
Molecular H2O promoted catalytic bicarbonate reduction with methanol into formate over Pd0.5Cu0.5/C under mild hydrothermal conditions†
Xiaoguang
Wang
a,
Yang
Yang
a,
Heng
Zhong
 *abc,
Tianfu
Wang
ac,
Jiong
Cheng
a and
Fangming
Jin
*abc
*abc,
Tianfu
Wang
ac,
Jiong
Cheng
a and
Fangming
Jin
*abc
aSchool of Environmental Science and Engineering, State Key Lab of Metal Matrix Composites, Shanghai Jiao Tong University, Shanghai 200240, China. E-mail: fmjin@sjtu.edu.cn; zhong.h@sjtu.edu.cn
bCenter of Hydrogen Science, Shanghai Jiao Tong University, Shanghai, 200240, China
cShanghai Institute of Pollution Control and Ecological Security, Shanghai 200092, P.R. China
First published on 19th November 2020
Abstract
Direct reduction of bicarbonate, a typical product of CO2 captured in alkaline solution, into value-added organics is one promising way to achieve a simplified and green CO2 capture and utilization process. In this work, a new strategy of bicarbonate reduction coupled with methanol oxidation into a dual formation of formate under mild hydrothermal conditions is reported. A 68% formate production efficiency based on the reductant methanol and nearly 100% selectivity of formate were obtained via a Pd0.5Cu0.5/C catalyst at 180 °C. An operando hydrothermal ATR-FTIR study proved that the bicarbonate was reduced by the in situ generated hydrogen from methanol, which was stepwise oxidized to formaldehyde and formic acid. Notably, DFT calculations and a qNMR study of the 13C and 2H (D) isotopic labelling revealed that H2O molecules not only supplied the hydrogen for bicarbonate reduction but also acted as an indispensable promoter to enhance the catalytic performance of Pd0.5Cu0.5/C for methanol activation.
1. Introduction
Reduction of CO2, a typical greenhouse gas, to useful chemicals has shown considerable environmental and economic benefits since the fixation of CO2 would significantly alleviate the associated environmental risks and the stress of fossil fuel depletion. Although the direct conversion of CO2 from industrial flue gas has been reported,1 CO2 generally needs to be captured from either the atmosphere or point sources usually by using alkaline absorbents before further utilization. This process transforms CO2 into HCO3− and/or CO32− (the final products are pH dependent).2,3 However, as shown in Fig. 1, the current technologies for CO2 conversion are mainly limited to gaseous CO2 transformation.4–6 Thus, the captured CO2 (in the form of HCO3− and/or CO32−) needs to be regenerated and released at a relatively high dissociation temperature of ca. 200–1000 °C and re-compressed to high-pressure CO2 gas before being subjected to these methods, which is not only complicated but also consumes a large amount of energy.7 On the other hand, HCO3− is the dominant substance (nearly 100%) among all the dissolved carbon species (CO2(aq), H2CO3, HCO3− and CO32−) in a CO2-saturated alkaline solution.8 If the captured CO2 could be converted directly into the form of HCO3−, not only would much energy be saved but also the process of CO2 capture and utilization would be greatly simplified. Therefore, the direct reduction of HCO3− could be considered as a promising green strategy for CO2 capture and utilization.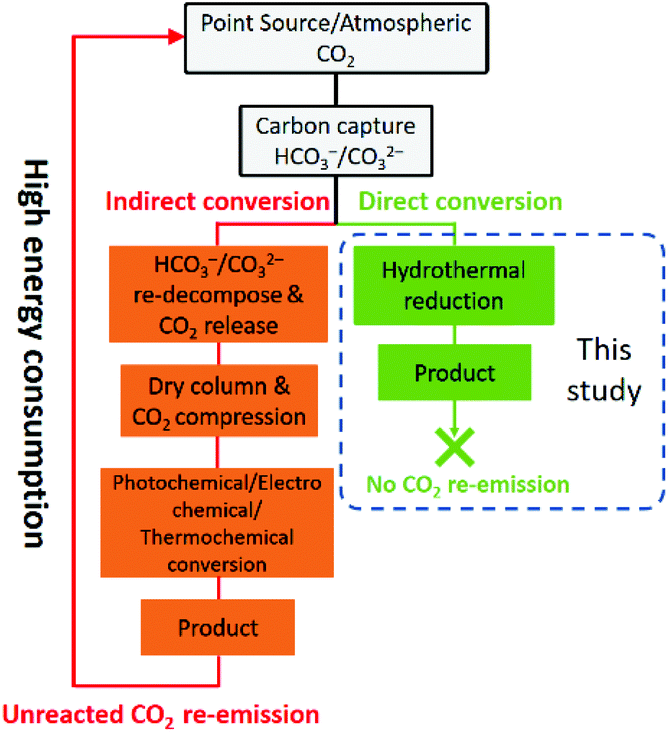 | ||
| Fig. 1 Scheme of CO2 capture and utilization – a comparison of previous methods and this study. | ||
Among the organics derived from CO2/HCO3−, formic acid is one of the most important products due to its versatile application in modern chemical industry. More importantly, formic acid with a 4.4 wt% hydrogen content has been recently regarded as a promising hydrogen carrier because of its stability, low toxicity, biodegradability, and convenience in storage and transportation.9,10 Current methods of formic acid production generally require fossil-based CO and extensive alkali, thus suffering from environmental risk and heavy reliance on fossil feedstock. Therefore, replacing CO with HCO3− in formic acid production, especially when driven by renewable energy, could be an ideal green and clean strategy.
For the CO2/HCO3− reduction, high-pressure H2 gas is usually an indispensable element, which is currently mainly derived from non-renewable natural gas and petroleum,11,12 resulting in high energy consumption and environmental problems. In addition, the use of high-pressure H2 gas involves energy consumption, storage, and transportation issues. On the other hand, biomass or their derivatives rich in reducing functional groups show great potential as renewable alternatives for molecular H2, by acting as active hydrogen donors through the pathway known as aqueous re-forming with the assistance of hydrothermal reactions.13,14 Meanwhile, a hydrothermal environment has been verified to show robustness for HCO3− reduction according to our previous works.15,16 Thus, with the in situ generated hydrogen from biomass hydrothermal re-forming, HCO3− reduction is expected to be achieved in a H2-free pathway under hydrothermal conditions.17,18
Methanol (CH3OH) has wide application in chemical reactions, and can be easily produced from renewable sources such as biomass.19 It has been reported that over 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 metric tons of methanol is annually produced from renewable resources in the United States.20 Given that the functional group of –OH is stored in methanol, it can serve as a clean, efficient and renewable hydrogen source, which makes it a high-potential hydrogen source for HCO3− reduction. More importantly, as shown in eqn (1), HCO3− reduction with methanol can achieve a dual formation of formic acid/formate simultaneously, indicating that good energy efficiency and high atomic economy can be obtained.
000 metric tons of methanol is annually produced from renewable resources in the United States.20 Given that the functional group of –OH is stored in methanol, it can serve as a clean, efficient and renewable hydrogen source, which makes it a high-potential hydrogen source for HCO3− reduction. More importantly, as shown in eqn (1), HCO3− reduction with methanol can achieve a dual formation of formic acid/formate simultaneously, indicating that good energy efficiency and high atomic economy can be obtained.
| CH3OH + 2HCO3− → HCOOH + 2HCOO− + H2O | (1) |
ΔrH⊖ (298 K) = −74.7 kJ mol−1; ΔrG⊖ (298 K) = −32.1 kJ mol−1
ΔrH⊖ (453 K) = −49.7 kJ mol−1; ΔrG⊖ (453 K) = −15.8 kJ mol−1
While the overall reduction of HCO3− with methanol into formate is thermodynamically favourable, a reasonable catalyst is expected to drive the reaction more efficiently. Pd-based materials are well-known hydrogenation catalysts due to their excellent hydrogen absorption capacity;21,22 however, the high cost and limited reserves have impeded their wide application. Diluting Pd with Earth-abundant materials is a feasible and facile strategy to decrease the cost of Pd-based catalysts and to further increase its catalytic performance and stability.23,24 Cu is one of the most extensively studied metal catalysts for CO2 hydrogenation.25 Also, Cu has been reported to be efficient in activating alcohols due to the enrichment of the atomic oxygen species at the copper surface and sub-/near-surface regions to form a copper suboxide phase during the reaction, which can effectively bind and activate the alcohol molecules.26,27 Therefore, combining the metallic Pd catalyst with Cu would probably be a logical strategy to catalyse the HCO3− reduction with methanol effectively.
In general, heterogeneous catalysts are the primary target for development due to their prominent role in catalysing the reaction. However, since the catalysis occurs at the interfacial sites, substances in liquid or gaseous phases can also affect the catalytic properties.28 In a typical hydrothermal reaction, water is generally considered to be an environmentally benign medium due to its unique inherent properties of high ionic product (Kw) and low dielectric constant at high temperature and under high pressure.29 However, beyond the well-known function as a solvent, there is evidence that water is indispensable for the hydrothermal conversion of biomass and HCO3− based on previous works,30,31 but the mechanism is still unknown.
Here, we propose a new route to HCO3− reduction into formate with methanol as the reductant under mild hydrothermal conditions. Methanol was oxidized into formate simultaneously, which led to a dual formation of formate. The results showed that HCO3− could be reduced efficiently at 180 °C on a Pd0.5Cu0.5/C catalyst. More importantly, H2O was found to not only serve as an environmentally benign solvent, but also act as a bridge for methanol activation and oxidation to produce H2 and thereby achieve HCO3− reduction. Also, immense efforts have been made in this research in exploring the role of H2O with the aid of 13C- and 2H-qNMR and DFT studies. The present study proposes a new and green method of direct reduction of bicarbonate with alcohols as renewable hydrogen sources to achieve the dual formation of formic acid/formate in one step. These results could provide new insights into developing a simplified and greener process for CO2 capture and utilization.
2. Experimental
2.1 Materials
Methanol (CH3OH, 99.9%), palladium chloride (PdCl2, 99.99%) and copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.99%) were purchased from Shanghai Macklin Biochemical Technology Co., Ltd. Sodium bicarbonate (NaHCO3, 99.9%) was purchased from Sinopharm Chemical Reagent Co., Ltd and was selected as the CO2 source, considering that NaHCO3 is the main product from trapping of CO2 in waste streams by alkaline solutions. Cabot Vulcan XC-72 carbon was used as the catalyst support. D2O (99.9%, Sinopharm Chemical Reagent Co.), NaH13![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O3 (99%, Sigma-Aldrich), H13OOH (≥95%, J Cambridge Isotope Laboratories Ltd) and CH313OOH (≥95%, J Cambridge Isotope Laboratories Ltd) were used for the determination of the formate source. All reagents were used without further purification. Deionized water was used throughout the study.
O3 (99%, Sigma-Aldrich), H13OOH (≥95%, J Cambridge Isotope Laboratories Ltd) and CH313OOH (≥95%, J Cambridge Isotope Laboratories Ltd) were used for the determination of the formate source. All reagents were used without further purification. Deionized water was used throughout the study.
2.2 Catalyst preparation
Pd0.5Cu0.5/C catalyst with a 7.5 wt% total loading was prepared by an impregnation method. First, a certain amount of PdCl2 was dissolved in 1 mol L−1 HCl to obtain a 0.2 mol L−1 H2PdCl4 solution. Then, 1.05 mL of 0.2 mol L−1 H2PdCl4 and 5 mL of 11.4 g L−1 Cu(NO3)2·3H2O solution were slowly added into a 100 mL flask and mixed with 0.4625 g of carbon powder under vigorous stirring. The pH of the mixture was increased to 10 by adding 0.5 mol L−1 NaOH solution. After stirring for 24 h, 10 mL of 2 mol L−1 NaBH4 was added dropwise into the mixture. After continuously stirring for another 12 h, the black suspension was filtered, washed with deionized water thoroughly and dried in a vacuum dryer to obtain the final Pd0.5Cu0.5/C catalyst. By keeping the total loading of metals (Pd and Cu) at 7.5 wt%, Pd–Cu catalysts with different molar ratios (Pd0.75Cu0.25/C, Pd0.66Cu0.33/C, Pd0.33Cu0.66/C and Pd0.25Cu0.75/C) were obtained; Pd/C and Cu/C were also prepared by using a similar method. The ICP-OES results of each catalyst are summarized in Table S1.†2.3 Experimental procedures
In this study, all experiments were conducted in a stainless steel (SUS 316) tubular reactor (3/8 inch o.d., 1 mm wall thickness) with an inner volume of 5.7 mL. The schematic drawing of the reactor system can be found in our previous research.32 A typical reaction procedure was as follows: 0.1 mol L−1 methanol, 1 mol L−1 NaHCO3 and 50 mg catalyst were loaded into the reactor. Then, the reactor was sealed and placed in a salt bath, which was preheated to a desired temperature. After a certain reaction time, the reactor was removed out of the salt bath and quickly submerged in a cold-water bath to quench the reaction. The liquid and solid samples were separated through a 0.22 μm syringe filter and collected for further analysis. The gas sample was collected by a water–gas replacement method in a measuring cylinder immersed in a saturated NaCl aqueous solution tank.In this study, the amount of formate obtained from HCO3− by one hydroxyl is used to evaluate the efficiency of NaHCO3 reduction with methanol (abbreviated as ‘formate production efficiency’). The detailed definition can be found in the ESI.†
2.4 Analytical methods
Liquid samples were analysed by high-performance liquid chromatography (HPLC), gas chromatography-mass spectrometry (GC-MS), gas chromatography-flame ionization detection (GC-FID) and 13C/2H-quantitative nuclear magnetic resonance (13C/2H-qNMR). Gas samples were characterized by GC-MS and quantified using a gas chromatography-thermal conductivity detector (GC-TCD). Solid samples were analysed using an X-ray diffractometer (XRD), an X-ray photoelectron spectrometer (XPS) and a transmission electron microscope (TEM). Operando hydrothermal ATR-FTIR was used to monitor the reaction process (Fig. S1†). The detailed analytical conditions can be found in the ESI.†3. Results and discussion
3.1 Characterization of catalysts
First, the Pd0.5Cu0.5/C catalyst was prepared and characterized. For comparison, a Pd/C catalyst was also synthesized by a similar procedure without adding the Cu precursor. The SEM image showed that nanoparticles (NPs) of ca. 100 nm were observed for the Pd0.5Cu0.5/C catalyst, which should be associated with the carbon support since similar morphologies were also observed for the Pd/C and carbon black particle samples (Fig. S2 and 3†). The TEM image of the Pd0.5Cu0.5/C catalyst showed that metal nanoparticles with an average diameter of 3.4 ± 0.3 nm were successfully dispersed on the carbon support (Fig. 2a). The composition of the NPs was verified by energy-dispersive X-ray spectroscopy (EDS) analysis, which clearly demonstrates that Cu and Pd atoms are randomly and homogeneously distributed in each NP (Fig. 2b–e), suggesting that the PdCu alloy was successfully formed. In a high-resolution TEM (HRTEM) image of a typical NP from Pd0.5Cu0.5/C (Fig. 2f), the measured lattice distance was 2.28 Å, which can be attributed to the 1/3(422) fringes of the PdCu alloy's face-centred cubic structure.33–35 In addition, the Pd nanoparticles had an average diameter of 2.6 nm as seen from the TEM image of Pd/C (Fig. S4†). From the XRD patterns (Fig. 2g), both Pd/C and Pd0.5Cu0.5/C showed a broad peak located at around 25°, which was attributed to the (002) plane of carbon with a hexagonal structure. The diffraction pattern of Pd/C showed a weak peak located at around 40°, which is attributed to the (111) plane of Pd's face-centred cubic (fcc) structure (ICDD 46-1043). On the other hand, Pd alloyed with Cu caused the diffraction peaks of Pd in the Pd0.5Cu0.5/C catalyst to shift slightly to higher 2θ values. The ICP-OES analysis of Pd0.5Cu0.5/C revealed that the weight percentage of Pd and Cu were 4.46% and 2.68%, respectively, which were close to the desired values (Pd 4.5 wt% and Cu 3.0 wt%). In brief, a carbon-supported PdCu alloy NP catalyst was successfully synthesized.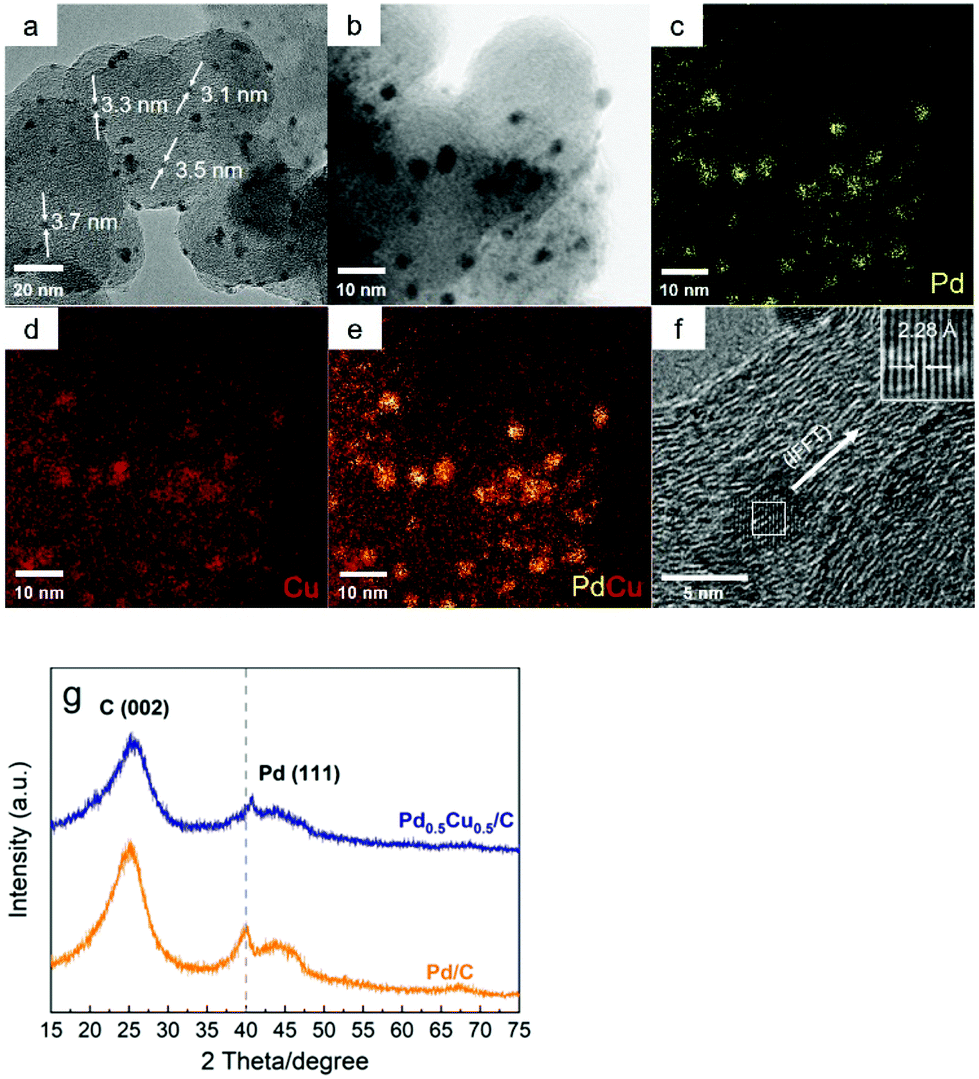 | ||
| Fig. 2 TEM image (a), TEM-EDS elemental mapping (b–e), HRTEM image (f), and XRD patterns (g) of the as-synthesized Pd0.5Cu0.5/C catalyst. | ||
3.2 HCO3− reduction with methanol over Pd0.5Cu0.5/C catalyst
The possibility of HCO3− reduction with methanol as the reductant and Pd0.5Cu0.5/C as the catalyst was investigated. The results showed that formate was successfully produced after the reaction as predicted, and the total formate concentration was 121.9 mmol L−1. For comparison, formic acid production from methanol alone was investigated in the absence of NaHCO3 (Table 1). However, only a negligible trace amount of formic acid was produced when CH3OH reacted alone. Furthermore, even after 1 mol L−1 NaOH was added to adjust the reaction pH, only 21.1 mol L−1 formate was produced without the addition of NaHCO3, which was much lower than that (121.9 mmol L−1) obtained with NaHCO3. These results indicated the participation of NaHCO3 in the formation of formate. It should be noted that formate and formic acid displayed a single peak in the HPLC analysis (Fig. S5†). Since both formic acid and formate were produced from the reaction of methanol with HCO3− (eqn (1)), 13C-qNMR measurement was performed to distinguish and quantify the NaHCO3-sourced formate and CH3OH-sourced formic acid. In a labelling experiment using NaH13O3 (Fig. S6†), a strong signal associated with H13OO− was observed at 171.2 ppm. The concentration of the NaH13O3-sourced formate was determined to be 67.3 mmol L−1 by adding 50 mmol L−1 CH313OOH as an internal standard. Based on the NaHCO3-sourced formate concentration determined by qNMR and the total formate concentration determined by HPLC, the quantification of NaHCO3-sourced formate and CH3OH-sourced formic acid is summarized in Table 1. The results revealed that the concentrations of the NaHCO3-sourced formate and CH3OH-sourced formic acid were 67.3 and 54.6 mmol L−1, respectively, which indicates that a dual formation of formate/formic acid from NaHCO3 and CH3OH was achieved. In addition, formaldehyde (HCHO) was also detected by GC-MS, suggesting that CH3OH was stepwise oxidized into formaldehyde and formic acid. It is encouraging that the selectivity of formate was nearly 100% although a trace amount of HCHO was detected in liquid samples. GC-TCD analysis was further performed on the gaseous samples, and only H2 was observed (Table 1). These results indicate that the coexistence of NaHCO3 and CH3OH is beneficial and necessary for the formation of formate.
| Liquid products (mmol L−1) | Gaseous products (mL) | |||
|---|---|---|---|---|
| Formate | HCHO | H2 | ||
| NaHCO3 sourced | CH3OH sourced | |||
| a Reaction conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaHCO3, 50% water filling, 50 mg Pd0.5Cu0.5/C, 180 °C, and 16 h. b 1 mol L−1 NaOH was added to simulate the alkalinity induced by NaHCO3. | ||||
| With NaHCO3 | 67.3 | 54.6 | Trace | 10.1 ml |
| w/o NaHCO3 | — | Trace | — | Trace |
| w/o NaHCO3b | — | 21.1 | Trace | 13.5 ml |
3.3 Reaction characteristics and catalyst stability
To enhance the production of formate from the NaHCO3 reduction with methanol, the effects of different reaction parameters such as reaction temperature, time (Fig. S7†) and catalyst amount (Fig. S8†) were investigated. The temperature profile shows that only a trace amount of formate was formed at 150 °C. Increasing the temperature significantly promoted the formation of formate, especially when the temperature exceeded 160 °C. On the other hand, when the temperature reached 180 °C, the formate production efficiency increased continuously with the extension of time. However, the reaction tended to slow down when the reaction time exceeded 16 h. At 200 °C, a high efficiency of 75% from NaHCO3 was obtained. The results of the effect of catalyst amount showed that increasing the catalyst amount obviously promoted the production of formate. However, when the catalyst amount reached 50 mg, further increase in the catalyst amount had no significant effect on the reaction efficiency. The stability of the Pd0.5Cu0.5/C catalyst was further examined. Although a slight decrease in the formate production efficiency was observed in the second cycle, further decline was not observed in the following five cycles (Fig. S9†). The Pd0.5Cu0.5/C catalyst after the reaction was collected and analysed by SEM, TEM and XRD (Fig. S10†). The results showed that the morphology and crystallinity of the Pd0.5Cu0.5/C catalyst had no obvious change after the reaction. However, the TEM analysis revealed that the PdCu alloy slightly aggregated after the reaction, and the NP size increased to ca. 4.5 nm, which probably caused the decrease in the formate production efficiency in the second cycle. The ICP analysis of the liquid sample after the reaction discovered no dissolved Pd or Cu species. These results indicated that the Pd0.5Cu0.5/C catalyst was relatively stable.3.4 Catalytic property and mechanism
Based on the detected intermediates of HCHO and H2 (Table 1), the overall process of the CH3OH reaction with NaHCO3 could be divided into two parts: hydrogen generation from methanol oxidation, which contains two steps (eqn (2) and (3)), and HCO3− hydrogenation to formate (eqn (4)).| CH3OH → HCHO + H2 | (2) |
| HCHO + H2O → HCOOH + H2 | (3) |
| H2 + NaHCO3 → HCOONa + H2O | (4) |
Then, the catalytic property of Pd0.5Cu0.5/C for these two parts was studied separately to explore the catalytic mechanism. First, H2 production from methanol oxidation was investigated in the presence and absence of the Pd0.5Cu0.5/C catalyst. For comparison, the results obtained from the Pd/C, Cu/C catalyst were also used. As summarized in Table 2, only a trace amount of H2 was produced in the absence of any catalyst and in the presence of carbon black (Table 2, entries 1 and 2). On the other hand, H2 generation was clearly promoted when Pd/C, Pd0.5Cu0.5/C and Cu/C were used, and the Pd0.5Cu0.5/C catalyst exhibited the best performance (Table 2, entries 3–5). The TON for hydrogen generation obtained on Pd0.5Cu0.5/C was 1.4 and 3.8 times higher than those obtained on Pd/C and Cu/C, respectively. These results indicate that Pd0.5Cu0.5/C could effectively promote methanol hydrothermal re-forming for hydrogen generation. Subsequently, NaHCO3 hydrogenation with ex situ gaseous H2 on the Pd0.5Cu0.5/C, Pd/C, and Cu/C was studied. The results showed that no formate was produced in the absence of any catalyst, and a significant increase in the production of formate appeared in the presence of the Pd0.5Cu0.5/C catalyst (Table S2†). The above results demonstrated that the Pd0.5Cu0.5/C catalyst played an irreplaceable part in both H2 generation from methanol and NaHCO3 hydrogenation.
| Entry | Catalyst | TON of hydrogen generationb (mol H2 per mol PdCu) | Formate production efficiencyc (%) |
|---|---|---|---|
| a H2 generation and formate production are two independent experiments. b Reaction conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaOH, 50% water filling, 50 mg catalysts, 180 °C, and 16 h. c Reaction conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaHCO3, 50% water filling, 50 mg catalysts, 180 °C, and 16 h. d Methanol was used as a solvent instead of water, and the other conditions are the same as mentioned in footnotes “b” and “c”. | |||
| 1 | None | Trace | Trace |
| 2 | Carbon black | Trace | Trace |
| 3 | Pd/C | 10.4 | 46.3 |
| 4 | Pd0.5Cu0.5/C | 14.3 | 67.6 |
| 5 | Cu/C | 3.8 | 15.2 |
| 6 | Pd0.75Cu0.25/C | 12.8 | 67.0 |
| 7 | Pd0.66Cu0.33/C | 13.1 | 67.5 |
| 8 | Pd0.33Cu0.66/C | 9.0 | 38.1 |
| 9 | Pd0.25Cu0.75/C | 7.6 | 22.2 |
| 10d | Pd0.5Cu0.5/C | Trace | Trace |
In addition, the effect of different Pd/Cu ratios (molar ratio) on hydrogen generation and formate production was tested (Table 2, entries 4, and 6–9). Pd0.5Cu0.5/C exhibited the highest catalytic activities in formate production, indicating that a 1:1 Pd/Cu ratio was the optimum condition (Table 2, entry 4). Notably, it was less sensitive for formate production when the molar ratio of Pd was higher than 50% (Table 2, entries 6 and 7). On the other hand, increasing the proportion of Cu could improve the efficiency of H2 generation; however, excessive Cu was unfavourable for formate production. Therefore, Pd is the core component in the PdCu alloy catalyst, and the introduction of Cu not only effectively reduced the cost of the precious metal but also improved hydrogen generation and further formate production.
The interaction between Pd and Cu should have an indispensable role in hydrogen generation and NaHCO3 hydrogenation. To further verify the property of the PdCu alloy, XPS analysis was conducted. Fig. 3 illustrates the XPS results of Pd0.5Cu0.5/C and Pd/C before the reaction. Compared to Pd/C, the Pd 3d peaks of PdCu alloy shifted to a lower binding energy. For Pd/C, the Pd 3d5/2 and Pd 3d3/2 peaks centred at 335.9 and 341.3 eV, respectively, while for Pd0.5Cu0.5/C, the position of the Pd 3d5/2 and Pd 3d3/2 peaks shifted to 335.3 and 340.6 eV, respectively. This is probably attributed to the smaller electron negativity of Cu as compared to that of Pd, which leads to an electron injection from Cu to Pd.36,37 The XPS result of Cu can be found in Fig. S11.† These results indicated that the incorporation of Cu could change the electronic structure of Pd, further leading to a downward shift of the Pd d-band centre.38 The shift of the d-band centre resulted in a decrease in the binding energy of hydrogen on the Pd surface, which positively improved the catalytic performance of Pd for hydrogen generation.39,40 On the other hand, when compared to the unalloyed state, the electronic structure of Pd modified by Cu doping caused the Pd d-band centre to be far away from its Fermi level.38,41 The farther the d-band centre of the transition metals to their Fermi level, the lower the adsorption energy on the transition metal surface for the C1 products.42,43 Thus, the formate adsorption enthalpy on the PdCu alloy surface became lower than that on pure Pd. As a result, when formate was formed on the PdCu alloy, it detached from the catalyst surface, leading to an enhanced formate production and selectivity of formate from HCO3− reduction. In brief, Pd should be the core reaction site for both the steps of hydrogen generation and formate production, while the introduction of Cu improved the Pd performance by altering the electronic structure of Pd.
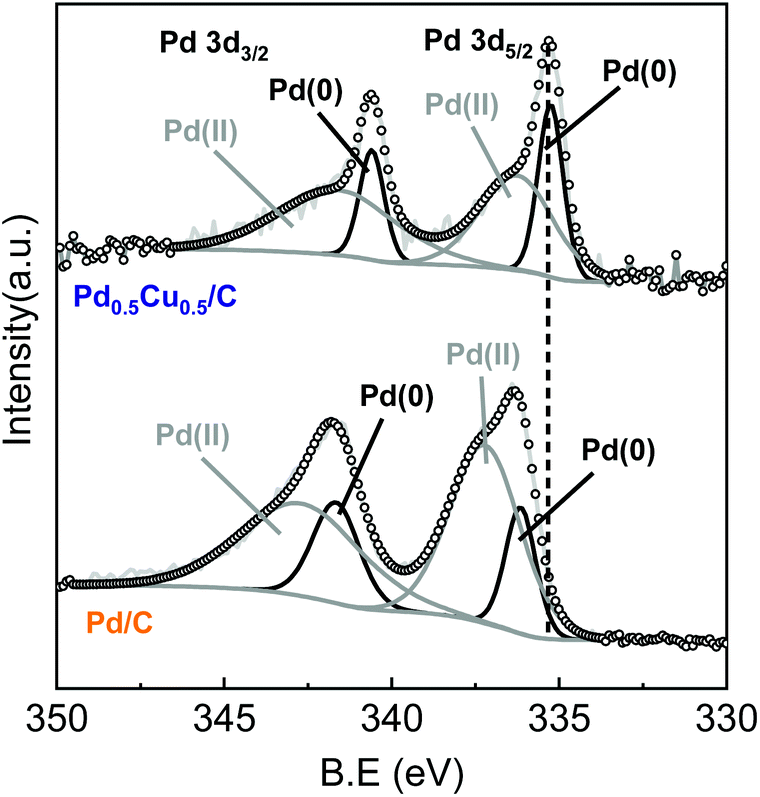 | ||
| Fig. 3 XPS spectra of Pd 3d in Pd0.5Cu0.5/C and Pd/C. | ||
3.5 Role of H2O
As shown in eqn (3), one obvious role of H2O was to react with HCHO to produce H2, indicating that H2O participated in the reaction as a hydrogen source. However, H2 and HCHO were hardly detected when methanol reacted with HCO3− in the absence of H2O (Table 2, entry 10), indicating that H2O was also indispensable for the first step of methanol activation (eqn (2)). To further verify the role of H2O and its mechanism, DFT calculations were performed on the process of hydrogen generation from methanol with or without H2O. The structures of the methanol molecule, the transition state without H2O, and the transition state with H2O were determined, which are shown in Fig. 4a, b, and c, respectively. The transition state structure exhibited a narrow angle of 1C–5O–6H in the absence of H2O (Fig. 4b), which was significantly bent (from 108.9° to 53.6°) as compared to that of the equilibrium structure of the isolated methanol (Fig. 4a). This result indicates that H2 generation from methanol without H2O requires overcoming a huge potential energy barrier. On the other hand, the 1C–5O–6H angle relaxed to 106.4° in the presence of H2O (Fig. 4c), which is nearly comparable to that of the isolated methanol. Moreover, the added H2O molecule (7O–8H–9H) behaves as a proton acceptor and formed a deformed H3O-cation-like conformation with the transition state atom 6H from the methanol molecule, which would drastically reduce the activation energy for the ionic water dissociation. As a result of these effects, the energy barrier of hydrogen generation from methanol with H2O addition was significantly reduced (from 378 kJ mol−1 to 220 kJ mol−1) compared to that in the absence of H2O (Fig. 4d), suggesting the critical role of H2O as a promoter in this process. Then, the corresponding reaction mechanism based on the calculation results is proposed in Fig. 4d. One H2O molecule acts as a bridge for methanol breaking down to produce one H2 and one HCHO. Thus, it is suggested that the H2 generated from methanol in eqn (2) should consist of one atom from methanol and one atom from H2O. Based on these results, eqn (2)–(4) could be rewritten as isotope-labelled forms (eqn (5)–(7)) for a better understanding of the mechanism, in which D2O and NaH13O3 were used instead of H2O and NaHCO3, respectively. This leads to the NaH13O3-sourced formate consisting of H13OO− and D13OO− in the ratio of 1:1.| CH3OH + D2O → HCHO + DH + DHO | (5) |
| HCHO + D2O → HCOOD + DH | (6) |
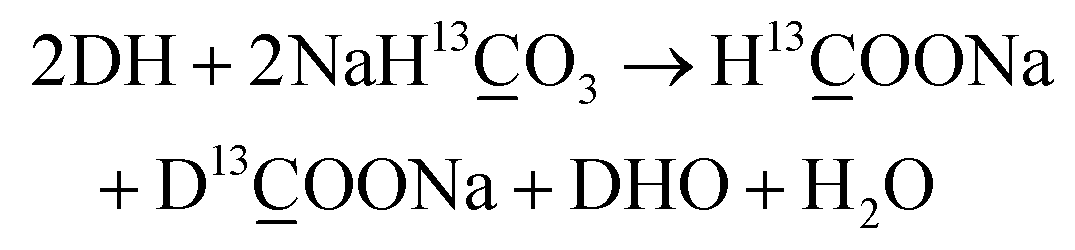 | (7) |
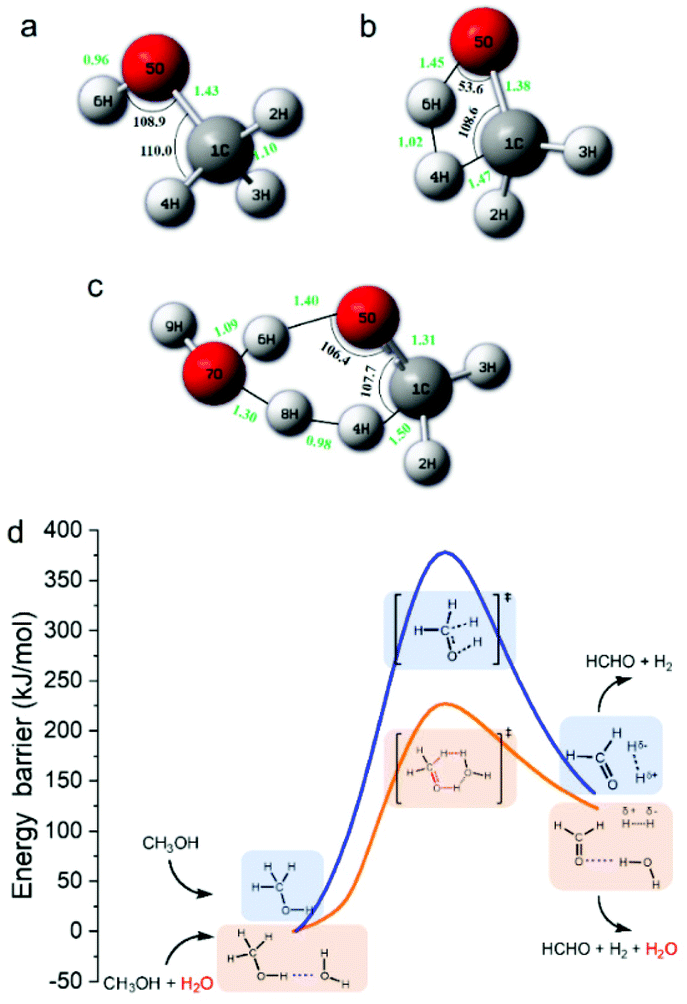 | ||
| Fig. 4 Transition states (a)–(c) and energy profiles (d) for methanol dissociation into H2 with or without H2O (a: methanol molecule; b: the transition state without H2O; c: the transition state with H2O; and d: the profile with H2O as the promoter is depicted in orange, while the profile without H2O is depicted in blue). | ||
To further confirm the DFT calculation results and the above assumption (eqn (5)–(7)), isotopic tracing reaction with D2O substituting H2O was conducted. In the meantime, NaH13O3 was used instead of NaHCO3 to trace the reaction path of carbon. It should be noted first that we confirmed that the H of –CH3 in CH3OH, –CH in HCHO or HCOO− would not undergo a hydrogen–deuterium exchange under hydrothermal conditions in an independent run (Fig. S12†). As displayed in Fig. 5a, the peak assigned to D13OO− was clearly observed in the D-qNMR spectrum, and it consisted of two parallel peaks, while the corresponding 13C-qNMR image showed that NaH13O3-sourced formate contained three parallel peaks, which was caused by the resonance effect between 13C and D (Fig. 5a and b). The COSY spectra of the liquid sample after NaH13O3 had reacted with methanol in D2O is also shown in Fig. 5c. These results confirmed that formate from NaHCO3 reduction carried D from D2O. Furthermore, the concentrations of D-labelled and 13C-labelled formate were determined with dimethyl sulfoxide-D6 and CH313OOH as the internal standards, respectively.44 The results showed that the final product of formate comprised of a nearly identical amount of H13OO− and D13OO− (Table 3), which was perfectly consistent with the DFT calculations and further confirmed the promoting effect of H2O in methanol activation.
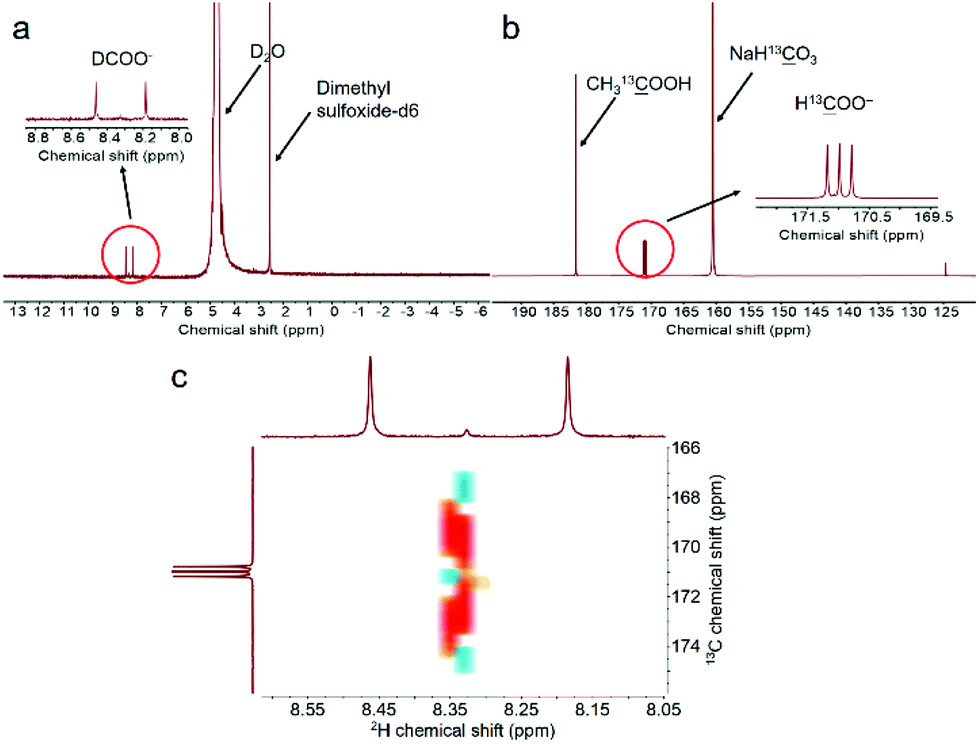 | ||
| Fig. 5 D-qNMR (a), 13C-qNMR (b) and COSY spectra (c) of the liquid sample after NaH13O3 reaction with methanol in D2O (reaction conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaH13O3, 50% D2O filling, 50 mg Pd0.5Cu0.5/C, 180 °C, and 16 h; dimethyl sulfoxide-D6 and CH313OOH were used as the internal standards to calculate the D-labelled and 13C-labelled formate concentrations, respectively). | ||
O3 reduction with methanol in D2Oa
| Formate concentration (mmol L−1) | NaH13O3 sourced |
CH3OH sourced | Total | |
|---|---|---|---|---|
| H13OO− |
D13OOH− |
|||
|
a Reaction conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaH13O3, 50% D2O filling, 50 mg Pd0.5Cu0.5/C, 180 °C, and 16 h.
|
||||
| 33.3 | 32.1 | 53.1 | 118.5 | |
In summary, the results of DFT and 13C/2H-qNMR clearly demonstrated that H2O served as a promoter for methanol activation in eqn (2). Thus, the total three roles of H2O in the process of HCO3− reduction with methanol can be summarized as follows: (1) acting as an indispensable promoter by lowering the reaction energy barrier of methanol activation into formaldehyde; (2) reacting with formaldehyde to release hydrogen; and (3) serving as a green solvent.
It should be noted that according to eqn (1), the ratio of CH3OH-sourced HCOOH to HCO3−-sourced HCOO− should be 1:2. However, our isotopic results showed that the actual ratio of CH3OH-sourced HCOOH to HCO3−-sourced HCOO− was ca. 1:1.2. These results suggested that in the reduction of HCO3− with CH3OH, the first part (eqn (2) and (3)) was easier to react than the second part (eqn (4)). Thus, the unreacted H2 product was detected in the gaseous phase.
3.6 Operando ATR-FTIR study
To obtain a full understanding of the reaction mechanism, an operando hydrothermal ATR-FTIR was further carried out to examine the transformation of the reactants. All the operando ATR-FTIR spectra were recorded as a subtraction spectrum with the initial state (25 °C, before starting the heating reaction) as the background. First, the spectra during the reaction with or without the catalyst were captured. As shown in Fig. 6a, the peaks at 1633 cm−1 and 1400 cm−1 are assigned to the asymmetrically and symmetrically adsorbed HCO3− (COO bond), respectively.45,46 The 1012 cm−1 peak can be ascribed to the C–O bond of the adsorbed CH3OH, and the vibration signals caused by the metal–O bond at 840 cm−1 and 713 cm−1 also confirmed the adsorption of methanol on the catalyst surface.47 Furthermore, the vibration peak due to the adsorption of reactants tended to weaken as the reaction proceeded, indicating that HCO3− and CH3OH were consumed for further reaction on the Pd0.5Cu0.5/C surface.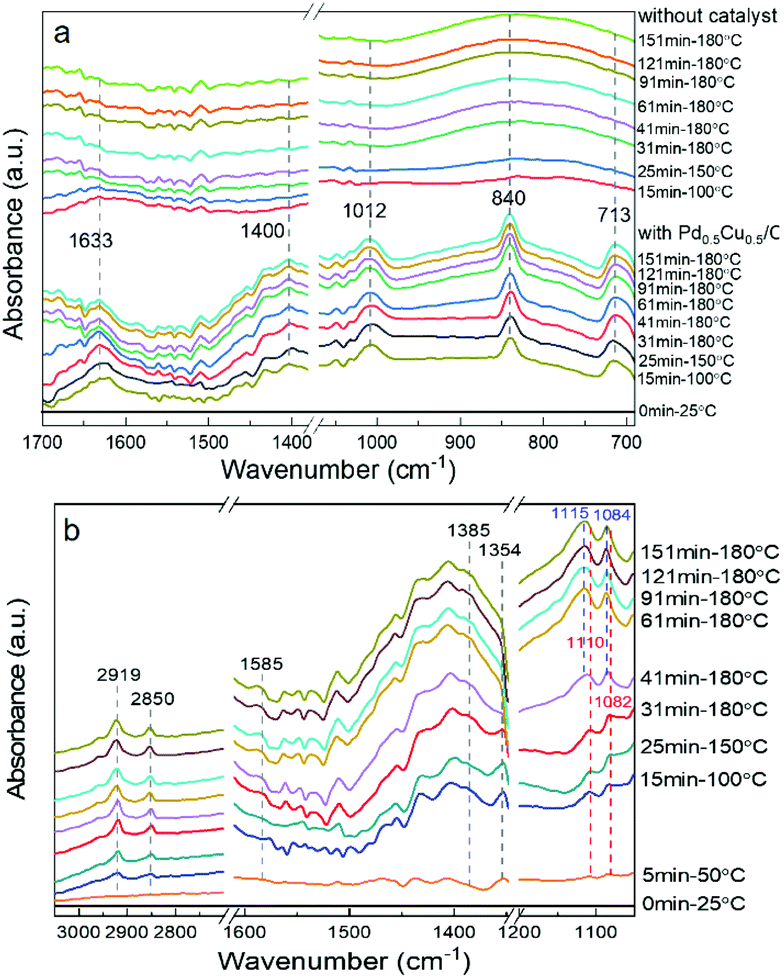 | ||
| Fig. 6 Operando ATR-FTIR subtraction spectra recorded during HCO3− reduction with methanol (all the spectra subtracted the spectrum of the initial state as the background; initial state: 0.1 mol L−1 CH3OH, 1 mol L−1 NaHCO3, 100% water filling, 5 mg Pd0.5Cu0.5/C, 25 °C; (a) ATR-FTIR spectra comparison with or without the catalyst; (b) temperature and time sequence spectra of the operando ATR-FTIR obtained with the Pd0.5Cu0.5/C catalyst. Test conditions: 0.1 mol L−1 CH3OH, 1 mol L−1 NaHCO3, 100% water filling, 5 mg Pd0.5Cu0.5/C). | ||
Then, as shown in Fig. 6b, upon heating the mixture of CH3OH and NaHCO3 in aqueous solution, the peaks located at 1585 cm−1 and 1385 cm−1, which represented the OCO asymmetric and symmetric stretches of HCOO−, appeared and intensified gradually with increasing temperature, and the peak of the C–H bond stretching vibration at 2850 cm−1 related to HCOO− also increased gradually. These results indicate that formate production rate was promoted with increasing temperature and time. The main formaldehyde bands are located at 2923, 2853, and 1354 cm−1 based on the previous report;48 however, the peaks at 2923 and 2853 cm−1 overlapped with the formate ν(C–H) bands in Fig. 6b, indicating that they were difficult to distinguish. On the other hand, the band at 1354 cm−1, belonging to ω(C–H) of formaldehyde, was clearly observed at 100 °C and its absorbance signal increased with the reaction temperature. Nevertheless, when temperature reached 180 °C, the formaldehyde vibration disappeared with prolonged time; this was probably attributed to the rapid transformation of formaldehyde to formate during the reaction, making it difficult to capture. Furthermore, the peaks at 1110 and 1082 cm−1, which could be assigned to the C–O bond of methanol, was blue-shifted along with the increase in reaction time. This was probably attributed to the methanol oxidation into formaldehyde, that is, the transformation from the C–O bond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond, causing the increase in the bonding energy of the C–O bond.
O bond, causing the increase in the bonding energy of the C–O bond.
3.7 Reaction mechanism
Based on the above research, a possible reaction mechanism of the HCO3− reaction with methanol in water on Pd0.5Cu0.5/C is proposed in Fig. 7. First, CH3OH (i) is oxidized into HCHO (iii) on the catalyst with H2 generation through the promotion effect of a H2O molecule. Then, the formed HCHO (iii) reacts with a second H2O molecule to produce another H2 and HCOOH (v). Thus, one CH3OH molecule produces two molecules of H2 with one H2O molecule as the promoter and another as the hydrogen source. Subsequently, the formed in situ H2 and HCO3− are activated by adsorbing on the Pd0.5Cu0.5/C surface ((vii) and (viii)), and then the activated H nucleophilic attacks the CO, leading to the formation of HCOO− ((ix) and (x)) through the cleavage of the C–OH bond.
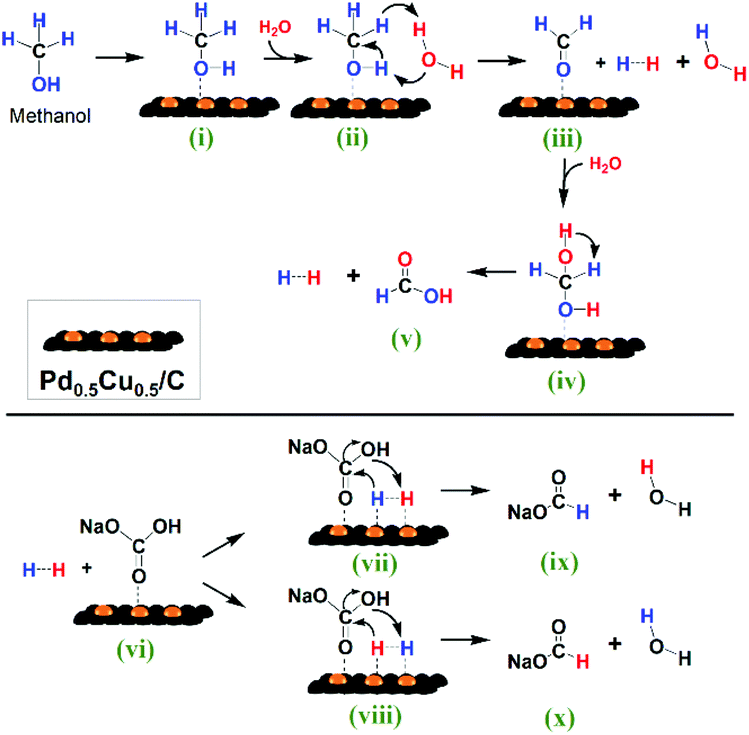 | ||
| Fig. 7 Reaction scheme of HCO3− reduction into formate with methanol in water on the Pd0.5Cu0.5/C catalyst. | ||
3.8 Universality research
The applicability of hydrothermal reduction of various inorganic carbon sources with different alcohols was examined. As summarized in Table 4, when gaseous CO2 was used as the starting material instead of bicarbonate, only a trace amount of formate was detected (Table 4, entry 2), suggesting it was difficult for CO2 gas to be directly converted in this system. However, when 1 mol L−1 NaOH was added, 48.6% formate production efficiency was successfully obtained (Table 4, entry 3), which should be attributed to the transformation of CO2 to HCO3− in NaOH solution. This result further suggests that a one-step CO2 capture and conversion reaction can be achieved by using the presented method. Furthermore, other carbonates and bicarbonates such as K2CO3, KHCO3, and Na2CO3 were also tested, and the formate production efficiency reached 87.3%, 70.5% and 83.4%, respectively (Table 4, entries 4–6). Notably, the hydrogenation of carbonates is generally more difficult than that of bicarbonates because the protonation of carbonate ions was considered to be inferior in aqueous solutions.49,50 To our delight, the carbonates showed better performance than the corresponding bicarbonates in the presented research. This is probably because the weak acidity of methanol was enhanced by the hydrothermal reaction, which then attacked the inert CO32− and made it more reactive than the ex situ HCO3−.| Entry | Alcohols | Carbon sourceb | Formate production efficiency (%) |
|---|---|---|---|
| a Reaction conditions: 0.1 mol L−1 alcohol, 1 mol L−1 HCO3− or CO32−, 50% water filling, 50 mg Pd0.5Cu0.5/C, 180 °C, and 16 h. b CO2: 2.5 MPa, others: 1 mol L−1. c 1 mol L−1 NaOH was added. | |||
| 1 | Methanol | — | Trace |
| 2 | Methanol | CO2 | Trace |
| 3c | Methanol | CO2 | 48.6 |
| 4 | Methanol | K2CO3 | 87.3 |
| 5 | Methanol | KHCO3 | 70.5 |
| 6 | Methanol | Na2CO3 | 83.4 |
| 7 | Methanol | NaHCO3 | 67.6 |
| 8 | Ethanol | NaHCO3 | 68.9 |
| 9 | 1-Propanol | NaHCO3 | 72.3 |
| 10 | 1-Butanol | NaHCO3 | 73.4 |
In addition, the activity of various alcohols used as a reductant for the hydrothermal reduction of HCO3− was examined. The results revealed that ethanol, 1-propanol and 1-butanol exhibited similar activity to that of methanol in the reduction of HCO3− to formate, while were themselves converted to their corresponding acids (Table 4, entries 8–10, Fig. S13†). Notably, the reactivity of alcohols slightly increased with the growth of the carbon chains and 1-butanol exhibited the best activity for HCO3− reduction. Based on the above results, the alcohol-bicarbonate system can be expected to be applied to the synergistic conversion of various hydroxyl-rich organics and CO2 sources.
4. Conclusions
In summary, we have developed an efficient strategy for HCO3− reduction with methanol as the hydrogen source on a Pd0.5Cu0.5/C catalyst under mild hydrothermal conditions. A 68% formate production efficiency and nearly 100% selectivity of formate from bicarbonate were obtained via a Pd0.5Cu0.5/C catalyst at 180 °C. Notably, the DFT calculations and qNMR studies confirmed that molecular H2O not only supplied hydrogen but also acted as an indispensable promoter to enhance the catalytic performance of Pd0.5Cu0.5/C for methanol activation. Furthermore, the operando ATR-FTIR revealed that the mechanism of methanol oxidation to generate hydrogen is CH3OH → HCHO → HCOOH, and HCO3− is reduced into formate with the in situ formed hydrogen. The present study provided a potential way towards simple and green one-step CO2 capture and utilization, which is of great interest in the practical application of CO2 conversion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the financial support of the National Key R&D Program of China (2018YFC0309800 and 2017YFC0506004), the National Natural Science Foundation of China (No. 21978170), the Natural Science Foundation of Shanghai (No. 19ZR1424800), the National Postdoctoral Program for Innovative Talent (No. BX20200208), the Oceanic Interdisciplinary Program of Shanghai Jiao Tong University (No. SL2020MS022), the Start-up Fund for Youngman Research at SJTU, and the Centre of Hydrogen Science, Shanghai Jiao Tong University, China.Notes and references
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J. M. Basset, Green Chem., 2016, 18, 3116–3123 RSC.
- D. W. Keith, G. Holmes, D. S. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS.
- S. Das, J. Pérez-Ramírez, J. Gong, N. Dewangan, K. Hidajat, B. C. Gates and S. Kawi, Chem. Soc. Rev., 2020, 49, 2937–3004 RSC.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- H. Zhong, K. Fujii and Y. Nakano, J. Electrochem. Soc., 2017, 164, F923–F927 CrossRef CAS.
- S. H. Kim, K. H. Kim and S. H. Hong, Angew. Chem., Int. Ed., 2014, 53, 771–774 CrossRef CAS.
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, J. Phys. Chem. C, 2015, 119, 55–61 CrossRef CAS.
- H. Zhong, M. Iguchi, M. Chatterjee, Y. Himeda, Q. Xu and H. Kawanami, Adv. Sustainable Syst., 2018, 2, 1700161 CrossRef.
- H. Zhong, M. Iguchi, M. Chatterjee, T. Ishizaka, M. Kitta, Q. Xu and H. Kawanami, ACS Catal., 2018, 8, 5355–5362 CrossRef CAS.
- R. Navarro, M. Sanchez-Sanchez, M. Alvarez-Galvan, F. Del Valle and J. Fierro, Energy Environ. Sci., 2009, 2, 35–54 RSC.
- S. Choi, T. C. Davenport and S. M. Haile, Energy Environ. Sci., 2019, 12, 206–215 RSC.
- G. W. Huber, J. Shabaker and J. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- F. Jin, Y. Gao, Y. Jin, Y. Zhang, J. Cao, Z. Wei and R. L. Smith Jr., Energy Environ. Sci., 2011, 4, 881 RSC.
- J. Duo, F. Jin, Y. Wang, H. Zhong, L. Lyu, G. Yao and Z. Huo, Chem. Commun., 2016, 52, 3316–3319 RSC.
- H. Zhong, L. Wang, Y. Yang, R. He, Z. Jing and F. Jin, ACS Appl. Mater. Interfaces, 2019, 11, 42149–42155 CrossRef CAS.
- H. Zhong, G. Yao, X. Cui, P. Yan, X. Wang and F. Jin, Chem. Eng. J., 2019, 357, 421–427 CrossRef CAS.
- M. Wang, M. Liu, J. Lu and F. Wang, Nat. Commun., 2020, 11, 1–9 Search PubMed.
- M. Liu, Y. Wang, X. Kong, R. T. Rashid, S. Chu, C.-C. Li, Z. Hearne, H. Guo, Z. Mi and C. J. Li, Chem, 2019, 5, 858–867 CAS.
- Z. Luo, Y. Ouyang, H. Zhang, M. Xiao, J. Ge, Z. Jiang, J. Wang, D. Tang, X. Cao and C. Liu, Nat. Commun., 2018, 9, 1–8 CrossRef.
- N. J. Johnson, B. Lam, B. P. MacLeod, R. S. Sherbo, M. Moreno-Gonzalez, D. K. Fork and C. P. Berlinguette, Nat. Mater., 2019, 18, 454–458 CrossRef CAS.
- J. D. A. Pelletier and J. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS.
- M. K. Samantaray, V. Delia, E. Pump, L. Falivene, M. Harb, S. O. Chikh, L. Cavallo and J. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS.
- S. Kattel, P. Liu and J. G. Chen, J. Am. Chem. Soc., 2017, 139, 9739–9754 CrossRef CAS.
- T. Schedel-Niedrig, M. Hävecker, A. Knop-Gericke and R. Schlögl, Phys. Chem. Chem. Phys., 2000, 2, 3473–3481 RSC.
- H. Bluhm, M. Hävecker, A. Knop-Gericke, E. Kleimenov, R. Schlögl, D. Teschner, V. I. Bukhtiyarov, D. F. Ogletree and M. Salmeron, J. Phys. Chem. B, 2004, 108, 14340–14347 CrossRef CAS.
- H. Duan, J. C. Liu, M. Xu, Y. Zhao, X. L. Ma, J. Dong, X. Zheng, J. Zheng, C. S. Allen, M. Danaie, Y. K. Peng, T. Issariyakul, D. Chen, A. I. Kirkland, J. C. Buffet, J. Li, S. C. E. Tsang and D. O’Hare, Nat. Catal., 2019, 2, 1078–1087 CrossRef CAS.
- F. Jin and H. Enomoto, Energy Environ. Sci., 2011, 4, 382–397 RSC.
- Y. Zhang, Z. Shen, X. Zhou, M. Zhang and F. Jin, Green Chem., 2012, 14, 3285–3288 RSC.
- L. Lu, H. Zhong, T. Wang, J. Wu, F. Jin and T. Yoshioka, Green Chem., 2020, 22, 352–358 RSC.
- F. M. Jin, A. Kishita, T. Moriya and H. Enomoto, J. Supercrit. Fluids, 2001, 19, 251–262 CrossRef CAS.
- N. Yang, Z. Zhang, B. Chen, Y. Huang, J. Chen, Z. Lai, Y. Chen, M. Sindoro, A. L. Wang and H. Cheng, Adv. Mater., 2017, 29, 1700769 CrossRef.
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2011, 6, 28 CrossRef CAS.
- W.-C. Cheong, C. Liu, M. Jiang, H. Duan, D. Wang, C. Chen and Y. Li, Nano Res., 2016, 9, 2244–2250 CrossRef CAS.
- Z. Guo, X. Kang, X. Zheng, J. Huang and S. Chen, J. Catal., 2019, 374, 101–109 CrossRef CAS.
- F. Pang, Z. Wang, K. Zhang, J. He, W. Zhang, C. Guo and Y. Ding, Nano Energy, 2019, 58, 834–841 CrossRef CAS.
- J. Fan, S. Yu, K. Qi, C. Liu, L. Zhang, H. Zhang, X. Cui and W. Zheng, J. Mater. Chem. A, 2018, 6, 8531–8536 RSC.
- G. W. Huber, J. W. Shabaker, S. T. Evans and J. A. Dumesic, Appl. Catal., B, 2006, 62, 226–235 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS.
- X. Yu, X. Tian and S. Wang, Surf. Sci., 2014, 628, 141–147 CrossRef CAS.
- G. C. Wang, Y. H. Zhou and J. Nakamura, J. Chem. Phys., 2005, 122, 044707 CrossRef.
- H. Ma, G. Wang, Y. Morikawa and J. Nakamura, Sci. China, Ser. B: Chem., 2009, 52, 1427–1433 CrossRef CAS.
- Y. Yang, H. Zhong, R. He, X. Wang, J. Cheng, G. Yao and F. Jin, Green Chem., 2019, 21, 1247–1252 RSC.
- A. Rodes, E. Pastor and T. Iwasita, J. Electroanal. Chem., 1994, 376, 109–118 CrossRef CAS.
- L. F. Liao, C. F. Lien, D. L. Shieh, M. T. Chen and J. L. Lin, J. Phys. Chem. B, 2002, 106, 11240–11245 CrossRef CAS.
- N. A. Anan, S. M. Hassan, E. M. Saad, I. S. Butler and S. I. Mostafa, Carbohydr. Res., 2011, 346, 775–793 CrossRef CAS.
- J. De Souza, S. Queiroz, K. Bergamaski, E. R. Gonzalez and F. C. Nart, J. Phys. Chem. B, 2002, 106, 9825–9830 CrossRef CAS.
- T. Wang, D. Ren, Z. Huo, Z. Song, F. Jin, M. Chen and L. Chen, Green Chem., 2017, 19, 716–721 RSC.
- J. Su, M. Lu and H. Lin, Green Chem., 2015, 17, 2769–2773 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02785e |
| This journal is © The Royal Society of Chemistry 2021 |