
HMF–glycerol acetals as additives for the debonding of polyurethane adhesives†
Sarah
Kirchhecker
a,
Andrea
Dell'Acqua
 a,
Astrid
Angenvoort
b,
Anke
Spannenberg
a,
Kenji
Ito
b,
Sergey
Tin
a,
Andreas
Taden
*b and
Johannes G.
de Vries
*a
a,
Astrid
Angenvoort
b,
Anke
Spannenberg
a,
Kenji
Ito
b,
Sergey
Tin
a,
Andreas
Taden
*b and
Johannes G.
de Vries
*a
aLeibniz Institut für Katalyse, e. V. Albert-Einstein-Strasse 29a, 18059 Rostock, Germany. E-mail: johannes.devries@catalysis.de
bHenkel AG & Co. KGaA, Henkel-Str. 67, 40589 Düsseldorf, Germany. E-mail: andreas.taden@henkel.com
First published on 14th December 2020
Abstract
Diols prepared via solvent-free acetalisation of hydroxymethylfurfural with glycerol were incorporated as additives into polyurethanes based on a bioderived polyether polyol. Moisture-cured as well as acrylate cross-linked films of these PUs were prepared. Both materials displayed excellent thermal stability and could be cleaved in acidic solutions. In particular the highly cross-linked film produced from the acrylate endcapped polyurethanes displayed a clear tuneability of the degradation behaviour according to the amount of acetal additive incorporated. This system has the potential to be used for the selective debonding of polyurethane-based adhesives at the end of their lifetime to facilitate the recycling of expensive components and raw materials from complex devices such as consumer electronics.
Introduction
There are growing concerns about the accumulation of plastic waste in the environment as well as the exhaustion of a range of natural resources. EU directives such as the circular economy action plan (The European Green Deal),1 aim to reduce the waste created by single use items and planned obsolescence of consumer products, stress the right to repair and promote recycling.Polyurethanes (PUs) are one of the major classes of polymers with a global production of over 22 million tons per year.2 They are used in many different areas, such as in the automotive and construction industry in the form of soft and rigid foams,3 coatings and adhesives,4 as well as in everyday consumer objects like mattresses. PUs are formed by the reaction of isocyanates with diols (hydroxy-terminated polyester or polyether chains = polyols). Depending on the number of functionalities of the two components, linear chains as well as highly cross-linked networks are possible.5
While thermoplastic polymers can be recycled by physical means such as repeat melt processing, this is not possible for cross-linked thermoset materials like PUs.6,7 Recently, there has been increasing research into the chemical recycling of polymers.8,9 Due to the different types of monomers and linkages in the cured product, the chemical recycling of PUs is more difficult than that of simple polymers, and is currently not practised. Nevertheless, some progress has been made in the application of transcarbamoylation reactions with glycols or alcohols, which break up PUs into soluble components that can be reused.10,11 However, currently there are still issues to be overcome, such as high reaction temperatures and the lower quality of recovered monomers due to contamination with the alcoholysing agents.12
One important area of application for PUs is in adhesives.4,13 The recycling of the PU adhesives itself is not important, as it only makes up a very small amount of the total material compared to the rest of the device (Fig. 1a). However, with more and more devices made from a range of expensive and rare raw materials being produced only for a very limited lifetime – such as smartphones14 – it is of utmost importance to develop adhesives that provide strong bonding when required, but which can be easily debonded after the device's lifetime has come to an end to facilitate the separation and replacement as well as subsequent recycling of components.15,16
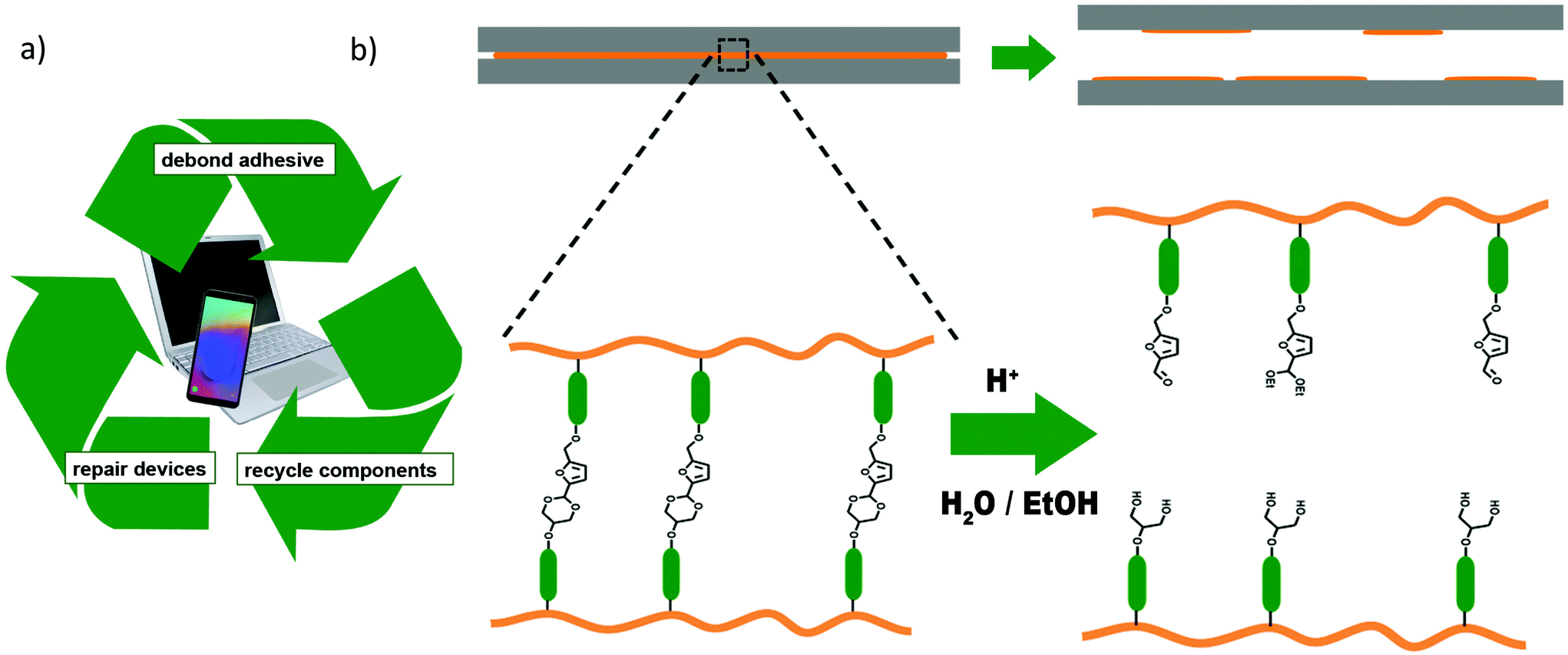 | ||
| Fig. 1 (a) Debonding of adhesives can enable the repair of devices by replacing broken parts and the recycling of expensive raw materials. (b) The debonding mechanism proposed in this work: adhesives containing cross-linked PUs with acetal diol additives can be cleaved by acid and aqueous solvent. | ||
Stimuli-responsive polymers17 are an active field of research, and especially in drug delivery,18 debonding on demand has been researched for a while. Several strategies for debonding and chemical recycling of thermosets have recently been investigated,19 including light-, electricity- and heat-triggered mechanisms, as well as physical debonding pathways, such as the inclusion of particles, which can be heated or expanded to break apart the two components.20 Other recent work employed fluoride ions as the debonding stimulus for adhesives,21 and PUs were cross-linked reversibly via a Diels–Alder reaction between pendant furan rings and short chain bismaleimide linkers,22,23 which can be reversed at higher temperatures.24 Poly-hydroxyurethanes based on acetals with reversible cross-linking under acidic conditions have also recently been reported.25 Recently several publications have demonstrated the use of bio-derived acetal diols as building blocks for epoxy resins, PUs and polyesters, which can be cleaved by the application of acid and solvents. Ma and co-workers reported several epoxy coatings based on an acetal diol synthesised from vanillin and pentaerythritol with different amines.26,27 These thermoset materials could be cleaved in acidic solutions of polar solvents. Very recently, the same group synthesised an acetal diol of vanillin and glycerol, which again was employed in an epoxy resin after derivatisation,28 as well as incorporated into a PU–carbon fibre composite.29 The materials could be cleaved in HCl in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v acetone:H2O at 50 °C. Zhang and co-workers synthesised a spiro acetal diol consisting of two molecules of 5-hydroxymethylfurfural (HMF) combined with pentaerythritol, which they employed as a monomer in both polyesters and polyurethane-ureas.30 The degradation behaviour of these polymers was not investigated.
:1 v/v acetone:H2O at 50 °C. Zhang and co-workers synthesised a spiro acetal diol consisting of two molecules of 5-hydroxymethylfurfural (HMF) combined with pentaerythritol, which they employed as a monomer in both polyesters and polyurethane-ureas.30 The degradation behaviour of these polymers was not investigated.
PU based adhesives can be tuned to a wide variety of applications. In fact, many of the areas specifically targeted for improvement in the European Green Deal, including consumer electronics, automobiles and packaging, use PU based adhesives.1
The possibility of adding a general, acid-triggered debonding-on-demand mechanism into existing materials and formulations would therefore be of huge benefit for industrial applications. In the following, we investigate the use of an acetal diol based on HMF and glycerol as an additive for this purpose (Fig. 1).
HMF is a platform chemical, which can be obtained from cellulose or sugars in the presence of an acidic catalyst.31 Glycerol is widely available as a side product from the production of biodiesel from vegetable oils.32 The acetalisation of furfural with glycerol has been reported by several groups using a range of different acid catalysts.33 A patent from 2008 claims the selective formation of the dioxolane (5-membered ring) isomer but the compound has not been commercialised.31 In general it is known that glycerol acetals and ketals always form a mixture of the dioxolane and dioxane products,34 with some catalysts being able to efficiently provide the kinetic dioxolane product before isomerisation occurs.35 Recently, Arias et al. reported the synthesis of HMF–glycerol acetals in excellent yield, as a mixture of four isomers, cis and trans isomers of dioxolanes and dioxanes, using solid acid catalysts at high dilution.36
For the purpose of this study, velvetol, a commercially available polyether polyol based on bioderived 1,3-propanediol was chosen as a model compound for the formation of PUs.37 However, we envisage the use of the additive to be applicable to a wide range of existing PUs to facilitate debonding. Therefore, in contrast to work discussed above, the acetal diol is added only in small amounts of up to 10 wt% of the polyol fraction, so as not to change the properties of the existing PU.
Experimental
Materials
HMF was purchased from AVA-Biochem BSL AG and stored under argon in the freezer. Glycerol (Alfa Aesar), MCM-41 (Sigma), isophorone diisocyanate (IDPI, Merck) and velvetol H2700 (poly(1,3-propanediol), 2600–2800 g mol−1, OH number 41, Alessa) were used as received without further purification.General method for the large scale synthesis of the mixture of cis/trans-2-(5-(hydroxymethyl)furan-2-yl)-1,3-dioxan-5-ol and cis-/trans-(5-(4-(hydroxymethyl)-1,3-dioxolan-2-yl)furan-2-yl)methanol (acetal diols)
HMF (15.8 g, 0.125 mol) and glycerol (18.3 mL, 0.250 mol) were stirred at 40 °C under vacuum (0.8–20 mbar) with 8 wt% MCM-41 (1.26 g) until the HMF was mostly converted (depending on the vacuum 6–24 h). After this time, the reaction mixture was dissolved in acetonitrile and the catalyst was removed by filtration. The catalyst was washed with ACN and EtOAc. The solvents were removed under reduced pressure and if necessary, the residue was stirred in a saturated solution of sodium bisulfite for 1.5 h to remove leftover HMF (this step is only needed if all acetal isomers are to be purified). The product was extracted from the aqueous phase with EtOAc four times. After solvent removal, the crude product was dissolved in DCM or EtOAc which led to the precipitation of one of the isomers as a white solid. This isomer was separated by filtration and the solvent was removed in vacuo. The remaining three isomers were purified by column chromatography using a gradient from 3:1 EtOAc:cyclohexane – 100% EtOAc as the eluent. The isomers were recombined. Typical isolated yield: 56%.
Mixture of 4 isomers
1H NMR (300 MHz, DMSO-d6) δH 6.48 (dd, J = 3.2, 0.4 Hz), 6.44 (dd, J = 3.2, 0.4 Hz), 6.38–6.33 (m), 6.29–6.22 (m) (2H), 5.90 (s), 5.79 (s), 5.57 (s), 5.44 (s) (1H), 5.23 (td, J = 5.8, 4.4 Hz, 1H), 4.98 (d, J = 4.2 Hz, 1H), 4.91 (td, J = 5.7, 3.0 Hz, 2H), 4.41–4.32 (m), 4.28–4.16 (m), 4.06–3.83 (m), 3.77 (dd, J = 7.9, 6.0 Hz), 3.71 (dd, J = 7.9, 6.2 Hz), 3.54–3.45 (m) (6H) ppm.13C NMR (75 MHz, DMSO-d6) δC 156.1 156, 155.1, 150.27, 150.1, 149.7, 109.9, 109.5, 107.9, 107.4, 107.3, 107.3, 97.2, 97.0, 94.8, 77.0, 76.3, 71.4, 71.1, 66.7, 66.4, 62.3, 61.7, 61.4, 55.7, 55.6 ppm.
ESI-MS (m/z): calculated for [M + Na]+ = 223.0582, found: 223.0583.
GC (area) = 99.0%.
trans-2-(5-(Hydroxymethyl)furan-2-yl)-1,3-dioxan-5-ol (trans-dioxane isomer) (1)
1H NMR (400 MHz, DMSO-d6) δH 6.34 (d, J = 3.2 Hz, 1H), 6.23 (d, J = 3.2 Hz, 1H), 5.44 (s, 1H), 5.24 (dd, J = 6.7, 5.0 Hz, 2H), 4.34 (d, J = 5.5 Hz, 2H), 4.12–4.03 (m, 2H), 3.69 (tq, J = 9.8, 4.9 Hz, 1H), 3.48–3.39 (m, 2H) ppm.13C NMR (75 MHz, DMSO-d6) δC 155.3, 149.6, 108.1, 107.3, 94.8, 71.4, 60.1, 55.6 ppm.
ESI-MS (m/z): calculated for [M + Na]+: 200.0582, found: 223.0580.
ATR-FT-IR: 3214, 2970, 2938, 2873, 1405, 1255, 1002, 795.
Elemental analysis for C9H12O5: expct, found: C: 54.00, 53.93; H: 6.04, 5.91.
Mp (DSC) = 104 °C.
Representative example of polyurethane synthesis type A (synthesis of 2–5%)
Velvetol 2700 (32.3 g, OH number 41) was dried at 90 °C under vacuum until there were no more bubbles visible (circa 1 h). The oil bath was removed and 1 (1.7 g, 5 wt% of polyols) was dissolved in anhydrous acetone and added to the velvetol under nitrogen. Enough solvent was added to form a homogeneous mixture. When the mixture was cooled to about 50 °C, isophorone diisocyanate (6.29 g, 1.4 eq.) was added followed by the catalyst (100 μl Borchi Kat 315). The progress of the reaction was followed by titration of the NCO number and terminated when the value was below 2%.Representative example of polyurethane synthesis type B (synthesis of 3–5%)
Velvetol 2700 (30.57 g, OH number 41) was dried at 90 °C under vacuum until there were no more bubbles visible (circa 1 h). The oil bath was removed and 1 (1.61 g, 5 wt% of polyols) was dissolved in anhydrous ethyl acetate and added to the velvetol under nitrogen. Ethyl acetate was added to form a homogeneous mixture. When the mixture was cooled to about 50 °C, isophorone diisocyanate (5.97 g, 1.4 eq.) was added followed by the catalyst (300 μl Borchi Kat 315) and the reaction was performed at 70 °C. The progress of the reaction was followed by titration of the NCO number until the value was below 2%. 4-Hydroxybutylacrylate (2.20 g, 0.015 mol) was added together with 100 μl Borchi Kat 315. The reaction was again followed by the NCO number until it was 0.03%, at which point the reaction was terminated and the solution was stored like this for further use.Polyurethane films
For the moisture-cured type A films 4, each prepolymer 2 (7.0 g) was dissolved in EtOAc (23 g) at room temperature and poured into a Teflon bowl. These were left in air for 14 days while the solvent evaporated, and cross-linking was achieved by reaction with moisture from the air. To ensure complete reaction of all NCO groups, the dishes with 4-0% and 4-5% were kept in a high moisture oven as preliminary experiments showed that curing took longer.For the acrylate cross-linked films 5 (type B) the PUs 3 (7 g) were dissolved in EtOAc (23 g) if not in solution already and 2 wt% (0.14 g) of TPO-L photoinitiator (ethyl(2,4,6-trimethylbenzoyl) phenyl phosphinate) was added. The films were poured into Teflon dishes and the solvent evaporated in the dark at 40 °C overnight. Then the films were cured by irradiation with 365 nm LED light in an LED oven for 1 minute.
NMR degradation experiment
To follow the degradation of the PUs by NMR, a sample of the PU was dissolved in 0.5 mL acetone-d6 in an NMR tube. After a spectrum was measured, 4.6 μL of conc. HCl was added and NMR spectra were recorded with one-minute intervals for 15 minutes.Film degradation experiments
Pieces of roughly equal thickness and size were cut from the six PU films and submerged in glass sample vials in solutions of NaOH, HCl, acetic acid of different concentrations (0.1, 1 and 5 M) in water, as well as a solution of HCl which was topped up to the required volume with an 8:2 mixture of EtOH:H2O at the same concentrations. Pure water was added as a control. The samples were stored at room temperature and inspected visually over a period of 14 days. Then samples were prepared in the same way of the films 5 in 5 M concentrations of HCl, acetic acid (AcOH), and HCl in EtOH:H2O and heated to 50 °C for a period of 1 week. The experiments in 5 M HCl in EtOH/H2O were performed in duplicate. A sample of similar thickness and weight (87–89 mg) was dissolved in 3 mL of the 5 M HCl in 8:2 EtOH:H2O solution and heated at 50 °C using a metal heating block on a hotplate until the solution was homogeneous by eye.
Afterwards, a sample was taken, and the liquid was evaporated to dryness on the rotavap. The residue was redissolved in THF for APC-GPC analysis.
Results and discussion
Synthesis of the acetal diol
As the high catalyst loading and high dilution employed in the report of Arias et al.36 are neither practical nor sustainable for large-scale synthesis, we decided to screen for a more scalable synthetic method (Table 1). In our catalyst testing, a commercially available version of MCM-41 performed best, although even under the same conditions as previously published, the highest yield we could achieve was 65% (Table 1, entry 1). This may be due to the higher Si:Al ratio (40 vs. 15) and hence lower acidity of the commercially available MCM-41 catalyst compared to the one used by Arias et al., which they prepared themselves. Performing the reaction in the melt with one equivalent excess of glycerol allowed the reaction to proceed with lower catalyst loading and at a much lower temperature of 40 °C (entries 2–4). Decreasing the catalyst loading by half from 20 to 10 wt% had a negligible effect on the yield (34 vs. 33%), while decreasing it further to 5 wt% lowered the yield to 30% (entry 4). Increasing the temperature to 60 °C gave a lower yield of 23% at nearly full conversion (entry 5). This is most probably due to the formation of humins from HMF in contact with the acid catalyst, a well know problem. While in other acetalisation reactions strong acid catalysts can be used without any problem, HMF only tolerates weakly acidic catalysts. Using the stronger heterogeneous acid catalyst Montmorillonite K10 (M K10) reduced the yield to only 17% at near complete conversion (entry 6) and using 1 mol% para-toluenesulfonic acid (PTSA) led to the immediate degradation of HMF under melt conditions (entry 7). Phosphoric acid, a medium strength homogeneous acid gave a lower yield of only 25% with a lot of degradation (entry 8). It was therefore also tested under Dean–Stark conditions as the higher dilution in organic solvents suppresses humins formation somewhat. However, use of phosphoric acid gave no desired product under these conditions (entry 9). It was mentioned by Arias et al. previously that the stirring rate has a large effect on the yield of the reaction as glycerol tends to adsorb to the catalyst.36 Under melt conditions at larger scale, magnetic stirring is in fact not possible anymore, due to the high viscosity of the reaction mixture.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Cat. loading [wt%] | Solvent | Temperature [°C] | Yield (conv.) [%] |
| Reaction conditions: Unless otherwise stated: 10 mmol HMF, 20 mmol glycerol, reduced pressure (0.8 mbar), 6 h.a 1:1 mixture of trifluorotoluene and acetonitrile, Dean–Stark, 24 h.b Stopped after 1 h due to degradation of HMF to humins.c Scale up (30 g) using mechanical stirrer.d nr = not recorded. |
|||||
| 1a | MCM-41 | 20 | TFT/ACN | 95 | 65 (72) |
| 2 | MCM-41 | 20 | Neat | 40 | 34 (88) |
| 3 | MCM-41 | 10 | Neat | 40 | 33 (90) |
| 4 | MCM-41 | 5 | Neat | 40 | 30 (78) |
| 5 | MCM-41 | 10 | Neat | 60 | 23 (98) |
| 6 | M K10 | 5 | Neat | 40 | 17 (98) |
| 7b | PTSA | 1 mol% | Neat | 40 | — (100) |
| 8 | H3PO4 | 5 mol% | Neat | 40 | 25 (85) |
| 9a | H3PO4 | 5 mol% | TFT/ACN | 95 | — (44) |
| 10c | MCM-41 | 8 | Neat | 40 | 56 (78) |
| 11c | MCM-41 | 5 | Neat | 40 | 49 (nr)d |

We therefore switched to mechanical stirring and found that the yield of the reaction changed quite drastically (entries 10 and 11). The yield increased from 33% to 55% and the conversion decreased slightly to 78% (entry 10). It appears that at larger scale the formation of humins is slowed down. Combined with the better mass transfer due to the mechanical stirring this is probably responsible for the higher yield. In most cases the reaction was performed for 6 h or overnight, with not much difference in yield. Conversion could be pushed to nearly 100% by leaving the reaction to run for longer time but with no increase in the yield of the desired products. It is possible that the yield can be increased with a better vacuum and therefore more efficient removal of water from the reaction, however, this was not possible with our set up. The catalyst was reused after thorough washing with ACN and ethyl acetate several times during scale up of the product with no significant change in yield (data not shown). Overall, we adhered to the principles of green chemistry by performing the synthesis in neat conditions, with a heterogeneous, easily reusable catalyst, and using the benign solvent ethyl acetate for workup as well as for the synthesis of the PUs.
The product of this reaction exists as four isomers, the dioxane and dioxolane products, each of which have cis and trans isomers. On workup of the reaction we discovered that one of the isomers is a crystalline solid, while the other three are viscous yellow oils at room temperature. Following the scaled-up reaction by 1H NMR (with a slightly lower vacuum of 20 mbar to slow down the reaction) showed that in the beginning there is a higher amount of the kinetic product, the dioxolanes, formed in nearly 50:50 cis:trans ratio. The ratio between the isomers changes over time in favour of the dioxanes (Fig. 2). This can be followed easily via the acetal peaks of the four isomers in the 1H-NMR spectrum.
 | ||
| Fig. 2 Crude 1H NMR spectra of the reaction between HMF and glycerol after 1, 2, and 2.5 days at 40 °C and 20 mbar (on average). Singlet peaks between 6.0 and 5.4 ppm correspond to the C–H peaks of the four isomers. From left to right: Dioxolanes, cis-dioxane, trans-dioxane. The decrease of the HMF peaks during the reaction is also visible, e.g. aldehyde peak at 9.55 ppm. | ||
The crystalline isomer (1) with a C–H peak at 5.45 ppm forms in the highest yield over time. After 2.5 days 70% of acetals are in the form of the crystalline isomer. It was isolated with an average overall yield of 54% from this mixture. Full NMR spectra of the different isomers are presented in the ESI (chapter S5).† Single crystal X-ray crystallography of 1 confirmed that it is the trans-dioxane isomer.37 The molecular structure is shown in Fig. 3 and the crystallographic data are given in chapter S6, ESI.† It was found that on prolonged storage the three non-crystalline isomers partially hydrolyse and isomerise to the crystalline isomer. The latter is more resistant to hydrolysis. In the recently published paper on vanillin glycerol acetals,28 the authors found that it was possible to force the formation of the crystalline dioxane acetal from vanillin and glycerol by the use of only a small excess of glycerol, which was rationalised by the fact that the crystalline isomer is less soluble in the glycerol and hence precipitates out. We also performed the synthesis with more and less equivalents of glycerol. However, in our case reducing the amount of glycerol to 1.5 equivalents led to a reduced yield of 49%, probably due to increased degradation of HMF caused by the higher concentration of HMF and acid, while a higher amount of glycerol (3 eq.) led to a reduced yield of 35% under the same conditions, probably due to the glycerol coating the catalyst and forming a gooey solid, preventing access for the HMF. We then also attempted to use a larger excess of glycerol (6 equivalents) as the solvent and performed the reaction at a higher temperature of 80 °C. While this prevented degradation of HMF to some extent, the reaction was also slower and there was no significant effect on the yield.
 | ||
| Fig. 3 Displacement ellipsoid plot (50% probability level) of 1. | ||
Due to its crystallinity, 1 can be easily separated from the mixture by crystallisation/precipitation. The other isomers are more difficult to separate, however it is possible to collect the pure cis-dioxane and the mixture of the two dioxolane isomers by column chromatography (see ESI S5† for NMRs). As the crystalline isomer is the main product of the reaction and also much easier to separate on a large, we decided to use it for the purpose of this study. However, the dioxane isomer contains a primary and a secondary alcohol group, while the dioxolanes have two primary –OH groups, which would be expected to react faster. To assess the difference in reactivities, we performed a kinetics study with a monofunctional isocyanate. The rate of reaction of 1 and the mixture of acetals remaining after crystallisation was compared via NMR (see ESI† chapter S3 for details). The reaction was slightly faster for the dioxolane-enriched mixture of acetals. This is expected due to the higher ratio of primary to secondary hydroxyl groups. However, the difference in rate was only small, so that the reaction was deemed fast enough to proceed with 1. Interestingly, there was no difference in reactivity of the primary and secondary alcohol detectable by monitoring the reaction with 1H-NMR (the primary alcohol group did not react first selectively).
Polyurethanes
For real life debonding applications, a small amount of acetal diol used as debonding trigger would be mixed into existing adhesive blends so as not to change the properties of the material too much. We therefore investigated two types of polyurethanes (PUs) with 0, 5 and 10 wt% of the polyol component exchanged for 1.The commercially available bio-derived polyether polyol velvetol was chosen as the polyol component for our model system. It was chosen for its superior hydrolytic stability compared to polyesters. In the first instance, we attempted to synthesise PUs with velvetol H2000. However, excessive gelling during and after the reaction made this impractical. We therefore switched to the highest available molecular weight, velvetol H2700. It still caused some issues with gelling, which could be suppressed by synthesising and storing the PU in ethyl acetate up until the pouring of the films. Gelling is assumed to be due to the slow crystallisation of the polyether chains. In commercial non-bio-derived polyethers there is always a mixture of PEG and PPG components, which inhibits this crystallisation. Two types of polyurethanes were synthesised (Scheme 1). For type A, velvetol with 0, 5 or 10 wt% of 1 was reacted with an excess of isophorone diisocyanate (IPDI) to give NCO-terminated prepolymers (2).
 | ||
| Scheme 1 Synthesis of linear polyurethanes and cross-linked films with 0–10 wt% incorporation of 1 in the polyol component. | ||
These PUs were then crosslinked by moisture curing for 14 days in air and/or a moisture cabinet where necessary to give urea-linked films 4. The type B series (3) was additionally end capped with 4-hydroxybutyl acrylate (4HBA) and cured radically under UV light using a photoinitiator (TPO-L, ethyl (2,4,6-trimethylbenzoyl) phenyl phosphinate) to give highly cross-linked films 5.
The PUs and films were analysed by NMR, IR, GPC, DSC and TGA. NMR spectra of the PUs before curing clearly show the incorporation of the acetal diol into the chain (ESI, S7†). IR analysis before and after curing confirmed the full conversion of NCO groups in type A by the complete disappearance of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch at just below 2300 cm−1 (ESI, S7.2†).
C stretch at just below 2300 cm−1 (ESI, S7.2†).
Thermal properties of the films were analysed by DSC and TGA (Fig. 4). As can be seen from Fig. 4a, the incorporation of the acetal diol into the PU chains in 4 suppressed the crystallisation of the velvetol segments, which is clearly visible in the sample without acetal additive (4-0%) on first heating (for the thermal analysis of pure velvetol see S10, ESI†). Apart from that, all other films show no phase transitions between 55 and 120 °C, indicating that the material is an amorphous solid in this temperature range. TGA analysis shows a slight decrease of thermal stability with increasing acetal content. The temperatures at 5% weight loss are given in Table 2. However, the thermal stability of the films is still very high for PUs. Even with 10% acetal diol added it is above 260 °C, confirming that the PUs are stable enough for a wide range of applications.
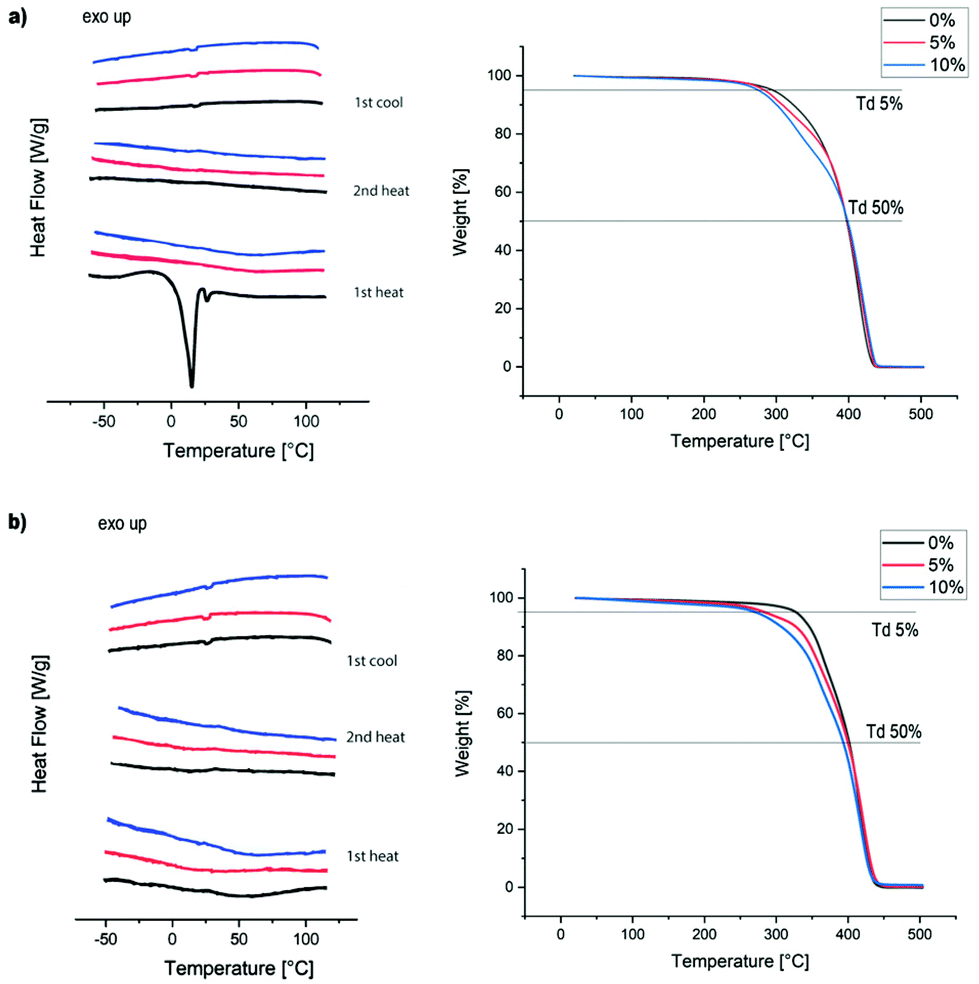 | ||
| Fig. 4 Thermal properties of PU films. (a) DSC and TGA of type A (4), (b) DSC and TGA traces of type B (5). | ||
Degradation experiments
To confirm the hypothesis that the acetal diol could be debonded by acid, we first studied the degradation of acrylate-endcapped PU 3-10% by NMR over time. Initially we added a 0.1 M HCl solution in D2O to the NMR sample, aiming to see the dissolution of fragments over time. However, the PU was too hydrophobic and floated on top of the solvent. No peaks were visible over a time scale of 10 minutes. Next, we used 0.1 M HCl in acetone-d6. The result is shown in Fig. 5. NMR spectra were recorded in one-minute intervals. Already during the two minutes it took to set up the experiment, the degradation started. After 15 minutes the acetal C–H peak had completely disappeared, while the HMF peaks grew visibly larger over time.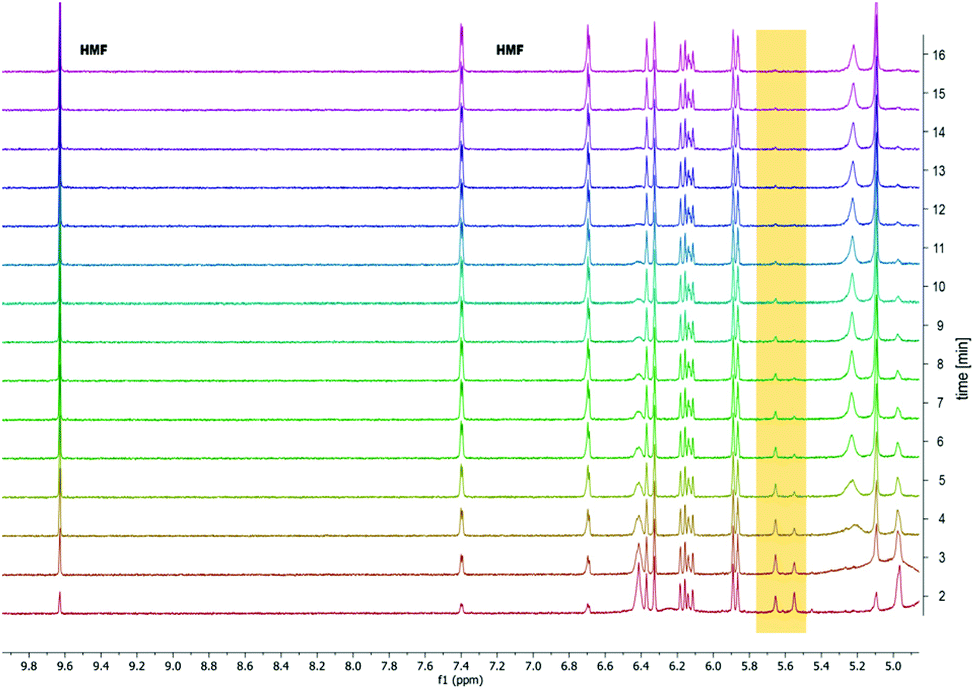 | ||
| Fig. 5 1H NMR degradation study of 3-10% over time in 0.1 M HCl in acetone showing the disappearance of the acetal peak at 5.55 ppm over time and appearance of the HMF aldehyde peak. | ||
Following these encouraging results, we tested the hydrolysis behaviour of the cured PU films under different debonding conditions. The films were cut into strips and submerged in solutions containing NaOH, HCl, AcOH or HCl in 8:2 (v/v) EtOH:H2O at different concentrations (0.1, 1 and 5 M), as well as pure water, and inspected visually over a period of 14 days at room temperature (see ESI S4† for photos). Additionally, the acrylate end-capped films were analysed in the 5 M acidic solutions at 50 °C. The films that visually ‘dissolved’ and those that were close to it were analysed by GPC. As expected, there was no change in the films’ structure for the samples in water and in basic solutions over the period of 14 days, demonstrating that the PUs are stable towards these conditions. Type A moisture-cured PUs 4 ‘dissolved’ in both 5 M aqueous HCl as well as in ethanolic HCl at all three concentrations within one day. There was no visible difference between the 0% and the acetal containing films, which shows that the bonds in these films are quite sensitive to acidic treatment and it is not necessary to include the acetal diol for debonding. Photos of all room temperature hydrolysis experiments can be found in the ESI.† In acetic acid the type A PUs only dissolved in the 5 M solution after 6 days. For the type B acrylate cross-linked films 5 the situation was quite different. These films proved to be very resistant to the aqueous acid treatment. Even in 5 M HCl no visible changes occurred in the film apart from a darkening in colour in the acetal containing samples. The results show how resistant these films are to degradation. The hydrophobic aliphatic backbone chains are likely to reduce the wettability of these materials compared to the polar urea linkages in the type A films, so that the aqueous acid cannot attack the polyurethane crosslinkers. However, the development of yellow to brown colour could be an indication of the formation and subsequent degradation of HMF. In 5 M HCl in 8:2 ethanol:water the films swelled a lot but did not disintegrate. Only the 10% acetal diol containing film 5-10% fully dissolved after 14 days at room temperature (Fig. 6a). Following on from this positive result, the three acrylate films 5 were retested in the 5 M acidic solutions at 50 °C. For the ethanolic HCl solution, the experiment was performed in duplicate. The results are shown in Fig. 6b and S4.2, ESI.† In each case 5-10% dissolved completely after 17 h, and 5-5% after 67 h–87 h depending on the size of film used. The 0% sample dissolved after 4–7 days at 50 °C. In 5 M aqueous HCl there was no dissolution at 50 °C after one week, again indicating that the organic solvent is necessary to swell the fairly hydrophobic films before the acid can attack the acetal bonds (ESI, S4.2†).
 | ||
| Fig. 6 PU samples before and after degradation test in 5 M HCl in 8:2 (v/v) ethanol:H2O. (a) Room temperature (b) 50 °C. | ||
To confirm full degradation of the dissolved films, GPC was measured for the samples of the fully ‘dissolved’ films, i.e.5-0%, 5-5%, 5-10% after the 5 M HCl/ethanol treatment at 50 °C and 5-10% after the same treatment at room temperature. The results are shown in Table 3. As it is not possible to dissolve the cross-linked films 5 in THF in order to measure GPC, the molecular weight of the end-capped PUs 3 are given as a comparison. The GPC results confirm the hydrolysis of the films into smaller segments. Although 5-0% eventually dissolved after one week, the fragments are larger than those for the acetal containing films 5-5% and 5-10%, indicating that acid-catalysed cleavage happens faster at the acetal moieties, while the urethane bonds react much slower. The values for the degraded film 5-10% are very similar for the experiment at room temperature and at 50 °C, indicating that the same reactions are taking place. IR spectra of the films 5 were compared to spectra of the degradation products (ESI, S8†). In samples 5-5% and 5-10% there is a peak at around 1045 cm−1, which is not present in film 5-0%. It is assigned to the C–O stretch of the acetal. After cleavage, this peak appears less pronounced in samples 5-5% and 5-10%, indicating that the acetal is cleaved. These data are in agreement with the recently published results on acetal containing epoxy coatings,26,27 which required 9:1 (v/v) acetone:H2O at 50 °C to cleave the material. In general, the stability of material 5-0% to weak aqueous acid and acetic acid is a useful property, since it excludes premature degradation during use. Addition of just 10% of the acetal trigger with respect to the polyol segment already resulted in a full degradation within 17 h. It is important to stress that these results have not been optimised for the organic solvent and a more polar solvent may cause an even faster degradation. Likewise, a slight increase in the amount of acetal trigger could speed up the degradation rate where needed.
| Sample | M w | M n | MP | PDI |
|---|---|---|---|---|
|
a Values taken from the main molecular weight peak of the PUs, which was in each case >90 area%.
b After hydrolysis at the indicated temperature in 5 M HCl in 8:2 ethanol:H2O.
|
||||
| 3-0% | 24000 |
11700 |
25000 |
2.1 |
| 5-0% (50 °C)b | 16900 |
3000 | 17900 |
5.6 |
| 3-5% | 21500 |
11000 |
22200 |
2.0 |
| 5-5% (50 °C)b | 11000 |
2400 | 11000 |
4.6 |
| 3-10% | 25600 |
13000 |
24100 |
2.0 |
| 5-10% (50 °C)b | 6400 | 1300 | 8300 | 4.9 |
| 5-10% (rt)b | 6700 | 1300 | 8400 | 5.1 |
Conclusions
We have demonstrated the incorporation of bio-derived HMF–glycerol acetal diols into polyurethanes as part of the polyol segment. Using a bio-derived polyether polyol, two types of PUs were synthesised and films containing 0, 5 and 10% of acetal diol were produced. The acetal diol was shown to be easily cleavable in acidic organic medium in less than 15 min at room temperature. The films containing the acetal diol additive were confirmed to have similar properties to the acetal-free PUs by DSC and TGA. Acidic treatment of the PU films led to very fast hydrolysis and dissolution of all urea cross-linked films. More interestingly, addition of only 5 wt% of acetal diol led to the disintegration of an acrylate cross-linked PU at 50° C in 5 M HCl in 8:2 EtOH/H2O. 10 wt% of acetal diol led to a reduction of the time it took for disintegration by more than half. These results demonstrate that the HMF–glycerol acetal diol can be used as an acid-labile degradation trigger in PU formulations. The additive can be used in low enough percentages, which are not expected to interfere with the material properties of the formulations. While we focused on the one crystalline isomer for the purpose of this study, the mixture of isomers could also be used in formulations that require additives with lower melting points or lower crystallinity. The more random structure generated by the mixture of isomers would help to prevent unwanted crystallisation, making this additive applicable to a range of different PUs. This system could find application in the debonding of adhesives after a device's lifetime for the facile recycling or exchange of components.
Abbreviations
| ACN | Acetonitrile |
| AcOH | Acetic acid |
| DCM | Dichloromethane |
| EtOAc | Ethyl acetate |
| EtOH | Ethanol |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK and AD are grateful to Henkel AG & Co for the financial support.Notes and references
- The European Green Deal, The European Commission, 2019. ( https://ec.europa.eu/info/sites/info/files/european-green-deal-communication_en.pdf).
- A. Cornille, R. Auvergne, O. Figovsky, B. Boutevin and S. Caillol, Eur. Polym. J., 2017, 87, 535–552 CrossRef CAS.
- N. V. Gama, A. Ferreira and A. Barros-Timmons, Materials, 2018, 11, 1841 CrossRef.
- L. A. Heinrich, Green Chem., 2019, 21, 1866–1888 RSC.
- G. Brereton, R. M. Emanuel Jr., R. Lomax, K. Pennington, T. Ryan, H. Tebbe, M. Timm, P. Ware, K. Winkler, T. Yuan, Z. Zhu, N. Adam, G. Avar, H. Blankenheim, W. Friederichs, M. Giersig, E. Weigand, M. Halfmann, F.-W. Wittbecker, D.-R. Larimer, U. Maier, S. Meyer-Ahrens, K.-L. Noble and H.-G. Wussow, Polyurethanes, in Ullmann's Encyclopedia of Industrial Chemistry, 2002, DOI:10.1002/14356007.a21_665.pub3.
- Recycling of Polyurethane Foams, ed. M. G. T. Sabu Thomas, A. V. Rane, K. Kanny and V. K. Abitha, William Andrew Publishing, 2018 Search PubMed.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7, 1579–1593 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- N. Asahi, K. Sakai, N. Kumagai, T. Nakanishi, K. Hata, S. Katoh and T. Moriyoshi, Polym. Degrad. Stab., 2004, 86, 147–151 CrossRef CAS.
- C. Molero, A. De Lucas and J. F. Rodríguez, Polym. Degrad. Stab., 2006, 91, 221–228 CrossRef CAS.
- T. Vanbergen, I. Verlent, J. De Geeter, B. Haelterman, L. Claes and D. De Vos, ChemSusChem, 2020, 13, 3835–3843 CrossRef CAS.
- W. Li, L. Bouzidi and S. S. Narine, Ind. Eng. Chem. Res., 2008, 47, 7524–7532 CrossRef CAS.
- S. M. Abdelbasir, S. S. M. Hassan, A. H. Kamel and R. S. El-Nasr, Environ. Sci. Pollut. Res., 2018, 25, 16533–16547 CrossRef.
- Z. Chen, M. Yang, Q. Shi, X. Kuang, H. J. Qi and T. Wang, Sci. Rep., 2019, 9, 1–9 CrossRef.
- A. Hutchinson, Y. Liu and Y. Lu, J. Adhes., 2017, 93, 737–755 CrossRef CAS.
- A. J. R. Amaral and G. Pasparakis, Polym. Chem., 2017, 8, 6464–6484 RSC.
- S. Binauld and M. H. Stenzel, Chem. Commun., 2013, 49, 2082–2102 RSC.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- C. Bandl, W. Kern and S. Schloegl, Int. J. Adhes. Adhes., 2020, 99, 1–19 CrossRef.
- T. S. Babra, M. Wood, J. S. Godleman, S. Salimi, C. Warriner, N. Bazin, C. R. Siviour, I. W. Hamley, W. Hayes and B. W. Greenland, Eur. Polym. J., 2019, 119, 260–271 CrossRef CAS.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520 CrossRef CAS.
- K. K. Oehlenschlaeger, N. K. Guimard, J. Brandt, J. O. Mueller, C. Y. Lin, S. Hilf, A. Lederer, M. L. Coote, F. G. Schmidt and C. Barner-Kowollik, Polym. Chem., 2013, 4, 4348–4355 RSC.
- K. K. Tremblay-Parrado and L. Avérous, ChemSusChem, 2020, 13, 238–251 CrossRef CAS.
- H. Matsukizono and T. Endo, J. Am. Chem. Soc., 2018, 140, 884–887 CrossRef CAS.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem. A, 2019, 7, 1233–1243 RSC.
- W. Yuan, S. Ma, S. Wang, Q. Li, B. Wang, X. Xu, K. Huang, J. Chen, S. You and J. Zhu, Eur. Polym. J., 2019, 117, 200–207 CrossRef CAS.
- B. Wang, S. Ma, Q. Li, H. Zhang, J. Liu, R. Wang, Z. Chen, X. Xu, S. Wang, N. Lu, Y. Liu, S. Yan and J. Zhu, Green Chem., 2020, 22, 1275–1290 RSC.
- B. Wang, S. Ma, X. Xu, Q. Li, T. Yu, S. Wang, S. Yan, Y. Liu and J. Zhu, ACS Sustainable Chem. Eng., 2020, 8, 11162–11170 CrossRef CAS.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S. H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- R. J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS.
- H. W. Tan, A. R. Abdul Aziz and M. K. Aroua, Renewable Sustainable Energy Rev., 2013, 27, 118–127 CrossRef CAS.
- B. L. Wegenhart, S. Liu, M. Thom, D. Stanley and M. M. Abu-Omar, ACS Catal., 2012, 2, 2524–2530 CrossRef CAS.
- J. Deutsch, A. Martin and H. Lieske, J. Catal., 2007, 245, 428–435 CrossRef CAS.
- C. Crotti, E. Farnetti and N. Guidolin, Green Chem., 2010, 12, 2225–2231 RSC.
- K. S. Arias, A. Garcia-Ortiz, M. J. Climent, A. Corma and S. Iborra, ACS Sustainable Chem. Eng., 2018, 6, 4239–4245 CrossRef CAS.
- CCDC 2022131† contains the supplementary crystallographic data for this paper.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2022131. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0gc04093b |
| This journal is © The Royal Society of Chemistry 2021 |