
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and evaluation of tryptophan analogues as tool compounds to study IDO1 activity†
Nicholas J.
Cundy
 a,
Roseanna K.
Hare
b,
Tina
Tang
c,
Andrew G.
Leach
d,
Thomas A.
Jowitt
e,
Omar
Qureshi
c,
John
Gordon
c,
Nicholas M.
Barnes
cf,
Catherine A.
Brady
cf,
Emma L.
Raven
g,
Richard S.
Grainger
*a and
Sam
Butterworth
*d
a,
Roseanna K.
Hare
b,
Tina
Tang
c,
Andrew G.
Leach
d,
Thomas A.
Jowitt
e,
Omar
Qureshi
c,
John
Gordon
c,
Nicholas M.
Barnes
cf,
Catherine A.
Brady
cf,
Emma L.
Raven
g,
Richard S.
Grainger
*a and
Sam
Butterworth
*d
aSchool of Chemistry, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK. E-mail: r.s.grainger@bham.ac.uk
bDivision of Infection, Immunity and Respiratory Medicine, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, M13 9PL, UK
cCelentyx Ltd, Birmingham Research Park, 97 Vincent Drive, Birmingham, B15 2SQ, UK
dDivision of Pharmacy and Optometry, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, M13 9PL, UK. E-mail: sam.butterworth@manchester.ac.uk
eWellcome Trust Centre for Cell Matrix Research, Faculty of Biology, Medicine and Health, University of Manchester, Manchester, M13 9PL, UK
fInstitute of Clinical Sciences, College of Medical and Dental Sciences, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
gSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
First published on 13th September 2021
Abstract
The metabolism of L-tryptophan to N-formyl-L-kynurenine by indoleamine-2,3-dioxygenase 1 (IDO1) is thought to play a critical role in tumour-mediated immune suppression. Whilst there has been significant progress in elucidating the overall enzymatic mechanism of IDO1 and related enzymes, key aspects of the catalytic cycle remain poorly understood. Here we report the design, synthesis and biological evaluation of a series of tryptophan analogues which have the potential to intercept putative intermediates in the metabolism of 1 by IDO1. Functionally-relevant binding to IDO1 was demonstrated through enzymatic inhibition, however no IDO1-mediated metabolism of these compounds was observed. Subsequent Tm-shift analysis shows the most active compound, 17, exhibits a distinct profile from known competitive IDO1 inhibitors, with docking studies supporting the hypothesis that 17 may bind at the recently-discovered Si site. These findings provide a start-point for development of further mechanistic probes and more potent tryptophan-based IDO1 inhibitors.
Introduction
IDO1 as an oncogenic target
Indoleamine-2,3-dioxygenase 1 (IDO1) is a heme-based dioxygenase enzyme that catalyses the oxidation of L-tryptophan 1 to N-formyl-L-kynurenine 2 (NFK) as the initial, rate-limiting step of the kynurenine pathway (Scheme 1).1–3 IDO1 is one of three dioxygenase enzymes: IDO1, IDO2 and TDO. Until 2007, IDO1 was referred to as IDO; subsequently, three groups independently identified a third dioxygenase, IDO2, and IDO was reclassified as IDO1.4–9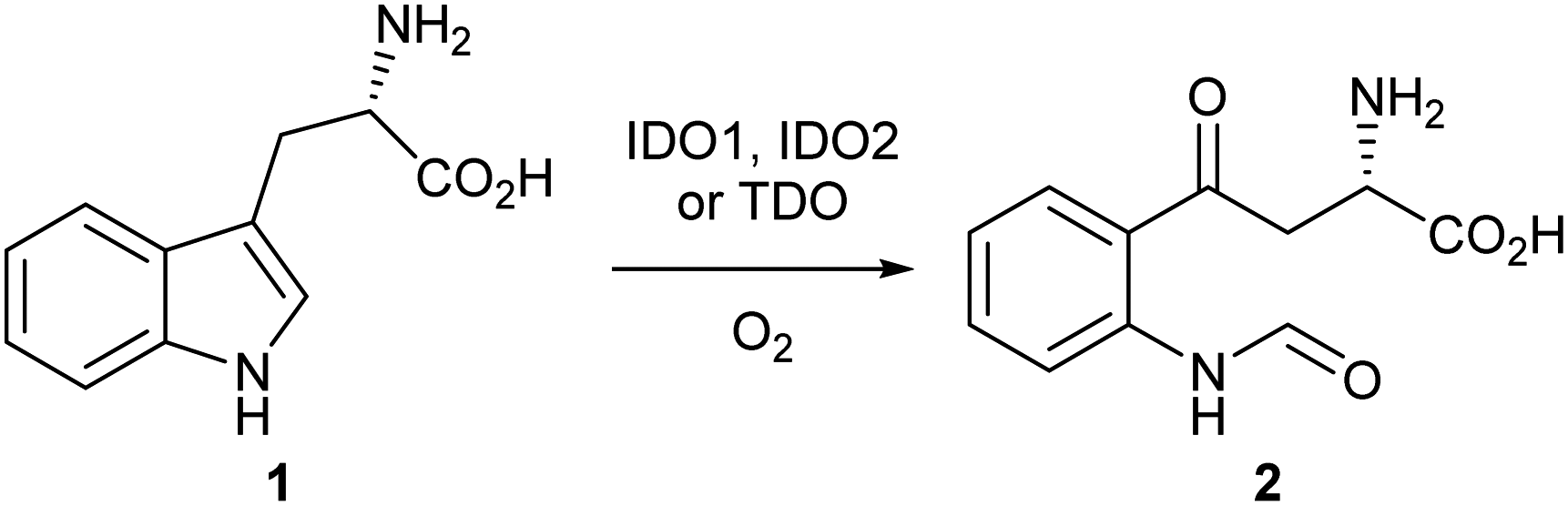 | ||
| Scheme 1 Dioxygenation of L-tryptophan 1 to N-formyl-L-kynurenine 2. | ||
In addition to its biosynthetic function, IDO1 has been demonstrated to serve an important role in the defence against intracellular viral and bacterial infection.10,11 In the event of pathogenic invasion, IDO1 upregulation leads to a depletion of L-tryptophan 1, an essential amino acid required for the proliferation of the attacking body, and produces anti-pathogenic metabolites preventing the spread of the invading species.12 Through the modulation of L-tryptophan concentrations, IDO1 also possesses a significant immunomodulatory potential.13,14
L-Tryptophan depletion affects the ability of immune cells with tryptophan sensitive checkpoints to proliferate and initiate an immune response, with natural killer (NK) and naïve T-cells shown to be particularly sensitive to L-tryptophan starvation.13,14 Downstream metabolites in the kynurenine pathway have also been implicated in IDO1's immunomodulatory potential by increasing rates of T-helper- and NK cell apoptosis.15
In a study of 25 tumours of varying origin, 15 demonstrated consistent expression of IDO1 across individual tumour samples and a further 4 tumour types displayed >25% IDO1 positive cells; over-expression of IDO1 in tumours is commonly associated with a poor patient prognosis.16–18 A seminal report by Van den Eynde demonstrated that inhibition of IDO1 leads to partial reversal of the immunosuppression afforded by IDO1 and significantly slowed tumour growth in mice.19 This report spurred intense investigation and discovery efforts in academic and industrial labs to find effective IDO1 inhibitors.20–23
Substrate analogue 1-methyl-1-D/L-tryptophan 3 (1-MT) was amongst the first examples of a reported IDO inhibitor. Indoximod 4 (1-methyl-1-D-typtophan), epacadostat 5 (INCB024360) and navoximod 6 (NLG919) are more recent examples of IDO1 inhibitors that have entered late stage clinical trials, however the therapeutic utility of these compounds remains to be fully established (Fig. 1).24–27
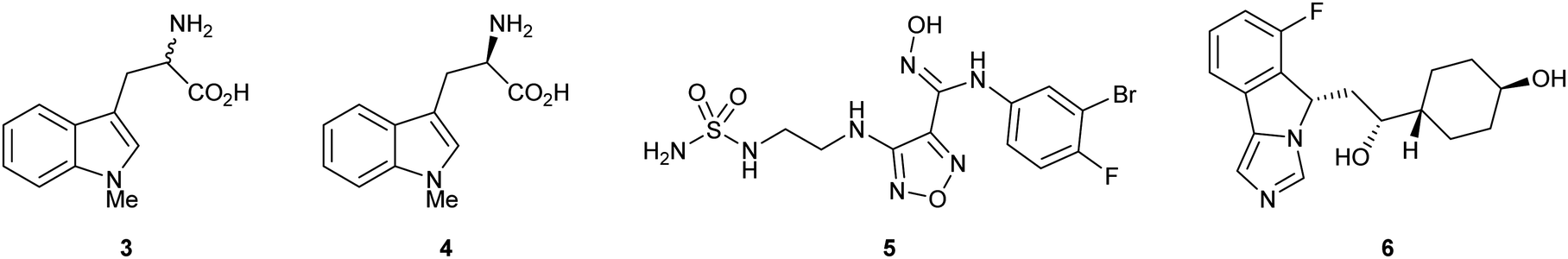 | ||
| Fig. 1 IDO inhibitor 1-MT 3 and clinically evaluated compounds indoximod 4, epacadostat 5 and navoximod 6. | ||
IDO1 mechanism of dioxygenation
The mechanism by which the dioxygenase family of enzymes metabolises 1 to 2 has been the subject of debate for over 60 years.28 Hayaishi's seminal report demonstrated that in an 18O2-enriched atmosphere, both oxygen atoms were incorporated into product 2.29 Alongside this discovery, Hayaishi proposed a dioxetane intermediate 7 which presumably degrades to give 2via a retro [2+2] cycloaddition (Scheme 2). Hamilton later challenged Hayaishi's dioxetane hypothesis, claiming the intermediate was too energetically strained; instead, Hamilton proposed the conversion of 1 to 2via a Criegee-type rearrangement giving intermediate 8 (Scheme 2).30 Hayaishi's dioxetane and Hamilton's Criegee-type rearrangement intermediates, although not based on experimental evidence, remained key to many subsequent mechanistic proposals of the dioxygenase family enzymes.31–33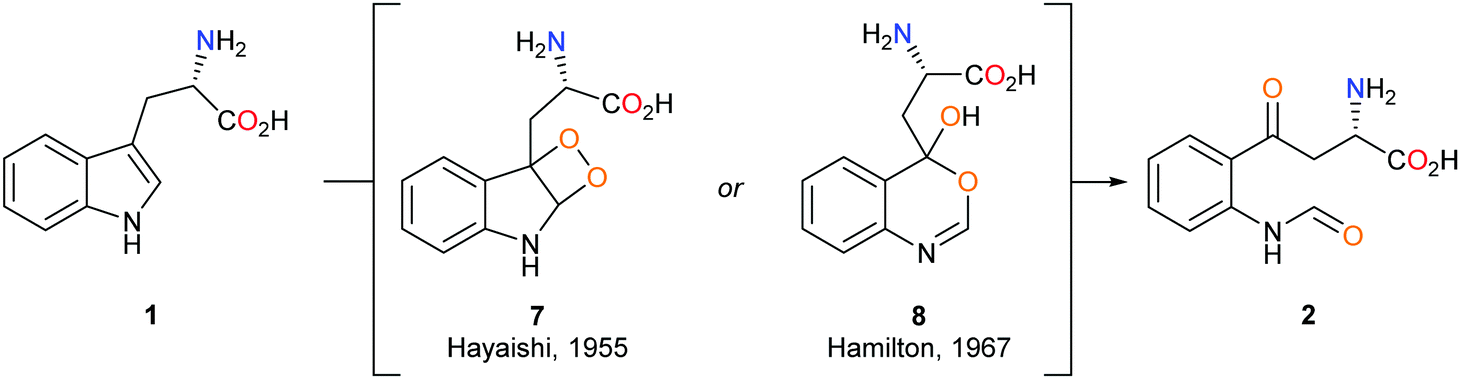 | ||
| Scheme 2 Hayashi's 7 and Hamilton's 8 proposed metabolic intermediates. | ||
Sono, Stocker and Chapman individually proposed the use of a basic residue within the active site, or molecular oxygen itself, to deprotonate the indole NH of 1 as part of IDO1s mode of action.31–33 Chemical interrogation of this hypothesis is not evident within the literature and it falls short when one considers the pKa of the indole NH (∼16.82 in H2O) and the lack of a sufficiently strong basic residue within the active site.34 The initial discovery of 1-MT 3 as an ‘inhibitor’ of IDO1 supported this theory based on its lack of an indole NH proton.24 However, later studies demonstrated that 1-MT is in fact a substrate and can be metabolised by IDO1.35–37
Since the discovery of IDO1 as an oncogenic target there has been a renewed interest in the mechanism of dioxygenation from both computational and experimental studies, with heme-intermediates of the dioxygenation being experimentally validated.38–44 Currently, IDO1 is thought to metabolise tryptophan 1via a (A) radical or (B) electrophilic-based mechanism and involve the formation of an epoxide intermediate and a ferryl iron species (Scheme 3).40–45
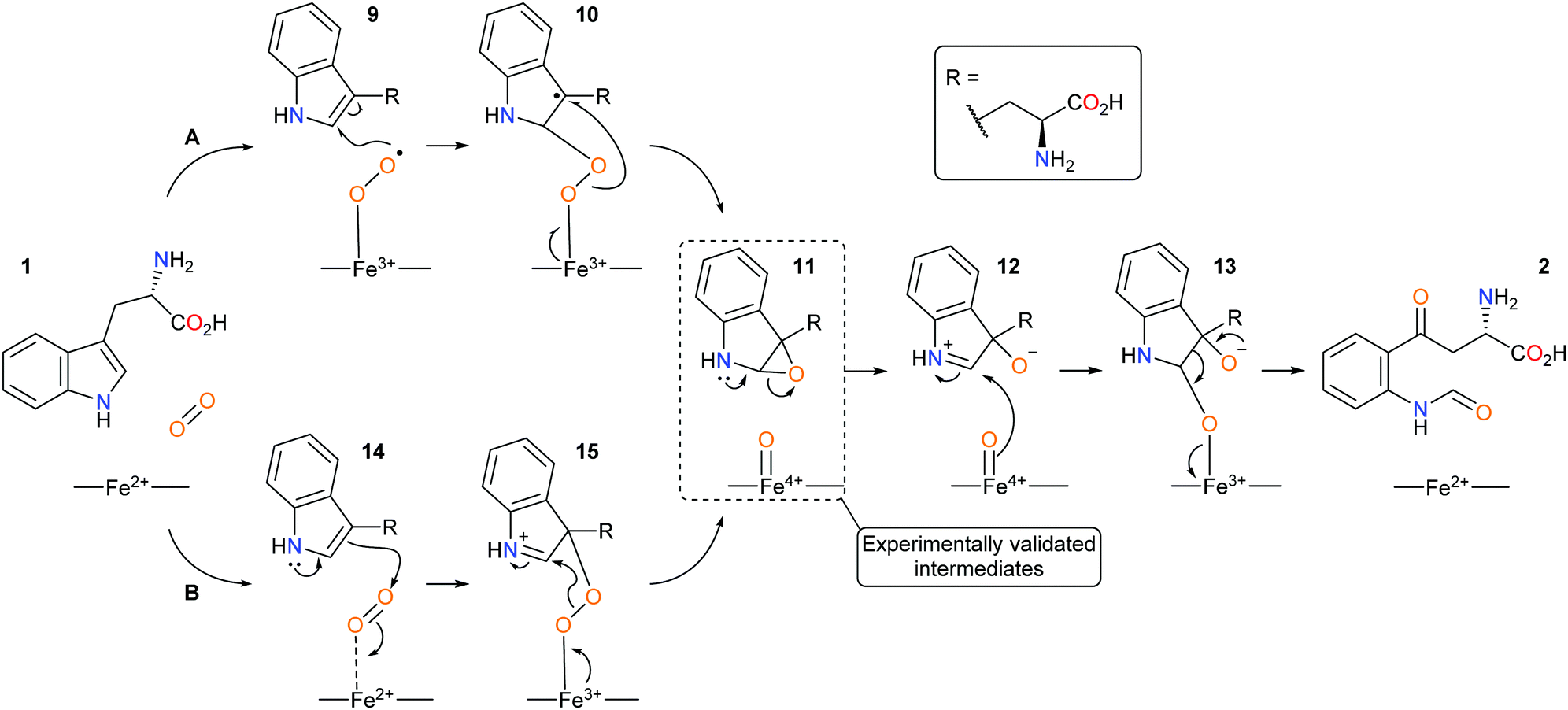 | ||
| Scheme 3 Proposed new mechanistic pathways of (A) radical and (B) electrophilic IDO1 mediated dioxygenation of L-tryptophan. | ||
Design rationale
We sought to design substrate mimics, 16–18, that have the potential to divert IDO1's dioxygenative mode of action with the hypothesis that these may be either mechanistic probes or potentially a novel class of inhibitors of IDO1 (Fig. 2).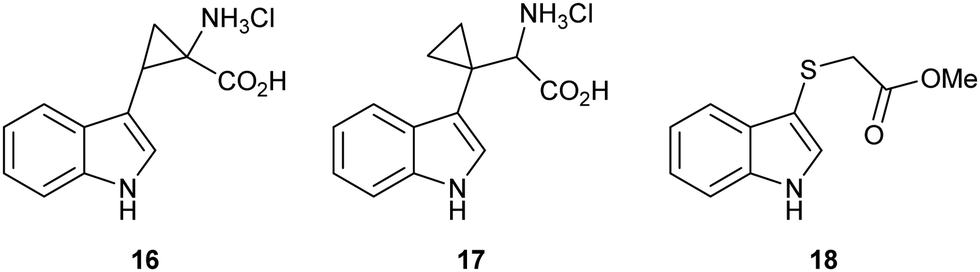 | ||
| Fig. 2 Proposed mechanistic probes of IDO1 16–18. | ||
Strategic placement of a cyclopropane in the α-position with respect to the proposed radical centres of mimics 16 and 17 allows for a potentially rapid ring opening and diversion of potential metabolic intermediates 19 and 21, respectively (Fig. 3A and B). This mechanism of action is seen in cyclopropane containing MAO inhibitors such as tranylcypromine.46 Ring opening of 19 and 21 would result in the formation of covalently bound intermediates 20 and 22. The ring opening of 19 results in the formation of stabilised captodative radical 20, giving a predicted enhancement in rate of ring opening. Further enzyme inactivation is plausible through a subsequent radical reaction with an active site residue. Compounds 16 and 17 were designed to explore this hypothesis, with evidence of ring-opened products or alkylated protein adducts supporting the role of radical intermediate 10 in the reaction mechanism.
 | ||
| Fig. 3 (A) Radical ring opening of cyclopropane 19 giving covalently bound inhibitor complex 20 and forming a captodative radical; (B) radical ring opening of cyclopropane 21 giving covalently bound inhibitor complex 22 and a reactive radical to interact further with the protein; (C) ring opening of epoxide 23via neighbouring group participation to give thionium 24 and a redox inactive protein; (D) loss of a thiolate anion over C–C bond cleavage of hemi-thioacetal 25 giving covalently bound inhibitor-probe complex 26. | ||
Radical clocks have long been used in mechanistic studies to investigate radical reactivity in organic compounds with the ring opening of a cyclopropylmethyl radical being amongst the fastest known.47–50 Cyclopropanes have similarly been utilised in the mechanistic study of enzyme metabolism. Newcombe successfully designed a cyclopropane-containing substrate mimic which could discriminate between cationic and radical reactivity, based on the metabolic products, to study the mechanism of cytochrome P-450 mediated hydroxylation.51,52
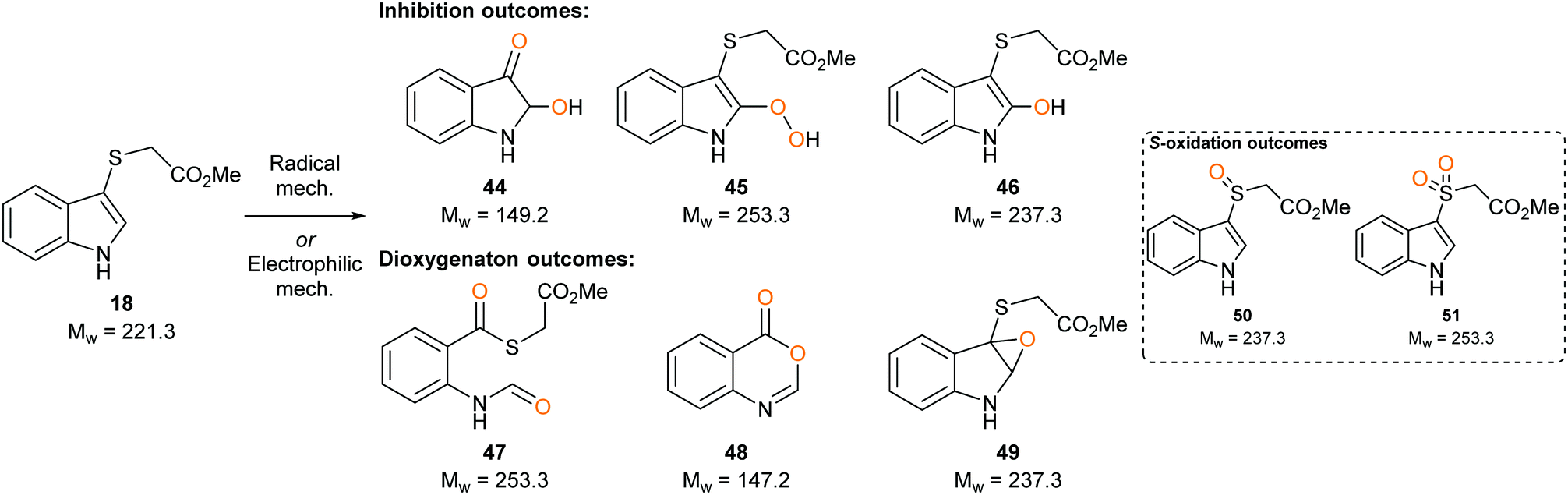
Sulfenylindole 18 was designed to intercept later-stage proposed intermediates, 11 and 13, of the dioxygenation mechanism (Fig. 3C and D). We hypothesised that α-epoxy sulfide 23 can disrupt a key metabolic intermediate via ring opening of the epoxide through neighbouring group participation to give thionium 24 and a redox inactive heme-centre. Alternatively, enzyme deactivation could be achieved through loss of a thiolate anion instead of C–C bond cleavage during the collapse of hemi-thioacetal 25 resulting in covalently bound indolone 26.
Results and discussion
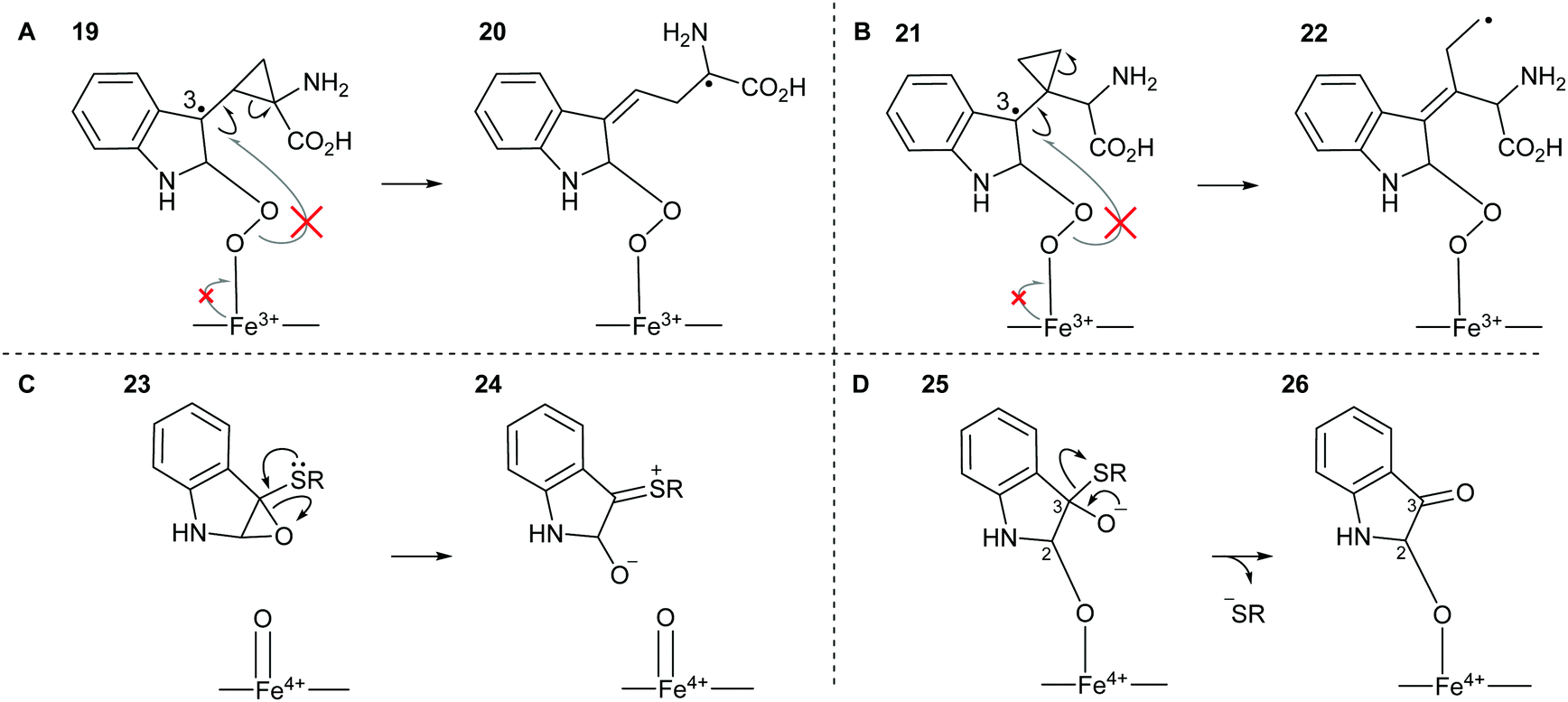
The potential for 16 and 17 to act as radical traps for IDO1 is supported by docking studies of both compounds into the active site of the tryptophan-bound IDO1 structure (PDB: 5WMV).53 Tryptophan re-docks into this structure with some flexibility in the orientation of the indole, however one of the lowest energy poses has the indole in a reactive conformation, which is effectively identical to the experimentally observed pose (Fig. 4A), while both 16 and 17 demonstrate a low-energy bound pose close to that of tryptophan (Fig. 4B and C), with similar docking score to the natural substrate. In the lowest energy poses for 16 and 17 the indole is positioned in a reactive geometry relative to the heme (Fig. 4D); in this position, the cyclopropane bond that would be expected to be cleaved is sufficiently close to an ideal 90° geometry, relative to the plane of the indole ring, suggesting ring-opening will be possible (16, 69° and 17, 87° – Fig. 4D).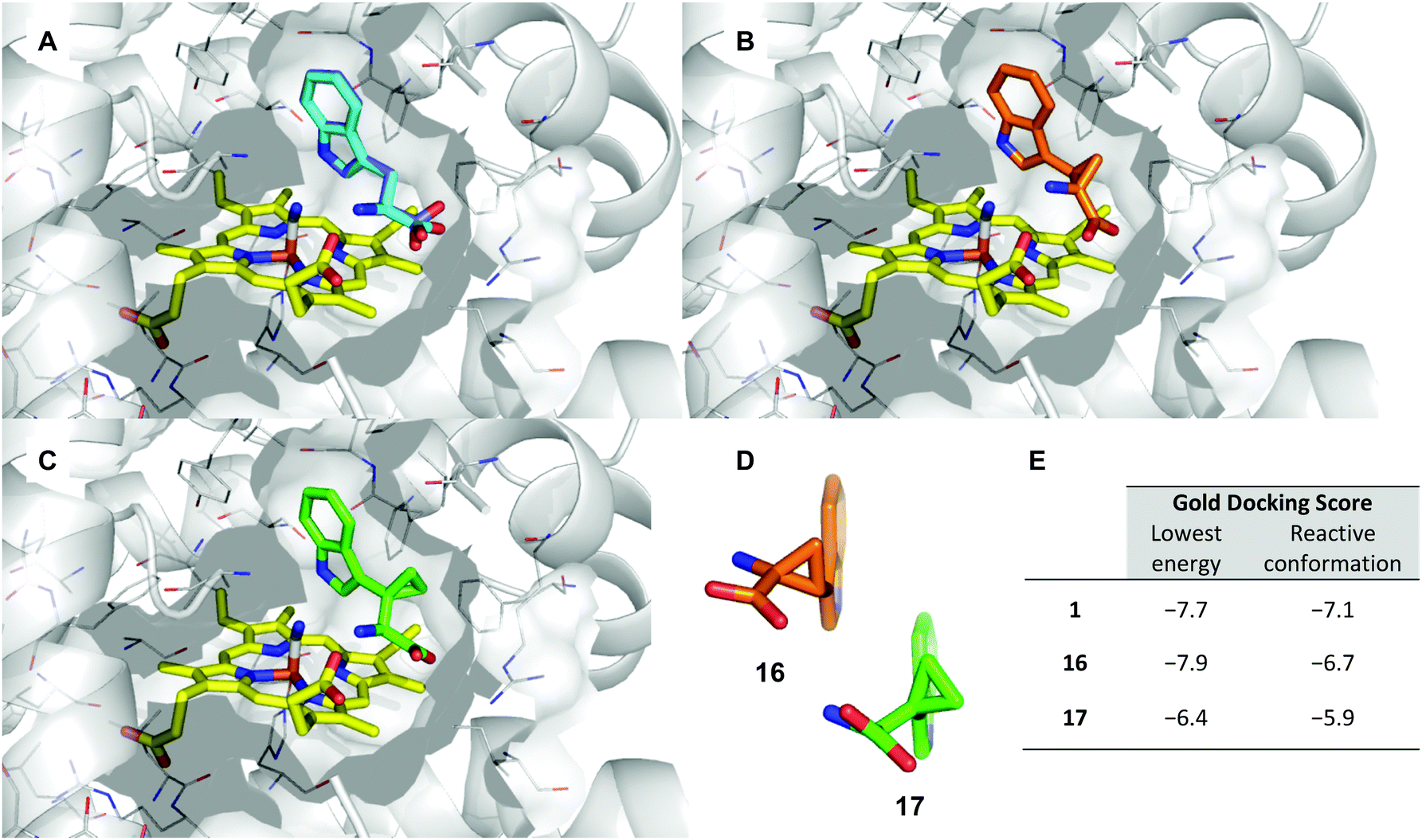 | ||
| Fig. 4 Representation of the IDO1 structure 5WMV, with IDO protein residues depicted in white cartoon/lines and active site surface in white, cut to reveal the CN-coordinated heme (yellow) and substrate binding sites; (A) experimental (cyan) and lowest energy docked (blue) pose of TRP. (B) Lowest energy docked pose of 16 (orange); (C) lowest energy docked pose of 17 (green); (D) projection of lowest energy docked poses of 16 and 17, showing the relationship between the indole ring and cyclopropane bonds; (E) gold docking scores for lowest energy pose and depicted poses with indole in a reactive conformation. Lower numbers are indicative of a more stable bound state. | ||
Synthesis of α,β-cyclopropyl tryptophan 16
Literature precedent exists for the preparation of α,β-cyclopropyl tryptophan 16, however the synthesis is cumbersome and relies on hazardous, inefficient chemistry.54 To access the required cyclopropane-core, 1,2-cyclopropane amino acids were synthesised via a 1,3-dipolar cycloaddition between an amino acrylate and an in situ generated diazo species.55,56Preliminary investigation of this route highlighted the need for an N-protecting group on the diazo precursor for successful application of the cyclopropanation chemistry; following a screen (see ESI,† Appendix 3.1), Boc was found to be the optimum protecting group. To enable efficient deprotection post-cyclopropanation, amino acrylate 34 was prepared from N-Boc serine 32. The desired cyclopropane core 30 could be accessed in excellent yield and high diastereoselectivity for the E-isomer. Following the cyclopropanation, deprotection gave substrate mimic 16 in excellent yield over six steps (Scheme 4).
 | ||
Scheme 4 Synthesis of α,β-cyclopropyl tryptophan-mimic 16. (a) POCl3, DMF, 0–50 °C, 2.5 h, 91% (27); (b) (Boc)2O, Et3N, DMAP, CH2Cl2, 23 °C, 18 h, 99% (28); (c) TsNHNH2, MeOH, 50 °C, 4 h, 99% (29); (d) (i) 29, Cs2CO3, BTAC, Dry PhMe 23 °C, 1.5 h, (ii) 34, 90 °C, 16 h, 91% (92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, E:Z) (30); (e) 10% Pd/C, MeOH, 23 °C, 0.75 h, 94% (31); (f) 4 M HCl in 1,4-dioxane, 23 °C, 23 h, 99% (16); (g) 32, Cs2CO3, DMF, 23 °C, 0.3 h, (ii) BnBr, 23 °C, 17.5 h, 78% (33); (h) (i) MsCl, DMAP, 0 °C, 0.25 h, (ii) Et3N, 0–23 °C, 16.5 h, 83% (34). :8, E:Z) (30); (e) 10% Pd/C, MeOH, 23 °C, 0.75 h, 94% (31); (f) 4 M HCl in 1,4-dioxane, 23 °C, 23 h, 99% (16); (g) 32, Cs2CO3, DMF, 23 °C, 0.3 h, (ii) BnBr, 23 °C, 17.5 h, 78% (33); (h) (i) MsCl, DMAP, 0 °C, 0.25 h, (ii) Et3N, 0–23 °C, 16.5 h, 83% (34). | ||
Difficulty in the isolation of pure 16 became apparent with the compound taking on an unexpected red colouration, despite no detectable impurities via1H-NMR analysis. Subjecting substrate mimic 16 to preparative HPLC eluted the compound-containing fractions as a colourless solution, however upon concentration of the eluent the sample took on a red colouration. We hypothesised that air-oxidation of the amine adjacent to the cyclopropane could result in the radical cation which could undergo a ring-opening process. Other groups have noted the instability of aryl cyclopropyl amines under aerobic conditions – the position of the amine adjacent to the cyclopropane was identified as key for radical-based product degradation.57 Determining the identity of the degradation product was beyond the scope of the investigation and the sample was taken forward and subjected to biochemical evaluation.
Synthesis of β,β′-cyclopropyl tryptophan 17
Synthesis of β,β′-cyclopropyl tryptophan substrate mimic 17 required the cyclodialkylation of 3-indole acetonitrile 35 to install the desired spirocyclic cyclopropane. Repetition of reported literature conditions failed to provide a productive outcome;58 judicious choice of di-halo ethane derivative and addition rate of LDA proved key to the success of the reaction and cyclopropyl nitrile 36 was accessed in excellent yield (see ESI,† Appendix 3.2). Subsequent DIBAL-H mediated reduction of nitrile 36 followed by a Strecker reaction of the resulting aldehyde 37 afforded 17 in moderate yield as the hydrochloride salt (Scheme 5).58,59 Inhibitor 17 was designed to overcome what was perceived to be the root of the instability of inhibitor 16: the presence of an amine directly attached to the cyclopropane. Upon isolation, inhibitor 17 proved to be stable to aerobic conditions.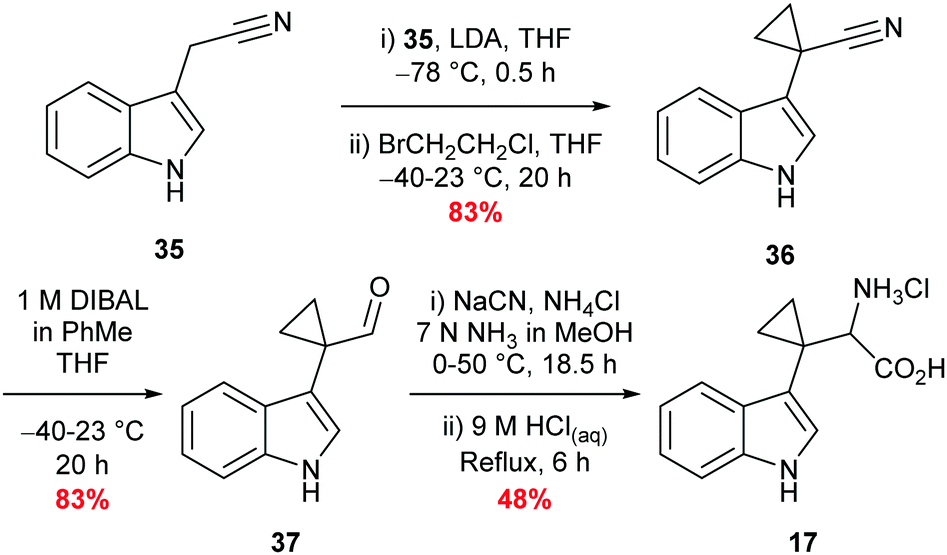 | ||
| Scheme 5 Synthesis of β,β′-cyclopropyl tryptophan-mimic 17. | ||
Synthesis of sulfenylindole 18
Sulfenylindole 18 was readily prepared in a single step via an iodine-mediated sulfylation of indole, accessing the desired material is excellent yield (Scheme 6).60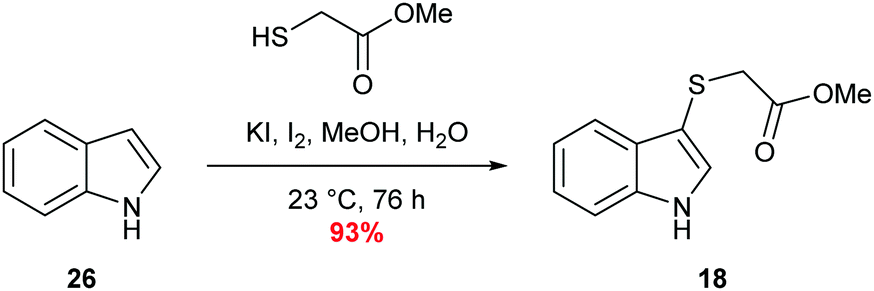 | ||
| Scheme 6 Synthesis of sulfenylindole 18. | ||
Biochemical evaluation
Compounds 16–18 were evaluated against recombinant human IDO1 mutant F164A. The F164A mutant represents a minor structural change in the active site topology with a ∼5-fold and 30-fold drop in Kd and Km, with respect to 1, when compared to wtIDO1.53 Importantly the F164A kcat. is comparable to wtIDO1 and, as a result, IDO1 F164A was deemed an appropriate mutant for mechanistic investigations.61LC-MS-based detection kinetic assay
To determine whether our compounds were metabolised by IDO1, we incubated IDO1 and substrate mimics 16–18 and analysed the post-assay media via LC-MS to detect any metabolic by-products. We hypothesised plausible metabolic by-products for 16 (38–41, Scheme 7), 17 (41–43, Scheme 7 below) and 18 (45–51, Scheme 8) and analysed the assay media for their presence (see ESI,† Appendix 3.3).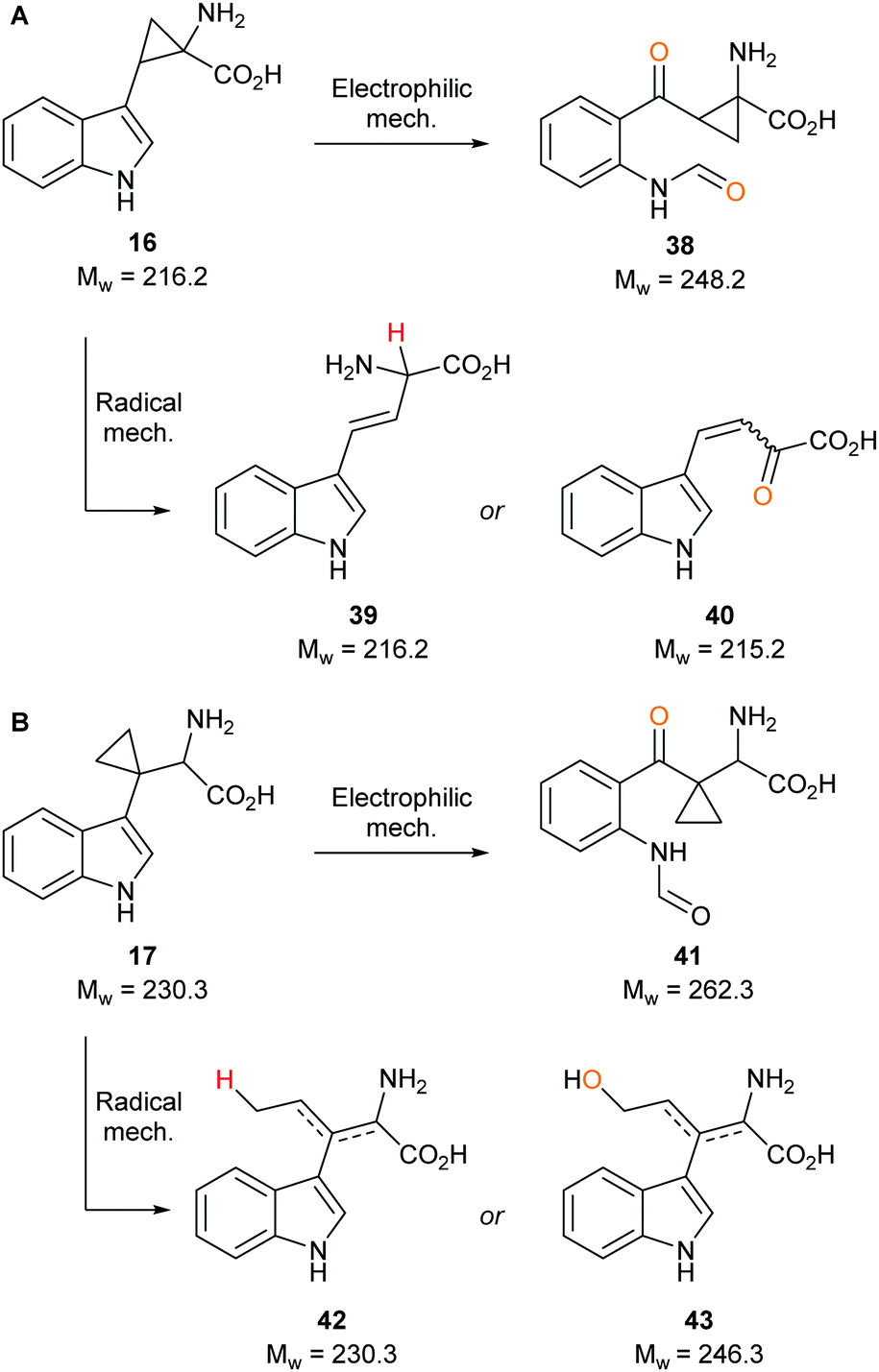 | ||
| Scheme 7 Plausible metabolic by-products of IDO1 mediated dioxygenation of tryptophan-mimics (A) 16 and (B) 17. | ||
 | ||
| Scheme 8 Plausible metabolic by-products of IDO1 mediated dioxygenation of sulfenylindole 18 metabolism. | ||
Our control experiments demonstrated clear turnover of 1 and 3 (see ESI,† Appendix 3.4) to their respective dioxygenated metabolites. When subjecting substrate mimics 16 or 17 to the assay, no masses corresponding to plausible metabolic products were observed (see ESI,† Appendix 3.5 and 3.6). One possible explanation for the lack of observed turnover is the lack of substrate mimic recognition by IDO1, while another could be that the substrate mimics are partially metabolised but effective inhibition is taking place and is inactivating IDO1 to further compound turnover.
Analysis of the metabolites of substrate mimic 18 identified a mass corresponding to the radical cation of peroxy intermediate 45 or thioester 47 (see ESI,† Appendix 3.7). A mass corresponding to deprotonated enol 46 or epoxide 49 (see ESI,† Appendix 3.8) was also observed. Alternatively, the observed masses could be a result of mono- or dioxidation of sulfide 18 to sulfoxide 50 or sulfone 51 (Scheme 8).
Due to a lack of convincing enzymatic turnover we sought to assess the binding of our compounds to IDO1 using both inhibition and Tm shift assays.
IDO1 inhibition assay
To understand the effects of compounds 16–18 on IDO1 they were pre-incubated with the active protein (5 μM) for 15 minutes (see ESI,† Appendix 3.8) prior to the addition of 1, with the formation of 2 (abs. = 321 nm) followed by UV-vis spectroscopy. A reduction in the rate of formation of 2versus the control was interpreted as inhibition of IDO1, with epacadostat 5 used as the positive control (Fig. 5).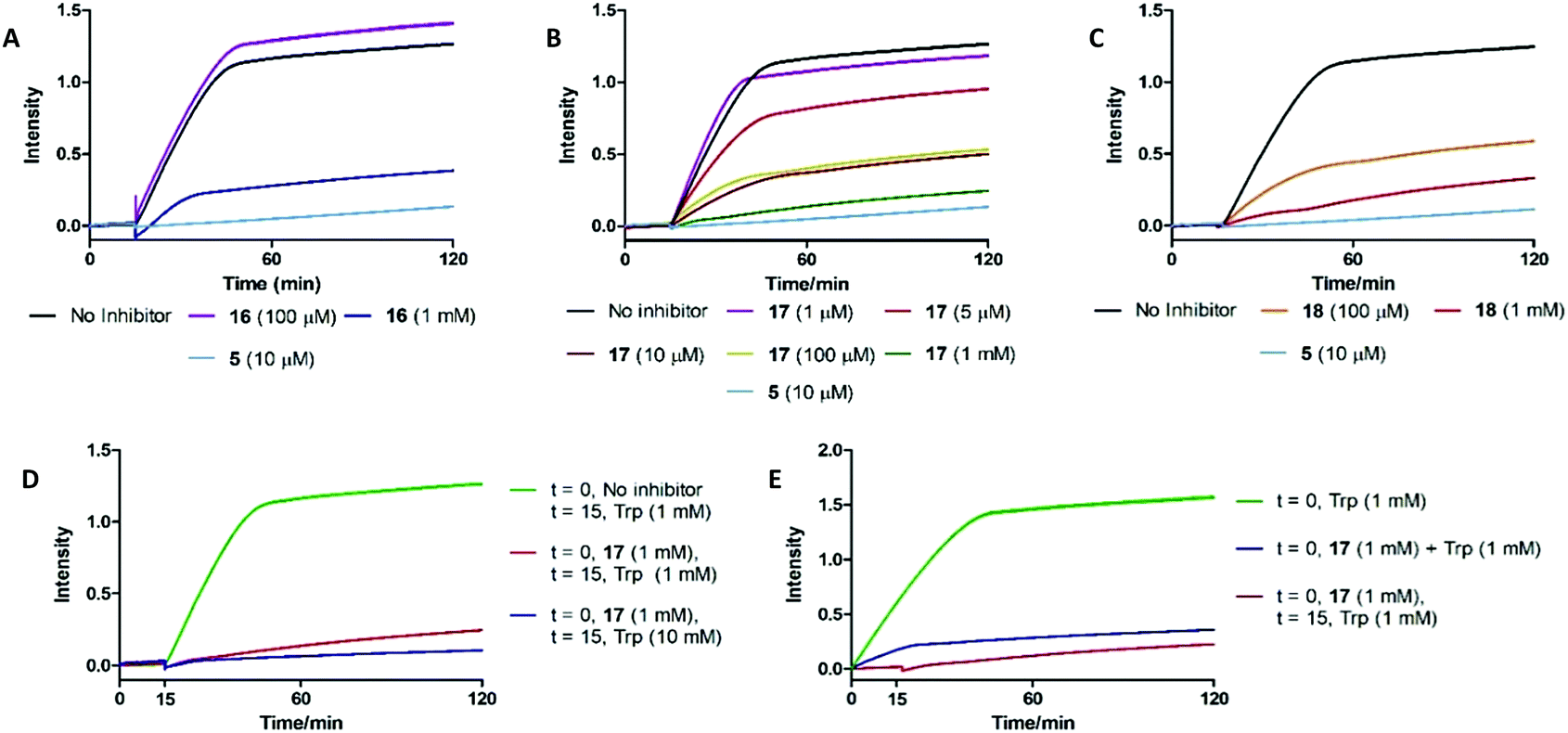 | ||
| Fig. 5 UV-based inhibition assay data for substrate mimics (A) 16, (B) 17 and (C) 18. (D) Out-competition and (E) co-incubation data for substrate mimic 17. | ||
Under these conditions high concentrations (1 mM) of 16–18 resulted in inhibition of IDO1 mediated production of 2. Reducing the dosed concentration of compound 16 saw a rapid loss in IDO1 inhibitory activity with the level of inhibition returning to control levels when dosed at 100 μM. Dosing compound 18 at 100 μM led to a minor alleviation in the inhibition afforded by the substrate mimic. Reduced detection of 2 in the inhibition experiments could therefore be a result of 18 acting as a competitive substrate. Inhibition studies of compound 17 however highlighted a significant reduction in levels of 2 at concentrations as low as 10 μM.
We developed two further assays to interrogate any time-dependency in substrate mimic 17's inhibitory action or the inability of compound 17 to be outcompeted by its natural substrate: key characteristics of an uncompetitive inhibition mechanism (Fig. 5). Co-administering 17 and 1 simultaneously led to an initial increase in the NFK produced, albeit at a reduced rate compared to the control, but after a period of 15 min the signal plateaued. To determine whether the 17 could be outcompeted, after a 15 min pre-incubation period of IDO1 and 17, 1 (10 mM) was introduced and the production of 2 was followed. No observation of additional 2 was observed suggesting 17 was not outcompeted by a higher concentration of Trp. Together, the data are suggestive of an uncompetitive inhibition mechanism. It is known that some classes of IDO1 inhibitors act by stabilising the heme-free apo state of the enzyme, which can be detected by loss of the characteristic IDO1-heme absorption band at 405 nm. No such change is observed on incubating 17 with IDO1, ruling out this mechanism of inhibition (see ESI,† Appendix 3.9).62
Thermal shift assay
Binding events between a ligand and a protein typically result in a stabilising interaction which is manifested in an altered melting point or Tm value, and this approach is now a widely accepted method of establishing protein–ligand interactions.63,64 In order to determine whether there is a binding event between IDO1 and 17, we subjected compound 17 to thermal shift analysis using 1-MT 3 and epacadostat 5 as positive controls for a binding interaction.Control experiments identified that at low IDO1:SYPRO (fluorescent dye) dilution ratios the protein was not stable, demonstrating a Tm ∼ 30 °C (Fig. 6). Increasing the IDO1:SYPRO dilution factor ratio gave a stable Tm value. Interestingly, we observed a change in fluorescence even in the absence of the SYPRO dye. As the instrument used utilises non-specific blue LED excitation (∼400–450 nm) which corresponds to the IDO-heme absorption band, we hypothesised that the fluorescence is a result of the release of fluorescent porphyrin from the heme-based enzyme upon heating.
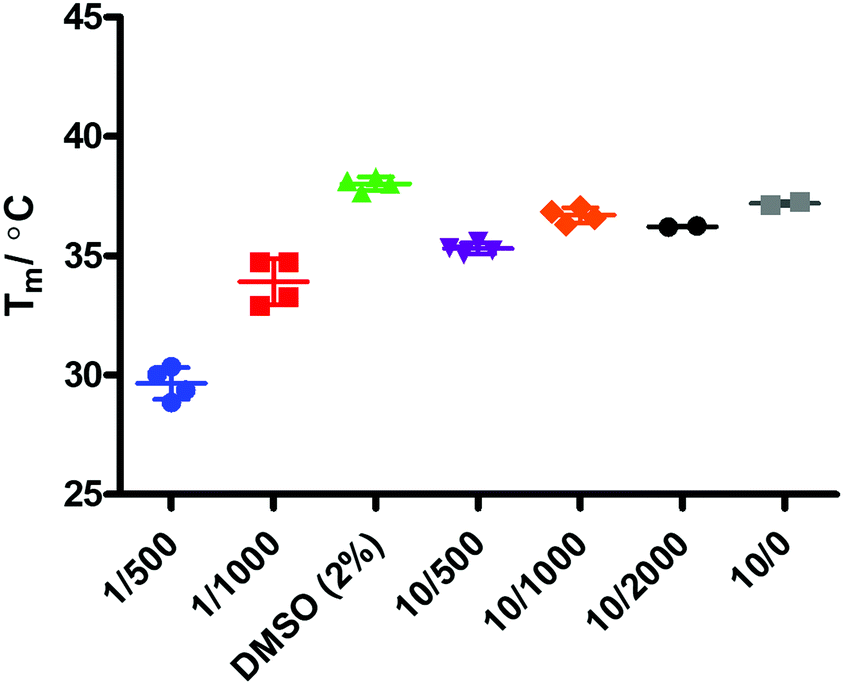 | ||
| Fig. 6 Controls – [IDO] μM/SYPRO dye dilution factor. | ||
Initial evaluation of the IDO1 Tm values in the presence of epacadostat 5 or 17 were performed in the presence of SYPRO dye (Fig. 7). We observed a significant shift in the Tm value when IDO1 and 5 were incubated, corresponding to a significant binding event as would be expected for this high affinity ligand. Unfortunately, when IDO1 and 1-MT 3 or cyclopropane 17 were incubated no shift in the protein Tm value was observed. The weak signal at lower concentrations of 5 and absence of shift for 3 suggest that the Tm of IDO is not sensitive to weaker ligands under these conditions.
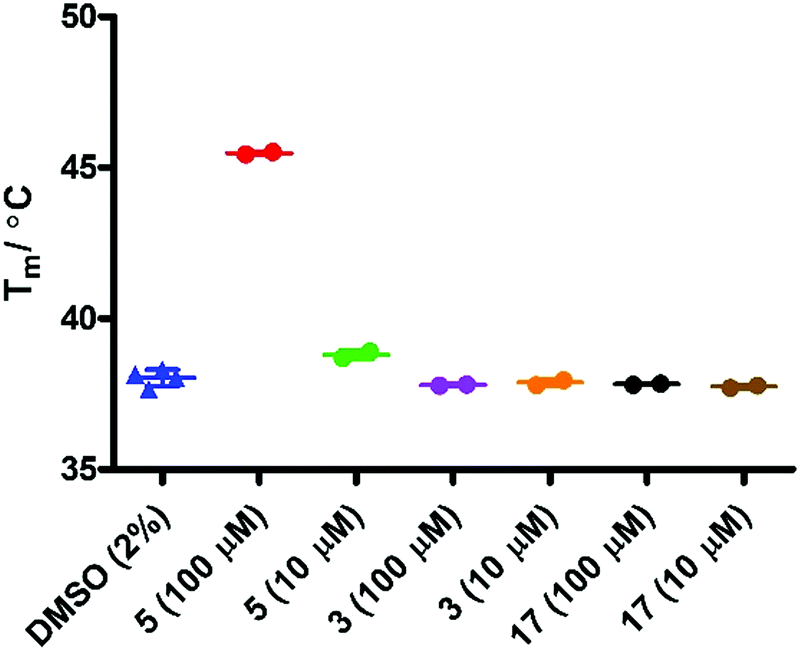 | ||
| Fig. 7 T m analysis in the presence of SYPRO dye (1/2000). | ||
In order to assess if removing SYPRO and following the change in heme signal alone would provide a more sensitive experiment, the experiment was replicated in the absence of the SYPRO dye and with higher concentrations of 3 and 17. Surprisingly this resulted in a significant change to the outcome of the experiment (Fig. 8). Incubation of IDO1 and epacadostat 5 or 1-MT 3 resulted in no change in the observed Tm value, whereas incubation of IDO1 and 17 saw a significant change in the Tm value at higher concentration. The conflicting data in these two experiments may be rationalised when considering the origin of the fluorescence. The SYPRO dye model changes in fluorescence as a result of the global change in the protein structure and the dye binding to the newly exposed hydrophobic surfaces. It would be expected that high affinity ligands with multiple binding interactions could afford significant stabilisation to an overall protein structure. In the absence of SYPRO, the fluorescence gain is a result of heme-environment changes alone suggesting any changes in Tm under these experimental parameters result from stabilisation of the region of the protein that binds the heme group.
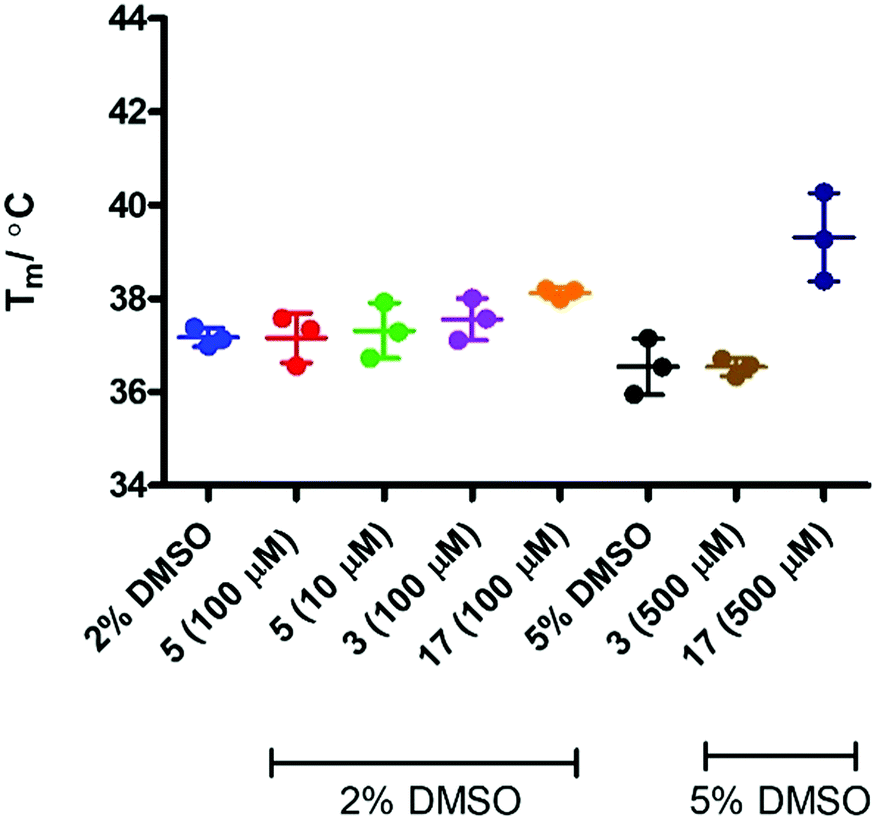 | ||
| Fig. 8 T m analysis in the absence of SYPRO dye. | ||
X-ray co-crystallographic studies of 5 have highlighted significant active site pocket interactions which could feasibly contribute to increased protein stability as observed in SYPRO-containing Tm assays.52
As the use of direct detection of IDO Tm is a novel assay format we sought an alternative biophysical method for the detection of the interaction between 17 and IDO-1. As seen in Fig. 9, this interaction can be detected by Microscale thermophoresis (MST) using labelled IDO1, with 17 demonstrating a Kd of 35 μM which is consistent with the inhibition data observed previously (Fig. 5b). Interestingly neither 1 nor 3 provide a significant signal in this model (see ESI,† Appendix 3.10), providing further evidence that 17 has a novel interaction/inhibition mechanism.
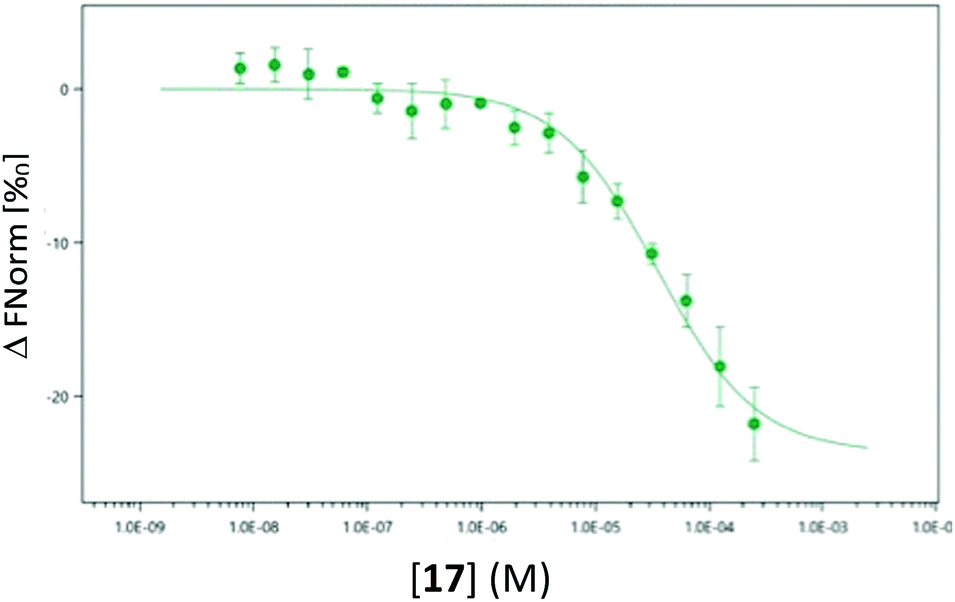 | ||
| Fig. 9 Microscale thermophoresis (MST) analysis of the dose-dependent interaction between 17 and IDO1, Kd 35.0 μM ± 6.96 μM. | ||
A 2017 study identified a secondary binding site of IDO1 (Si) which, when bound, inhibits turnover of 1.52 It was demonstrated that at high concentrations of 1, IDO1 was significantly inhibited by its natural substrate through suspected binding to the Si site – subsequent crystallographic data identified inhibitors bound to the proximal-side of the heme-centre, effecting inhibition via an altered active site topology. Based on binding to the Si site, substrate mimic 17 is not predicted to have stabilising active site pocket interactions, but it is predicted to have a significant interaction with the heme-centre.
Given the data demonstrating a mode of inhibition of 17 that is clearly distinct from the control inhibitors tested, we then considered the possibility that 17 may act by binding at this site. Docking of 17 into the Si site suggests the cyclopropane ring would be tolerated, with the key interactions made by TRP retained and docking scores comparable between tryptophan and 17, as well as 17 at the S site (Table 1 and Fig. 10).
| Gold docking score | |||
|---|---|---|---|
| S site lowest | Si site lowest | Si site shown | |
| 1 | −7.7 | −7.2 | −7.0 |
| 17 | −6.4 | −7.0 | −6.4 |
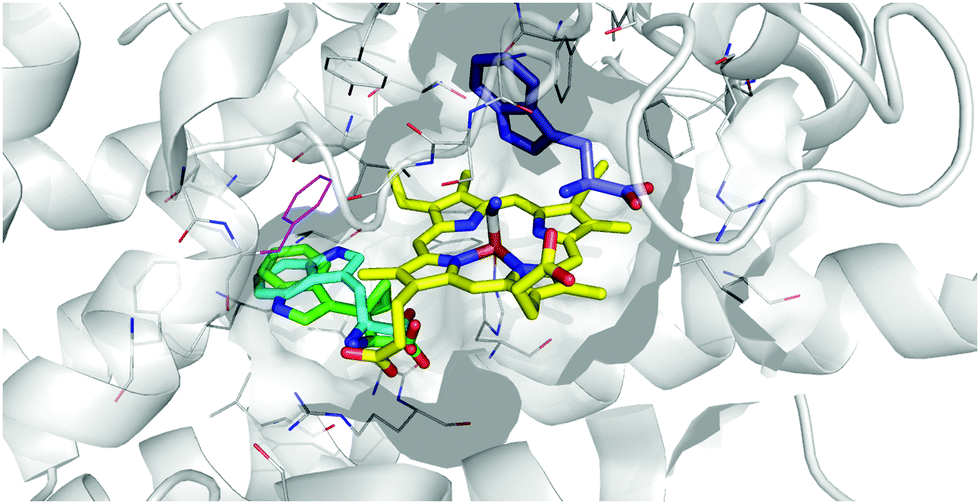 | ||
| Fig. 10 Representation of the IDO1 structure 5MWV, with IDO protein residues depicted in white cartoon/lines and active site surface in white, cut to reveal the CN-coordinated heme (yellow) and substrate binding sites. Docked pose of tryptophan at the S site (blue) and Si site (cyan) are overlaid with 17 (green), with F270 which moves to form the Si site depicted in magenta. | ||
Conclusions
A series of IDO1 mechanistic probes 16–18 were designed based on putative intermediates in the IDO1-mediated metabolism of L-tryptophan, with docking studies supporting their potential to interact with IDO1 with a tryptophan-like binding pose. These compounds were successfully synthesised and characterised, however α,β-cyclopropyl tryptophan 16 showed unexpected instability which limited its use in subsequent biological studies.Incubation of compounds 16–18 with IDO1 did not result in the observation of established epoxide or di-carbonyl metabolites, however there was also no evidence of metabolites by diversion of putative enzymatic intermediates. Given 16–18 are able to inhibit IDO1-mediated metabolism of L-tryptophan these results suggest compounds 16–18 do not act as simple active-site substrate mimics and therefore our findings do not provide further clarification of the IDO1 mechanism.
More detailed studies on the inhibitory activity of β,β′-cyclopropyl tryptophan 17 demonstrated an uncompetitive inhibition mechanism. Tm studies on IDO1 incubated with epacadostat 5 detected denaturation using the fluorescent dye SYPRO, showing clear stabilisation of the protein which is not replicated by 17. However it was also observed that the denaturation of IDO1 can be directly monitored under dye-free conditions based on change in heme fluorescence. In this experimental set up 17 shows a significant temperature shift, consistent with stabilisation of the heme environment independent of overall protein folding. Our UV-data demonstrated that co-incubation of IDO1 and 17 did not displace heme and an orthogonal MST assay confirmed that target engagement with inhibitor 17 could be achieved. The lack of target engagement of known active site ligands, 1 or 3, in further MST assays strongly suggests that 17 adopts a distinct binding mode from active site (S site) ligands. These findings, alongside the evidence of uncompetitive inhibition, led to the hypothesis that 17 binds at the recently-discovered Si site, which is supported by subsequent docking studies.
Author contributions
NJC, RH, TT, AGL, OQ, JG, NMB, CAB, ER, RSG and SB designed the research, analysed data and reviewed and approved the final manuscript. NJC performed the synthesis, carried out the LC-MS and IDO1 inhibition assays and co-wrote the manuscript with RSG and SB. RH conducted Tm assays, TAJ performed the MST assays, SB conducted the heme-UV assays and TT verified IDO1 activity. AGL conducted molecular docking.Conflicts of interest
NJC was a PhD student sponsored by Celentyx Ltd. TT, OQ, CAB are employees of Celentyx Ltd with share options in the company. JG and NMB are Directors of Celentyx Ltd with shareholdings in the company. There are no other conflicts to declare.Acknowledgements
The authors would like to thank Dr C S Le Duff, Dr C W Tsang and Dr P Ashton for analytical support and Celentyx Ltd and EPSRC (EP/N509590/1) for funding (CASE award to NJC).Notes and references
- Y. Kotake, Hoppe-Seyler's Z. Physiol. Chem., 1936, 243, 237–265 CrossRef CAS.
- C. Itagaki and Y. Nakayama, Hoppe-Seyler's Z. Physiol. Chem., 1941, 270, 83–88 Search PubMed.
- O. Hayaishi and R. Y. Stanier, J. Bacteriol., 1951, 62, 691–709 CrossRef CAS PubMed.
- S. Yamamoto and O. Hayaishi, J. Biol. Chem., 1967, 242, 5260–5266 CrossRef CAS.
- F. Hirata, O. Hayaishi, T. Tokuyama and S. Senoh, J. Biol. Chem., 1974, 249, 1311–1313 CrossRef CAS.
- O. Hayaishi, Acta Vitaminol. Enzymol., 1975, 29, 17–20 CAS.
- R. Metz, J. B. Duhadaway, U. Kamasani, L. Laury-Kleintop, A. J. Muller and G. C. Prendergast, Cancer Res., 2007, 67, 7082–7087 CrossRef CAS PubMed.
- H. J. Yuasa, M. Takubo, A. Takahashi, T. Hasegawa, H. Noma and T. Suzuki, J. Mol. Evol., 2007, 65, 705–714 CrossRef CAS PubMed.
- H. J. Ball, A. Sanchez-Perez, S. Weiser, C. J. Austin, F. Astelbauer, J. Miu, J. A. McQuillan, R. Stocker, L. S. Jermiin and N. H. Hunt, Gene, 2007, 396, 203–213 CrossRef CAS PubMed.
- R. Yoshida, Y. Urade, M. Tokuda and O. Hayaishi, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 4084–4086 CrossRef CAS PubMed.
- R. Yoshida and O. Hayaishi, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3998–4000 CrossRef CAS PubMed.
- S. V. Schmidt and J. L. Schultze, Front. Immunol., 2014, 5, 384 Search PubMed.
- D. H. Munn, E. Shafizadeh, J. T. Attwood, I. Bondarev, A. Pashine and A. L. Mellor, J. Exp. Med., 1999, 189, 1363–1372 CrossRef CAS PubMed.
- G. K. Lee, H. J. Park, M. Macleod, P. Chandler, D. H. Munn and A. L. Mellor, Immunology, 2002, 107, 452–460 CrossRef CAS PubMed.
- G. Frumento, R. Rotondo, M. Tonetti, G. Damonte, U. Benatti and G. B. Ferrara, J. Exp. Med., 2002, 196, 459–468 CrossRef CAS PubMed.
- M. Platten, W. Wick and B. J. Van den Eynde, Cancer Res., 2012, 72, 5435–5440 CrossRef CAS PubMed.
- L. Hornyák, N. Dobos, G. Koncz, Z. Karányi, D. Páll, Z. Szabó, G. Halmos and L. Székvölgyi, Front. Immunol., 2018, 9, 151 CrossRef PubMed.
- L. Brochez, I. Chevolet and V. Kruse, Eur. J. Cancer, 2017, 76, 167–182 CrossRef CAS PubMed.
- C. Uyttenhove, L. Pilotte, I. Theate, V. Stroobant, D. Colau, N. Parmentier, T. Boon and B. J. Van den Eynde, Nat. Med., 2003, 9, 1269–1274 CrossRef CAS PubMed.
- U. F. Röhrig, S. R. Majjigapu, P. Vogel, V. Zoete and O. Michielin, J. Med. Chem., 2015, 58, 9421–9437 CrossRef PubMed.
- G. C. Prendergast, W. P. Malachowski, J. B. DuHadaway and A. J. Muller, Cancer Res., 2017, 77, 6795–6811 CrossRef CAS PubMed.
- J. E. Cheong, A. Ekkati and L. Sun, Expert Opin. Ther. Pat., 2018, 28, 317–330 CrossRef CAS PubMed.
- B. J. van den Eynde, N. van Baren and J.-F. Baurain, Annu. Rev. Cancer Biol., 2020, 4, 241–256 CrossRef.
- S. G. Cady and M. Sono, Arch. Biochem. Biophys., 1991, 291, 326–333 CrossRef CAS.
- E. Soliman, T. Neuger, E. C. Dees, R. D. Harvey, H. Han, R. Ismail-Khan, S. Minton, N. N. Vahanian, C. Link, D. M. Sullivan and S. Antonia, Oncotarget, 2014, 5, 8136–8146 CrossRef PubMed.
- C. Jochems, M. Fantini, R. I. Fernando, A. R. Kwilas, R. N. Donahue, L. M. Lepone, I. Grenga, Y. S. Kim, M. W. Brechbiel, J. L. Gulley, R. A. Madan, C. R. Heery, J. W. Hodge, R. Newton, J. Schlom and K. Y. Tsang, Oncotarget, 2016, 7, 37762–37772 CrossRef PubMed.
- A. Nayak-Kapoor, Z. Hao, R. Sadek, R. Dobbins, L. Marshall, N. N. Vahanian, W. Jay Ramsey, E. Kennedy, M. R. Mautino, C. J. Link, R. S. Lin, S. Royer-Joo, X. Liang, L. Salphati, K. M. Morrissey, S. Mahrus, B. McCall, A. Pirzkall, D. H. Munn, J. E. Janik and S. N. Khleif, J. Immunother. Cancer, 2018, 6, 61 CrossRef PubMed.
- E. L. Raven, J. Biol. Inorg. Chem., 2017, 22, 175–183 CrossRef CAS PubMed.
- O. Hayaishi, S. Rothberg, A. H. Mehler and Y. Saito, J. Biol. Chem., 1957, 229, 889–896 CrossRef CAS.
- G. A. Hamilton, Adv. Enzymol. Relat. Areas Mol. Biol., 1969, 32, 55–96 CAS.
- M. Sono, M. P. Roach, E. D. Coulter and J. H. Dawson, Chem. Rev., 1996, 96, 2841–2888 CrossRef CAS PubMed.
- A. C. Terentis, S. R. Thomas, O. Takikawa, T. K. Littlejohn, R. J. W. Truscott, R. S. Armstrong, S. R. Yeh and R. Stocker, J. Biol. Chem., 2002, 277, 15788–15794 CrossRef CAS PubMed.
- S. J. Thackray, C. G. Mowat and S. K. Chapman, Biochem. Soc. Trans., 2008, 36, 1120–1123 CrossRef CAS PubMed.
- G. Yagil, Tetrahedron, 1967, 23, 2855–2861 CrossRef CAS.
- S. J. Thackray, C. Bruckmann, J. L. R. Anderson, L. P. Campbell, R. Xiao, L. Zhao, C. G. Mowat, F. Forouhar, L. Tong and S. K. Chapman, Biochemistry, 2008, 47, 10677–10684 CrossRef CAS PubMed.
- N. Chauhan, S. J. Thackray, S. A. Rafice, G. Eaton, M. Lee, I. Efimov, J. Basran, P. R. Jenkins, C. G. Mowat, S. K. Chapman and E. L. Raven, J. Am. Chem. Soc., 2009, 131, 4186–4187 CrossRef CAS PubMed.
- C. Lu, Y. Lin and S.-R. Yeh, J. Am. Chem. Soc., 2009, 131, 12866–12867 CrossRef CAS PubMed.
- L. W. Chung, X. Li, H. Sugimoto, Y. Shiro and K. Morokuma, J. Am. Chem. Soc., 2008, 130, 12299–12309 CrossRef CAS PubMed.
- L. W. Chung, X. Li, H. Sugimoto, Y. Shiro and K. Morokuma, J. Am. Chem. Soc., 2010, 132, 11993–12005 CrossRef CAS PubMed.
- A. Lewis-Ballester, D. Batabyal, T. Egawa, C. Y. Lu, Y. Lin, M. A. Marti, L. Capece, D. A. Estrin and S. R. Yeh, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17371–17376 CrossRef CAS PubMed.
- S. Yanagisawa, K. Yotsuya, Y. Hashiwaki, M. Horitani, H. Sugimoto, Y. Shiro, E. H. Appelman and T. Ogura, Chem. Lett., 2010, 39, 36–37 CrossRef CAS.
- R. M. Davydov, N. Chauhan, S. J. Thackray, J. L. R. Anderson, N. D. Papadopoulou, C. G. Mowat, S. K. Chapman, E. L. Raven and B. M. Hoffman, J. Am. Chem. Soc., 2010, 132, 5494–5500 CrossRef CAS PubMed.
- J. Basran, I. Efimov, N. Chauhan, S. J. Thackray, J. L. Krupa, G. Eaton, G. A. Griffith, C. G. Mowat, S. Handa and E. L. Raven, J. Am. Chem. Soc., 2011, 133, 16251–16257 CrossRef CAS PubMed.
- A. Lewis-Ballester, F. Forouhar, S.-M. Kim, S. Lew, Y. Wang, S. Karkashon, J. Seetharaman, D. Batabyal, B.-Y. Chiang, M. Hussain, M. A. Correia, S.-R. Yeh and L. Tong, Sci. Rep., 2016, 6, 35169–35182 CrossRef CAS PubMed.
- I. Efimov, J. Basran, S. J. Thackray, S. Handa, C. G. Mowat and E. L. Raven, Biochemistry, 2011, 50, 2717–2724 CrossRef CAS PubMed.
- T. Malcomson, K. Yelekci, M. Teresa Borrello, A. Ganesan, E. Semina, N. De Kimpe, S. Mangelinckx and R. R. Ramsay, FEBS J., 2015, 282, 3190–3198 CrossRef CAS PubMed.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- M. Newcomb, Tetrahedron, 1993, 49, 1151–1176 CrossRef CAS.
- B. Roschek, K. A. Tallman, C. L. Rector, J. G. Gillmore, D. A. Pratt, C. Punta and N. A. Porter, J. Org. Chem., 2006, 71, 3527–3532 CrossRef CAS PubMed.
- M. Newcomb, C. C. Johnson, M. B. Manek and T. R. Varick, J. Am. Chem. Soc., 1992, 114, 10915–10921 CrossRef CAS.
- M. Newcomb, M. H. Le Tadic-Biadatti, D. L. Chestney, E. S. Roberts and P. F. Hollenberg, J. Am. Chem. Soc., 1995, 117, 12085–12091 CrossRef CAS.
- S. Y. Choi, P. E. Eaton, P. F. Hollenberg, K. E. Liu, S. J. Lippard, M. Newcomb, D. A. Putt, S. P. Upadhyaya and Y. Xiong, J. Am. Chem. Soc., 1996, 118, 6547–6555 CrossRef CAS.
- A. Lewis-Ballester, K. N. Pham, D. Batabyal, S. Karkashon, J. B. Bonanno, T. L. Poulos and S.-R. Yeh, Nat. Commun., 2017, 8, 1693 CrossRef PubMed.
- D. Donati, A. Garzon-Aburbeh, B. Natalini, C. Marchioro and R. Pellicciari, Tetrahedron, 1996, 52, 9901–9908 CrossRef CAS.
- V. K. Aggarwal, E. Alonso, I. Bae, G. Hynd, K. M. Lydon, M. J. Palmer, M. Patel, M. Porcelloni, J. Richardson, R. A. Stenson, J. R. Studley, J.-L. Vasse and C. L. Winn, J. Am. Chem. Soc., 2003, 125, 10926–10940 CrossRef CAS PubMed.
- C. Zhu, J. Li, P. Chen, W. Wu, Y. Ren and H. Jiang, Org. Lett., 2016, 18, 1470–1473 CrossRef CAS PubMed.
- A. Blackburn, D. M. Bowles, T. T. Curran and H. Kim, Synth. Commun., 2012, 42, 1855–1863 CrossRef CAS.
- D. C. Horwell, M. J. McKiernan and S. Osborne, Tetrahedron Lett., 1998, 39, 8729–8732 CrossRef CAS.
- P. W. Blackby, M. de Kort, M. Enthoven, P. S. Hinchcliffe, C. Paulie, C. M. Timmers and S. Verkiak, NL. Pat., WO2013041458, 2013 Search PubMed.
- P. R. Reid, T. M. Bridges, D. J. Sheffler, H. P. Cho, L. M. Lewis, E. Days, J. S. Daniels, C. K. Jones, C. M. Niswender, C. D. Weaver, P. J. Conn, C. W. Lindsley and M. R. Wood, Bioorg. Med. Chem. Lett., 2011, 21, 2697–2701 CrossRef CAS PubMed.
- N. Chauhan, J. Basran, S. A. Rafice, I. Efimov, E. S. Millett, C. G. Mowat, P. C. E. Moody, S. Handa and E. L. Raven, FEBS J., 2012, 279, 4501–4509 CrossRef CAS PubMed.
- W. M. Kazmierski, B. Xia, J. Miller, M. De la Rosa, D. Favre, R. M. Dunham, Y. Washio, Z. Zhu, F. Wang, M. Mebrahtu, H. Deng, J. Basilla, L. Wang, G. Evindar, L. Fan, A. Olszewski, N. Prabhu, C. Davie, J. A. Messer,and and V. Samano, J. Med. Chem., 2020, 63(7), 3553–3562 CrossRef PubMed.
- M. W. Pantoliano, E. C. Petrella, J. D. Kwasnoski, V. S. Lobanov, J. Myslik, E. Graf, T. Carver, E. Asel, B. A. Springer, P. Lane and F. R. Salemme, J. Biomol. Screening, 2001, 6, 429–440 CrossRef CAS PubMed.
- C. J. Layton and H. W. Hellinga, Biochemistry, 2010, 49, 10831–10841 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, 1H-NMR, 13C-NMR, LRMS and HRMS; UV-vis, LC-MS and MST assay data. See DOI: 10.1039/d0cb00209g |
| This journal is © The Royal Society of Chemistry 2021 |