crystalline than the rest. Simple filtration of the resulting suspensions was sufficient in this case to provide good yields of pure (exo-) products 3k–m (43–52%). Moreover, the adduct 3l could be obtained in a much-improved yield (82%), simply by performing the reaction in more concentrated conditions (see ESI† for details). Notably, the reactions are clean and the filtrate containing unreacted starting materials can readily be recycled. Such crystallization from the crude reaction mixture did not occur with higher homologous esters (entries 4–6), necessitating chromatographic purification to isolate the adducts. Most likely some cycloreversion occurs in the process, leading to erosion of the final yield. Finally, 2-furamides were included in the substrate scope and also proved to be surprisingly reactive dienes (entries 7–9). The water soluble dimethylamide adduct 3r was formed in a comparable yield to the parent compound 3a under similar conditions (Table 2, entry 2), whereas adduct precipitation likely pushed conversions above 90% for products 3q and 3s by favourably shifting the thermodynamic equilibrium. Thus, the ester and amide derivatives of 2-furoic acid also show good reactivity towards maleimides and the aqueous protocol generally allows for a facile synthesis and isolation of the corresponding adducts. Again, the reactions feature high stereoselectivity towards the exo diastereoisomer (typically >12:1, with the exception of adduct 3s).
To the best of our knowledge, none of the adducts 3 have been previously synthesized and characterized. In particular, while acknowledging the fact that the reactive diene is not FDCA itself but its Na salt, we would like to highlight the distinct nature of this adduct (Table 3) among the typical structures of furan DA adducts.44,45
Follow-up chemistry
In addition to their facile preparation, Diels–Alder adducts 3 represent valuable synthons for further synthetic elaboration (Scheme 2). To complement the broad synthetic scope of our methodology, diversification at the X- and R2-positions in 3 (Scheme 2) can also be readily achieved by straightforward transformations of title adducts 3a and 3l: ethyl ester 3n can be obtained by esterification of 3a in the presence of SOCl2 (90% yield), whereas alkylation of 3l with benzyl bromide afforded 3t in 88% yield. Aromatization of the 7-oxanorbornene core by dehydration is a particularly interesting (and extensively studied) transformation,45 as it provides access to renewable aromatics based on the carbohydrate fraction of lignocellulosic biomass. For the substrates studied here, the presence of an electron-withdrawing substituent at the bridghead position in 3 is anticipated to hinder (typically) acid-mediated dehydration. Nonetheless, preliminary experimentation indicated that 33 wt% HBr in AcOH46 is a suitable acid for the conversion of 3a to phthalimide derivative 4 (66% isolated yield). Acidic hydrolysis of 4 produces in high yield hemimellitic acid 5, an aromatic tricarboxylic acid with potential application in the polymer and lubricant industry.47,48 Finally, catalytic hydrogenation towards oxanorbornane derivative 6 was also facile.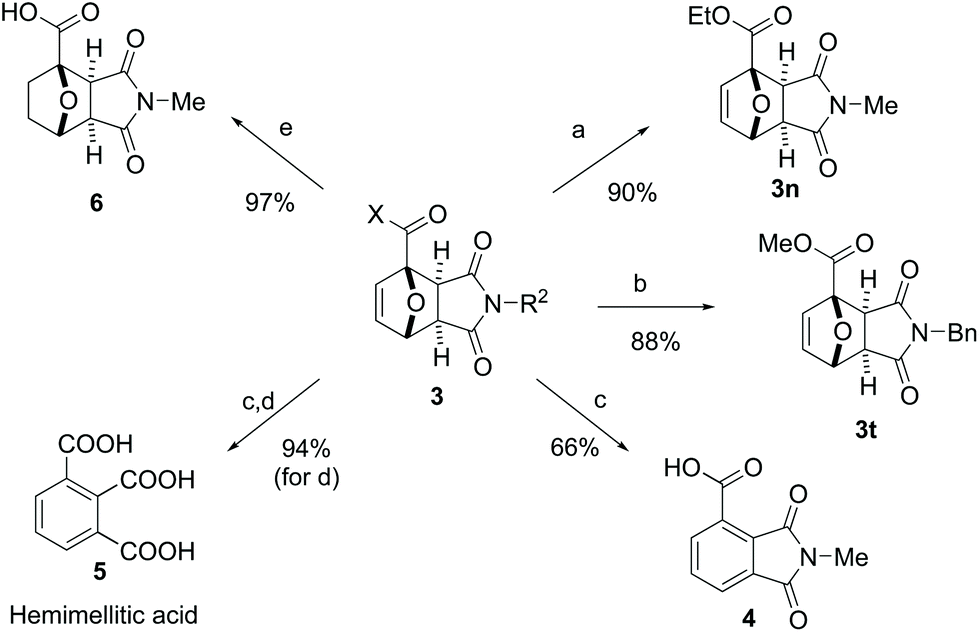 | ||
| Scheme 2 Follow-up chemistry starting with adduct 3. Reagents and conditions: a. SOCl2, EtOH, rt (from 3a); b. BnBr, K2CO3, DMF, rt (from 3l); c. 33 wt% HBr in AcOH, rt to 60 °C (from 3a); d. 35% HCl, 100 °C (from 4); e. H2, Pd/C, rt (from 3a). | ||
Mechanistic understanding
The above shows that furan carboxylic acid derivatives undergo surprisingly efficient Diels–Alder couplings to maleimides in aqueous solution. This result challenges the widely accepted idea that the diene scope in furan Diels–Alder chemistry is limited to electron-rich derivatives. While the nature of the furan substituent impacts the cycloaddition kinetics in a relatively predictable manner (the stronger the electron-withdrawing effect, the slower the DA reaction), we would like to note that moderate thermal activation can provide sufficient acceleration for seemingly unreactive inputs such as the 2-furoic acid derivatives used here; this is particularly true when water is used as solvent. The effect water has on the reaction can have multiple causes and may differ for the furoic acids on the one hand and the esters and amides on the other, depending on their physical characteristics. The hydrophobic effect49 is evidently relevant for the biphasic reactions, but may not play a decisive role for the highly water-soluble 2-furoic acid substrates. To probe the operation of the hydrophobic effect in this case, we studied the title reaction between 1a and maleimide 2a in the presence of salting-in and salting-out reagents (Table 5). The effect of salt additives on the rate of aqueous Diels–Alder reactions has been extensively studied.50 In general, salting-out reagents (e.g. simple inorganic salts like NaCl and CaCl2) lead to rate enhancements, whereas additives that disturb hydrophobic interactions like guanidinium (Gn) salts characteristically retard conversion.51 In addition, enhanced hydrophobic interactions are typically associated with an increased preference for the endo configuration of the adduct (the more compact geometry). Both of these hallmarks of the hydrophobic effect are consistently augmented with increasing additive concentration. Data in Table 5 summarizes the effect of additives on the rate of the cycloaddition between 1a and 2a (0.2 M). The observed yield and stereochemistry are generally consistent with the expected trends, illustrating that hydrophobic effect is a relevant influence in this system.|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Additive | Amount | exo-3a, % | endo-3a, % | Total, % | endo selectivity, % |
| General procedure: 2-Furoic acid 1a (0.5 mmol), (additive) and N-methyl maleimide 2a (1.5 equiv.) stirred in H2O (0.2 M) at 50 °C for 6 h; yields determined from the 1H-NMR ratios of product isomers and starting material.a GnCl = guanidinium chloride. | ||||||
| 1 | None | n/a | 39 | 5 | 44 | 13 |
| 2 | NaCl | 1 M | 42 | 5 | 47 | 12 |
| 3 | NaCl | 2 M | 43 | 7 | 50 | 16 |
| 4 | NaCl | 4 M | 43 | 9 | 52 | 22 |
| 5 | CaCl2 | 2 M | 50 | 9 | 59 | 18 |
| 6 | GnCla | 2 M | 37 | 4 | 41 | 11 |
In addition, hydrogen bonding is likely to also play an important role in our system, for instance by preferentially stabilizing the transition state (and product) over the addends.37 For example, the electron density around the oxygen atom in the furan ring expectedly changes significantly during the reaction since aromaticity is lost as the reaction proceeds; moreover, in the product (and transition state), both oxygen lone pairs can serve as hydrogen bond acceptors. In addition, the engagement of the COOH group as H-bond donor in interactions with surrounding water molecules increases the electron density around the furan ring and thus the rate of the DA coupling. Indeed, the addition of base is the extreme case of this, i.e. leading to complete proton transfer, as illustrated by the observations that 2-furoate salts already react with maleimides at ambient temperatures and that the bis-Na salt of FDCA is, counter to expectation, a reactive diene. It is important to also note that in base-mediated reactions the thermodynamics is favourably impacted as well: adduct 3a is roughly 4 times more acidic than the starting 2-furoic acid 1a (ΔpKa approx. 0.6, see ESI†), which supplies an additional −3.6 kJ mol−1 to the 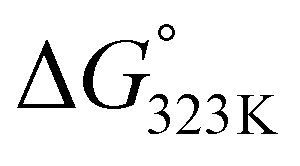 , sufficient to render the reaction essentially irreversible (>95% equilibrium conversion, see Table 2, entry 4 vs. entry 2).
, sufficient to render the reaction essentially irreversible (>95% equilibrium conversion, see Table 2, entry 4 vs. entry 2).
When 2-furoic acid esters are used as dienes, the system is no longer homogeneous and the reactions proceed ‘on-water’. Hydrophobic interactions and hydrogen bonding with water molecules at the interface play an activating role here, together with the high local concentration effect (in neat conditions for example, the reaction also proceeds readily, but conversion is hampered by the poor solubility of 2a in methyl furoate). In addition, the yield of adduct formed, i.e. the ultimate efficiency of the reaction, is definitely impacted by the crystallization of the product; this improves kinetics by reducing the rate of the back reaction and pushes the conversion beyond the solution equilibrium. Indeed, in all examples where product crystallization occurred, increased conversions were obtained (e.g.Table 4, entries 1, 7 and 9).
Finally, furamides show comparable behaviour to the parent furoic acids, as the aqueous reactions commence homogeneously at 50 °C. In terms of kinetics, furamides are likely somewhat more reactive than furoic acids, while the tendency of the corresponding adducts to crystallize out of aqueous solution is more pronounced. For instance, with unsubstituted 2-furamide precipitation was observed within minutes (at 50 °C), benefiting conversion. In the absence of product crystallization (entry 8, Table 4, adduct 3r) the efficiency of the reaction is lower and quite comparable to the case of parent furoic acid 1a under similar conditions (entry 2, Table 2).
Conclusions
Herein we showcase the successful use of biomass-derived 2-furoic acids, esters and amides as dienes in Diels–Alder cycloadditions. Thus, a variety of novel DA adducts could be selectively obtained following a green synthetic protocol involving the use of renewable feedstock, aqueous solvent, mild conditions, and non-chromatographic purification. The DA couplings proceed surprisingly efficiently with these readily available dienes, which represents an important expansion of the current scope of furan DA reactions to include underrepresented electron-poor derivatives. Some opportunities for downstream diversification of the adducts into valuable chemical products, including substituted bio-based aromatics, is also demonstrated. Expansion of the dienophile scope beyond maleimides52 is currently underway in our laboratories.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by The Netherlands Organization for Scientific Research (NWO LIFT grant 741.018.408). Dr T. N. Ran and Dr J. T. B. H. Jastrzebski (Utrecht University) are acknowledged for technical assistance. We thank Dr J. Sastre Torano (Utrecht University) for performing the ESI-MS measurements and E. C. Monkcom for the ToC artwork.Notes and references
- O. Diels and K. Alder, Chem. Ber., 1929, 62, 554–562 CrossRef.
- C. E. Puerto Galvis, L. Y. Vargas Méndez and V. V. Kouznetsov, Chem. Biol. Drug Des., 2013, 82, 477–499 CrossRef CAS PubMed.
- J. Plumet and S. Roscales, Heterocycles, 2015, 90, 741–810 CrossRef.
- M. Gregoritza and F. P. Brandl, Eur. J. Pharm. Biopharm., 2015, 97, 438–453 CrossRef CAS PubMed.
- M. Vauthier, L. Jierry, J. C. Oliveira, L. Hassouna, V. Roucoules and F. B. Gall, Adv. Funct. Mater., 2019, 1806765, 1–16 Search PubMed.
- H. Sun, C. P. Kabb, M. B. Sims and B. S. Sumerlin, Prog. Polym. Sci., 2019, 89, 61–75 CrossRef CAS.
- C. D. Spicer, E. T. Pashuck and M. M. Stevens, Chem. Rev., 2018, 118, 7702–7743 CrossRef CAS PubMed.
- C. S. Schindler and E. M. Carreira, Chem. Soc. Rev., 2009, 38, 3222–3241 RSC.
- R. C. Boutelle and B. H. Northrop, J. Org. Chem., 2011, 76, 7994–8002 CrossRef CAS PubMed.
- V. Froidevaux, M. Borne, E. Laborbe, R. Auvergne, A. Gandini and B. Boutevin, RSC Adv., 2015, 5, 37742–37754 RSC.
- A. D. Pehere, S. Xu, S. K. Thompson, M. A. Hillmyer and T. R. Hoye, Org. Lett., 2016, 18, 2584–2587 CrossRef CAS PubMed.
- I. Scodeller, S. Mansouri, D. Morvan, E. Muller, K. de Oliveira Vigier, R. Wischert and F. Jérôme, Angew. Chem., Int. Ed., 2018, 57, 10510–10514 CrossRef CAS PubMed.
- R. W. Foster, L. Benhamou, M. J. Porter, D. K. Bučar, H. C. Hailes, C. J. Tame and T. D. Sheppard, Chem. – Eur. J., 2015, 21, 6107–6114 CrossRef CAS PubMed.
- F. Gaviña, A. M. Costero, P. Gil, B. Palazón and S. V. Luis, J. Am. Chem. Soc., 1981, 103, 1797–1798 CrossRef.
- J. J. Pacheco and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 8363–8367 CrossRef CAS PubMed.
- M. Orazov and M. E. Davis, Chem. Sci., 2016, 7, 2264–2274 RSC.
- J. J. Pacheco, J. A. Labinger, A. L. Sessions and M. E. Davis, ACS Catal., 2015, 5, 5904–5913 CrossRef CAS.
- J. K. Ogunjobi, T. J. Farmer, C. R. McElroy, S. W. Breeden, D. J. MacQuarrie, D. Thornthwaite and J. H. Clark, ACS Sustainable Chem. Eng., 2019, 7, 8183–8194 CrossRef CAS.
- A. Z. Kikri, J.-M. Ha, Y.-K. Park, H. Lee, J. D. Suh and J. Jae, Catal. Today, 2020, 351, 37–43 CrossRef.
- B. Wang, G. J. M. Gruter, M. A. Dam and R. M. Kriegel, WO2014/065657, 2014.
- E. M. Serum, S. Selvakumar, N. Zimmermann and M. P. Sibi, Green Chem., 2018, 20, 1448–1454 RSC.
- C. Xu, E. Paone, D. Rodríguez-Padrón, R. Luque and F. Mauriello, Chem. Soc. Rev., 2020, 49, 4273–4306 RSC.
- K. I. Galkin, E. A. Krivodaeva, L. V. Romashov, S. S. Zalesskiy, V. V. Kachala, J. V. Burykina and V. P. Ananikov, Angew. Chem., Int. Ed., 2016, 55, 8338–8342 CrossRef CAS PubMed.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, ChemSusChem, 2016, 9, 2736–2748 CrossRef CAS PubMed.
- S. Dutta and N. S. Bhat, Biomass Convers. Biorefin., 2020 DOI:10.1007/s13399-020-01042-z.
- J. Pang, M. Zheng, R. Sun, A. Wang, X. Wang and T. Zhang, Green Chem., 2016, 18, 342–359 RSC.
- A. E. Settle, L. Berstis, N. A. Rorrer, Y. Roman-Leshkóv, G. T. Beckham, R. M. Richards and D. R. Vardon, Green Chem., 2017, 19, 3468–3492 RSC.
- J. Ax and G. Wenz, Macromol. Chem. Phys., 2012, 213, 182–186 CrossRef CAS.
- C. Ninh and C. J. Bettinger, Biomacromolecules, 2013, 14, 2162–2170 CrossRef CAS PubMed.
- H. Y. Lee and S. H. Cha, Macromol. Res., 2017, 25, 640–647 CrossRef CAS.
- K. S. Byun, W. J. Choi, H. Y. Lee, M. J. Sim, S. H. Cha and J. C. Lee, RSC Adv., 2018, 8, 39432–39443 RSC.
- K. C. Koehler, A. Durackova, C. J. Kloxin and C. N. Bowman, AIChE J., 2012, 58, 3545–3552 CrossRef CAS.
- C. S. Lancefield, B. Fölker, R. C. Cioc, K. Stanciakova, R. E. Bulo, M. Lutz, M. Crockatt and P. C. A. Bruijnincx, Angew. Chem., 2020, 59, 23480–23484 CrossRef CAS PubMed.
- R. C. Cioc, M. Lutz, E. A. Pidko, M. Crockatt, J. C. van der Waal and P. C. A. Bruijnincx, Green Chem., 2021, 23, 367–373 RSC.
- A. Shrinidhi, ChemistrySelect, 2016, 1, 3016–3021 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS PubMed.
- S. Otto, W. Blokzijl and J. B. F. N. Engberts, J. Org. Chem., 1994, 59, 5372–5376 CrossRef CAS.
- J. C. Van Der Waal, S. Thiyagarajan, H. C. Genuino, E. De Jong, J. Van Haveren, B. M. Weckhuysen, P. C. A. Bruijnincx and D. S. Van Es, ChemSusChem, 2015, 8, 3052–3056 CrossRef PubMed.
- S. Thiyagarajan, H. C. Genuino, J. C. Van Der Waal, E. De Jong, B. M. Weckhuysen, J. Van Haveren, P. C. A. Bruijnincx and D. S. Van Es, Angew. Chem., Int. Ed., 2016, 55, 1368–1371 CrossRef CAS PubMed.
- H. C. Genuino, S. Thiyagarajan, J. C. van der Waal, E. de Jong, J. van Haveren, D. S. van Es, B. M. Weckhuysen and P. C. A. Bruijnincx, ChemSusChem, 2017, 10, 277–286 CrossRef CAS PubMed.
- F. A. Kucherov, K. I. Galkin, E. G. Gordeev and V. P. Ananikov, Green Chem., 2017, 19, 4858–4864 RSC.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS PubMed.
- T. Kitanosono and S. Kobayashi, Chem. – Eur. J., 2020, 26, 9408–9429 CrossRef CAS PubMed.
- C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179–14233 CrossRef CAS.
- F. A. Kucherov, L. V. Romashov, G. M. Averochkin and V. P. Ananikov, ACS Sustainable Chem. Eng., 2021, 9, 3011–3042 CrossRef CAS.
- M. G. Van Campen and J. R. Johnson, J. Am. Chem. Soc., 1933, 55, 430–431 CrossRef CAS.
- P. D. Giorgi, S. H. Soo-Tang, S. Antoniotti and J. C. van der Waal, ChemistrySelect, 2017, 2, 10766–10770 CrossRef CAS.
- M. Crockatt and J. H. Urbanus, WO2017/146581, 2017.
- D. C. Rideout and R. Breslow, J. Am. Chem. Soc., 1980, 102, 7816–7817 CrossRef CAS.
- A. Kumar, Chem. Rev., 2001, 101, 1–19 CrossRef CAS PubMed.
- A. Kumar, U. D. Phalgune and S. S. Pawar, J. Phys. Org. Chem., 2002, 15, 131–138 CrossRef CAS.
- We observed that the imide motif in 2a (and 4) is much more prone to ring opening that in the product 3a. For instance, imide 3a is fully stable in 35% HCl at 100 °C. This suggests that the fused succinimide ring in 3a has less strain than the flat structures (all sp2 atoms in the ring) 2a and 4. Thus, upon DA cycloaddition with 2a, strain energy is presumably released, which would be an important contributor to the reaction overall thermodynamics. This might explain (in part) why maleimides are such efficient dienophiles.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc01535d |
| This journal is © The Royal Society of Chemistry 2021 |