
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promising nanomaterials in the fight against malaria
Livia
Neves Borgheti-Cardoso†
 abc,
María
San Anselmo†
d,
Elena
Lantero
abc,
Alexandre
Lancelot
d,
José Luis
Serrano
d,
Silvia
Hernández-Ainsa
de,
Xavier
Fernàndez-Busquets
*abc and
Teresa
Sierra
*d
abc,
María
San Anselmo†
d,
Elena
Lantero
abc,
Alexandre
Lancelot
d,
José Luis
Serrano
d,
Silvia
Hernández-Ainsa
de,
Xavier
Fernàndez-Busquets
*abc and
Teresa
Sierra
*d
aNanomalaria Group, Institute for Bioengineering of Catalonia (IBEC), The Barcelona Institute of Science and Technology, Baldiri Reixac 10-12, ES-08028 Barcelona, Spain
bBarcelona Institute for Global Health (ISGlobal, Hospital Clínic-Universitat de Barcelona), Rosselló 149-153, ES-08036 Barcelona, Spain
cNanoscience and Nanotechnology Institute (IN2UB), University of Barcelona, Martí i Franquès 1, ES-08028 Barcelona, Spain. E-mail: xfernandez_busquets@ub.edu
dInstituto de Nanociencia y Materiales de Aragón (INMA), Dep. Química Orgánica-Facultad de Ciencias, CSIC-Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: tsierra@unizar.es; tsierra@ctq.csic.es
eARAID Foundation, Government of Aragón, Zaragoza 50018, Spain
First published on 19th August 2020
Abstract
For more than one hundred years, several treatments against malaria have been proposed but they have systematically failed, mainly due to the occurrence of drug resistance in part resulting from the exposure of the parasite to low drug doses. Several factors are behind this problem, including (i) the formidable barrier imposed by the Plasmodium life cycle with intracellular localization of parasites in hepatocytes and red blood cells, (ii) the adverse fluidic conditions encountered in the blood circulation that affect the interaction of molecular components with target cells, and (iii) the unfavorable physicochemical characteristics of most antimalarial drugs, which have an amphiphilic character and can be widely distributed into body tissues after administration and rapidly metabolized in the liver. To surpass these drawbacks, rather than focusing all efforts on discovering new drugs whose efficacy is quickly decreased by the parasite's evolution of resistance, the development of effective drug delivery carriers is a promising strategy. Nanomaterials have been investigated for their capacity to effectively deliver antimalarial drugs at local doses sufficiently high to kill the parasites and avoid drug resistance evolution, while maintaining a low overall dose to prevent undesirable toxic side effects. In recent years, several nanostructured systems such as liposomes, polymeric nanoparticles or dendrimers have been shown to be capable of improving the efficacy of antimalarial therapies. In this respect, nanomaterials are a promising drug delivery vehicle and can be used in therapeutic strategies designed to fight the parasite both in humans and in the mosquito vector of the disease. The chemical analyses of these nanomaterials are essential for the proposal and development of effective anti-malaria therapies. This review is intended to analyze the application of nanomaterials to improve the drug efficacy on different stages of the malaria parasites in both the human and mosquito hosts.
 Livia Neves Borgheti-Cardoso | Livia Neves Borgheti-Cardoso graduated in Pharmacy-Biochemistry at the University of São Paulo (USP), Brazil. She got her Master (2012) and PhD (2016) degrees in Science at the USP with one-year as a PhD visitor at the University Medical Center Utrecht, The Netherlands, working on the development of organogels for the treatment of skin cancer by gene therapy. She received the Honorable Mention Award of the 2017 Capes Thesis Award, Brazil. In 2017 she joined the Nanomalaria Group as a Marie Skłodowska-Curie Actions fellow, with a research project based on the exploration of extracellular vesicles as a drug delivery system. She is currently a postdoctoral researcher at the Biomimetic Systems for Cell Engineering Group, IBEC. |
 María San Anselmo | María San Anselmo got her degree in Biochemistry at University of Zaragoza in 2014. In 2015 she studied for a Master degree in Molecular and Cellular Biology and enjoyed a research grant in Taiwan. She joined the Liquid Crystals and Polymers group in 2016, where she is developing her PhD in Chemistry at INMA in the field of dendritic nanocarriers for biomedical applications. |
 Elena Lantero | Elena Lantero is a Biotechnology graduate from the Universitat de València (2013), with a Master degree in Nanostructured Materials for Nanotechnology Applications obtained at the Universidad de Zaragoza (2015). She is currently finishing her PhD in the Nanomalaria Group focused on targeting strategies against Plasmodium, which has included two three-month research stays (2018 and 2019) at the Institute of Hygiene and Tropical Medicine (IHMT) in Lisbon. |
 Alexandre Lancelot | Alexandre Lancelot received his PhD in Chemistry in 2017 at the University of Zaragoza (Spain) where he studied dendritic materials for applications in nanomedicine. Nowadays, he is a postdoctoral researcher at the Autonomous University of Barcelona (Spain). His research interests focus on organic adhesives and coatings using green chemistry principles. |
 José Luis Serrano | José Luis Serrano has been a Professor of Organic Chemistry (University of Zaragoza) since 1996. He is director of the research group “Liquid Crystals and Polymers” (CLIP), which was recognized as Group of Excellence by the Government of Aragon. In 2017 he was awarded the highest prize of the Russian LC Society (Sodruzhestvo) “Frederiks medal” for the outstanding results in the field of Chemistry of Liquid Crystals. https://liquidcrystals.unizar.es/. |
 Silvia Hernández-Ainsa | Silvia Hernández-Ainsa completed her PhD in Chemistry in 2011 at the University of Zaragoza (Spain) in the area of dendritic materials. She then joined the University of Cambridge (UK) to work on nanopores for sensing and on DNA nanotechnology. Since 2017 she is ARAID researcher working at INMA (CSIC-University of Zaragoza). Her current research focuses on the development of DNA-based structures and other organic materials for applications in nanobiotechnology. |
 Xavier Fernàndez-Busquets | Xavier Fernàndez-Busquets graduated in Biochemistry at the Universitat Autònoma de Barcelona (1986), where he also obtained his PhD in the field of Molecular Biology (1992). Between 1992 and 2001 he held several postdoctoral positions, among which are those at the Friedrich Miescher Institut (Novartis AG, Basel) and at the Woods Hole Marine Biological Laboratory (Massachusetts, US), where he worked on the evolutionary origins of specific cell adhesion and started his training in Nanotechnology. He obtained in 2001 a 5 year tenure track Ramón y Cajal position at the Universitat de Barcelona where he led a group focused on nanotechnological approaches to amyloid diseases and malaria. He became in 2006 Associate Researcher at the Institute for Bioengineering of Catalonia (IBEC) and since 2010 he is Head of the Nanomalaria Group (IBEC/Barcelona Institute for Global Health, ISGlobal). http://www.fernandezbusquets.eu. |
 Teresa Sierra | Teresa Sierra received her PhD in Chemistry from the University of Zaragoza (Spain) and then she did a postdoctoral stay at the University of Colorado at Boulder, working on ferroelectric liquid crystals for NLO with Prof. D. M. Walba. Currently, she holds a position as Scientific Researcher at the INMA (CSIC-University of Zaragoza). Her research interests focus on exploiting liquid crystalline organization to attain functional materials and on dendritic materials for biomedical applications. |
1 Introduction
As defined by the World Health Organization (WHO), malaria is one of the infectious diseases of poverty (DoP) of global health importance in tropical and subtropical regions.1 In 2018, 93% of the malaria cases occurred in the WHO African Region, followed by 3.4% in the South-East Asian Region, and 2.1% in the Eastern Mediterranean Region.2 Although the number of infected patients and deaths caused by malaria has decreased from 251 million cases and 585![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths in 2010 to 228 million cases and 405000 deaths in 2018,2 eradication is still beyond reach, and the search for accessible and effective prevention and curing strategies along with significant investments is still a need to make it feasible.
000 deaths in 2010 to 228 million cases and 405000 deaths in 2018,2 eradication is still beyond reach, and the search for accessible and effective prevention and curing strategies along with significant investments is still a need to make it feasible.
In this context, nanoscience and nanotechnology are well placed to bring innovative solutions to the enormous challenge of winning the fight against DoP.3 Nanomaterials, which have been extensively focused on cancer-related biomedical applications for many years, are being intensively investigated to implement strategies to prevent and cure malaria, with the aim of overcoming the problems arising with conventional therapies.4–6 Furthermore, as a vector borne parasitic disease, malaria can be faced at several stages of the infection process, from the mosquito vector to the infected erythrocytes, and these constitute different challenges to generate novel antimalarial strategies. Most efforts are addressed towards targeted drug-delivery systems, mostly including known antimalarial drugs. When developing anti-infective therapeutics, two essential objectives have to be fulfilled: delivering sufficiently high local doses that will rapidly eliminate the pathogen without allowing for it to evolve resistance, and doing so with overall doses sufficiently low so not to have side effects on the human host. Both goals can be simultaneously achieved through targeted drug delivery strategies that make use of nanocarriers (NCs).7,8 Many NCs have the capacity to encapsulate different types of drugs and of being functionalized with ligands targeting them to the desired cell type. In addition, if the NCs are loaded with large numbers of drug molecules to be delivered together, their antiparasitic effect will be potentiated and they will have a higher chance to quickly kill the infectious agent. NCs can be made having long shelf half-lives, low unspecific toxicity, large drug loading capacity, extended blood circulation times and safe elimination mechanisms.
In addition to therapeutic applications, nanomaterials could be ideally adapted to the development of new prevention strategies. Considering the difficulties in obtaining an efficient malaria vaccine,9 NCs are suited to be functionalized with several antigens, which might dramatically boost the prospects of obtaining a stronger immune response than with the current classical vaccination approaches.
Mosquito vector control constitutes an essential issue for malaria prevention since it can be highly effective to prevent transmission. The use of nets treated with insecticides and indoor spraying are the most frequent protection measures. However, the mosquito can be also reached through nanomaterials. The antimicrobial activity of metals such as silver, gold or platinum can be enhanced in biosynthesized nanostructured particles, which show up as a potential prevention weapon to hamper malaria transmission.10,11
This review collects the research done on different types of nanomaterials which can be adapted to implement promising antimalarial tools with therapeutic or prophylactic roles. Before discussing the most relevant results in the literature, this review includes sections dedicated to the main characteristics of malaria infection and those challenges that nanomaterials face to constitute effective weapons against malaria.
2 Malaria
Malaria is a parasitic disease caused by Plasmodium spp. Among the more than 120 species in this genus, five of them (Plasmodium falciparum, Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, and Plasmodium knowlesi) cause malaria in humans, P. falciparum and P. vivax being those involved in global malaria.12,13P. falciparum is the species responsible for the most severe and potentially fatal form of malaria, whereas P. vivax has dormant liver stage parasites (hypnozoites) that can trigger a fully-fledged infection after months or even years remaining in a quiescent intrahepatocytic state.4Plasmodium is an intracellular parasite with a complex life cycle shared between the human host and a mosquito vector from the Anopheles genus. When an infected mosquito bites a human, Plasmodium sporozoites (Fig. 1.1) that reside in the salivary glands are released into the blood stream and reach the liver, where they infect hepatocytes and multiply inside them (Fig. 1.2). Subsequently, parasite forms called merozoites are released into the blood, where in less than a minute14 they invade red blood cells (RBCs) and mature inside them. Ring stages grow into trophozoites as they feed on hemoglobin, and later divide their nuclei to form schizonts, from which the next generation of merozoites egresses to reinvade new RBCs, thus perpetuating the so-called intraerythrocytic cycle (Fig. 1.3). These stages of the parasite are responsible for all malaria symptoms, such as periodic fevers resulting from the immune response to the parasite,15 or more severe conditions such as placental malaria16 or capillary occlusion,17 which are derived from parasite sequestration in capillaries18 and from a phenomenon called rosetting, i.e. the adhesion between parasitized RBCs (pRBCs) and naïve RBCs to form cell aggregates termed rosettes.
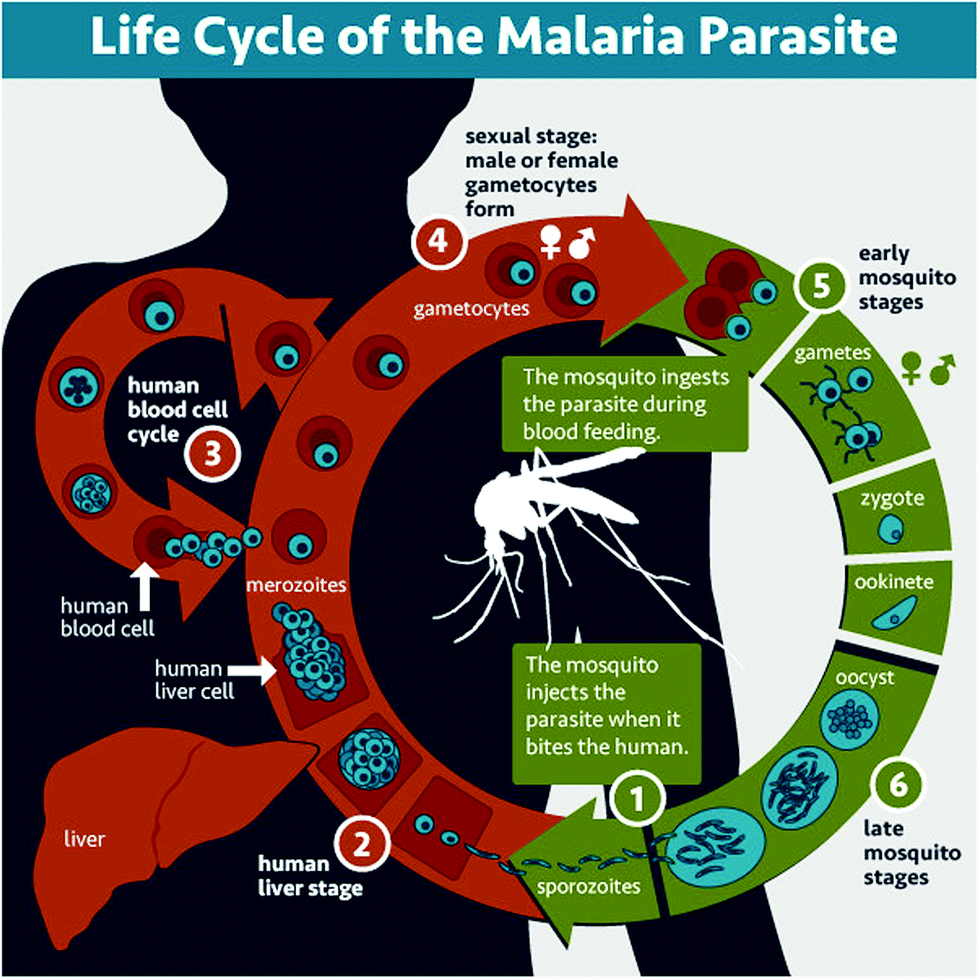 | ||
| Fig. 1 Life cycle of Plasmodium. In a mosquito bite, sporozoites are injected (1), invade hepatocytes and divide inside them (2). Then merozoites are released into the blood and invade erythrocytes, growing and reinvading new RBCs in the intraerythrocytic cycle (3). Some merozoites differentiate into gametocytes (4), which are taken in another mosquito blood meal, and egress from the pRBC, forming the gametes that fuse into the zygote (5). The zygote develops into an ookinete that traverses the mosquito midgut to form an oocyst (6) that will grow until releasing the infective sporozoites. Image courtesy of the National Institute of Allergy and Infectious Diseases (https://www.niaid.nih.gov/diseases-conditions/malaria-parasite). | ||
A small number of pRBCs differentiate in each cycle towards the formation of gametocytes (Fig. 1.4),19 sexual forms of the parasite that sequester in the bone marrow until they mature and return to the circulation. When removed with a mosquito blood meal, gametocytes egress from the pRBC and become gametes. Fertilization produces a zygote (Fig. 1.5) that becomes a motile ookinete, which leaves the blood bolus to reach and cross the mosquito midgut endothelium, where it differentiates into an oocyst (Fig. 1.6) that grows to spawn sporozoites. These close the cycle by travelling through the haemolymph towards the mosquito salivary glands, where they are ready for new inoculation into a human host (Fig. 1.1).
3 Challenges for drug delivery
3.1 Drugs
There is currently a large variety of antimalarial drugs (Fig. 2), but their application is often limited due to the development of resistance. In this regard, the WHO encourages the use of combination therapy, where two or more medicines with different mechanisms of action are used together, which helps to prevent or delay drug resistances.20 In this section, we present the most commonly used antimalarial drugs and briefly describe their distinct characteristics as well as precise anti-plasmodial use.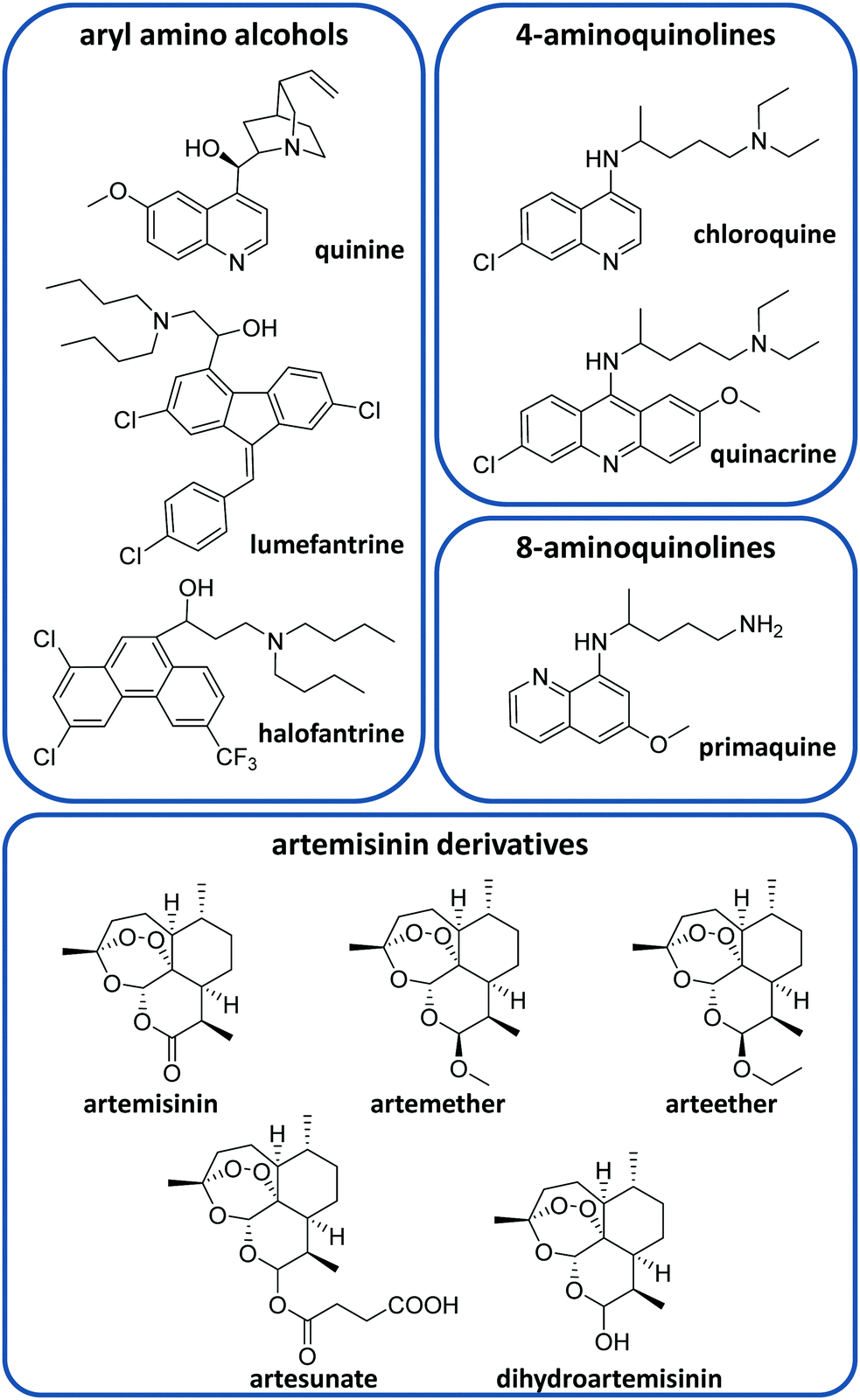 | ||
| Fig. 2 Some antimalarial drugs discussed in this review. | ||
Quinine (QN) was the first purified compound used to treat malaria and has been the most common and effective option to date.21 This drug is active against all Plasmodium spp. schizonts and the gametocytes of P. vivax, P. malariae and P. ovale but not against those of P. falciparum.20,22 Due to its reported resistance, QN is not currently used as the leading treatment but it is still employed for severe malaria.20,21 QN is also recommended together with clindamycin in the treatment of uncomplicated malaria during the first trimester of pregnancy.20,23 This drug belongs to the aryl amino alcohol pharmacophore-type like lumefantrine, mefloquine, halofantrine or quinidine. Lumefantrine is another widely used drug, which is not administered alone but in combination with artemether (an artemisinin-derivative) to treat uncomplicated P. falciparum malaria in multiple doses.20 Due to its high lipophilicity, it requires being taken up within a fatty diet, which can represent a problem due to the difficulty in poor populations of having continuous access to such a meal.20 Mefloquine is commonly used as a chemoprophylactic for all types of malaria and in combination with artesunate (ATS) to treat uncomplicated malaria.20
Other common active compounds against malaria include the 4-aminoquinolines (e.g. chloroquine, quinacrine, hydroxychloroquine, piperaquine, and amodiaquine) and the 8-aminoquinolines (primaquine, tafenoquine, and pamaquine). Chloroquine (CQ) is well absorbed from the gastrointestinal tract after oral administration. This drug is used to treat uncomplicated malaria caused by P. vivax, P. malariae and P. ovale in areas without CQ resistance.20 Primaquine (PQ) is the most remarkable drug belonging to the 8-aminoquinolines. It shows activity against P. vivax and P. ovale hypnozoites and it is generally administered together with artemisinin derivatives or CQ.20 Like other 8-aminoquinoline-type drugs, primaquine can entail risk for patients with glucose-6-phosphate dehydrogenase deficiency, which is unfortunately very common in malaria-endemic areas.24
On the other hand, ATS, artemether, arteether and dihydroartemisinin are the most common artemisinin derivatives employed to treat malaria through artemisinin-based combination therapy (ACT).21 The most frequent ACT drug combinations to treat uncomplicated malaria are artemether/lumefantrine, ATS/amodiaquine, dihydroartemisinin/piperaquine, ATS/mefloquine and ATS/sulfadoxine-pyrimethamine.25 Atovaquone/proguanil is a non-artemisinin-based combination therapy that is commonly employed for the prophylaxis of malaria and can be also used to treat CQ-resistant malaria.20,21
3.2 Antimalarial drug distribution
As the erythrocytic stage is responsible for all symptoms and pathologies of malaria, it is an obvious target for the administration of drugs.26 The bioavailability of antiplasmodial drugs is highly dependent on the administration route, the parenteral route being used for severe malaria, while oral delivery is the optimal strategy for uncomplicated malaria in order to maximize patient adherence to treatment. Some drugs like amodiaquine27 and all artemisinin derivatives except ATS28 have poor oral bioavailability, with below 35% of the ingested drug reaching the blood stream for most of them, thus requiring high dosages that could result in toxic side effects.A second challenge for proper treatment is presented by the pharmacokinetic profile of antimalarial compounds, which has led to the implementation of combination therapies where, as explained above, two drugs are delivered together, one of them being usually an artemisinin derivative. In general, artemisinin derivatives have short half-lives (t1/2)29,30 and therefore the accompanying drug is usually long-lived (t1/2 > 4 days) in order to achieve complete clearance of the parasite. This approach is, however, a matter of debate, because the time during which one of the drugs is present might be long enough to stimulate resistance evolution.31 Although CQ has an excellent absorption and circulation time profile,32 its widespread use in the past triggered the emergence of resistance.33 As a result, CQ is not widely used as first line treatment anymore, being only recommended in areas where resistances are not described in non-falciparum malaria.34
Antimalarials targeting the intraerythrocytic cycle have to face the challenge of RBC penetration. Mature erythrocytes do not endocyte,35 and drugs have to be passively absorbed to penetrate them. Although it is possible to efficiently load large amounts of drugs on RBCs ex vivo, it requires harsh manipulation such as hypotonic conditions.36 Drugs targeting RBCs should be hydrophobic to enhance their penetrability (the front-line drug artemisinin and its derivatives fulfill this requirement37), although hydrophobicity results in poor pharmacokinetics and off-target delivery, leading to accumulation in non-targeted tissues or interaction with plasma proteins.28,29
Loading drugs into RBCs and using these as vascular carriers is an attractive alternative because these cells have a large life span in the blood (of up to 120 days) and a large size, which prolongs the circulation time and permits the encapsulation of large amounts of drugs. Erythrocyte drug transporters limit extravasation, in addition to being biocompatible and safely removed from the body through natural mechanisms.38–40 As shown by Wilson et al., RBCs pretreated with the antimalarials halofantrine, lumefantrine, piperaquine, amodiaquine and mefloquine retained them inside, inhibiting the growth of the parasites that were invading drug-containing RBCs.38
3.3 Drugs reaching Plasmodium
Targeting Plasmodium in its liver phase represents an attractive prophylactic approach that would prevent the progress of the disease and its ensuing symptoms and disability caused by erythrocytic stages. Moreover, the administration of antimalarial drugs specifically to the liver could also be useful for P. vivax therapy. However, PQ is the only approved drug for hypnozoites, with the drawbacks that were discussed in Section 3.1.Administration to Plasmodium early erythrocytic stages would be an interesting strategy to allow drugs a longer time to eliminate the parasite before it completes its intraerythrocytic development. However, delivering active compounds to RBCs infected with Plasmodium early stages is a challenging task, because the permeability of pRBCs to drugs remains low until ca. six hours after invasion.40,41
To reach their molecular targets inside pRBCs, antimalarial drugs must cross at least 3 membranes (the erythrocyte membrane, the parasitophorous vacuole and the parasite membrane),42 plus one extra lipid bilayer for those compounds having their target inside organelles such as the food vacuole or the apicoplast. Lipophilic drugs are able to cross these membranes through passive diffusion, whereas other compounds are carried inside the parasite by available channels or transporters.42 During Plasmodium intraerythrocytic infection and maturation, significant alterations in the structural and functional characteristics of RBCs are observed.43,44 Namely, Plasmodium changes the lipid and protein composition of the host cell plasma membrane in order to take up nutrients from the blood circulation, dispose of metabolic waste, and create and maintain electrochemical ion gradients.26,44 Around 10 to 20 h post-invasion, these alterations result in the formation of new permeation pathways, which allow the entry into parasitized RBCs of particles up to diameters of 70 nm.26,44 One transporter that is exported by the parasite to the pRBC membrane is the plasmodial surface anion channel, which contributes to nutrient acquisition for the intracellular parasites.45 Variation or differences in expression of this channel can be a source of potential resistances to the drugs relying on it.46–48
For those drugs able to cross the erythrocyte membrane, further penetration through the pasaritophorous vacuole or parasite plasma membranes seems to occur with more facility, as both have high capacity for permeating many solutes,42 possibly to facilitate intake of nutrients into the parasite. Penetration and a long residence time in certain target organelles are also important for several drugs to exert their antiparasitic action. In the case of CQ, the main resistance mechanism consists of increasing drug efflux from the digestive vacuole,49 where CQ blocks the heme detoxification pathway.50 Mutations or gene duplications in transporter proteins can produce resistance to quinolines.49,51–53 Other biological features of Plasmodium can significantly reduce the drug amounts reaching target cells, thus favoring the evolution of resistance. In the murine malaria model Plasmodium berghei, spleen-sequestered pRBCs were less sensitive to artemisinin,54 likely because the limited diffusion of drugs into the organ resulted in final doses reaching the parasite that were much smaller than those found in the blood circulation.
4 Nanomaterials against malaria
As discussed in the following sections, nanomaterials and nanotechnology offer good opportunities for the development of customized nanocarriers capable of trapping antimalarial drugs as well as exerting targeted delivery. We include below several subsections in which antimalarial nanomaterials of different nature are discussed. Namely, nanocarriers built with lipids, polymers and dendrimers as well as inorganic NPs are presented. In the final subsection, we describe some other molecular platforms that do not belong to the previous categories but also display interesting potential for innovative antimalarial strategies.4.1 Liposomes
Liposomes are artificial lipid bilayer vesicles containing an aqueous core formed by the self-assembly of phospholipids in aqueous media.4,55–57 Since their description in 1965, they have been extensively and successfully used as a drug delivery system. Due to their dual hydrophilic/hydrophobic nature, they can encapsulate water-soluble drugs in the aqueous core and lipophilic compounds in the phospholipid bilayer.4,57 Liposomes are versatile NCs with diverse properties depending on the size, phospholipid composition (Fig. 3), surface charge, and functionalization with diverse ligands.58,59 This versatility permits modulating their structure to protect drugs from chemical degradation, increase their solubility, prolong and sustain drug release, reduce toxicity and target cells specifically.60–62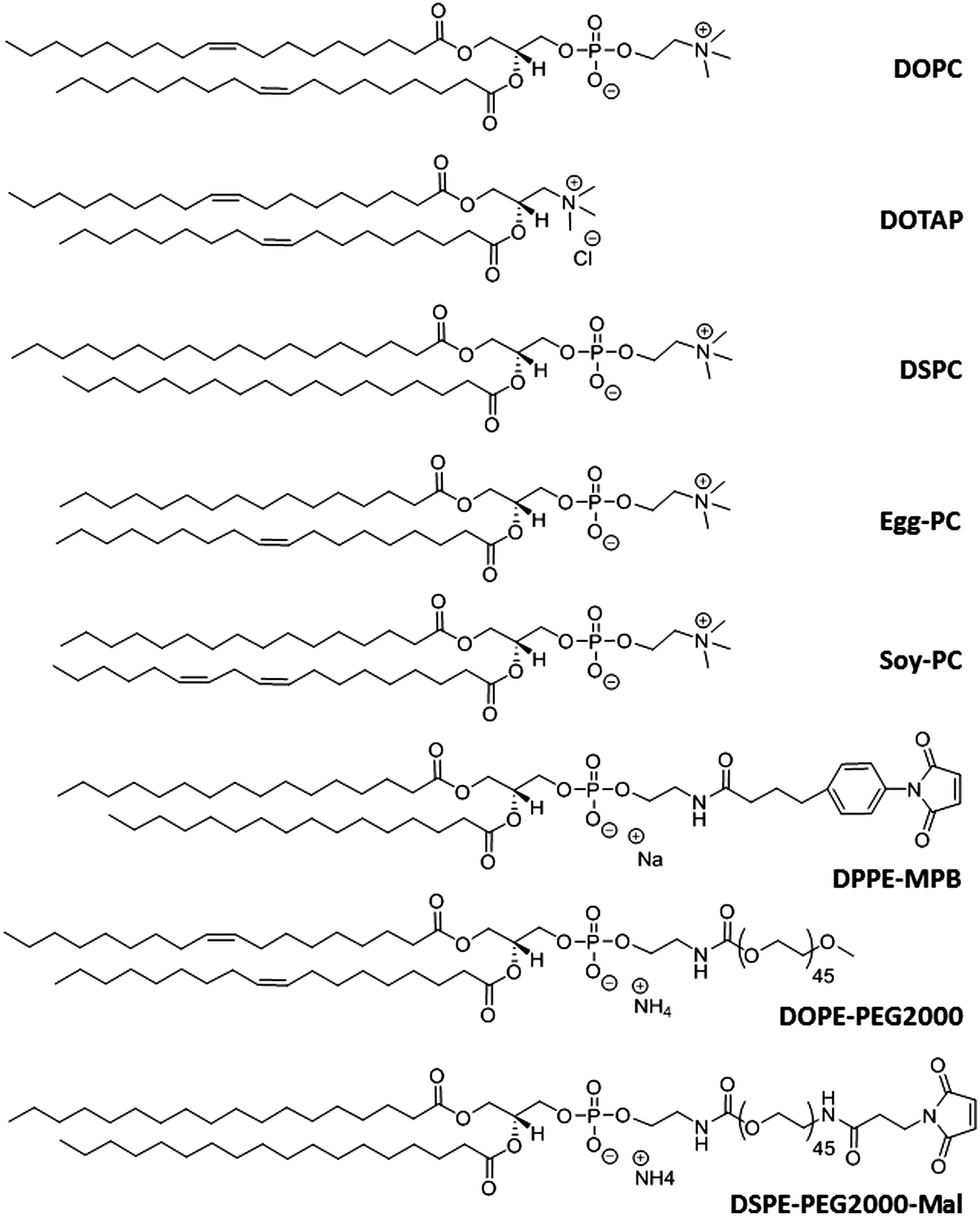 | ||
| Fig. 3 Chemical structure of some phospholipids used in liposome formulation for malaria therapy. 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), L-α-phosphatidylcholines (Egg-PC and Soy-PC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidophenyl)butyramide] (DPPE-MPB), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DOPE-PEG2000), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-Mal). | ||
Liposomes have been successfully administered by parenteral routes, but their oral administration is hampered by several obstacles such as instability in the gastrointestinal tract and difficulties in crossing biomembranes. By modifying the compositions of the lipid bilayers and adding polymers or certain ligands, both the stability and permeability of liposomes can be significantly improved for oral drug delivery.63 For instance, Coma-Cros et al. used the anionic copolymer Eudragit® S100 to assemble liposomes incorporating curcumin and containing either hyaluronan (Eudragit-hyaluronan liposomes) or the water-soluble dextrin Nutriose® FM06 (Eudragit-nutriosomes). As a result, curcumin incorporated into Eudragit-nutriosomes enhanced its antimalarial activity upon oral administration to Plasmodium yoelii-infected mice. Eudragit-nutriosomes presented better performance than Eudragit-hyaluronan liposomes due to the greater stability of the former, as shown by the vesicle behavior in gastrointestinal fluids.64
Several studies have tried to target liposomes encapsulating PQ to the liver stage of malaria.65 In P. berghei infections in mice, liposomes could decrease the toxicity of parenteral PQ delivery, which allowed for the administration of higher doses and resulted in the cure of the animals following a single intravenous injection.66 Tissue distribution studies demonstrated that PQ encapsulated in liposomes accumulated mainly in the liver and spleen and not so much in the kidneys, lungs, heart, or brain, being eliminated more slowly than its free counterparts.67,68 Although peptide- and amino acid-conjugated derivatives of PQ encapsulated in liposomes did not increase the therapeutic effect in comparison to free PQ,66 they presented a reduction of toxicity.68 In another study, liposomes loaded with antimalarial drugs (especially PQ) were engineered to contain a galactosyl glycolipid that specifically targeted a galactose-binding lectin in hepatocytes. These experiments showed a remarkable phenomenon: drug-free liposomes had a curative effect on sporozoite-induced murine malaria, likely due to the competition of liposomes and sporozoites for the same receptor.69 In addition, considering the reduced doses of PQ required to achieve a curative effect, it was estimated that the efficacy of PQ was enhanced more than 46000 times by its encapsulation into liposomes containing the galactosyl glycolipid.70
To specifically target the liver in vivo, Longmuir et al. formulated a liposome bearing a 19-amino acid sequence present at the surface of the P. berghei circumsporozoite protein. This liposome successfully reached the liver following intravenous administration in mice.71 Despite these promising results, liver-targeted-liposomes have not progressed towards a strategy to fight malaria, probably because of the lack of sufficiently specific markers for Plasmodium-infected hepatocytes.43 As a consequence, most current studies using liposomes to deliver antimalarial drugs target the erythrocytic stage.
Essentially, liposomes can be developed to deliver drugs to the pathogen's erythrocytic forms at three different times: (i) before parasite infection, (ii) to RBCs infected with Plasmodium early stages, and (iii) to RBCs infected with Plasmodium late stages. The administration of a drug for each of these phases has its own advantages and challenges. Regardless of the time when liposomes will deliver the drug, the lack of endocytic processes in RBCs is one of the main obstacles to be dealt with. In a proof of concept study, quantum dots encapsulated in liposomes allowed the visualization of a homogeneous distribution of liposome content within P. falciparum-infected RBCs. This indicated that the liposomal cargo entered the cell through fusion of the liposome bilayer with the cell plasma membrane.72 It has been reported that the fusion of liposomes with mammalian cells is significantly dependent on the lipid composition and fluidity of the liposome membrane.44 For instance, the efficacy of stearylamine in inhibiting the growth of P. falciparum was reduced when incorporated in sphingomyelin liposomes compared to either egg phosphatidylcholine (Egg-PC) or soy phosphatidylcholine (Soy-PC) liposomes. This probably occurred due to reduced fusion/interaction of pRBCs with rigid sphingomyelin-containing liposomes compared to fluid Egg-PC or Soy-PC liposomes.44 Thus, to favor fusion events, it is advisable to formulate liposomes with relatively fluid lipid bilayers, although this incurs the risk of leaking for small drugs encapsulated in the liposomal aqueous core. On the other hand, liposomes formulated with saturated lipids retain drugs better because they have less fluid bilayers, but at the cost of decreased membrane merging with target cells.39 In both cases, the amount of delivered drug will get reduced, and a balance must be found to limit drug leakage while allowing lipid bilayer fusion events.
Antibodies have been successfully used to target pRBCs.43,73–76 Antibody-targeted liposomes containing on their surface F(ab′)2 fragments of a mouse monoclonal antibody raised against P. berghei-infected mouse erythrocytes significantly increased the therapeutic efficacy of CQ, showing that target-specific liposomes can cure CQ-resistant malarial infections.73 Urbán et al. also observed an improvement of up to tenfold in the therapeutic effect of CQ when delivered inside liposomes covalently functionalized with oriented, specific half-antibodies against P. falciparum late form-infected RBCs.43 Further studies showed that this same targeted liposome similarly improved the in vitro efficacy of fosmidomycin, another antimalarial drug.74 Immunoliposomes formulated using as a targeting molecule a polyclonal antibody against the NTS-DBL1α N-terminal domain of P. falciparum erythrocyte membrane protein 1 (PfEMP1) and encapsulating lumefantrine were also used to specifically recognize and disrupt rosettes while simultaneously eliminating those pRBCs forming them.76 As shown by these studies, antibody-functionalized liposomes can discriminate pRBCs from non-infected RBCs and specifically deliver antimalarial drugs to pRBCs. However, some issues limit the use of antibodies as targeting molecules in antimalarial therapy, such as the elevated cost of production,77 the high immunogenicity unless genetically engineered humanized forms are used, and the potential decrease in targeting efficiency due to the variability of Plasmodium proteins exposed on pRBC surfaces.26
Past research has paid little attention to the development of liposomes to deliver a drug to RBCs before Plasmodium infection; however, this strategy could represent an interesting prophylactic approach that deserves further study. Moles et al. developed an immunoliposome bearing antibodies against the RBC surface protein glycophorin A that targeted all RBCs, both naïve and Plasmodium-infected.39 In addition to targeting the parasite, this approach permitted loading of drugs into RBCs prior to infection. To improve the targeting properties, a lipid bearing a 2 kDa polyethylene glycol (PEG) linker terminated with a maleimide group (Mal) was included in the liposome formulation aiming to facilitate antigen access to targeting antibodies (Ab) and to enhance the coupling reaction sterically hindered in the original immunoliposome by the polar heads of surrounding lipids. Three types of liposomes (LP) were designed and the linker was used for the oriented binding of half-antibodies through their free thiols (LP-PEG-Mal-Ab) or of whole antibodies through their carbohydrate (CHO) moieties present in antibody Fc regions (LP-PEG-Mal-CHO-Ab), and for non-oriented binding through primary amino groups on Ab amino acid residues (LP-PEG-Mal-NH2-Ab). The type of binding influenced the cell targeting efficiency (LP-PEG-Mal-NH2-Ab ≫ LP-PEG-Mal-CHO-Ab ≫ LP-PEG-Mal-Ab).39 These glycophorin A-targeted liposomes loaded with CQ were highly efficient in transferring the drug into RBCs, as demonstrated by a complete inhibition of in vitro parasite growth after only 15 min of incubation with the immunoliposomized drug, whereas after the same time an equal amount of free CQ had no observable effect on the parasite viability. In this scenario, the parasite encountered the drug immediately after entering the host cell, which drastically compromised its survival capacity.39
Although the administration of active compounds to RBCs is a promising prophylactic strategy, some requirements have to be considered, including (i) determination of a maximum immunoliposome concentration in vivo to avoid RBC agglutination,39 (ii) restriction to drugs that do not interfere with RBC physiology,78 and (iii) use of liposome formulations that will merge with the RBC before the cell is removed by the spleen due to alterations induced by liposomes adsorbed on its surface.39
Since only after around 24 h post-invasion will Plasmodium export a substantial number of receptors and transporters to the host cell plasma membrane, the design of liposomes specifically targeted to early blood stages has remained remarkably elusive.40 Most published studies report the development of liposomes for delivering an active compound to late parasite stages,26,39,43,60,73–76,79–84 when significant alterations in the structural and functional characteristics of RBCs are observed.43,44 These liposomes have been formulated to deliver a high amount of drug to the targeted site to compensate for the short time available to kill the parasite before its egress. Different strategies can be used alone or in combination to guarantee an optimal therapeutic drug dose (as depicted in Fig. 4), including approaches to improve the encapsulation efficiency,39,75,83 to efficiently target RBCs infected with Plasmodium late stages26,43,73–76,84 and to increase the circulation time of liposomes.80–82 In addition, combination therapies contemplating the simultaneous administration of two or more active compounds have also been proposed.60,79 The encapsulation efficiency in liposomes of many antimalarial drugs can be increased using active loading techniques based on pH gradients.39,75,83 Drugs with amphiphilic nature such as CQ and PQ, which depending on the solution pH present different degrees of protonation, can be efficiently encapsulated using a pH gradient.75,83 The majority of antimalarial drugs under development are lipophilic with poor plasma solubility and large biodistribution volumes and, consequently, with low accumulation in RBCs.57,75 Active encapsulation in liposomes using a citrate-buffered pH gradient allowed for the stable incorporation of hydrophobic drugs containing weakly basic ionizable moieties, which accumulate in the inner liposome membrane leaflet. Aryl amino alcohol- and 4-aminoquinoline-type drugs were efficiently delivered to P. falciparum-infected RBCs upon encapsulation into liposomes targeting all RBCs, inhibiting parasite growth in vitro and exhibiting considerable improvements in drug activity when compared to the free administered drug after only 15 min of exposure to P. falciparum cultures.75
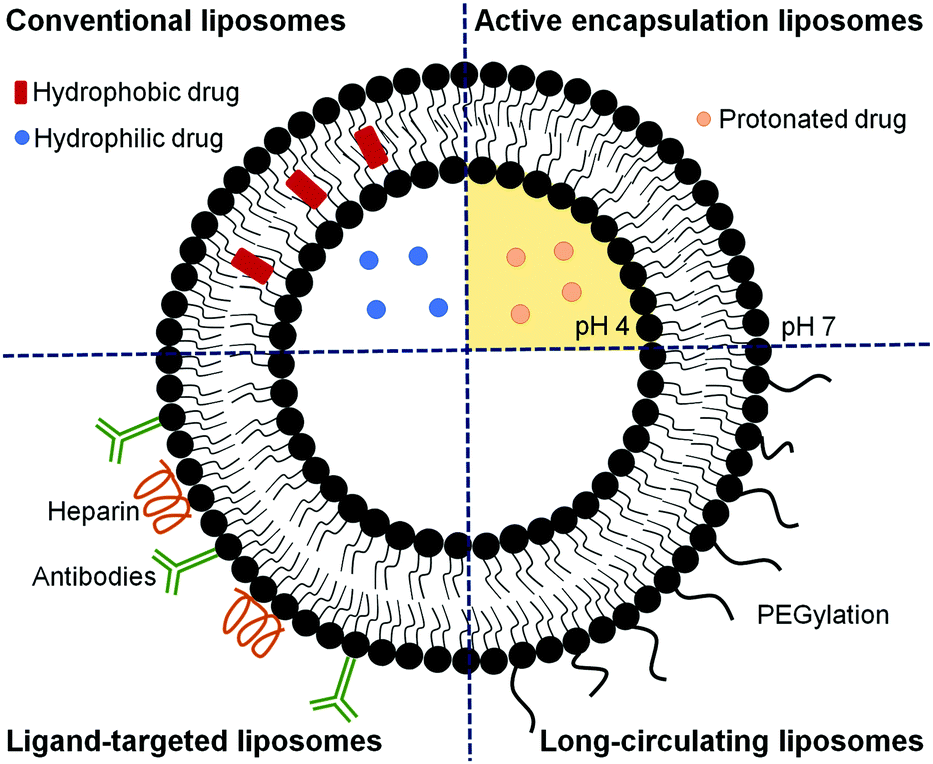 | ||
| Fig. 4 Schematic representation of liposomes formulated using different strategies aiming at improving the therapeutic efficacy of encapsulated drugs. Conventional liposomes are formed by phospholipids and cholesterol and can encapsulate hydrophilic drugs in their aqueous core and hydrophobic compounds in their phospholipid bilayer. Active encapsulating liposomes hold a pH gradient that can improve the loading of drugs with amphiphilic nature, which, depending on the pH, are found in their protonated or deprotonated forms. Ligand-targeted liposomes are developed using specific pRBC ligands such as antibodies or heparin and promote the delivery of high drug doses with few side effects. Long-circulating liposomes can be formulated by modifying the surface of liposomes with PEG to enhance the blood residence time. | ||
As interesting alternatives to antibody-mediated targeting, certain glycosaminoglycans like heparin, heparan sulfate and chondroitin sulfate84 are being recognized as one of the main pRBC-binding molecules.26 Heparin bound to liposomes has dual activity as a pRBC targeting molecule acting mainly on trophozoites of some PfEMP1-expressing lines,85 and on schizont stages,86 and as an antimalarial drug it blocks merozoite invasion of erythrocytes.87 In addition, heparin is significantly less expensive to obtain than monoclonal antibodies, resulting in heparin-liposomes having a cost about ten times lower than immunoliposomes of similar targeting activity.84 In addition, and most importantly, no resistance to heparin as an antimalarial drug has been described so far.87 Furthermore, it has been shown that the anticoagulant activity of heparin when covalently bound to liposomes was substantially reduced.84 Heparin used at non-anticoagulating concentrations electrostatically adsorbed onto positively charged liposomes enhanced threefold the activity of encapsulated PQ in in vitro P. falciparum cultures.
Heparin-related polysaccharides such as heparan sulfate88,89 and chondroitin sulfate90 could also be used as targeting molecules and, compared with heparin, they have the advantage of having a much lower anticoagulant activity.84 Marques et al. used single-molecule force spectroscopy to measure the binding forces between chondroitin 4-sulfate and pRBCs or non-infected RBCs deposited on poly-L-lysine-coated glass slides.84 Although specificity of adhesion of these polysaccharides to pRBCs has been observed, challenges related to the attachment of a ligand on the liposome surface in a reproducible manner under good manufacturing practice conditions should be considered and stability requirements have to be optimized.91
Long-circulating liposomes can considerably enhance the blood residence time, improve the release profile of the drug at the site of infection, and facilitate a better interaction with infected RBCs, resulting in the inhibition of parasite growth to a higher extent.4,80 It has been shown that the interaction of PEG-grafted liposomes with pRBCs depends on the density of the polymer and the optimal chain length.80 Rajendram et al. demonstrated that the circulation life of the drug monensin can be modulated using PEG-functionalized lipids and liposomes modified with different densities of distearoyl phosphatidylethanolaminemethoxy-PEG 2000 (DSPE-mPEG-2000), which enhanced the antimalarial efficacy of liposomal formulations compared with that of free monensin. This study also reported that the coadministration of stearylamine, monensin, and artemisinin with 5% DSPE-mPEG-2000 could completely eliminate the parasite burden.80 It has also been shown that the antiplasmodial activity of stearylamine in liposomes has the maximum effect when delivered through Soy-PC liposomes. The inhibition of parasite growth was regulated by the chain length of the alkyl group and the density of stearylamine in liposomes. In addition, the incorporation of either cholesterol or DSPE-mPEG-2000 in Soy-PC-stearylamine liposomes improved their efficacy.44 Liposomes were also used to improve the short half-life and poor bioavailability of artemisinin81 and its derivatives, such as ATS.82 Artemisinin loaded in conventional and PEG-containing liposomes showed a much longer blood circulation time than free artemisinin, being detectable 3 and 24 h after administration, respectively, while free artemisinin was rapidly cleared from the plasma and barely detected 1 h after administration.81 Hydrophobic ATS was conjugated with hydrophilic glycerophosphorylcholine as a linker to synthesize an ATS-based dimeric conjugate. The conjugate was assembled into liposomes with a high drug loading of 77.6%, exhibiting a longer blood half-life and superior in vitro antimalarial activity compared to free ATS and conventional ATS-loaded liposomes, without causing hemolysis.82
The simultaneous administration of several drugs usually improves their separate antimalarial effects,60,92 and liposomes, due to their structure, are ideally suited for such combination therapies at the nanoscale.77,84 Curcuminoids, which are poorly water-soluble compounds with promising antimalarial activity, were loaded into liposomes for intravenous administration and combined with the intramuscular administration of α/β artemether, which is known to have a short half-life in the body. This combination therapy was able not only to cure P. berghei-infected mice but also prevented recrudescence.60 Nevertheless, combined drugs exhibit different pharmacokinetic profiles and are often eliminated from the circulation at different rates, which decreases the treatment efficacy. However, if drugs are delivered encapsulated together, they will be removed from the organism simultaneously with their carrier, thus sustaining constant relative ratios throughout the treatment. Based on this concept, a glycophorin A-targeted immuno-PEG-liposome was formulated to encapsulate the hydrophilic antimalarial pyronaridine in its aqueous core and the lipophilic compounds domiphen bromide or atovaquone in its lipid bilayer. When tested in in vitro P. falciparum cultures, this strategy offered antiparasitic activities significantly higher than when the drugs were administered together in their free forms.79
Despite the considerable therapeutic advantages of liposomes, their clinical translation to antimalarial treatments has not progressed as quickly as the encouraging positive preliminary results predicted. The main issues behind this delay are pharmaceutical manufacturing limitations, government regulations and intellectual property.93
4.2 Polymers
This section includes relevant studies reported in recent years focused on polymeric nanostructures developed for drug delivery systems, polymer-based prodrugs and also prophylactic tools.94 The last sub-section is devoted to dendrimers as a unique family of hyperbranched polymers with special features for drug conjugation.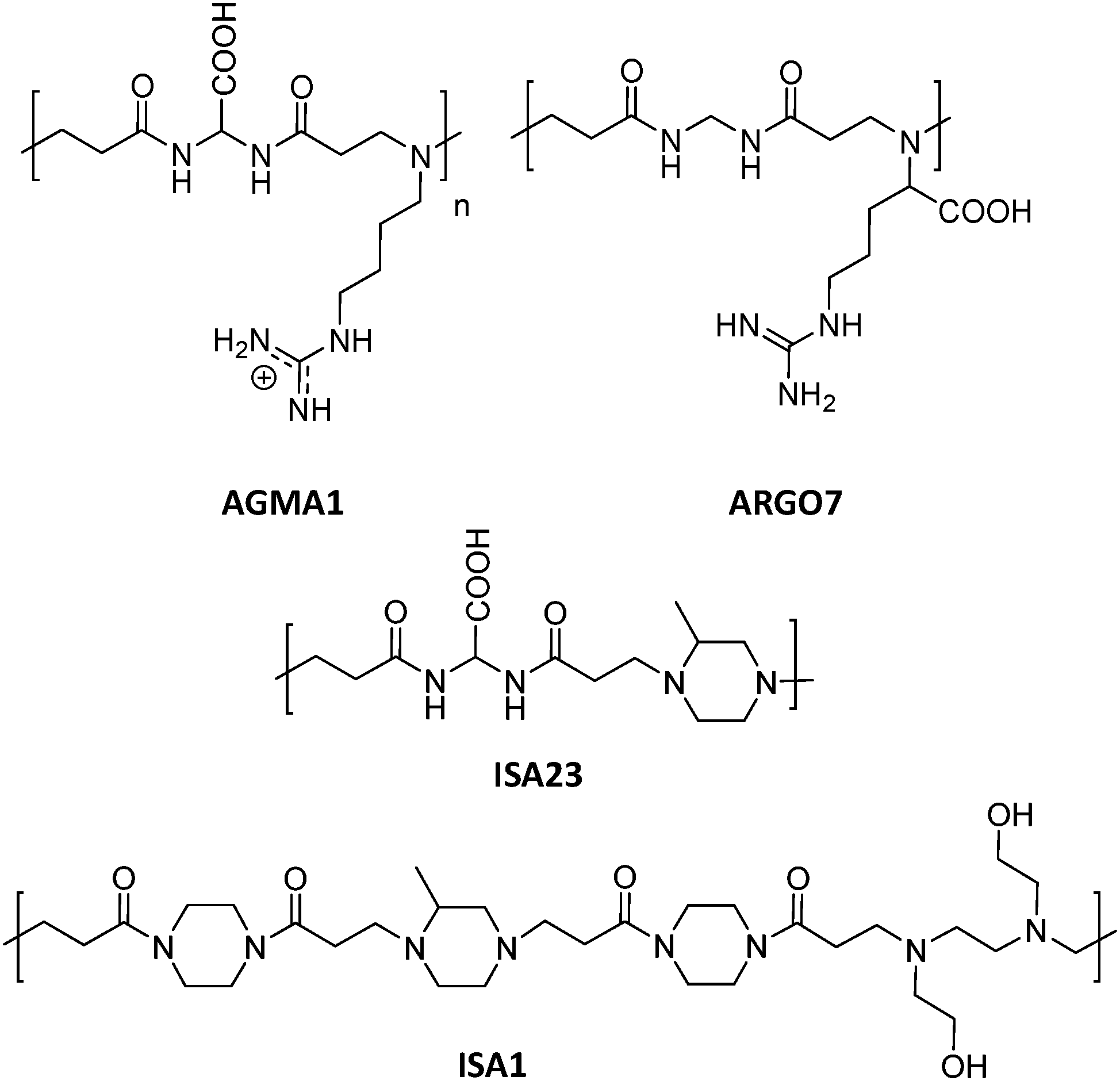 | ||
| Fig. 5 Chemical structures of AGMA1, ISA23, ISA1 and ARGO7.95,96 | ||
Biodegradable synthetic polymers such as poly(D,L-lactic-co-glycolic acid) (PLGA), polyalkylcyanoacrylates or poly(D,L-lactic acid) have been broadly used to prepare polymeric NPs.6,98 Some studies with PLGA NPs included the synergistic co-delivery of CQ and the antibiotic azithromycin99 (the most potent antimalarial macrolide widely studied in combination with other antimalarial drugs to improve their efficacy100) and the co-encapsulation of ATS and curcumin101 as promising combination therapies for malaria.
Another possibility explored is the use of nanocapsules composed of an oil core surrounded by a polymeric membrane. One example is found in the work of Haas et al., where poly(ε-caprolactone) (PCL) and polysorbate 80 (also called Tween 80) nanocapsules were used to deliver QN.102 Similarly, the antimalarial halofantrine was encapsulated into PCL nanocapsules, which resulted in an improvement of the cardiovascular parameters frequently altered by this drug.103 In this line, Velasques and co-workers developed polysorbate 80-coated PCL nanocapsules to co-encapsulate QN and curcumin, increasing the efficacy against P. falciparum at the same time that the cytotoxicity was reduced.104
A series of three hydroxypropylmethylcellulose (HPMC) polymers with different grades of acetyl (A) and succinyl (S) functionalization (HPMC-AS, Fig. 6), and the amphiphilic block copolymer PCL-block-poly(ethylene glycol) (PCL-b-PEG) were tested to improve the oral bioavailability of the synthetic antimalarial artefenomel mesylate (or OZ439). Specifically, the drug was coupled with an anionic species to form a hydrophobic ion pair complex,105 in an encapsulation process that was done through a scalable and cheap method called flash nano precipitation. Polymers with higher succinate contents displayed larger negative charge, whereas the introduction of acetyl moieties resulted in more hydrophobicity. Polymers with larger percentages of either succinate or acetyl did not show enhanced encapsulation efficiency, suggesting that both hydrophobic and ionic interactions (succinate groups on HPMC-AS) were required for stability. This was also demonstrated by the lack of stability provided by the non-charged amphiphilic PCL-b-PEG. Release experiments were done mimicking oral administration and in vivo conditions. These polymer-based NPs exhibited a sustained OZ439 release profile and significantly higher drug bioavailability compared to that of the unencapsulated drug.
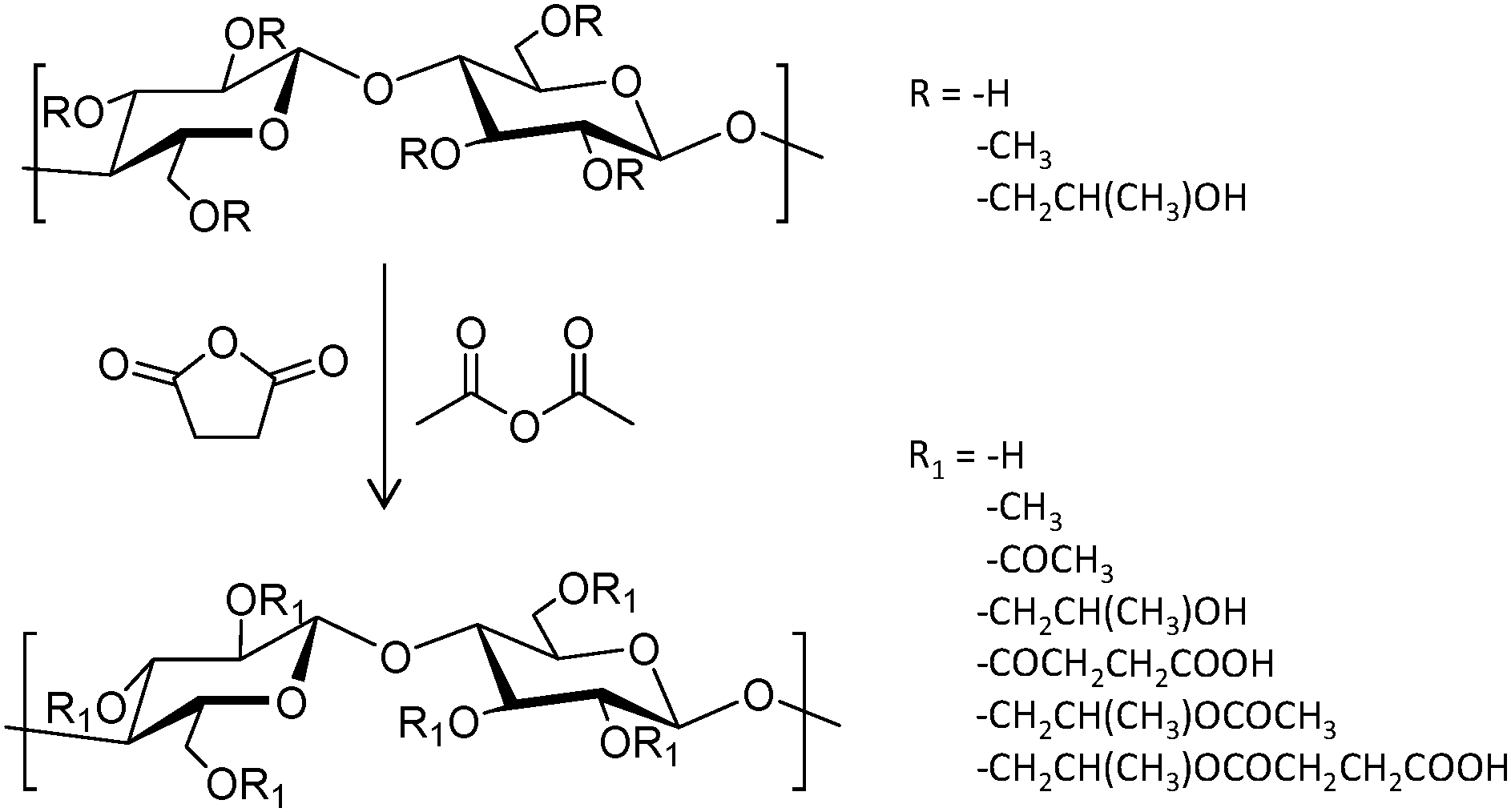 | ||
| Fig. 6 General reaction scheme to convert HPMC polymers to HPMC-AS through the addition of acetyl and succinyl groups.105 | ||
Antisense oligodeoxynucleotides (ODN) represent an alternative to traditional antimalarial drugs. However, their low stability in plasma can hamper their therapeutic function. The potential of the positively charged chitosan polymer to stabilize ODN through the formation of polyplexes was explored by Foger et al., enabling ODN to show optimal interference with the expression of Plasmodium topoisomerase II, an enzyme necessary for parasite division after invasion of human erythrocytes.106 Further studies employed this natural aminoglucopyran to covalently conjugate artemisinin (Fig. 7), obtaining more soluble derivatives.107 This original strategy consisting of the covalent bonding of the drug to a polymer carrier was proposed in 1975 by Helmut Ringsdorf.108 Generally, an enzyme or pH-sensitive linker is employed to enable drug release once in the intracellular environment.24
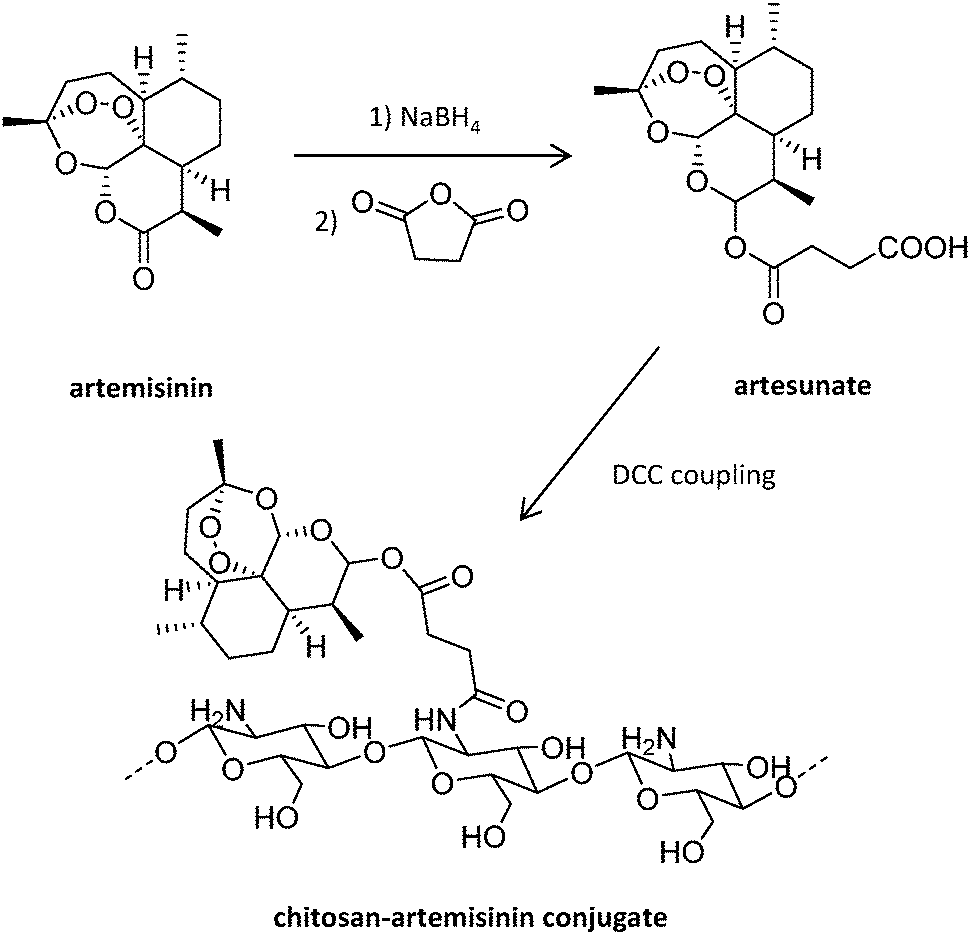 | ||
| Fig. 7 Synthesis of chitosan-artemisinin derivatives.107 | ||
Despite the scarce exploration of this strategy in the field of infectious diseases caused by a parasite, its successful use for leishmaniasis treatment109,110 encouraged its application to malaria since both pathologies share many features.24 PQ represents the most widely studied antimalarial drug within the polymer–drug conjugation approach. Already in 1986, PQ modified with a pendant cysteine group was linked through a disulfide bond to a protein carrier containing (pyridyldithio)propionate groups.111 More recently, PQ was also conjugated to poly-γ-glutamic acid (PGA) modified with a synthetic trivalent glyco-ligand (TriGalNAc) through an amide linkage, obtaining a TriGalNAc-PGA-PQ conjugate.112
This covalent approach has been also used for the conjugation of more than one drug onto the same polymer, a desirable strategy for combination therapy. An example is found in Kumar and co-workers’ poly(organophosphazenes), an inorganic polymer designed as a nanocarrier for the co-delivery of PQ and dihydroartemisinin.113,114 Firstly, the polymeric backbone of poly(dichlorophosphazene) was functionalized with various substituents like isopropyl alcohol, 4-hydroxyacetanilide, 4-hydroxybenzaldehyde and ethyl 4-aminobenzoate, providing a wide variety of physicochemical properties. Then, the remaining unsubstituted polymer backbone was substituted stepwise with the two drugs to provide the polymer-linked combined antimalarial drug conjugates. All the prepared NPs showed negative surface charge (between −21 and −32 mV) and a diameter between 128 and 179 nm, and their antimalarial activity was improved compared to the standard drug combination.
Polymer NCs have been also investigated to improve the action of the antimalarial drug atovaquone, creating long-acting injectable formulations. Bakshi et al. prepared a library of solid drug NPs containing atovaquone and a binary combination of surfactant and polymer, including e.g. poly(ethylene oxide)80-block-poly(propylene oxide)27-block-poly(ethylene oxide)80 (Pluronic F68), poly(vinylpyrrolidone) (PVP K30), HPMC, polyvinyl alcohol (PVA), and PEG.115 The most promising chemoprophylactic activity was shown for two nanoformulations containing the nonionic hydrophilic polymers PVP K30 and PVA.
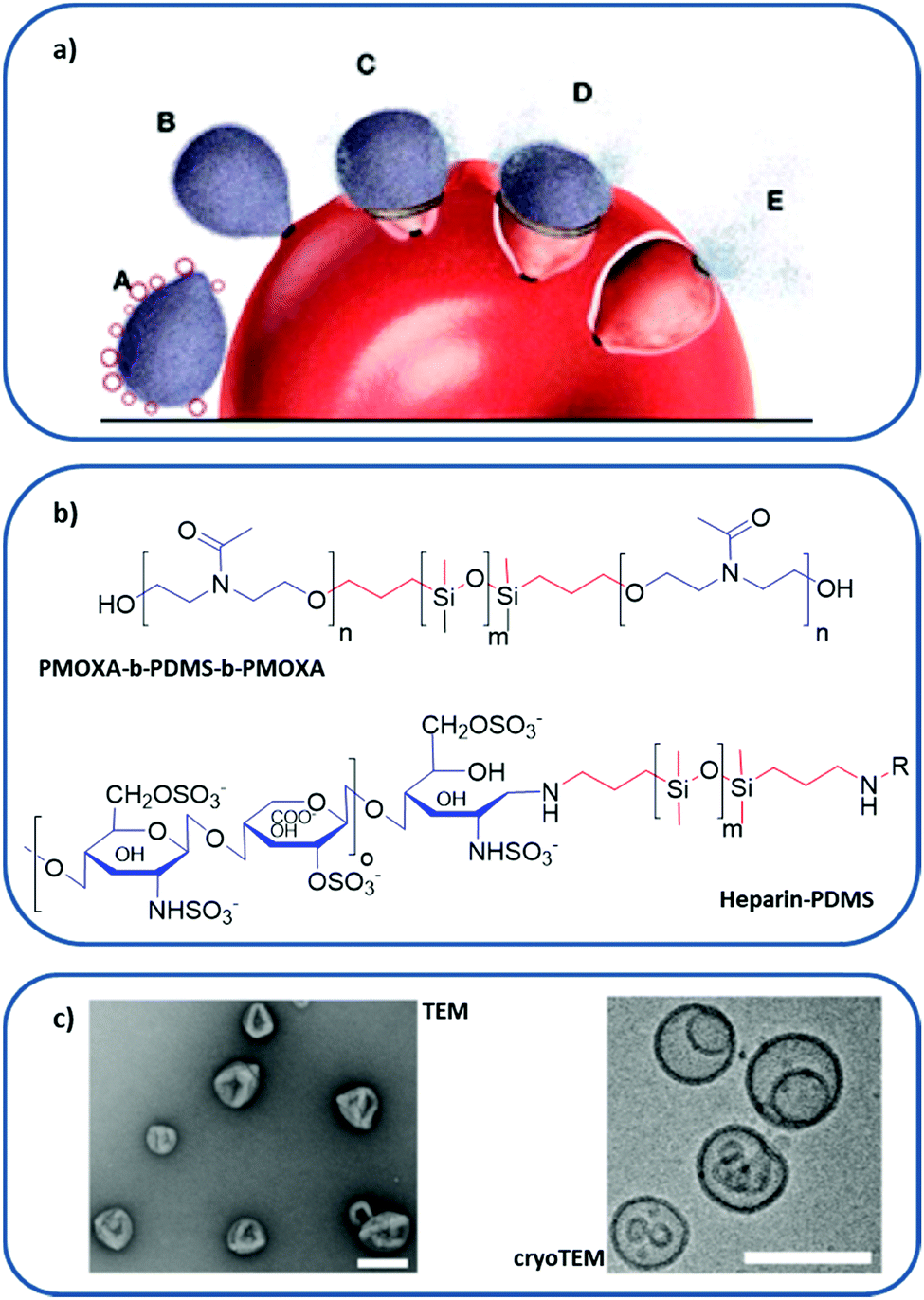 | ||
| Fig. 8 Polymer-based RBC membrane nanomimic concept and characteristics: (a) schematic representation of the nanomimic concept. In step A, fresh nanomimic structures (red circles) block merozoites (gray oval structures) before they enter new RBCs (steps B, C, D, and E). (b) Chemical structure of PMOXA-b-PDMS-b-PMOXA and PDMS–heparin, where hydrophilic domains are colored in blue and hydrophobic cores in red. (c) TEM and cryo-TEM images of the nanomimics; scale bars: 200 nm. (a and c) reprinted (adapted) with permission from Najer et al.;116 copyright (2014) American Chemical Society. | ||
The design of nanomimics involved the initial synthesis of two different block copolymers: poly(2-methyl-2-oxazoline)-block-poly(dimethylsiloxane)-block-poly(2-methyl-2-oxazoline) (PMOXA-b-PDMS-b-PMOXA), which forms a polymersome, and the PDMS–heparin block copolymer, with the chain of the receptor heparin as a hydrophilic domain (Fig. 8b). Then, the nanomimic 3D structure (Fig. 8c) was obtained by bulk rehydration of a mixture of PMOXA-b-PDMS-b-PMOXA with 25% (w/w) PDMS–heparin. The resulting self-assembled structure exposed most of the heparin part outwards, making it accessible to bind parasite ligands involved in the initial attachment to host cells. These nanomimics have displayed a therapeutic effect without the need for any other substance by blocking invasion of host RBCs by the P. falciparum merozoite. Furthermore, they can also boost immune responses due to the increased number of free merozoites circulating, thus acting as vaccine adjuvants.
In a further study,117 the antimalarial activity of different nanomimics constructed by varying the number or length of heparin molecules on their surface was evaluated. Specifically, their interaction with a well-known heparin-binding protein on the surface of merozoites, P. falciparum merozoite surface protein 1, was measured, showing high binding affinity. These findings involving malaria parasite receptor molecules are very relevant for the optimization of future nanomimic therapies for infectious diseases.
An example of using hydrogels as antimalarial nanocarriers is the gum acacia-based hydrogels as a controlled dual-drug delivery system for 4-aminoquinolines and curcumin.119 The drug release profile of this system showed dependence on the degree of crosslinking of the hydrogel network and exhibited different pharmacokinetics for both drugs, which may contribute to overcoming undesirable drug resistance. Another study developed by Aderibigbe120 comprised soy protein isolate-carbopol-polyacrylamide-based hydrogels, which exhibited pH sensitivity and were biodegradable after drug release. In this case, the hydrogels were loaded with CQ diphosphate and curcumin and their release mechanism was also appropriate as dual drug delivery systems. More recently, Dawre et al.121 formulated biodegradable hydrogels formed in situ taking advantage of the rapid phase transition of lyotropic liquid crystals in an environment that mimics intracellular conditions. A mixture of oleic acid, dibutyl sebacate and polysorbate 80 composed the lyotropic liquid crystalline preconcentrates, into which the antimalarials arteether and lumefantrine were dissolved. The addition of the biodegradable copolymer PLGA allowed a higher than threefold enhancement of the hydrogel strength and sustained release of the drugs. In addition, modified in vivo Peter's four day suppressive antimalarial assay122 and a clinical simulation study revealed a rapid reduction in parasitemia levels with no recrudescence and complete mice survival when using this efficacious therapeutic system.
In 2005, a poly-L-lysine (PLL) dendrimer (Fig. 9a) modified with PEG was first employed to improve the solubility of lipophilic artemether in water, boosting at the same time its controlled release in vitro.124 Dendrimers coated with sugars, commonly called glycodendrimers, were later used to encapsulate CQ and PQ. A galactose-coated polypropylene imine dendrimer125 and galactose-coated PLL126 could encapsulate PQ and CQ, respectively, enhancing their blood circulation time in vitro and in vivo. Chondroitin sulfate A- and PEG-coated PLL increased the stability of CQ in blood as well as its circulation time in vivo.127 In all cases, the addition of sugar moieties at the dendrimer's periphery led to higher drug encapsulation levels while reducing the toxicity of the nanocarriers.
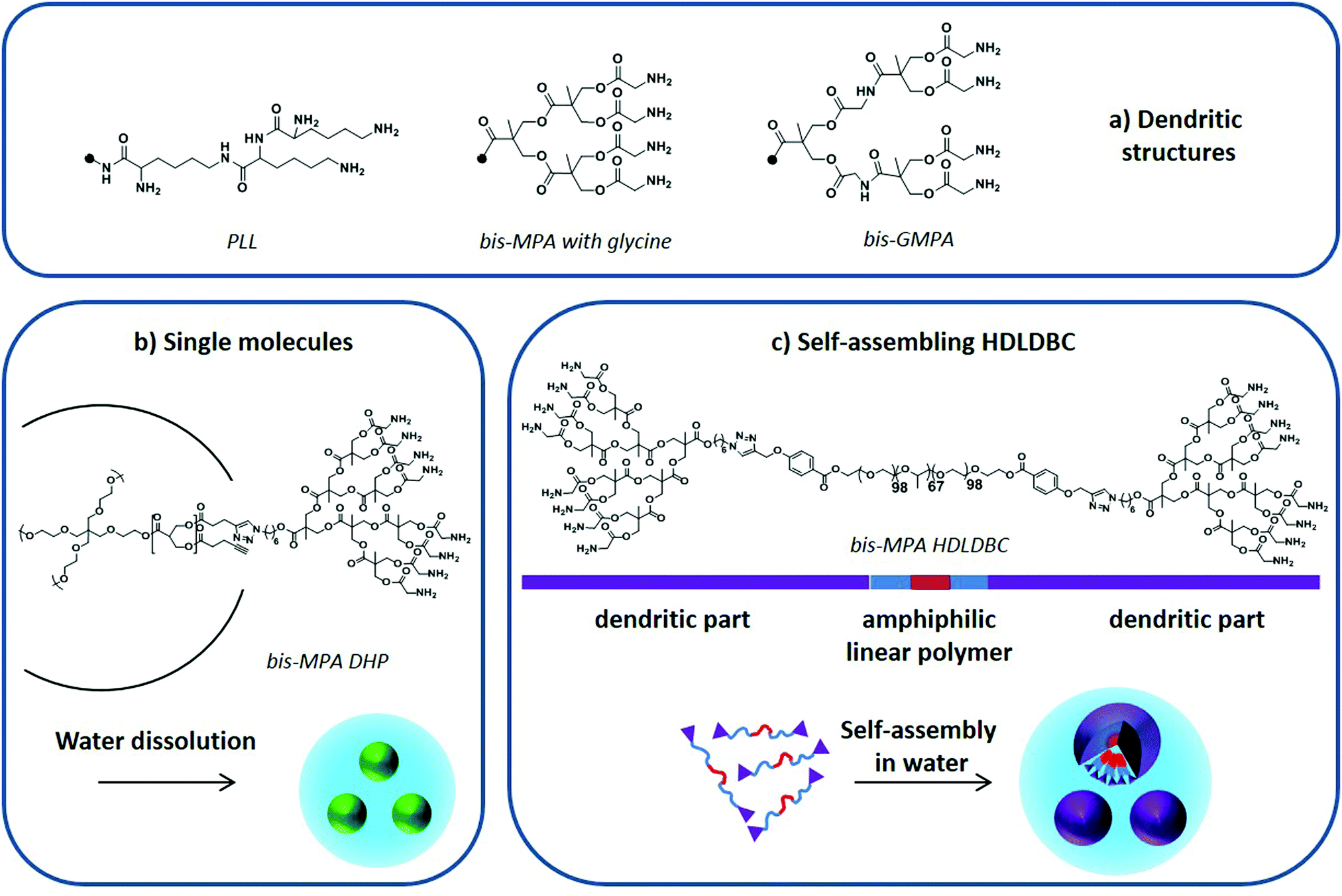 | ||
| Fig. 9 (a) PLL, bis-MPA and bis-GMPA dendritic structures, (b) DHP “single molecule” dissolution in water, and (c) amphiphilic HDLDBC based on Pluronic F-127 and bis-MPA dendron self-assembly in water. Adapted from Movellan et al.129 and Martí Coma-Cros et al.128 | ||
Dendrimers based on a biocompatible and biodegradable 2,2-bis(hydroxymethyl)propionic acid (bis-MPA) skeleton modified with glycine moieties at their periphery (Fig. 9a) have also been used to encapsulate antimalarial drugs. Both single macromolecules, i.e. dendronized hyperbranched molecules (DHP) (Fig. 9b),128 and self-assembled amphiphilic structures, i.e. hybrid dendritic–linear–dendritic block copolymers (HDLDBC) (Fig. 9c) and Janus dendrimers, have been used.129 They all showed high drug loading capacities and excellent targeting towards pRBCs. Moreover, the amphiphilic HDLDBCs, which are based on linear Pluronic F-127 and bis-MPA dendrons, could enhance the activity of CQ and PQ in vitro. A similar amphiphilic HDLDBC based on Pluronic F-127 and a novel dendritic structure based on poly(esteramide) 2,2-bis(glycyloxymethyl)propionic acid (bis-GMPA in Fig. 9a) has been described to encapsulate CQ, PQ and quinacrine. It displayed high drug loading capacity and good targeting towards pRBCs while showing a similar drug activity after encapsulation.128
Artemisinin has been also covalently linked to 1st generation dendrimers leading to trimeric and tetrameric molecules. The trimeric dendrimer showed a higher drug activity in vitro than the free artesumic acid precursor and slightly lower than free dihydroartemisinin, whereas the tetrameric dendrimer exhibited a lower drug activity. The lower activity of the tetrameric dendrimer was related to its high lipophilicity, which might limit its solubility in the culture media of malaria parasites.130
4.3 Inorganic NPs
The research on NPs of inorganic composition as components of potential treatments for malaria infection has received increasing attention in recent years. However, it is still an area that deserves further research in order to understand the mechanism of action of different NPs and the stage of infection in which they can be effective to prevent or cure malaria. Most of the studies selected from recent years deal with NPs that have active participation in the prevention or cure of malaria. Such is the case of the metallic NPs and iron oxide NPs described below. In addition, inorganic NPs of mesoporous silica composition have been tested as effective carriers for traditional antimalarial drugs.The activity of AgNPs is greatly influenced by their morphological and physical features such as, among others, size, shape and aggregation propensity. Although the control of these aspects during synthesis is not yet fully mastered, the benefits of biological processes to prepare AgNPs are broadly accepted. Indeed, the synthesis of metal NPs from plant extracts has become an eco-friendly and cost-effective process, and provides different types of reducing and capping agents to yield stable AgNPs. A comprehensive review by Benelli et al. gathers the biosynthesis of NPs from plant extracts, and how this type of process brings the great potential of such nanosystems to the fight against the vectors of many infectious diseases such as malaria.133 An important aspect of this research is that different plants can be classified as reducing and capping agents to prepare active metal NPs. A significant amount of work has been done in India to develop medical products from Indian medicinal plants, including nanotechnology-based ones.
Mosquitocidal activity of metallic NPs has been described at different stages of the insect's life cycle with increased pesticide efficacy compared to plant-based preparations. Thus, the half lethal concentration, LC50, of synthesized AgNPs estimated for eggs, larvae, pupae and adults has been found to be between 1 and 40 μg ml−1, which is a much lower value than that observed for the corresponding plant extracts.133 AgNPs have been mainly applied as larvicides but are also active as ovicides, adulticides, and pupicides. It is important to remark that they do not show toxicity for non-target organisms.134 Other effects of AgNPs are to reduce mosquito longevity and fecundity as well as to act as a lure and kill approach. This has been observed for AgNPs synthesized from a plant that provides capping with metabolites that are attractive for a kind of mosquito.135 In addition to Ag, other metal compositions including Au, Pd, Co, Ni, Cu and also ZnO and TiO2 have shown toxicity against the mosquito larval stage, features that could be conveniently exploited to prevent malaria.11
The utilization of noble metal-based NPs as antimalarial agents has been less described. Leaf extracts of Andrographis paniculata allowed the synthesis of AgNPs with diameter around 55 nm, which showed antiplasmodial activity.136 More recently, the same authors described the great potential of fern extracts from Indian plants to synthesize AgNPs with promising inhibitory activity against CQ-resistant P. falciparum.137
Similarly to AgNPs, biosynthetic procedures from leaf extracts also permitted the synthesis of PdNPs that decreased significantly (by 78%) the parasite growth in mice infected with P. berghei, with a half maximal inhibitory concentration (IC50) value of 16.44 mg Kg−1 body weight.138 With respect to the synthesis, leaf aqueous extracts of Eclipta prostrata were employed, providing a green and cheap process to prepare PdNPs stable in solution with an average size of 27 nm and promising antimalarial activity.
Au-N-LK3 AuNPs with sizes between 5 and 50 nm and prepared from the marine actinobacteria Streptomyces spp. delayed parasitemia by 6% at day 8 of infection, and increased survival from 50 to 85% in in vivo experiments with P. berghei-infected mice.139 The role attributed to Au-N-LK3 is related to the response of the organism to parasite infection, helping to regulate the presence of blood factors that play an important role in the immune response to malaria infection. In particular, the development of severe malaria alters the concentration of TNF-α (tumor necrosis factor α) and TGF-β1 (transforming growth factor-β1). In a similar way to CQ, Au-N-LK3 inhibits the production of TNF-α, whose concentration increases in blood upon infection.
Interestingly, such low pH is encountered in the interior of the parasite food vacuole and this favors the specificity of such ATA-IONP-fortified ATS towards Plasmodium during the trophozoite stage (Fig. 10). These results provide the possibility of overcoming problems related to artemisinin-resistant malaria parasites. Furthermore, in vivo studies in P. berghei-infected mice showed that a tenfold lower dose compared to free ATS was enough to decrease parasitemia by 80% on day 7 of infection. A combination of 30 mg Kg−1 ATS and 5 mg Kg−1 ATA-IONPs showed the same antimalarial action as 60 mg Kg−1 of the free drug.
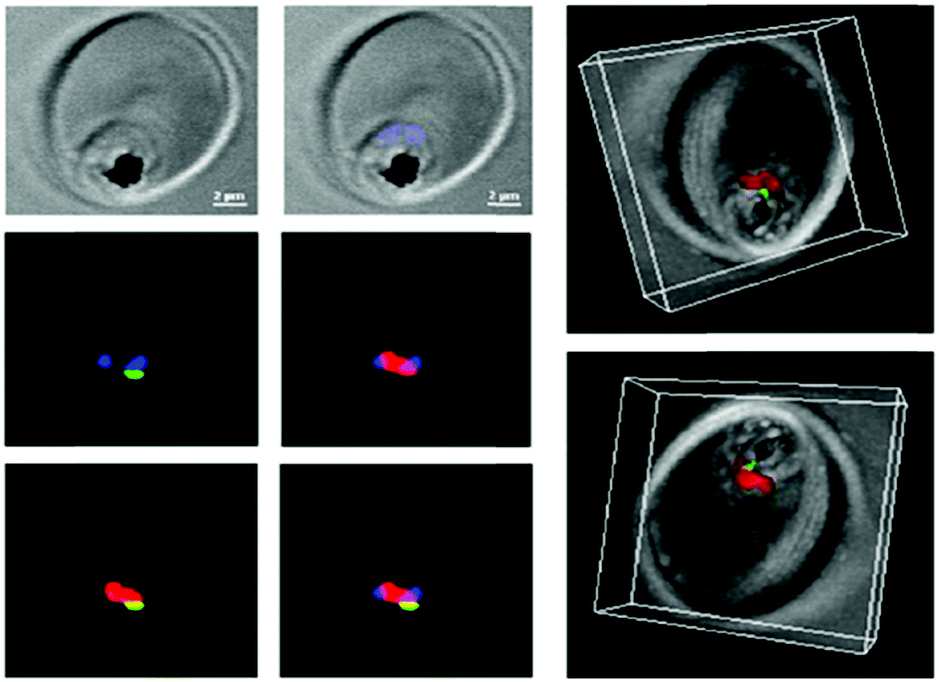 | ||
| Fig. 10 Confocal microscopy was used to explore the localization of fluorescein-labeled ATA-IONPs, which appear in green within the food vacuole compartment of Plasmodium marked by LysoTracker Red. The nucleus of Plasmodium is labeled by Hoechst in blue. Figure reprinted (adapted) with permission from Kannan et al.;140 copyright (2019) Elsevier. | ||
4.4 Miscellaneous
This section includes some other recently explored molecular platforms that, although they do not fit within the previous classification, have shown interesting antimalarial properties either on their own or acting as antimalarial drug carriers and hence their use is considered promising for future malaria therapeutic strategies.Due to its excellent biocompatibility,148 GO has also been explored for its active role in preventing RBC infection by Plasmodium. Lim et al. have proposed a two-pronged action mechanism of GO nanosheets, which are able to inhibit significantly the invasion of RBCs by P. falciparum parasites.149 First, GO nanosheets constitute a physical barrier against parasite infection by capturing merozoites present in blood, thus impeding their adhesion to RBCs. Second, electrostatic and π–π interactions impart GO nanosheets with a strong affinity for many biomolecules, among which are the nutrients required for parasite growth; their removal from blood delays the intraerythrocytic maturation of Plasmodium, and hence reduces its viability.
5 Future perspectives and conclusions
After one hundred years of using classical therapeutic (drugs) and prophylactic (vaccines) approaches that have not succeeded in eradicating malaria, nanotechnology and nanomaterials can offer new opportunities to build customized tools for prophylactic and therapeutic purposes that bring new solutions in different stages of malaria pathophysiology.Issues concerning drug solubility, biostability, toxicity and uncontrolled pharmacokinetics can be overcome with adequately designed drug NCs based on liposomes, polymers or dendrimers. Furthermore, drug resistance evolves when one cannot deliver to the parasites a sufficiently high dose to kill them rapidly. This happens more easily when the drug is not specifically targeted to the pathogen and therefore it distributes throughout the body, which imposes a maximum overall dose that does not trigger undesirable side effects. NCs carrying binding molecules for specific receptors present only on cells infected by the malaria parasite, represent a highly promising option.
For most malaria cases, to be treated in endemic regions with poor or inexistent healthcare infrastructure, and in order to favor patient compliance, oral delivery will be the preferred option and polymer-based NCs can be made apt for this. However, for severe malaria cases where the patient is usually hospitalized, or in other contexts such as in a future malaria eradication campaign where the disease will be entrenched in small contained areas, intravenous administration can be reasonably considered. In these cases, liposomal NCs are an interesting alternative since they have a long track record of successful clinical applications in a wide variety of diseases.
Targeting specificity to infected cells is a key issue that can be also faced with NCs. Since they can operate at the molecular level, nanomaterials can be more precise in reaching a particular drug target, or they can be designed to present several antigens to immune cells for the production of a new generation of better vaccines.
Another possibility for prevention is attacking the mosquito vector. In this respect, mainly metal-based NPs but also liposome-based ones have been shown to be successful to prevent malaria infection.
The main concern when using nanomaterials is related to their production cost, which is an issue to consider when dealing with pathologies like malaria, where the vast majority of cases are in poverty-ridden areas. Pathologies that are not prominent in high income regions have little chance of attracting industrial interest since the large investment in product development will not be recovered as easily as when selling drugs to treat carcinomas, Alzheimer's disease or obesity. However, severe malaria is such a serious life-threatening condition and eradication is such a long sought-after goal that in these cases arguing economic reasons would be out of place. Examples of ways to overcome this obstacle exist, such as the current commitment of a few big pharma companies to devote part of their budget to infectious disease research, but more public and private economic efforts are needed. Some of the basic research results obtained in this field are highly promising, but it is necessary to capture the interest of clinicians and establish collaborations with the medical sector in order to adapt this new technology to the requirements of its deployment as a treatment for people.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This research was supported by grants RTI2018-094579-B-I00, PCIN-2017-100, PGC2018-097583-B-I00, PGC2018-093761-B-C31, CTQ2017-90596-REDT (Ministerio de Ciencia, Innovación y Universidades, Spain, which included FEDER funds), 2017-178 (ERA-NET Cofund EURONANOMED), 2017-SGR-908 (Generalitat de Catalunya, Spain), and E47_20R (Gobierno de Aragón-FSE). ISGlobal and IBEC are members of the CERCA Programme, Generalitat de Catalunya, and both acknowledge support from the Spanish Ministry of Science, Innovation and Universities through the “Centro de Excelencia Severo Ochoa 2019–2023” Program (CEX2018-000806-S). This research is part of ISGlobal's Program on the Molecular Mechanisms of Malaria, which is partially supported by the Fundación Ramón Areces. LNB-C was supported by the European Commission under Horizon 2020's Marie Skłodowska-Curie Actions COFUND scheme (712754) and by the Severo Ochoa programme of the Spanish Ministry of Science and Competitiveness, SEV-2014-0425 (2015–2019). MSA thanks the Ministerio de Ciencia, Innovación y Universidades, for her grant (BES-2016-078774). We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- TDR Strategy 2018–2023, https://www.who.int/tdr/publications/about-tdr/strategy/strategy-2018-23/en/.
- World Malaria Report 2019. Accessed May 2020, https://www.who.int/publications-detail/world-malaria-report-2019.
- G. A. Islan, M. Durán, M. L. Cacicedo, G. Nakazato, R. K. T. Kobayashi, D. S. T. Martinez, G. R. Castro and N. Durán, Nanopharmaceuticals as a solution to neglected diseases: is it possible?, Acta Trop., 2017, 170, 16–42 Search PubMed.
- N. S. Santos-Magalhães and V. C. F. Mosqueira, Nanotechnology applied to the treatment of malaria, Adv. Drug Delivery Rev., 2010, 62, 560–575 Search PubMed.
- P. Urbán and X. Fernàndez-Busquets, Nanomedicine against malaria, Curr. Med. Chem., 2014, 21, 605–629 Search PubMed.
- E. Dennis, V. A. Peoples, F. Johnson, R. K. Bibbs, D. Topps, A. Bopda-Waffo and M. T. Coats, Utilizing nanotechnology to combat malaria, J. Infect. Dis. Ther., 2015, 3, 229 Search PubMed.
- S. Tripathy and S. Roy, A review of age-old antimalarial drug to combat malaria: efficacy up-gradation by nanotechnology based drug delivery, Asian Pac. J. Trop. Med., 2014, 7, 673–679 Search PubMed.
- N. Puttappa, R. S. Kumar, G. Kuppusamy and A. Radhakrishnan, Nano-facilitated drug delivery strategies in the treatment of Plasmodium infection, Acta Trop., 2019, 195, 103–114 Search PubMed.
- R. S. Oakes and C. M. Jewell, A plug-and-play approach for malaria vaccination, Nat. Nanotechnol., 2018, 13, 1096–1097 Search PubMed.
- K. Rahman, S. U. Khan, S. Fahad, M. X. Chang, A. Abbas, W. U. Khan, L. Rahman, Z. Ul Haq, G. Nabi and D. Khan, Nano-biotechnology: a new approach to treat and prevent malaria, Int. J. Nanomed., 2019, 14, 1401–1410 Search PubMed.
- H. Barabadi, Z. Alizadeh, M. T. Rahimi, A. Barac, A. E. Maraolo, L. J. Robertson, A. Masjedi, F. Shahrivar and E. Ahmadpour, Nanobiotechnology as an emerging approach to combat malaria: a systematic review, Nanomedicine, 2019, 18, 221–233 Search PubMed.
- R. Carter and K. N. Mendis, Evolutionary and historical aspects of the burden of malaria, Clin. Microbiol. Rev., 2002, 15, 564–594 Search PubMed.
- A. Amir, F. W. Cheong, J. R. De Silva, J. W. K. Liew and Y. L. Lau, Plasmodium knowlesi malaria: current research perspectives, Infect. Drug Resist., 2018, 1145–1155 Search PubMed.
- G. E. Weiss, B. S. Crabb and P. R. Gilson, Overlaying molecular and temporal aspects of malaria parasite invasion, Trends Parasitol., 2016, 32, 284–295 Search PubMed.
- C. R. S. Garcia, R. P. Markus and L. Madeira, Tertian and quartan fevers: temporal regulation in malarial infection, J. Biol. Rhythms, 2001, 16, 436–443 Search PubMed.
- J. D. Smith, J. A. Rowe, M. K. Higgins and T. Lavstsen, Malaria's deadly grip: cytoadhesion of Plasmodium falciparum-infected erythrocytes, Cell. Microbiol., 2013, 15, 1976–1983 Search PubMed.
- A. Rowe, J. Obeiro, C. I. Newbold and K. Marsh, Plasmodium falciparum rosetting is associated with malaria severity in Kenya, Infect. Immun., 1995, 63, 2323–2326 Search PubMed.
- A. Muthusamy, R. N. Achur, V. P. Bhavanandan, G. G. Fouda, D. W. Taylor and D. C. Gowda, Plasmodium falciparum-infected erythrocytes adhere both in the intervillous space and on the villous surface of human placenta by binding to the low-sulfated chondroitin sulfate proteoglycan receptor., Am. J. Pathol., 2004, 164, 2013–2025 Search PubMed.
- G. A. Josling, K. C. Williamson and M. Llinás, Regulation of sexual commitment and gametocytogenesis in malaria parasites, Annu. Rev. Microbiol., 2018, 72, 501–519 Search PubMed.
- World Health Organization, Guidelines for the Treatment of Malaria, 3rd edn, WHO Press, Geneva, Switzerland, 2015 Search PubMed.
- E. G. Tse, M. Korsik and M. H. Todd, The past, present and future of anti-malarial medicines, Malar. J., 2019, 18, 93 Search PubMed.
- J. Achan, A. O. Talisuna, A. Erhart, A. Yeka, J. K. Tibenderana, F. N. Baliraine, P. J. Rosenthal and U. D’Alessandro, Quinine, an old anti-malarial drug in a modern world: role in the treatment of malaria, Malar. J., 2011, 10, 144 Search PubMed.
- S. J. Rogerson, Management of malaria in pregnancy, Indian J. Med. Res., 2017, 146, 328–333 Search PubMed.
- S. Mvango, W. M. R. Matshe, A. O. Balogun, L. A. Pilcher and M. O. Balogun, Nanomedicines for malaria chemotherapy: encapsulation vs. polymer therapeutics, Pharm. Res., 2018, 35, 237 Search PubMed.
- C. Nsanzabana, Resistance to artemisinin combination therapies (ACTs): do not forget the partner Drug!, Trop. Med. Infect. Dis., 2019, 4, 26 Search PubMed.
- J. Marques, E. Moles, P. Urbán, R. Prohens, M. A. Busquets, C. Sevrin, C. Grandfils and X. Fernàndez-Busquets, Application of heparin as a dual agent with antimalarial and liposome targeting activities toward Plasmodium-infected red blood cells, Nanomedicine, 2014, 10, 1719–1728 Search PubMed.
- M. A. Ali, M. A. Penny, T. A. Smith, L. Workman, P. Sasi, G. O. Adjei, F. Aweeka, J.-R. Kiechel, V. Jullien, M. J. Rijken, R. McGready, J. Mwesigwa, K. Kristensen, K. Stepniewska, J. Tarning, K. I. Barnes and P. Denti, Population pharmacokinetics of the antimalarial amodiaquine: a pooled analysis to optimize dosing, Antimicrob. Agents Chemother., 2018, 62, e02193 Search PubMed.
- V. Navaratnam, S. M. Mansor, N.-W. Sit, J. Grace, Q. Li and P. Olliaro, Pharmacokinetics of artemisinin-type compounds, Clin. Pharmacokinet., 2000, 39, 255–270 Search PubMed.
- P. Newton, Y. Suputtamongkol, P. Teja-Isavadharm, S. Pukrittayakamee, V. Navaratnam, I. Bates and N. White, Antimalarial bioavailability and disposition of artesunate in acute falciparum malaria, Antimicrob. Agents Chemother., 2000, 44, 972–977 Search PubMed.
- J. L. Das, S. Rulisa, P. J. De Vries, P. F. Mens, N. Kaligirwa, S. Agaba, J. Tarning, M. O. Karlsson and T. P. C. Dorlo, Population pharmacokinetics of artemether, dihydroartemisinin, and lumefantrine in Rwandese pregnant women treated for uncomplicated Plasmodium falciparum malaria, Antimicrob. Agents Chemother., 2018, 62, e00518 Search PubMed.
- H. Liu, H. Zhou, T. Cai, A. Yang, M. Zang and J. Xing, Metabolism of piperaquine to its antiplasmodial metabolites and their pharmacokinetic profiles in healthy volunteers, Antimicrob. Agents Chemother., 2018, 62, e00260 Search PubMed.
- S. Krishna and N. J. White, Pharmacokinetics of quinine, chloroquine and amodiaquine: clinical implications, Clin. Pharmacokinet., 1996, 30, 263–299 Search PubMed.
- J.-F. O. Trape, The public health impact of chloroquine resistance in Africa, Am. J. Trop. Med. Hygene, 2001, 64, 12–17 Search PubMed.
- N. J. White, Sulfadoxine-pyrimethamine for the treatment of malaria, Trans. R. Soc. Trop. Med. Hyg., 1991, 85, 556–557 Search PubMed.
- D. M. Harmening, V. C. Hughes and C. Del Toro, Clinical Hematology and Fundamentals of Hemostasis, F. A. Davis Company, Philadelphia, 1997, pp. 54–70 Search PubMed.
- C. H. Villa, A. C. Anselmo, S. Mitragotri and V. Muzykantov, Red blood cells: supercarriers for drugs, biologicals, and nanoparticles and inspiration for advanced delivery systems, Adv. Drug Delivery Rev., 2016, 106, 88–103 Search PubMed.
- T. Dai, W. Jiang, Z. Guo, Y. Xie and R. Dai, Comparison of in vitro/in vivo blood distribution and pharmacokinetics of artemisinin, artemether and dihydroartemisinin in rats, J. Pharm. Biomed. Anal., 2019, 162, 140–148 Search PubMed.
- D. W. Wilson, C. Langer, C. D. Goodman, G. I. McFadden and J. G. Beeson, Defining the timing of action of antimalarial drugs against Plasmodium falciparum, Antimicrob. Agents Chemother., 2013, 57, 1455–1467 Search PubMed.
- E. Moles, P. Urbán, M. B. Jiménez-Díaz, S. Viera-Morilla, I. Angulo-Barturen, M. A. Busquets and X. Fernàndez-Busquets, Immunoliposome-mediated drug delivery to Plasmodium-infected and non-infected red blood cells as a dual therapeutic/prophylactic antimalarial strategy, J. Controlled Release, 2015, 210, 217–229 Search PubMed.
- E. Moles and X. Fernàndez-Busquets, Loading antimalarial drugs into noninfected red blood cells: an undesirable roommate for Plasmodium, Future Med. Chem., 2015, 7, 837–840 Search PubMed.
- K. Kirk, Membrane transport in the malaria-infected erythrocyte, Physiol. Rev., 2001, 81, 495–537 Search PubMed.
- H. P. De Koning, A. Kaddoumi, P. Hsiao, S. A. Desai, K. Basore, Y. Cheng, A. K. Kushwaha and S. T. Nguyen, How do antimalarial drugs reach their intracellular targets?, Front. Pharmacol., 2015, 6, 1–7 Search PubMed.
- P. Urbán, J. Estelrich, A. Cortés and X. Fernàndez-Busquets, A nanovector with complete discrimination for targeted delivery to Plasmodium falciparum-infected versus non-infected red blood cells in vitro, J. Control. Release, 2011, 151, 202–211 Search PubMed.
- G. M. Hasan, N. Garg, E. Dogra, R. Surolia and P. C. Ghosh, Inhibition of the growth of Plasmodium falciparum in culture by stearylamine-phosphatidylcholine liposomes, J. Parasitol. Res., 2011, 2011, 1–9 Search PubMed.
- A. D. Pillai, W. Nguitragool, B. Lyko, K. Dolinta, M. M. Butler, S. T. Nguyen, N. P. Peet, T. L. Bowlin and S. A. Desai, Solute restriction reveals an essential role for clag3-associated channels in malaria parasite nutrient acquisition, Mol. Pharmacol., 2012, 82, 1104–1114 Search PubMed.
- A. Alkhalil, J. V. Cohn, M. A. Wagner, J. S. Cabrera, T. Rajapandi and S. A. Desai, Plasmodium falciparum likely encodes the principal anion channel on infected human erythrocytes, Blood, 2004, 104, 4279–4286 Search PubMed.
- A. Alkhalil, A. D. Pillai, A. A. B. Bokhari, A. B. Vaidya and S. A. Desai, Complex inheritance of the plasmodial surface anion channel in a Plasmodium falciparum genetic cross, Mol. Microbiol., 2009, 72, 459–469 Search PubMed.
- S. Mira-Martínez, N. Rovira-Graells, V. M. Crowley, L. M. Altenhofen, M. Llinás and A. Cortés, Epigenetic switches in clag3 genes mediate blasticidin S resistance in malaria parasites, Cell. Microbiol., 2013, 15, 1913–1923 Search PubMed.
- R. E. Martin, R. V. Marchetti, A. I. Cowan, S. M. Howitt, S. Bröer and K. Kirk, Chloroquine transport via the malaria parasite's chloroquine resistance transporter, Science, 2009, 325, 1680–1682 Search PubMed.
- K. Bachhawat, C. J. Thomas, N. Surolia and A. Surolia, Interaction of chloroquine and its analogues with heme: an isothermal titration calorimetric study, Biochem. Biophys. Res. Commun., 2000, 276, 1075–1079 Search PubMed.
- M. Henry, S. Briolant, A. Zettor, S. Pelleau, M. Baragatti, E. Baret, J. Mosnier, R. Amalvict, T. Fusai, C. Rogier and B. Pradines, Plasmodium falciparum Na+/H+ exchanger 1 transporter is involved in reduced susceptibility to quinine, Antimicrob. Agents Chemother., 2009, 53, 1926–1930 Search PubMed.
- R. N. Price, A.-C. Uhlemann, A. Brockman, R. Mcgready, E. Ashley, L. Phaipun, R. Patel, K. Laing, S. Looareesuwan, N. J. White, F. Nosten and S. Krishna, Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number, Lancet, 2004, 364, 438–447 Search PubMed.
- A. B. S. Sidhu, A. Uhlemann, S. G. Valderramos, J. Valderramos, S. Krishna and D. A. Fidock, Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin, J. Infect. Dis., 2006, 194, 528–535 Search PubMed.
- R. S. Lee, A. P. Waters and J. M. Brewer, A cryptic cycle in haematopoietic niches promotes initiation of malaria transmission and evasion of chemotherapy, Nat. Commun., 2018, 9, 1–9 Search PubMed.
- G. Gregoriadis, Drug entrapment in liposomes, FEBS Lett., 1973, 36, 292–296 Search PubMed.
- A. S. Abu Lila and T. Ishida, Liposomal delivery systems: design optimization and current applications, Biol. Pharm. Bull., 2017, 40, 1–10 Search PubMed.
- R. Nisini, N. Poerio, S. Mariotti, F. De Santis and M. Fraziano, The multirole of liposomes in therapy and prevention of infectious diseases, Front. Immunol., 2018, 9, 155 Search PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Liposome: classification, preparation, and applications, Nanoscale Res. Lett., 2013, 8, 102 Search PubMed.
- M. S. Mufamadi, V. Pillay, Y. E. Choonara, L. C. Du Toit, G. Modi, D. Naidoo and V. M. K. Ndesendo, A review on composite liposomal technologies for specialized drug delivery, J. Drug Delivery, 2011, 2011, 939851 Search PubMed.
- N. P. Aditya, G. Chimote, K. Gunalan, R. Banerjee, S. Patankar and B. Madhusudhan, Curcuminoids-loaded liposomes in combination with arteether protects against Plasmodium berghei infection in mice, Exp. Parasitol., 2012, 131, 292–299 Search PubMed.
- N. Maurer, D. B. Fenske and P. R. Cullis, Developments in liposomal drug delivery systems, Expert Opin. Biol. Ther., 2001, 1, 923–947 Search PubMed.
- F. S. Nas, A. Yahaya and M. Ali, Application of liposomes nanoparticles in the treatment of malaria: a mini review, J. Biotechnol. Biores., 2018, 1, JBB.000510 Search PubMed.
- H. He, Y. Lu, J. Qi, Q. Zhu, Z. Chen and W. Wu, Adapting liposomes for oral drug delivery, Acta Pharm. Sin. B, 2019, 9, 36–48 Search PubMed.
- E. M. Coma-Cros, A. Biosca, E. Lantero, M. L. Manca, C. Caddeo, L. Gutiérrez, M. Ramírez, L. N. Borgheti-Cardoso, M. Manconi and X. Fernàndez-Busquets, Antimalarial activity of orally administered curcumin incorporated in Eudragit®-containing liposomes, Int. J. Mol. Sci., 2018, 19, 131 Search PubMed.
- C. R. Alving, Liposomes as drug carriers in leishmaniasis and malaria, Parasitol. Today, 1986, 2, 101–107 Search PubMed.
- P. Pirson, R. F. Steiger, A. Trouet, J. Gillet and F. Herman, Primaquine liposomes in the chemotherapy of experimental murine malaria, Ann. Trop. Med. Parasitol., 1980, 74, 383–391 Search PubMed.
- P. Pirson, R. Steiger and A. Trouet, The disposition of free and liposomally encapsulated antimalarial primaquine in mice, Biochem. Pharmacol., 1982, 31, 3501–3507 Search PubMed.
- A. Trouet, P. Pirson, R. Steiger, M. Masquelier, R. Baurain and J. Gillet, Development of new derivatives of primaquine by association with lysosomotropic carriers, Bull. W. H. O., 1981, 59, 449–458 Search PubMed.
- C. R. Alving, I. Schneider, G. M. Swartz and E. A. Steck, Sporozoite-induced malaria: therapeutic effects of glycolipids in liposomes, Science, 1979, 205, 1142–1144 Search PubMed.
- C. R. Alving, Therapeutic potential of liposomes as carriers in leishmaniasis, malaria, and vaccines, in Targeting of Drugs, ed. G. Gregoriadis, J. Senior and A. Trouet, Plenum Press, New York, 1982, pp. 337–353 Search PubMed.
- K. J. Longmuir, R. T. Robertson, S. M. Haynes, J. L. Baratta and A. J. Waring, Effective targeting of liposomes to liver and hepatocytes in vivo by incorporation of a Plasmodium amino acid sequence, Pharm. Res., 2006, 23, 759–769 Search PubMed.
- A. Pujol, P. Urbán, C. Riera, R. Fisa, I. Molina, F. Salvador, J. Estelrich and X. Fernàndez-Busquets, Application of quantum dots to the study of liposome targeting in leishmaniasis and malaria, Int. J. Theor. Appl. Nanotechnol., 2014, 2, 1–8 Search PubMed.
- M. Owais, G. C. Varshney, A. Choudhury, S. Chandra and C. M. Gupta, Chloroquine encapsulated in malaria-infected erythrocyte-specific antibody-bearing liposomes effectively controls chloroquine-resistant Plasmodium berghei infections in mice, Antimicrob. Agents Chemother., 1995, 39, 180–184 Search PubMed.
- P. Urbán, J. Estelrich, A. Adeva, A. Cortés and X. Fernàndez-Busquets, Study of the efficacy of antimalarial drugs delivered inside targeted immunoliposomal nanovectors, Nanoscale Res. Lett., 2011, 6, 620 Search PubMed.
- E. Moles, S. Galiano, A. Gomes, M. Quiliano, C. Teixeira, I. Aldana, P. Gomes and X. Fernàndez-Busquets, ImmunoPEGliposomes for the targeted delivery of novel lipophilic drugs to red blood cells in a falciparum malaria murine model, Biomaterials, 2017, 145, 178–191 Search PubMed.
- E. Moles, K. Moll, J. H. Ch’ng, P. Parini, M. Wahlgren and X. Fernàndez-Busquets, Development of drug-loaded immunoliposomes for the selective targeting and elimination of rosetting Plasmodium falciparum-infected red blood cells, J. Controlled Release, 2016, 241, 57–67 Search PubMed.
- X. Fernàndez-Busquets, Toy kit against malaria: magic bullets, LEGO, Trojan horses and Russian dolls, Ther. Delivery, 2014, 5, 1049–1052 Search PubMed.
- I. L. Lisovskaya, I. M. Shcherbachenko, R. I. Volkova and F. I. Ataullakhanov, Clotrimazole enhances lysis of human erythrocytes induced by t-BHP, Chem. – Biol. Interact., 2009, 180, 433–439 Search PubMed.
- A. Biosca, L. Dirscherl, E. Moles, S. Imperial and X. Fernàndez-Busquets, An ImmunoPEGliposome for targeted antimalarial combination therapy at the nanoscale, Pharmaceutics, 2019, 11, 341 Search PubMed.
- V. Rajendran, S. Rohra, M. Raza, G. M. Hasan, S. Dutt and P. C. Ghosh, Stearylamine liposomal delivery of monensin in combination with free artemisinin eliminates blood stages of Plasmodium falciparum in culture and P. berghei infection in murine malaria, Antimicrob. Agents Chemother., 2016, 60, 1304–1318 Search PubMed.
- B. Isacchi, S. Arrigucci, G. La Marca, M. C. Bergonzi, M. G. Vannucchi, A. Novelli and A. R. Bilia, Conventional and long-circulating liposomes of artemisinin: preparation, characterization, and pharmacokinetic profile in mice, J. Liposome Res., 2011, 21, 237–244 Search PubMed.
- M. Ismail, L. Ling, Y. Du, C. Yao and X. Li, Liposomes of dimeric artesunate phospholipid: a combination of dimerization and self-assembly to combat malaria, Biomaterials, 2018, 163, 76–87 Search PubMed.
- S. Ibrahim, T. Tagami and T. Ozeki, Effective-loading of platinum-chloroquine into PEGylated neutral and cationic liposomes as a drug delivery system for resistant malaria parasites, Biol. Pharm. Bull., 2017, 40, 815–823 Search PubMed.
- J. Marques, J. J. Valle-Delgado, P. Urbán, E. Baró, R. Prohens, A. Mayor, P. Cisteró, M. Delves, R. E. Sinden, C. Grandfils, J. L. de Paz, J. A. García-Salcedo and X. Fernàndez-Busquets, Adaptation of targeted nanocarriers to changing requirements in antimalarial drug delivery, Nanomedicine, 2017, 13, 515–525 Search PubMed.
- A. Barragan, D. Spillmann, P. G. Kremsner, M. Wahlgren and J. Carlson, Plasmodium falciparum: molecular background to strain-specific rosette disruption by glycosaminoglycans and sulfated glycoconjugates, Exp. Parasitol., 1999, 91, 133–143 Search PubMed.
- S. Glushakova, B. L. Busse, M. Garten, J. R. Beck, R. M. Fairhurst, D. E. Goldberg and J. Zimmerberg, Exploitation of a newly-identified entry pathway into the malaria parasite-infected erythrocyte to inhibit parasite egress, Sci. Rep., 2017, 7, 12250 Search PubMed.
- M. J. Boyle, J. S. Richards, P. R. Gilson, W. Chai and J. G. Beeson, Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites, Blood, 2010, 115, 4559–4568 Search PubMed.
- A. M. Vogt, A. Barragan, Q. Chen, F. Kironde, D. Spillmann and M. Wahlgren, Heparan sulfate on endothelial cells mediates the binding of Plasmodium falciparum-infected erythrocytes via the αDBL1 domain of PfEMP1, Blood, 2003, 101, 2405–2411 Search PubMed.
- A. M. Vogt, G. Winter, M. Wahlgren and D. Spillmann, Heparan sulphate identified on human erythrocytes: a Plasmodium falciparum receptor, Biochem. J., 2004, 381, 593–597 Search PubMed.
- A. Salanti, M. Dahlbäck, L. Turner, M. A. Nielsen, L. Barfod, P. Magistrado, A. T. R. Jensen, T. Lavstsen, M. F. Ofori, K. Marsh, L. Hviid and T. G. Theander, Evidence for the involvement of VAR2CSA in pregnancy-associated malaria, J. Exp. Med., 2004, 200, 1197–1203 Search PubMed.
- D. J. A. Crommelin, P. van Hoogevest and G. Storm, The role of liposomes in clinical nanomedicine development. What now? Now what?, J. Controlled Release, 2020, 318, 256–263 Search PubMed.
- P. B. Memvanga, R. Coco and V. Préat, An oral malaria therapy: curcumin-loaded lipid-based drug delivery systems combined with β-arteether, J. Controlled Release, 2013, 172, 904–913 Search PubMed.
- L. Sercombe, T. Veerati, F. Moheimani, S. Y. Wu, A. K. Sood and S. Hua, Advances and challenges of liposome assisted drug delivery, Front. Pharmacol., 2015, 6, 286 Search PubMed.
- Z. Mhlwatika and B. A. Aderibigbe, Polymeric nanocarriers for the delivery of antimalarials, Molecules, 2018, 23, 2527 Search PubMed.
- P. Urbán, J. J. Valle-Delgado, N. Mauro, J. Marques, A. Manfredi, M. Rottmann, E. Ranucci, P. Ferruti and X. Fernàndez-Busquets, Use of poly(amidoamine) drug conjugates for the delivery of antimalarials to Plasmodium, J. Controlled Release, 2014, 177, 84–95 Search PubMed.
- E. M. Coma-Cros, A. Biosca, J. Marques, L. Carol, P. Urbán, D. Berenguer, M. C. Riera, M. Delves, R. E. Sinden, J. J. Valle-Delgado, L. Spanos, I. Siden-Kiamos, P. Pérez, K. Paaijmans, M. Rottmann, A. Manfredi, P. Ferruti, E. Ranucci and X. Fernàndez-Busquets, Polyamidoamine nanoparticles for the oral administration of antimalarial drugs, Pharmaceutics, 2018, 10, 225 Search PubMed.
- P. Urbán, E. Ranucci and X. Fernàndez-Busquets, Polyamidoamine nanoparticles as nanocarriers for the drug delivery to malaria parasite stages in the mosquito vector, Nanomedicine, 2015, 10, 3401–3414 Search PubMed.
- S. Alven and B. Aderibigbe, Combination therapy strategies for the treatment of malaria, Molecules, 2019, 24, 3601 Search PubMed.
- N. Jawahar, U. K. Baruah and V. Singh, Co-delivery of chloroquine phosphate and azithromycin nanoparticles to overcome drug resistance in malaria through intracellular targeting, J. Pharm. Sci. Res., 2019, 11, 33–40 Search PubMed.
- P. J. Rosenthal, Azithromycin for malaria?, Am. J. Trop. Med. Hyg., 2016, 95, 2–4 Search PubMed.
- O. Oyeyemi, O. Morenkeji, F. Afolayan, K. Dauda, Z. Busari, J. Meena and A. Panda, Curcumin-artesunate based polymeric nanoparticle; antiplasmodial and toxicological evaluation in murine model, Front. Pharmacol., 2018, 9, 562 Search PubMed.
- S. E. Haas, C. C. Bettoni, L. K. de Oliveira, S. S. Guterres and T. Dalla Costa, Nanoencapsulation increases quinine antimalarial efficacy against Plasmodium berghei in vivo, Int. J. Antimicrob. Agents, 2009, 34, 156–161 Search PubMed.
- E. A. Leite, A. Grabe-Guimarães, H. N. Guimarães, G. L. L. Machado-Coelho, G. Barratt and V. C. F. Mosqueira, Cardiotoxicity reduction induced by halofantrine entrapped in nanocapsule devices, Life Sci., 2007, 80, 1327–1334 Search PubMed.
- K. Velasques, T. R. Maciel, A. H. de Castro Dal Forno, F. E. G. Teixeira, A. L. da Fonseca, F. de P. Varotti, A. R. Fajardo, D. S. de Ávila and S. E. Haas, Co-nanoencapsulation of antimalarial drugs increases their in vitro efficacy against Plasmodium falciparum and decreases their toxicity to Caenorhabditis elegans, Eur. J. Pharm. Sci., 2018, 118, 1–12 Search PubMed.
- H. D. Lu, K. D. Ristroph, E. L. K. Dobrijevic, J. Feng, S. A. McManus, Y. Zhang, W. D. Mulhearn, H. Ramachandruni, A. Patel and R. K. Prud’homme, Encapsulation of OZ439 into nanoparticles for supersaturated drug release in oral malaria therapy, ACS Infect. Dis., 2018, 4, 970–979 Search PubMed.
- F. Föger, W. Noonpakdee, B. Loretz, S. Joojuntr, W. Salvenmoser, M. Thaler and A. Bernkop-Schnürch, Inhibition of malarial topoisomerase II in Plasmodium falciparum by antisense nanoparticles, Int. J. Pharm., 2006, 319, 139–146 Search PubMed.
- D. Xiao, B. Yang, X. M. Yang, D. Yi, X. L. Liao, J. Yang and C. Z. Gao, Synthesis of water soluble chitosan-artemisinin conjugate, Asian J. Chem., 2013, 25, 4637–4639 Search PubMed.
- H. Ringsdorf, Structure and properties of pharmacologically active polymers, J. Polym. Sci., Polym. Symp., 1975, 51, 135–153 Search PubMed.
- J. Golenser, S. Frankenburg, T. Ehrenfreund and A. J. Domb, Efficacious treatment of experimental leishmaniasis with amphotericin B-arabinogalactan water-soluble derivatives, Antimicrob. Agents Chemother., 1999, 43, 2209–2214 Search PubMed.
- S. Nicoletti, K. Seifert and I. H. Gilbert, N-(2-hydroxypropyl)methacrylamide–amphotericin B (HPMA–AmB) copolymer conjugates as antileishmanial agents, Int. J. Antimicrob. Agents, 2009, 33, 441–448 Search PubMed.
- J. Hofsteenge, A. Capuano, R. Altszuler and S. Moore, Carrier-linked primaquine in the chemotherapy of malaria, J. Med. Chem., 1986, 29, 1765–1769 Search PubMed.
- N. Tomiya, J. G. Jardim, J. Hou, R. Pastrana-Mena, R. R. Dinglasan and Y. C. Lee, Liver-targeting of primaquine-(poly-γ-glutamic acid) and its degradation in rat hepatocytes, Bioorg. Med. Chem., 2013, 21, 5275–5281 Search PubMed.
- S. Kumar, R. K. Singh, R. S. R. Murthy and T. R. Bhardwaj, Synthesis and evaluation of substituted poly(organophosphazenes) as a novel nanocarrier system for combined antimalarial therapy of primaquine and dihydroartemisinin, Pharm. Res., 2015, 32, 2736–2752 Search PubMed.
- S. Kumar, R. K. Singh, R. Sharma, R. S. R. Murthy and T. R. Bhardwaj, Design, synthesis and evaluation of antimalarial potential of polyphosphazene linked combination therapy of primaquine and dihydroartemisinin, Eur. J. Pharm. Sci., 2015, 66, 123–137 Search PubMed.
- R. P. Bakshi, L. M. Tatham, A. C. Savage, A. K. Tripathi, G. Mlambo, M. M. Ippolito, E. Nenortas, S. P. Rannard, A. Owen and T. A. Shapiro, Long-acting injectable atovaquone nanomedicines for malaria prophylaxis, Nat. Commun., 2018, 9, 315 Search PubMed.
- A. Najer, D. Wu, A. Bieri, F. Brand, C. G. Palivan, H.-P. Beck and W. Meier, Nanomimics of host cell membranes block invasion and expose invasive malaria parasites, ACS Nano, 2014, 8, 12560–12571 Search PubMed.
- A. Najer, S. Thamboo, J. T. Duskey, C. G. Palivan, H.-P. Beck and W. Meier, Analysis of molecular parameters determining the antimalarial activity of polymer-based nanomimics, Macromol. Rapid Commun., 2015, 36, 1923–1928 Search PubMed.
- J. Kopeček, Hydrogel biomaterials: a smart future?, Biomaterials, 2007, 28, 5185–5192 Search PubMed.
- B. Aderibigbe, E. Sadiku, J. Jayaramudu and S. S. Ray, Controlled dual release study of curcumin and a 4-aminoquinoline analog from gum acacia containing hydrogels, J. Appl. Polym. Sci., 2015, 132, 41613 Search PubMed.
- B. A. Aderibigbe and Z. Mhlwatika, Dual release kinetics of antimalarials from soy protein isolate-carbopol-polyacrylamide based hydrogels, J. Appl. Polym. Sci., 2016, 133, 43918 Search PubMed.
- S. Dawre, S. Pathak, S. Sharma and P. V. Devarajan, Enhanced antimalalarial activity of a prolonged release in situ gel of arteether-lumefantrine in a murine model, Eur. J. Pharm. Biopharm., 2018, 123, 95–107 Search PubMed.
- W. Peters, Drug resistance in Plasmodium berghei Vincke and Lips, 1948. III. Multiple drug resistance, Exp. Parasitol., 1965, 17, 97–102 Search PubMed.
- R. M. Kannan, E. Nance, S. Kannan and D. A. Tomalia, Emerging concepts in dendrimer-based nanomedicine: from design principles to clinical applications, J. Intern. Med., 2014, 276, 579–617 Search PubMed.
- D. Bhadra, S. Bhadra and N. K. Jain, Pegylated lysine based copolymeric dendritic micelles for solubilization and delivery of artemether, J. Pharm. Pharm. Sci., 2005, 8, 467–482 Search PubMed.
- D. Bhadra, A. K. Yadav, S. Bhadra and N. K. Jain, Glycodendrimeric nanoparticulate carriers of primaquine phosphate for liver targeting, Int. J. Pharm., 2005, 295, 221–233 Search PubMed.
- P. Agrawal, U. Gupta and N. K. Jain, Glycoconjugated peptide dendrimers-based nanoparticulate system for the delivery of chloroquine phosphate, Biomaterials, 2007, 28, 3349–3359 Search PubMed.
- D. Bhadra, S. Bhadra and N. K. Jain, PEGylated peptide dendrimeric carriers for the delivery of antimalarial drug chloroquine phosphate, Pharm. Res., 2006, 23, 623–633 Search PubMed.
- E. M. Coma-Cros, A. Lancelot, M. San Anselmo, L. N. Borgheti-Cardoso, J. J. Valle-Delgado, J. L. Serrano, X. Fernàndez-Busquets and T. Sierra, Micelle carriers based on dendritic macromolecules containing bis-MPA and glycine for antimalarial drug delivery, Biomater. Sci., 2019, 7, 1661–1674 Search PubMed.
- J. Movellan, P. Urbán, E. Moles, J. M. de la Fuente, T. Sierra, J. L. Serrano and X. Fernàndez-Busquets, Amphiphilic dendritic derivatives as nanocarriers for the targeted delivery of antimalarial drugs, Biomaterials, 2014, 35, 7940–7950 Search PubMed.
- T. Fröhlich, F. Hahn, L. Belmudes, M. Leidenberger, O. Friedrich, B. Kappes, Y. Couté, M. Marschall and S. B. Tsogoeva, Synthesis of artemisinin-derived dimers, trimers and dendrimers: investigation of their antimalarial and antiviral activities including putative mechanisms of action, Chem. – Eur. J., 2018, 24, 8103–8113 Search PubMed.
- W. J. Youngs, A. R. Knapp, P. O. Wagers and C. A. Tessier, Nanoparticle encapsulated silver carbene complexes and their antimicrobial and anticancer properties: a perspective, Dalton Trans., 2012, 41, 327–336 Search PubMed.
- M. Rai, A. P. Ingle, P. Paralikar, I. Gupta, S. Medici and C. A. Santos, Recent advances in use of silver nanoparticles as antimalarial agents, Int. J. Pharm., 2017, 526, 254–270 Search PubMed.
- G. Benelli, F. Maggi, R. Pavela, K. Murugan, M. Govindarajan, B. Vaseeharan, R. Petrelli, L. Cappellacci, S. Kumar, A. Hofer, M. R. Youssefi, A. A. Alarfaj, J. S. Hwang and A. Higuchi, Mosquito control with green nanopesticides: towards the One Health approach? A review of non-target effects, Environ. Sci. Pollut. Res., 2018, 25, 10184–10206 Search PubMed.
- G. Benelli, Plant-mediated biosynthesis of nanoparticles as an emerging tool against mosquitoes of medical and veterinary importance: a review, Parasitol. Res., 2016, 115, 23–34 Search PubMed.
- G. Benelli and M. Govindarajan, Green-synthesized mosquito oviposition attractants and ovicides: towards a nanoparticle-based “lure and kill” approach?, J. Cluster Sci., 2017, 28, 287–308 Search PubMed.
- C. Panneerselvam, S. Ponarulselvam and K. Murugan, Potential anti-plasmodial activity of synthesized silver nanoparticle using Andrographis paniculata Nees (Acanthaceae), Arch. Appl. Sci. Res., 2011, 3, 208–217 Search PubMed.
- C. Panneerselvam, K. Murugan, M. Roni, A. T. Aziz, U. Suresh, R. Rajaganesh, P. Madhiyazhagan, J. Subramaniam, D. Dinesh, M. Nicoletti, A. Higuchi, A. A. Alarfaj, M. A. Munusamy, S. Kumar, N. Desneux and G. Benelli, Fern-synthesized nanoparticles in the fight against malaria: LC/MS analysis of Pteridium aquilinum leaf extract and biosynthesis of silver nanoparticles with high mosquitocidal and antiplasmodial activity, Parasitol. Res., 2016, 115, 997–1013 Search PubMed.
- G. Rajakumar, A. A. Rahuman, I. M. Chung, A. V. Kirthi, S. Marimuthu and K. Anbarasan, Antiplasmodial activity of eco-friendly synthesized palladium nanoparticles using Eclipta prostrata extract against Plasmodium berghei in Swiss albino mice, Parasitol. Res., 2015, 114, 1397–1406 Search PubMed.
- L. Karthik, G. Kumar, T. Keswani, A. Bhattacharyya, B. P. Reddy and K. V. B. Rao, Marine actinobacterial mediated gold nanoparticles synthesis and their antimalarial activity, Nanomedicine, 2013, 9, 951–960 Search PubMed.
- D. Kannan, N. Yadav, S. Ahmad, P. Namdev, S. Bhattacharjee, B. Lochab and S. Singh, Pre-clinical study of iron oxide nanoparticles fortified artesunate for efficient targeting of malarial parasite, EBioMedicine, 2019, 45, 261–277 Search PubMed.
- A. Mehmood, H. Ghafar, S. Yaqoob, U. F. Gohar and B. Ahmad, Mesoporous silica nanoparticles: a review, J. Dev. Drugs, 2017, 6, 1000174 Search PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Mesoporous silica nanoparticles in target drug delivery system: a review, Int. J. Pharm. Invest., 2015, 5, 124–133 Search PubMed.
- S. A. Amolegbe, H. Ohmagari, K. Wakata, H. Takehira, R. Ohtani, M. Nakamura, C. Yu and S. Hayami, Synthesis of mesoporous materials as nano-carriers for an antimalarial drug, J. Mater. Chem. B, 2016, 4, 1040–1043 Search PubMed.
- S. A. Amolegbe, Y. Hirano, J. O. Adebayo, O. G. Ademowo, E. A. Balogun, J. A. Obaleye, A. U. Krettli, C. Yu and S. Hayami, Mesoporous silica nanocarriers encapsulated antimalarials with high therapeutic performance, Sci. Rep., 2018, 8, 378 Search PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, A new family of mesoporous molecular sieves prepared with liquid crystal templates, J. Am. Chem. Soc., 1992, 114, 10834–10843 Search PubMed.
- P. Y. Chen, M. Zhang, M. Liu, I. Y. Wong and R. H. Hurt, Ultrastretchable graphene-based molecular barriers for chemical protection, detection and actuation, ACS Nano, 2018, 12, 234–244 Search PubMed.
- C. J. Castilho, D. Li, M. Liu, Y. Liu, H. Gao and R. H. Hurt, Mosquito bite prevention through graphene barrier layers, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 18304–18309 Search PubMed.
- J. Shi and Y. Fang, Biomedical applications of graphene, in Graphene: Fabrication, Characterizations, Properties and Applications, ed. Z. Hongwei, X. Zhiping, X. Dan, and Y. Fang, Academic Press, Cambridge, MA, USA, 2017, pp. 215–232 Search PubMed.
- Kenry, Y. B. Lim, M. H. Nai, J. Cao, K. P. Loh and C. T. Lim, Graphene oxide inhibits malaria parasite invasion and delays parasitic growth in vitro, Nanoscale, 2017, 9, 14065–14073 Search PubMed.
- S. S. Braga, Cyclodextrins: emerging medicines of the new millennium, Biomolecules, 2019, 9, 801 Search PubMed.
- I. E. Crandall, W. A. Szarek, J. Z. Vlahakis, Y. Xu, R. Vohra, J. Sui and R. Kisilevsky, Sulfated cyclodextrins inhibit the entry of Plasmodium into red blood cells: implications for malarial therapy, Biochem. Pharmacol., 2007, 73, 632–642 Search PubMed.
- A. G. Balducci, E. Magosso, G. Colombo, F. Sonvico, N. A. K. Khan, K. H. Yuen, R. Bettini, P. Colombo and A. Rossi, Agglomerated oral dosage forms of artemisinin/β-cyclodextrin spray-dried primary microparticles showing increased dissolution rate and bioavailability, AAPS PharmSciTech, 2013, 14, 911–918 Search PubMed.
- Y. Hao, S. Chen, L. Tian, Z. Yang and G. Huang, Inclusion complex of artemether with 2-hydroxypropyl-β-cyclodextrin for the treatment of malaria: preparation, characterization and evaluation, MOJ Bioequivalence Bioavailability, 2017, 3, 145–151 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |