
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhosphinate esters as novel warheads for activity-based probes targeting serine proteases†
Jan Pascal
Kahler
 a and
Steven H. L.
Verhelst
*ab
a and
Steven H. L.
Verhelst
*ab
aLaboratory of Chemical Biology, Department of Cellular and Molecular Medicine, KU Leuven, Herestraat 49, Box 802, 3000 Leuven, Belgium. E-mail: steven.verhelst@kuleuven.be
bLeibniz Institute for Analytical Sciences ISAS, Otto-Hahn-Strasse 6b, 44227 Dortmund, Germany. E-mail: steven.verhelst@isas.de
First published on 9th July 2021
Abstract
Activity-based protein profiling enables the specific detection of the active fraction of an enzyme and is of particular use for the profiling of proteases. The technique relies on a mechanism-based reaction between small molecule activity-based probes (ABPs) with the active enzyme. Here we report a set of new ABPs for serine proteases, specifically neutrophil serine proteases. The probes contain a phenylphosphinate warhead that mimics the P1 amino acid recognized by the primary recognition pocket of S1 family serine proteases. The warhead is easily synthesized from commercial starting materials and leads to potent probes which can be used for fluorescent in-gel protease detection and fluorescent microscopy imaging experiments.
Introduction
Activity-based protein profiling (ABPP) is a powerful technique that allows for profiling of the active fraction of a given enzyme. It has been particularly useful for the study of proteases, because these enzymes are tightly regulated by post-translational processes.1–3 ABPP relies on activity-based probes (ABPs), small molecules that react covalently with the active enzyme in a mechanism-based manner. They incorporate a reactive group, the so-called “warhead”, and a detection tag such as a fluorophore or a handle allowing for biorthogonal chemistry. These two parts are generally connected by a linker which can be exploited to confer selectivity (Fig. 1a). For proteases, which recognize amino acid side chains around the scissile bond of their substrates with different pockets adjacent to the active site (Fig. 1b), substrate specificity information may be utilized for the design of ABPs with selectivity towards a certain protease or protease family.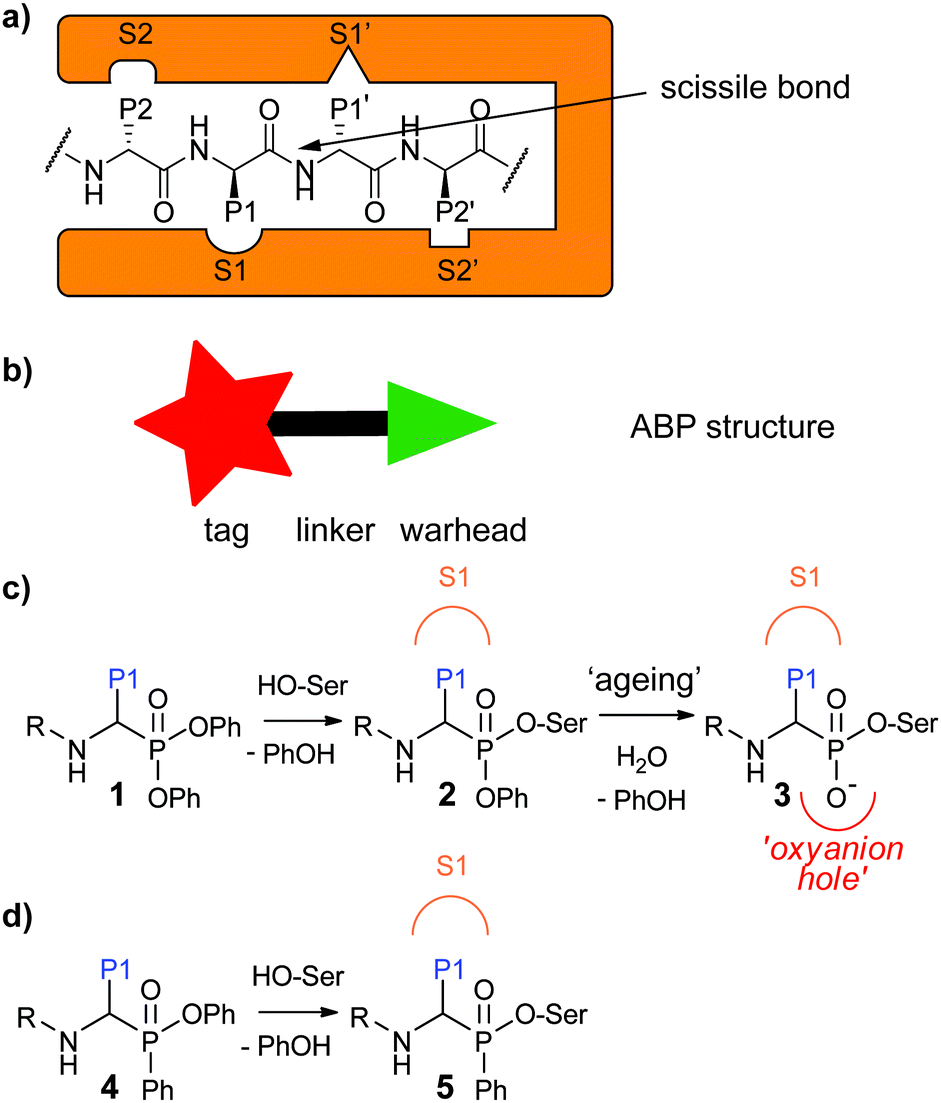 | ||
| Fig. 1 (a) Proteases recognize their substrates by specific pockets in which the individual amino acid side chains fit. The amino acids are called P1, P2,…Pn in N-terminal and P1′, P2′,…Pn′ in C-terminal direction from the scissile bond. The pockets recognizing the individual amino acids in the protease are named S1, S2,…Sn and S1′, S2′,…Sn′. (b) Schematic representation of an ABP displaying the warhead, tag and linker. (c) Diphenyl phosphonates react with the active site serine with phenoxy acting as a leaving group. This is followed by a slow ageing process in which the second phenoxy group leaves the molecule. The resulting oxygen sits in the oxyanion hole. (d) A phenyl phosphinate reacts with a serine protease with phenoxy acting as a leaving group. | ||
Aberrant protease activity contributes to the development of many diseases, for example cancer,4,5 Alzheimer's disease,6 chronic obstructive pulmonary disease (COPD)7 and cystic fibrosis.8 ABPs enable to profile and/or localize defective protease activity regulation within human disease and thereby identify novel drug targets or diagnostic markers. Some examples of ABP applications in this regard include profiling of serine proteases in inflammation,9 localizing cysteine cathepsins in pancreatic cancer10 and detection of paracaspase activity in lymphoma subtypes.11,12
Serine proteases are the largest group of proteases. They use an active site serine residue for attack on the scissile peptide bond. Many serine reactive electrophiles such as isocoumarins,13,14 benzoxazinones,15 phosphoramidates16 and oxolactams17 have been used as warhead for serine protease ABPs. Most applied however, are α-aminoalkyl diphenyl phosphonates.18,19 Since these phosphonates bind to the serine protease in a substrate-like manner, substrate specificity information can directly be used to design selective probes. This has been done, for example, to design selective probes for the human neutrophil serine proteases (NSPs): neutrophil elastase (NE),20 proteinase 3 (PR3),21 cathepsin G (CatG)20,21 and neutrophil serine protease 4 (NSP4).21 Probes for these enzymes have been used to investigate their spatial distribution,21 their activity enhancement by lactoferrin22 and their stereochemical selectivity towards the diphenyl phosphonate electrophile.23 In addition, they have been applied in profiling of NE in inflammatory bowel disease24 and neutrophil extracellular traps,25 and of CatG in immune cells.26
Recently, we devised a short synthetic route towards phosphinate esters (Fig. 1c for general structure, Scheme 1a for synthesis) and showed that a phenyl substituent on the phosphorus led to superior covalent inhibition compared with diphenyl phosphonate counterparts.27 We reasoned that phenylphosphinate esters may also be used as novel ABP warheads for serine proteases. Here, we report the synthesis of phosphinate-based ABPs and their application in the detection and imaging of neutrophil serine proteases in human primary cells.
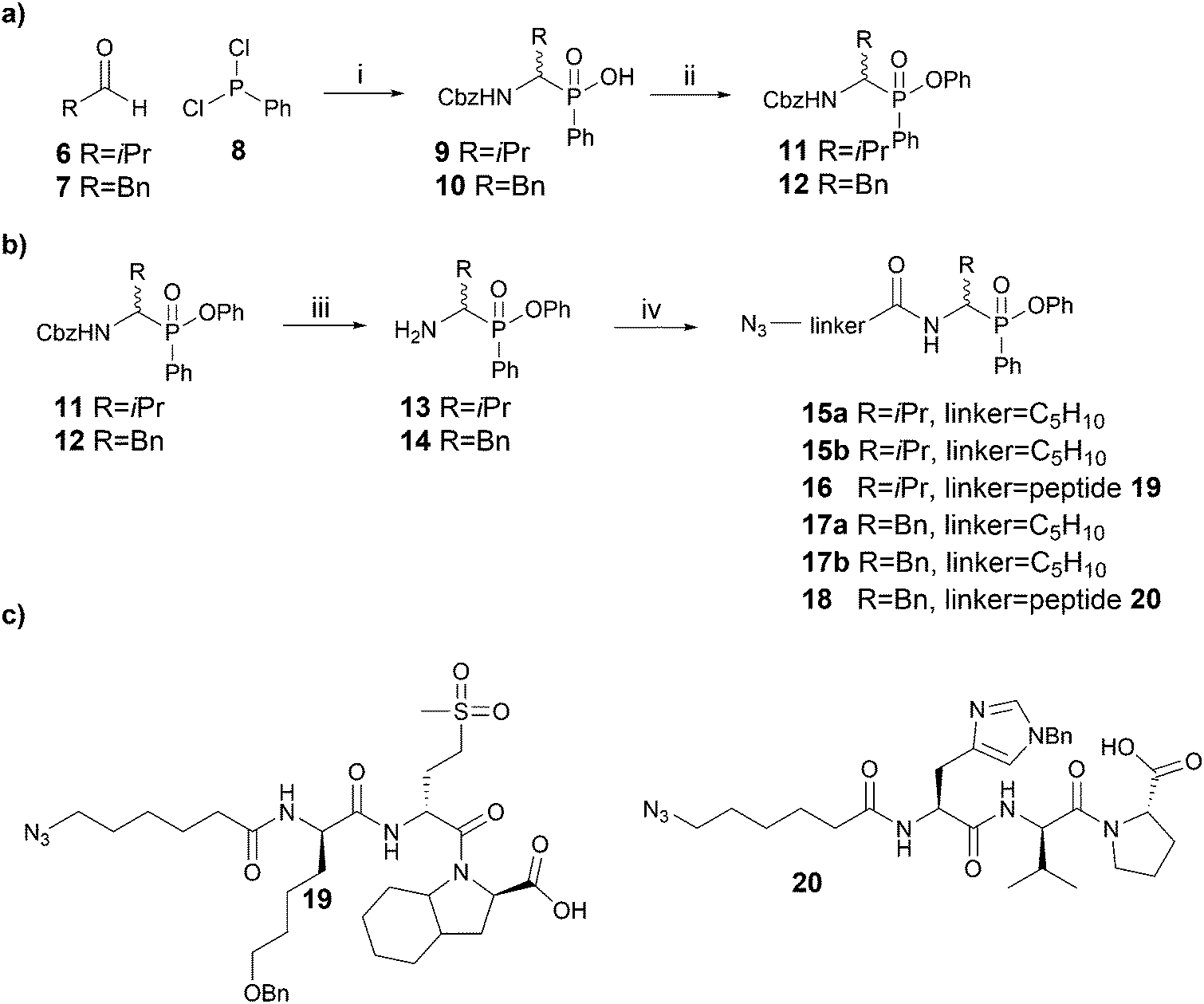 | ||
| Scheme 1 Synthesis of phosphinate ABPs. (a) Synthesis of Cbz-protected inhibitors as previously described. (b) Synthesis of novel phosphinate ABPs incorporating azido hexanoic acid or a tripeptide as linker. (c) Structures of the tripeptides employed as linker in P4 probes. Reagents and conditions: (i) CbzNH2, acetonitrile, reflux, aqueous workup; (ii) phenol, DIC, DMAP, toluene, 80 °C, 4 h; (iii) 33% HBr/AcOH, rt, 1 h; (iv) N3-linker-OH, HATU, DIPEA, DMF, rt, overnight. | ||
Results and discussion
Since Cbz-protected phenylphosphinate analogues of phenylalanine (PhePhP) and valine (ValPhP) showed good inhibitory activity against cathepsin G (CatG) and NE, we used PhePhP and ValPhP as warheads for novel NE and CatG ABPs. We designed two different sets of probes. The first set, to which we will refer as P1 probes, only includes the P1 position. The second set contains the P4–P1 positions by incorporation of a peptide linker developed to give high activity for the respective protease of interest.28,29 We refer to these probes as P4 probes. The probes were constructed in a four-step reaction sequence. First, the Cbz-protected phosphinates 11 and 12 were synthesized in two steps as previously reported (Scheme 1a).27 In brief, benzyl carbamate, dichlorophenyl phosphine 8 and the desired aldehyde were heated in dry acetonitrile. The pure acids 9 and 10, obtained after aqueous workup, were then coupled in a Steglich-type esterification with phenol. Next, the Cbz was removed using 33% hydrobromic acid in acetic acid, yielding the free amines 13 and 14 in sufficient purity for the following amide coupling. For our P1 series, an azidohexanoic acid linker was coupled. Gratifyingly, we were able to separate the diastereomers of the resulting phosphinate esters 15a/b and 17a/b, which allowed us to investigate the influence of the stereochemistry on activity and selectivity (see below). For the P4 probes 16 and 18, the azido-hexanoic acid conjugated tripeptides 19 and 20, synthesized on chlorotrityl resin, were coupled to the amine function of 13 and 14, respectively. Unfortunately, we were not able to obtain single diastereomers of the peptidic probes by HPLC purification and therefore used the mixture of stereoisomers for biochemical experiments.Initially, we tested general activity and selectivity of probes on a set of purified proteases: NE, PR3, CatG (all human), chymotrypsin (ChT) and trypsin (Try, both bovine). All enzymes were in an active state, as they displayed reactivity against the general serine hydrolase probe FP-Rhodamine (FP-Rh) (Fig. 2a). After incubation of the proteases with the phenylphosphinate probes, labelled proteases were visualized by introduction of a TAMRA-fluorophore by copper(I)-catalysed azide-alkyne cycloaddition (CuAAC). P4 ABP 18, containing a tetrapeptide optimized for CatG, labels both CatG and ChT, but not NE and PR3. Reactivity against ChT is not surprising in view of the broad selectivity of ChT. P4 ABP 16, containing a tetrapeptide designed for NE, did not only react with NE and the closely related PR3, but also with CatG and ChT – despite the presence of the suboptimal valine P1 residue. This reactivity is likely a combination of (1) the extended recognition element that may bind in the active site cleft of CatG and ChT, (2) the higher reactivity of the phosphinate warhead in comparison with diphenyl phosphonates29 and (3) the relatively high concentration of the probes used in this experiment. In contrast, the P1 probes show strict selectivity for their intended target enzymes: NE and PR3 are exclusively labelled by ValPhP15, while CatG and ChT are labelled by PhePhP17. In addition, none of the probes reacted with trypsin, further underlining their selectivity. The potency of the ABPs was determined in a titration series of the P4 probes (Fig. 2b and c) and the P1 probes (Fig. 2d and e). Both P4 probes 16 and 18 were able to detect their target proteases at the lowest probe concentration of 40 nM: probe 16 labeled NE and PR3, and probe 18 labeled CatG (Fig. 2b and c). Note that the enzyme concentrations were 284 nM and 226 nM for CatG and NE respectively, meaning that the ABPs reacted stoichiometrically, underlining their potency.
 | ||
| Fig. 2 Labelling potency and selectivity of ABPs on purified proteases. (a) Determination of probe selectivity (1 μM for 17a, 15a/b, 16 and FP-Rh and 500 nM for 17b and 18) on related proteases NE and PR3 as well as CatG and ChT along with trypsin as negative control. General serine hydrolase probe FP-Rh is included as positive control. (b) Titration of CatG with 18. Competition by the general serine protease inhibitor DCI reveals that labelling is activity-dependent. (c) Titration of NE and PR3 with 16. Competition by the general serine protease inhibitor DCI reveals that labelling is activity-dependent. (d) Titration of CatG and ChT with diastereomers of 17. (e) Titration of NE and PR3 with diastereomers of 15. (f) Schematic view of the stereochemical attack on the carbonyl of the substrate (left) and proposed model of reaction with the phosphoryl of the phosphinate (middle and right), with either a match or a mismatch with the Re-face attack. | ||
Because we were able to separate the diastereomers of the P1 probes 15 and 17, we determined the potency of labelling of both diastereomers. Generally, the P1 probes exhibited a slightly lower labelling capacity than the P4 probes. This is in line with expectations, because of the shorter peptide element for interaction with the protease recognition pockets. Most interesting are the different labelling capacities of diastereomers a and b (Fig. 2d, e and Fig. S1, ESI†). Whereas this effect is modest for probe 15, it is very pronounced in the case of probe 17, where diastereomer b shows superior labelling of CatG and ChT compared with diastereomer a which only gives faint labelling, even at the highest probe concentration. We presume that compounds with an R-configuration at the α-atom to the phosphorus represent the active compounds, as these correspond to the natural L-configuration of amino acids (Fig. 2f).23 This has also been shown in various crystal structures of diphenyl phosphonates in covalent complex with various serine proteases.30,31 Hence, it suggests that the difference in reactivity is caused by the configuration at the phosphorus centre. Because the reaction between the active site serine and the phosphinate ester occurs with an SN2p mechanism, the leaving group must be opposite of the incoming oxygen nucleophile, in a similar fashion as the Re-face attack on the substrate carbonyl (Fig. 2f). A mismatch in the configuration will therefore negatively affect the attack onto the electrophile and lead to a decreased potency.
Next, we went on to assess the labelling capacity of the probes in a complex proteome. To this end, we incubated lysates of the human promyeloblast cell line HL-60 with the general serine protease inhibitor DCI or with vehicle, followed by probes 15–18 and eventually performed CuAAC for target visualization (Fig. 3a and b). P4 Probes 18 and 16 as well as P1 probe 17b displayed selective labelling of a protein band around 25 kDa which was competed by prior incubation with DCI. The labelling pattern of the general serine hydrolase probe FP-rhodamine highlights the selectivity of our ABPs, since it reacted with various other hydrolases in the lysate. Compound 17b is the only P1 probe that showed labelling in these lysates (Fig. 3a), in accordance with the results on purified enzymes, which revealed 17b as the most potent P1 probe (Fig. 2d). To further exemplify the value of our probes we proceeded to label NSPs in lysates of freshly isolated primary human neutrophils (Fig. 3c and d). Here, all probes displayed clear and intense labelling of proteins corresponding to molecular weights of NSPs. We then tested the probes’ cell permeability. Freshly isolated human neutrophils of healthy volunteers were incubated with ABPs 15–18 at two different concentrations. After treatment, cells were washed and lysed, and the lysate was subjected to CuAAC. Analysis of labelling by SDS-PAGE showed good labelling of two protein bands of similar molecular weight around 30 kDa by PhePhP probes 17b and 18. The ValPhP probes gave rise to a fluorescent protein band at the same molecular weight at 10 μM concentration but only P4 probe 16 showed a strong band and was capable of labelling the protein at 1 μM concentration (Fig. 3e). Having cell permeability of the probes established, we wanted to demonstrate their applicability in fluorescence microscopy. To this end, freshly isolated neutrophils from healthy volunteers were treated with probes 16 and 18. After fixation and permeabilization of the cells, a TAMRA-dye was introduced by in situ copper-mediated ‘click’-chemistry. In an additional experiment, cells were treated with inhibitors for NE or CatG prior to probe incubation.
 | ||
| Fig. 3 Labelling of NSPs in complex proteomes. (a) Lysates of the HL-60 cell line reveal activity-based labelling of one predominant band by probes 17b and 18 (500 nM), but multiple bands with FP-Rh (1 μM). (b) Lysates of the HL-60 cell line lead to activity-based labelling of one band by probe 16 and multiple by FP-Rh (all probes at 1 μM, except 16 at (500 nM)). (c) Neutrophil lysates, spiked (+) or not spiked (−) with purified human CatG are labelled well by probes 17 (500 nM) and 18 (500 nM) as well as FP-Rh (1 μM) as positive control. (d) Neutrophil lysates (lane 1–4) as well as purified NE (lane 5) and PR3 (lane 6) are labelled by 15 (1 μM) and 16 (500 nM) or FP-Rh as control (1 μM). (e) All ABPs show some degree of NSP labelling in live cells and are thus cell permeable to a certain degree. FP-Rh included as control. | ||
Microscopic images show punctuate red staining in probe-treated neutrophils indicating cytoplasmic granules. Vehicle-treated neutrophils exhibited almost no red staining (Fig. 4 and Fig. S2, S3, ESI†), illustrating low background from the fluorophore used in the click reaction. Moreover, incubation with an inhibitor prior to probe treatment competes labelling, indicating that the staining is caused by active protease. In all cases DAPI staining of the cells illustrates the polymorphonuclear character of the nucleus, confirming successful neutrophil isolation. Altogether these data reveal specific labelling of NSPs by probes 16 and 18 within live neutrophils and demonstrate the use of phenylphosphinates as new warhead for the design of ABPs applicable for fluorescence microscopy.
 | ||
| Fig. 4 Labelling of neutrophils with ABP 18, subsequent ‘click’-reaction with TAMRA and fluorescent microscopy. Depicted are representative images showing two neutrophil cells. Clear labelling can be seen in the probe-treated cells (middle panels) in comparison with the dmso vehicle-treated ones (upper panels). Additionally, treatment of neutrophils with a CatG inhibitor prior to probe treatment diminishes labelling by ABP 18 (lower panels). Histograms that show the distribution of brightness in the TAMRA channel also reveal a clear difference between the probe treated samples in comparison with vehicle and inhibitor control. Brightness of DAPI channel images was adjusted for better representation. | ||
Conclusions
We have incorporated a phenylphosphinate warhead into novel ABPs targeting serine proteases. They display good to excellent labelling capacities, with probes 16, 17b and 18 being able to react stoichiometrically with target proteases. Interestingly, diastereomers of probes 15 and 17 exhibited clearly different potencies towards their target, which was most pronounced for ABP 17. We attribute this difference to the stereochemistry around the phosphorus atom, on which the leaving group in one stereoisomer is better located for attack by the serine nucleophile than in the other.The obtained probes are cell permeable and can therefore be used to label NSPs in situ. We illustrated this application by performing fluorescent microscopy with subsequent in situ click-chemistry on fixed and permeabilized human primary neutrophils, which displayed clear and activity-dependent labelling.
We believe that the phosphinate scaffold offers several distinct advantages over the closely related diphenyl phosphonates. First, the carbon substituent at the phosphorus can potentially be exploited for an increase in selectivity. To achieve this, the phenyl substituent on the phosphorus would need to be derivatized with, for example, a carboxylic acid group, which can then be coupled to amino acids reflecting primed site preferences. Second, the presence of a single phenoxy leaving group in phenylphosphinates would easily enable the development of quenched fluorescent ABPs (qABPs) by appending a dark quencher to the phenoxy substituent. Currently, the only warhead scaffold that allows for the synthesis of peptide-like serine protease qABPs is a mixed alkyl-aryl phosphonate.32 As the synthesis of phosphinates is more straightforward, this warhead therefore represents a valuable addition to the narrow chemical toolbox for serine protease qABPs.
We expect that the here described ABPs will find use in the detection of neutrophils and NSPs, for example in inflammatory diseases or within the tumour microenvironment. Moreover, cell impermeable derivatives may be used to localize extracellular NSPs after excretion by activated neutrophils during inflammatory reactions.
Obviously, the use of the phenylphosphinate warhead is not limited to the NSPs. By adjusting the side chain substituent on the phosphinate building block as well as the peptide element, phenylphosphinate ABPs may be directed towards other classes of serine proteases, as has previously been achieved with diphenyl phosphonates, such as the granzymes33 or the kallikreins.34 This makes the phosphinate warhead a versatile tool for future development of ABPs tailored towards elucidation of the roles of serine proteases in physiological and pathological processes.
Ethical statement
Experiments involving the use of human neutrophils were reviewed and approved by the UZ/KU Leuven ethics committee before being performed (study number S62598). Healthy volunteers were informed about the risks of their participation in the study and consented before blood being taken. Blood was taken by trained MDs.Author contributions
S. H. L. V. and J. P. K. developed the project and designed the compounds. J. P. K. synthesized compounds, performed biochemical experiments, acquired microscopic images and wrote the manuscript. S. H. L. V. revised the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
We acknowledge funding by the FWO (project grant G0D8617N and PhD fellowship 1S18520N to JPK), the Ministerium für Kultur und Wissenschaft des Landes Nordrhein-Westfalen, the Regierende Bürgermeister von Berlin–inkl. Wissenschaft und Forschung, and the Bundesministerium für Bildung und Forschung. Images were recorded on a Zeiss LSM 880 – Airyscan (Cell and Tissue Imaging Cluster CIC), supported by Hercules AKUL/15/37_GOH1816N and FWO G.0929.15 to Pieter Vanden Berghe, University of Leuven. We thank Ines Verlinden and Wouter Vankrunkelsven for help with acquiring blood samples of healthy donors.Notes and references
- S. Chakrabarty, J. P. Kahler, M. A. T. van de Plassche, R. Vanhoutte and S. H. L. Verhelst, Curr. Top. Microbiol. Immunol., 2019, 420, 253–281 CAS.
- L. E. Sanman and M. Bogyo, Annu. Rev. Biochem., 2014, 83, 249–273 CrossRef CAS PubMed.
- S. Serim, U. Haedke and S. H. L. Verhelst, ChemMedChem, 2012, 7, 1146–1159 CrossRef CAS PubMed.
- O. C. Olson and J. A. Joyce, Nat. Rev. Cancer, 2015, 15, 712–729 CrossRef CAS PubMed.
- J. Cathcart, A. Pulkoski-Gross and J. Cao, Genes Dis., 2015, 2, 26–34 CrossRef PubMed.
- B. De Strooper, Physiol. Rev., 2010, 90, 465–494 CrossRef CAS PubMed.
- K. Hoenderdos and A. Condliffe, Am. J. Respir. Cell Mol. Biol., 2013, 48, 531–539 CrossRef CAS PubMed.
- A. Roghanian and J. M. Sallenave, J. Aerosol Med. Pulm. Drug Delivery, 2008, 21, 125–144 CrossRef CAS PubMed.
- L. E. Edgington-Mitchell, N. Barlow, L. Aurelio, A. Samha, M. Szabo, B. Graham and N. Bunnett, Bioorg. Med. Chem. Lett., 2017, 27, 254–260 CrossRef CAS PubMed.
- J. A. Joyce, A. Baruch, K. Chehade, N. Meyer-Morse, E. Giraudo, F. Y. Tsai, D. C. Greenbaum, J. H. Hager, M. Bogyo and D. Hanahan, Cancer Cell, 2004, 5, 443–453 CrossRef CAS PubMed.
- A. C. Eitelhuber, O. Vosyka, D. Nagel, M. Bognar, D. Lenze, K. Lammens, F. Schlauderer, D. Hlahla, K. P. Hopfner, G. Lenz, M. Hummel, S. H. Verhelst and D. Krappmann, Chem. Biol., 2015, 22, 129–138 CrossRef CAS PubMed.
- J. Hachmann, L. E. Edgington-Mitchell, M. Poreba, L. E. Sanman, M. Drag, M. Bogyo and G. S. Salvesen, Chem. Biol., 2015, 139–147 CrossRef CAS PubMed.
- C. M. Kam, A. S. Abuelyaman, Z. Z. Li, D. Hudig and J. C. Powers, Bioconjugate Chem., 1993, 4, 560–567 CrossRef CAS PubMed.
- U. Haedke, M. Gotz, P. Baer and S. H. L. Verhelst, Bioorg. Med. Chem., 2012, 20, 633–640 CrossRef CAS PubMed.
- J. Yang, R. J. Mendowicz and S. H. L. Verhelst, ChemBioChem, 2021, 1–5 Search PubMed.
- U. R. Haedke, S. C. Frommel, F. Hansen, H. Hahne, B. Kuster, M. Bogyo and S. H. Verhelst, ChemBioChem, 2014, 15, 1106–1110 CrossRef CAS PubMed.
- E. F. P. Ruivo, L. M. Gonçalves, L. A. R. Carvalho, R. C. Guedes, S. Hofbauer, J. A. Brito, M. Archer, R. Moreira and S. D. Lucas, ChemMedChem, 2016, 2037–2042 CrossRef CAS PubMed.
- Z. Pan, D. A. Jeffery, K. Chehade, J. Beltman, J. M. Clark, P. Grothaus, M. Bogyo and A. Baruch, Bioorg. Med. Chem. Lett., 2006, 16, 2882–2885 CrossRef CAS PubMed.
- A. S. Abuelyaman, J. C. Powers, D. Hudig and S. L. Woodard, Bioconjugate Chem., 1994, 5, 400–405 CrossRef CAS PubMed.
- F. Zou, M. Schmon, M. Sienczyk, R. Grzywa, D. Palesch, B. O. Boehm, Z. L. Sun, C. Watts, R. Schirmbeck and T. Burster, Anal. Biochem., 2012, 421, 667–672 CrossRef CAS PubMed.
- P. Kasperkiewicz, M. Poreba, S. J. Snipas, S. J. Lin, D. Kirchhofer, G. S. Salvesen and M. Drag, PLoS One, 2015, 10, e0132818 CrossRef PubMed.
- S. Eipper, R. Steiner, A. Lesner, M. Sienczyk, D. Palesch, M. E. Halatsch, E. Zaczynska, C. Heim, M. D. Hartmann, M. Zimecki, C. R. Wirtz and T. Burster, PLoS One, 2016, 11, e0151509 CrossRef PubMed.
- A. C. Schulz-Fincke, M. Blaut, A. Braune and M. Gütschow, ACS Med. Chem. Lett., 2018, 9, 345–350 CrossRef CAS PubMed.
- B. M. Anderson, D. P. Poole, L. Aurelio, G. Z. Ng, M. Fleischmann, P. Kasperkiewicz, C. Morissette, M. Drag, I. R. van Driel, B. L. Schmidt, S. J. Vanner, N. W. Bunnett and L. E. Edgington-Mitchell, Sci. Rep., 2019, 9, 13295 CrossRef PubMed.
- P. Kasperkiewicz, A. Hempel, T. Janiszewski, S. Kołt, S. J. Snipas, M. Drag and G. S. Salvesen, J. Biol. Chem., 2020, 295, 17624–17631 CrossRef CAS PubMed.
- R. Schroeder, R. Grzywa, C. R. Wirtz, M. Sienczyk and T. Burster, Anal. Biochem., 2020, 588, 113488 CrossRef CAS PubMed.
- J. P. Kahler, S. Lenders, M. A. T. Van De Plassche and S. H. L. Verhelst, ACS Med. Chem. Lett., 2020, 11, 1739–1744 CrossRef CAS PubMed.
- P. Kasperkiewicz, M. Poreba, S. J. Snipas, H. Parker, C. C. Winterbourn, G. S. Salvesen and M. Drag, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 2518–2523 CrossRef CAS PubMed.
- P. Kasperkiewicz, Y. Altman, M. D’Angelo, G. S. Salvesen and M. Drag, J. Am. Chem. Soc., 2017, 139, 10115–10125 CrossRef CAS PubMed.
- P. Hof, I. Mayr, R. Huber, E. Korzus, J. Potempa, J. Travis, J. C. Powers and W. Bode, EMBO J., 1996, 15, 5481–5491 CrossRef CAS PubMed.
- B. C. Lechtenberg, P. Kasperkiewicz, H. Robinson, M. Drag and S. J. Riedl, ACS Chem. Biol., 2015, 10, 945–951 CrossRef CAS PubMed.
- S. Serim, P. Baer and S. H. L. Verhelst, Org. Biomol. Chem., 2015, 13, 2293–2299 RSC.
- S. Mahrus and C. S. Craik, Chem. Biol., 2005, 12, 567–577 CrossRef CAS PubMed.
- T. Kryza, N. Bock, S. Lovell, A. Rockstroh, M. L. Lehman, A. Lesner, J. Panchadsaram, L. M. Silva, S. Srinivasan, C. E. Snell, E. D. Williams, L. Fazli, M. Gleave, J. Batra, C. Nelson, E. W. Tate, J. Harris, J. D. Hooper and J. A. Clements, Mol. Oncol., 2020, 14, 105–128 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and supplemental data figures. See DOI: 10.1039/d1cb00117e |
| This journal is © The Royal Society of Chemistry 2021 |